

Background and Aims:
Cholestatic liver diseases, including primary sclerosing cholangitis, are characterized by periportal inflammation with progression to hepatic fibrosis and ultimately cirrhosis. We recently reported that the thioredoxin antioxidant response is dysregulated during primary sclerosing cholangitis. The objective of this study was to examine the impact of genetic and pharmacological targeting of thioredoxin reductase 1 (TrxR1) on hepatic inflammation and liver injury during acute cholestatic injury.
Approach and Results:
Primary mouse hepatocytes and intrahepatic macrophages were isolated from 3-day bile duct ligated (BDL) mice and controls. Using wildtype and mice with a liver-specific deletion of TrxR1 (TrxR1LKO), we analyzed the effect of inhibition or ablation of TrxR1 signaling on liver injury and inflammation. Immunohistochemical analysis of livers from BDL mice and human cholestatic patients revealed increased TrxR1 staining in periportal macrophages and hepatocytes surrounding fibrosis. qPCR analysis of primary hepatocytes and intrahepatic macrophages revealed increased TrxR1 mRNA expression following BDL. Compared with sham controls, BDL mice exhibited increased inflammation, necrosis, and increased mRNA expression of pro-inflammatory cytokines, fibrogenesis, the NLRP3 inflammatory complex, and increased activation of NFkB, all of which were ameliorated in TrxR1LKO mice. Importantly, following BDL, TrxR1LKO induced periportal hepatocyte expression of Nrf2-dependent antioxidant proteins and increased mRNA expression of basolateral bile acid transporters with reduced expression of bile acid synthesis genes. In the acute BDL model, the TrxR1 inhibitor auranofin (10 mg/kg/1 d preincubation, 3 d BDL) ameliorated BDL-dependent increases in Nlrp3, GsdmD, Il1β, and TNFα mRNA expression despite increasing serum alanine aminotransferase, aspartate aminotransferase, bile acids, and bilirubin.
Conclusions:
These data implicate TrxR1-signaling as an important regulator of inflammation and bile acid homeostasis in cholestatic liver injury.
KEY POINTS
Thioredoxin reductase 1 (TrxR1) is upregulated in hepatocytes and macrophages in human and murine cholestasis.
Hepatocyte-specific deletion of TrxR1 ameliorates neutrophil infiltration during bile duct ligation (BDL).
Hepatocyte-specific deletion of TrxR1 reduces hepatocyte necrosis following BDL.
Hepatocyte-specific deletion of TrxR1 ameliorates a proinflammatory cytokine expression following BDL.
TrxR1 is an important mediator of NFkB activation during cholestasis.
INTRODUCTION
The cholestatic liver disease accounts for ~9% of adult and 43% of pediatric liver transplants in the US.1 Cholangiopathies include primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and pediatric biliary atresia (BA) and are characterized by extensive periportal inflammation and ductular reaction with fibrosis and obstruction of the biliary system.2 A key component of cholestatic liver disease is extensive oxidative stress which has been proposed to contribute to overall hepatic injury.3,4
By its ability to regulate protein dithiol/disulfide status and deliver reducing power to key antioxidant enzymes, the cytosolic thioredoxin reductase system [comprised of reduced NADPH, thioredoxin-1 (Trx1), TrxR1, and thioredoxin interacting protein 1 (Txnip)] is an important modulator of cellular redox status.5 Recently, the Trx1 pathway has been linked to signaling the production of proinflammatory cytokines by the NLR family pyrin domain-containing-3 (Nlrp3) inflammasome in both cell culture models as well as murine models of chronic hepatic inflammation.6–9 In murine models, liver-specific ablation of TrxR1 (TrxR1LKO) results in the activation of the nuclear factor erythroid 2-related factor 2 (NRF2) antioxidant response pathway.10,11 We have recently reported increased expression of Trx1 and TrxR1 in human PSC liver tissue as well as in Mdr2KO mice, an established mouse model of fibrosing cholangiopathies.3,4,12
Recent studies have shown that the Nlrp3 complex [Nlrp3, apoptosis-associated speck-like (ASC, PYD, And CARD Domain-Containing) (PYCARD), Gasdermin D and caspase-1] is an important contributor to hepatic inflammation. Upon activation of the Nlrp3 inflammasome, cleavage and maturation of pro-IL1β and pro-IL18 by caspase 1 results in inflammatory-induced hepatocyte death (pyroptosis), which promotes fibrogenesis.13 Nlrp3 inflammasome activation plays an important role in the pathogenesis of chronic liver diseases including cholestasis.14,15 Following BDL, increased hepatocyte concentrations of bile constituents result in apical membrane rupture and the release of damage-associated molecular patterns which induce macrophage and Nlrp3 activation.13,16 Inhibition of Nlrp3 inflammasome activation by Nlrp3 deletion or by pharmacologically targeting the inflammasome complex (Nlrp3, Caspase 1) significantly reduced BDL-dependent fibrosis and biochemical markers of liver injury.16–19 In contrast, data have emerged suggesting that deletion of Nlrp3 may result in increased injury during short-term BDL but attenuates liver injury in long-term BDL17 supporting time-dependent effects. Recent reports link TrxR1 signaling and regulation of the Nlrp3 inflammasome. Inhibition of TrxR1 activity by the FDA-approved drug auranofin inhibits Nlrp3 activation in cultured macrophages and ameliorates hepatic injury in murine models of fibrosis.7–9,20
The objective of this study was to examine the effect of hepatocyte-specific deletion of TrxR1 (TrxR1LKO) on hepatic inflammation, inflammasome activation, and injury during acute BDL-induced cholestatic injury. Our results support a role for TrxR1 in the regulation of hepatic inflammation and bile acid signaling during acute cholestatic liver injury.
METHODS
Human sample procurement
All use of human samples was approved by the Institutional Review Board of the University of Colorado Denver (Exempt). For human studies, to determine the status of TrxR1 localization during cholestasis, paraffin-embedded and frozen hepatic tissue explants from end-stage PSC (ages 23–62), PBC (ages 35–62), and BA patients (ages 0.5–5 y) (N=6/condition, Table S1, http://links.lww.com/HC9/A50), and liver from normal organ donors, were procured during liver transplantation, and obtained from the University of Minnesota Liver Tissue Cell Distribution Center (NIH Contract #HHSN276201200017C) as described.3,4 Informed consent for research was obtained from each patient, and the study protocol conformed to the Ethical Guidelines of the 1975 Declaration of Helsinki. The research protocol was reviewed and approved by the Ethic Committees of the National Institutes of Health, and the regional committees for medical and health research ethics at the University of Minnesota. No donor organs were obtained from executed prisoners or other institutionalized individuals.
Murine sample procurement
TrxR1LKO mice were homozygous for a functional wildtype (WT) floxed allele of the gene encoding TrxR1 (Txnrd1 fl/fl , JAX #028283)21 and either hemizygous or homozygous for AlbCre (JAX #003574).10 Cre-driven recombination of this allele generates a TrxR1 protein-null allele, and TrxR1LKO livers are devoid of all detectable TrxR1 protein and activity.10,21,22 Mice were subjected to BDL or sham (laparotomy only) surgeries as described.23 After 3 days, mice were euthanized and tissue and blood were harvested.12,24 All mouse surgeries and harvests were performed between 9:30 and 11:30 am to minimize circadian variations. Whole livers were excised and weighed. Liver tissue was fixed in 10% neutral buffered formalin, paraffin-embedded for histological and immunohistochemical (IHC) analyses. The remaining hepatic tissue flash frozen in liquid nitrogen for western blotting and qRT-PCR. For auranofin studies, mice received auranofin (#A6733, SIGMA, St. Louis, MO) at a dose of 10 mg/kg/i.p. ×4 day (1 d preincubation/3 d during BDL). After 3 days, mice were euthanized and processed as described above. All animal protocols were approved by the Montana State University (#2019-50-97, 2021-118-01) or the University of Colorado (IACUC protocol #00000879) Institutional Animal Care and Use Committee and were performed in accordance with published National Institutes of Health guidelines. To ensure rigor and reproducibility, all samples were randomly coded and blindly analyzed. All studies involving animal experiments conformed with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.25
Western blotting
Western blotting was performed using 10–40 μg of liver extract protein and primary antibodies listed in Table S2 (http://links.lww.com/HC9/A50) as previously described.12 All exposures were normalized using glyceraldehyde-3-dehydrogenase (GAPDH) expression.
Histological and IHC evaluation
Formalin-fixed tissue was analyzed following embedding, sectioning at 5 µm, and stained with hematoxylin and eosin or picrosirius red. All immunohistochemistry was performed using the antibodies listed in Table S1 (http://links.lww.com/HC9/A50) as described.12,26 For IHC staining, except for TrxR1 staining (Tris pH 9.0), and F4/80 staining [DAKO target Antigen Retrieval Solution (Dako, Carpinteria, CA)], heat-induced antigen retrieval was performed in Citrate pH 6.0 buffer (SIGMA). For light microscopy, staining was visualized using 3, 3′-diaminobenzidine (SK4105, Vector Laboratories, Newark, CA) and either a horse anti-rabbit (MP-7401, Vector Laboratories) or a goat anti-rat (MP-7444, Vector Laboratories) secondary antibody. For fluorescent microscopy, staining was visualized using a donkey anti-Rat IgG (H+L) Alexa Fluor 488 conjugate and a goat anti-Mouse Alexa Fluor 594 conjugate (Invitrogen, Thermofisher, Waltham, MA) secondary antibodies. IHC staining of CK7 was quantified with the Slidebook program version 6.0 (Intelligent Imaging Innovations, Denver, Colorado). Histologic images were captured on an Olympus BX51 microscope equipped with a four-megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics). All images were cropped and assembled using Photoshop CS2 (Adobe Systems Inc.; Mountain View, CA) and quantified under blinded conditions using Slidebook version 6.0.
Quantitative RT-PCR
qRT-PCR was done using TaqMan probes (Table S3, http://links.lww.com/HC9/A50) purchased from Applied Biosystems (Foster City, CA) as previously described.27
Isolation of primary intrahepatic mononuclear cells (ihMNCs)
Primary ihMNCs (90%–95% pure macrophages) were isolated from hepatic tissue extracted from WT (n=3) and BDL (n=3) livers as described.28 Following isolation, cells were lysed, mRNA extracted, and qRT-PCR performed as previously described.
Isolation and treatment of primary mouse hepatocytes
Primary mouse hepatocytes were isolated from fresh liver tissue as described.28 Following isolation, cells were plated into 6 well plates. After 24 hours, cells were washed twice in PBS and then preincubated with auranofin (2 μM) in DMEM plus pen/strep and 10% bovine serum for 1 hour. Following preincubation, TNF-alpha (TNFα) (10 ng/ml)/lipopolysaccharide (LPS) (100 ng/mL) was added to the medium, and the cells were allowed to incubate for an additional 4 hours. After 4 hours, cells were washed twice in ice-cold PBS, lysed, and mRNA extracted.
Isolation and treatment of bone marrow-derived macrophages (BMDM)
BMDM were isolated as described.29 After 2 weeks, cells were washed twice in PBS and then preincubated with auranofin (2 μM) in DMEM plus penicillin/streptomycin and 10% bovine serum for 1 hour. Following preincubation, TNFα (10 ng/mL)/LPS (100 ng/ml) was added to the medium, and the cells were allowed to incubate for an additional 4 hours. After 4 hours, cells were washed twice in ice-cold PBS, lysed, and mRNA extracted.
Biochemical assessment of liver injury
Plasma was separated by centrifugation at 5000 rpm for 5 minutes at 4°C and was assayed for alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP) activity, total serum bilirubin, and total serum bile acids as described.27
Quantification of serum IL-1β concentrations
The mouse IL-1β/IL-1F2 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) was used according to the manufacturer’s instructions to detect serum mouse IL-1β.
Statistical analysis
The data are presented as means± SEM. Comparisons between WT and BDL were performed by Student T tests, and comparisons between multiple groups were completed using 1-way ANOVA. Statistical significance was set at p < 0.05. Prism 5 for Windows (GraphPad Software, San Diego, CA) was used to perform all statistical tests.
RESULTS
Hepatic expression of TrxR1 is increased in macrophages in human and murine cholestasis
We have previously shown that expression of TrxR1 is increased and localized in periportal hepatocytes as well as macrophages in human PSC tissue.3 Expression was examined in hepatic tissue by western blotting (Trx1, TrxR1) and tissue sections (TrxR1) from human end-stage BA and PBC patients. From the western blots, expression of Trx1 and TrxR1 was significantly increased in PBC and BA replicating previous data in PSC (Figure 1A, B).3 From immunohistochemistry, in normal human liver, TrxR1 staining was evident in hepatocytes (Figure 1C, yellow arrows) as well as in scattered macrophages within the sinusoids (Figure 1C, blue arrows). In cholestatic tissue, TrxR1 staining was increased in periportal hepatocytes as well as in macrophages supporting the upregulation of TrxR1 during human cholestasis. To further validate the cell type specificity of TrxR1 in human cholestasis, the colocalization of TrxR1 and the pan macrophage marker CD68 was examined using fluorescent microscopy. TrxR1 expression (blue arrows) was elevated in periportal hepatocytes (Figure 1D). Interestingly, colocalization of TrxR1 was present in periportal CD68+ macrophages (yellow arrows) but not in CD68+ macrophages outside of the periportal region (red arrows).
FIGURE 1.
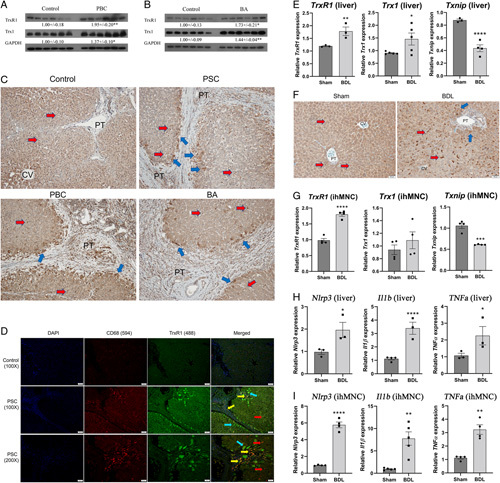
Increased TrxR1 expression in human cholestasis. Western blot analyses of Trx1 and TrxR1 protein expression in humans. (A) PBC and (B) BA. Expression was quantified using ImageJ and normalized to GAPDH expression. Values are mean ± SEM. *p < 0.05, **p < 0.01. (C) Immunohistochemical staining for TrxR1 in representative paraffin-embedded formalin-fixed tissue sections obtained from the liver from normal and end-stage PSC, PBC, and BA patients (N=4 per condition, ×200). Blue arrows, TrxR1-overexpressing hepatic macrophages; yellow arrows, TrxR1-overexpressing hepatocytes. (D) Colocalization of TrxR1 with CD68+ macrophages in human PSC. Control and PSC liver were analyzed immunohistochemically using a rabbit polyclonal antibody directed against TrxR1, a mouse monoclonal antibody directed against the macrophage marker CD68 followed by Alexa Fluor 594 conjugated anti-mouse and Alexa Fluor 488-conjugated anti-rabbit secondary antibodies. Slides were examined using fluorescent microscopy (blue arrows=TrxR1 positive hepatocytes, red arrows=CD68+ macrophages, yellow arrows=colocalization), and nuclei were visualized by DAPI (N=4 per condition, ×200). (E) mRNA expression of Trx1, TrxR1, and Txnip in hepatic tissue isolated from 3-day Sham/BDL mice. mRNA was normalized to Hprt expression. N=3–4 per condition. (F) Formalin-fixed, paraffin-embedded liver sections from Sham and BDL WT mice were immunostained for TrxR1 (blue arrows=TrxR1 positive hepatocytes, red arrows=F4/80 positive macrophages) (N=3 per condition, ×200). (G) mRNA expression of Trx1, TrxR1, and Txnip in purified ihMNCs. N=4 per condition, mRNA was normalized to Hprt expression. (H and I) mRNA expression of the Nlrp3 inflammasome and proinflammatory cytokines is increased following BDL. mRNA expression of Nlrp3, Il1b, TNFa in (H). Hepatic tissue isolated from Sham and BDL mice. (I) Purified ihMNCs isolated from indicated mice. N=3–4/condition. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Abbreviations: BA, biliary atresia; BDL, bile duct ligation; CV, central vein; PBC, primary biliary cholangitis; PT, portal triad; TrxR1, thioredoxin reductase 1; WT, wildtype.
Upregulation of Trx1 expression has been reported following BDL30 and we recently reported increased expression of the Trx1/TrxR1 pathway in liver tissue isolated from the Mdr2KO cholestatic model.12 To determine the impact of acute cholestasis on TrxR1 signaling, mice were subjected to Sham or BDL and sacrificed after 3 days. Hepatic tissue was isolated and mRNA expression of the Trx1, TrxR1, and Txnip was performed. Compared with Sham, mRNA expression of TrxR1 and Trx1 was significantly increased but Txnip expression was suppressed following BDL (Figure 1E). TrxR1 expression was then examined by immunohistochemistry. In the sham controls, TrxR1 staining was panlobular with a few TrxR1 positive macrophages (Figure 1F arrows). In the BDL mice, prominent TrxR1 staining was present in macrophages along with a slight increase in staining in periportal hepatocytes. To further explore the effect of BDL on Trx signaling in intrahepatic macrophages, primary ihMNC, ~95% macrophages,28 were purified from each group and mRNA expression was examined. In ihMNCs, expression of TrxR1 was increased but Txnip was suppressed following BDL (Figure 1G), consistent with the TrxR1 immunohistological data.
The Nlrp3 inflammasome has been shown to be upregulated in human cholestasis as well as following murine BDL.15 To validate Nlrp3 inflammasomes upregulation during BDL, qRT-PCR was used. mRNA analysis revealed increased expression of Nlrp3, Il1b, and TNFα in hepatic tissue and in ihMNCs isolated from 3D BDL mice (Figure 1H, I).
Hepatocyte-specific deletion of TrxR1 decreased BDL-induced necrosis and expression of fibrotic genes
To further examine the contribution of TrxR1 in regulating inflammation during cholestatic injury, Sham/BDL was performed in WT and hepatocyte-specific TrxR1 deletion mice (TrxR1LKO).10 In the Sham controls, there were no significant differences in necrosis, fibrosis, and or ductular proliferation between WT and TrxR1LKO (Figure 2A, B). When comparing WT BDL and TrxR1LKO BDL, no differences were present with respect to fibrosis or ductal proliferation. Extensive necrosis was evident in WT BDL mice but unexpectedly, in TrxR1LKO BDL, necrosis was significantly suppressed. At 3 days post-BDL, although overt fibrosis is not yet evident, the genes that regulate fibrogenesis (TGFβ, Col1a1, TIMP1, and MMP9) are upregulated supporting the initiation of fibrosis. To gain further insight into the effect of TrxR1LKO on initiation of BDL-induced fibrosis, qRT-PCR of profibrotic genes was performed; TrxR1LKO significantly ameliorated BDL-induced increased expression of profibrotic genes (Figure 2C).
FIGURE 2.
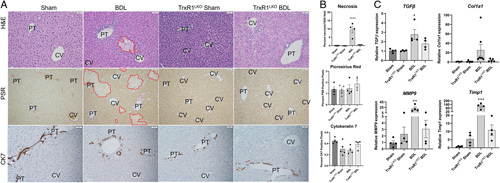
TrxR1LKO significantly decreases BDL-induced hepatic necrosis and expression of fibrogenesis genes. (A) Hematoxylin and eosin (H&E), picrosirius red (PSR), and Cytokeratin 7 (CK7) immunostaining of representative liver sections from 3-day Sham/BDL treated WT or TrxR1LKO mice. Areas of hepatocellular necrosis are outlined in red, N=4 per condition, ×200. (B) Quantification of histology, values are mean± SEM. (C) mRNA expression of fibrogenesis genes TGFβ, Col1A1, TIMP1, and MMP9 in liver tissue isolated from indicated conditions. N=4 per condition, mRNA was normalized to Hprt expression. Other abbreviations as in Figure 1. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Hepatocyte-specific deletion of TrxR1 inhibits neutrophil infiltration following BDL-induced liver injury
Kupffer cells and neutrophils play an important role in BDL-induced cholestatic injury.31,32 To determine whether hepatocyte-specific loss of TrxR1 impacted inflammatory cell infiltrates during acute cholestasis, tissue sections were stained for myeloperoxidase (neutrophils), F4/80 (macrophages), CD3 (T-lymphocytes), and B220 (B-lymphocytes) and quantified (Figure 3A, B). In Sham controls deletion of TrxR1 resulted in a significant decrease in CD3+ cells but no other significant differences were evident. From the quantification, compared with Sham controls, BDL induced significant increases in F4/80+ macrophages, neutrophils, and CD3+ lymphocytes but B220+ cells were suppressed. Compared with WT BDL, neutrophil, but not macrophage or lymphocyte infiltration was significantly suppressed in TrxR1LKO.
FIGURE 3.
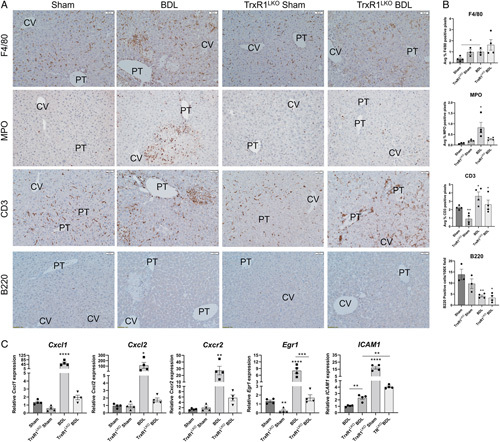
TrxR1LKO significantly reduces neutrophil but not T-lymphocyte or macrophage infiltration following BDL. (A) Immunohistochemical analysis of F4/80+ macrophages, myeloperoxidase (MPO)+ neutrophils, CD3+ lymphocytes, and B220+ plasma B cells in liver sections isolated from indicated conditions, N=4 per condition, ×200. Other abbreviations as in Figure 1. (B) Quantification of inflammatory cell infiltrates. (C) mRNA expression of neutrophil recruitment and regulatory genes (Cxcl1, Cxcl2, Cxcr2, Egr1, and ICAM1) in hepatic tissue isolated from indicated conditions. N=4 per condition, mRNA was normalized to Hprt expression. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, **** p <0.0001.
In the liver, neutrophil infiltration is regulated in part by the chemokines Cxcl1, Cxcl2, and the chemokine receptor Cxcr2 as well as through ICAM1 upregulation by the transcription factor EGR1.33 Changes in chemokine and chemokine receptor expression was examined using qPCR. Compared with WT BDL, mRNA expression of Cxcl1, Cxcl2, Cxcr2, Egr1, and ICAM1 were all markedly suppressed in TrxR1LKO, consistent with the reduced neutrophil infiltration (Figure 3C).
Hepatocyte-specific deletion of TrxR1 suppressed BDL-induced mRNA expression of the Nlrp3 inflammasome complex and proinflammatory cytokines but does not affect Nlrp3 inflammasome activation
We next hypothesized that decreased expression of proinflammatory cytokines may be contributing to reduced inflammation present in TrxR1LKO livers. mRNA expression of the Nlrp3 inflammasome and proinflammatory cytokines was assessed in hepatic tissue from each group. In the WT groups, BDL significantly upregulated mRNA expression of the inflammasome (Nlrp3, ASC, GsdmD), proinflammatory cytokines (Il1b, Il1rn, Il18, TNFα, TNFiap3, and NOS2) and IL-6 signaling (Il6, Socs1, Socs3) (Figure 4A). Interestingly in the Sham TrxR1LKO, expression of Il1b, Il6 was suppressed whereas Il1rn was increased. There were no significant differences in the expression of any of the other genes examined. Remarkably, apart from GsdmE, in the TrxR1LKO, following BDL, upregulation of the inflammasome and all markers of inflammation were ameliorated. Gasdermin E was not elevated in the BDL group but was increased in the TrxR1LKO BDL group. During cholestasis, following Nlrp3 activation, pro-Caspase 1, and pro-IL-1b all undergo proteolytic cleavage and activation.13 To further explore the effect of TrxR1LKO on inflammasome activation, cleavage of pro-Caspase 1 and pro-IL-1b was evaluated by western blotting and quantified (Figure 4B). From the quantification, compared with their respective Sham controls, expression of pro-Caspase 1 decreased in both WT and TrxR1LKO BDL, but no differences were evident in pro-IL-1β expression. To further delineate the effect of TrxR1LKO on IL-1β secretion, serum concentrations were determined (Figure 4C). TrxR1LKO suppressed BDL-induced serum concentrations of IL-1β. Recent evidence suggests TrxR1-mediated inhibition of Nlrp3 activation occurs through inhibition of NFkB-dependent expression of downstream targets.34 During activation, the NFkB-p65 is phosphorylated on Ser536 and undergoes nuclear translocation.35 Using anti-phosphoSer536 NFkB-p65 antibodies, phosphorylation of NFkB-p65 was examined in each group. Compared with Sham controls, NFkB-p65 phosphorylation was increased following BDL in WT mice but was markedly suppressed in the TrxR1LKO BDL group (Figure 4D). Thus, suppression of NFkB signaling may be responsible for the inhibition of Nlrp3 in the TrxR1LKO BDL group.
FIGURE 4.
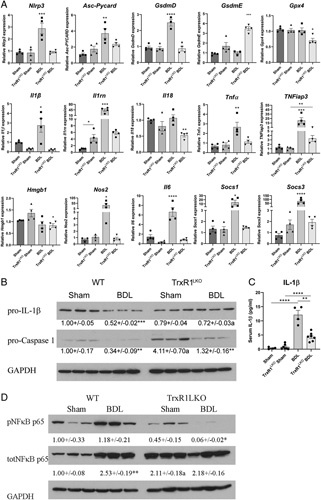
TrxR1LKO significantly reduces hepatic mRNA expression of Nlrp3 inflammasomes and proinflammatory cytokines/chemokines following BDL. qRT-PCR analysis of mRNA expression. (A) mRNA expression of genes representative of NLRP3 inflammasomes and ferroptosis (Nlrp3, ASC, GsdmD, GsdmE, Gpx4) and proinflammatory cytokines (Il1b, Il1rn, Il18, TNFa, TNFiap, Hmgb1, Nos2, Il6, Socs1, Socs3). (B) Western analysis of pro-IL-1β and pro-Caspase 1 in hepatic tissue from indicated conditions. Expression was quantified using ImageJ and normalized to GAPDH expression. (C) Quantification of peripheral serum IL-1β concentrations. N=3–7 per condition, values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with sham control; a p < 0.01 compared with WT under same condition. (D) Western blot analyses of protein expression of NFkB phosphorylation in the liver from indicated conditions. Expression was quantified using ImageJ and normalized to GAPDH expression. Values are mean ± SEM. *p < 0.05, **p < 0.01 compared with sham control.
Abbreviations: BDL, bile duct ligation; TrxR1, thioredoxin reductase 1; WT, wildtype.
Hepatocyte-specific deletion of TrxR1 does not impact BDL-induced liver injury
The effect of TrxR1LKO on serum markers of hepatic injury following BDL was examined. In WT mice, BDL caused increased ALT, AST, ALP, total bilirubin, and serum bile acids (Figure 5). Compared with WT BDL controls, TrxR1LKO BDL mice exhibited a significant increase in serum bile acids but no differences in the other liver injury markers were evident.
FIGURE 5.
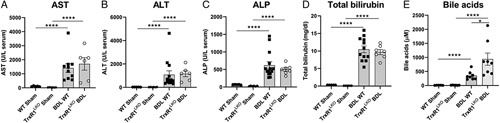
TrxR1LKO does not affect serum biochemical markers of liver injury in 3 day-BDL mice. Serum concentrations of: (A) AST, (B) ALT, (C) ALP. (D) Total bilirubin and (E) total bile acids. N=4–8 per condition, values are mean ± SEM. ****p < 0.0001.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BDL, bile duct ligation; TrxR1, thioredoxin reductase 1; WT, wildtype.
Hepatocyte-specific deletion of TrxR1 upregulates the NRF2 antioxidant response following BDL
We have shown that NRF2 antioxidant responses are upregulated in TrxR1LKO mice.10 To determine the status of the NRF2-antioxidant response following BDL in TrxR1LKO, tissue samples were analyzed for mRNA and protein expression of selected NRF2 target genes. Following BDL, compared with Sham controls, mRNA expression of NAD(P)H quinone oxidoreductase-1 (Nqo1) and Heme oxygenase-1 (Hmox-1) was significantly increased but expression of glutamate-cysteine ligase catalytic subunit (Gclc) was suppressed (Figure 6A). Hepatospecific deletion of TrxR1 increased mRNA expression of Nqo1 and Gclc which was maintained following BDL. Interestingly, Hmox-1 mRNA expression was not increased in Sham TrxR1LKO liver but was significantly increased following BDL. Examining protein expression (Figure 6B), compared with Sham controls, BDL increased expression of carbonyl reductase 3 (CBR3), glutathione-S-transferase-mu (GSTµ), HO-1, NQO1 but suppressed Gclc. Deletion of TrxR1 resulted in a significant upregulation of all NRF2 targets examined, but only GSTμ was further increased following BDL.
FIGURE 6.
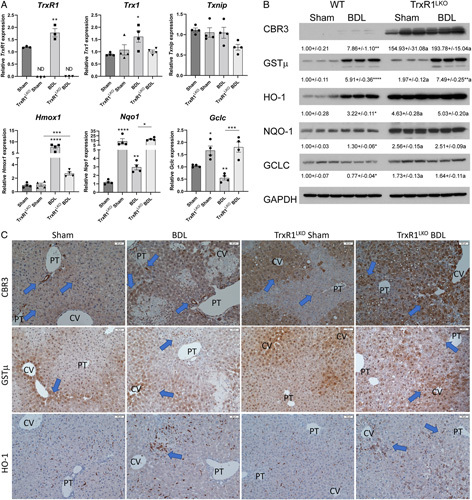
TrxR1LKO increases periportal expression NRF2 antioxidant responses following 3 day-BDL. (A) qRT-PCR analysis of Txn1, TrxR1, Txnip, Hmox-1, Nqo1, and Gclc in hepatic tissue from mice with indicated conditions. mRNA was normalized to Hprt expression. (B) Western blot analyses of protein expression of CBR3, GSTµ, HO-1, NQO-1, and GCLC in the liver from indicated conditions. Expression was quantified using ImageJ and normalized to GAPDH expression. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with sham control; a p < 0.05 compared with WT under same condition. (C) Immunohistochemical analysis of hepatic CBR3, GSTµ, and HO-1. Blue arrows represent increased staining. Other abbreviations as in Figure 1. N=4/condition, ×200 magnification. Blue arrows indicate the area of increased staining.
In the liver, NRF2 expression is primarily in hepatocytes surrounding the central vein.26 To further examine Nrf2 signaling, IHC analysis of CBR3, GSTµ, and HO-1 was assessed using liver sections from WT or TrxR1LKO Sham and BDL mice (Figure 6C). In the WT Sham controls, weak staining of CBR3 was evident in hepatocytes surrounding the central vein with higher staining in cholangiocytes as well as a few scattered macrophages (arrows). Following BDL, CBR3 staining increased in hepatocytes as well as few scattered macrophages around necrotic tissue. Examining TrxR1LKO, in Shams, compared with WT, CBR3 staining was markedly elevated in hepatocytes surrounding the central vein. Interestingly, BDL induced an expansion of CBR3 staining/expression into periportal hepatocytes. Examining GSTµ in sham mice, expression was present in panlobular nuclei with cytosolic hepatocyte staining primarily surrounding the central vein. Apart from staining present in infiltrating inflammatory cells, GSTµ staining was not notably different in WT versus BDL. No differences were evident when comparing Sham controls in WT and TrxR1LKO. Following BDL, GSTµ staining expanded into periportal hepatocytes in a checkerboard pattern. In WT Sham and BDL, HO-1 staining was almost exclusively co-localized to macrophages. Comparing Shams, staining was not noticeably different in TrxR1LKO but was increased in scattered hepatocytes surrounding the central vein following BDL.
Hepatocyte-specific deletion of TrxR1 upregulates basolateral bile acid transporters during acute cholestasis
Our current and previous data support the upregulation of NRF2 antioxidant responses in TrxR1LKO.10 Previous research has shown that the basolateral bile acid transporters (the ATP-binding cassette family C, ABCC, proteins) Abcc3 and Abcc4 are in part regulated by NRF2.36 In the Sham mice, TrxR1LKO significantly suppressed both cytochrome P450 (CYP) family members Cyp7a1 and Cyp8b1 supporting possible effects on bile acid synthesis but did not affect sodium-dependent bile acid co-transporter (NTCP, Slc10a1). In WT mice, expression of Cyp7a1, Cyp8b1, and Slc10a1, were all significantly suppressed following BDL but compared with TrxR1LKO BDL there were no significant differences. Interestingly, expression of basolateral efflux transporters Abcc3 and Abcc4 were increased in the Sham TrxR1LKO mice when compared with WT but only Abcc4 remained increased following BDL (Figure 7A). Expression of Cyp7a1 is in part, regulated through FGF15/19 binding to FGFR4 resulting in c-Jun N-terminal kinase (Jnk) phosphorylation and activation.37 The status of Jnk activation was assessed in both genotypes. From the western blot, in Sham controls, phosphorylation of Jnk-p55 was increased in the TrxR1LKO group (Figure 7B). Following BDL, both genotypes exhibited significant increases in Jnk phosphorylation/activation compared with Sham controls supporting this mechanism for Cyp7a1 suppression.
FIGURE 7.
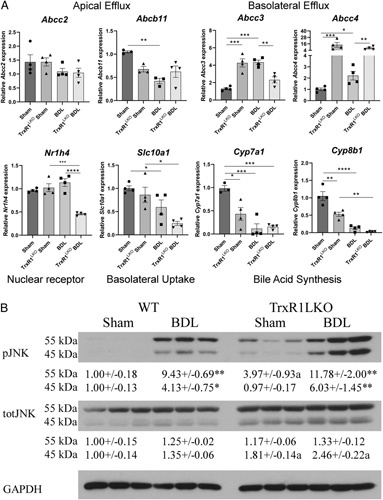
TrxR1LKO induces expression of bile acid synthesis and Abcc4 but not apical bile acid efflux pumps following 3-day BDL. qRT-PCR analysis of Abcc2, Abcb11, Abcc3, Abcc4, Nr1h4, Slc10a1, Cyp7a1, and Cyp8b1 in hepatic tissue isolated from indicated conditions. mRNA was normalized to Hprt expression, values are mean ± SEM. * p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) Western blot analyses of protein expression of Jnk phosphorylation in liver from indicated conditions. Expression was quantified using ImageJ and normalized to GAPDH expression. Values are mean ± SEM. *p < 0.05, **p < 0.01, a p < 0.01 compared with WT under same condition.
Abbreviations: BDL, bile duct ligation; WT, wildtype.
Pharmacological inhibition of TrxR1 inhibits NLRP3 activation in hepatic mononuclear cells
The thioredoxin pathway has come to the forefront as a significant mediator of inflammation through its ability to regulate the NLRP3 inflammasome complex.6 In cell lines, inhibition of TrxR1 prevented NLRP3 activation and production of proinflammatory cytokines.9,20 To determine the effect of pharmacologic TrxR1 inhibition in hepatocytes and hepatic macrophages, primary mouse hepatocytes and intrahepatic mononuclear cells (~95% macrophages) were purified from WT mouse livers. Cells were plated and incubated with the TrxR1 inhibitor auranofin (2 μM) or vehicle for 30 minutes followed by stimulation with TNFα (10 ng/mL) + LPS (100 ng/mL). After 4 hours, cells were harvested and mRNA expression of Nlrp3, Il1b, and TrxR1 was examined. Combined treatment with TNFα+LPS significantly increased mRNA expression of Nlrp3, Il1b in both primary hepatocytes and ihMNCs (Figure 8A, B). Preincubation with auranofin increased TrxR1 and suppressed TNF+LPS-induced Nlrp3 and Il1b expression, supporting a TrxR1-dependent role in the regulation of NLRP3 and Il-1β. Similar results were evident in BMDM undergoing the same treatments (Figure S1, http://links.lww.com/HC9/A49).
FIGURE 8.
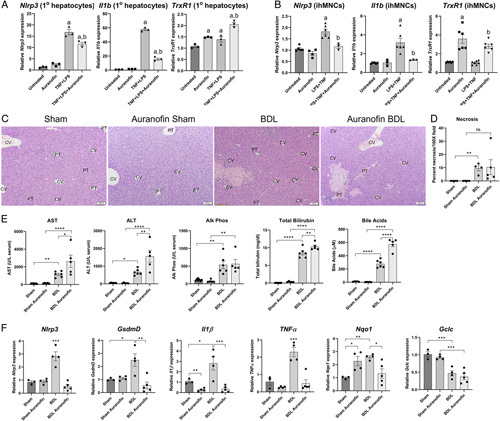
Pharmacological inhibition of TrxR1 prevents LPS + TNFα-induced expression of Nlrp3 and Il1b in cultured cells. Cells were incubated with LPS + TNFα (100 ng/mL + 10 ng/mL for 4 h) +/− the TrxR1 inhibitor auranofin (2 µM preincubation for 1 h). mRNA was isolated and expression of Nlrp3, Il1b, and TrxR1 measured by qPCR. (A) Primary mouse hepatocytes. (B) Intrahepatic mononuclear cells (ihMNCs). mRNA was normalized to Hprt expression, values are mean ± SEM. a p < 0.05 compared with untreated, b p < 0.05 compared with LPS+TNFα. Pharmacological targeting of TrxR1 prevents BDL-dependent increases in proinflammatory cytokines but does not ameliorate hepatic injury following BDL. WT mice were subjected Sham/BDL with or without auranofin for 4 days (10 mg/kg/i.p., 1 day pretreatment). (C) Hematoxylin staining of liver sections isolated from each group (N=4 per condition, ×100 magnification). (D) Quantification of necrosis present in each group. (E) Serum concentrations of AST, ALT, alkaline phosphatase, total bilirubin, and total bile acids in each group (N=4–6 per condition). (F) mRNA expression of Nlrp3, GsdmD, Il1β, TNFα, Nqo1 and Gclc in hepatic tissue isolated from each group. N=3–5 per condition, mRNA was normalized to Hprt expression. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BDL, bile duct ligation; CV, central vein; LPS, lipopolysaccharide; PT, portal triad; TrxR1, thioredoxin reductase 1.
The TrxR1 inhibitor auranofin has been shown to ameliorate hepatic injury in murine NASH.7 To determine the effects of TrxR1 inhibition in acute cholestasis, mice were treated with auranofin (10 mg/kg i.p., N=5/group) for 4 days (1D preincubation followed by either Sham surgery or BDL for 3D), sacrificed and liver injury assessed. Hematoxylin and eosin-stained tissue sections were evaluated, and the percent area of necrosis was quantified (Figure 8C, D). Interestingly, no significant differences in necrosis were present when comparing the BDL and auranofin BDL groups. Examining serum biochemical parameters of injury, compared with BDL, treatment with auranofin resulted in an increase in serum AST, ALT, total bilirubin, and bile acids but did not change serum ALP (Figure 8E). In our cell culture experiments, auranofin inhibited Nlrp3 expression. To determine the effect of auranofin in mice, mRNA expression of Nrlp3, GsdmD, Il1b, TNFα, and Nqo1 was examined in liver tissue isolated from each group. Treatment with auranofin-suppressed BDL induced increases in Nlrp3, GsdmD, Il1β, TNFα mRNA (Figure 8F). Examining Nrf2 activation, in the Sham controls, auranofin increased Nqo1 but not Gclc expression. Following BDL, Nqo1 expression decreased and Gclc was not significantly different when the BDL group was compared with BDL plus auranofin (Figure 8F). In summary, these data support the hypothesis that inhibition of TrxR1 by auranofin represses Nlrp3 expression and provide evidence that the Nlrp3 inflammasome exerts a protective response during early cholestasis.
DISCUSSION
Increased oxidative stress has been linked to the extent of liver injury in both human and murine models of cholestasis.3,4,12,38 Recent evidence has shown that the TrxR1/Trx1 pathway is upregulated during cholestasis and that diminishing oxidative stress has protective effects on cholestatic liver injury in animal models.38–40 In the present study, we examined the impact of hepatospecific deletion of TrxR1 on acute cholestatic liver injury and inflammation. Although the TrxR1/Trx1 system provides reducing power to peroxiredoxins, the major cellular ROS scavenger, as well as to other cytoprotective reductase enzymes,41 unexpectedly TrxR1 deletion ameliorated histologic hepatocellular necrosis and downregulated the expression of fibrogenesis genes following acute cholestatic injury. Likely accounting for this, TrxR1LKO potently induced NRF2 antioxidant responses, which were associated with inhibition of neutrophil infiltration, suppression of NFkB activation, decreased mRNA expression of the NLRP3 inflammasome complex, and release of pro-inflammatory cytokines. Taken together, these results suggest that TrxR1-signaling is more important as a mediator of hepatic injury and inflammation during cholestasis than TrxR1-reducing power, per se, as a source of antioxidant activity.
In the liver, NRF2 regulates cytoprotective processes including antioxidant systems, drug metabolism phase-2 conjugases, and drug metabolism phase-3 exporters, including critical transporters involved in basolateral bile acid transport.36,42 Through oxidation of KEAP1, NRF2 activation is regulated by cellular redox processes. Enzymatically, by its ability to reduce protein cysteine disulfides, the Trx-signaling pathway is a critical regulator of cellular redox homeostasis. We have shown that pharmacological inhibition or genetic deletion of TrxR1 activates NRF2-dependent antioxidant responses but does not induce oxidative stress.11 Furthermore, an active TrxR1 is not necessary to maintain normal redox homeostasis in part due to redox redundancy that is present from the normal production of glutathione which can substitute for thioredoxin as a reductant of oxidized cysteines.10,43 Our previous data have shown that despite Nrf2 activation, oxidative stress is only increased when both the glutathione reductase (GSR) and the thioredoxin pathways are deleted.26 In agreement with previous data, we found that Nrf2 responses are activated by BDL and that they are further increased in the TrxR1LKO groups.44
There are conflicting data about the role of Nrf2 in cholestatic injury. Constitutive Nrf2 activation by deletion of Keap1 activates Nrf2-antioxidant responses thereby diminishing necrotic cellular injury and serum bilirubin levels in cholestasis, but it does not affect overall serum biochemical evidence of injury.38 Pharmacological induction of Nrf2 by sulforaphane decreased BDL-induced fibrosis with no effect on biochemical evidence of liver injury, whereas an alternative Nrf2 activator (Oltipraz) increased injury, but decreased fibrosis as evidenced by reduced hepatic hydroxyproline concentrations.45,46 Herein, following BDL, data obtained from TrxR1LKO mice are consistent with Nrf2 genetic ablation models suggesting that pharmacological targeting of Nrf2, as compared with genetic deficiencies, may have broader effects and further supporting the role of TrxR1 in regulating Nrf2 responses. Although the mechanism is not clear, genetic ablation of Nlrp3 results in increased liver injury following acute BDL.17 When TrxR1LKO and auranofin-treated BDL groups are compared, increased biochemical injury is present in the auranofin BDL group but not the TrxR1LKO BDL group supporting cell-specific effects. Importantly, hepatospecific ablation of TrxR1 did not increase injury, but global pharmacological inhibition resulted in increased injury supporting the role of macrophages in cholestatic injury. Elevated injury is correlated with decreased mRNA expression of the Nlrp3 complex as well as proinflammatory cytokines. This supports a role of TrxR1 in regulating the inflammasome and a role of Nlrp3 in acute early cholestatic injury. Interestingly, TrxR1LKO groups exhibited a robust activation of Nrf2 targets even during BDL, whereas auranofin had no effect on Nqo1 and did not restore Gclc expression following BDL. These results provide evidence that the effect of auranofin on Nlrp3 may be Nrf2 independent. Furthermore, increased hepatic injury in the absence of a robust Nrf2 activation in the BDL auranofin group supports a role for Nrf2 in mitigating BDL-induced liver injury.
A number of reports have supported the infiltration of peribiliary proinflammatory macrophages in the pathogenesis of human PSC.47 In PSC, we found that expression of TrxR1 was increased predominantly in periportal hepatocytes and periportal CD68+ Kupffer cells but not in Kupffer cells that reside in the centrilobular region supporting a TrxR1-specific role in inflammasome activation. During cholestasis, bile acids initiate NLRP3 inflammasome activation in hepatocytes resulting in the release of proinflammatory cytokines.48 This results in proinflammatory crosstalk between hepatocytes and macrophages. Based on the observed decrease in NFkB activation, we hypothesize that in TrxR1LKO mice, hepatocyte Nlrp3 activation is suppressed. This prevents normal hepatocyte/macrophage crosstalk which occurs due to hepatocytes’ production of proinflammatory cytokines.
Activation of Nrf2 impacts bile acid secretion and uptake by hepatocytes, presumably through increased expression of the apical canalicular bile salt export pump and suppression of bile acid uptake through basolateral NTCP. In addition, there may be increased serum bile acid export through expression of the basolateral transporters ABCC3 and ABCC4. By contrast, in TrxR1LKO BDL livers, apical bile salt export pump expression is decreased while basolateral ABCC4 expression is increased. Importantly, the export of bile salts by ABCC4 also requires GSH and GST-mediated conjugation for effective bile acid transport.49 NRF2 induces increased expression of GCLC resulting in increased GSH synthesis, which in turn promotes hepatocyte basolateral bile salt export into the blood. Furthermore, activation of NRF2 negatively influences bile acid synthesis.50 This is consistent with the suppressed expression of Cyp7a1 and Cyp8b1 in Sham TrxR1LKO livers that is further decreased following BDL. Taken together, these data support the role of TrxR1 in regulating hepatocellular bile acid export through basolateral transporters as well as bile acid synthesis.
In WT mice, IHC analysis of NRF2 demonstrates zone 3 nuclear localization in hepatocytes which corresponds to increased expression of NRF2-target genes including CRB3 and GSTμ.26 In TrxR1LKO livers, we have reported that basal NRF2 nuclear localization is panlobular.26 In the TrxR1LKO liver, in the absence of injury, periportal expression of downstream NRF2 targets such as CBR3 and GSTµ is not significant. Following BDL, a dramatic increase in periportal (zone 1) CBR3 and GSTμ staining is evident. Current data validate that TrxR1LKO results in induction of zone 3 expression of NRF2 targets at baseline, but that upon initiation of a hepatic insult that periportal transcription of NRF2-dependent targets is activated.
The NLRP3 inflammasome is an important contributor to cholestatic liver injury. Inhibition of inflammasome activity by MCC950 significantly reduced hepatocellular necrosis, neutrophil infiltration, and fibrosis after 7 days of BDL in mice. Moreover, the deletion of NLRP3 caused a switch in cell death to apoptosis and necroptosis in BDL mice.17 Data presented herein suggest that, although inflammasome activation is present in 3-day BDL mice as evidenced by cleavage of Caspase 1, the expression of proinflammatory cytokines and fibrogenesis genes is suppressed by TrxR1LKO supporting a cytokine-dependent role in the induction of fibrosis. Concurrently, NFkB phosphorylation and nuclear localization being suppressed thereby suppressing the activation of proinflammatory cytokines. In vivo data are further supported by the in vitro data demonstrating that inhibition of TrxR1 by auranofin in cultured primary hepatocytes, BMDMs, and ihMNCs and macrophage cell lines results in suppression of Il1b mRNA.20 This is in contrast to a recent report showing that, by preventing downstream NLRP3 signaling using caspase 1-knockout mice, BDL led to alternative activation of macrophages which contributed to increased fibrosis but a decrease in overall liver injury.15 Our data suggest that, through hepatocyte activation of Nrf2 in BDL mice, TrxR1LKO increased hepatocellular basolateral bile acid export and reduced macrophage activation (demonstrated by decreased chemokine, proinflammatory cytokines and inflammatory cell infiltrates), despite failure to prevent NLRP3 activation (as shown by cleavage of Caspase 1).
In conclusion, this study demonstrates that a hepatocyte-specific genetic deficiency of TrxR1 abrogates the Nlrp3-dependent inflammatory response and mitigates hepatocellular necrosis following bile duct ligation. In contrast, pharmacologic TrxR1 inhibition still abrogates the inflammatory response but concurrently increases hepatic injury supporting a macrophage-specific role in the mitigation of early acute cholestatic injury. Overall, data herein support TrxR1-signaling as an important regulator of inflammation and bile acid homeostasis in cholestatic liver injury.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
C.T.S.: conceptualization; writing—original draft. C.T.S., A.L.A., D.J.O., C.G.M., and R.C.N.: data collection. C.T.S., D.J.O., E.E.S., and R.J.S.: formal analysis. E.E.S. and R.J.S.: funding acquisition. E.E.S., N.B., and R.J.S.: resources. All authors: writing—review & editing; final approval.
CONFLICT OF INTEREST
R.J.S. consults for Albireo Pharma and Mirum Pharma. For the remaining authors none declared.
Footnotes
Funding information This project was supported in part by NIH grant UL1TR002535 (awarded to R.J.S.) and grants R56DK123738, R21OD026444, and R21AG055022 (awarded to E.E.S.). Its contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Abbreviations: ABCC, ATP-binding cassette family C proteins; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BDL, bile duct ligation; CBR3, carbonyl reductase-3; GAPDH, glyceraldehyde-3-dehydrogenase; GCLC, glutamate-cysteine ligase catalytic subunit; GST, glutathione S-transferase; HO-1, heme oxygenase 1; Hprt, hypoxanthine-guanine phosphoribosyl transferase-1; ihMNC, intrahepatic mononuclear cells; Jnk, c-Jun N-terminal kinase; LPS, lipopolysaccharide; NLRP3, NLR family pyrin domain-containing-3; NQO1, NAD(P)H-quinone oxidoreductase-1; NRF2, nuclear factor erythroid 2-related factor 2; NTCP, sodium-dependent bile acid co-transporter; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis; qRT-PCR; quantitative reverse transcriptase-PCR; TNFiap3, TNFα-interacting protein-3; TNFα, TNF-alpha; Trx1, thioredoxin-1; TrxR1, thioredoxin reductase-1; Txnip, Trx-interacting protein.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Colin T. Shearn, Email: COLIN.SHEARN@CUANSCHUTZ.EDU.
Aimee L. Anderson, Email: Aimee.anderson@cuanschutz.edu.
Colin G. Miller, Email: CMiller67601@gmail.com.
Reed C. Noyd, Email: reednoyd@gmail.com.
Nata Balasubramaniyan, Email: Nata.Bala@ucdenver.edu.
David J. Orlicky, Email: David.orlicky@cuanschutz.edu.
Edward E. Schmidt, Email: eschmidt@montana.edu.
Ronald J. Sokol, Email: ronald.sokol@childrenscolorado.org.
REFERENCES
- 1.Kim WR, Lake JR, Smith JM, Schladt DP, Skeans MA, Harper AM, et al. OPTN/SRTR 2016 annual data report: liver. Am J Transplant. 2018;18(suppl 1):172–253. [DOI] [PubMed] [Google Scholar]
- 2.Giordano DM, Pinto C, Maroni L, Benedetti A, Marzioni M. Inflammation and the gut-liver axis in the pathophysiology of cholangiopathies. Int J Mol Sci. 2018;19:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen DR, Orlicky DJ, Roede JR, Shearn CT. Aberrant expression of redox regulatory proteins in patients with concomitant primary sclerosing cholangitis/inflammatory bowel disease. Exp Mol Pathol. 2018;105:32–6. [DOI] [PubMed] [Google Scholar]
- 4.Shearn CT, Orlicky DJ, Petersen DR. Dysregulation of antioxidant responses in patients diagnosed with concomitant primary sclerosing cholangitis/inflammatory bowel disease. Exp Mol Pathol. 2018;104:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones DP, Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab. 2010;12(suppl 2):116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muri J, Thut H, Feng Q, Kopf M. Thioredoxin-1 distinctly promotes NF-kappaB target DNA binding and NLRP3 inflammasome activation independently of Txnip. Elife. 2020;9:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwangbo H, Kim MY, Ji SY, Kim SY, Lee H, Kim GY, et al. Auranofin attenuates non-alcoholic fatty liver disease by suppressing lipid accumulation and NLRP3 inflammasome-mediated hepatic inflammation in vivo and in vitro. Antioxidants (Basel). 2020;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HY, Choi YJ, Kim SK, Kim H, Jun DW, Yoon K, et al. Auranofin prevents liver fibrosis by system Xc-mediated inhibition of NLRP3 inflammasome. Commun Biol. 2021;4:824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wall SB, Li R, Butler B, Burg AR, Tse HM, Larson-Casey JL, et al. Auranofin-mediated NRF2 induction attenuates interleukin 1 beta expression in alveolar macrophages. Antioxidants (Basel). 2021;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suvorova ES, Lucas O, Weisend CM, Rollins MF, Merrill GF, Capecchi MR, et al. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4:e6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cebula M, Schmidt EE, Arner ES. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid Redox Signal. 2015;23:823–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shearn CT, Fennimore B, Orlicky DJ, Gao YR, Saba LM, Battista KD, et al. Cholestatic liver disease results increased production of reactive aldehydes and an atypical periportal hepatic antioxidant response. Free Radic Biol Med. 2019;143:101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Dong R, Zheng S. Roles of the inflammasome in the gutliver axis (review). Mol Med Rep. 2019;19:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luan J, Ju D. Inflammasome: a double-edged sword in liver diseases. Front Immunol. 2018;9:2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai SY, Ge M, Mennone A, Hoque R, Ouyang X, Boyer JL. Inflammasome is activated in the liver of cholestatic patients and aggravates hepatic injury in bile duct-ligated mouse. Cell Mol Gastroenterol Hepatol. 2019;9:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai SY, Boyer JL. Bile infarcts: new insights into the pathogenesis of obstructive cholestasis. Hepatology. 2019;69:473–475. [DOI] [PubMed] [Google Scholar]
- 17.Frissen M, Liao L, Schneider KM, Djudjaj S, Haybaeck J, Wree A, et al. Bidirectional role of NLRP3 during acute and chronic cholestatic liver injury. Hepatology. 2021;73:1836–1854. [DOI] [PubMed] [Google Scholar]
- 18.Qu J, Yuan Z, Wang G, Wang X, Li K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int Immunopharmacol. 2019;70:147–155. [DOI] [PubMed] [Google Scholar]
- 19.Gong Z, Zhou J, Zhao S, Tian C, Wang P, Xu C, et al. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget. 2016;7:83951–83963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isakov E, Weisman-Shomer P, Benhar M. Suppression of the pro-inflammatory NLRP3/interleukin-1beta pathway in macrophages by the thioredoxin reductase inhibitor auranofin. Biochim Biophys Acta. 2014;1840:3153–3161. [DOI] [PubMed] [Google Scholar]
- 21.Bondareva AA, Capecchi MR, Iverson SV, Li Y, Lopez NI, Lucas O, et al. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43:911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rollins MF, van der Heide DM, Weisend CM, Kundert JA, Comstock KM, Suvorova ES, et al. Hepatocytes lacking thioredoxin reductase 1 have normal replicative potential during development and regeneration. J Cell Sci. 2010;123:2402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balasubramaniyan N, Devereaux MW, Orlicky DJ, Sokol RJ, Suchy FJ. Up-regulation of miR-let7a-5p leads to decreased expression of ABCC2 in obstructive cholestasis. Hepatol Commun. 2019;3:1674–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, et al. Hepatocyte hyperproliferation upon liver-specific co-disruption of thioredoxin-1, thioredoxin reductase-1, and glutathione reductase. Cell Rep. 2017;19:2771–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NCRRGW Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLoughlin MR, Orlicky DJ, Prigge JR, Krishna P, Talago EA, Cavigli IR, et al. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc Natl Acad Sci U S A. 2019;116:11408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El Kasmi KC, Anderson AL, Devereaux MW, Balasubramaniyan N, Suchy FJ, Orlicky DJ, et al. Interrupting tumor necrosis factor-alpha signaling prevents parenteral nutrition-associated cholestasis in mice. JPEN J Parenter Enteral Nutr. 2022;46:1096–1106. [DOI] [PubMed] [Google Scholar]
- 28.El Kasmi KC, Vue PM, Anderson AL, Devereaux MW, Ghosh S, Balasubramaniyan N, et al. Macrophage-derived IL-1beta/NF-kappaB signaling mediates parenteral nutrition-associated cholestasis. Nat Commun. 2018;9:1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El Kasmi KC, Anderson AL, Devereaux MW, Vue PM, Zhang W, Setchell KD, et al. Phytosterols promote liver injury and Kupffer cell activation in parenteral nutrition-associated liver disease. Sci Transl Med. 2013;5:206ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeMorrow S, Francis H, Gaudio E, Ueno Y, Venter J, Onori P, et al. Anandamide inhibits cholangiocyte hyperplastic proliferation via activation of thioredoxin 1/redox factor 1 and AP-1 activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G506–G519. [DOI] [PubMed] [Google Scholar]
- 31.Sato K, Hall C, Glaser S, Francis H, Meng F, Alpini G. Pathogenesis of kupffer cells in cholestatic liver injury. Am J Pathol. 2016;186:2238–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konishi T, Schuster RM, Goetzman HS, Caldwell CC, Lentsch AB. Cell-specific regulatory effects of CXCR2 on cholestatic liver injury. Am J Physiol Gastrointest Liver Physiol. 2019;317:G773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raninga PV, Di Trapani G, Vuckovic S, Tonissen KF. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-small ka, Cyrillicbeta inhibition. Cell Cycle. 2016;15:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. J Biol Chem. 2005;280:34538–47. [DOI] [PubMed] [Google Scholar]
- 36.Aleksunes LM, Slitt AL, Maher JM, Augustine LM, Goedken MJ, Chan JY, et al. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol. 2008;226:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu C, Wang F, Jin C, Huang X, McKeehan WL. Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J Biol Chem. 2005;280:17707–14. [DOI] [PubMed] [Google Scholar]
- 38.Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, et al. Nrf2 counteracts cholestatic liver injury via stimulation of hepatic defense systems. Biochem Biophys Res Commun. 2009;389:431–6. [DOI] [PubMed] [Google Scholar]
- 39.Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. 2017;67:549–58. [DOI] [PubMed] [Google Scholar]
- 40.Barikbin R, Neureiter D, Wirth J, Erhardt A, Schwinge D, Kluwe J, et al. Induction of heme oxygenase 1 prevents progression of liver fibrosis in Mdr2 knockout mice. Hepatology. 2012;55:553–62. [DOI] [PubMed] [Google Scholar]
- 41.Arner ES. Focus on mammalian thioredoxin reductases—important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790:495–526. [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Zheng Q, Chen Z. The Nrf2 pathway in liver diseases. Front Cell Dev Biol. 2022;10:826204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Locy ML, Rogers LK, Prigge JR, Schmidt EE, Arner ES, Tipple TE. Thioredoxin reductase inhibition elicits Nrf2-mediated responses in Clara cells: implications for oxidant-induced lung injury. Antioxid Redox Signal. 2012;17:1407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aleksunes LM, Slitt AL, Maher JM, Dieter MZ, Knight TR, Goedken M, et al. Nuclear factor-E2-related factor 2 expression in liver is critical for induction of NAD(P)H:quinone oxidoreductase 1 during cholestasis. Cell Stress Chaperones. 2006;11:356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh CJ, Kim JY, Min AK, Park KG, Harris RA, Kim HJ, et al. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. Free Radic Biol Med. 2012;52:671–82. [DOI] [PubMed] [Google Scholar]
- 46.Weerachayaphorn J, Luo Y, Mennone A, Soroka CJ, Harry K, Boyer JL. Deleterious effect of oltipraz on extrahepatic cholestasis in bile duct-ligated mice. J Hepatol. 2014;60:160–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O’Hara SP, et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol. 2018;69:676–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song M, Chen Z, Qiu R, Zhi T, Xie W, Zhou Y, et al. Inhibition of NLRP3-mediated crosstalk between hepatocytes and liver macrophages by geniposidic acid alleviates cholestatic liver inflammatory injury. Redox Biol. 2022;55:102404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology. 2003;38:374–84. [DOI] [PubMed] [Google Scholar]
- 50.Liu J, Lickteig AJ, Zhang Y, Csanaky IL, Klaassen CD. Activation of Nrf2 decreases bile acid concentrations in livers of female mice. Xenobiotica. 2021;51:605–15. [DOI] [PubMed] [Google Scholar]