

Abstract
Dual catalytic systems involving photocatalytic activation and transition metal-catalyzed steps have enabled innovative approaches to the construction of carbon–carbon and carbon–heteroatom bonds. However, the mechanistic complexity of the dual catalytic processes presents multiple challenges for understanding of the roles of divergent catalytic species that can impede the development of future synthetic methods. Here, we report a dual catalytic process that enables the previously inaccessible, broad-scope, direct conversion of carboxylic acids to aromatic sulfones―centrally important carbonyl group bioisosteric replacements and synthetic intermediates―by a tricomponent decarboxysulfonylative cross-coupling with aryl halides. Detailed mechanistic and computational studies revealed the roles of the copper catalyst, base, and halide anions in channeling the acridine/copper system via a distinct dual catalytic manifold. In contrast to the halide-free decarboxylative conjugate addition that involves cooperative dual catalysis via low-valent copper species, the halide counteranions divert the decarboxysulfonylative cross-coupling with aryl halides through a two-phase, orthogonal relay catalytic manifold, comprising a kinetically coupled (via antithetical inhibitory and activating roles of the base in the two catalytic cycles), mechanistically discrete sequence of a photoinduced, acridine-catalyzed decarboxylative process and a thermal copper-catalyzed arylative coupling. The study underscores the importance of non-innocent roles of counteranions and key redox steps at the interface of catalytic cycles for enabling previously inaccessible dual catalytic transformations.
Keywords: acridines, carboxylic acids, decarboxylation, photocatalysis, sulfones
INTRODUCTION
The past decade has witnessed a rapid increase in the complexity of photocatalytic systems that have evolved to combine multiple mechanistically distinct types of intertwined catalytic cycles.1 The new photoinduced multicatalytic reactions have enabled a variety of functionalizations that harness the diverse reactivity of radical and excited state species interfaced through a variety of single electron transfer and two-electron processes. However, the growing complexity of the emerging multicatalytic systems has posed significant challenges to mechanistic understanding of the underlying processes. In particular, the factors that determine the interactions between multiple catalytic cycles and the roles of divergent relay and cooperative multicatalytic modes remain underexplored.
Sulfones are centrally important synthetic intermediates,2 pharmacophores,3 and functional linchpins in materials science applications (Scheme 1.A).4 Their notable metabolic and physicochemical stability, as well as a distinctive chemical reactivity imparted by the sulfonyl group have resulted in a growing number of applications that require facile and rapid synthetic access to structurally diverse sulfone-containing molecules.5 Given the structural and electronic similarity between the sulfonyl and the carbonyl groups, sulfones have emerged as efficient bioisosteric replacements for reactive carboxylic acids and other carbonyls.6 Notably, nearly one half of the approved sulfone drugs feature a sulfonyl group bearing a (hetero)aryl and an alkyl substituent, underscoring the importance of the alkyl (hetero)aryl sulfones.3b
Scheme 1.
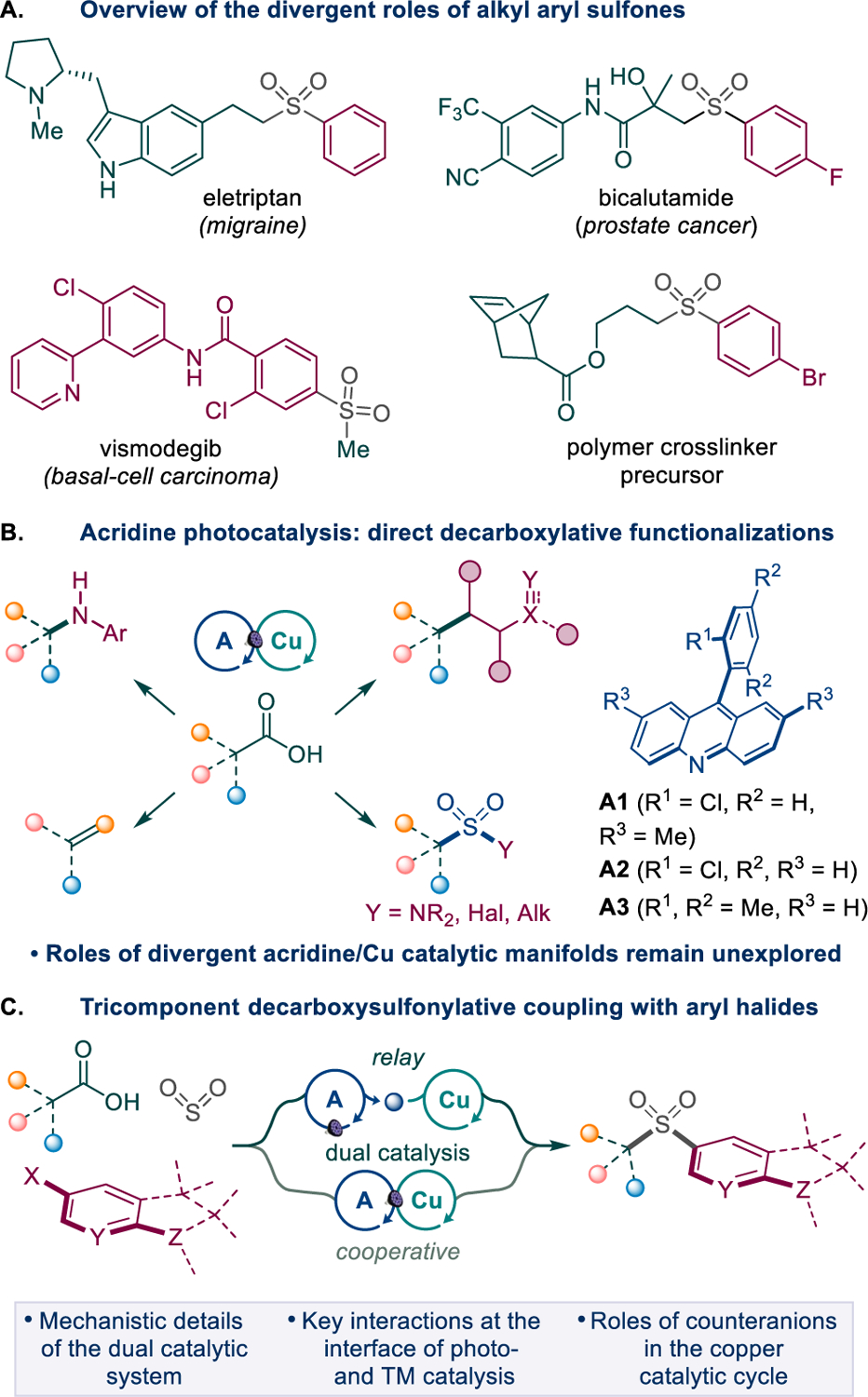
Decarboxysulfonylative cross-coupling of aryl halides and carboxylic acids
Photocatalytic direct decarboxylative functionalization has opened new directions in synthetic methodology by enabling one-step functional group interconversions between abundant and structurally diverse carboxylic acids and other valuable functionalities without the need for preactivation of the carboxylic group.7–9 However, photoactivation of the carboxylic group towards decarboxylation poses significant challenges with respect to the scope of the acids and the diversity of carbon–carbon and carbon–heteroatom bond-forming reactions, due to the combination of the acidity of the carboxylic group that may interfere with other catalytic processes and the resistance of acids to oxidative cleavage (Eox > 2.0 V vs SCE)10 or O–H hydrogen atom transfer (BDE 112 kcal/mol).11
We recently described a new class of photocatalysts for visible light-induced direct decarboxylative functionalization of carboxylic acids that enabled a one-step access to several important functionalities and provided insights into the mechanistic underpinnings of complex dual and triple catalytic systems (Scheme 1.B).8 The reaction scope of the new photocatalytic system has also recently been successfully expanded to the synthetically useful hydrazone additions and perfluoropyridinethiolation by the Dilman group.9 Mechanistic studies showed that the photocatalytic decarboxylation takes place in the excited state of the acridine–carboxylic acid hydrogen bond complex without prior dissociation to acridinium carboxylate via a proton-coupled electron transfer (PCET) process.8a,e Importantly, the acridine photocatalytic cycle could be readily interfaced with several transition metal-catalyzed (e.g., Co8a and Cu8b–d) processes, and the mechanisms of the catalyst turnover were specific and dependent on the mode of interaction between the acridine and the metal-catalyzed processes.
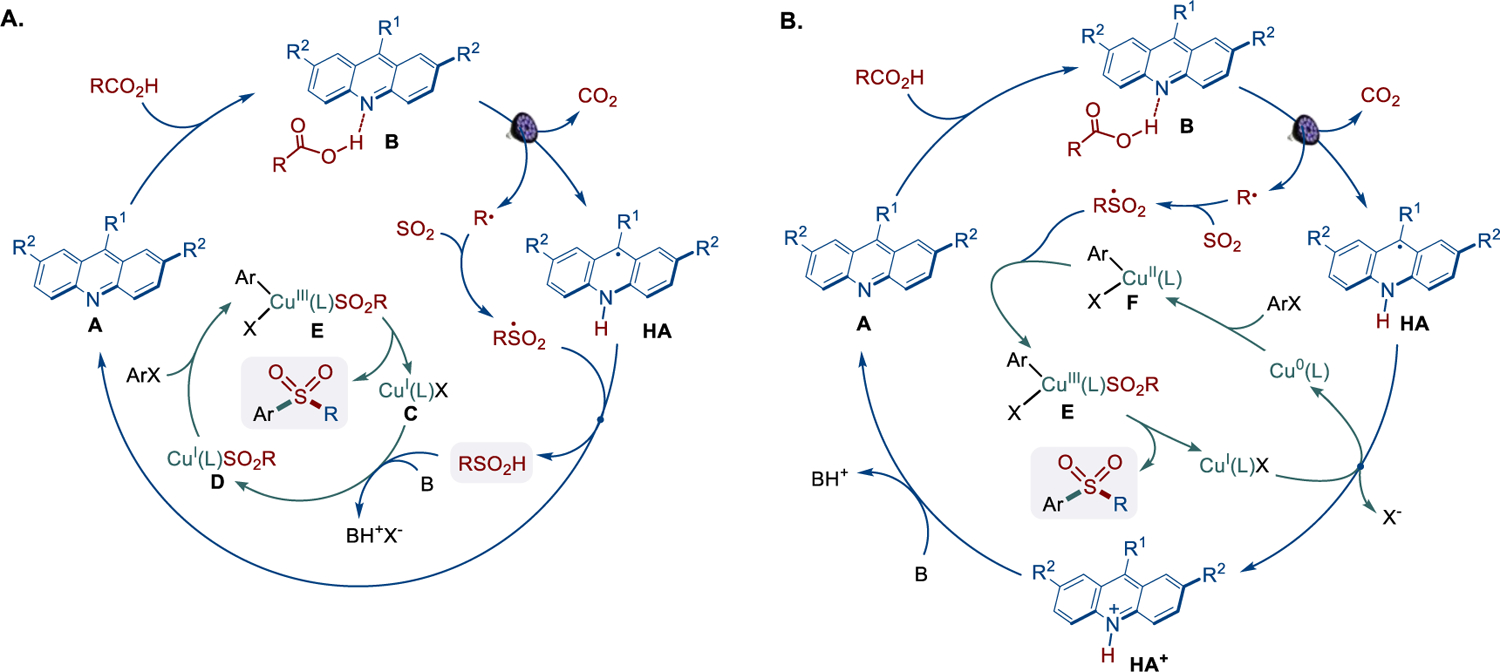
Our previous studies showed that the alkyl radical generated in the acridine photocatalytic cycle can be readily intercepted by sulfur dioxide, producing a sulfonyl radical.8d We therefore hypothesized that the sulfonyl radical can subsequently be engaged in a cross-coupling with aryl halides, resulting in net arylative decarboxysulfonylation (Scheme 1.C). If developed, this method would for the first time enable a direct one-step conversion of carboxylic acids and (hetro)aryl halides that do not readily undergo nucleophilic aromatic substitution (SNAr)12 to aromatic sulfones. The reaction would allow for a rapid access to broad chemical space, given the ready availability and structural diversity of these two reactant classes, while by-passing preactivation and other auxiliary functional group interconversions.13 Our earlier work on the direct decarboxylative C–S/C–N coupling reactions indicated that copper was compatible with the acridine photocatalytic cycle and facilitated the transfer of electrophiles on the sulfonyl group.8d We envisioned that the sulfonyl radical could produce sulfinic acid by a hydrogen abstraction from the acridinyl radical HA, emerging from the photoinduced proton-coupled electron transfer (PCET)-enabled decarboxylation in complex B (path a, Scheme 2.A). The base-mediated anion exchange could then convert CuI complex C to sulfinate intermediate D that can undergo an oxidative addition with an aryl halide via intermediate E, followed by reductive elimination, giving rise to the sulfone product in an overall orthogonal relay dual catalytic process.14 While the CuI/CuIII mechanism is consistent with the roles of the copper catalysis in facilitating aryl–heteroatom bond-forming reactions,15 our mechanistic studies of the acridine/copper dual catalytic conjugate addition reactions also showed that the acridinyl radical may produce Cu0 species channeling the reaction via a divergent catalytic cycle within a cooperative dual catalytic system.8c Acridinyl radical-mediated formation of Cu0 species could also enable an alternative mechanism for the aromatic decarboxysulfonylation that proceeds through an oxidative addition of the aryl halide to the Cu0 intermediate, producing arylcopper species F (path b, Scheme 2.B). Subsequent cross-termination with the sulfonyl radical would lead to copper intermediate E with ensuing generation of the sulfone product by reductive elimination. Given the mechanistic dichotomy observed in the acridine/copper dual catalytic systems, further mechanistic investigation is necessary to clarify the influence of various factors that favor each pathway in order to leverage the synthetic potential of direct decarboxylation methodologies.
Scheme 2.
Mechanistic pathways of the Dual Catalytic Decarboxysulfonylation
We report herein the development of a dual catalytic acridine/copper decarboxysulfonylation that allows for direct conversion of carboxylic acids to alkyl (hetero)aryl sulfones by a cross coupling with aryl halides. Mechanistic studies point to key roles of halide anions in determining the operative mechanism in the copper catalytic cycle and how it is interfaced with the acridine photocatalytic system in a two-phase orthogonal relay catalytic manifold, comprising a photocatalytic decarboxylative process and a copper-catalyzed arylative cross-coupling.
RESULTS AND DISCUSSION
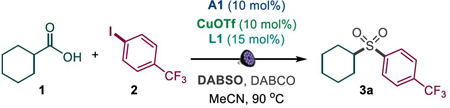
Optimization studies revealed that the tricomponent decarboxysulfonylative cross-coupling of carboxylic acid 1 with aryl iodide 2 occurs readily in the dual catalytic system of acridine photocatalyst A1 and diamine L1-ligated copper(I) triflate, with DABCO as a basic additive and either DABSO (DABCO·(SO2)2)16 (Table 1, entry 1) or potassium metabisulfite (entry 2) as stable sulfur dioxide donors, in acetonitrile at 100 °C and purple LED light (λ = 400 nm), providing sulfone 3a in 97 and 91% yields. Both catalysts as well as the visible light irradiation were essential for achieving the desired transformation (entry 3). DABCO was also necessary for an efficient conversion to sulfone 3a. Other acridines, e.g., A2 and A3, were less suitable, while the optimal temperature was 90 °C (entries 5–7). Notably, a range of other photocatalytic systems, including N-methylacridinium salts, 4CzIPN, as well as iridium, and ruthenium complexes, did not produce the sulfone product (Table S1), pointing to the key role of acridine photocatalysis in enabling the decarboxysulfonylative cross-coupling. Other copper catalyst precursors were less catalytically active (e.g., entries 8 and 9). Significant structural effects were also observed for the diamine ligand. Desmethyl ligand L2 resulted in a lower yield, indicating that a N-methyl group was necessary for the promotion of the reaction (entry 10). Interestingly, while the vicinally disubstituted diamine ligand L3 was also ineffective, the unsubstituted ligand L4 provided the product in 91% yield with DABSO and 88% with potassium metabisulfite (entries 11, 12). Finally, acetonitrile emerged as the optimal solvent (entry 15). The acridine/copper dual catalyzed decarboxylative conjugate addition reaction proceeded with a quantum yield of 0.06.
Table 1.
Reaction conditions for the photocatalytic direct decarboxylative tricomponent coupling of carboxylic acids and aryl iodidesa
| ||
|---|---|---|
| Entry | Change from optimal conditions | Yield, %b |
| 1 | none | 97 (82b) |
| 2 | K2S2O5 instead of DABSO at 100°C | 91 (82b) |
| 3 | no light or A1 or CuOTf | 0 |
| 4 | No DABCO | 45 |
| 5 | A2 instead of A1 | 73 |
| 6 | A3 instead of A1 | 77 |
| 7 | 80°C | 78 |
| 8 | CuBr instead of CuOTf·½PhMe | 76 |
| 9 | CuI instead of CuOTf·½PhMe | 64 |
| 10 | L2 instead of L1 | 41a |
| 11 | L3 instead of L1 | 59 |
| 12 | L4 instead of L1 | 91 |
| 13 | L4 instead of L1 with K2S2O5 | 88 |
| 15 | PhCH3 instead of MeCN | 19 |
Reaction conditions: carboxylic acid (0.3 mmol), DABSO (0.36 mmol) or K2S2O5 (0.36 mmol), aryl iodide (0.6 mmol), A1 (10 mol%), CuOTf·½PhMe (10 mol%), L1 (15 mol%), MeCN (3 mL), LED light (400 nm), 90 °C (for DABSO) or 100 °C (for K2S2O5), 14 h. Yield was determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as an internal standard.
Isolated yield.
DABSO = O2S–N(CH2CH2)3N–SO2.

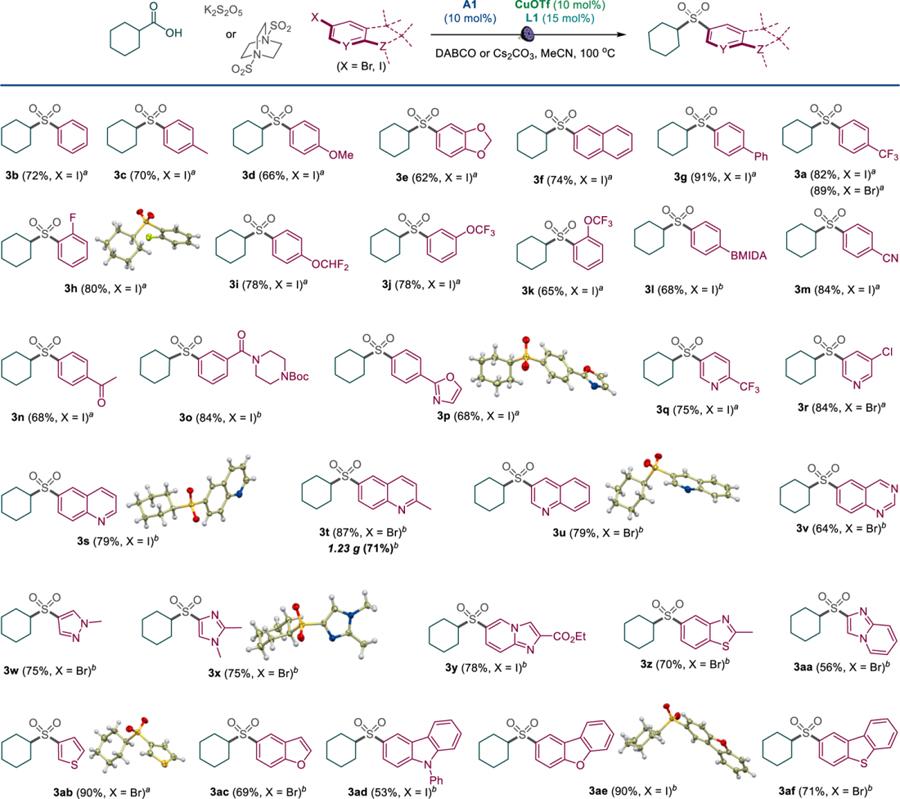
The scope of the aromatic coupling partners was evaluated next with carboxylic acid 1 as a substrate (Table 2). Aryl iodides bearing electron-donating alkyl and alkoxy groups were converted to sulfones 3b-3e in good yields. Sulfones 3f-3g bearing polycyclic aromatic groups were also formed in 74–91% yields. The medicinally relevant fluorine-containing groups were similarly well-tolerated (3a, 3h-3k). Notably, the reaction performed well with both aryl bromides and iodides (e.g., 3a). Other suitable coupling partners included aryl halides bearing the MIDA-protected boryl group, as well as cyano, keto, amide, carbamate and 2-oxazolyl groups (3m-3p). The scope of the new sulfone synthesis can be extended to a wide array of heterocyclic halides. Nitrogenous heterocycles of the pyridine, quinoline, and quinazoline series provided the corresponding sulfone products in good yields (3q-3v). Similarly efficient transformations were also observed in the pyrazole, imidazole, and imidazopyridine series (3w-3y). Other heterocyclic motifs can also be readily accessed, including benzothiazole, thiophene, benzofuran, carbazole, dibenzofuran, and dibenzothiophene (3z-3af). The reaction can be performed either with the carboxylic acid (method A) or the aryl halide (method B) as a limiting reagent. While the former procedure can be carried out with potassium metabisulfite in the presence of DABCO, the latter one showed optimal performance with DABSO and cesium carbonate.
Table 2.
Scope of haloarenes in the direct tricomponent decarboxysulfonylative cross-coupling
|
Reaction conditions for method A: carboxylic acid (0.3 mmol), K2S2O5 (0.36 mmol), aryl iodide (0.6 mmol), A1 (10 mol%), CuOTf·½PhMe (10 mol%), L1 (15 mol%), DABCO (0.21 mmol), MeCN (3 mL), LED light (400 nm), 100 °C, 14 h.
Reaction conditions for method B: aryl iodide (0.3 mmol), carboxylic acid (0.6 mmol), DABSO (0.72 mmol), A1 (10 mol%), CuOTf·½PhMe (10 mol%), L1 (15 mol%), Cs2CO3 (0.45 mmol), MeCN (4.5 mL), LED light (400 nm), 90 °C, 14 h. BMIDA = B(O2CCH2)2NMe.
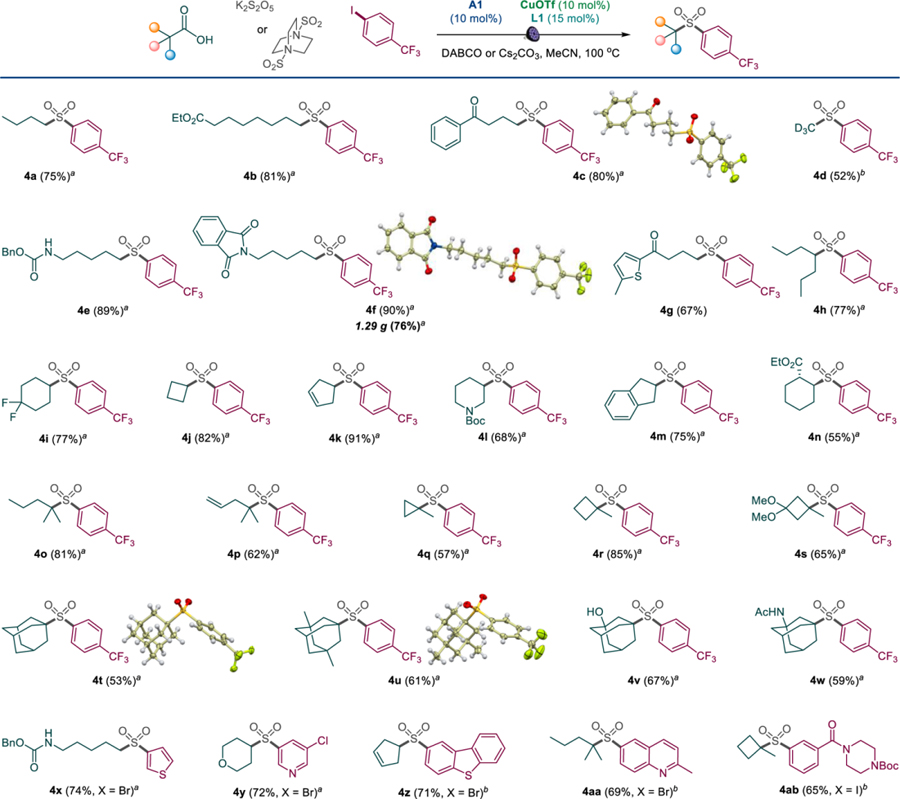
The scope of the carboxylic acids was examined next (Table 3). Primary alkylcarboxylic acids bearing ester, ketone, carbamate, phthalimide and thiophene groups reacted smoothly and produced a range of diversely substituted sulfones 4a-4g.
Table 3.
Scope of carboxylic acids in the direct tricomponent decarboxysulfonylative cross-coupling
|
Method A.
Method B. For reaction conditions, see Table 2.
The use of deuterated methyl groups to improve metabolic stability of drug candidates has become an established strategy in medicinal chemistry.17 To this end, the decarboxysulfonylation reaction can be used for a straightforward access to deuterated methyl sulfones (e.g., 4d) from the readily available d4-acetic acid. Sulfones 4h-4n derived from acyclic and cyclic secondary alkylcarboxylic acids, including strained and unsaturated rings (4j, 4k) were also readily produced. Notably, sulfone 4n was obtained as a single trans-diastereomer, underscoring the effect of the substrate-induced stereoselectivity. Diverse tertiary alkylcarboxylic acids were similarly well-tolerated (4o-4w), both in the acyclic and cyclic series and without any negative effects of the ring strain. Furthermore, a series of substituted adamantine-derived sulfones, including unprotected alcohol and amide-substituted 4v and 4w, were also accessed. The tricomponent decarboxysulfonylative cross-coupling can also be carried out with a range of combinations of diverse acids and arene and heteroarene coupling partners without any detriment to the reaction efficiency (4x-4ab), pointing to the applicability of the new method in a variety of structural settings.
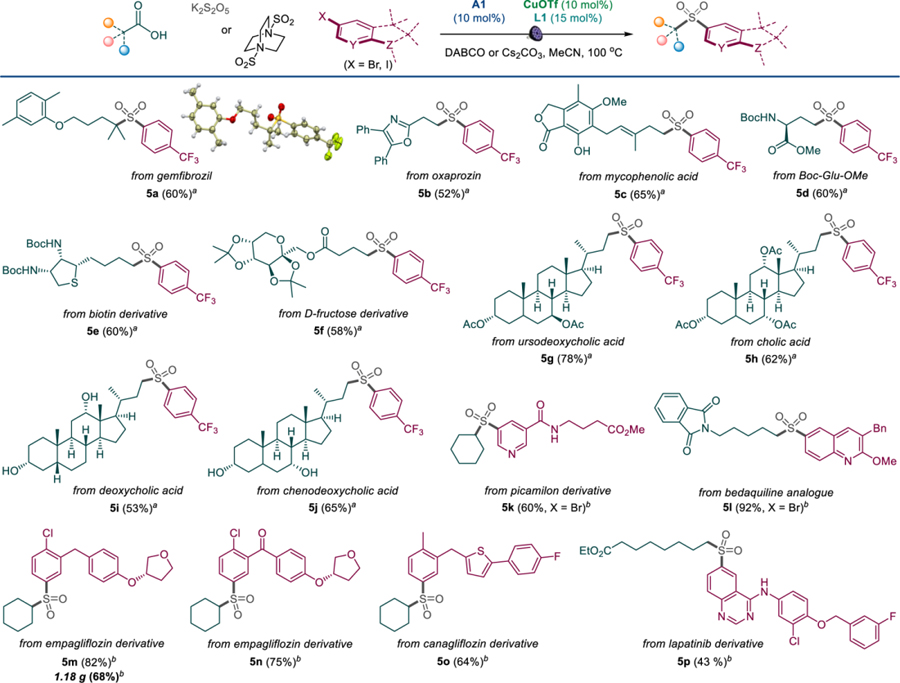
The scope and functional group tolerance of the reaction was further examined with more structurally complex substrates comprising natural products and active pharmaceutical ingredients (Table 4). Antihyperlipidemic gemfibrozil (5a), anti-inflammatory oxaprozin (5b), and immunosuppressive mycophenolic acid (5c) were converted to the corresponding sulfones. Similarly, the γ-sulfone analogue of glutamic acid (5d), as well as sulfones 5e and 5f derived from biotin and D-fructose were accessed using the new method. Bile acids were also suitable substrates (5g-5j). Likewise, halogen-containing derivatives and precursors of anxiolytic picamilon (5k), antituberculotic bedaquiline (5l), antidiabetic empagliflozin (5m, 5n) and canagliflozin (5o), as well as antineoplastic lapatinib (5p) all produced the desired sulfones. Furthermore, the reaction was amenable to scale-up, affording gram quantities of sulfone products (3t, 4f, 5m). The diversity of the structural classes and functional groups in the substrates indicate that the tricomponent decarboxysulfonylative cross-coupling exhibits broad chemoselectivity that is suitable for a wide range of synthetic applications.
Table 4.
Arylsulfonylation of natural products and pharmaceuticals by the direct tricomponent decarboxysulfonylative cross-coupling
|
Method A.
Method B. For reaction conditions, see Table 2. Aryl iodides were used unless otherwise specified.
In order to gain insight into the thermodynamic and kinetic feasibility of the CuI/CuIII (path a) and Cu0/CuI/CuIII (path b) catalytic systems, DFT calculations were carried out. For path a, our prior studies established the thermodynamic facility of the acridine-catalyzed formation of sulfinic acids by trapping the intermediate alkyl radical with sulfur dioxide and the following hydrogen atom transfer from the intermediate acridinyl radical to the sulfonyl radical8d (Scheme 2.A). Building on this mechanistic analysis, attention was turned to the copper catalytic cycle (Figure 1.A). The studies revealed that the halide–sulfinate anion exchange in CuI intermediate 6 is endergonic by 4.9 kcal/mol. Subsequent oxidative addition of aryl bromide 7 to sulfinate intermediate 8 proceeds over a barrier of 27.6 kcal/mol (TSA) with an overall barrier of 32.5 kcal/mol from intermediate 6 that is consistent with the thermal activation required to effect the dual catalytic process.
Figure 1.
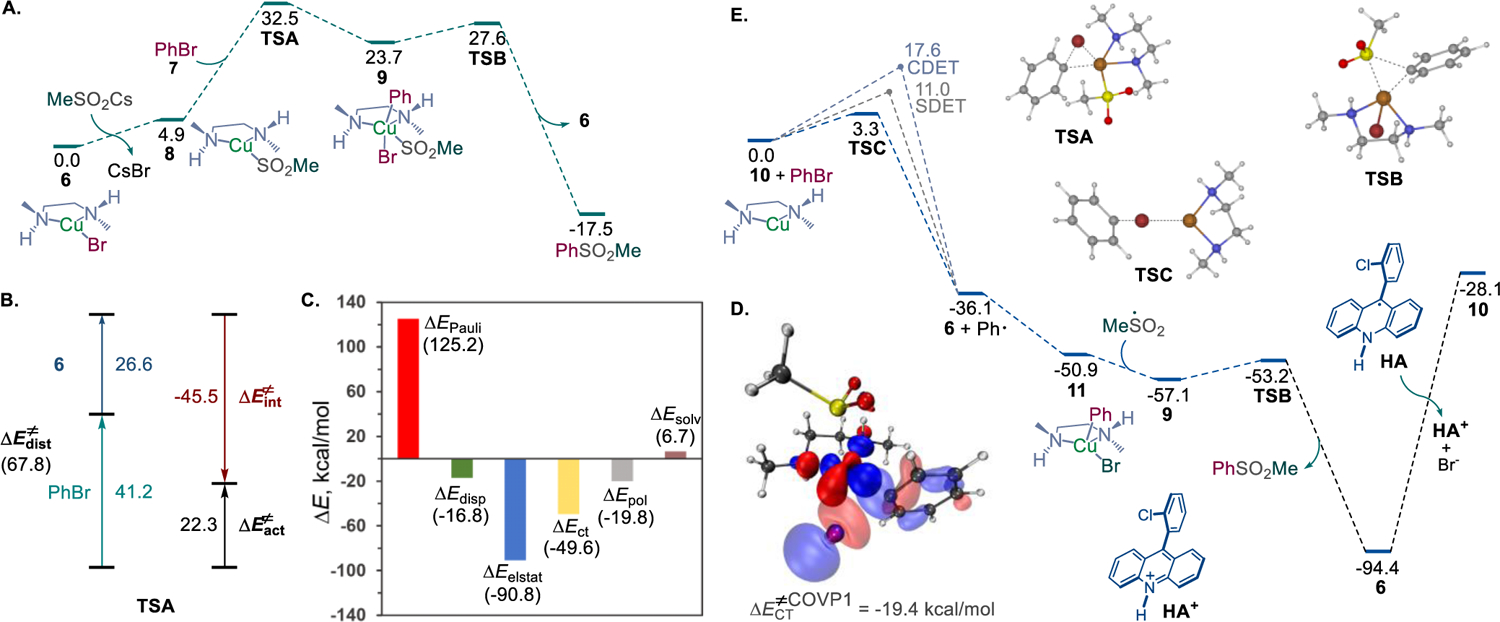
Computational studies of the dual catalytic decarboxysulfonylation. A. Computed Gibbs free energy profile for path a, ΔG, kcal/mol. B. Activation strain model analysis for the oxidative addition to complex 8. C. Energy decomposition analysis for TSA. D. The most significant complementary occupied-virtual pair (COVP) for TSA. E. Computed Gibbs free energy profile for path b, ΔG, kcal/mol.
Distortion/interaction activation strain model (AIM)18 analysis points to the distortion of the aryl halide as the major contributor to the overall distortion energy in transition state TSA (Figure 1.B). Furthermore, an energy decomposition analysis based on absolutely localized molecular orbitals (ALMO-EDA(solv))19 indicates that the substantial Pauli (steric) repulsion is primarily compensated by strong electrostatic and charge transfer interactions with minor contributions from polarization and dispersion (Figure 1.C). Furthermore, complementary occupied-virtual pairs (COVP)20 analysis shows that the most significant charge transfer takes place between the HOMO-1 orbital of the copper complex and the LUMO+2 of aryl halide (Figure 1.D). Collectively, these results point to the key contribution of the energetic cost of breaking the strong aryl–halogen bond and the important role of the ligand in stabilizing transition state TSA. Following the oxidative addition, the resulting intermediate 9 undergoes strongly exergonic reductive elimination that is more kinetically facile than the reversal to sulfinate 8 and aryl halide, driving the reaction forward and affording the sulfone product with concomitant regeneration of CuI catalyst 6. These results support the thermodynamic and kinetic feasibility of the sequential dual catalytic pathway (path a).
The acridine/Cu0/CuI/CuIII catalytic pathway (path b) was examined next (Figure 1.E). Given the previously established efficiency of the generation of the sulfonyl radical,8d overall feasibility of the path b depends on the energetic parameters of the Cu-catalytic cycle and the key redox step that regenerates both the acridine and the Cu catalysts. Due to the strongly reducing character of the Cu0 intermediate 10, the reaction with the aryl halide via a halogen atom transfer was both exergonic and readily kinetically accessible (TSC). Stepwise (SDET) and concerted (CDET) dissociative electron transfer pathways were also accessible but less kinetically favorable. Subsequent trapping of the aryl radical by CuI species 6 resulted in the exergonic formation of CuII intermediate 11, that upon an exergonic cross-termination with the sulfonyl radical, afforded intermediate 9. The ensuing reductive elimination delivered CuI intermediate 6 that would need to be reduced by acridinyl radical HA to complete both catalytic cycles.
This step, however, was revealed to be highly endergonic (ΔG = 66.3 kcal/mol), pointing to a prohibitively thermodynamically unfavorable redox process that contrasted with the facility of the preceding sequence. This result is consistent with the very low reduction potentials reported for copper(I) halide complexes (Ered <–2.5 V vs SCE),21 making the reduction by acridinyl radical HA (Eox = −0.66 V) thermodynamically unfavorable.22 These results underscore the importance of the redox steps at the intersection of the catalytic cycles1a,b,8c and highlight the differences in the redox properties of the more readily reducible cationic CuI/Cu0 systems with labile counteranions (e.g., tetrafluoroborate) and the substantially more stable copper(I) halide systems that may lead to distinctive mechanistic behavior in dual catalytic reactions. Comparing the mechanisms of the direct decarboxylative conjugate addition catalyzed by a cationic CuI complex with a labile tetrafluoroborate anion8c with path b of the arylative decarboxysulfonylation, it is evident that the halide plays a key role in diverting the reaction from path b by forestalling the key catalyst turnover redox step in the otherwise kinetically and thermodynamically favorable pathway.
To gain experimental support for the computationally derived pathway, mechanistic and kinetic studies were carried out for the decarboxysulfonylative coupling and the individual catalytic cycles (i.e., the acridine-catalyzed sulfinic acid formation and the copper-catalyzed cross coupling). Addition of TEMPO led to the suppression of the decarboxysulfonylation and formation of the alkyl radical trapping product 12, indicating that the reaction involves the decarboxylation-derived alkyl radical (Figure 2.A). By contrast, no aryl-TEMPO 13 product was observed.23 Similarly, a reaction with aryl iodide 14 produced sulfone 15 without the cyclization that involves the pendant allyl group (i.e., formation of sulfone 16, Figure 2.B). These results suggest that the aryl radical is not involved in the sulfonylation step, pointing to the oxidative addition as the C–X bond activation pathway.
Figure 2.
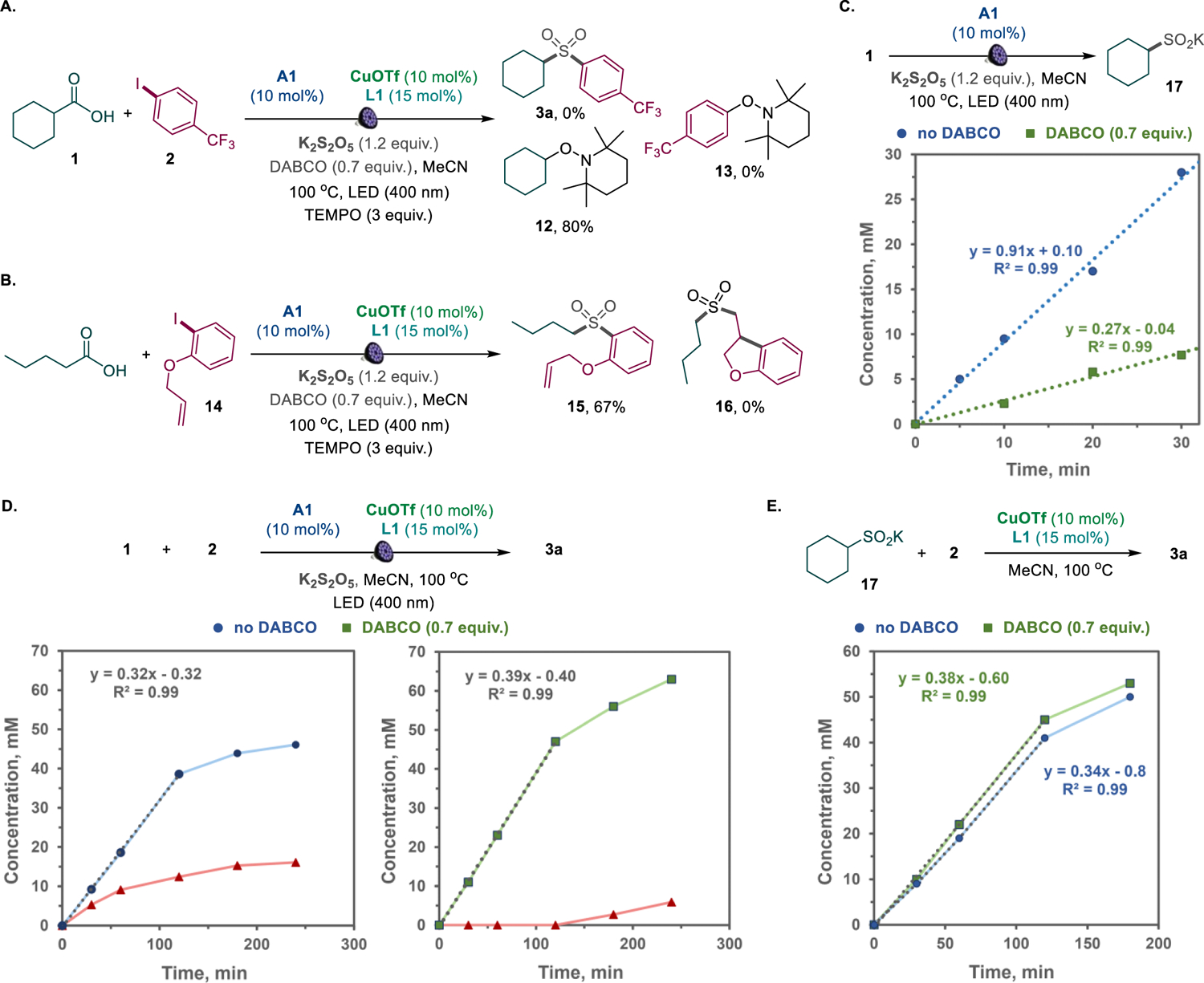
Kinetic profile of the dual catalytic decarboxysulfonylation. A. Radical trapping experiment with TEMPO. B. Cyclization experiment with aryl iodide 14. C. Kinetics of the photoinduced decarboxylative sulfination of acid 1 at 100 °C with (green squares) and without (blue dots) added DABCO. D. Kinetics of the decarboxysulfonylation (blue dots) and the accumulation of sulfinate salt 17 (red triangles) during the decarboxysulfonylation. E. Kinetic profile of the cross-coupling of sulfinate 17 and aryl iodide 2. The experiments were carried out under the method A reaction conditions with or without irradiation as specified in each reaction scheme.
Interestingly, the acridine-catalyzed sulfinic acid production rate was 2.8 times higher than the overall rate of the sulfone production in the decarboxysulfonylative dual catalytic system (Figure 2.C,D, no DABCO). Despite the substantially faster sulfinic acid production, no accumulation of the sulfinic acid was observed in the initial stages of the decarboxysulfonylative process in the presence of DABCO (Figure 2.D, with DABCO).
Further studies revealed that the Cu-catalyzed cross coupling of intermediate sulfinate salt 17 with the aryl halide proceeded at the same rate as the decarboxysulfonylation (Figure 2.D,E). These results suggest that the overall rate of the decarboxysulfonylation is controlled by the Cu-catalyzed sulfinate‒aryl halide cross-coupling, while the rate of the acridine-catalyzed sulfinic acid production is dampened under the optimal decarboxysulfonylation conditions to be better aligned with the Cu-catalyzed process. This observation is supported by the accumulation of the sulfinate in the dual catalytic process in the absence of DABCO in contrast to the DABCO-mediated reaction (Figure 2.D). We hypothesized that DABCO may play a dual role of the promoter of the Cu-catalyzed step (Figure 2.E) as a basic proton shuttle (i.e., B in Scheme 2.A) and an inhibitor of the acridine-catalyzed step. This result was expected to be in line with our previous studies that demonstrated the inhibitory effect of added amine bases on the acridine-catalyzed decarboxylative reactions.8a–c Indeed, when the acridine-catalyzed sulfinic acid formation was carried out in the presence of DABCO to match the decarboxysulfonylation conditions, the reaction rate decreased and was close to the rate of the decarboxysulfonylation (Figure 2.C). The identical rates of the Cu-catalyzed sulfinate‒aryl halide coupling in the presence of a stoichiometric amount of the sulfinate and the decarboxysulfonylation proceeding with low free sulfinate concentrations also suggested that the Cu-catalyzed coupling was zero order in the sulfinate. Kinetic studies based on the variable time normalization analysis24 (VTNA) and the initial rates method revealed that the Cu-catalyzed sulfinate‒aryl halide coupling was indeed zero order in the sulfinate, as well as DABCO, and first order in both the Cu catalyst and the aryl halide (Figure 3). This result is congruent with the oxidative addition as the rate-limiting step, in line with the computationally derived path a mechanism. Taken together, the computational and experimental studies indicate that the decarboxysulfonylative cross-coupling of carboxylic acids with aryl halides proceeds by an orthogonal relay dual catalytic process, comprising a photocatalytic formation of the sulfinate intermediate in the acridine photocatalytic cycle and a copper catalyzed cross-coupling of the sulfinate intermediate with aryl halide channeled via path a, i.e. through an oxidative addition of aryl halide to a copper(I) sulfinate complex with subsequent reductive elimination en route to the aryl sulfone product.
Figure 3.
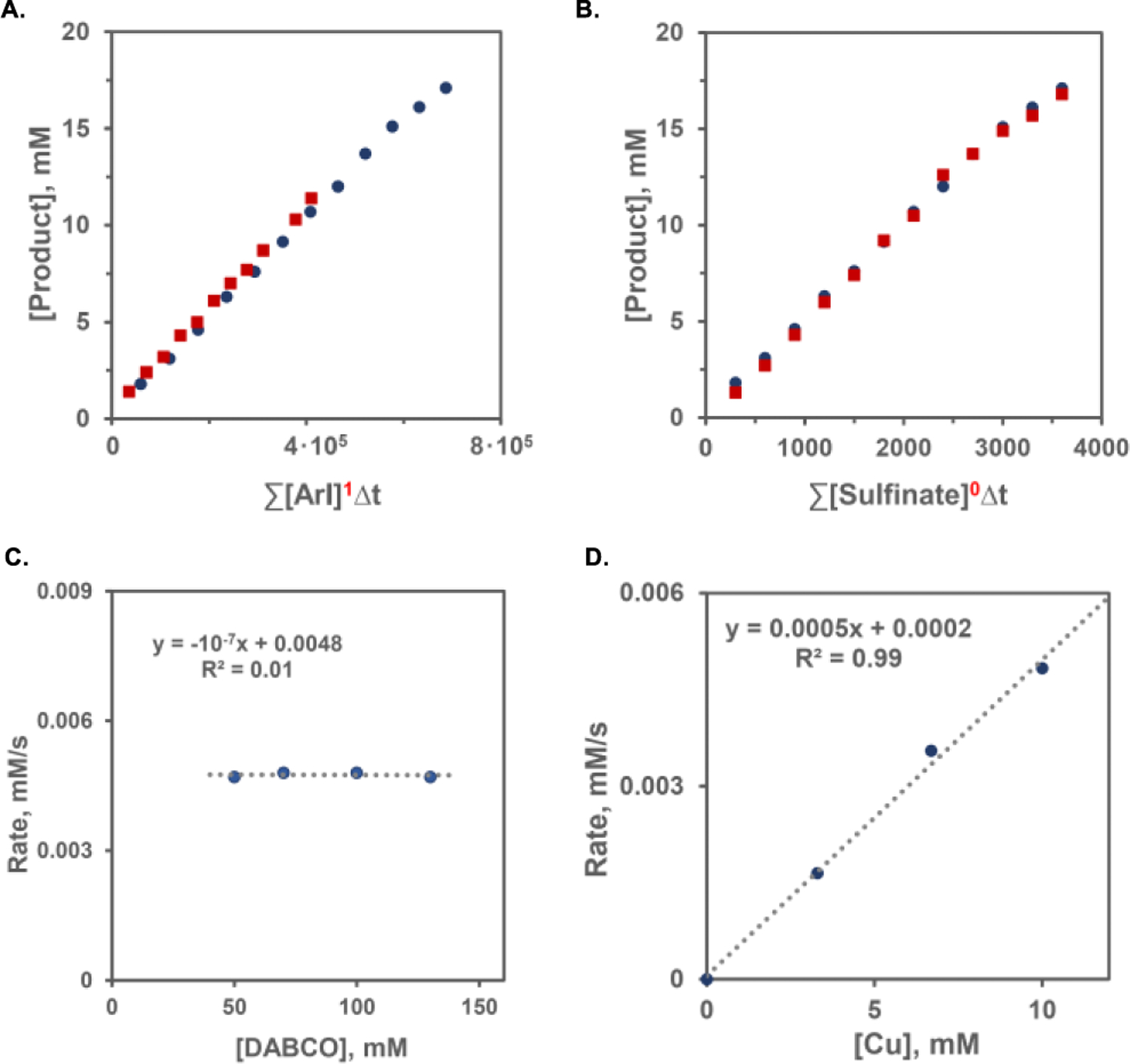
Kinetic studies of the copper-catalyzed cross-coupling of aryl iodide 2 and sulfinate 17. A. Order in aryl iodide 2. B. Order in sulfinate 17. C. Order in DABCO. D. Order in the copper catalyst. The experiments were carried out according to method A in the absence of the acridine photocatalyst and without the irradiation.
CONCLUSION
In summary, we have developed a visible light-induced, dual catalytic, direct decarboxysulfonylative cross-coupling of carboxylic acids with aryl halides. The reaction enables the previously inaccessible construction of alkyl aryl sulfones directly from carboxylic acids without preactivation and has a broad scope with respect to the acid and aryl halide coupling partners, providing a route for the direct interconversion of the bioisosteric carboxylic and sulfonyl groups. Mechanistic and computational studies revealed that the acridine/copper dual catalytic system facilitates the tricomponent decarboxysulfonylative coupling by an orthogonal relay catalytic pathway that is distinct from the previously described decarboxylative conjugate addition system, underscoring the import roles of halide anions in channeling the dual catalytic system via a divergent process that comprises acridine-catalyzed decarboxylative generation of an alkyl radical followed by the formation of an alkylsulfinate intermediate and the ensuing copper-catalyzed sulfinate cross-coupling with aryl halides. The studies highlight the mechanistic versatility of the acridine/copper dual catalytic system and point to new opportunities in catalytic carbon–heteroatom bond formation.
Supplementary Material
ACKNOWLEDGMENT
Financial support by NIGMS (GM134371) and NSF (CHE-2102646) is gratefully acknowledged. The UTSA NMR and X-ray crystallography facilities were supported by the NSF (CHE-1625963 and CHE-1920057). The authors acknowledge the Texas Advanced Computing Center (TACC) and the Extreme Science and Engineering Discovery Environment (XSEDE) for providing computational resources.
Footnotes
The authors declare no competing financial interest.
Supporting Information
Experimental and spectral details for all new compounds and all reactions reported. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122, 1485–1542. [DOI] [PubMed] [Google Scholar]; (b) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hossain A; Bhattacharyya A; Reiser O Science 2019, 364, eaav9713; [DOI] [PubMed] [Google Scholar]; (d) Cheng W-M; Shang R ACS Catal. 2020, 10, 9170–9196; [Google Scholar]; (e) Cheung KPS; Sarkar S; Gevorgyan V Visible Light-Induced Transition Metal Catalysis. Chem. Rev 2022, 122, 1543–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Patai S The Chemistry of Sulfinic Acids, Esters and their Derivatives; John Wiley & Sons, Ltd: New Jersey, 1990. [Google Scholar]; (b) Patai S; Rapport Z The Chemistry of Sulfonic Acids, Esters and their Derivatives; John Wiley & Sons: New York, 1991. [Google Scholar]; (c) Aissa C Mechanistic Manifold and New Developments of the Julia–Kocienski Reaction. Eur. J. Org. Chem 2009, 1831–1844. [Google Scholar]; (d) García-Domínguez A; Müller S; Nevado C Nickel‐Catalyzed Intermolecular Carbosulfonylation of Alkynes via Sulfonyl Radicals. Angew. Chem., Int. Ed 2017, 56, 9949–9952. [DOI] [PubMed] [Google Scholar]; (e) Burke EG; Gold B; Hoang TT; Raines RT; Schomaker JM Fine-Tuning Strain and Electronic Activation of Strain-Promoted 1,3-Dipolar Cycloadditions with Endocyclic Sulfamates in SNO-OCTs. J. Am. Chem. Soc 2017, 139, 8029–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nguyen VT; Dang HT; Pham HH; Nguyen VD; Flores-Hansen C; Arman HD; Larionov OV Highly Regio-and Stereoselective Catalytic Synthesis of Conjugated Dienes and Polyenes. J. Am. Chem. Soc 2018, 140, 8434–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ratushnyy M; Kamenova M; Gevorgyan V A Mild Light-Induced Cleavage of the S–O Bond of Aryl Sulfonate Esters Enables Efficient Sulfonylation of Vinylarenes. Chem. Sci 2018, 9, 7193–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Ariki ZT; Maekawa Y; Nambo M; Crudden CM Preparation of Quaternary Centers via Nickel-Catalyzed Suzuki-Miyaura Cross-Coupling of Tertiary Sulfones. J. Am. Chem. Soc 2018, 140, 78–81. [DOI] [PubMed] [Google Scholar]; (i) Nambo M; Yim JC-H; Freitas LBO; Tahara Y; Ariki ZT; Maekawa Y; Yokogawa D; Crudden CM Modular Synthesis of α-Fluorinated Arylmethanes via Desulfonative Cross-Coupling. Nat. Commun 2019, 10, 4528. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Hell SM; Meyer CF; Misale A; Sap JBI; Christensen KK; Willis MC; Trabanco AA; Gouverneur V Hydrosulfonylation of Alkenes with Sulfonyl Chlorides under Visible Light Activation. Angew. Chem., Int. Ed 2020, 59, 11620‒11626. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Chu X-Q; Ge D; Cui Y-Y; Shen Z-L; Li C -J. Desulfonylation via Radical Process: Recent Developments in Organic Synthesis. Chem. Rev 2021, 121, 12548–12680. [DOI] [PubMed] [Google Scholar]; (l) Nambo M; Maekawa Y; Crudden CM Desulfonylative Transformations of Sulfones by Transition-Metal Catalysis, Photocatalysis, and Organocatalysis. ACS Catal. 2022, 12, 3013–3032. [Google Scholar]
- (3).(a) Ilardi EA; Vitaku E; Njardarson JT Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem 2014, 57, 2832–2842. [DOI] [PubMed] [Google Scholar]; (b) Scott KA; Njardarson JT Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top. Curr. Chem 2018, 376, 5. [DOI] [PubMed] [Google Scholar]
- (4).(a) Kausar A; Zulfiqar S; Sarwar MI Recent Developments in Sulfur-Containing Polymers. Polym. Rev 2014, 54, 185–267. [Google Scholar]; (b) Bell WK; Rawlings BM; Long BK; Webb RC; Keitz BK; Haußling L; Willson CG J. Polym. Sci., Part A: Polym. Chem 2014, 52, 2769–2775. [Google Scholar]
- (5).For recent approaches to sulfones, see:; (a) Emmett EJ; Hayter BR; Willis MC Palladium‐Catalyzed Synthesis of Ammonium Sulfinates from Aryl Halides and a Sulfur Dioxide Surrogate: A Gas‐and Reductant‐Free Process. Angew. Chem. Int. Ed 2014, 53, 10204–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deeming AS; Russell CJ; Hennessy AJ; Willis MC DABSO-Based, Three-Component, One-Pot Sulfone Synthesis. Org. Lett 2014, 16, 150–153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Johnson MW; Bagley SW; Mankad NP; Bergman RG; Mascitti V; Toste FD Application of Fundamental Organometallic Chemistry to the Development of a Gold‐Catalyzed Synthesis of Sulfinate Derivatives. Angew. Chem., Int. Ed 2014, 53, 4404–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shavnya A; Coffey SB; Smith AC; Mascitti V Org. Lett 2013, 15, 6226–6229. [DOI] [PubMed] [Google Scholar]; (e) Margraf N; Manolikakes G One-Pot Synthesis of Aryl Sulfones from Organometallic Reagents and Iodonium Salts. J. Org. Chem 2015, 80, 2582–2600. [DOI] [PubMed] [Google Scholar]; (f) Shavnya A; Hesp KD; Mascitti V; Smith AC Palladium‐Catalyzed Synthesis of (Hetero) Aryl Alkyl Sulfones from (Hetero) Aryl Boronic Acids, Unactivated Alkyl Halides, and Potassium Metabisulfite. Angew. Chem., Int. Ed 2015, 54, 13571–13575. [DOI] [PubMed] [Google Scholar]; (g) Chen Y; Willis MC Copper (I)-Catalyzed Sulfonylative Suzuki–Miyaura Cross-Coupling. Chem. Sci 2017, 8, 3249–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gong X; Ding Y; Fan X; Wu J Synthesis of β‐Keto Sulfones via Coupling of Aryl/Alkyl Halides, Sulfur Dioxide and Silyl Enolates through Metal‐Free Photoinduced C–X Bond Dissociation. Adv. Synth. Catal 2017, 359, 2999–3004. [Google Scholar]; (i) Griffiths RJ; Chung Kong W; Richards SA; Burley GA; Willis MC; Talbot EPA Oxidative β-C–H Sulfonylation of Cyclic Amines. Chem. Sci 2018, 9, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) He J; Chen G; Zhang B; Li Y; Chen JR; Xiao WJ; Liu F; Li C Catalytic Decarboxylative Radical Sulfonylation. Chem 2020, 6, 1149–1159. [Google Scholar]; (k) Li Y; Chen S; Wang M; Jiang X Sodium Dithionite‐Mediated Decarboxylative Sulfonylation: Facile Access to Tertiary Sulfones. Angew. Chem., Int. Ed 2020, 59, 8907–8911. [DOI] [PubMed] [Google Scholar]; (l) Jin S; Haug GC; Trevino R; Nguyen VD; Arman HD; Larionov OV Photoinduced C(sp3)–H Sulfination Empowers a Direct and Chemoselective Introduction of the Sulfonyl Group. Chem. Sci 2021, 12, 13914–13921. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Sarver PJ; Bissonnette NB; MacMillan DWC Decatungstate-Catalyzed C(sp3)−H Sulfinylation: Rapid Access to Diverse Organosulfur Functionality. J. Am. Chem. Soc 2021, 143, 9737–9743. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Granados A; Cabrera-Afonso MJ; Escolano M; Badir SO; Molander GA Thianthrenium-enabled sulfonylation via electron donor-acceptor complex photoactivation. Chem Catal. 2022, 2, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Patani GA; LaVoie EJ Bioisosterism: A Rational Approach in Drug Design. Chem. Rev 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]; (b) Smith DA Metabolism, Pharmacokinetics and Toxicity of Functional Groups; Royal Society of Chemistry: London, United Kingdom, 2010, pp 99–167. [Google Scholar]
- (7).(a) Kautzky JA; Wang T; Evans RW; MacMillan DWC Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Till NA; Smith RT; MacMillan DWC Decarboxylative Hydroalkylation of Alkynes. J. Am. Chem. Soc 2018, 140, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sun X; Chen J; Ritter T Catalytic Dehydrogenative Decarboxyolefination of Carboxylic Acids. Nat. Chem 2018, 10, 1229–1233. [DOI] [PubMed] [Google Scholar]; (d) Cartwright KC; Tunge JA Decarboxylative Elimination of N-Acyl Amino Acids via Photoredox/Cobalt Dual Catalysis. ACS Catal. 2018, 8, 11801–11806. [Google Scholar]; (e) Cartwright KC; Lang SB; Tunge JA Photoinduced Kochi Decarboxylative Elimination for the Synthesis of Enamides and Enecarbamates from N-Acyl Amino Acids. J. Org. Chem 2019, 84, 2933–2940. [DOI] [PubMed] [Google Scholar]; (f) Faraggi TM; Li W; MacMillan DWC Decarboxylative Oxygenation via Photoredox Catalysis. Isr. J. Chem 2020, 60, 410–415. [Google Scholar]; (g) Li J; Huang CY; Han JT; Li C-J Development of a Quinolinium/Cobaloxime Dual Photocatalytic System for Oxidative C–C Cross-Couplings via H2 Release. ACS Catal. 2021, 11, 14148–14158. [Google Scholar]; (h) Li QY; Gockel SN; Lutovsky GA; DeGlopper KS; Baldwin NJ; Bundesmann MW; Tucker JW; Bagley SW; Yoon TP Decarboxylative Cross-Nucleophile Coupling via Ligand-to-Metal Charge Transfer Photoexcitation of Cu(II) Carboxylates. Nat. Chem 2022, 14, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review, see:; (i) Shang R; Liu L Transition metal-catalyzed decarboxylative cross-coupling reactions. Sci. China Chem 2011, 54, 1670–1687; [Google Scholar]; (j) Kitcatt DM; Nicolle S; Lee A-L Direct Decarboxylative Giese Reactions. Chem. Soc. Rev 2022, 51, 1415–1453. [DOI] [PubMed] [Google Scholar]
- (8).(a) Nguyen VT; Nguyen VD; Haug GC; Dang HT; Jin S; Li Z; Flores-Hansen C; Benavides B; Arman HD; Larionov OV Alkene Synthesis by Photocatalytic, Chemoenzymatically-Compatible Dehydrodecarboxylation of Carboxylic Acids and Bio-mass. ACS Catal. 2019, 9, 9485–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen VT; Nguyen VD; Haug GC; Vuong NT; Dang HT; Arman HD; Larionov OV Visible‐Light‐Enabled Direct Decarboxylative N‐Alkylation. Angew. Chem., Int. Ed 2020, 59, 7921–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dang HT; Haug GC; Nguyen VT; Vuong NTH; Arman HD; Larionov OV Acridine Photocatalysis: Insights into the Mechanism and Development of a Dual Catalytic Direct Decarboxylative Conjugate Addition. ACS Catal. 2020, 10, 11448–11457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nguyen VT; Haug GC; Nguyen VD; Vuong NTH; Arman HD; Larionov OV Photocatalytic Decarboxylative Amidosulfonation Enables Direct Transformation of Carboxylic Acids to Sulfonamides. Chem. Sci 2021, 12, 6429–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nguyen VT; Haug GC; Nguyen VD; Vuong NTH; Karki GB; Arman HD; Larionov OV Functional Group Divergence and the Structural Basis of Acridine Photocatalysis Revealed by Direct Decarboxysulfonylation. Chem. Sci 2022, 13, 4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Dmitriev IA; Levin VV; Dilman AD Boron Chelates Derived from N-Acylhydrazones as Radical Acceptors: Photocatalyzed Coupling of Hydrazones with Carboxylic Acids. Org. Lett 2021, 23, 8973–8977. [DOI] [PubMed] [Google Scholar]; (b) Zubkov MO; Kosobokov MD; Levin VV; Dilman AD Photocatalyzed Decarboxylative Thiolation of Carboxylic Acids Enabled by Fluorinated Disulfide. Org. Lett 2022, 24, 2354–2358. [DOI] [PubMed] [Google Scholar]; See also:; (c) Zubkov MO; Kosobokov MD; Levin VV; Kokorekin VA; Korlyukov AA; Hu J; Dilman AD A novel photoredox-active group for the generation of fluorinated radicals from difluorostyrenes. Chem. Sci 2020, 11, 737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sawyer DT; Sobkowiak A; Roberts JL Electrochemistry for Chemists, 2nd ed.; Wiley: New York, 1995; pp 125. [Google Scholar]
- (11).Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (12).Nguyen VD; Nguyen VT; Haug GC; Dang HT; Arman HD; Ermler WC; Larionov OV Rapid and Chemodivergent Synthesis of N-Heterocyclic Sulfones and Sulfides: Mechanistic and Computational Details of the Persulfate-Initiated Catalysis. ACS Catal. 2019, 9, 4015–4024. [Google Scholar]
- (13).Crossley SWM; Shenvi RA A Longitudinal Study of Alkaloid Synthesis Reveals Functional Group Interconversions as Bad Actors. Chem. Rev 2015, 115, 9465–9531. [DOI] [PubMed] [Google Scholar]
- (14).Inamdar SM; Shinde VS; Patil NT Enantioselective Cooperative Catalysis. Org. Biomol. Chem 2015, 13, 8116–8162. [DOI] [PubMed] [Google Scholar]
- (15).(a) Ma D; Cai Q Copper/Amino Acid Catalyzed Cross-Couplings of Aryl and Vinyl Halides with Nucleophiles. Acc. Chem. Res 2008, 41, 1450–1460. [DOI] [PubMed] [Google Scholar]; (b) Jones GO; Liu P; Houk KN; Buchwald SL Computational Explorations of Mechanisms and Ligand-Directed Selectivities of Copper-Catalyzed Ullmann-Type Reactions. J. Am. Chem. Soc 2010, 132, 6205–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Amal Joseph PJ; Priyadarshini S Copper-Mediated C–X Functionalization of Aryl Halides. Org. Process Res. Dev 2017, 21, 1889–1924. [Google Scholar]; (d) Bhunia S; Pawar GG; Kumar SV; Jiang Y; Ma D Selected copper-based reactions for C-N, C-O, C-S, and C-C bond formation. Angew. Chem., Int. Ed 2017, 56, 16136–16179. [DOI] [PubMed] [Google Scholar]; (e) Chen Z; Jiang Y; Zhang L; Guo Y; Ma. D. Oxalic Diamides and tert-Butoxide: Two Types of Ligands Enabling Practical Access to Alkyl Aryl Ethers via Cu-Catalyzed Coupling Reaction. J. Am. Chem. Soc 2019, 141, 3541–3549. [DOI] [PubMed] [Google Scholar]; See also:; Cabrera-Afonso MJ; Lu Z-P; Kelly CB; Lang SB; Dykstra R; Gutierrez O; Molander GA Engaging sulfinate salts via Ni/photoredox dual catalysis enables facile Csp2–SO2R coupling. Chem. Sci 2018, 9, 3186–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Woolven H; González-Rodríguez C; Marco I; Thompson AL; Willis MC DABCO-Bis(Sulfur Dioxide), DABSO, as a Convenient Source of Sulfur Dioxide for Organic Synthesis: Utility in Sulfonamide and Sulfamide Preparation. Org. Lett 2011, 13, 4876–4878. [DOI] [PubMed] [Google Scholar]
- (17).(a) Gant TG; Using deuterium in drug discovery: leaving the label in the drug. J. Med. Chem 2014, 57, 3595–3611; [DOI] [PubMed] [Google Scholar]; (b) Pirali T; Serafini M; Cargnin S; Genazzani AA Applications of Deuterium in Medicinal Chemistry. J. Med. Chem 2019, 62, 5276–5297. [DOI] [PubMed] [Google Scholar]
- (18).Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction‐Activation Strain Model. Angew. Chem., Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Horn PR; Sundstrom EJ; Baker TA; Head-Gordon M Unrestricted Absolutely Localized Molecular Orbitals for Energy Decomposition Analysis: Theory and Applications to Intermolecular Interactions Involving Radicals. J. Chem. Phys 2013, 138, 134119–134132. [DOI] [PubMed] [Google Scholar]; (b) Horn PR; Mao Y; Head-Gordon M Probing Non-Covalent Interactions with a Second-Generation Energy Decomposition Analysis Using Absolutely Localized Molecular Orbitals. Phys. Chem. Chem. Phys 2016, 18, 23067–23079. [DOI] [PubMed] [Google Scholar]
- (20).Khaliullin RZ; Bell AT; Head-Gordon M Analysis of Charge Transfer Effects in Molecular Complexes Based on Absolutely Localized Molecular Orbitals. J. Chem. Phys 2008, 128, 184112. [DOI] [PubMed] [Google Scholar]
- (21).Romanov AS; Becker CR; James CE; Di D; Credgington D; Linnolahti M; Bochmann M Copper and Gold Cyclic (Alkyl)(amino)carbene Complexes with Sub-Microsecond Photoemissions: Structure and Substituent Effects on Redox and Luminescent Properties. Chem. Eur. J 2017, 23, 4625–4637. [DOI] [PubMed] [Google Scholar]
- (22).The redox step was also highly kinetically unfavorable, with the barriers to the concerted and stepwise electron transfers of 74.8 and 95.7 kcal/mol.
- (23).For examples of trapping aryl radicals with TEMPO, see:; (a) Iwata Y; Tanaka Y; Kubosaki S; Morita T; Yoshimi Y Chem. Commun 2018, 54, 1257–1260; [DOI] [PubMed] [Google Scholar]; (b) Xu Z-J; Liu X-Y; Zhu M-Z; Xu Y-L; Yu Y; Xu H-R; Cheng A-X; Lou H-X Org. Lett 2021, 23, 9073–9077. [DOI] [PubMed] [Google Scholar]
- (24).Burés J Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem., Int. Ed 2016, 55, 16084–16087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.