

Abstract
Accurate somatic mutation detection from single-cell DNA sequencing (scDNA-seq) is challenging due to amplification-related artifacts. To reduce this artifact burden, an improved amplification technique, primary template-directed amplification (PTA), was recently introduced. We analyzed whole-genome sequencing data from 52 PTA-amplified single neurons using SCAN2, a new genotyper we developed to leverage mutation signatures and allele balance in identifying somatic single-nucleotide variants (SNVs) and small insertions and deletions (indels) in PTA data. Our analysis confirms an increase in non-clonal somatic mutation in single neurons with age, but revises the estimated rate of this accumulation to be 16 SNVs per year. We also identify artifacts in other amplification methods. Most importantly, we show that somatic indels increase by at least 3 indels per year per neuron and are enriched in functional regions of the genome such as enhancers and promoters. Our data suggest that indels in gene regulatory elements have a significant effect on genome integrity in human neurons.
Introduction
Although somatic mutation has been studied extensively in cancer, investigation into the abundance, patterns, and effects of somatic mosaicism in non-neoplastic tissues has only recently begun1–6. Unlike tumor tissue in which somatic mutations of interest are shared by large clones, somatic mutations in normal tissues are typically shared by relatively few cells and are hence difficult to detect. Recent studies have circumvented the technical difficulty of detecting rare somatic mutations by ultradeep sequencing of very small tissue samples3,7, exploiting naturally occurring genetically homogenous clones8, or clonal expansion of cells in vitro5,9,10.
Another strategy for detecting somatic mosaic mutations is to directly sequence DNA from a single cell. Single-cell DNA sequencing (scDNA-seq) is capable of detecting the rarest somatic mutations (i.e., mutations private to a single cell) and can also provide information about cell lineage through shared somatic mutations2,11. This strategy is especially useful for examining post-mitotic cells such as neurons. A major challenge, however, is the difficulty of amplifying the genome of a single cell accurately and evenly prior to sequencing. For example, multiple displacement amplification (MDA)12, a popular amplification method for detecting point mutations, produces non-uniformity across the genome13 and often amplifies homologous alleles of diploid cells at different rates, leading to allelic imbalance14. These amplification artifacts pose substantial challenges for identifying mutations from short-read sequencing data—especially mutations that are non-clonal and thus cannot be confirmed in other cells. We previously used LiRA15, a read-level phasing strategy, to filter artifacts in MDA samples and discovered an age-associated increase in somatic mutations in human neurons6; however, this approach was limited to analyzing mutations within a few hundred base pairs of single nucleotide polymorphisms (SNPs), making it adequate for estimating the overall mutational spectrum and burden in a sample, but not for other analyses. Another method, SCAN-SNV14, could find SNVs over more of the genome by estimating local allelic imbalance, but it was optimized for MDA-amplified data.
A new single-cell amplification method called primary template-directed amplification (PTA) reduces amplification-associated artifacts by dampening the exponential nature of isothermal MDA16. Indeed, our comparison below of single neurons amplified by both the MDA and PTA protocols from the prefrontal cortices of the same individuals shows that PTA substantially improves upon MDA. Despite PTA’s improvements, the resulting data still require specialized single-cell mutation calling, as conventional bulk-oriented somatic SNV (sSNV) analysis based on the Genome Analysis Toolkit’s (GATK) best practices can yield an order of magnitude more false positives (FPs) than there are mutations in some non-neoplastic cells (~0.9 FPs per megabase10). We therefore developed SCAN2 (Single Cell ANalysis 2), a genotyper that augments the SCAN-SNV model of allelic imbalance with a novel mutation signature17 approach to increase sSNV detection sensitivity. Furthermore, SCAN2 enables analysis of somatic indels from single-cell DNA sequencing data for the first time. Applied to PTA data, SCAN2 detects somatic SNVs in scDNA-seq data with ~60-fold fewer FPs per megabase than conventional GATK calling and >5-fold fewer FPs than other single-cell SNV genotypers. Importantly, unlike phylogenetic or population genetics-based genotypers18,19, SCAN2 is not fundamentally limited to detecting shared mutations and can thus recover non-clonal, private mutations such as those that occur in post-mitotic cells. Using SCAN2 and PTA, we produced a catalog of 20,090 somatic SNVs and 2,714 somatic indels from 52 healthy human neurons. Our catalog confirms a previously discovered age-related SNV signature6 (with a slightly revised rate of accumulation) and reveals an enrichment of somatic mutations—particularly indels—in transcribed genes and brain-specific regulatory elements.
Results
PTA improves amplification quality and reduces artifacts
Using PTA, we amplified the genomes of 52 single neurons from the prefrontal cortex (PFC) of 12 neurotypical individuals and sequenced to 30–60X, including 15 neurons from 5 neurotypical individuals from another study20 (Figure 1a, Supplementary Table 1). 75 single neurons from 11 of the 17 individuals were previously amplified by MDA6, providing a direct comparison between the two protocols. Despite being sequenced to lower depth, PTA-amplified neurons showed several favorable characteristics compared to MDA-amplified cells, including substantial reduction in coverage variability and allelic dropout across the genome (Figure 1b–d). Regions of allelic imbalance were generally not reproduced between PTA amplifications with the exception of neurons from a single subject (4638) (Extended Data Figure 1). Surprisingly, large-scale somatic copy number mutations (>5 Mb, Methods) were detected in only two of the 52 PTA neurons (Supplementary Note and Supplementary Figure 1), in contrast to the previous reports of pervasive copy number alterations in human neurons, especially in young individuals21,22.
Figure 1. Improved large-scale amplification characteristics of PTA compared to MDA.

a. Study design. Single neurons were collected from the prefrontal cortex (PFC) of brains of 17 individuals ranging in age from infantile to elderly. Single neurons were amplified by either PTA or MDA and then sequenced to high coverage. Created with BioRender.com. b. Representative copy number profiles for bulk (top), MDA-amplified (middle) and PTA-amplified (bottom) genomes. c. MAPD (median absolute pairwise difference) for MDA-amplified and PTA-amplified neuronal genomes from the same individuals; lower values indicate better performance. The average MAPDs of MDA (0.75) and PTA (0.21) correspond to an average fluctuation in read depth between neighboring 50 kb windows of 68% and 14%, respectively. Boxplot whiskers, the furthest outlier <=1.5 times the interquartile range from the box; box, 25th and 75th percentiles; centre bar, median. n=17 bulk samples, n=52 PTA neurons, n=75 MDA neurons. d. Allele balance for germline heterozygous SNPs measures the evenness of amplification between homologous alleles in a diploid cell. Each line corresponds to one single cell or bulk sample. Values near 0.5 indicate balanced amplification of homologous alleles; values near 0 or 1 indicate complete dropout of one allele. On average, 71% of each PTA genome was balanced (allele balance between 0.3–0.7) compared to only 39% of each MDA genome.
Amplification also creates artifactual SNVs (on the order of 10,000 per MDA amplification23) and indels, typically by spontaneous DNA damage or polymerase errors. The majority of artifacts occur late in the amplification reaction and, as a result, are not present on all sequencing reads derived from one haplotype. This leads to improper read phasing with nearby SNPs and inconsistent variant allele fractions (VAFs), enabling genotypers such as LiRA and SCAN-SNV to filter the majority of late artifacts. Early artifacts, especially those that occur prior to amplification (e.g., during cell lysis), can be more difficult to identify since they are present on a larger fraction of reads. The most severe case, which we previously described15 and refer to as single-strand dropout (SSD), occurs when no sequencing reads from the pre-artifact haplotype are present.
Haploid male X chromosomes provide an opportunity to measure the rate of SSD artifacts: because both true mutations and SSD artifacts should have near-100% VAF, a systematic excess of near-100% VAF putative mutations in MDA compared to PTA neurons from the same individual would imply presence of SSD artifacts. Using a simple genotyping approach (Methods), we found a median excess of 15 somatic SNVs and 3.7 somatic indels in MDA X chromosomes (Figure 2a–b, Supplementary Figure 2), corresponding to about 550 SNV and 136 indel SSD artifacts per MDA-amplified genome.
Figure 2. PTA identifies MDA-induced artifacts.
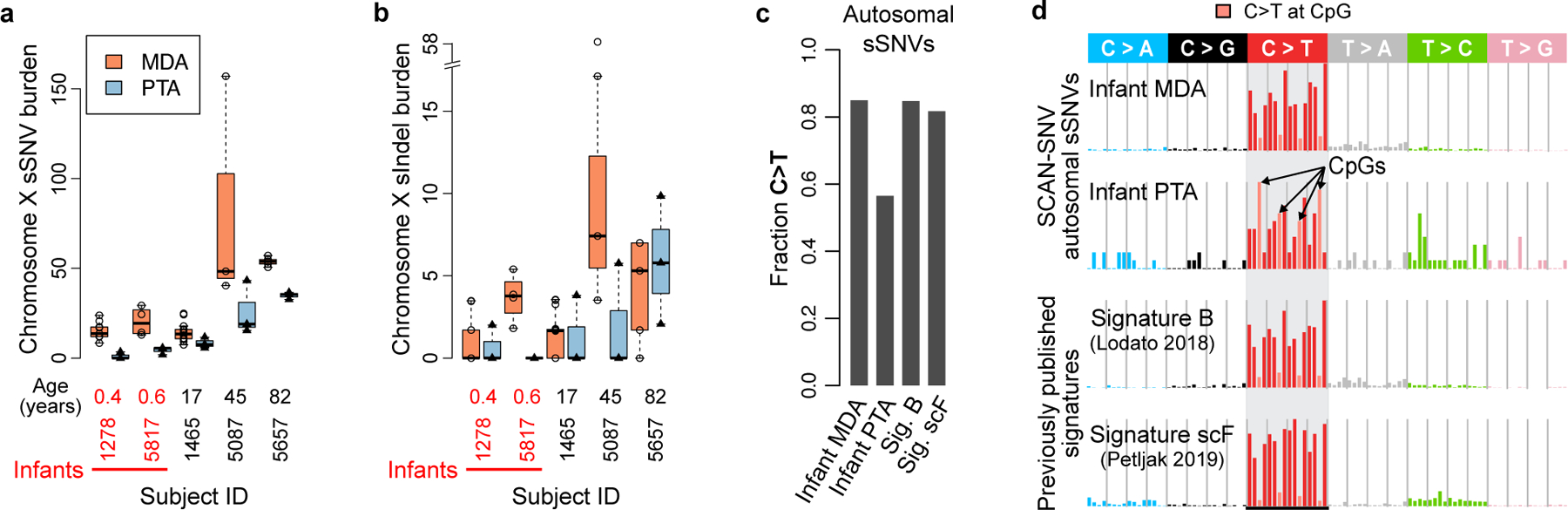
a-b. Sensitivity-adjusted somatic SNV (sSNV) (a) and indel (b) burdens per X chromosome for 5 male subjects with both MDA and PTA-amplified neurons. Boxplot whiskers, the furthest outlier <=1.5 times the interquartile range from the box; box, 25th and 75th percentiles; centre bar, median; n=16 PTA neurons and n=39 MDA neurons. c. Fraction of C>Ts among SCAN-SNV sSNV calls in infant neurons and two previously published signatures. d. Mutation spectra of SCAN-SNV sSNVs across 13 MDA infant neurons, 6 PTA infant neurons, the C>T rich Signature B reported in Lodato et al, 2018 and the MDA artifact Signature scF reported by Petljak et al, 2019. Light red bars denote C>Ts that occur at CpG sites. Boxplot whiskers, the furthest outlier <=1.5 times the interquartile range from the box; box, 25th and 75th percentiles; centre bar, median.
Analysis of autosomes, which includes both SSD and other MDA arifacts, identified a C>T-dominated MDA artifact signature. We focused on infant neurons, which contain the fewest age-related mutations and thus are expected to contain the highest proportion of MDA artifacts. sSNVs from MDA-amplified infant neurons were ~10-fold more abundant (282 vs. 26 sSNVs per neuron) compared to those from PTA-amplified neurons despite similar detection sensitivity, enriched for C>T mutations (85% vs. 59%; Figure 2c) and resembled two signatures previously reported to be associated with technical artifacts (Signature B6 and Signature scF24; Figure 2d). Analysis by LiRA produced similar patterns (cosine similarity 0.988). Although PTA sSNVs were also primarily C>T, preference for CpG contexts (similar to COSMIC SBS1) suggests true somatic mutations acquisition during developmental mitoses rather than an artifactual origin. Nevertheless, raw mutation counts indicate a much lower burden of SNV artifacts compared to MDA.
SCAN2: detecting somatic SNVs and indels in PTA single cells
SCAN2 builds upon SCAN-SNV, a single-cell somatic SNV genotyper that creates a genome-wide, position-specific model of allelic amplification imbalance by integrating local allele balance information indicated by the VAFs of heterozygous germline SNPs. Inspired by the characteristic mutation signature of SNV artifacts in MDA, SCAN2 incorporates signature analysis as a novel source of information to further identify mutations that could not otherwise be confidently distinguished from artifacts by VAF alone. The approach operates in two passes (Figure 3a, Methods). First, the signature of true mutations is learned by “VAF-based” calling (which determines if candidate mutation VAFs are consistent with local estimates of allele imbalance) with stringent calling thresholds. If individual cells do not provide a sufficient number of mutations to estimate the true signature, several single cells subject to the same mutational processes (SCAN2 provides a test of this assumption, see Methods) can be combined. Second, the newly learned true mutation spectrum is compared against a universal PTA artifact signature that we have identified (Supplementary Note and Supplementary Figure 3) and candidate mutations rejected in the first pass may be rescued if they are unlikely to have originated from the artifact signature (Figure 3b; see Supplementary Figure 4 for examples of signatured-based artifact likelihood estimation).
Figure 3. SCAN2 mutation signature-based calling approach for somatic SNVs and indels.
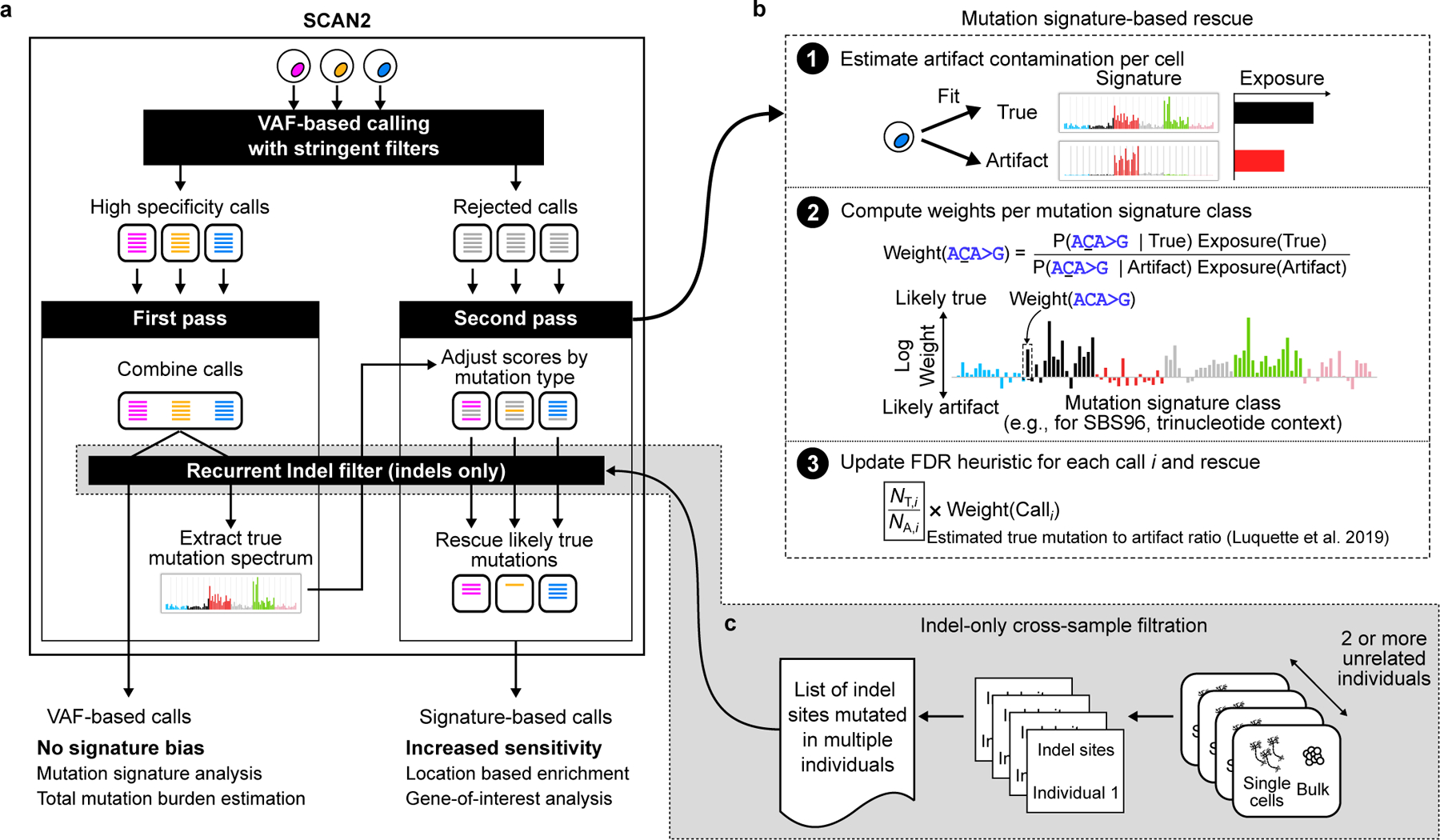
Overview of SCAN2 workflow using somatic SNV spectra for demonstration; 83-channel indel spectra are used for somatic indel analysis. a. SCAN2’s two-pass mutation signature-based calling, in which mutation signatures from high-specificity calls are used to rescue likely true mutations from the rejected call set. Mutations may be combined across cells exposed to the same mutation processes to increase the number of VAF-based calls used in extracting the true mutation signature. This may not be necessary for cells with very high mutation burden. b. Candidate somatic mutations are rescored separately for each single cell given the true mutation signature learned in panel (b). The likelihood of being generated by the true signature is computed for each mutation class (96-dimensional “SBS96” for SNVs and 83-dimensional “ID83” for indels). This likelihood acts as a prior for a previously described heuristic that estimates the number of true mutations (NT,i) and artifacts (NA,i) with characteristics similar to mutation candidate i. c. For indel calling only, recurrent artifacts are further removed by a cross-sample list of sites where indels are observed across cells from multiple unrelated individuals.
SCAN2 performance was assessed using both simulated data (synthetic diploid X chromosomes; see Methods) and a kindred single-cell system. Varying mutation burden levels were used in both assessments since it can strongly influence FDR. For high mutation burdens (e.g., germline variant detection), a genotyper’s FDR may appear low since true variants (annotated SNPs and individual-specific variants in germline analysis) greatly outnumber artifacts; however, the same genotyper may produce unacceptable FDRs when artifacts outnumber mutations, as is the case at the low mutation burdens typical of healthy human cells (e.g., 0.1–1.0 sSNVs/Mb5,6,9,10, Supplementary Note and Supplementary Figure 5).
On simulated sSNVs, SCAN2 outperformed SCAN-SNV by increasing sensitivity by ~82% (46% vs. 25%) while maintaining similar FDR (8.6% vs. 9.5%) (Extended Data Figure 2a,b). SCAN2 also outperformed two other single-cell SNV genotypers (Monovar18 and SCcaller23) by several-fold reduction of FDR (Extended Data Figure 2c,d). For SCAN2’s signature-based rescue, near-maximal performance was achieved when 500–1000 mutations were available for learning the mutation signature of true sSNVs (Extended Data Figure 2e,f) and SCAN2’s increased sensitivity was maintained for sSNV simulations using various COSMIC signatures (range of cosine similarity to the PTA SNV artifact signature: 0.06–0.871, Extended Data Figure 2g–i).
Kindred single cell systems further confirmed SCAN2’s low FDR. In typical kindred cell analyses, somatic mutations called in one kindred cell are validated if they are also present in other kindred cells or bulk sequencing of the kindred clone. However, some true somatic mutations are private and would not validate by this approach, resulting in an overestimated FDR. We therefore used crossbred mouse embryonic stem cell (mESC) lines, which have greatly increased SNP density (~10-fold greater than human SNP rates), to enable LiRA analysis across more of the genome and provide an alternative mutation validation metric. Two mESC clones were created and one was treated with aristolochic acid I (AAI) to induce a high burden of sSNVs with a known signature (SBS22) (Methods). We sequenced four PTA-amplified single cells and one clonal bulk from each clone for performance assessment. On the untreated clone, SCAN2 recovered 23% more sSNVs than SCAN-SNV (32% vs. 26%) with FDR between 9%−32% depending on how false positives were defined (Methods, Extended Data Figure 3a,b). SCAN2 recovered 28% more sSNVs than SCAN-SNV on the AAI-treated clone (52% vs. 41%) and clearly recovered the aristolochic acid signature (Extended Data Figure 3c). On AAI-treated cells, both SCAN2 and SCAN-SNV achieved FDR ≈ 1%, which is expected due to the high sSNV rate induced by AAI.
A major advance in SCAN2 is the ability to identify somatic indels from scDNA-seq data. Indel detection uses a modified sSNV pipeline, offering both VAF-based and signature-based calling, but depends on an additional filter to remove recurrent indel artifacts (Figure 3c). While it is rare for a particular sSNV artifact to occur twice in the same amplification, processes that generate artifactual indels (e.g., polymerase stutter25 and microhomology-mediated chimera formation26) occur more frequently in certain genomic regions and can therefore recur, leading to inflated artifact VAFs. This effect can be further exacerbated by ambiguities that cause different indels to look alike (e.g., in a homopolymer such as AAAAA, a single base deletion of any of the five As would appear identically in sequencing data). To remove these recurrent indel artifacts, SCAN2 builds a list of sites which contain indel-supporting reads in single cells from multiple individuals; somatic indel candidates overlapping these sites are rejected.
We first adapted other methods to detect indels in simulated data, but found impractically high error rates: naïve application of SCAN-SNV to indels yielded 19.9% sensitivity but 61%−85% of calls were false positives; GATK HaplotypeCaller with Variant Quality Score Recalibration (using criteria similar to SCAN2 to remove germline indels, see Methods) recovered 57% of indels but with >99% FDR. Even when adding SCAN2’s recurrent indel filter to GATK HaplotypeCaller, the FDR remains high at 54%−90%. Only SCAN2 was able to achieve high specificity: 33.6% (16.9% using only VAF-based calls) of spike-in indels were recovered with mean FDR <2% Extended Data Figure 4a–c). In contrast to sSNVs, indel properties such as their length often affect detection sensitivity; indeed, we found reduced sensitivity for indels in homopolymers and tandem repeats of >4 units (Extended Data Figure 4d,e).
The effects of various SCAN2 filtering steps on sSNV and indel calling are provided in Supplementary Figure 6.
Nonclonal somatic SNV accumulation in aging human neurons
SCAN2 is also able to predict the genome-wide somatic mutation burden per cell by adjusting for somatic detection sensitivity and the fraction of the genome accessible to analysis (Methods). SCAN2 accurately predicted the number of spike-in mutations in the simulated datasets used in our performance assessment (Supplementary Figure 7). By fitting a linear model to SCAN2 somatic SNV burden estimates from the 52 PTA-amplified neurons, we estimate that 16.5 sSNVs accumulate per year in the autosomes of human neurons (Figure 4a). LiRA, which predicts genome-wide mutations burdens using a smaller set of very high confidence sSNVs, predicted a similar rate of 17 sSNVs per year, helping to validate SCAN2’s approach (Extended Data Figure 5). De novo signature analysis of VAF-based sSNVs from PTA-amplified neurons confirmed Signature A, an aging-associated signature we previously recovered from MDA-amplified neurons6 (Supplementary Figure 8). Notably, no signature resembling Signature B was extracted from de novo analysis of our PTA sSNVs. Importantly, our filters, which require sSNVs be undetectable in matched bulk, remove most clonal somatic mutations that occur during nervous system development. Thus, the intercept of our aging trend underestimates the somatic mutation burden at birth.
Figure 4. SCAN2 VAF-based somatic SNVs and indels in aging human neurons.

a. Genome-wide extrapolated accumulation rate of somatic SNVs in PTA- (triangles) and MDA- (circles) amplified single human neurons. Colors represent 17 individuals. b. Genome-wide extrapolated rate of somatic indel accumulation. c. Age-related increase of somatic insertions and deletions called from PTA neurons; raw counts are reported, not sensitivity-adjusted genome-wide rates. d. Distribution of somatic indel lengths from PTA neurons. e. Raw mutation spectrum of somatic indels. f. Exposures to COSMIC ID signatures calculated by least squares fitting. Exposures were corrected by normalizing indel counts by ID83 channel-specific sensitivity (Extended Data Figure 4f) before fitting. g. Association of ID4, a signature of unknown aetiology, with neuron age; P-value: two-sided t-test for correlation=0. Trend lines in a-c and g: mixed effects linear regressions to account for multiple points being derived from the same individual.
We previously estimated a yearly increase of ~23 sSNVs per year in a larger cohort of MDA neurons using LiRA6. Using the 74 MDA neurons in this study, SCAN2 estimated 31 sSNVs per year in MDA neurons. De novo signature extraction recovered both Signatures A and B from the combined set of MDA and PTA sSNVs. We hypothesized that if the difference in MDA and PTA accumulation rates were due to Signature B-like MDA artifacts, then its removal from MDA neurons should result in sSNV accumulation rates more consistent with PTA neurons. Indeed, after subtracting the Signature B-like exposure from MDA neurons, SCAN2’s yearly accumulation rate estimate decreased from 31 sSNVs/year to 22 sSNVs/year and removal of a strong elderly outlier (subject 5219) further decreased the rate to 19 sSNVs/year, more closely matching that of PTA neurons (Supplementary Note and Supplementary Figure 9). Taken together, these observations provide compelling evidence that sSNVs accumulate in human neurons at a rate closer to 16 sSNVs/year with a Signature A-like pattern and further confirms that MDA artifacts can be largely attributed to Signature B.
Characteristics of somatic indels in single human neurons
SCAN2 identified 1,541 indels from the 52 PTA-amplified neuronal genomes using VAF-based calling. Somatic indels increased with age by ~3 somatic indels per neuron per year (Methods, Figure 4b), which is similar to rates observed in several mitotically active cell types8–10,27. However, our rate likely represents a lower bound on indel accumulation owing to lower sensitivity for indels of varying length and repeat content. Deletions accumulated 3.3-fold faster than insertions (Figure 4c) and indel sizes ranged from −29 bp to +17 bp (Figure 4d). As was the case for sSNVs, MDA yielded a higher accumulation rate estimate of 6.0 somatic indels/year and we again attribute this to MDA artifacts (Supplementary Figure 10a). 7 out of 75 MDA neurons contained an exceptionally high number of indel calls characterized by single base insertions in homopolymers of length 3 or greater (Supplementary Figure 10b–e). Due to the added artifacts, MDA indels were not included in subsequent analyses.
De novo mutation signature extraction yielded only a single spectrum (Figure 4e) that was broadly similar to spectra from dividing cells9,10,27 but with a greater burden of deletions (Extended Data Figure 6a–e). Fitting the aggregate indel spectrum to the COSMIC indel catalog produced 6 indel signatures with >5% contribution; however, the COSMIC catalog is relatively new and may not contain the ID signatures relevant to neurons. Two of the four ID signatures described as clock-like, ID5 and ID8 (Figure 4f, Extended Data Figure 6f), were detected. The absence of the two other clock-like signatures, ID1 and ID2, is consistent with the proposed etiology involving DNA replication, which cannot be active in post-mitotic neurons. However, our analysis of indel sensitivity on simulated data indicated that lack of ID1 and ID2 could also be explained by low sensitivity that uniquely impacts these signatures (Extended Data Figure 4f). The most prevalent signature was ID4, a deletion-rich signature observed in several cancer types but with unknown mechanism. Surprisingly, ID4 is more strongly correlated with age in neurons than the clock-like signatures ID5 and ID8 (Figure 4g, Extended Data Figure 6g,h); correlation with age = 0.82, 0.42 and 0.69, for ID4, ID5 and ID8, respectively). ID3 was recently detected in normal bronchial epithelium27, especially in smokers, and also shows correlation with age in neurons (correlation = 0.60). The remainder of the detected signatures (ID9 and ID11) contribute similar numbers of mutations as ID3 but are less well-correlated with age.
Neuronal SNVs and indels are enriched in regulatory elements
The increased sensitivity of SCAN2’s mutation signature-based approach is particularly advantageous when quantifying somatic mutation enrichment in genomic regions of interest. Using mutation signatures, SCAN2 recovered approximately 36% more somatic SNVs (20,090 vs. 14,748) and 76% more somatic indels (2,714 vs. 1,541) from PTA neurons compared to VAF-based calls. Only a handful of neurons showed evidence of deviation from the batch-wide sSNV and indel signatures (P < 0.05 for 3/52 and 2/52 neurons for SNV and indel signatures, respectively; statistical test described in Supplementary Note and Supplementary Figure 11). To estimate enrichment levels in genomic regions, background mutation rates were determined by randomly permuting somatic mutations across regions of genome accessible to SCAN2 (Methods, Extended Data Figure 7).
Spurred by reports of transcriptional strand bias in neuronal SNVs (particularly T>C mutations in ATN trinucleotide contexts2,6), we first compared neuronal somatic mutation density to gene expression levels from 54 tissues in GTEx (Methods). In genic regions, there was a significant positive relationship between mutation burden (both sSNV and indel) and gene expression specifically for brain tissues (Figure 5a,b), with the most expressed decile containing a ~15% increase in sSNV and a 50–100% increase in indel mutation density. Among genic mutations, there were more than twice as many high impact (determined by SnpEff28) somatic indels than sSNVs, despite sSNVs outnumbering indels 8:1 (Figure 5c). Indels were also strongly enriched in the 10% of the genome with the highest evolutionary conservation, with an overrepresentation of 42% (Figure 5d).
Figure 5. Enrichment of neuronal mutations in functionally active genomic regions with tissue- and cell-type specificity.
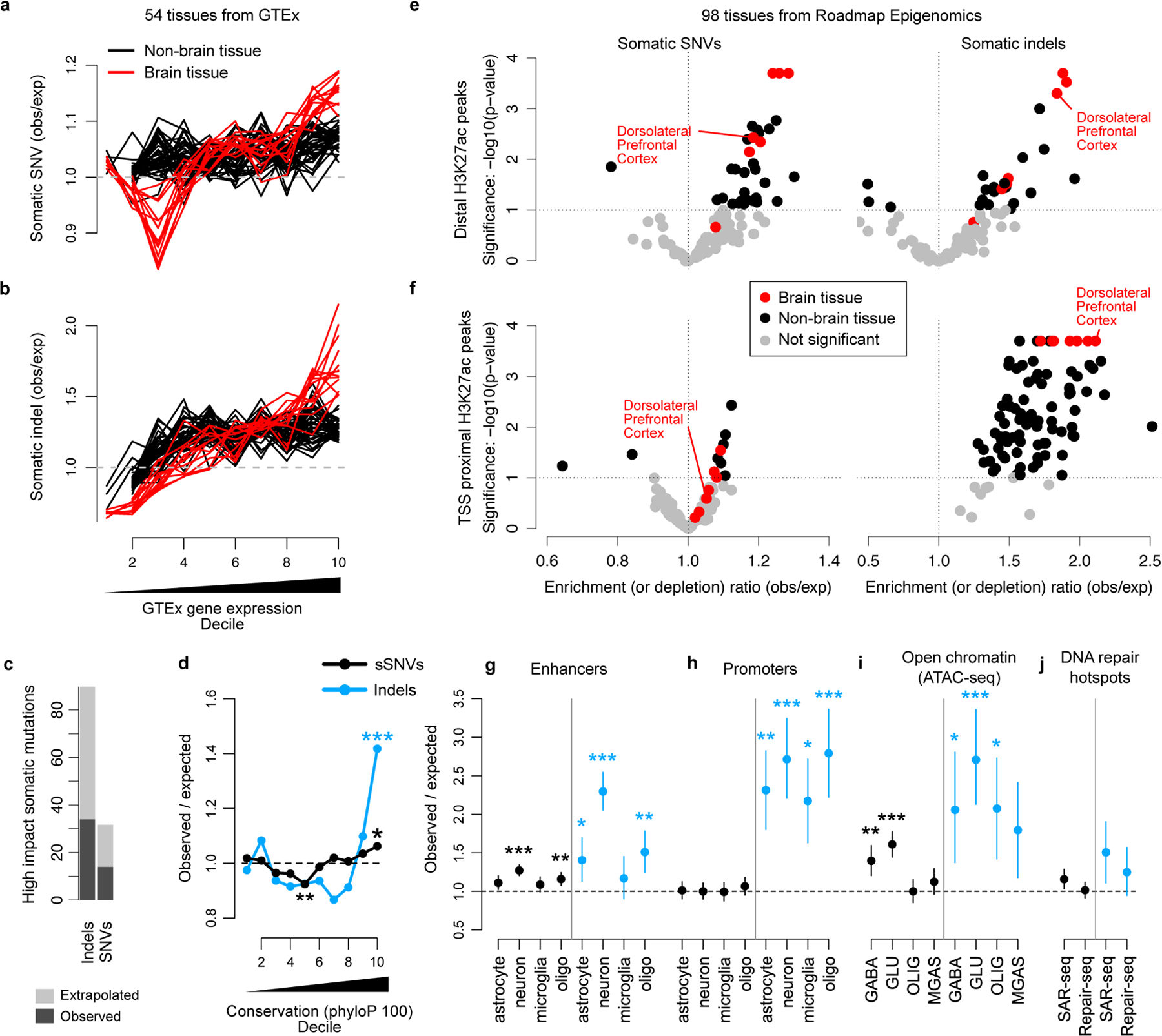
a-b. sSNV (a) and somatic indel (b) enrichment compared to local gene expression levels measured by GTEx. Each line corresponds to one GTEx tissue type; tissues from primary brain specimens are always shown in red. c. The number of high impact (classified HIGH by SnpEff; includes several severely protein altering effects such as stop gains, stop losses and frameshifts) sSNVs and somatic indels detected by SCAN2’s signature-based approach (dark grey) and extrapolation to autosomes (light grey). d. Mutation enrichment compared to local sequence conservation. e-f. Enrichment analysis of neuronal mutations in H3K27ac peaks from 98 Roadmap Epigenomics tissues. H3K27ac peaks are classified according to whether they are within 2 kb of an H3K4me3 peak in the same tissue (f, TSS proximal) or not (e, distal). Distal peaks are interpreted as intergenic enhancers. g-j. Mutation enrichment analysis of several datasets. Dorsolateral prefrontal cortex is shown since it most closely matches the neurons sequenced in this study. Cell-type specific enhancers (g) and promoters (h) from Nott et al. 2019; cell-type specific open chromatin regions (OCRs) measured by ATAC-seq from Hauberg et al. 2020 (i); DNA repair hotspots measured in induced human neurons (j) reported by Wu et al. 2021 (SAR-seq) and Reid et al. 2021 (Repair-seq). GABA, GABAergic neurons; GLU, glutamatergic neurons; OLIG, oligodendrocytes; MGAS, microglia and astrocytes. Error bars (g-j): 95% bootstrapping C.I. with n=104 bootstrap samplings; centre point: observed mutation count divided by the mean mutation count over bootstrap samplings. * - P < 0.01, ** - P < 0.001, *** P < 0.0001 by two-sided permutation test (Methods) without multiple hypothesis correction.
The large number of somatic SNVs and indels identified using PTA and SCAN2 allow the analysis of both mutation types in relation to promoters29 and promoter-distal enhancers30, which have been recently reported to show elevated levels of DNA damage, DNA repair, and double-stranded breaks in neurons29–31. Enhancers and promoters were defined using H3K27ac and H3K4me3 ChIP-seq peaks from the Roadmap Epigenomics Project (98 tissues and cell lines32; Methods). A significant enrichment in transcription start site (TSS)-distal enhancers was detected for both SNVs (~30% increase, ~1.3 observed/expected) and indels (~80% increase, ~1.8 observed/expected), and, critically, the most significant enrichments were seen in primary brain tissue (Figure 5e). Near active TSSs, only somatic indels showed evidence of enrichment and it was not tissue specific (Figure 5f). Chromatin states32—which offer alternative definitions of promoters and enhancers based on a combination of chromatin marks from ChIP-seq signals—showed similar patterns, with indel enrichment in active TSSes (ChromHMM annotation: 1_Tss) and non-genic enhancers (7_Enh; Extended Data Figure 8). In agreement with our GTEx analysis, chromatin state analysis also revealed enrichment for SNVs and indels in weakly transcribed regions (5_TxWk), a state which often covers the bodies of transcribed genes. Strong depletion was observed for indels in inactive chromatin states such as heterochromatin (9_Het) and Polycomb repressed regions (14_ReprPcWk), while minor depletions were found for sSNVs in heterochromatin.
Remarkably, both sSNVs and indels showed highly significant (sSNVs and indels: P < 10−4) enrichment in neuronal enhancers (Figure 5g) but reduced or marginal significance in enhancers active in non-neuronal cell types (sSNVs: P = 0.0005, 0.017, 0.071; indels: P = 0.0009, 0.007, 0.255 for oligodendrocytes, astrocytes and microglia, respectively). Promoter and enhancer elements active in several brain-specific cell types were obtained from a study of FACS-purified neurons, microglia, oligodendrocytes and astrocytes33. Mutation enrichment levels in these cell type-specific regulatory elements were similar to those estimated from H3K27ac peak analysis, with SNVs and indels increased by 27% and 129%, respectively. Consistent with Roadmap Epigenomics data, indels but not SNVs were enriched in promoters and did not show a preference for cell type (Figure 5h).
Analysis of open chromatin regions (OCRs) derived from ATAC-seq of flow-sorted GABAergic and glutamatergic neurons, oligodendrocytes, microglia and astrocytes34 provided further evidence of preferential mutation accumulation in regulatory elements. sSNVs were strongly enriched in neuron-specific OCRs while indel enrichment was strong but less tissue specific (Figure 5i).
Finally, we measured enrichment of mutations in the DNA repair hotspots recently reported by Wu et al. and Reid et al. (refs. 30 and 29). Enhancer-associated hotspots30 were enriched for somatic indels (51% increase; 95% CI [11%, 90%], P = 0.02) but no enrichment was found in promoter-associated hotspots29 (Figure 5j). sSNVs were also enriched in enhancers but with marginal significance. Notably, all enrichments presented in Figure 5 remained robust when reanalyzed with higher minimum sequencing depth requirements, providing further evidence that local differences in sensitivity do not explain our observations (Extended Data Figure 9).
Discussion
Our analyses of PTA-generated single-neuron genome sequencing represent a major advance in single-cell DNA-sequencing technology and provide insight into the mutagenic processes of long-lived human neurons. Direct comparison of PTA- and MDA-amplified neurons from the same brain sample identified MDA artifacts, confirmed the signature of age-related somatic SNVs and refined the estimated yearly accumulation rate of sSNVs in post-mitotic human neurons. Further, SCAN2 analysis of 52 PTA neurons provided mutation density profiles that, when compared against a variety of data modalities (gene expression, ChIP-seq, ATAC-seq, evolutionary conservation and coding sequence impact), provided consistent signals of mutation enrichment in functional regions of the genome. Most strikingly, the increased enrichment level of indels in brain-specific regulatory regions suggests that somatic indels may interfere with neuronal regulatory programs. For example, DNA breaks in the promoters of early-response genes triggered by neuronal activity31,35 may be responsible for some of these indels and, if true, the associated indels may be especially deleterious.
Both PTA and SCAN2 were pivotal in enabling these findings. While PTA itself is a significant improvement over MDA, genotypers tuned for low mutation burdens remain critical for analysis of healthy cells. SCAN2’s key advantages over other tools are the ability to detect somatic indels and its use of multi-sample information (e.g., mutational signatures) to enhance sensitivity for non-shared sSNVs and indels genome-wide. Indeed, compared to LiRA and SCAN-SNV in this cohort of 52 PTA neurons, SCAN2 recovered 533% and 36% more sSNVs, respectively, and is the only tool designed to detect indels. For optimal SCAN2 performance, cells combined for mutation signature-based rescue should be subject to the same mutational processes (SCAN2 provides a statistical test to help discover strong violations of this assumption) and, for some analyses (e.g., de novo mutation signature extraction or fitting), it may be more appropriate to use SCAN2’s VAF-based calls to avoid signature-related biases. When analyzing somatic mutation density in small genomic regions (e.g., within promoter or enhancer regions), we recommend correcting for local differences in nucleotide content to account for signature-related biases in the SCAN2 calls, as done in this study using permutations.
The rates and signatures of SNV and indel mutations we report are in line with results from two recent studies using orthogonal technologies. META-CS, a single-cell amplification technique that tags Watson and Crick strands, reported an increase of ~16 sSNVs per year in neurons36. NanoSeq, a single molecule consensus sequencing method for bulk DNA, estimated 17.1 sSNVs and 2.5 indels per year37. Our study additionally provides unprecedented power to analyze the distribution of somatic mutations in human neurons by detecting >6-fold more sSNVs than the META-CS study (~20,000 vs. ~3,000) and ~4-fold more sSNVs and indels than the NanoSeq study (~20,000 sSNVs and ~2,700 indels vs. ~5,000 sSNVs and 600 indels for this study and NanoSeq, respectively). Furthermore, the majority of the human genome is accessible to PTA while other technologies can be more limited (restriction enzyme-based NanoSeq is limited to ~29% of the genome37). This difference in genome coverage may explain discrepancies in findings: for example, the NanoSeq study only found an association between indel burden—not sSNV burden—and transcription levels and found a weak sSNV enrichment rather than depletion in heterochromatic regions.
Our study establishes a methodology for somatic mutation detection from scDNA-seq of PTA amplified whole genomes. In particular, our approach can analyze genomes with low mutation burden and in cases where somatic mutations may not be shared by multiple cells. We anticipate that our methodology will enable a wide range of studies, including somatic mutation analysis of neurons from individuals with neurodegenerative diseases, further characterization of mutations caused by exposures to mutagenic compounds, and measuring the efficiency and accuracy of CRISPR editing at the single cell level.
Methods
Human tissue, case selection and ethical approval
Postmortem frozen human tissues were obtained from the NIH Neurobiobank at the University of Maryland Brain and Tissue Bank (UMBTB). Tissue collection and distribution for research and publication was conducted according to protocols approved by the University of Maryland Institutional Review Board (for UMBTB: 00042077), and after provision of written authorization and informed consent. Research on these de-identified specimens and data was performed at Boston Children’s Hospital with approval from the Committee on Clinical Investigation (S07–02-0087 with waiver of authorization, exempt category 4). and processed according to an IRB-approved protocol at Boston Children’s Hospital. Consent was obtained by the NIH Neurobiobank. Non-disease neurotypical individuals had no clinical history of neurologic disease and were selected to represent a range of ages from infancy to older adulthood.
Isolation of single neuronal nuclei for single-cell whole genome sequencing
Single neuronal nuclei were isolated using fluorescence-activated nuclear sorting (FANS) for NeuN, as described previously6,38. Briefly, nuclei were prepared from unfixed frozen human brain tissue, previously stored at −80°C, in a dounce homogenizer using a chilled tissue lysis buffer (10mM Tris-HCl, 0.32M sucrose, 3mM Mg(Oac)2, 5mM CaCl2, 0.1mM EDTA, 1mM DTT, 0.1% Triton X-100, pH 8) on ice. Tissue lysates were carefully layered on top of a sucrose cushion buffer (1.8M sucrose 3mM Mg(Oac)2, 10mM Tris-HCl, 1mM DTT, pH 8) and ultra-centrifuged for 1 hour at 30,000 x g. Nuclear pellets were incubated and resuspended in ice-cold PBS supplemented with 3mM MgCl2, filtered (40 μm pore size), then stained with Alexa Fluor 488-conjugated anti-NeuN antibody (Millipore MAB377X). Large neuronal nuclei were then subjected to FANS, one nucleus per well into 96-well plates.
Single nucleus whole genome amplification by primary template-directed amplification (PTA)
Isolated single neuronal nuclei were lysed and their genomes amplified using PTA, a recently developed method that pairs an isothermal DNA polymerase with a termination base16. PTA reactions were performed using the ResolveDNA EA Whole Genome Amplification Kit (formerly SkrybAmp EA WGA kit) (BioSkryb, Durham, NC), using the manufacturer’s protocol. Briefly, single nuclei were sorted into wells containing 3 μL Cell Buffer pre-chilled on ice, then alkaline lysed on ice with MS Mix, mixed at 1400rpm, then neutralized with SN1 Buffer. SDX buffer was then added to the neutralized nuclei followed by a brief incubation at room temperature. Reaction-Enzyme Mix were added, then the amplification reaction was carried out for 10 hrs. at 30°C, followed by enzyme inactivation at 65°C for 3 min. Amplified DNA was then cleaned up using AMPure beads, and yield determined by the picogreen method (Quant-iT dsDNA Assay Kit, ThermoFisher). Samples were subjected to quality control by multiplex PCR for 4 random genomic loci as previously described6, and by Bioanalyzer for fragment size distribution. Amplified genomes demonstrating positive amplification for all 4 loci were then prepared for Illumina sequencing. The majority of the PTA scWGS neuron experiments described here were performed specifically for this report, and they are supplemented with experiments from aged individuals described elsewhere20, as indicated in Supplementary Table 1.
Library preparation for scWGS
Libraries were made following a modified KAPA HyperPlus Library Preparation protocol provided in the ResolveDNA EA Whole Genome Amplification protocol. Briefly, end repair and A-tailing were performed for 500 ng of amplified DNA. Adapter ligation was then performed using the SeqCap Adapter Kit (Roche, 07141548001). Ligated DNA was cleaned up using AMPure beads and amplified through an on-bead PCR amplification. Amplified libraries were selected for 300–600 bp size using AMPure beads. Libraries were subjected to quality control using picogreen and Tapestation HS D1000 Screen Tape (Agilent PN 5067–5584) before sequencing. Single cell genome libraries were sequenced on the Illumina NovaSeq platform (150 bp x 2) at 30X except for subjects 1278 (HiSeq, 60X) and 1465 (NovaSeq, 60X). Illumina reads were aligned to the human reference with decoy sequence GRCh37d5 (hs37d5) using bwa mem.
Kindred mouse embryonic stem cell clones
Pluripotent mESCs on a C57BL/6J x SPRET/Ei F1 background were grown on feeders and maintained in N2B27 media supplemented with the glycogen synthase kinase-3 inhibitor, CHIR99021 (Axon Medchem, 1386, 3 μM), the MEK/ERK inhibitor PD0325901 (Axon Medchem, 1408, 0.4 μM), and mouse leukemia inhibitory factor (LIF) at 1000 U/ml referred to as to 2i + LIF media. mESCs were treated (or not) with aristolochic acid I 50 uM (AAI, Sigma A5512) for 48 hours, and subsequently disaggregated into single cells and plated at limiting dilution. Single cell clones were picked after one week, allowed to expand for another week to provide enough DNA for bulk sequencing and single cells were sorted for PTA. Single cells, clones and the initial mESC line were sequenced to 30x on the Illumina NovaSeq platform (150 bp x 2) and aligned to GRCm38 using bwa mem.
Single-cell amplification quality metrics
Median absolute pairwise differences (MAPD) were computed by estimating copy number in bins of size 50 kb following ref. 39; subsequently, . Copy number profiles in Figure 1b were produced using Ginkgo40 with variable bin size 100 kb and pseudoautosomal regions masked. Allele balance distributions were computed for each neuron by rounding single-cell VAFs to 3 decimal places at all heterozygous SNP sites used to train the SCAN2 allele balance model and then applying R’s density function.
Genome-wide allelic imbalance analysis
Phased training hSNPs for each cell (located in path/to/SCAN2_output/ab_model/[single_cell]/hsnps.tab) were mapped to 1 kb non-overlapping tiles across autosomes from GRCh37d5. The allele balance for tile i containing hSNPs is , where is the number of reads supporting haplotype k. The heatmap in Extended Data Figure 1e was produced by pheatmap with default parameters on the correlation matrix of A vectors.
Comparison of MDA and PTA somatic mutation calls
Both MDA- and PTA-amplified neurons were available for 5 male subjects. For X chromosome analysis, GATK HaplotypeCaller (v3.8.1) was run in joint mode across all samples (bulk, PTA and MDA) for each individual using dbSNP 147_b37_common_all_20160601 and parameters --dontUseSoftClippedBases -rf BadCigar -mmq60. GVCF joint calling was not used because information can be lost compared to providing all BAMs to the same instance of HaplotypeCaller. Pseudoautosomal regions were excluded. The resulting VCF was filtered for mutations using GATK SelectVariants -selectType SNP -selectType INDEL -restrictAllelesTo BIALLELIC -env -trimAlternates. Somatic SNVs and indels in single cells were called separately using the following criteria: VAF > 90%, single cell depth > median(single cell depth), 0 alternate reads in bulk, bulk depth > 10 and absence from dbSNP. A set of germline SNPs and indels for estimating sensitivity was defined by sites with bulk VAF > 90%, bulk depth > median(bulk depth) and no more than 2 reference reads in bulk. For each single cell, somatic sensitivity was approximated as the fraction of these germline sites passing the somatic filters (except 0 alternate reads in bulk and absence from dbSNP). The final estimated number of mutations was calculated by (#corrected calls) = (#somatic mutations called) / (estimated sensitivity).
For the autosomal sSNV comparison in infant neurons, SCAN-SNV commit 5905707 was run on all MDA, PTA and bulk data for subjects 1278 and 5817 separately (Supplementary Table 2). SCAN-SNV was run with --target-fdr=0.01 and the same external data as in SCAN2 analysis of single human neurons.
Somatic indel detection with SCAN-SNV
To adapt SCAN-SNV for indel calling, SCAN-SNV commit 5905707 was first run (with the same calling parameters and data resources as SCAN2) to fit the AB model for each synthetic diploid (SD). Somatic indel candidate loci were identified by requiring a sum of 2 or more mutation supporting reads across the 63 SDs, single-cell read depth >= 10; depth >=10, 0 mutation supporting reads and a 0/0 GATK genotype string in the matched synthetic bulk. Loci present in dbSNP v147_common were further excluded. Local AB at each somatic indel candidate was estimated by SCAN-SNV’s infer.gp function with chunk=1 and flank=1e5. All SCAN-SNV statistical tests and filters for sSNVs were applied to indel candidates with a target FDR of 0.01.
Somatic indel detection with GATK HaplotypeCaller
GATK HaplotypeCaller was run jointly on all synthetic diploids (SDs) and the matched synthetic bulk with the same parameters as in section Somatic mutation calling on male X chromosomes. For each SD, an indel VCF was created by running GATK SelectVariants with -selectType INDEL -select ‘vc.isBiallelic()’ -env -trimAlternates and removing any indel with a nocall (./.) in either the synthetic bulk or SD being analyzed. GATK VQSR was then run using recommended parameters: VariantRecalibrator was first run with –mode INDEL –maxGaussians 4 -resource:mills,known=false,training=true,truth=true,prior=12 Mills_and_1000G_gold_standard.indels.b37.vcf -resource:dbsnp,known=true,training=false,truth=false,prior=2 dbsnp_147_b37_common_all_20160601.vcf followed by ApplyRecalibration with -mode INDEL –-ts_filter_level 90.0. To remove germline and clonal mutations, candidate indels must be supported by 0 reads in bulk and >2 reads in the single cell; >10 reference bulk reads and >= 10 total reads in the single cell; and must not be present in dbSNP.
Synthetic diploid X chromosome simulations
Synthetic diploid (SDs) X chromosomes14 were used to assess the performance of SCAN2 and other callers. SDs are created by merging chromosome X reads from two male single cells (or matched bulks) from different subjects. This recreates allelic amplification imbalance and preserves real amplification artifacts. 9 SDs with 30x mean depth were generated by making all pairings of the 3 PTA cells from subjects 1278 and 5817 and by downsampling the reads in each BAM to ~15x. The youngest subjects (0.4 and 0.6 years old) were chosen to minimize the number of endogenous somatic mutations. Endogenous mutations were identified by applying GATK HaplotypeCaller v3.8 jointly to the 9 SDs, 6 original PTA BAMs and 2 matched bulks using the same parameters as in Somatic mutation calling on male X chromosomes. An additional HaplotypeCaller run with -mmq 1 was also performed. Sites satisfying the following filters in the original, full depth PTA BAMs were considered endogenous somatic mutations: VAF >= 90% and <2 reference reads; depth >= 5 in the single cell, depth > 10 in the matched bulk and no mutation supporting reads in bulk in either the mapping quality 60 or mapping quality 1 runs. A single cluster of sSNVs identified by these filters at chrX:77471371–77471423 caused by clipped alignment was manually excluded. No endogenous indels were identified.
Each SD received a burden of 10, 25, 50, 100, 250, 500 and 1000 SNV and indel spike-ins, for a total of 63 SDs. Random spike-in positions were uniformly sampled from chrX excluding assembly gaps (https://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/gap.txt.gz), 5 bp windows centered on each non-reference site reported by GATK in subject 1278 or 5817 and 5 bp windows centered on all sites in dbSNP v147 common. Somatic SNV spikeins following COSMIC signatures SBS1, SBS11, SBS12, SBS16, SBS19, SBS2, SBS23, SBS3, SBS30, SBS32, SBS4, SBS5, SBS54, SBS6, SBS7b, SBS88 and SBS9 were created by generating batches of SNVs and downsampling to match the signature being simulated. This process was iterated until the desired number of spike-ins was generated. SDs with COSMIC signatures were only created with burden=1,000 SNVs. Somatic indel spike-in candidates further required random lengths; candidates were generated and classified (by first left-aligning indels by bcftools norm and then using SigProfilerMatrixGenerator41 to determine ID83 status) until >1000 candidates were obtained for each ID83 class. Somatic indel spikeins were further required to be >150 bp from the nearest indel spikein candidate to prevent crowding in repetitive tracts. SNV and indel spikeins were not allowed to overlap. SCAN2 was run jointly on the set of 63 SDs, 6 full-depth PTA BAMs, 2 matched bulks and 1 synthetic bulk with the same parameters used in the analysis of single neurons. Sensitivity was calculated as the fraction of known spike-ins called; any call not in the endogenous sSNV or spike-in sets was considered a false positive. Due to the ambiguous nature of indel representation, indel calls were considered matches to known spike-ins if either: (1) the calls matched the spike-in indel exactly or (2) the called indel was the correct length and was located exactly 1 bp away from the spike-in location.
To better approximate real-world performance, SD candidate mutations were combined with autosomal somatic mutation candidates from single cell 5817PFC-A before analysis with SCAN-SNV and SCAN2. This allows the NT/NA FDR heuristics to be computed on a full genome of data, which should better reflect real-world performance.
SNV calling with Monovar
Monovar commit 7b47571 was used and somatic SNVs were called following the authors’ protocol18. BAMs were input to samtools mpileup version 1.9 with options -BQ0 -d10000 -q 40, which was piped into the monovar.py script with options -p 0.002 -a 0.2 -t 0.05 -m 2 as recommended by the authors. To determine whether SNVs were somatic or germline, samtools was run with the same options on matched bulk data. Somatic SNVs were determined by the following filters: Monovar’s single cell genotype must not match ./. or 0/0; single cell depth >= 10 with at least 3 mutation supporting reads; bulk depth >= 6 and <= 1 mutation supporting read; and single cell VAF ≥ 10% for sSNVs with >100 depth or VAF ≥ 15% for sSNVs with depth between 20 and 100. Finally, sSNVs were filtered if any other call occurred within 10 bp.
SNV calling with SCcaller
SCcaller version 1.1 was run following the authors recommendations. BAMs were converted to pileups using samtools version 1.3.1 with the option -C50 and hSNPs were defined using dbSNP version 147 common. Single cell somatic SNVs were called by applying SCcaller’s -a varcall, -a cutoff and reasoning v1.0 script in sequence with default parameters. As recommended on SCcaller’s Github README, passing somatic mutations were required to have VAF > 1/8, filter status = PASS, bulk status = refgenotype and must not have been observed in dbSNP. The standard calling parameter is α = 0.05, while the stringent calling parameter is α = 0.01.
SNV calling with LiRA
LiRA version 1f4cab4 was run following instructions on Github. The joint VCF produced internally by SCAN2 (/path/to/scan2/gatk/hc_raw.mmq60.vcf) for each individual was supplied as the input VCF to LiRA. All samples were processed as male to restrict calls to the autosomes and to use a single genome size for burden estimation. Current LiRA versions use a genome size of G=6.349 for males, so LiRA burden estimates were multiplied by 5.845/6.349 to match the autosomal extrapolation presented here and in ref. 6. LiRA burden estimates retrieved from Supplementary Table S5 of ref. 6 did not require similar correction.
Kindred mESC analysis
LiRA version 3bc0ae1 was used with the global option reference_identifier GRCm38 and the –-force flag to lira varcall following the authors’ instructions. SCAN2 commit d8edd85 was configured with scan2 config --target-fdr 0.01 --callable-regions True --gatk gatk3_joint --score-all-sites --parsimony-phasing (Supplementary Note). SCAN2 data sources were: reference genome GRCm38, the SHAPEIT2 1000 genomes reference panel (ignored by –-parsimony-phasing), and a custom dbSNP database of SPRET_EiJ sites from mgp.v5.merged.snps_all.dbSNP142.vcf from https://www.sanger.ac.uk/data/mouse-genomes-project/. One aim of the kindred analysis was to approximate real-world SCAN2 performance in human cells with a non-simulated truth set. It was therefore necessary to reduce the high SNP density of cross-bred mice (~33 million SNPs/genome) to avoid an overly accurate AB model. The SCAN2 pipeline was manually halted after rule training_hsnps_helper and the output files path/to/scan2/abmodel/[sample]/hsnps.{tab,vcf} containing training hSNPs were downsampled to ~2 million randomly sites by R’s sample function. The SCAN2 pipeline was then restarted.
For FDR calculations using the standard kindred approach, sSNVs were considered true mutations if and only if they satisfied any of: VAF >=20% in the kindred clone bulk; >=5 reads in another kindred cell; or >=1 read in >=2 other kindred cells. For LiRA-based FDR, sites with UNLINKED status were removed and FDR was defined as the fraction of sites with status FILTERED_FP.
For sensitivity calculation, a truth set of clonal sSNVs was constructed separately for each clone using the following criteria: at least 10 reference reads and no mutation supporting reads in the initial mESC population bulk; 50% <= VAF < 100% and depth >= 10 in the kindred clone being analyzed; and VAF = 0 in the other kindred clone. A total of 130 clonal SNVs were identified in the untreated clone and 17,002 SNVs were detected in the AAI clone. Reported sensitivities are the mean fraction of clonal sSNVs recovered across the 4 cells from each clone.
SCAN2 analysis of single human neurons
SCAN2 version 0.9 was run separately for each of the 17 subjects; for each subject, all MDA, PTA and bulk samples were provided to SCAN2. Non-default parameters to SCAN2 were: --abmodel-chunks=4, --abmodel-samples-per-chunk=5000, --target-fdr=0.01 –somatic-indels –-somatic-indel-pon path/to/filter.rda. SCAN2 data resources: human reference genome GRCh37d5, SHAPEIT2 phasing panel 1000GP_Phase3 and dbSNP version 147_b37_common_all_20160601. All following scan2 commands used SCAN2 v1.0. The cross sample filter (–-somatic-indel-pon) was generated by scan2 makepanel with all 128 MDA and PTA single cells and 17 bulks supplied via the –-bam flag. Mutations from all 52 PTA samples were combined and supplied to scan2 rescue –-rescue-target-fdr 0.01. MDA calls were not included in signature-based rescue. Two neurons were excluded from analysis: MDA neuron 5087pfc-Rp3C5, due to high mutation burden (both in ref. 6 and here), and PTA neuron 4638-Neuron-4, due to a very low mutation burden.
Per-cell total mutation burdens were computed separately for sSNVs and indels using mutburden.R (SCAN2 0.9). Current versions of SCAN2 compute burdens automatically. Yearly mutation accumulation rates were derived from a mixed-effects linear model to account for subject-specific effects. Mixed-effects model fitting was performed separately for sSNVs and indels using the lme442 R package with the command lmer(age ~ total_burden + (1|subject)). total_burden refers to the SCAN2 total burden estimate for each neuron.
De novo signature extraction was performed by SigProfiler43 on VAF-based calls from PTA neurons only, which produced a single signature for both sSNVs and indels. Fits to COSMIC indel signatures used COSMIC version 3, signatures ID1–17. For the discovery of active signatures in Figure 4f, all 1,541 VAF-based indels were combined and exposures to each of the 17 signatures were estimated by least squares (lsqnonneg from the pracma R package). For correlation of signature exposure with age, indels from each cell were kept separate. For indels, differing sensitivities among the ID83 channels were corrected before lsqnonneg by dividing by the channel-specific sensitivities derived from synthetic diploid X chromosomes (Extended Data Figure 4f).
Functional impact of point mutations
The severity of somatic SNV and indel mutations reported in Figure 5c were derived from SnpEff29 version 4.3t using the hg19 database. Duplicate and clustered mutations were removed as described in Enrichment analysis of somatic mutations. High impact mutations were those annotated as HIGH in the first reported ANN field. Extrapolation from called mutations to the expected number over the PTA cohort was obtained by dividing mutation counts by the cohort-wide sensitivity estimates of 48.7% for sSNVs and 46.2% for somatic indels.
Enrichment analysis of somatic mutations
To prevent regions with localized artifacts from driving functional impact or enrichment signals, duplicate mutation calls (i.e., exact recurrence of a mutation) were either removed or downsampled to 1 call. For duplicate calls occurring in >1 subject, all instances were removed; for duplicate mutations in >1 neuron from the same subject (1.1% of sSNVs, 0% of indels), 1 occurrence was arbitrarily retained. An additional 57 sSNV calls were removed due to duplicates observations (not SCAN2 calls) in >1 subject with target.fdr < 50%. Clustered mutations (any mutation within 50 bp of another mutation in a single neuron; 1.5% of sSNVs, 4% of indels) were also removed.
Permutation testing was used to generate the expected number of somatic mutations for enrichment analysis. Permutations with matching mutational signatures to the neuronal set were generated by scan2 permtool (SCAN2 v1.0) with default parameters. For each mutation set S consisting of NS mutations, 10,000 permutation sets Pi of size NS mutations each were generated. The positions of permuted mutations were uniformly selected from the subset of the single neuron genome (in which the corresponding mutation in S was called) with single cell depth >5 for sSNVs (>=10 for indels). Permuted mutations were then downsampled to match the SBS96 spectrum (or ID83 spectrum for indels) of S. This step controls for the expected signature bias of SCAN2 rescued calls and nucleotide content bias in the genomic region of interest. Enrichment over any genomic region R is the number of R-overlapping mutations in S divided by the average number of R-overlapping mutations from the 10,000 permuted datasets Pi. A two-sided P-value is calculated by counting the number of permutation sets with greater absolute log fold-change than observed. Confidence intervals for enrichment estimates are computed by bootstrapping the observed mutation set and computing enrichment as described above 10,000 times. To analyze enrichments with higher sequencing depth cutoffs D (Extended Data Figure 9), mutations in S with depth <D were removed and permutation locations were further restricted to the subset of each single neuron genome with depth >=D.
Genomic covariates for enrichment analysis
GTEx expression values for 54 tissues was downloaded from https://storage.googleapis.com/gtex_analysis_v8/rna_seq_data/GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_median_tpm.gct.gz. Gene coordinates were obtained from https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gtf.gz and isoforms were collapsed into a single record using https://github.com/broadinstitute/gtex-pipeline/tree/master/gene_model/collapse_annotation.py.
GRCh37d5 autosomes were tiled with 1 kb non-overlapping windows and the average read depth across the 52 PTA cells was computed. Windows with mean depth < 6 or mean depth in the top 2.5% of windows were removed. The remaining windows were assigned a genic coverage-weighted TPM value of the gene overlapping the window multiplied by the fraction of the window covered by the gene. If multiple genes overlap a region, the gene with highest expression is used. Windows that were <80% covered by genes were removed and considered intergenic. Finally, windows were ranked into deciles by their genic coverage-weighted TPM values and windows within each decile were merged to create 10 regions.
H3K4me3 and H3K27ac narrowPeak files for the 98 epigenomes with H3K27ac data were downloaded from the Roadmap Epigenomics Project server. H3K27ac peaks were classified as TSS-proximal if they occurred within 2 kb of an H3K4me3 peak from the same epigenome; otherwise they were considered TSS-distal. ChromHMM 15-state mnemonic BED files were downloaded from the Roadmap Epigenomics Project server for 127 epigenomes. For each of the 15 ChromHMM states, a single, merged region was created. Brain samples were defined as those with ANATOMY=BRAIN and TYPE=PrimaryTissue. The phyloP 100-way track was downloaded from the UCSC genome browser in BigWig format; average phyloP scores were computed over the same 1 kb tiling used for GTEx expression analysis, including removal of low and high depth windows, using the UCSC bigWigAverageOverBed v2 program. Bins were then ranked into deciles by average phyloP score and windows within each decile were merged to create 10 regions. Cell-type specific enhancer and promoter regions33 were extracted from Supplementary Table 5 tabs Astrocyte enhancers, Astrocyte promoters, etc.. Enhancer or promoter regions were merged within each cell type to produce 2 regions per cell type. Open chromatin regions for GABA, GLU, OLIG and MGAS from dorsolateral prefrontal cortex (DLPFC)34 were downloaded from https://bendlj01.u.hpc.mssm.edu/ggoma/. SAR-seq DNA repair hotspots30 were downloaded from GEO (GSE167257, GSE167257_SARseq_iNeuron_OverlapRep123.peaks.bed.gz); Repair-seq peaks29 were obtained from Supplementary Table S1 of ref. 28.
Extended Data
Extended Data Fig. 1. Allele balance is not generally correlated between PTA amplifications.

a. Genome-wide allele balance (binned in 100 kb windows) for 3 typical PTA cells from the same individual. b. Allele balance for cells in (a) plotted against each other. c-d. Allele balance averaged across the cohort of 52 PTA cells (c) or 75 MDA cells (d); i.e., each point represents the average allele balance for a single 100 kb window. A small number of regions show consistent allelic imbalance across many amplifications (arrows). e. Correlation of allele balance profiles between all pairs of PTA cells. Correlation is generally low; cells from the same individual show slightly higher correlations; and a single individual (4638) shows the strongest correlation.
Extended Data Fig. 2. SCAN2 performance on simulated sSNVs.

sSNVs were simulated using the synthetic diploid (SD) X chromosome approach (Methods). Sensitivity is the fraction of known spike-ins recovered and false positives (FPs) are defined as calls that are neither known spike-ins nor somatic mutations endogenous to the haploid X chromosomes used to create each SD. Each point in a-d represents a single SD simulation with 10–250 spike-ins. a-b. Comparison of SCAN2 and SCAN-SNV sensitivity (a; lines are R loess() fits) and false discovery rates (b; lines are linear regression fits to FDR ~ 1/mutations per Mb). c-d. Comparison to other single cell SNV genotypers. c. Sensitivity vs. false positives per megabase of analyzed sequence. d. False discovery rate vs. the number of spike-ins per megabase. Lines are parameterized by mean sensitivity S and false positive rate per megabase F measured across all points: FDR = F / (F + xS). SCcaller standard uses a calling threshold of α = 0.05 while stringent calling uses α = 0.01. e-f. Performance of SCAN2 mutation signature-based rescue as a function of the number of sSNVs available for learning the true mutation signature. Sensitivity (e) and false discovery rate (f) are shown relative to the sensitivity or false discovery rate of the same SD simulation using the maximum sSNV catalog of 4,666 sSNVs. was added to all quantities to avoid division by zero. Solid lines are fitted by R’s loess() function. g. Effect of mutation signature of spike-ins on SCAN2 sensitivity. Each point is the average sensitivity of 9 SD simulations with 1000 spike-ins from a single COSMIC SBS signature. Mutation signatures are characterized by their similarity to the PTA SNV artifact signature. Solid line: linear regression on all points except PTAerr. SBS30 (h) is the most similar COSMIC signature to the PTA SNV artifact signature (i).
Extended Data Fig. 3. Mutation spectra of SCAN2 and LiRA calls on kindred mouse ESC cells.
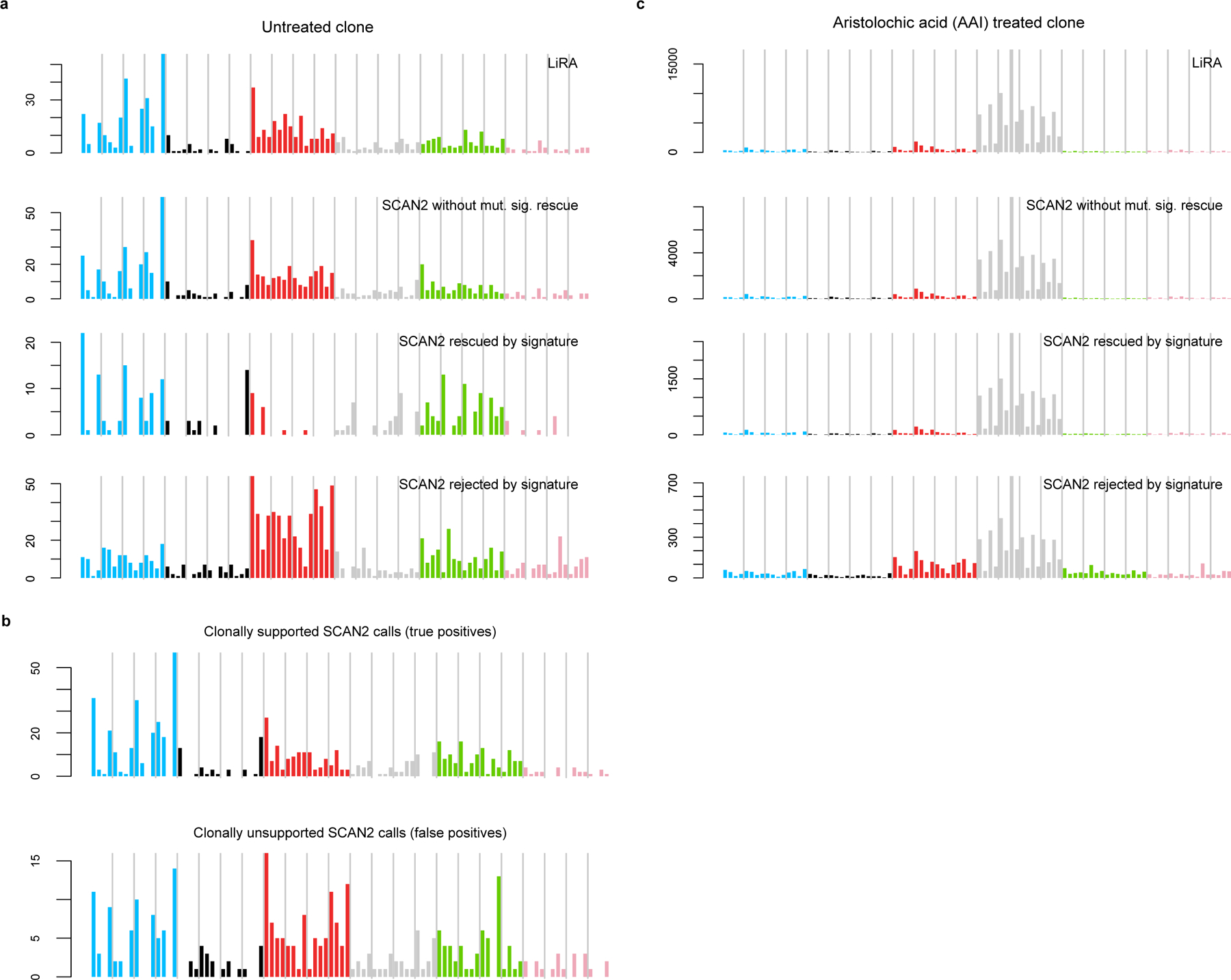
a-b. SBS96 signatures of somatic SNVs called in 4 single cells from the untreated clone. C>A mutations (blue peaks) are characteristic of COSMIC SBS18 and the mutation signature of SNVs acquired during clonal expansion5. These peaks persist in the clonally unsupported SNVs (b), suggesting that the method for classifying true positives is overly conservative. c. Signatures for SNVs called in the 4 single cells taken from an aristolochic acid (AAI)-treated clone.
Extended Data Fig. 4. SCAN2 performance on simulated somatic indels.
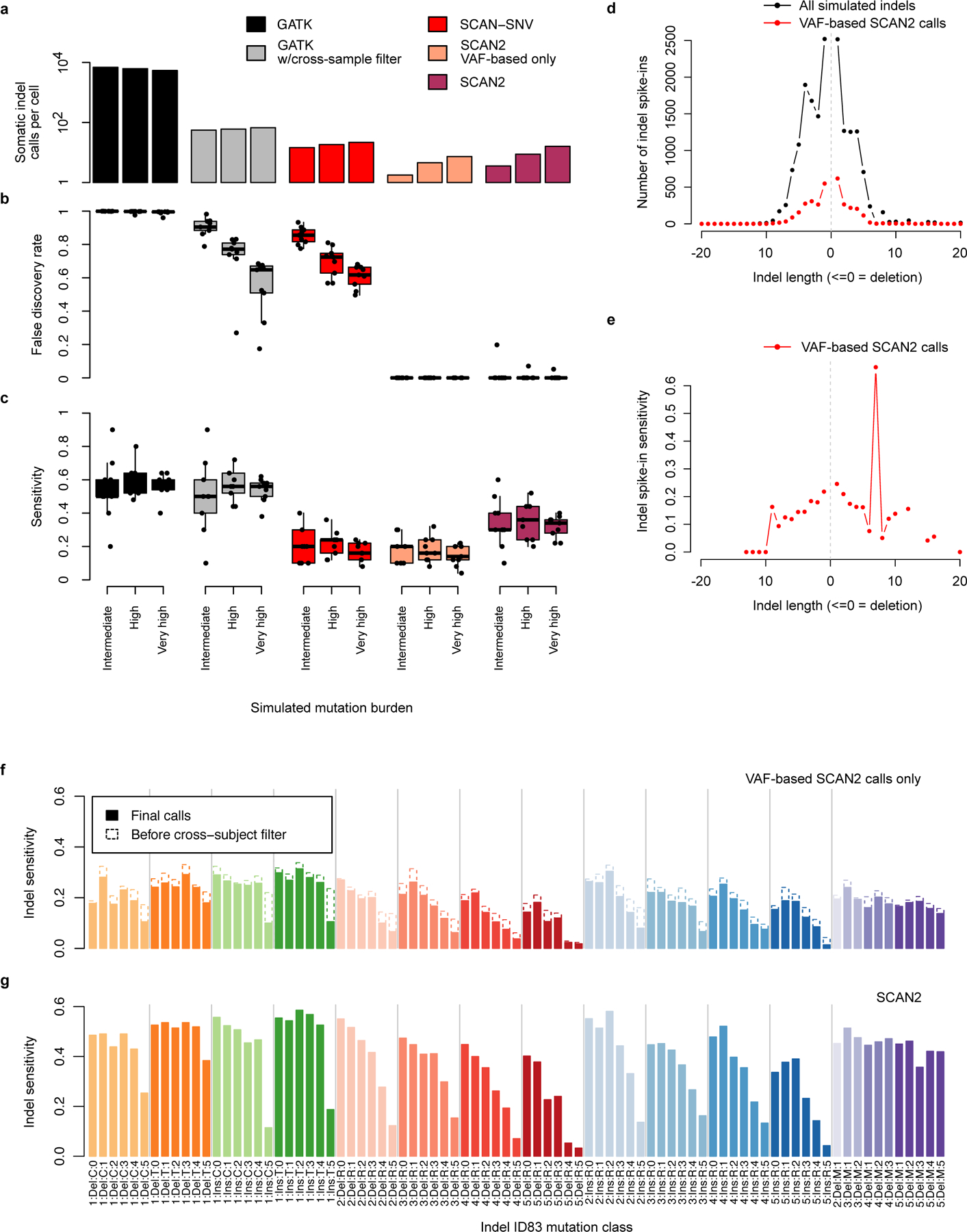
a-c. SCAN2 and other callers were applied to simulated indels using the synthetic diploid (SD) X chromosome spike-in approach (Methods). SDs received 10, 25 or 50 indel spike-ins each, which correspond, respectively, to genome-wide rates of approximately 170 (intermediate), 430 (high) and 850 (very high) somatic indels. Performance was measured by the average number of indels called per SD (a), the fraction of false positives per indel call set (b) and the fraction of spike-ins recovered (c). Tested methods were SCAN2 (with and without signature-based rescue), GATK HaplotypeCaller, GATK HaplotypeCaller with filtration by SCAN2’s cross-sample recurrent artifact filter and an adaptation of SCAN-SNV’s somatic SNV discovery approach to indels. Boxplot whiskers, the furthest outlier <=1.5 times the interquartile range from the box; box, 25th and 75th percentiles; centre bar, median; n=9 SDs per boxplot. d. Distribution of indel lengths among all simulated indels (black) and VAF-based SCAN2 indel calls (red). e. Spike-in indel sensitivity by length for VAF-based SCAN2 calls. f. Sensitivity for VAF-based SCAN2 indel calling stratified by the 83-dimensional indel classification scheme used by COSMIC indel signatures (ID83). Dotted outlines: sensitivity before applying cross-subject filtration. g. ID83-stratified indel sensitivity for SCAN2 calls with signature-based rescue.
Extended Data Fig. 5. Comparison of SCAN2 and LiRA sSNV calls on human neurons.
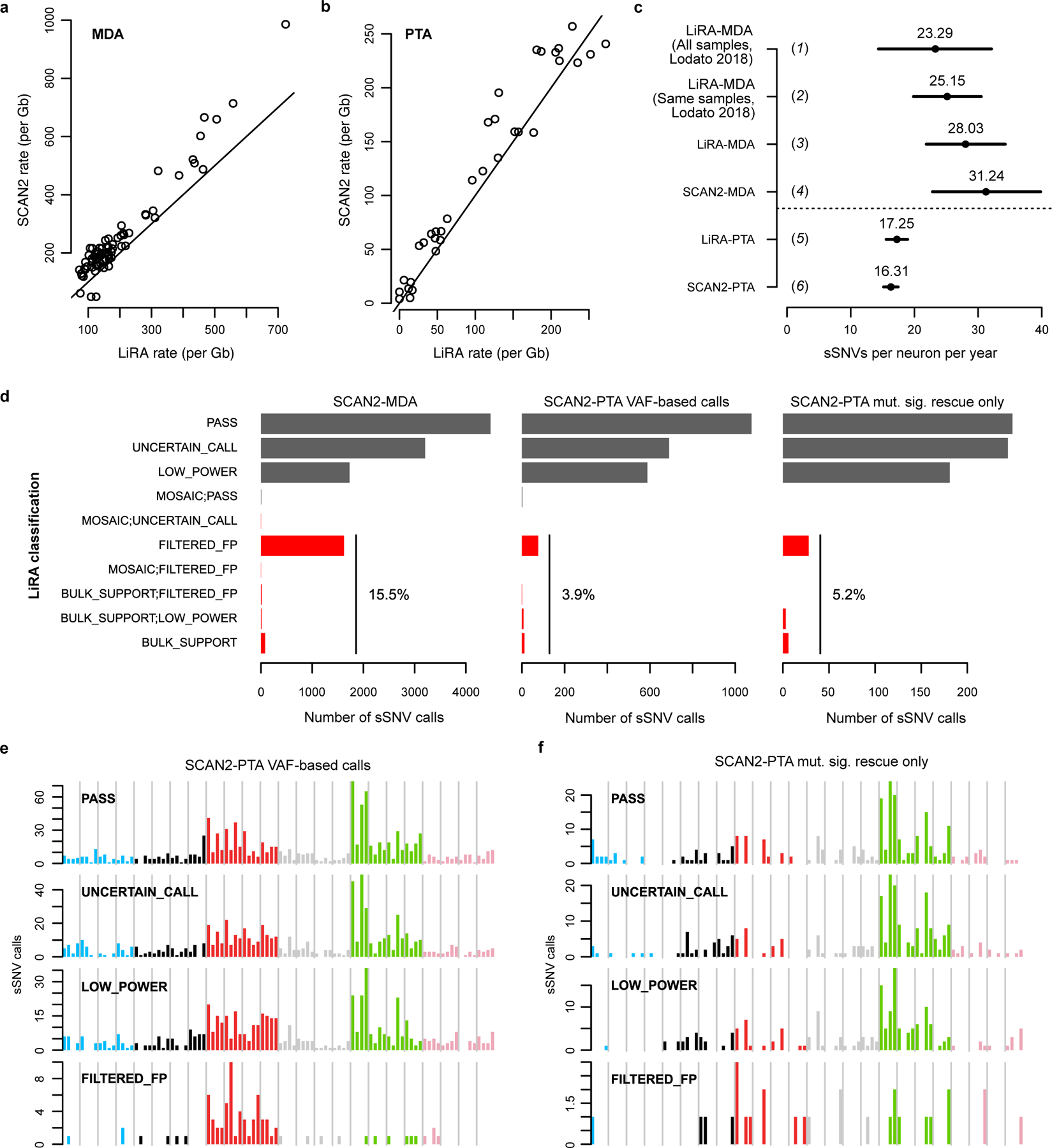
Single human neurons were previously analyzed by LiRA15, a specific but lower sensitivity approach for calling somatic SNVs. a-b. SCAN2 and LiRA extrapolations for the total (not called) sSNV burden per diploid Gb of human sequence from MDA- (a) and PTA-amplified (b) single neurons. Solid lines: y=x. c. Linear regression estimates for the number of sSNVs accumulated per neuron per year from several sources and analyses. Horizontal bars represent 95% C.I.s produced by confint applied to an lmer fit by the lme4 R package; centre points from fixef applied to the same fits. (1) LiRA rates taken from ref. 6, which used a larger set of n=91 MDA-amplified PFC neurons; (2) LiRA rates taken from ref. 6 using n=73 of the 75 MDA-amplified PFC neurons from subjects analyzed in this study (the two excluded neurons are 5087pfc-Rp3C5, an extreme outlier, and 4638-MDA-14); (3) rerun of LiRA on n=74 MDA-amplified neurons in (2) using the same input provided to SCAN2; (4) SCAN2 on n=74 MDA-amplified neurons; (5) LiRA on n=34 PTA-amplified neurons from donors also analyzed in ref. 6 (N.B. LiRA’s higher rate estimate in (c) occurs despite lower burden estimations in (b) due to differences in model intercepts: SCAN2 intercept=95.83, LiRA intercept=17.63); (6) SCAN2 on all n=52 PTA-amplified neurons generated here. d. LiRA classification of SCAN2 calls where reads linked to nearby germline heterozygous SNPs are available (black: likely true sSNVs, red: possible false positives). PASS is the highest quality LiRA class. UNCERTAIN and LOW_POWER indicate lack of linking reads to make a confident call, but no evidence of artifactual status is detected. All other classes (red) are interpreted as false positives. Percentages show the fraction of all false positive classes among SCAN2 calls. e-f. Raw mutation spectra for SCAN2 calls without (e) and with mutation signature-based calling (f) SCAN2 calls stratified by LiRA classification. The similarities between PASS and the two lower quality UNCERTAIN_CALL and LOW_POWER classes suggest that the majority of UNCERTAIN_CALL and LOW_POWER SCAN2 calls are true mutations. Confident false positives (FILTERED_FPs) possess a C>T dominated signature with lack of C>Ts at CpGs.
Extended Data Fig. 6. Somatic indels mutation spectra in human neurons and other cells.
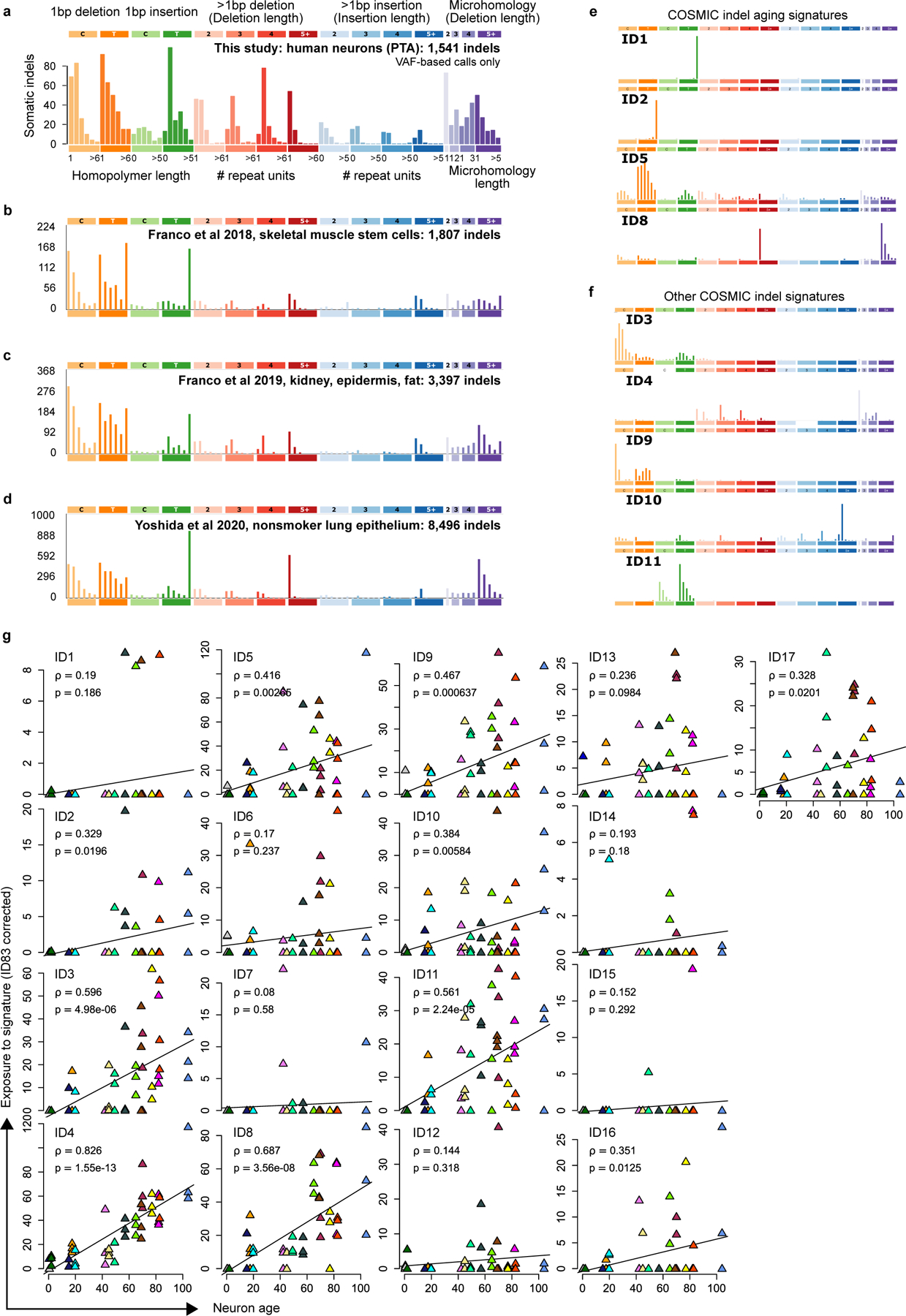
a. Spectrum of 1541 indels from PTA neurons from this study, same as Figure 4e. b-e. Somatic indel spectra from other studies: clonally expanded single skeletal muscle stem cells (b), clonally expanded single kidney (excluding hypermutated kidney cells, designated KT2 in the original study), epidermis and fat cells (c) and clonally expanded bronchial epithelial cells from children and never-smokers (d). e. COSMIC signatures with clock-like or age-associated annotations. f. Non-aging COSMIC signatures with >5% contribution to single neurons. g. Per-neuron COSMIC signature fits, corrected for ID83 sensitivity (Methods). Correlation (ρ) between age and exposure and P-value of two-sided t-test for correlation=0 (p) are shown for each COSMIC signature. P-values were not adjusted for multiple comparisons. Colors correspond to subject IDs as shown in Figure 4. Note that y-axes are not the same scale.
Extended Data Fig. 7. PTA sensitivity over genomic regions for SNVs and indels.
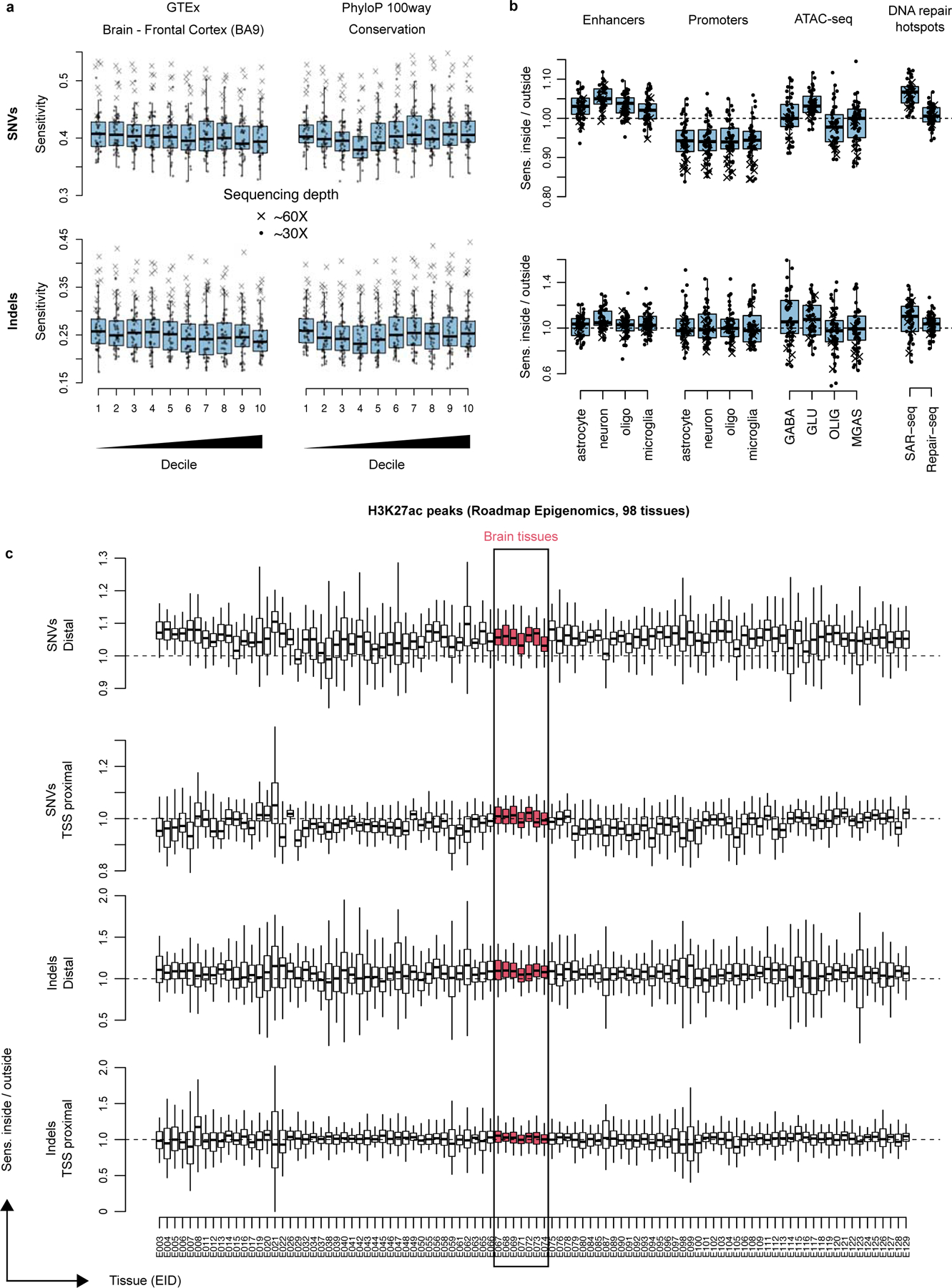
a. Absolute sensitivity for spatial measurements that divide the genome into roughly equally sized deciles (median GTEx expression for a single tissue type, brain BA9 prefrontal cortex, and phyloP 100way conservation). b-c. Relative sensitivities: sensitivity inside of the tested region divided by sensitivity of the complemented region. Enhancers and promoters from Nott et al. 2019, ATAC-seq from Hauberg et al. 2020, DNA repair hotspots from Wu et al. 2021 and Reid et al. 2021, H3K27ac peaks from Roadmap Epigenomics. Each point represents one PTA neuron; crosses represent the 7 PTA neurons sequenced to 60x, circles represent 30x depth samples. Boxplot whiskers, the furthest outlier <=1.5 times the interquartile range from the box; box, 25th and 75th percentiles; centre bar, median.
Extended Data Fig. 8. ChromHMM states and neuronal mutations.
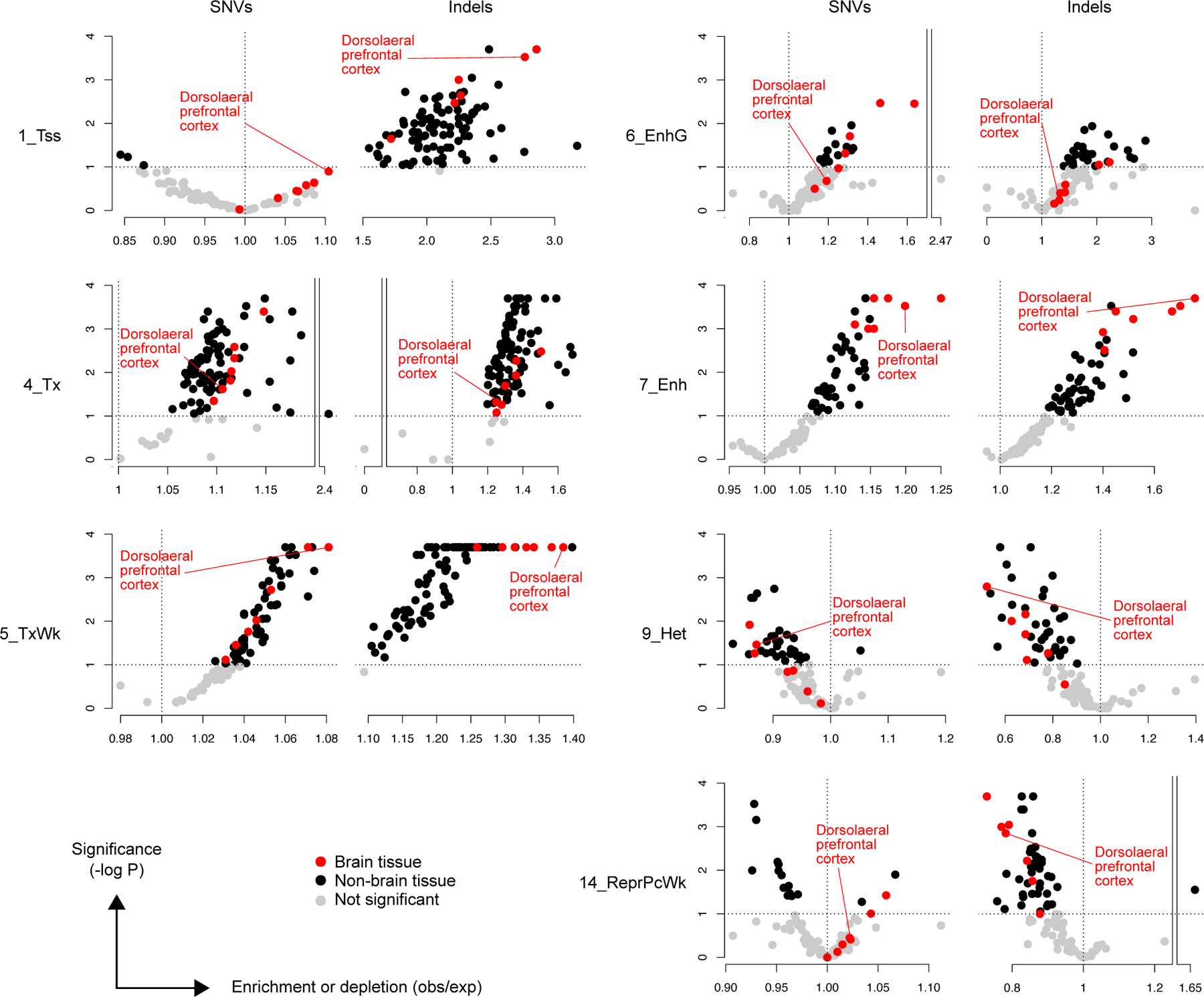
Enrichment analysis of ChromHMM states from 127 tissues from the Roadmap Epigenomics Project. Active regions include 1_Tss, 4_Tx, 5_TxWk, 6_EnhG and 7_Enh; inactive states include 9_Het and 14_ReprPCWk. Red points, brain tissue regardless of significance level; black points, non-brain tissue; grey points, enrichment not significant at the P < 0.1 level. No correction for multiple hypothesis testing was applied.
Extended Data Fig. 9. Patterns of mutation enrichment persist at increasing sequencing depth thresholds.
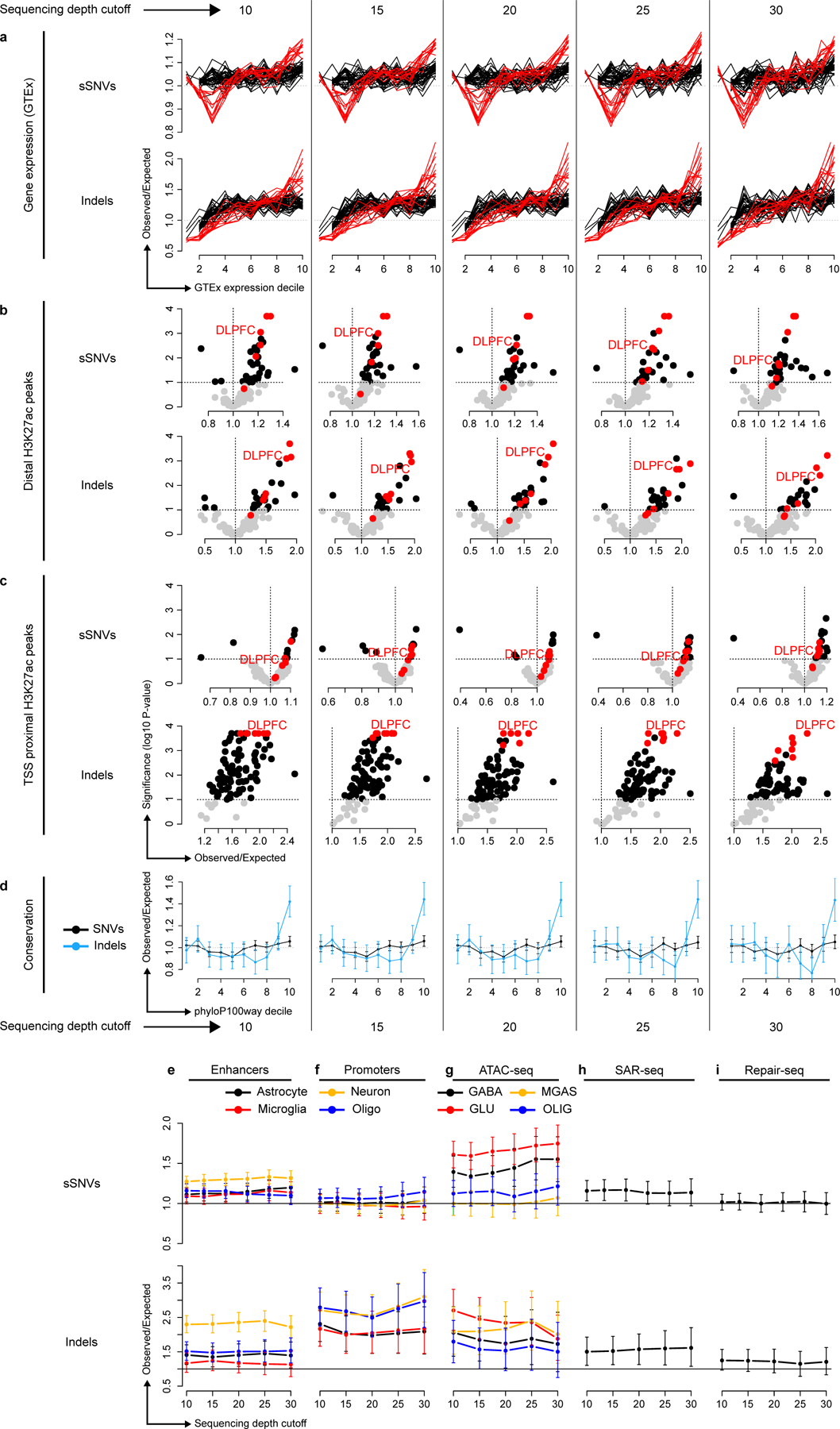
Analyses presented in Figure 5 rerun using mutations supported by at least 10, 15, 20, 25 and 30 reads; permutations used for enrichment analysis are also restricted to the subset of the genome with the corresponding sequencing depth. GABA, GABAergic neurons; GLU, glutamatergic neurons; OLIG, oligodendrocytes; MGAS, microglia and astrocytes. Error bars: 95% bootstrapping confidence intervals. For panels a-d, each plot presents an analysis at one depth cutoff; for panels e-i, each plot contains the full range of depth cutoffs, as indicated on the x-axis. Error bars in d-i represent bootstrap 95% C.I.s using n=10,000 bootstrap samples; centre points are the observed mutation count divided by the mean mutation count of the bootstrap samples.
Supplementary Material
Acknowledgements
We thank R.S. Hill, Ronald Mathieu and Lakshmi (Sahithi) Cheemalamarri at the Boston Children’s Hospital & Harvard Stem Cell Institute Flow Cytometry Research Facility, the Research Computing group at Harvard Medical School, and the Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center (IDDRC) Molecular Genetics Core for assistance. Human tissue was obtained from the NIH Neurobiobank at the University of Maryland, and we thank the donors and families for their invaluable contributions for the advancement of science. This work was supported by the Bioinformatics and Integrative Genomics training grant (L.J.L., T32HG002295), K08 AG065502 (M.B.M.), T32 HL007627 (M.B.M.), the Brigham and Women’s Hospital Program for Interdisciplinary Neuroscience through a gift from Lawrence and Tiina Rand (M.B.M.), the donors of the Alzheimer’s Disease Research program of the BrightFocus Foundation A20201292F (M.B.M.), the Doris Duke Charitable Foundation Clinical Scientist Development Award 2021183 (M.B.M.), PRMRP Discovery Award W81XWH2010028 (Z.Z.), the Edward R. and Anne G. Lefler Center postdoctoral fellowship (Z.Z.), R00 AG054748 (M.A.L.), R01 AG070921 (C.A.W.), R01NS032457, U01MH106883 (P.J.P. and C.A.W.), and the Allen Discovery Center program, a Paul G. Allen Frontiers Group advised program of the Paul G. Allen Family Foundation (C.A.W.). C.A.W. is an Investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing interests. The authors declare the following competing interests: C. G. is Director and cofounder and J. W. is CEO and cofounder of Bioskryb, Inc., the manufacturer of PTA kits used in this study. C.A.W. is a consultant for Maze Therapeutics (cash, equity), Third Rock Ventures (cash), and Flagship Pioneering (cash), none of which have any relevance to this study. The remaining authors declare no competing interests.
Code availability
SCAN2 is available for download at https://github.com/parklab/SCAN2. Additional scripts used in this study are available at https://github.com/parklab/SCAN2_PTA_paper_2022 and Zenodo44.
Statistics and Reproducibility
No statistical method was used to predetermine sample size. All 4 PTA neurons from brain 1465 were excluded from copy number analyses, but no other PTA neurons were excluded from any analysis. One MDA neuron from a previous study6 (5087pfc-Rp3C5) was excluded from most analyses due to high mutation burden; one PTA neuron from this study (4638-Neuron-4) was excluded due to a very low mutation burden. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Data availability
All MDA-amplified single neurons and matched bulks listed in Supplementary Table 2 were downloaded from dbGaP, identifier phs001485.v1.p1. Only neurons from the pre-frontal corteces from individuals for which additional PTA data were generated were used. Raw sequencing read data for PTA-amplified human neurons can be downloaded from dbGaP, identifier phs001485.v3.p1. PTA-amplified mESC kindred cells and bulks can be downloaded from the NCBI Sequence Read Archive, identifier PRJNA832209.
References
- 1.Poduri A, Evrony GD, Cai X & Walsh CA Somatic Mutation, Genomic Variation, and Neurological Disease. Science 341, 43–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lodato M et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martincorena I et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaiswal S et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blokzijl F, de Ligt J, Jager M et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lodato M et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martincorena I et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee-Six H et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Franco I et al. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat Commun 9, 800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franco I, Helgadottir HT et al. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biology 20, 285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodworth MB, Girskis KM, & Walsh CA Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat Rev Genet 18, 230–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evrony G, Lee E, Park PJ & Walsh CA Resolving rates of mutation in the brain using single-neuron genomics. eLife 5, e12966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang CZ, Adalsteinsson VA, Francis J, Cornils H, Jung J, Maire C, Ligon KL, Meyerson M & Love JC Calibrating genomic and allelic coverage bias in single-cell sequencing. Nat Commun 6, 6822 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luquette LJ et al. Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat Commun 10, 3908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohrson C et al. Linked-read analysis identifies mutations in single-cell DNA sequencing data. Nat Genet 51, 749–754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Pena V, Natarajan S et al. Accurate Genomic Variant Detection in Single Cells with Primary Template-Directed Amplification. Proc. Natl. Acad. Sci. U.S.A 118, e2024176118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zafar H, Wang Y, Nakhleh L, Navin N & Chen K Monovar: single-nucleotide variant detection in single cells. Nat Meth 13, 505–507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singer J, Kuipers J, Jahn K and Beerenwinkel N Single-cell mutation identification via phylogenetic inference. Nat Commun 9, 5144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller MB, Huang AY, Kim J, Zhou Z, et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604, 714–722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McConnell MJ, Lindberg MR, Brennand KJ, et al. Mosaic copy number variation in human neurons. Science 342, 632–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chronister WD, Burbulis IE, Wierman MB, et al. Neurons with Complex Karyotypes Are Rare in Aged Human Neocortex. Cell Rep 26, 825–835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X, Zhang L, Milholland B, et al. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods 14, 491–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petljak M et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 176, 1282–1294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gymrek M PCR-free library preparation greatly reduces stutter noise at short tandem repeats. bioRxiv doi: 10.1101/043448 (2016). [DOI] [Google Scholar]
- 26.Lasken RS, Stockwell TB Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnology 7, doi: 10.1186/1472-6750-7-19 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida K et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cingolani P, Platts A, Wang LL, Coon, M. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid D et al. Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science 372, 91–94 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu W et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madabhushi R et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roadmap Epigenomics Consortium, Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nott et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauberg M et al. Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons. Nat Commun 11, 5581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alt FW, Schwer B DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 71, 158–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xing D et al. Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl. Acad. Sci 118, e2013106118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abascal F et al. Somatic mutation landscapes at single-molecule resolution. Nature 593, 405–410 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Evrony GD, Cai X, Lee E, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baslan T, Kendall J, Rodgers L, et al. Genome-wide copy number analysis of single cells. Nat Protoc 7, 1024–1041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garvin T, Aboukhalil R, Kendall J, et al. Interactive analysis and assessment of single-cell copy-number variations. Nat Methods 12, 1058–1060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergstrom EN, Huang MN, Mahto U et al. SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics 20, 685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bates D, Mächler M, Bolker B and Walker S Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software 67, 1–48 (2015). [Google Scholar]
- 43.Alexandrov L (2020). SigProfiler (https://www.mathworks.com/matlabcentral/fileexchange/38724-sigprofiler), MATLAB Central File Exchange. Retrieved January 1, 2020.
- 44.Luquette L (2022). SCAN2_PTA_paper_2022. Zenodo, doi: 10.5281/zenodo.6532827. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All MDA-amplified single neurons and matched bulks listed in Supplementary Table 2 were downloaded from dbGaP, identifier phs001485.v1.p1. Only neurons from the pre-frontal corteces from individuals for which additional PTA data were generated were used. Raw sequencing read data for PTA-amplified human neurons can be downloaded from dbGaP, identifier phs001485.v3.p1. PTA-amplified mESC kindred cells and bulks can be downloaded from the NCBI Sequence Read Archive, identifier PRJNA832209.