

Abstract
Thrombocytopenia is a common complication in sepsis and is associated with higher mortality. Activated platelets express CD62P, which facilitates platelet-leukocyte aggregate (PLA) formation and contributes to thrombocytopenia in sepsis. We have reported that thrombocytopenia in murine sepsis is partly attributable to TLR7 signaling, but the underlying mechanism is unclear. In the current study, we tested the hypothesis that TLR7 mediates platelet activation and PLA formation during sepsis. In vitro, whole blood from WT mice treated with loxoribine, a TLR7 agonist, exhibited a dose-dependent increase in activated platelets compared to the control (PBS with 0.05% DMSO) or loxoribine-treated TLR7−/− whole blood. In a murine model of sepsis, there was a significant increase in platelet activation and PLA formation 24 hours after cecal ligation and puncture (CLP) as evidenced by double positive expression of CD41+/CD62P+ and CD45+/CD62P+, respectively. The sepsis-induced PLA formation was significantly attenuated in TLR7−/− mice. Finally, in ex-vivo experiments, plasma isolated from septic mice induced WT platelet activation, but such effect was significantly attenuated in platelets deficient of TLR7. These findings demonstrate a pivotal role of TLR7 signaling in platelet activation and PLA formation during bacterial sepsis.
Keywords: Innate immunity, platelet leukocyte aggregates, platelets, sepsis, TLR7, Toll-like receptor 7
Introduction
Sepsis is defined as a dysregulated host response to infection resulting in systemic inflammation, coagulation activation, and organ failure. Sepsis-induced thrombocytopenia is a common complication in sepsis and is associated with poor prognosis and higher mortality [1,2]. Numerous studies have demonstrated the impact of platelet counts on morbidity in sepsis, and the degree of thrombocytopenia has been confirmed to be a predictor of sepsis mortality [3,4]. Platelets are known to be versatile effectors of coagulation functioning to localize and amplify the coagulation response at sites of vascular injury. It has become increasingly clear, however, that blood platelets are also a critical mediator of inflammation, and express innate immune pattern recognition Toll-like receptors (TLR), which play a pivotal role in inflammation-mediated coagulopathy during sepsis [5].
We have previously demonstrated [6] that sepsis-induced thrombocytopenia, systemic inflammation, and coagulopathy were partly attributable to TLR7, a single-stranded RNA sensor [7–9]. However, the mechanism by which TLR7 signaling mediates platelet activation and thrombocytopenia in bacterial sepsis remains unknown. Platelet alpha granules contain up to 300 different types of proteins and polypeptides, which serve to supplement thrombin generation and mediate inflammation [10]. Platelet activation and alpha degranulation will lead to the translocation of CD62P (P-selectin), a cell adhesion molecule, to the platelet membrane to facilitate platelet-leukocyte aggregate (PLA) formation [10]. This is followed by a rapid sequestration of activated platelets from circulation and contributes to thrombocytopenia [5,11]. In the current study, we tested the hypothesis that TLR7 stimulation is sufficient to induce platelet activation and mediates PLA formation during bacterial sepsis.
Materials and methods
Animals
All animal experiments were approved by the Institutional Animal Use and Care Committee of the University of Maryland and performed using Wild-type (WT) C57BL/6 J and Toll-like receptor 7 deficient (TLR7−/−) male mice aged between 10 and 18 weeks. TLR7−/− mice (Tlr7tm1Flv/J, stock No. 008380) on the C57BL/6 N background were originally purchased from Jackson laboratory and cross-bred with C57BL/6 J mice for at least 10 generations. C57BL/6 J mice have a naturally occurring five exon deletion that results in the absence of Nnt (Nicotinamide (NAD) nucleotide transhydrogenase) gene and differentiates it from C57BL/6 N mice [12]. Our TLR7−/− mice also had a separate natural deletion of the Nnt gene, which verified their C57BL/6 J background (data not shown). All mice were housed in the pathogen-free animal care facility for at least one week prior to experimentation and all received the same pathogen-free diet with free access to water. Mice were kept in temperature-controlled rooms with 12-hour light–dark cycles.
Cecal ligation and puncture model of polymicrobial sepsis
Polymicrobial sepsis was induced using the cecal ligation and puncture technique as previously described [6,13]. Briefly, mice were anesthetized with ketamine (100 mg/kg) and xylazine (4 mg/kg) solution. A laparotomy was performed, and the cecum was ligated below the ileocecal valve at 1.5 cm from the cecal tip. A through and through puncture of the cecum was made using an 18-gauge needle and 2–3 mm of fecal matter was extruded before placing the cecum back into the abdomen and closing the peritoneum and skin. Sham mice underwent laparotomy only. All sham and CLP mice received post-operative 0.03 ml/g of prewarmed normal saline and single administrations of incisional and subcutaneous bupivacaine (3 mg/kg) and buprenorphine (1 mg/kg), respectively. Rectal temperatures were measured at 4 or 24 hours later. Of note, simple randomization was used to assign animals, and the operators (B.W., J. Z.) were blinded to the strain information.
Blood and plasma collection
At 4- or 24-hour post-surgery, whole blood was collected via cardiac puncture and transferred to microcentrifuge tubes containing 100 μL of 3.2% sodium citrate anticoagulant at a volume ratio of 1:8 (anticoagulant: blood). Complete blood counts were performed using the Coulter Ac●T diff2 Hematology Analyzer (Beckman Coulter). For select experiments, blood from WT mice 24-hour post-surgery was processed into plasma via centrifugation. Blood was first spun at 1000 x g at room temperature for 10 minutes and supernatants transferred to new Eppendorf tube and centrifuged at 1000 x g at 4°C for 10 minutes. Supernatants were transferred to new Eppendorf tube and finally centrifuged at 10,000 x g at 4°C for 10 minutes and resultant plasma was stored at −80° C for future experiments.
Platelet isolation from whole blood
Whole blood was mixed gently with 100 μL of 3.2% sodium citrate at a ratio of 1:8 (anticoagulant: blood). Two-hundred microliters of CGS-EDTA buffer (100 mM NaCl, 8.5 mM Tris, 8.5 mM Dextrose, 1 mM EDTA) was added to the sample, mixed gently, and the blood was centrifuged at room temperature for 8 minutes at 50 g. The supernatant plus 200 uL of the top layer of red blood cells were transferred into new microcentrifuge tube and centrifuged at 100 g for 5 minutes. Without touching the sedimented red cell layer, the platelet-rich plasma (PRP) layer was collected and pooled from two to three mice into a new microcentrifuge tube. The PRP was centrifuged at 800 g for 8 minutes, after which the supernatant was removed, and the platelet pellet was washed gently once with CGS-EDTA buffer. Platelets were then resuspended in Tyrode’s buffer (134 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 Na2HPO4, 10 mM HEPES, 1 mM MgCl2, 5 mM Dextrose and 3 mg/ml of bovine serum albumin (BSA)) to a concentration of approximately 2 × 105 platelets/μL. To determine the purity of platelet preparation, isolated mouse platelets were stained with PE-anti-CD41 (BD Biosciences PMG-561850; Clone MWReg30) antibody for platelets and PerCpCy5.5-anti-CD45 (BioLegend BLD-103131; Clone 30-F11) antibody for leukocytes. Leukocyte contamination in isolated platelet suspension as compared to unstained sample was found to be approximately 1 in every 10,000 cells or 0.01% (data not shown).
TLR7 expression in circulating blood cells
To evaluate the intracellular expression of TLR7 in peripheral blood cells, whole blood was collected from age-matched WT and TLR7−/− male mice. Red blood cells were lysed with RBC lysis buffer (150 mM ammonium chloride, 10 mM potassium carbonate, and 0.1 mM EDTA) prior to viability dye (BioLegend Zombie NIR™) staining and treatment with an Fc receptor blocking agent. To identify specific blood cell populations, we performed surface staining with PerCP Cy5.5-anti-CD45 for leukocytes and FITC-anti-CD41 (BD Biosciences PMG-553848; Clone MWReg30) for platelets. Cells were then fixed and permeabilized using Invitrogen™ eBioscience™ Intracellular Fixation & Permeabilization buffer set, and then incubated with PE-anti-TLR7 (monoclonal antibody (mAB) BD Biosciences PMG-565557; Clone A94B10). The percentage of cells positive for TLR7 was analyzed using BD LSR II Flow Cytometer and FlowJo V10.8 software.
The efficiency of intracellular protein staining was validated by AF647 anti-beta actin (BLD-643810; Clone 2F1-1) (a highly conserved cytoskeletal protein) staining in blood cells with and without permeabilization. Further, to test the performance and ensure the specificity of the TLR7 antibody, bone marrow cells from both WT and TLR7−/− mice were first stained with PerCP CY5.5-anti-CD45 and AF647-anti-CD19 (BLD-115522; Clone 6D5) to identify the leukocyte and B-cell populations, fixed and permeabilized, and finally stained with PE-anti-TLR7. To assess the nonspecific background staining of the anti-TLR7 antibody with and without permeabilization, blood cells collected from WT mice first underwent surface staining, fixation, and permeabilization as above, and then stained with either PE-anti-TLR7 or PE IgG1, κ isotype control (BD Biosciences 554680). In each of the above experiments, the percentage of cells positive for TLR7 was analyzed using BD LSR II Flow Cytometer and FlowJo V10.8 software (Fig. S1–S3).
Detecting platelet activation and PLA formation
Blood was collected from WT and/or TLR7−/− mice at 4- or 24-hour post-sham or CLP surgery. Whole blood or isolated platelets from WT and/or TLR7−/− mice were treated with 0.05% DMSO in PBS, 10 μg/ml of the TLR2 agonist Pam3CSK4 (P3C-Enzo Life Sciences ALX-165-066-M002), or the TLR7 agonist loxoribine (InvivoGen tlrl-lox) at the indicated concentrations for 15 minutes at 37°C. Loxoribine supplied as a lyophilized powder was reconstituted with 50% DMSO in PBS and then diluted with PBS to 100x working solution containing 5% DMSO. After adding to the platelet suspension, the final DMSO concentration became 0.05% in both ligand and control (w/o loxoribine) groups. Isolated platelets from WT and TLR7−/− mice were treated with pooled plasma from WT sham or CLP mice at the indicated concentrations. In this experiment, the operator (B.W.) was blinded to strain information. In each of the above, platelet activation and PLA formation were tested with flow cytometry as stated below.
Blood was stained with PerCP CY5.5-anti-CD45, PE-anti-CD41, and AF647-anti-CD62P (BD Biosciences PMG-563674; Clone Rb40.34), and then treated with BD Phosflow™ Lyse/Fix Buffer. Platelets were stained with anti-CD41 and anti-CD62P antibodies and then treated with 2% paraformaldehyde in 1:1 v/v ratio for fixation. Data were recorded in the BD LSR II Flow Cytometer and analyzed with FlowJo V10.8 software. Leukocytes were identified as CD45 positive events and platelets as CD41 positive events. The activation status of platelets was determined by the increased expression of CD62P. Platelet activation and activated PLA were determined based on forward (FSC) and side (SSC) scatter properties and double positive expression of CD41+/CD62P+ and CD45+/CD62P+ cells, respectively. Gating strategies are shown in the supplemental figures: in vitro whole blood (Fig. S4), in vivo whole blood (Fig. S5), and in vitro/ex vivo isolated platelets (Fig. S6).
Statistical analysis
GraphPad Prism 9 software was used for statistical analysis. All data are presented as mean ± SD. Data were tested for Gaussian distribution and parametric or non-parametric tests were applied accordingly. For comparison between two groups, unpaired Student’s t-test or Mann–Whitney test was used according to data distribution. For comparison among more than two groups, statistical significance was determined with one-way ANOVA or with Brown-Forsythe and Welch ANOVA according to data distribution with post-hoc Dunnett or Sidak for multiple comparison test. P values <.05 were considered to be statistically significant.
Results
Quantification of platelet TLR7 expression by flow cytometry
Peripheral blood cells from WT mice underwent intracellular staining with anti-TLR7 to quantitate protein expression. The TLR7 antibody demonstrated both reliable performance and strong specificity in a series of quality control experiments (Fig. S1–S3). Using flow cytometry, blood cells were gated on CD41+CD45− for platelets, CD45+CD41− for leukocytes, and double positive CD41+CD45+ for spontaneous PLA (Figure 1a). TLR7 expressing leukocytes served as an internal positive control. Compared to TLR7−/− mice, WT mice exhibited TLR7-expressing leukocytes (2.4 ± 3.8% vs 17.5 ± 6.9%, p = .03) and platelets (1.3 ± 0.7% vs 3.7 ± 1.1%, p = .0040) (Figure 1b,c). Spontaneous PLA formation is a common consequence of whole blood processing and handling [14]. Gating for double positive cells CD45+CD41+ revealed population of PLA which demonstrated a higher percentage of TLR7 protein expression compared to leukocytes and platelets and was significantly higher than TLR7−/− mice (3.6 ± 5.5% vs 29.3 ± 11.4% p = .0019) (Figure 1c). These results quantify the extent to which circulating murine platelets express TLR7 protein under basal conditions as detected by flow cytometry and reaffirm prior studies that have reported on platelet TLR7 expression [15,16].
Figure 1.
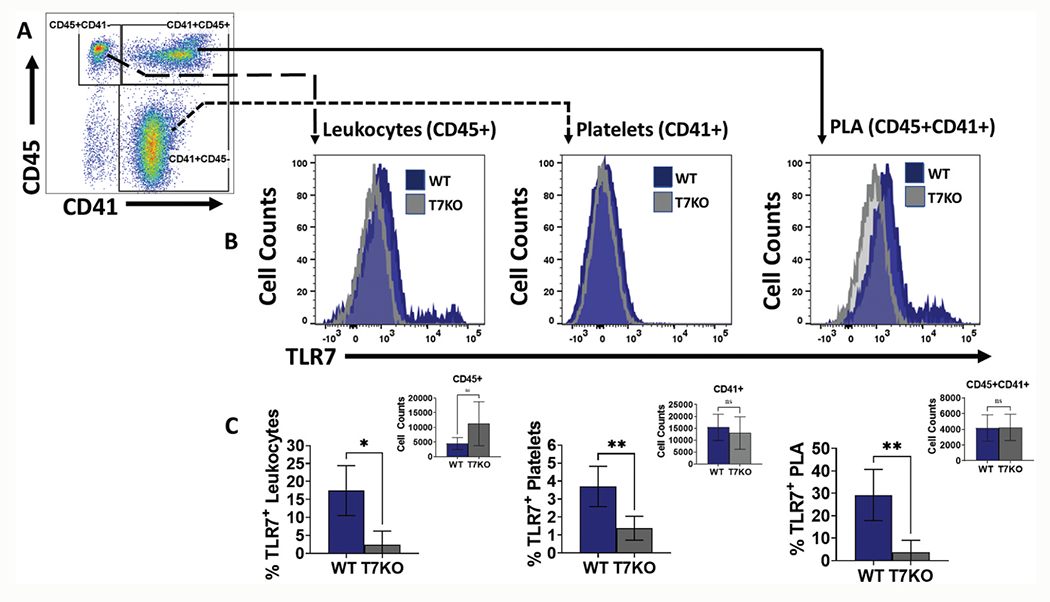
Circulating platelets express TLR7. TLR7 expression was tested in both blood leukocyte and platelets. (a) Representative gating strategy of platelets (CD41+), leukocytes (CD45+), and platelet-leukocyte aggregates-PLA (CD45+CD41+) populations. (b) Representative histograms of TLR7 expression in WT (blue) and TLR7−/− (gray) leukocytes, platelets, and PLA. (c) Percentage of TLR7 positive population. Each bar represents mean ± SD of 5 mice/group of the percentage of TLR7-positive leukocytes (left panel), platelets (middle panel), and PLA (right panel) in circulation. T7KO = TLR7-deficient mice. *p< .05, **p< .01.
TLR7 agonist loxoribine induces platelet activation and PLA formation in vitro
We next evaluated whether stimulation with known TLR7 agonist loxoribine induced platelet activation as measured by surface CD62P (P-selectin) expression. CD62P is a marker of activated platelets and is rapidly mobilized to the surface to facilitate platelet binding with leukocytes. WT blood cells treated with loxoribine demonstrated a dose-dependent increase in CD62P-positive platelets (CD41+CD62P+) (Figure 2a) and PLA (CD45+CD62P+) (Figure 2b). In order to determine the extent of platelet activation in the absence of leukocytes, we treated isolated WT platelets with 1 mM of loxoribine or 10 μ/ml of P3C and demonstrated a statistically significant increase in CD62P+ platelets compared to control (0.05% DMSO in PBS) (Figure 2c), indicating direct effect of the TLR ligands on platelet activation. To determine the necessity of TLR7 in loxoribine-induced platelet activation and PLA formation, we treated blood cells of TLR7−/− mice. As shown in Figure 2d,e, blood cells from TLR7−/− mice treated with loxoribine showed no differences in platelet activation or PLA formation when compared to controls (Figure 2d,e). Of note, WT blood treated with loxoribine showed a significant increase in CD62P+ platelets (30.7 ± 10.8% vs 43.8 ± 2.4%, p = .003) and CD62P+ leukocytes (30.9 ± 4.4% vs 39.5 ± 2.4%, p = .001) above that of TLR7−/− samples (Figure 2d,e). These data not only confirmed a previous study [15] that TLR7 agonist loxoribine can induce PLA formation in whole blood but also demonstrated that loxoribine induced platelet CD62P expression via TLR7 signaling in whole blood and in isolated platelets.
Figure 2.
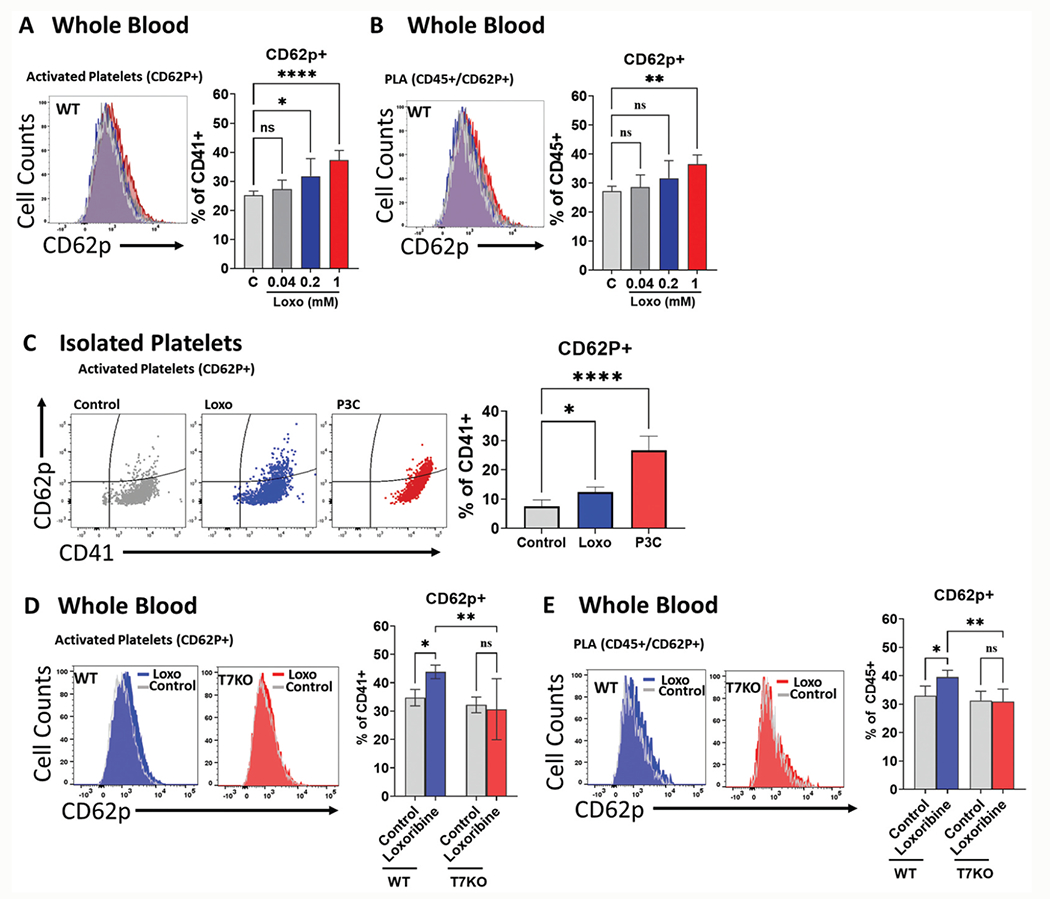
Loxoribine activates platelets in a dose dependent manner and response is dependent on TLR7. Whole blood from WT mice were treated with loxoribine at concentrations of 0.04, 0.2, and 1 mM at 37°C for 15 minutes or platelets from mice were treated with 1 mM of loxoribine under the same conditions. Activated platelets or PLA formation were analyzed by flow cytometry. Activated platelets were defined as the percentage of CD62P positive platelets over total platelets (CD62P+CD41+/CD41+x100%). Activated PLA was defined as the percentage of CD62P positive leukocytes over leukocytes double positive for CD45 and CD41 (CD62P+CD45+/CD45+CD41+x100%). (a) TLR7 activation led to increased platelet activation and (b) PLA formation in WT blood cells in a dose-dependent manner. (c) Mouse platelets treated with 1 mM of loxoribine had increased CD62P+ platelets compared to control. P3C was used as a positive control. (d-e) Blood from WT mice or T7KO mice were treated with 1 mM of loxoribine and analyzed as above. (d) Absence of TLR7 reduced activated platelets (d) and PLA formation (e). P3C = Pam3CSK4, T7KO = TLR7 deficient mice. Bars represent the mean ± SD of 2 replicate experiments run with triplicate samples: *p< .05, **p< .01, ****p< .0001.
In vivo platelet activation and PLA formation in sepsis
Platelets form heterotypic aggregates with leukocytes (PLA) via engagement of CD62P of activated platelets with leukocyte P-selectin glycoprotein ligand-1 (PSGL-1). In sepsis, the ongoing formation of PLA may contribute to reduced platelet number due to adhesion of platelets with leukocytes and peripheral sequestration within the microvasculature [17]. Consistent with our previous work [6], wild-type septic mice exhibited hypothermia, leukopenia, and a decrease in platelet counts at 4 and 24 hours after CLP, although the decrease in platelet counts did not reach statistical significance until 24 hours (Figure 3a). Whole blood was collected and stained with antibodies against platelet CD41, platelet activation marker CD62P and leukocyte marker CD45, as noted above. At 4 hours, flow cytometry revealed an increase in circulating CD62P+ platelets (6.4 ± 4.2% vs 2.3 ± 1.1%, p = .0071) with concomitant increase in average mean fluorescence intensity (MFI) for CD62P in septic mice compared to sham mice (2365 ± 744 vs 1621 ± 332, p= .0017; data not shown). The increase in circulating CD62P+ platelets in CLP mice compared to sham mice persisted at 24 hours, although the results did not achieve statistical significance (3.1 ± 2.2% vs 1.6 ± 0.8%, p = .1309) (Figure 3b,c). Further, at 24 hours circulating CD62P positive platelets quantified by flow cytometry were significantly lower compared to the early 4-hour time point in CLP mice. The platelet count determined by automated cell counter decreased by 26% from 4 to 24 hours post CLP procedure (798.7 x103/μL vs 587.6 x103/μL, Figure 3a). Interestingly, circulating PLA in CLP mice remained significantly higher than sham mice at both 4 (8.9 ± 4.7% vs 3.5 ± 1.9%, p= .0021) and 24 hours (8.6 ± 6.8% vs 2.6 ± 1.5%, p= .0296) (Figure 3d,e). Taken together, these data suggest that early platelet activation is followed by PLA formation and the decrease in systemic platelet counts at 24 hours may in part reflect continued PLA formation.
Figure 3.
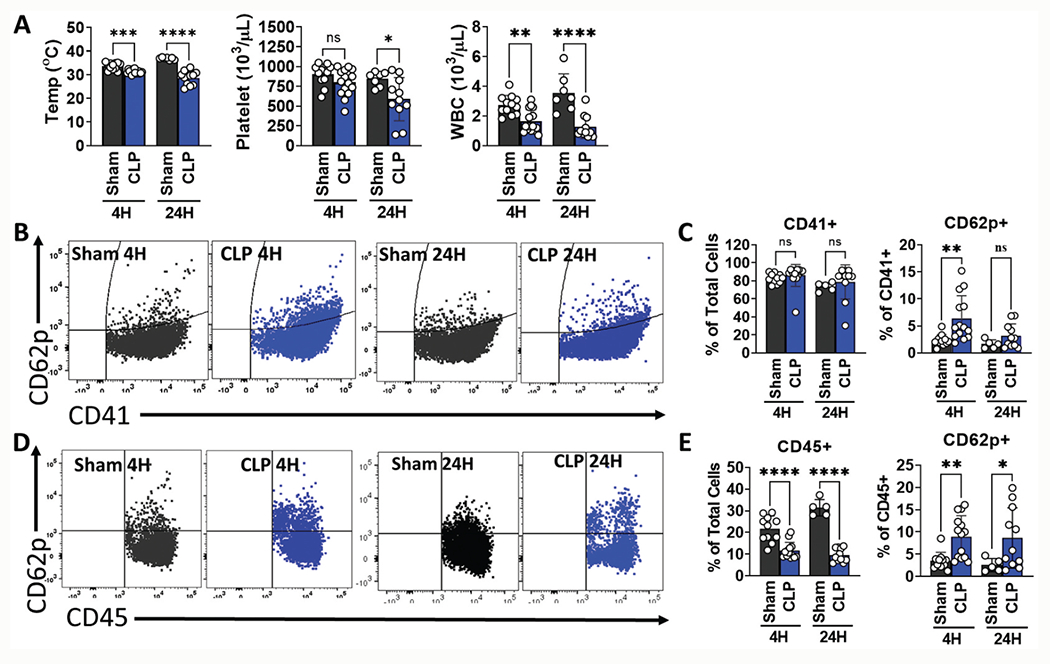
Septic wild type (WT) mice have early increase in circulating platelet-leukocyte aggregates that persists at 24 hours. WT sham or CLP surgery was performed, and blood samples collected at 4 (early) and 24 (late) hours. Activated platelets were defined as the percentage of CD62P positive platelets over total platelets (CD62P+CD41+/CD41+x100%). Platelet-leukocyte aggregates were defined as the percentage of CD62P positive leukocytes over total leukocytes (CD62P+CD45+/CD45+x100%). (a) At both the early and late time points, septic mice had decreased temperatures and WBC counts compared to sham mice. Platelets in septic mice were lower at 4 hours, but this difference did not achieve statistical significance until 24 hours post CLP. (b) and (c) Circulating platelets were activated at the early stage (4 hours) of CLP. (b) Representative flow cytometry dot plots in sham and CLP mice. (c) Percent of CD62P+ platelets in sham and CLP mice at 4 and 24 hours. (d) and (e) Platelet-leukocyte aggregates were formed at 4 hours in septic mice and persisted at 24 hours indicating continuous formation of activated PLA (d) Representative flow cytometry dot plots in sham and CLP mice. (e) Percent of CD62P+ leukocytes in sham and CLP mice at 4 and 24 hours. *p< .05, **p< .01, ***p< .001, ****p< .0001.
Impact of TLR7 signaling on PLA formation in sepsis
To determine the impact of TLR7 signaling on sepsis-induced thrombocytopenia and PLA formation, we collected whole blood from WT and TLR7−/− mice at 4 and 24 h after CLP and examined activated platelets and PLA by flow cytometry. There was no difference in activated platelets at 4 hours between TLR7−/− and WT septic mice (Figure 4a). There was no significant difference in temperatures (31.7 ± 1.6 °C vs 30.5 ± 1.02 °C, p= .054) or platelet counts (761.8 ± 138 x103/μL vs 795.3 ± 95.3 x103/μL, p= .495) (Figure 4b) between TLR7−/− and WT septic mice at 4 hours. At 24 hours, compared to WT mice, TLR7−/− mice had significantly higher temperatures (33.9 ± 2.04 °C vs 29.2 ± 4.97 °C, p= .043), and platelet counts were overall better preserved in TLR7−/− mice although the difference did not achieve statistical significance (750 ± 164.3 x103/μL vs 593.5 ± 247.7 x103/μL, p= .2002) (Figure 4c). Importantly, TLR7−/− septic mice demonstrated decreased circulating PLA at both 4 hours (5.7 ± 1.9% vs 9.99 ± 4.9%, p = .0093) and 24 hours (4.8 ± 1.5% vs 7.5 ± 2.6%, p = .0412) compared to WT mice (Figure 4d,e). These data indicate that the loss of TLR7 signaling results in attenuated PLA formation in bacterial sepsis.
Figure 4.
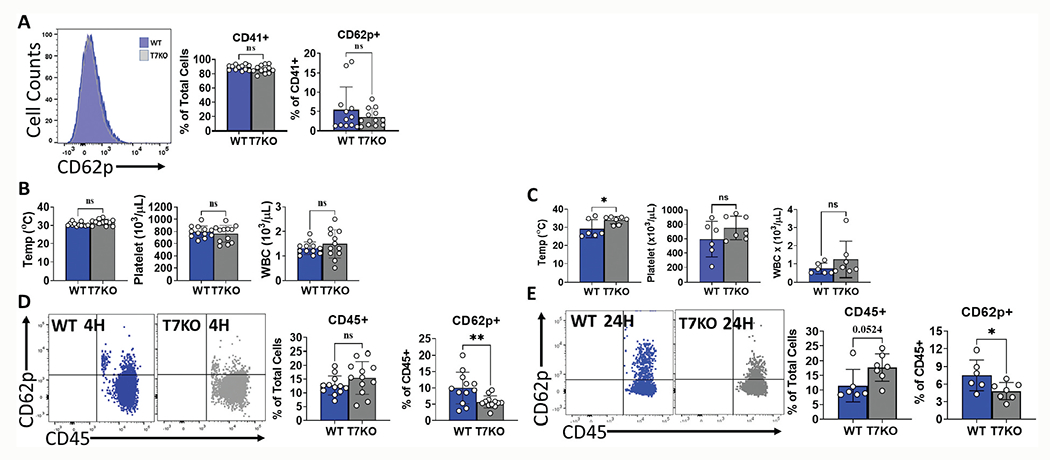
TLR7 deficiency in sepsis attenuates early and late PLA formation. WT and T7KO mice underwent CLP surgery. Whole blood was collected at 4 and 24 hours later and cell counts were performed using automated cell counter. Activated platelets were analyzed using flow cytometry and defined as the percentage of CD62P positive platelets over total platelets (CD62P+CD41+/CD41+x100%). Platelet-leukocyte aggregates was defined as the percentage of CD62P positive leukocytes over total leukocytes (CD62P+CD45+/CD45+x100%). (a) Deficiency of TLR7 did not impact the percentage of activated platelets by CLP at 4 hours. Left panel is the representative flow cytometry histograms. (b) There was no difference in temperature, platelet counts, or WBC counts between WT and T7KO mice at 4 hours; (c) Temperatures were significantly lower in WT mice at 24 hours after CLP vs T7KO mice. (d) Absence of TLR7 reduced PLA at 4 hours after CLP; representative flow cytometry dot plots and corresponding bar graph shown. (e) Absence of TLR7 reduced PLA at 24 hours post CLP; representative flow cytometry dot plots and corresponding bar graph shown. T7KO = TLR7 deficient mice. *p< .05, **p< .01.
Septic plasma induces platelet activation via TLR7 signaling
Sepsis is characterized by systemic inflammation driven by mediators derived from pathogens (i.e., PAMPs) or injured host cells (i.e., DAMPs). We tested the hypothesis that plasma from septic mice induces platelet activation via a TLR7-dependent mechanism. Plasma was pooled from sham and CLP WT mice for each experimental set. Isolated WT platelets treated with WT septic plasma demonstrated a dose-dependent increase in CD62P+ platelets above that of platelets treated with sham plasma (Figure 5a,b). Notably, TLR7−/− platelets treated with 40% septic plasma had a significantly lower response compared to WT platelets (7.3 ± 2.04% vs 10.8 ± 2.2%, p < .0001) (Figure 5c,d) indicating that the septic plasma-induced CD62P expression was partially dependent on TLR7 signaling in platelets. In contrast, P3C, a TLR2 ligand known to activate platelets [18], induced CD62P expression to the same degree in WT and TLR7−/− platelets (Figure 5c,d). These data support the premise of circulating plasma mediator(s) that signals in part via platelet-TLR7 and contributes to platelet CD62P expression in murine sepsis (Figure 5e).
Figure 5.
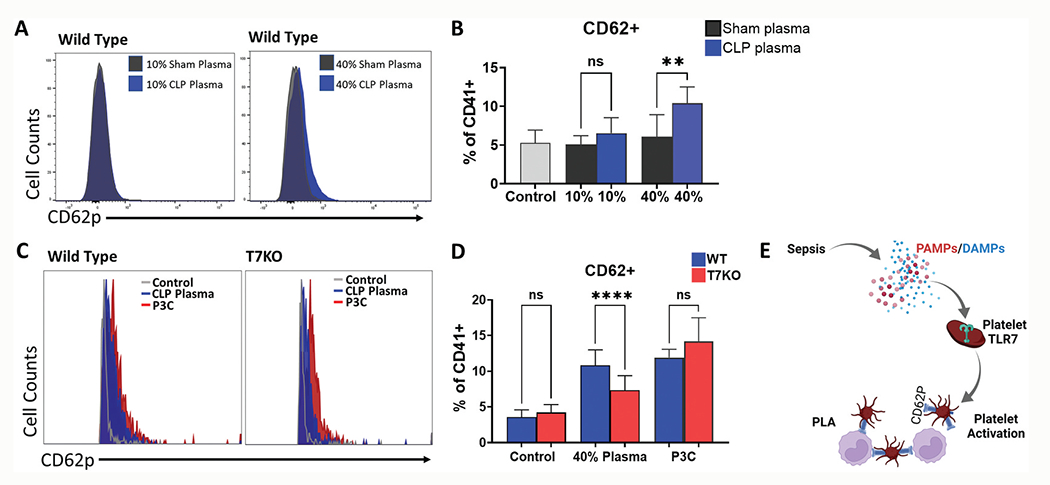
Septic plasma activates platelets and response is attenuated in platelets deficient of TLR7. Mice underwent sham (n = 2) or CLP surgery (n = 4) and 24 hours later whole blood was collected and processed to plasma. Platelets (n = 2) were isolated from whole blood of WT mice and treated with 10% or 40% (v/v) sham or CLP plasma. In the control group, same volume of Tyrode’s buffer was added. Activated platelets were analyzed using flow cytometry and defined as the percent of CD62P+ platelets over total platelets (CD62P+CD41+/CD41+x100%). (a) Representative flow cytometry histograms in WT platelets and (b) Corresponding bar graphs. Platelets isolated from whole blood of WT (n = 2) and T7KO (n = 2) mice were treated with 40% WT CLP plasma (n = 4) and CD62P+ platelets were quantified as above. In the control group, same volume of Tyrode’s buffer was added. (c) Representative flow cytometry histograms in WT and T7KO platelets treated with CLP plasma; (d) CLP plasma induced platelet activation in WT platelets and the response was attenuated in T7KO platelets. Samples treated with P3C (10 μg/ml) served as a positive control. (e) In sepsis, microbial infection leads to release of PAMPs and/or DAMPs that signals via platelet-TLR7 and contributes to downstream platelet activation and PLA formation. Created with BioRender.com. P3C = Pam3CSK4, T7KO = TLR7 deficient mice. WT = Wild-type. Bars represent the mean ± SD of 2 replicate experiments run with triplicate samples, **p< .01, ****p< .0001.
Discussion
We made several notable key findings in the study. Direct treatment with TLR7 ligand in whole blood or isolated platelets was sufficient to activate platelets as determined by CD62P expression in vitro. In vivo, compared to WT mice, TLR7−/− mice had attenuated PLA formation following CLP. Finally, the treatment of isolated platelets with WT septic plasma induced a dose-dependent increase in CD62P+ platelets and this effect was attenuated in TLR7−/− platelets. Together, these data suggest an important role of TLR7 signaling in platelet activation and PLA formation in bacterial sepsis.
Studies have previously reported on TLR7 gene expression and semi-quantitative TLR7 protein expression in platelets using Western blotting or immunofluorescence microscopy [15,16]. Employing WT and TLR7−/− mice and flow cytometry, our study specifically quantified TLR7 protein expression in circulating platelets of whole blood. A few other studies have used intracellular staining and flow cytometry to detect endosomal-based TLR protein expression in platelets [19–21]. Evaluation of TLR3 expression in human platelets demonstrated a small percentage of platelets expressing surface (7.97% ± 2.27) and intracellular (15.85% ± 7.01) TLR3 protein under basal conditions [19]. We did not detect any significant cell surface expression of TLR7 protein (Fig. S3), and this supports prior reports that TLR7 protein is confined to the intracellular endosomal compartment of isolated platelets [15,16]. We did, however, detect that approximately 3–5% of circulating platelets express TLR7 protein under basal conditions, much lower than the 15–20% in leukocytes. Platelets are anucleate fragments of megakaryocytes and contain only a fraction of intracellular material from their parent cells, and therefore this result was not completely unexpected. Importantly, we utilized TLR7−/− mice to assure the specificity of the TLR7 antibody and the resultant signal; this is especially critical given the low frequency of TLR7 positive platelets detected and the nonspecific binding that may be associated with commercially available antibodies. TLR7−/− mice showed minimal levels of nonspecific staining across PLA, leukocytes, and platelets. Using this approach, our study quantifies TLR7 protein expression in a small fraction of circulating platelets and reaffirms prior reporting on platelet-TLR7 expression.
Our data further demonstrate an important role of TLR7 in platelet activation by mediating platelet CD62P expression and PLA formation during bacterial sepsis. Treatment with the TLR7 agonist loxoribine induced PLA formation in a TLR7-dependent manner reaffirming prior studies that evaluated PLA formation based on platelet marker CD41 [15]. Moreover, we further demonstrated the impact of TLR7 signaling on CD62P expression both on platelets and within PLA following loxoribine treatment. CD41 or glycoprotein (GP) IIb is constitutively expressed on the platelet surface, whereas CD62P is a glycoprotein stored within platelet alpha (α) granules and translocated to the surface only upon platelet activation. Platelet CD62P then facilitates platelet–leukocyte interactions via P-selectin glycoprotein ligand-1 (PSGL-1). In our study, it’s presence on the platelet surface indicates its release from alpha granules following stimulation with TLR7 agonist loxoribine and further demonstrates the functionality of platelet-TLR7.
Leukocyte contribution to platelet activation and CD62P expression is a confounder when using whole blood to assess platelet activation. Prior work showed that loxoribine-treated platelets mixed with untreated neutrophils resulted in PLA formation [15]. The study concluded that the activation of platelet-TLR7 was the primary signaling event as no PLA formation occurred when mixing loxoribine treated neutrophils with untreated platelets, implying that it was the activation of platelet-TLR7 that drove PLA formation [15]. In our study, we demonstrated TLR7 agonist loxoribine induced CD62P protein expression in isolated platelets. Notably, the fold increase in CD62P+ platelets above the control samples was similar for both whole blood and isolated platelets (approximate 1.5-fold increase), although the absolute values differed by approximately 20%. This observed difference may be due to centrifugation leading to spontaneous activation of platelets and release of CD62P or the impact of reduced platelet numbers in isolates versus whole blood. However, taken together, these data do suggest that loxoribine is sufficient to activate TLR7 in platelet isolates as determined by CD62P expression in vitro.
We noted an early but small increase in circulating CD62P+ platelets in WT septic mice at 4 hours, but by 24 hours we no longer detected a difference when compared to sham. Prior work demonstrated that IP injection of S. pyogenes into female Balb/c mice neither induced an early (5 hours) or late (18 hours) change in CD62P positivity on platelets [22]. Our early detection of CD62P+ platelets may be secondary to differences in sepsis model (i.e., single bolus and exogenous source versus indolent and endogenous infection) which could contribute to different in vivo responses and platelet activation kinetics. Furthermore, our increased PLA seen at 4 hours, with concurrent increase in CD62P+ platelets, may be indicative of the rapid interactions between activated platelets and leukocytes in vivo. For instance, a prior study was able to detect large PLAs just 1 hour after IP injection with loxoribine, and by 2 hours, the vast majority of platelets within the sample were associated with neutrophils as PLA [15]. In our studies, we were no longer able to detect circulating CD62P+ platelets at 24 hours, but PLA formation persisted even at 24 hours indicating that as sepsis progresses, platelets and leukocytes will invariably continue to form PLA. Taken together, the data suggest that early platelet activation and CD62P expression is followed rapidly by platelet–leukocyte interactions and PLA formation. We speculate that the reduction in platelet counts by 24 hours in septic mice is in part the result of sequestration of platelets via formation of PLA.
TLR7 signaling plays a role in thrombocytopenia in bacterial sepsis, as we recently reported [6], but the underlying mechanism is not clear. In the current study, we found that TLR7−/− septic mice had attenuated PLA formation at both early and late time points compared with WT septic mice. We speculate the reduced PLA formation in part contributes to better preserved platelet counts in TLR7−/− septic mice. A similar role in PLA formation has been demonstrated for TLR2 following infection with P. gingivalis [23]. Interestingly, the intraperitoneal injection of lipopolysaccharide, a TLR4 agonist, resulted in thrombocytopenia and increased fibrinogen adhesion, but failed to induce platelet CD62P expression in vitro [24]. It is likely that multiple TLR signaling pathways synergistically contribute to platelet activation and subsequent PLA formation through various modulations of platelet functions. Notably, our study found that WT platelets, upon exposure to septic plasma, demonstrated an increased surface expression of CD62P, while TLR7−/− platelets exhibited an attenuated response. Treatment with TLR2 ligand induced similar responses in both WT and TLR7−/− platelets indicating the attenuation was specific to the absence of TLR7. Taken together, these data suggest that circulating plasma mediator(s) in septic mice activate platelets in part via a TLR7-dependent mechanism and that TLR7 signaling mediates PLA formation in sepsis.
Sepsis is characterized by an increase in proinflammatory mediators known as DAMPs (danger associated molecular patterns), endogenous biomolecules released from injured tissues and cells capable of triggering host inflammation [25]. One such DAMP is extracellular RNA. In a binding study using TLR7 crystal structure and synthetic short single-stranded (ss) RNA, Zhang, et al., found that TLR7 activation involves synergistic binding of guanosine and ssRNA [26]. Further, in our prior studies, endogenous circulating RNA is increased in the plasma of both septic humans and animals, and approximately 70% was identified as micro(mi)RNA [27]. miRNAs are short, single stranded (ss), non-coding RNAs involved in post-transcriptional regulation but have been shown capable of triggering release of procoagulant and inflammatory factors in a TLR7-dependent manner [6,27,28]. Exogenous human miRNA precursor mimics have also been shown to induce platelet activation, resulting in a six-fold increase in CD62P expression compared to controls and stimulating platelet aggregation in the presence of low-dose thrombin in vitro [29]. Based on these and the findings of our current study, we speculate on a potential role for guanosine and circulating miRNAs as endogenous TLR7 activators driving platelet activation and PLA formation in bacterial sepsis.
There are a few limitations in the current study. First, platelet activation following septic plasma treatment was only partially abolished in TLR7-deficient platelets given that other mediators (e.g., serotonin, ADP, thromboxane) in septic plasma may contribute to platelet activation via alternative signaling pathways. However, the use of TLR7-deficient mice did allow us to determine whether the activation response induced by septic plasma was mediated in part by TLR7. Further, a low level of leukocyte contamination was present in isolated platelet preparations, and therefore a minor contribution from leukocyte-mediated platelet activation cannot be ruled out. However, we speculate that the level of such effect is minimal given leukocytes represented <0.02% of the cell population in isolated platelets. Second, we did not identify the plasma effectors in sepsis that trigger platelet activation via TLR7-dependent pathway. TLR7 is a sensor for guanosine and single-stranded RNA [7,26]. The natural candidates would be plasma RNA such as miRNAs. These plasma RNA in septic mice are biologically active and capable of activating host innate immune response [27,28,30,31]. In addition, our study was limited to male animal samples only. Given known sex differences in human sepsis outcomes [32,33], future study of sex difference in murine sepsis models will be needed. Finally, we only used CD62P expression to define platelet activation. There exist several different markers of platelet activation such as integrin αIIbβ3 (Glycoprotein (GP)2b3a) or platelet factor 4 to evaluate platelet activities that would be important for our future studies.
In summary, TLR expression in circulating blood platelets offers new avenues for understanding the bridge between inflammation and coagulation responses in sepsis and the high incidence of associated thrombocytopenia. As illustrated in Figure 5e, the current study demonstrates that circulating platelets in mice express functional TLR7 that impacts platelet activation in sepsis. Our data highlight the importance of platelet-TLR7 signaling and offer a novel mechanism for platelet activation in bacterial sepsis. A clearer understanding of the mechanism behind platelet depletion in sepsis is essential for disease management and targeted therapeutic interventions.
Supplementary Material
Acknowledgements
We would like to acknowledge Dr. Magali Fontaine of the University of Maryland School of Medicine for project discussion. We would also like to acknowledge the University of Maryland School of Medicine Center for Innovative Biomedical Resources, Flow Cytometry Core– Baltimore, Maryland.
Funding
This work was supported in part by the National Institutes of Health (NIH), United States, grants K08HL153784 (B.W.), R35GM124775 (L. Z.), and R35GM140822 (W.C.), by Mentored Research Training Grant (2019) from the Foundation in Anesthesia Education and Research (B. W.), and by Frontiers in Anesthesia Research Award from International Anesthesia Research Society (W.C.)
Footnotes
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2022.2107627
References
- 1.Lyons PG, Micek ST, Hampton N, Kollef MH. Sepsis-associated coagulopathy severity predicts hospital mortality. Crit Care Med. 2018;46(5):736–742. doi: 10.1097/CCM.0000000000002997 [DOI] [PubMed] [Google Scholar]
- 2.Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. 2010;2(3):e2010024. doi: 10.4084/mjhid.2010.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Stoppelaar SF, van ‘t Veer C, van der Poll T. The role of platelets in sepsis. Thromb Haemost. 2014;112(4):666–677. doi: 10.1160/TH14-02-0126 [DOI] [PubMed] [Google Scholar]
- 4.Al Saleh K, AlQahtani RM. Platelet count patterns and patient outcomes in sepsis at a tertiary care center: beyond the APACHE score. Medicine. 2021;100(18):e25013–e. doi: 10.1097/MD.0000000000025013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assinger A, Schrottmaier WC, Salzmann M, Rayes J. Platelets in sepsis: an update on experimental models and clinical data. Front Immunol. 2019;10(1687). doi: 10.3389/fimmu.2019.01687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams B, Neder J, Cui P, Suen A, Tanaka K, Zou L, and Chao W. Toll-like receptors 2 and 7 mediate coagulation activation and coagulopathy in murine sepsis. J Thromb Haemost. 2019;17(10):1683–1693. doi: 10.1111/jth.14543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, and Flavell RA. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101(15):5598–5603. doi: 10.1073/pnas.0400937101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z, Ohto U, Shibata T, Taoka M, Yamauchi Y, Sato R, Shukla NM, David SA, Isobe T, Miyake K, et al. Structural analyses of toll-like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. 2018;25(12):3371–81.e5. doi: 10.1016/j.celrep.2018.11.081 [DOI] [PubMed] [Google Scholar]
- 9.Jian W, Gu L, Williams B, Feng Y, Chao W, Zou L. Toll-like receptor 7 contributes to inflammation, organ injury, and mortality in murine sepsis. Anesthesiology. 2019;131(1):105–118. doi: 10.1097/ALN.0000000000002706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–189. doi: 10.1016/j.blre.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, Lordkipanidzé M, and Nash GB. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets. 2015;26(6):507–520. doi: 10.3109/09537104.2015.1064881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman HC, Hugill A, Dear NT, Ashcroft FM, Cox RD. Deletion of nicotinamide nucleotide transhydrogenase: a new quantitative trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes. 2006;55(7):2153–2156. doi: 10.2337/db06-0358 [DOI] [PubMed] [Google Scholar]
- 13.Toscano MG, Ganea D, Gamero AM. Cecal ligation puncture procedure. J Vis Exp. 2011;(51):2860. doi: 10.3791/2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding SA, Din JN, Sarma J, Jessop A, Weatherall M, Fox KA, andNewby D. Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: methodological considerations. Thromb Haemost. 2007;98(2):451–456. doi: 10.1160/TH06-11-0654 [DOI] [PubMed] [Google Scholar]
- 15.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, and Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124(5):791–802. doi: 10.1182/blood-2013-11-536003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, Garvy BA, Myint T, and and Whiteheart SW. Platelets endocytose viral particles and are activated via TLR (Toll-Like Receptor) signaling. Arterioscler Thromb Vasc Biol. 2020;40(7):1635–1650. doi: 10.1161/ATVBAHA.120.314180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vardon-Bounes F, Ruiz S, Gratacap M-P, Garcia C, Payrastre B, Minville V. Platelets are critical key players in sepsis. Int J Mol Sci. 2019;20(14):3494. doi: 10.3390/ijms20143494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, and Schattner M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res. 2014;133(2):235–243. doi: 10.1016/j.thromres.2013.11.028 [DOI] [PubMed] [Google Scholar]
- 19.Anabel A-S, Eduardo P-C, Pedro Antonio H-C, Carlos S-M, Juana N-M, Honorio T-A, Nicolás V-S, and Sergio Roberto A-R. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum Immunol. 2014;75(12):1244–1251. doi: 10.1016/j.humimm.2014.09.013 [DOI] [PubMed] [Google Scholar]
- 20.Thon JN, Peters CG, Aslam R, Rowley J, Weyrich AS, Semple JW, Flaumenhaft RC, and Italiano JE. The functional role of TLR9 in human platelets. Blood. 2011;118(21):366. doi: 10.1182/blood.V118.21.366.366 [DOI] [Google Scholar]
- 21.D’Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, Yin H, and Schattner M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J Thromb Haemostasis. 2015;13(5):839–850. doi: 10.1111/jth.12842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurley SM, Lutay N, Holmqvist B, Shannon O. The dynamics of platelet activation during the progression of streptococcal sepsis. Plos One. 2016;11(9):e0163531. doi: 10.1371/journal.pone.0163531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE, et al. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009;104(3):346–354. doi: 10.1161/CIRCRESAHA.108.185785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andonegui G, Kerfoot SM, McNagny K, Ebbert KVJ, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106(7):2417–2423. doi: 10.1182/blood-2005-03-0916 [DOI] [PubMed] [Google Scholar]
- 25.Ito T. PAMPs and DAMPs as triggers for DIC. J Intensive Care. 2014;2(1):67. doi: 10.1186/s40560-014-0065-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Ohto U, Shibata T, Krayukhina E, Taoka M, Yamauchi Y, Tanji H, Isobe T, Uchiyama S, Miyake K, et al. Structural analysis reveals that toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity. 2016;45(4):737–748. doi: 10.1016/j.immuni.2016.09.011 [DOI] [PubMed] [Google Scholar]
- 27.Wang S, Yang Y, Suen A, Zhu J, Williams B, Hu J, Chen F, Kozar R, Shen S, Li Z, et al. Role of extracellular microRNA-146a-5p in host innate immunity and bacterial sepsis. iScience. 2021;24(12):103441. doi: 10.1016/j.isci.2021.103441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu J, Feng Y, Jeyaram A, Jay SM, Zou L, Chao W. Circulating plasma extracellular vesicles from septic mice induce inflammation via microRNA- and TLR7-Dependent mechanisms. J Immunol. 2018;201(11):3392–3400. doi: 10.4049/jimmunol.1801008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaslavsky A, Adams M, Cao X, Yamaguchi A, Henderson J, Busch-Østergren P, Udager A, Pitchiaya S, Tourdot B, Kasputis T, et al. Antisense oligonucleotides and nucleic acids generate hypersensitive platelets. Thromb Res. 2021;200:64–71. doi: 10.1016/j.thromres.2021.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou L, He J, Gu L, Shahror RA, Li Y, Cao T, Wang S, Zhu J, Huang H, Chen F, et al. Brain innate immune response via miRNA-TLR7 sensing in polymicrobial sepsis. Brain Behav Immun. 2022;100:10–24. doi: 10.1016/j.bbi.2021.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou L, Feng Y, Xu G, Jian W, Chao W. Splenic RNA and MicroRNA mimics promote complement factor B production and alternative pathway activation via innate immune signaling. J Immunol. 2016;196(6):2788–2798. doi: 10.4049/jimmunol.1502106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakr Y, Elia C, Mascia L, Barberis B, Cardellino S, Livigni S, Fiore G, Filippini C, and Ranieri V. The influence of gender on the epidemiology of and outcome from severe sepsis. Crit Care. 2013;17(2):R50. doi: 10.1186/cc12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angele MK, Pratschke S, Hubbard WJ, Chaudry IH. Gender differences in sepsis: cardiovascular and immunological aspects. Virulence. 2014;5(1):12–19. doi: 10.4161/viru.26982 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.