

Abstract
Background and Objectives
Missense variants of the valosin-containing protein (VCP) gene cause a progressive, autosomal dominant disease termed VCP multisystem proteinopathy (MSP1). The disease is a constellation of clinical features including inclusion body myopathy (IBM), Paget disease of bone (PDB), frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS), typically reported at a frequency of 90%, 42%, 30%, and 9%, respectively. The Hispanic population is currently underrepresented in previous reports of VCP myopathy. We expand our genotype-phenotype studies in 5 Hispanic families with the c.476G>A, p.R159H VCP variant.
Methods
We report detailed clinical findings of 11 patients in 5 Hispanic families with the c.476G > A, p.R159H VCP variant. In addition, we report frequencies of the main manifestations in 28 additional affected members of the extended family members. We also compared our findings with an existing larger cohort of patients with VCP MSP1.
Results
FTD was the most prevalent feature reported, particularly frequent in females. PDB was only seen in 1 patient in contrast to the earlier reported cohorts. The overall frequency of the different manifestations: myopathy, PDB, FTD, and ALS in these 5 families was 39%, 3%, 72%, and 8%, respectively. The atypical phenotype and later onset of manifestations in these families resulted in a noticeable delay in the diagnosis of VCP disease.
Discussion
Studying each VCP variant in the context of ethnic backgrounds is pivotal in increasing awareness of the variability of VCP-related diseases across different ethnicities, enabling early diagnosis, and understanding the mechanism for these genotype-phenotype variations.
Heterozygous missense variants in the valosin-containing protein (VCP) gene are associated with autosomal dominant, progressive adult‐onset disorder also termed multisystem proteinopathy (MSP1). It is characterized by inclusion body myopathy (IBM), Paget disease of bone (PDB), and frontotemporal dementia (FTD). Related clinical presentations include amyotrophic lateral sclerosis (ALS), Parkinson disease, cardiomyopathy, and Charcot-Marie-Tooth disease.1 Here, we report 5 unrelated Hispanic families identified carrying the same c.476G>A; R159H variant in exon 5 of the VCP gene (NM_007126.5) (Figure 1). The most common finding among these families was FTD followed by myopathy. The high intra- and interfamilial variation in the phenotype1,2 made diagnosis and genotype-phenotype correlations challenging.
Figure 1. Pedigrees of 4 Hispanic Families.

The R159H variant was confirmed in all 11 patients described, but not in the 28 family members who are obligate carriers of the variant. The location of the shading in the figure indicates the different manifestations of the disease within the family. Upper right = inclusion body myopathy; lower right = Paget disease of bone; lower left = expression of frontotemporal dementia; upper left = ALS. ALS = amyotropic lateral sclerosis. The arrow indicates the proband.
IBM is characterized by progressive weakness and atrophy of pelvic and shoulder girdle muscles.3 IBM presents with proximal upper and lower limb weakness, followed by scapular winging and occasionally atrophy of distal muscles.4 In VCP-associated IBM, the onset of muscle weakness usually starts in people in their 30s and 40s. Ultimately, muscle weakness progresses to involve respiratory and cardiac muscles, and patients die from respiratory or cardiac failure typically in their 40s–60s.2 Histologically, IBM is characterized by cytoplasmic rimmed vacuoles containing protein aggregates of tau, amyloid, and TAR DNA-binding protein 43 (TDP-43).3,5 These proteins are typically found in ubiquitinated inclusions in the brains of patients with neurodegenerative diseases. In muscle, TDP-43 is normally found in the nucleus, but in this disease, TDP-43 is also found primarily in ubiquitinated inclusions in sarcoplasm.3,5 Laboratory findings include elevated serum creatine kinase (CK). Clinical testing includes EMG, which can show myopathic and neuropathic changes, typical of ALS,2 and nerve conduction studies (NCSs), which can reveal Charcot-Marie Tooth changes.
FTD is an early-onset dementia that is most diagnosed between the ages of 45 and 64 years6,7; however, in VCP disease, it is associated with an earlier age at onset. Previous studies have shown that cortical regions in patients with VCP-associated FTD contained TDP-43 intranuclear ubiquitinated inclusions.5 FTD typically has a lower penetrance, developing in only 30% of patients in VCP disease.6 FTD is classified into 3 clinical variants: behavioral variant (bvFTD), which is associated with executive deficits; nonfluent variant primary progressive aphasia with deficits in speech and grammar; and semantic-variant primary progressive aphasia, which is a disorder of semantic knowledge and naming.7
PDB is a skeletal disease resulting from overactive osteoclasts and osteoblasts leading to increased bone resorption. Serum alkaline phosphatase levels are usually elevated in active PDB.8
ALS is a motor neuron disease associated with degeneration of upper and lower motor neurons typically causing death within a few years of onset due to respiratory failure. ALS and FTD commonly have TDP-43 aggregates in affected tissues, which are also found in muscle of patients with VCP-related myopathy.9
There are currently over 50 missense VCP variants that cause disease.9 Previous studies have shown some correlations between the specific variant and the phenotype seen in VCP-associated disease.6,10
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the University of California Irvine Institutional Review Board (#2007-5832). All patients provided approved consent for the studies. The study is listed under ClinicalTrials.gov (Identifier: NCT01353430).
Statistical Analysis
Using SPSS 28.0 for Windows, we performed a cross-table analysis of the clinical features in symptomatic and asymptomatic individuals in our cohort with the R159H variant compared with the bigger cohort of 187 patients with all VCP variants.6 Statistical significance using the Pearson χ2 test for categorical variables and the t test for continuous variables was demonstrated when a p value of <0.05 was obtained. Survival time was analyzed using the Kaplan-Meier survival method.
Histologic Staining
Hematoxylin and eosin (H&E) and immunohistochemistry staining was performed on muscle and brain tissues using standard protocols.5,15
Data Availability
Anonymized data will be shared by request from any qualified investigator.
Results
Clinical Reports
Medical records of 11 individuals from the 5 Hispanic families were reviewed. Clinical findings and diagnostic studies are summarized in Table 1.
Table 1.
Clinical and Laboratory Data of Affected Individuals With the VCP R159H Variant

Family 1
Case 1
Patient III:3 is a 78-year-old woman who received genetic testing because of her brother's diagnosis of VCP-associated IBM caused by the R159H VCP variant.11 The behavioral changes were noted at age 71 years, including cognitive decline, weight loss, and personality change such as anger issues, and acute psychosis suggestive of FTD. Brain MRI displayed mild, chronic small vessel ischemic changes (Figure 2), and CT of the head 4 months later showed some dilatation in ventricular size of the frontal horns indicating progression of the disease. As the FTD progressed, she experienced increased aggression, obsessive behavior, and mania, struggled with short-term and long-term memory, and required assistance in feeding and toileting.
Figure 2. Brain Imaging Findings in a 71-Year-Old Woman With Dementia (Case 1).

(A) Brain MRI axial T2 image showing slight prominence of the frontal horns of the lateral ventricle. Few nonspecific foci of increased T2 signal in the periventricular and subcortical white matter of the frontal lobes were noted (partially shown) without remarkable cortical atrophy. (B) Brain CT axial image performed 4 months later shows a slight increase in lateral ventricular size.
She had a notable family history of dementia and myopathy in several family members, with only 1 individual in this family being diagnosed with PDB (Figure 1 and Table 1). The ethnic background is Hispanic and Native American.
Case 2
Patient III:15 is a 58-year-old man with progressive muscle weakness beginning at age 52 years, at which time red-rimmed vacuoles were identified on muscle biopsy. At age 55 years, he developed a wide-based gait with L>R bilateral foot drop for which he used an ankle foot orthosis and a walker 2 years later with increased difficulty with fine motor tasks, dressing and bathing, increased fatigue, hip, and shoulder pain.
Case 3
Patient III:17 is a 65-year-old woman with progressive memory loss and diminished speech associated with hypersomnia during the day. She had rapid progression over a period of 1 month, becoming very confused with everyday tasks and displaying marked anomia. At age 67 years, she had a hemorrhagic infarct in the right frontal and temporal region of the brain and developed seizures. She was thought to have vascular dementia until she was identified with the familial variant. She had worsening cognitive function until she died at age 71 years.
Family 2
Case 4
The proband of this family (IV:1) with Mexican and Native American ancestry started to notice stumbling at age 51 years, and muscle weakness with difficulty walking starting at age 57 years. He had proximal muscle weakness involving the lower extremities and interosseus muscles of the hands with a distally predominant large fiber sensory loss in the lower extremities. Biopsy of the left biceps muscle was performed at age 58 years. There was a paucity of type 2 muscle fibers, scattered red-rimmed vacuoles, and fat cells5 (Figure 3).
Figure 3. Muscle Biopsy Findings of Case 4.

H&E-stained section (A) shows fiber size variability including angular and roungulated fibers, along with nonspecific abnormalities including mildly increased numbers of internalized nuclei, and a basophilic tinged regenerative/degenerative fiber (lower middle of image A). Engel-Gomori trichrome–stained sections (B and C) demonstrate muscle fibers with peripherally located vacuoles with red rimming (red-rimmed vacuoles), see arrows in C. Immunohistochemical staining with antibodies against LC-3 (D) and TDP-43 (E) demonstrates positivity within vacuoles (E, arrows). In addition to vacuolar staining, the autophagic vacuolar marker LC-3 also demonstrates increased granular sarcoplasmic staining within fibers (D, arrows). Alkaline phosphatase reactivity (F) highlights a rare regenerative/degenerative fiber and shows normal, nonincreased staining within endomysial and perimysial connective tissue. Cytochrome oxidase (COX) reaction displays normal variation between fiber types and the presence of increased numbers of COX-negative fibers (G, arrows). Immunohistochemical staining for MHC-1 (H) demonstrates no upregulation of MHC-1 (a possible distinguishing feature from other vacuolar myopathies such as sporadic inclusion body myositis [sIBM], which typically shows diffuse sarcoplasmic upregulation of MHC-1 immunostaining). The muscle biopsy also showed mild neuropathic changes, including angulated fibers (see A and B) that consisted of both type 1 and type 2 fibers (not shown). Mild fiber type grouping, consistent with denervation followed by reinnervation, was appreciated by ATPase staining at pH = 4.3 (I) for fiber type determination. The right half of photomicrograph (I), outlined by arrows, demonstrates mild fiber type grouping evidenced by expanded areas of darkly staining type 1 fibers along with mild loss of checkerboard appearance. The left half of photomicrograph (I) demonstrates a more regular checkerboard appearance.
At age 58 years, the patient was diagnosed with sporadic inclusion body myositis (sIBM) and chronic inflammatory demyelinating polyneuropathy based on NCSs, muscle biopsy, increased CSF protein, and elevated immunoglobulin (Ig) E, IgA, IgM, and complement component 1q immune complex. Immunohistochemical staining of muscle (Figure 3H) did not demonstrate diffuse sarcoplasmic upregulation of major histocompatibility complex class 1 (MHC-1) immunostaining12 typically associated with sIBM. He was started on duloxetine HCL typically used to treat anxiety, fibromyalgia, and chronic muscle pain without improvement of his symptoms. A trial of levocarnitine and other mitochondrial supplements was also not beneficial. At age 60 years, he was started on a trial of intravenous immunoglobulin, which may have provided a slight improvement in motor strength, which was apparent retrospectively when he discontinued the treatment.
Because of the beneficial effects of sirolimus in a VCP preclinical model,13,14 the patient was started empirically on a trial of sirolimus (Rapamycin) at age 58 years. However, the patient developed an exacerbation of his diverticulitis, in addition to hair loss, and the trial was discontinued. At age 61 years, he had progressive weakness, muscle atrophy, and fasciculations, bilateral foot drop, diminished reflexes, and started using a walker to ambulate. Vibration sense was diminished in his toes, and he had numbness in the left arm and foot. Worsening of muscle strength necessitated the use of a wheelchair. He also reported dysphagia. At age 63 years, he complained of nighttime paresthesia, painful leg cramps, and orthopnea. He was bedridden for 2 years before he died at age 68 years.
On further review of family history, the patient's mother was diagnosed with dementia and died at age 63 years, most likely from FTD. Other family members were diagnosed with myopathy, FTD, and ALS. DNA analysis was positive for the c.476G>A, p.R159H variant in the VCP gene.
Case 5
The proband's 73-year-old sister (IV:6) had memory impairment at age 68 years and was placed on donepezil by her primary care provider. Neurologic assessment revealed a score of 14/30 on the Montreal Cognitive Assessment with poor fund of knowledge and unawareness of current events. At that time, she had normal muscle tone and strength in all extremities. At age 70 years, she had a syncopal episode, and CT of the brain revealed bilateral cerebral atrophy, which was most prominent in the temporal regions. At age 71 years, she had an unsteady gait requiring the use of a transfer chair. At age 72 years, she had an unwitnessed fall, and CT of the brain revealed atrophy of the left frontal and temporal lobes (figure 4). Her condition deteriorated with anomia and inability to perform activities of daily living (ADLs) until she died at age 73 years.
Figure 4. Brain Imaging Findings in a 72-Year-Old Woman With Frontotemporal Dementia (Case 5).

CT of the brain (A and B, axial and C, coronal images) revealed cortical atrophy mainly of the left frontal and temporal lobes and dilation of the third and lateral ventricles, left more than right.
Family 3
Case 6
This 63-year-old woman (IV:2) started showing progressive functional decline with social withdrawal at age 56 years. She has a history of migraines, partial complex seizures, and myoclonus. At age 57 years, she had a generalized tonic-clonic seizure followed by additional cognitive and behavioral changes. Shortly thereafter, she began using words imprecisely, having compulsive behaviors and sleep disturbance. At age 57 years, she had multiple hospitalizations for odd behavior and decreased responsiveness. She also displayed unsteady gait, dysphagia, perseverative speech, tremulous hands, and hand clapping. Neuropsychiatric testing showed some frontal dysfunction and impairment of executive function, but with some sparing of delayed recall. Her symptoms progressed to include delusional thinking, poor judgment, and difficulty managing instrumental ADLs. At age 59 years, brain MRI showed global volume loss and mild enlargement of the third and lateral ventricles (figure 5). EEG revealed seizures originated in the left frontal cortex, and prion disease was suspected. A brain biopsy of the left frontal cortex reported no evidence of encephalitis or Creutzfeldt-Jakob disease.
Figure 5. Brain Imaging Findings in a 63-Year-Old Woman With Progressive Functional Decline (Case 6).

Brain MRI coronal T2 images at ages 58 and 62 years (A and B) demonstrating progressive cortical atrophy in the frontotemporal region with progressive dilatation of the lateral and 3rd ventricles. Brain MRI midline sagittal FLAIR T1 images at ages 58 and 62 years (C and D) showing interval severe thinning and atrophy of the corpus callosum and atrophic changes in the midbrain and posterior fossa.
At age 60 years, she developed aphasia with echolalia, aberrant motor behavior, and dysphagia. Family history was positive for early-onset dementia referred to as AD with seizures in her mother, maternal uncle, and maternal cousin. Genetic testing for autosomal dominant disorders including PSEN1, PSEN2, and APP was negative. Whole-exome sequencing (WES) only detected a heterozygous pathogenic VCP variant c.476C>A (p.R159H). Subsequent histologic analysis of the brain biopsy revealed increased TDP-43 staining with nuclear to cytoplasmic translocation of TDP-4315 (figure 6).
Figure 6. Brain Biopsy Pathology Findings of Case 6.
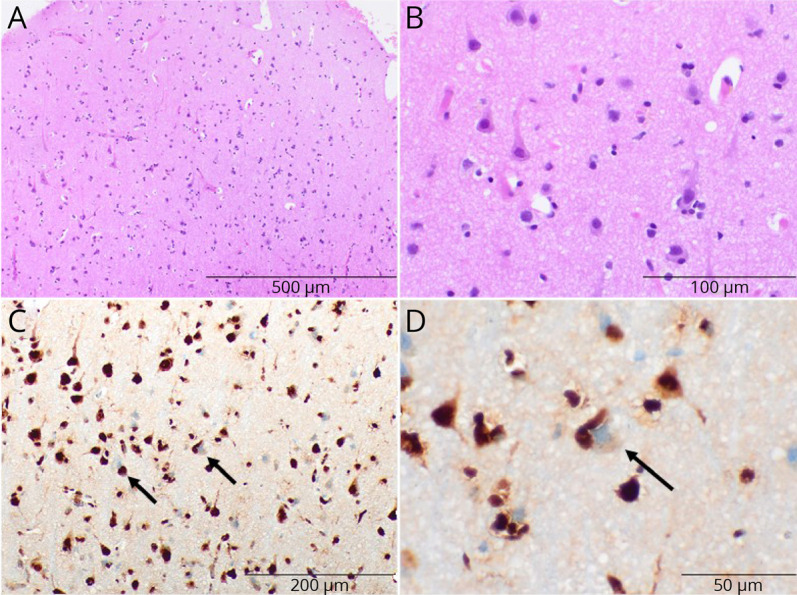
(A and B) Hematoxylin and eosin–stained sections show the neocortex with normal neuronal lamination and populations and no appreciable neuron dropout or neuronal inclusions. (C and D) Total TDP-43 staining shows many neurons with nuclear to cytoplasmic translocation of TDP-43 (arrows).
At age 63 years, in addition to continued cognitive decline, the patient developed parkinsonian features with masked facies, decreased blink rate, short steppage gate, and hunched posture. She was started on carbidopa-levodopa.
Family 4
Case 7
The 27-year-old proband (III:1) of this Hispanic family started having progressively worsening balance and memory impairment starting at age 22 years. She reported episodes of loss of consciousness, olfactory hallucinations, and feelings of sudden déjà vu since childhood. Brain MRI at age 22 years showed minimal nonspecific T2/fluid attenuated inversion recovery hyperintensities in subcortical white matter, which were attributed to her migraine. Her electroencephalogram was unremarkable. At age 25 years, she noticed an increased frequency of falls and weakness in her legs for which she started using a cane for ambulation. She also reported confusion, worsening of her memory impairment, increased fatigue associated with excessive daytime sleepiness, and obstructive sleep apnea attributed to obesity, managed with continuous positive airway pressure. Physical examination showed normal tone, bulk, and 5/5 strength in all 4 extremities. Reflexes were diminished in all 4 extremities with bilateral Babinski sign.
At age 26 years, her laboratory results showed elevated liver enzymes, and she was diagnosed with nonalcoholic steatohepatitis. VCP variants are occasionally associated with hepatic steatosis.16 At that time, her neuropsychological evaluation indicated mild difficulties on some delayed tasks of memory, visual spatial skills, and working memory.
At age 27 years, she reports ongoing weakness in both arms and legs. Genetic testing using a comprehensive myopathy panel revealed the c.476G>A (p.R159H) VCP variant. Her family history was significant for her mother and 2 maternal uncles with myopathy, memory problems, and impulsive behavior and a maternal cousin with ALS.
Case 8
The 61-year-old proband's mother (II:2) started having muscle weakness at age 50 years. Sequence analysis of the VCP gene confirmed the pathogenic c.476G>A (p.R159H) variant. At age 60 years, she continues to have progressive muscle weakness in bilateral upper and lower extremities, tingling and numbness in the face and hands, fatigue, swallowing difficulty, and gait abnormalities. She also reports progressive memory loss, trouble finding words, and learning new skills. Physical examination at age 61 years shows weakness of bilateral upper and lower extremities with distal muscle atrophy of the left foot more than the right foot, absent reflexes throughout, decreased sensations to light touch and pinprick in the lower extremities, and a Trendelenburg gait.
Case 9
This male patient (III:7) aged 55 years started having progressive gait abnormalities at age 47 years, and 1 year later, because of falls, he started using a cane for ambulation. Physical examination at age 50 years revealed motor weakness in bilateral lower extremities with right foot drop and Trendelenburg gait. At age 51 years, he was diagnosed with ALS and treated with riluzole for 6 months without any improvement.
Case 10
This male patient (II:9) aged 66 years started having motor weakness in both legs at age 60 years and was diagnosed with ALS. He was managed with physical therapy for 6 months; however, his condition continued to deteriorate. Currently, he has poor balance, right foot drop, and worsening bilateral lower extremity motor weakness, necessitating the use of a cane to ambulate.
Family 5
Case 11
A 61-year-old Hispanic woman of Mexican ethnic background has a history of asymmetric weakness in the distal upper extremities started at age 40 years, with rapid progression of weakness and atrophy at age 49 years diagnosed as multifocal motor neuropathy. Electrodiagnostic studies, however, did not show conduction block, and antiganglioside antibodies were negative. She was treated with intravenous immunoglobulin with no objective improvement; however, there has been no progression of her symptoms either. Genetic testing with WES revealed the pathogenic p.R159H VCP variant. Family history is unavailable because she was adopted.
Summary of Results and Comparison With Previous Reported Cases
We report detailed clinical findings in 11 patients (4M/7F) at a mean age/range of 64.7/27–78 years in 5 unique families of Hispanic origin with the R159H variant (Table 1). We noted that the age at onset of clinical features of FTD (average 64.2 ± 6.3 years) trended later compared with the previously reported age at onset in the entire cohort of 187 individuals (average 56.1 ± 13.7 years) (p = 0.11). The frequency of myopathy was 64%, lower than the previously reported 90% (p = 0.008).6 Myopathy age of onset (average 46.9 ± 12.8 years) was comparable to the previously reported cohort (average 43.4 ± 7.7 years).6 There were no cases of PDB vs previously reported incidence of 42% (p = 0.007) in the general VCP population.6 The frequency of dementia in the 11 patients was 46% vs previously reported 30% (p = 0.261) and ALS frequency at 18% vs 9% as reported previously (p = 0.280) in the larger cohort.6
In addition, we reviewed the pedigrees of these families and identified an additional 28 affected members who are obligate carriers of the variant for whom only data on the main manifestations were available. We analyzed the frequencies of the different manifestations in the complete cohort (N = 39) (Table 2). The most striking feature was that patients had a higher frequency of dementia of 72% compared with previous reports of 30% in the cohort of 186 individuals (p < 0.001).6 We found that patients had a lower frequency of myopathy of 39% compared with previous reports of 90% (p < 0.001).6 Of interest, we found a very low incidence of PDB, only present in 1 individual (3%), compared with the reported incidence of 42% (p < 0.001). We did not find a difference in the frequency of ALS compared with the main cohort (Table 2). We compared the frequency and age at onset of FTD of the R159 variant with those in the larger cohort (R159H/C) and found similar results. Of those individuals with the R159H variant, 3/5 (60%) from family 2 in the current study had dementia at a mean age of 66 years, and 7/10 (70%) individuals with the R159C variant had dementia at a mean age of 60 years. We reviewed sex differences and found that FTD in females with this variant was higher (85%) than FTD in females with all other VCP variants reported previously (39%) (p < 0.001) (unpublished analysis of data6).
Table 2.
Proportion of Each Feature in Patients With the VCP R159H Variant Compared With All VCP Variants6

We analyzed the influence of phenotype on survival in the current cohort of 11 patients with the R159H variant; the mean survival was 72.5 years (standard error 1.82, 95% confidence interval 68.93–76.07). This indicates a milder phenotype associated with the R159H variant, in contrast to a previous study showing that the mean survival for the affected individuals with 10 different allelic variants was 62.53 years (standard error 0.94, 95% confidence interval 60.68–64.39).10 The average life span after diagnosis of myopathy and Paget was 18 and 19 years, respectively. Dementia led to rapid progression of the disease with an average life span of 6 years.10
Because large families with a particular phenotype might drive associations, a proband only analysis was performed of the 5 Hispanic families with the R159H variant. Findings were compared with data from 187 patients with 15 different VCP variants.6 Proband analysis of 5 patients (1 M/4 F) was at a mean age/range of 59.4/range 27–78 years. The average age at onset of clinical features of FTD was 64 ±11.3 years, in contrast to the previously reported age at onset (average 56.1 ± 13.7 years) (p = 0.49). The frequency of myopathy was 60%, lower than the previously reported 90% (p = 0.035). The age at onset of myopathy (average 39.7 ± 17.5 years) was comparable to the previously reported cohort (average 43.4 ± 7.7 years). There were no cases of PDB in these 5 individuals vs the previously reported incidence of 42% (p = 0.06) in the general VCP population. The frequency of dementia was 40% vs previously reported 30% (p = 0.61) and ALS frequency at 0% vs 9% as reported previously (p = 0.50).
Although 1 Hispanic patient from our study was also included in the cohort of 187 individuals (case 4 from family 2), it is highly unlikely that the data would be skewed as a result of including this individual. We did not adjust for multiple testing, and therefore, these results should be considered exploratory.
Discussion
We report 5 unrelated Hispanic families with the p.R159H VCP variant. Of interest, among the ancient population of the Basque, 2 families carried the c.476 G>A; p.R159H variant in the VCP gene.17 Haplotype analysis was not performed on the subjects; however, thorough genealogical examinations of these 5 families did not indicate that they shared a common ancestor. Because the variant has been identified in the ancient Basque population, we speculate that this variant could be the result of a founder effect.
The analysis was performed by comparing 11 and 28 patients from 5 Hispanic families (all with the R159H variant) to 187 patients with 15 different VCP variants (R155H, R155C, R155P, R191Q, R159C, R159H, L198W, R95G, R93C, A232E, N387H, G97E, A160P, G128A, and M158I).6 The only genotype-phenotype association was IBM where variant group R159C was found to have a later age at onset of muscle weakness (57 years) compared with L198W (37 years), R155H (43 years), R155P (43 years), or R155C (38 years) (p ≤ 0.04). Otherwise, this study did not find differences between the genotypes.6
Among our 5 families, the incidence of FTD is high, with 72% of the family members showing FTD phenotypes, which is significantly higher than previously reported.6 FTD was mainly seen in females, and only 1 of them had myopathy, suggesting either a genotype-phenotype correlation or that this variant may be considered milder in the Hispanic patients. This is supported by survival analysis in our 11 patients with the R159H variant. The mean survival was 72.5 years in contrast to the previously reported mean survival of 62.53 years for affected individuals with 10 other VCP variants.10 Notably, homozygous p.R159H variants were reported in a Belgian male from a consanguineous family18 with myopathy at an early age of 29 years and a markedly elevated CK value (1,138 U/L) and PDB. This again suggests a milder phenotype associated with this variant because VCP variant homozygosity had previously been thought to be incompatible with life in view of our studies in the R155H VCP homozygous mouse model.19
We found a higher incidence of dementia in this cohort. The most pronounced early symptoms of bvFTD in our subjects included personality changes, disinhibition, apathy, anger issues, increased aggression, and obsessive behavior.7 One patient (6) also showed stereotyped behaviors, including repetitive hand clapping and echolalia.7 FTD features also included diminished speech and anomia, disorientation to people, places, and time, and difficulties with word finding and comprehension (most noted in 3, 5, and 8).
Of interest, neuroimaging revealed findings consistent with FTD demonstrating cortical atrophy predominance in the frontal and temporal lobes and ventricular dilatation. Case 5 revealed asymmetric findings more pronounced on the left side of the brain.
Generalized and myoclonic seizures occur in FTD at higher rates than in the general population.20,21 One case (6) had a history of partial complex, myoclonic, and generalized tonic-clonic seizure followed by additional cognitive and behavioral changes. Interestingly, she was the only patient with Parkinson disease (9%) similar to prior reports of 4% of patients with MSP1.6
We found a lower incidence of myopathy (39%) in these 5 families compared with 90% in the overall VCP population. Because the main cause of death in VCP disease is respiratory failure or cardiomyopathy secondary to the involvement of respiratory muscles and heart,2 the lower incidence of myopathy might contribute to prolonged living and consequently a higher chance of developing FTD. The incidence of ALS is similar to previously reported.
PDB was diagnosed in only 1 patient among our 5 Hispanic families. The p.R159H variant reported in other families from different backgrounds was reviewed for comparison11,22-25 (eTable 1, links.lww.com/NXG/A569). The overall incidence of PDB among those families was about 33%, which is significantly higher than 3% seen in our 5 Hispanic families, but lower than 42% overall incidence of PDB in VCP disease. A possible explanation would be variation in the penetrance of different ethnic backgrounds.26-29 Further studies are needed to identify the mechanism of a possible protective effect this variant might have for PDB. Of interest, the frequencies of FTD and myopathy in the other reported families are 70% and 39%, respectively. These results are comparable to our cohort in whom the frequencies of FTD and myopathy are 72% and 39%, respectively. We and others have previously reported families with no evidence of confirmed PDB carrying the p.R159C and R159S variants.3,30,31
Overall, this report expands the current genotype-phenotype variations in VCP disease to include individuals with a Hispanic ethnic background. This study highlights the high prevalence of FTD in Hispanic females with the p.R159H VCP variant associated with a later age at symptom onset and a lower incidence of IBM and PDB compared with other ethnic backgrounds. One limitation is the lack of detailed analyses and confirmed diagnosis of FTD on deceased individuals reported in our cohort. Historically, they appear to have had manifestations of FTD rather than Alzheimer disease.
Acknowledgment
The authors thank the patients, their families, health care providers, and numerous collaborators and researchers for their generous contribution to this work. They also thank Eiman Abayazed Abdoalsadig and Merwah Hamid for their assistance with the pedigrees.
Glossary
- ADLs
activities of daily living
- ALS
amyotrophic lateral sclerosis
- bvFTD
behavioral variant
- FTD
frontotemporal dementia
- H&E
hematoxylin and eosin
- IBM
inclusion body myopathy
- MCH‐1
major histocompatibility complex class 1
- MSP 1
multisystem proteinopathy
- NCSs
nerve conduction studies
- PDB
Paget disease of bone
- sIBM
sporadic inclusion body myositis
- TDP‐43
TAR DNA-binding protein 43
- VCP
valosin-containing protein
- WES
whole-exome sequencing
Appendix. Authors
Study Funding
This work was funded by the NIH (Grant AR050236 [VK]), the Institute of Clinical and Translational Science (ICTS), University of California, Irvine, and The National Center for Advancing Translational Sciences (NCATS) (Grant TR001414).
Disclosure
The authors have nothing to disclose. Full disclosures provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Evangelista T, Weihl CC, Kimonis V, et al. 215th ENMC international workshop VCP-related multi-system proteinopathy (IBMPFD) 13-15 November 2015, Heemskerk, The Netherlands. Neuromuscul Disord. 2016;26(8):535-547. doi: 10.1016/j.nmd.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nalbandian A, Donkervoort S, Dec E, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget's disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci. 2011;45(3):522-531. doi: 10.1007/s12031-011-9627-y. [DOI] [PubMed] [Google Scholar]
- 3.Al-Tahan S, Al-Obeidi E, Yoshioka H, et al. Novel valosin-containing protein mutations associated with multisystem proteinopathy. Neuromuscul Disord. 2018;28(6):491-501. doi: 10.1016/j.nmd.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Palmio J, Sandell S, Suominen T, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord. 2011;21(8):551-555. doi: 10.1016/j.nmd.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79(10):1186-1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Obeidi E, Al-Tahan S, Surampalli A, et al. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet. 2018;93(1):119-125. doi: 10.1111/cge.13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672-1682. doi: 10.1016/S0140-6736(15)00461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. 2008;1782(12):744-748. doi: 10.1016/j.bbadis.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Korb MK, Kimonis VE, Mozaffar T. Multisystem proteinopathy: where myopathy and motor neuron disease converge. Muscle Nerve. 2021;63(4):442-454. doi: 10.1002/mus.27097. [DOI] [PubMed] [Google Scholar]
- 10.Mehta SG, Khare M, Ramani R, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet. 2013;83(5):422-431. doi: 10.1111/cge.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haubenberger D, Bittner RE, Rauch-Shorny S, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology. 2005;65(8):1304-1305. doi: 10.1212/01.wnl.0000180407.15369.92. [DOI] [PubMed] [Google Scholar]
- 12.Sundaram C, Uppin MS, Meena AK. Major histocompatibility complex class I expression can be used as a diagnostic tool to differentiate idiopathic inflammatory myopathies from dystrophies. Neurol India. 2008;56(3):363-367. doi: 10.4103/0028-3886.43457. [DOI] [PubMed] [Google Scholar]
- 13.Ching JK, Elizabeth SV, Ju JS, Lusk C, Pittman SK, Weihl CC. mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet. 2013;22(6):1167-1179. doi: 10.1093/hmg/dds524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nalbandian A, Llewellyn KJ, Nguyen C, Yazdi PG, Kimonis VE. Rapamycin and chloroquine: the in vitro and in vivo effects of autophagy-modifying drugs show promising results in valosin containing protein multisystem proteinopathy. PLoS One. 2015;10(4):e0122888. doi: 10.1371/journal.pone.0122888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann M, Mackenzie IR, Cairns NJ, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66(2):152-157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 16.Kimonis V. Inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. University of Washington, Seattle; 2007. [Google Scholar]
- 17.Massó JFM, Zarranz JJ, Otaegui D, López de Munain A. Neurogenetic disorders in the Basque population. Ann Hum Genet. 2015;79(1):57-75. doi: 10.1111/ahg.12088. [DOI] [PubMed] [Google Scholar]
- 18.De Ridder W, Azmi A, Clemen CS, et al. Multisystem proteinopathy due to a homozygous p.Arg159His VCP mutation: a tale of the unexpected. Neurology. 2020;94(8):e785‐e796. doi: 10.1212/WNL.0000000000008763. [DOI] [PubMed] [Google Scholar]
- 19.Nalbandian A, Llewellyn KJ, Kitazawa M, et al. The homozygote VCP(R155H/R155H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS One. 2012;7(9):e46308. doi: 10.1371/journal.pone.0046308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beagle AJ, Darwish SM, Ranasinghe KG, La AL, Karageorgiou E, Vossel KA. Relative incidence of seizures and myoclonus in Alzheimer's disease, dementia with lewy bodies, and frontotemporal dementia. J Alzheimers Dis. 2017;60(1):211-223. doi: 10.3233/JAD-170031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnaldi D, Donniaquio A, Mattioli P, et al. Epilepsy in neurodegenerative dementias: a clinical, epidemiological, and EEG study. J Alzheimers Dis. 2020;74(3):865-874. doi: 10.3233/JAD-191315. [DOI] [PubMed] [Google Scholar]
- 22.Papadimas GK, Paraskevas GP, Zambelis T, et al. The multifaceted clinical presentation of VCP-proteinopathy in a Greek family. Acta Myol. 2017;36(4):203-206. [PMC free article] [PubMed] [Google Scholar]
- 23.Koppers M, van Blitterswijk MM, Vlam L, et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(4):837.e7–837.e13. doi: 10.1016/j.neurobiolaging.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Ramos-Campoy O, Antonell A, Falgàs N, et al. Screening of dementia genes by whole-exome sequencing in Spanish patients with early-onset dementia: likely pathogenic, uncertain significance and risk variants. Neurobiol Aging. 2020;93:e1-e9. doi: 10.1016/j.neurobiolaging.2020.02.008. [DOI] [PubMed] [Google Scholar]
- 25.van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology. 2009;73(8):626-632. doi: 10.1212/WNL.0b013e3181b389d9. [DOI] [PubMed] [Google Scholar]
- 26.Corral-Gudino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, Ralston S. Epidemiology of Paget's disease of bone: a systematic review and meta-analysis of secular changes. Bone. 2013;55(2):347-352. doi: 10.1016/j.bone.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 27.Vallet M, Ralston SH. Biology and treatment of Paget's disease of bone. J Cell Biochem. 2016;117(2):289-299. doi: 10.1002/jcb.25291. [DOI] [PubMed] [Google Scholar]
- 28.Selby PL, Davie MW, Ralston SH, Stone MD. Guidelines on the management of Paget's disease of bone*. Bone. 2002;3131(33):437366-437373. doi: 10.1016/s8756-3282(02)00817-7. [DOI] [PubMed] [Google Scholar]
- 29.Mautalen C, Pumarino H, Blanco MC, González D, Ghiringhelli G, Fromm G. Paget's disease: the South American experience. Semin Arthritis Rheum. 1994;23(4):226-227. doi: 10.1016/0049-0172(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 30.Chan N, Le C, Shieh P, et al. Valosin-containing protein mutation and Parkinson's disease. Parkinsonism Relat Disord. 2012;18(1):107-109. doi: 10.1016/j.parkreldis.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Wong TH, Pottier C, Hondius DC, et al. Three VCP mutations in patients with frontotemporal dementia. J Alzheimers Dis. 2018;65(4):1139-1146. doi: 10.3233/JAD-180301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.