

Abstract
Background
Kearns‐Sayre syndrome (KSS) is a rare multisystem mitochondrial disorder characterized by onset before 20 years of age and a typical clinical triad: progressive external ophthalmoplegia, pigmentary retinopathy and cardiac conduction anomalies. In most cases KSS is caused by spontaneous heteroplasmic single large‐scale mitochondrial DNA (mtDNA) deletions. Long‐range polymerase chain reaction (LR‐PCR), next generation sequencing (NGS) and multiplex ligation‐dependent probe amplification (MLPA) are the most widely applied methods for the identification of mtDNA deletions. Here, we report the case of 20‐year‐old male who presented with classic Kearns‐Sayre syndrome, confirmed by novel 5,9 kb mtDNA deletion.
Methods and results
LR‐PCR and MLPA methods were applied to identify the mitochondrial DNA deletion for the patient, but the results were conflicting. Molecular analysis using primer walking and Sanger sequencing identified a novel 5888 base pairs mtDNA deletion (NC_012920.1:m.6069_11956del) with CAAC nucleotides repeat sequence at the breakpoints.
Conclusion
Our study enriched the mtDNA variation spectrum associated with KSS and demonstrated the importance of choosing relevant molecular genetic methods.
Keywords: Kearns‐Sayre syndrome, KSS, mitochondrial disorder, single large‐scale mitochondrial DNA deletion syndromes
Novel 5,9 kb mtDNA deletion was identified for a 20‐year‐old male who presented with classic Kearns‐Sayre syndrome. Different molecular genetic methods were applied to identify the mitochondrial DNA deletion, but obtaining conflicting research results demonstrated the importance of choosing relevant molecular genetic methods.
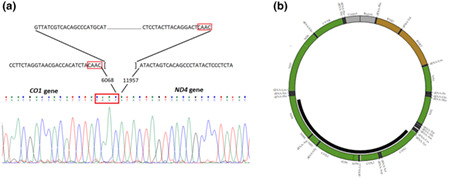
1. INTRODUCTION
Kearns‐Sayre syndrome is a very rare multisystem mitochondrial disorder that occurs before 20 years of age and is characterized by a typical clinical triad: progressive external ophthalmoplegia with ptosis, pigmentary retinopathy and cardiac conduction anomalies including heart block (Guo et al., 2020; Luca et al., 2020). Other clinical manifestations may also include muscle weakness, neurological dysfunction, such as cerebellar ataxia, impaired intellectual and cognitive function, sensorineural hearing loss and neuropathy, various endocrine abnormalities, nephropathy and dental anomalies (Gross‐Jendroska et al., 1992; Guo et al., 2020; Maceluch & Niedziela, 2006).
The exact prevalence of the disorder is unknown, but some studies have reported the prevalence of 0.8–3 cases per 100,000 individuals (Kisilevsky et al., 2020; Leal et al., 2016; Luca et al., 2020).
In 90% of cases KSS is caused by spontaneous heteroplasmic single large‐scale mitochondrial DNA (mtDNA) deletions occurring in the germ‐cell level during embryonic development and ranging from 1.1 to 10 kb (Kisilevsky et al., 2020; Leal et al., 2016). Rarely point mutations, single nucleotide deletions, mtDNA duplications as well as deletions, or multiple mtDNA deletions and nuclear gene defects predisposing to multiple deletions are identified as causing KSS (Pitceathly et al., 2012; Yamashita et al., 2008). MtDNA rearrangements usually affect genes encoding respiratory chain proteins and large number of various tRNAs. These rearrangements impair oxidative phosphorylation resulting reduced energy production in mitochondria and leading to dysfunction of many tissues, especially those with high energy demand (Maceluch & Niedziela, 2006; Yamashita et al., 2008). The most common deletion identified in KSS is a 4977 base pairs deletion (NC_012920.1:m.8483_13459del), but 140–150 different mtDNA deletions have been associated with KSS (Goldstein & Falk, 2019; Ruiz‐Pesini et al., 2010).
Historically molecular testing of mtDNA deletions was performed by Southern hybridization using a probe composed of the whole mitochondrial sequence or of the D‐loop region. However, PCR methods are most widely used allowing quick and sensitive diagnosis. Long‐range PCR covering almost the whole mtDNA, short PCR towards common deletion, real time PCR or sequencing technologies (Sanger and NGS) are used in genetic laboratories, depending on their potential. Multiplex ligation probe‐dependent amplification (MLPA) assay was also introduced to diagnostic and research laboratories (Mayorga et al., 2016; Tońska et al., 2012).
Here, we report the case of 20‐year‐old male who presented with classic Kearns‐Sayre syndrome. Molecular analysis identified a novel 5888 base pairs mtDNA deletion (NC_012920.1:m.6069_11956del) with CAAC nucleotides repeat sequence at the breakpoints.
2. PATIENT AND METHODS
2.1. Ethical compliance
DNA sample and data collection were performed in accordance with the regulation issued by the Vilnius Regional Biomedical Research Ethics Committee of Lithuania. Written informed consent was obtained from the patient and his mother prior to inclusion.
2.2. Clinical evaluation of the patient
The patient, a 20‐year‐old male, with clinically diagnosed Kearns‐Sayre syndrome was referred to clinical geneticist for molecular diagnosis. Since the age of 3, he had been experiencing visual symptoms—photophobia and nystagmus with slowly progressive deteriorating vision. At the age of 8, pigmentary retinopathy was diagnosed. At the age of 6, he developed a third grade atrioventricular (AV) block, and a year later a pacemaker was implanted due to two episodes of syncope within a week. Sensorineural hearing loss has been diagnosed at the same time. He had a poor appetite and was under‐weight for his age. Furthermore, he suffered with exercise intolerance. At the age of 12, he developed tremor and progressive cerebellar ataxia. He lost the ability to walk and became wheelchair‐bound since the age of 14. His mother reported that he was diagnosed with hypocalcaemia, hypokalemia, hypogonadism and decreased vitamin D levels in early childhood. None of his family members had similar symptoms. On examination, he was wheelchair‐dependent, had a thin body habitus and short stature. Bilateral ophthalmoplegia, ptosis, nystagmus and torticollis were present. Muscle strength examination revealed bilateral symmetrical lower limbs muscle weakness (Medical Research Council [MRC] grade 2) while proximal and distal muscles strength of the upper limbs was near normal. The deep tendon reflexes were reduced and bilateral ankle reflexes were absent. Sensory examination was normal. Laboratory tests revealed recurrent elevated blood lactate levels (6.8–1.7 mM/L, normal range 0.7–2.6 mM/L) and elevated CK levels (247 U/L, normal range 25–195 U/L). Blood amino acids analysis showed normal levels. T2‐weighted brain MRI revealed symmetric high signal intensity lesions in basal ganglia, cerebellar dentate nuclei and in subcortical white matter. Unfortunately, this patient deceased shortly after in the course of pneumonia due to respiratory failure.
2.3. Molecular genetic analysis
Molecular genetic testing for the patient was performed using total DNA extracted from peripheral blood samples using a standard phenol–chloroform extraction method. Long‐range polymerase chain reactions (LR‐PCR) were performed to detect mtDNA deletion using Phusion™ High‐Fidelity DNA Polymerase (Thermo Fisher Scientific, Vilnius, Lithuania) kit and two pairs of primers. Primers 5′‐GATTAACCCAAGTCAATAGAA‐3′ (mtDNA nucleotide positions 909–930) and 5′‐GTGAAGTATAGTACGGATGCT‐3′ (15805–15825) were used to amplify PCR product of 14917 bp in length. The initial denaturation step for 3 min at 98°C was followed by amplification for 35 cycles of denaturation at 98°C for 20 s, annealing at 63°C for 45 s, and extension at 72°C for 10 min, and then finally an extension 72°C for 9 min. Primers 5′‐CGGTATGCACTTTTAACAGTC‐3′ (411–431) and 5′‐GTTGGTATCCTAGTGGGTGAG‐3′ (16261–16281) were used to amplify the PCR product of 15871 bp in length, covering almost the whole mtDNA. The conditions for the second PCR reaction were as follows: initial denaturation at 98°C for 3 min; 35 cycles of denaturation at 98°C for 20 s, annealing at 64°C for 45 s, and extension at 72°C for 10 min; and a final extension at 72°C for 10 min. PCR products were separated on 0.6% agarose gel and visualized with ethidium bromide.
Multiplex ligation‐dependent probe amplification (MLPA) method using Mitochondria Salsa MLPA Kit P125 (MRC‐Holland) was used to confirm mtDNA deletion for the patient. The MLPA kit contains 32 probes that hybridize to a number of different mitochondrial sequences and 5 mutation‐specific probes for the frequent point substitutions, including: m.3243A>G, m.3460G>A; m.8344A>G; m.11778G>A and m.14484T>C. Capillary electrophoresis of probe amplification products was performed on an ABI 3130xl DNA analyzer (Applied Biosystems). Peak plots were visualized and normalized, and the dosage ratios were calculated using Coffalyser.Net. Samples from healthy controls were included in each assay.
Finally, primer walking was performed using the set of eight different primer pairs to identify the deletion breaking points. The fusion point was identified by DNA sequencing using PCR and sequencing primers 5′‐CACGCTACTCCTACCTATCTC‐3′ (5468–5488) and 5′‐CGATGAACAGTTGGAATAGGT‐3′ (12743–12763).
3. RESULTS
Long‐range polymerase chain reaction using two different pairs of primers revealed about 6‐kb heterozygous deletion of mtDNA for the patient (Figure 1).
FIGURE 1.
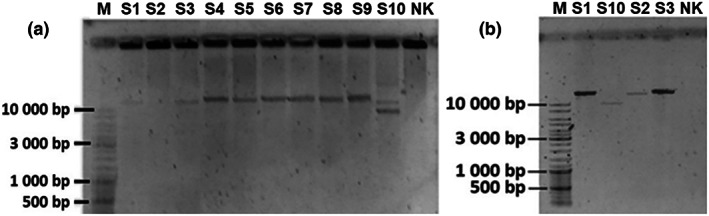
(a) The results of LR‐PCR using the 1st pair of primers (mtDNA nucleotide positions 909–930 and 15,805–15,825). PCR product size is 14,917 bp. (b) The results of LR‐PCR using the 2nd pair of primers (mtDNA nucleotide positions 411–431 and 16,261–16,281). PCR product size is 15,871 bp. M, molecular size standard, NK, negative control, S1–S10, samples of tested individuals, S10, patient with KSS
In order to confirm mtDNA deletion and to predict the range of the deletion, MLPA analysis was performed. Surprisingly, MLPA results were within the normal ranges even after repeating test using different DNA amounts (i.e., 1, 4, 10 ng) (Figure 2).
FIGURE 2.
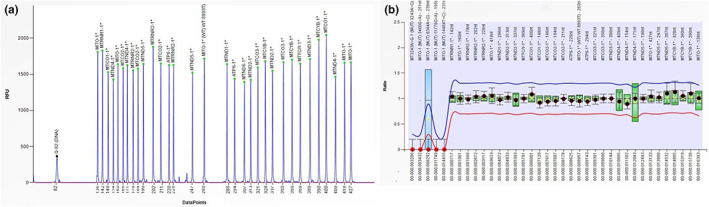
The results of MLPA analysis of the patient. (a) 32 probes in the test sample show normal peaks. (b) Normalized data shows that all probes in the test sample have normal copy number as compared to the reference samples
Finally, molecular analysis using primer walking and Sanger sequencing let us to identify exact breaking points of the deletion and a novel 5888 bp mtDNA deletion (NC_012920.1:m.6069_11956del) was confirmed for the proband (Figure 3). The breakpoints were in MT‐CO1 (MIM #516030) and MT‐ND4 (MIM #516003) genes with CAAC nucleotides repeat sequence at the breakpoints. The deletion region also encompassed protein‐coding genes MT‐CO2 (MIM #516040), MT‐ATP8 (MIM #516070), MT‐ATP6 (MIM #516060), MT‐CO3 (MIM #516050), MT‐ND3 (MIM #516002), MT‐ND4L (MIM #516004) and tRNA‐coding genes MT‐TS1 (MIM #590080), MT‐TD (MIM #590015), MT‐TK (MIM #590060), MT‐TG (MIM #590035), MT‐TR (MIM #590005).
FIGURE 3.
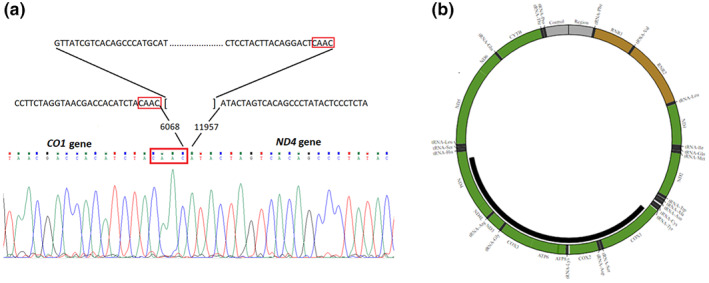
(a) Sanger sequencing identified a 5888 bp mtDNA deletion NC_012920.1:m.6069_11956del; (b) The deletion region also encompassed 8 protein‐coding genes and 5 tRNA‐coding genes (http://mitobreak.portugene.com/cgibin/Mitobreak_submit.cgi?Species=Homsap&Breaktype=Del&5Breakpoint=6068&3Breakpoint=11957+&junction=&submit=Submit+rearrangement)
After molecular genetic testing, mtDNA deletion was not identified for the healthy mother of the patient.
4. DISCUSSION
Patients with Kearns‐Sayre syndrome manifest with multisystem and heterogenous clinical phenotypes because of oxidative phosphorylation impairment and reduced energy production.
Based on the classic phenotype of KSS, including progressive external ophthalmoplegia and pigmentary retinopathy, which could lead to ptosis, and accompanying other characteristic symptoms, a present patient was clinically diagnosed with KSS in his teens. However, the clinical suspicion of KSS in the early stages of the disease may be complicated due to overlapping phenotypes. The occurrence of multisystem presentation in our patient was observed before the age of 10 and this is consistent with reported data (Anteneová et al., 2020; Mancuso et al., 2015).
Single large‐scale deletions of mtDNA are usually identified in the blood DNA samples for the patients with Kearns‐Sayre syndrome. Deletions vary in size and location on the mitochondrial genome in different individuals, although the most reported deletion (NC_012920.1:m.8470_13446del4977), is identified in more than a third of patients with KSS and involves 4977 bp and 12 mitochondrial genes (Sabella‐Jiménez et al., 2020; Shemesh & Margolin, 2018). The molecular investigation revealed novel de novo 5888 bp mtDNA deletion (NC_012920.1:m.6069_11956del) for our patient. The deletion region involved several genes encoding enzymes in the mitochondrial respiratory chain: MT‐ND3, MT‐ND4 and MT‐ND4L genes encode subunits of Complex I (NADH: ubiquinone oxidoreductase), MT‐CO1, MT‐CO2 and MT‐CO3 encode subunits of Complex IV (cytochrome c oxidase), while MT‐ATP6 and MT‐ATP8 genes encode subunits of Complex V (ATP synthase). Five tRNA‐coding genes (MT‐TS1, MT‐TD, MT‐TK, MT‐TG, MT‐TR) were also encompassed by deletion region. This kind of deletion disturb the function of the mitochondria leading to respiratory deficiency of these mitochondria and causing a dysfunction of highly oxidative tissues.
In patients with mtDNA deletions, about 60%–69% of deletions are flanked by short, directly repeated sequences between 4 and 13 bp, known as Class I deletions, 30% of deletions are flanked by imperfect repeat sequences, known as Class II deletions, and the remaining patient‐derived deletions are flanked by nonrepetitive sequences, known as Class III deletions (Fontana & Gahlon, 2020; Nissanka et al., 2019). In our patient, two direct repeats of CAAC nucleotides were identified at the breakpoints, one of which is retained whereas the other is lost during deletion formation, suggesting a common class I deletion.
According to the current recommendations, a variety of methods could be used for the testing of mtDNA rearrangements including long‐range PCR, Southern blotting, quantitative PCR and next generation sequencing / whole genome sequencing methodologies (Mavraki et al., 2020). Despite the multiplicity of the methods used for the detection of mitochondrial DNA deletions, they are still causing diagnostic problems, including sensitivity, DNA quality, time and cost efficiency. Tońska et al. (2012) compared four molecular methods (Southern hybridization, PCR, long‐range PCR and MLPA), the results were not completely consistent and demonstrated that MLPA method was less sensitive compared to PCR‐based methods. The authors concluded that MLPA is a useful method, but some of the deletions may be missed, especially when present at low heteroplasmy levels below 40%. Another study suggested that as low amounts as 1 ng of DNA sample should be used for MLPA reaction to get higher sensitivity (Mayorga et al., 2016). Although several authors reported successfully identified deletions (Liu et al., 2021) and duplications (Sabella‐Jiménez et al., 2020) using MLPA method, in our case, the results of the test were negative using different DNA dilutions. Conversely, long‐range PCR method detected heteroplasmic deletion using two different pairs of PCR primers. Although the method cannot assess the level of heteroplasmy and determine the breakpoints of mtDNA deletion, the long‐range PCR method is the most commonly used routine methodology (Mavraki et al., 2020). In our case, the method was sensitive enough to identify a large‐scale mtDNA deletion for KSS patient in a DNA sample extracted from blood. Nowadays, next generation sequencing (NGS) based methods displaced traditional genetic tools and can effectively analyze mtDNA. Nevertheless, some studies show that LR‐PCR may be more sensitive in the detection of large‐scale mtDNA deletions compared to NGS.
In conclusion, we report the novel mtDNA deletion of 5888 bp; notably, this alteration resulted in a classic phenotype of the Kearns‐Sayre syndrome. These findings enriched the variant spectrum of mtDNA. Moreover, we highlight the importance to use alternative diagnostic methods in case of negative results of molecular genetic testing for mtDNA deletions. In our case, the long‐range PCR method proved to be more sensitive in identifying large mtDNA deletions compared to MLPA method. However, additional methods should be applied for determination of mtDNA rearrangement breakpoints and quantitation of heteroplasmy level as this knowledge may be useful for prognosis and clinical interpretation.
AUTHOR CONTRIBUTIONS
Study concepts and design: B.B., K.G., D.B. Clinical information collection: B.B. Data acquisition: K.G., D.B. Data analysis/interpretation: K.G., D.B., L.A. Manuscript preparation: K.G., B.B. Manuscript editing/revision/review: L.A., D.B., A.U. Principal investigator: A.U. All authors confirmed the manuscript.
FUNDING INFORMATION
The study was supported by the Taiwan‐Latvian‐Lithuanian Collaboration Project ‘Functional model for the mitochondrial disease evaluation and biomarker development’ (grant from Research Council of Lithuania, TAP LLT‐02/2015).
CONFLICT OF INTEREST
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACKNOWLEDGMENTS
We are very thankful to the family for taking part in this study.
Grigalionienė, K. , Burnytė, B. , Balkelienė, D. , Ambrozaitytė, L. , & Utkus, A. (2023). Kearns‐Sayre syndrome case. Novel 5,9 kb mtDNA deletion. Molecular Genetics & Genomic Medicine, 11, e2059. 10.1002/mgg3.2059
REFERENCES
- Anteneová, N. , Kelifová, S. , Kolářová, H. , Vondráčková, A. , Tóthová, I. , Lišková, P. , Magner, M. , Zámečník, J. , Hansikova, H. , Zeman, J. , Tesařová, M. , & Honzík, T. (2020). The phenotypic spectrum of 47 Czech patients with single, large‐scale mitochondrial DNA deletions. Brain Sciences, 10(11), 766. 10.3390/brainsci10110766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana, G. A. , & Gahlon, H. L. (2020). Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Research, 48(20), 11244–11258. 10.1093/nar/gkaa804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, A. , & Falk, M. J. (2019). Mitochondrial DNA deletion syndromes. In Adam M. P., Everman D. B., Mirzaa G. M., Pagon R. A., & Wallace S. E. (Eds.), GeneReviews®. University of Washington. https://www.ncbi.nlm.nih.gov/books/NBK1203/ [PubMed] [Google Scholar]
- Gross‐Jendroska, M. , Schatz, H. , McDonald, H. R. , & Johnson, R. N. (1992). Kearns‐Sayre syndrome: A case report and review. European Journal of Ophthalmology, 2(1), 15–20. [DOI] [PubMed] [Google Scholar]
- Guo, L. , Wang, X. , & Ji, H. (2020). Clinical phenotype and genetic features of a pair of Chinese twins with Kearns–Sayre syndrome. DNA and Cell Biology, 39(8), 1449–1457. 10.1089/dna.2019.5010 [DOI] [PubMed] [Google Scholar]
- Kisilevsky, E. , Freund, P. , & Margolin, E. (2020). Mitochondrial disorders and the eye. Survey of Ophthalmology, 65(3), 294–311. 10.1016/j.survophthal.2019.11.001 [DOI] [PubMed] [Google Scholar]
- Leal, M. , Dhoble, C. , Lee, J. , Lopez, D. , & Menéndez, L. S. (2016). A rare case of Kearns‐Sayre syndrome in a 17‐year‐old Venezuelan male with bilateral ptosis as the initial presentation. Oxford Medical Case Reports, 2016(3), 34–36. 10.1093/omcr/omw007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, R. , Mo, G. L. , & Song, Y. Z. (2021). Identification of a novel large deletion of the mitochondrial DNA in an infant with Pearson syndrome: A case report. Translational Pediatrics, 10(1), 204–208. 10.21037/tp-20-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca, P. , Alessia, G. , Camilla, R. E. M. , Antonio, N. , Diego, M. , Federica, D. , Daria, D. , Rosalba, C. , Carlo, D. V. , & Daniela, L. (2020). Spinal cord involvement in Kearns‐Sayre syndrome: A neuroimaging study. Neuroradiology, 62(10), 1315–1321. 10.1007/s00234-020-02501-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceluch, J. A. , & Niedziela, M. (2006). The clinical diagnosis and molecular genetics of kearns‐sayre syndrome: a complex mitochondrial encephalomyopathy. Pediatric Endocrinology Reviews, 4(2), 117–137. [PubMed] [Google Scholar]
- Mancuso, M. , Orsucci, D. , Angelini, C. , Bertini, E. , Carelli, V. , Comi, G. P. , Donati, M. A. , Federico, A. , Minetti, C. , Moggio, M. , Mongini, T. , Santorelli, F. M. , Servidei, S. , Tonin, P. , Toscano, A. , Bruno, C. , Bello, L. , Ienco, E. C. , Cardaioli, E. , … Siciliano, G. (2015). Redefining phenotypes associated with mitochondrial DNA single deletion. Journal of Neurology, 262(5), 1301–1309. 10.1007/s00415-015-7710-y [DOI] [PubMed] [Google Scholar]
- Mavraki, E. , Labrum, R. , Sergeant, K. , Alston, C. L. , Woodward, C. , Smith, C. , Watson EL, Polke J, Taylor RW, Fratter C (2020). Best practice guidelines for the molecular diagnosis of mitochondrial disease. Ratified .
- Mayorga, L. , Laurito, S. R. , Loos, M. A. , Eiroa, H. D. , de Pinho, S. , Lubieniecki, F. , Arroyo, H. A. , Pereyra, M. F. , Kauffman, M. A. , & Roqué, M. (2016). Mitochondrial DNA deletions detected by multiplex ligation‐dependent probe amplification. Mitochondrial DNA Part A, 27(4), 2864–2867. 10.3109/19401736.2015.1053132 [DOI] [PubMed] [Google Scholar]
- Nissanka, N. , Minczuk, M. , & Moraes, C. T. (2019). Mechanisms of mitochondrial DNA deletion formation. Trends in Genetics, 35(3), 235–244. 10.1016/j.tig.2019.01.001 [DOI] [PubMed] [Google Scholar]
- Pitceathly, R. D. S. , Rahman, S. , & Hanna, M. G. (2012). Single deletions in mitochondrial DNA–molecular mechanisms and disease phenotypes in clinical practice. Neuromuscular Disorders, 22(7), 577–586. 10.1016/j.nmd.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Pesini, E. , Lott, M. T. , Procaccio, V. , Poole, J. , Brandon, M. C. , Mishmar, D. , Yi, C. , Kreuziger, J. , Baldi, P. , & Wallace, D. C. (2010). MITOMAP: A human mitochondrial genome database. http://www.mitomap.org
- Sabella‐Jiménez, V. , Otero‐Herrera, C. , Silvera‐Redondo, C. , & Garavito‐Galofre, P. (2020). Mitochondrial DNA deletion and duplication in Kearns‐Sayre Syndrome (KSS) with initial presentation as Pearson Marrow‐Pancreas Syndrome (PMPS): Two case reports in Barranquilla, Colombia. Molecular Genetics & Genomic Medicine, 8(11), e1509. 10.1002/mgg3.1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh, A. , & Margolin, E. (2018). Kearns sayre syndrome. In StatPearls. StatPearls Publishing. [PubMed] [Google Scholar]
- Tońska, K. , Piekutowska‐Abramczuk, D. , Kaliszewska, M. , Kowalski, P. , Tańska, A. , Bartnik, E. , Pronicka, E. , & Krajewska‐Walasek, M. (2012). Molecular investigations of mitochondrial deletions: evaluating the usefulness of different genetic tests. Gene, 506(1), 161–165. 10.1016/j.gene.2012.06.081 [DOI] [PubMed] [Google Scholar]
- Yamashita, S. , Nishino, I. , Nonaka, I. , & Goto, Y. I. (2008). Genotype and phenotype analyses in 136 patients with single large‐scale mitochondrial DNA deletions. Journal of Human Genetics, 53(7), 598–606. 10.1007/s10038-008-0289-8 [DOI] [PubMed] [Google Scholar]