

Abstract

Dicarbofunctionalization is an important efficient synthetic technique for adding two chemical moieties across an alkene. Here, a novel method of reductive dicarbofunctionalization has been developed using a single alkenyl triflate as the electrophile, combined with an unactivated alkene. The reaction does not require an external auxiliary and proceeds with complete regioselectivity.
Introduction
Alkene dicarbofunctionalization has emerged as a powerful synthetic method for the modular synthesis of complex molecules.1 One advantage of dicarbofunctionalization is that the reaction utilizes alkenes, which are relatively simple starting materials that have versatile reactivities and are abundant. During dicarbofunctionalization, two carbon entities are simultaneously added across the double bond in a single step. This allows for significant increases in the chemical and economic efficiency in the synthesis of complex molecules. Many strategies have demonstrated the addition of new carbon moieties across an alkene, including redox-neutral,2 oxidative,3 and reductive4 dicarbofunctionalization (Scheme 1).
Scheme 1. Strategies for Dicarbofuctionalization.

Redox-neutral dicarbofunctionalization employs two different moieties, a nucleophile and an electrophile, respectively, to form the desired product (Scheme 1A). Generally, organohalides and triflates serve as the electrophile, whereas many organometallic reagents have been used as the nucleophile. Examples of redox-neutral dicarbofunctionalization occurred by tethering one of the coupling partners to the alkene. More recently, intermolecular dicarbofunctionalization of alkenes has gained popularity as the substrate and reaction scope is increased relative to their tethered counterpart.1a−1c,5 However, intermolecular dicarbofunctionalization is entropically less favorable and can suffer from poor regioselectivity. Consequently, strong directing groups have been employed to control regioselectivity to a varying degree of success.6 Unfortunately, the use of directing groups increases the complexity of the synthetic route, as additional synthetic steps are required to install and remove the directing group.
In addition to redox-neutral processes, oxidative dicarbofunctionalization reactions have received substantial consideration.3a This method employs only nucleophiles, generally organometallic reagents as the sole carbon source, rather than a combination of a nucleophile and an electrophile (Scheme 1B).1a,3b,7 For example, Sigman and Urkalan applied tributylphenyltin and tributylvinyltin to difunctionalize alkenes wherein oxygen served as an oxidant.3d Other oxidants, such as benzoquinone and TEMPO, have also been employed for oxidative diarylation of ethers and glycals with boronic acids.8
An alternative, yet equally powerful, method is reductive alkene dicarbofunctionalization in which electrophiles serve as a carbon source. Many electrophiles are abundant and commercially available, which provide an alluring substitute to many less available nucleophiles. Prior to 2008, only a few existing studies of cross-electrophile coupling were available; however, since then, electrophilic cross-coupling has been thoroughly examined for the development of reductive alkene dicarbofunctionalization reactions (Scheme 1C).4d,4,9 For example, Nevado and co-workers demonstrated Ni-catalyzed reductive alkene alkylarylation using t-butyl and aryl iodides with the addition of TDAE as a reductant (Scheme 2A).4a Koh and co-workers developed regioselective alkene dialkylation utilizing an 8-aminoquinoline tethered alkene and redox-active esters (Scheme 2A,B).6i Diao has performed reductive diarylation of vinylarenes with aryl bromides using zinc as the reductant.10 Despite these advances, existing methods are limited in achieving broad substrate scope with the use of simple, unactivated alkenes.
Scheme 2. Selected Examples of Reductive Dicarbofunctionalization and This Work.

The development of reductive dicarbofunctionalization across alkenes is complicated by regio- and site-selectivity issues.6j These reactions have been limited to aryl-alkylation4a,4 and acyl-alkylation11 using aryl iodides or acyl chlorides with perfluoroalkyl or tertiary alkyl iodides. Another limitation of the reductive coupling approach is the multitude of undesired products that can form, including regioisomers and cross-electrophile coupled products.9a,9,12 In some instances, a directing group has been employed to overcome these challenges.6j,13 Herein, we demonstrate an intermolecular reductive dicarbofunctionalization reaction utilizing alkenyl triflates as the sole electrophile to create two new carbon–carbon bonds (Scheme 2B). Moreover, only the 1,3-regioisomer is observed for the dialkenylated product, demonstrating a high degree of regioselectivity. To our knowledge, reductive, regioselective dicarbofunctionalization across an unactivated alkene using a single alkenyl triflate as the electrophile without an external auxiliary moiety has not been achieved.
Results and Discussion
We began our studies for dialkenylation of γ,δ-alkenyl α-cyano esters with cyclohexenyl triflates in the presence of reductants shown to facilitate reductive difunctionalization. When using zinc and manganese as reductants, the reaction only generated product 2 in approximately 20% yield (Table 1, entries 2 and 3). Acidic hydrolysis and the X-ray crystallographic analysis of the hydrolyzed product (2a) confirmed the regiochemistry of the product as β,δ-dialkenylation (Scheme 3). TDAE increased the yield to 43% (Table 1, entry 4). Diethylzinc, which has previously been shown to serve as a reductant,9h did not yield a product (Table 1, entry 6). Addition of phenethylzinc as a reductant promoted β,δ-dialkenylation of γ,δ-alkenyl α-cyano ester 1 in NMP, N-methyl-2-pyrrolidone, at 80 °C in 58% yield (Table 1, entry 7). Unfortunately, multiple side products were observed in addition to the expected product (2). We hypothesized that promoting β-H elimination from the alkylzinc reagent would reduce side products and increase yield of the desired dialkenylated product. We changed the reductant to phenpropylzinc bromide, which contains more β-H’s. The use of this different alkylzinc promoted β-H elimination, while suppressing homocoupling and cross-coupling, and the yield of the dialkenylated product was increased to 79% (Table 1, entry 8).
Table 1. Reaction Optimization Conditionsa.

| entry | deviation in conditions | yield (%) |
|---|---|---|
| 1 | standard conditions | 79 (63%) |
| 2 | 2 equiv Zn(0) | 21 |
| 3 | 2 equiv Mn(0) | 24 |
| 4 | 2 equiv TDAE | 43 |
| 5 | no reductant | <1 |
| 6 | diethylzinc bromide | <1 |
| 7 | phenethylzinc bromide | 58 |
| 8 | no additives | 49 |
| 9 | dioxanylzinc bromide | 56 |
| 10 | cyclopropylzinc bromide | 59 |
| 11 | tert-butylzinc bromide | 46 |
| 12 | 1-ethylpropylzinc bromide | 70 |
| 13 | DMF instead of NMP | 66 |
| 14 | DMSO instead of NMP | 70 |
| 15 | toluene instead of NMP | 25 |
| 16 | THF instead of NMP | 29 |
| 17 | MeCN instead of NMP | 0 |
0.1 mmol scale reactions in 0.5 mL of solvent.1H NMR yields using pyrene as an internal standard. The value in parentheses is the isolated yield [diastereomeric ratio (dr) = 1:1] from 0.5 mmol. The dr was determined by 1H NMR of the crude reaction mixture.
Scheme 3. Hydrolysis and Confirmation of Regioselectivity of Dialkeynylation Reaction.

Based on our previous work,14 the inclusion of KPF6, ZnCl2, and pyridine was necessary to achieve the high yields obtained for this reaction. In their absence, the yield decreased to 49% (Table 1, entry 8). KPF6, a non-coordinating counteranion, could increase the concentration of cationic Ni necessary for the addition of a vinyl triflate, ZnCl2 could activate the triflate, while pyridine could act as a ligand to nickel.14 Additionally, the impact of various solvents and alkylzinc reagents was examined, resulting in a range of yields between 0 and 70% (Table 1, entries 9–17).
After the reaction conditions were optimized, we began to explore the scope of β,δ-dialkenylation of γ,δ-alkenyl α-cyanocarboxylic esters with various alkenyl triflates (Scheme 4). The reaction proceeds well with cyclic triflates, such as cyclopentenyl triflate and cyclohexenyl triflate (2 and 4). The reaction also tolerates sterics on the alkenyl triflate. For example, 4-tert-butylcyclohexenyl triflate and a camphor-derived triflate afforded the corresponding of β,δ-dialkenylated products (9 and 7) in moderate yields. A ketal triflate (10), which bears an additional functional handle, was also well tolerated in the reaction. The reaction generated products 11 and 12 in good yields with α-cyano ethyl esters containing internal γ,δ-alkenes. The compatibility of this dialkenylation reaction with different alkene-based substrates was also investigated. The reaction was successful when an γ,δ-alkenyl α-cyano amide and γ,δ-alkenyl α-cyano isopropyl ester were used (13–15). Additionally, the reaction yielded the β,δ-dialkenylated product when α-allyl malononitrile was used as an alkene substrate (16), illustrating the broader scope of the reaction.
Scheme 4. Scope with Alkenes and Alkenyl Triflates.

Isolated from 0.5 mmol. Phenpropylzinc bromide (1.5 equiv) and vinyl triflate (2 equiv) were used. dr was determined by 1H NMR of the crude reaction mixture. The value in parentheses is 1H NMR yield using pyrene as an internal standard.
Isolated from 1.0 mmol.
dr was determined by 1H NMR of the isolated product.
Functional group tolerance was assessed to further understand the robustness of this dialkenylation reaction.15 We performed our standard reaction in the presence of one molar equivalent of various additives with each possessing a different structural motif or chemical functionality. We analyzed the percent yield of the reaction and the amount of starting material remaining in the presence of these additives by 1H NMR. Table 2 demonstrates the tolerance of various functional groups and heterocycles, respectively. Esters, ketones, and amines were well tolerated, while alcohols, nitriles, and aldehydes were less tolerated. Biologically active compounds such as coumarin, carvone, and camphor were also well tolerated. Additionally, vinyl arenes were compatible with the dialkenylation reaction. The nitro group was not compatible, giving no yield of the dialkenylated product. In regard to heterocycle compatibility, it appears that oxygen is very well tolerated, nitrogen is moderately tolerated, and sulfur is not tolerated at all. Overall, the majority of functional groups were either very well tolerated or moderately tolerated by the reaction, demonstrating the robustness of this reaction.
Table 2. Examination of the Functional Group and Heterocycle Compatibilityabc.
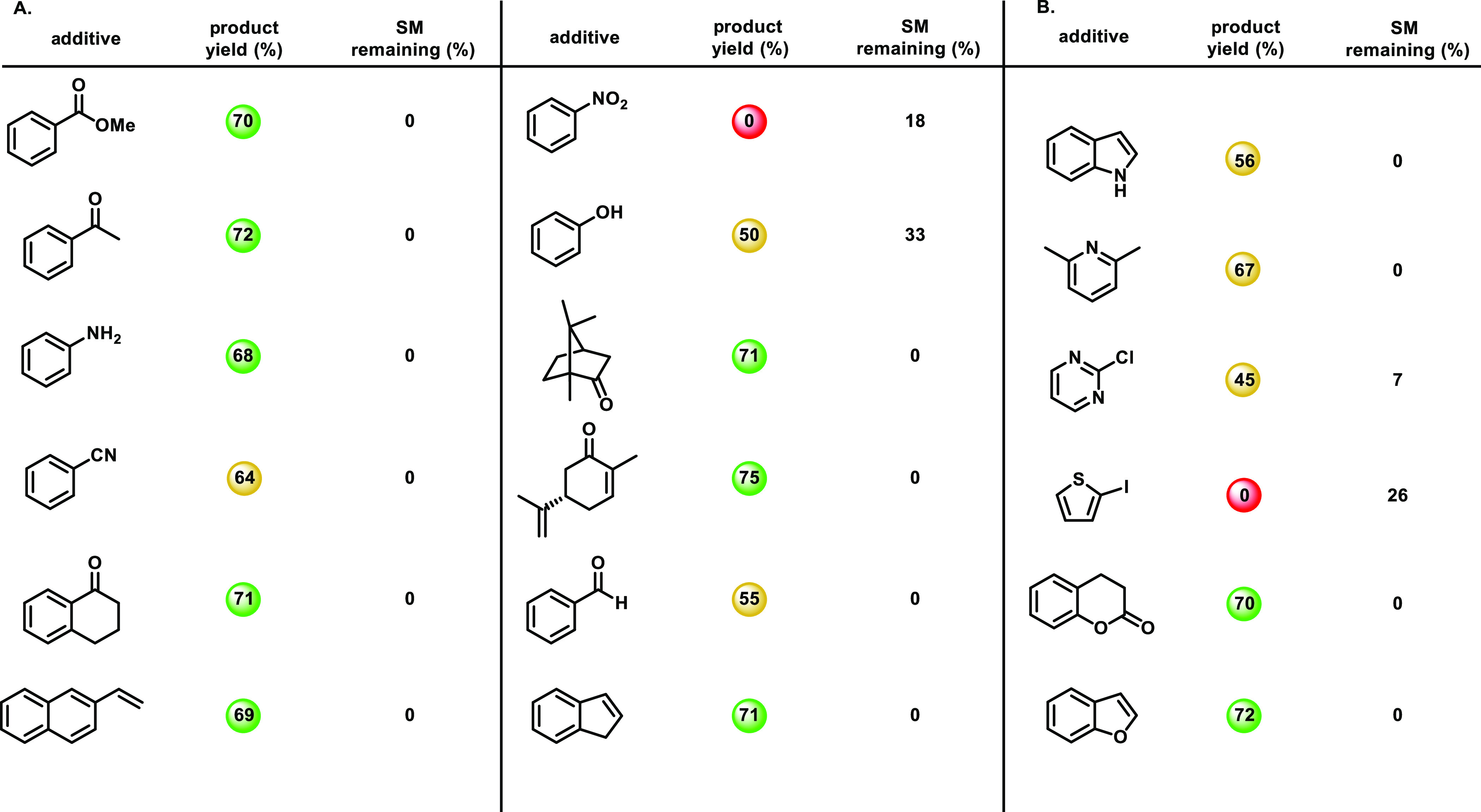
Color coding as follows: green (above 66%), yellow (34–66%), and red (below 34%).
All 1H NMR yields were determined using pyrene as an internal standard.
0.1 mmol reactions in 0.5 mL solvent.
A sensitivity screen was also conducted in order to assess the reproducibility of this dialkenylation reaction.16 We examined the influence of multiple parameters including the concentration of catalyst and organozinc reagent, temperature, and reaction time. The altered parameters were chosen as they include random errors as well as systematic errors that could occur during reaction setup.16 The percent deviation from the yield obtained under standard conditions was calculated and illustrated as a black line in the radar diagram shown in Figure 1.
Figure 1.

Sensitivity assessment of the dialkenylation reaction illustrated as a color-coded radar diagram.
The black line around the light green ring indicates low sensitivity and represents the 0% deviation line. Any deviation of the black line from the light green perimeter toward red zones corresponds to a higher degree of sensitivity to the given reaction parameter. As illustrated in Figure 1, the reaction is robust in regards to the majority of reaction parameters, including the amount of catalyst, organozinc, and length of reaction time. Moreover, the black line remains consistent around the 0% deviation ring for over half of the investigated parameters. The only weakly influencing parameter was the change in reaction temperature, which produced a slight deflection of the black line toward the 25% deviation limit. Therefore, this dialkenylation reaction is reproducible under a multitude of reaction conditions. This demonstrates that this reaction may be employed to increase the chemical and economic efficiency of existing reactions, which require the addition of two electrophiles across a double bond, independently of the existing reaction conditions.
Conclusions
Overall, we were able to successfully develop a Ni-catalyzed alkene dialkenylation reaction via a reductive difunctionalization method. We employ single alkenyl triflates as the electrophilic coupling partner. This reaction utilizes a variety of unactivated alkenes that do not require a separate auxiliary for metal coordination and exploits alkylzinc as a reductant to generate a completely regioselective 1,3-dialkenylated product. Furthermore, the reaction is compatible with internal alkenes, as well as terminal, and a variety of electronically and sterically different alkenyl triflates. Additionally, it was observed that the reaction is compatible with various functional groups, including those present in biologically active compounds, as well being reproducible under many conditions.
Experimental Section
General Information
All reactions were set up inside a nitrogen-filled glove box, and all chemicals were handled under a nitrogen atmosphere unless otherwise stated. All glassware and vials were dried in an oven before use. Anhydrous solvents (DMF, DMSO, NMP, THF, toluene, and MeCN) were obtained from Sigma-Aldrich and used without further purification. Ni(cod)2 was purchased from Strem Chemicals. 1H (400 MHz) and 13C NMR (101 MHz) spectra were recorded on a Bruker instrument and internally referenced to the residual solvent signals of CDCl3 for 1H and 13C NMR at 7.26 and 77.16 ppm, respectively. The chemical shifts for NMR and the coupling constants (J) are reported in parts per million (ppm). The following conventions are used for multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublets; and dt, doublet of triplets. High-resolution mass spectra of new compounds were recorded at the University of Georgia and Indiana University. All NMR spectra were collected at the Pennsylvania State University. Infrared (IR) spectra were recorded on a Bruker ATR-IR at the Pennsylvania State University, and νmax is reported in cm–1.
General Procedure for Reaction Screening
In a glove box, in a 1 dram vial, alkylzinc(II) bromide (0.15 mmol, 0.78 M in THF, 192.3 mL) was dried under vacuum. After an hour, Ni(cod)2 (2.8 mg, 0.010 mmol, 10 mol %), ZnCl2 (13.6 mg, 0.10 mmol), KPF6 (36.8 mg, 0.2 mmol), pyridine (8.1 μL, 0.10 mmol), ethyl 2-cyanopent-4-enoate (15.3 mg, 0.10 mmol), and cyclohex-1-en-1yl trifluoromethanesulfonate (46.0 mg, 0.20 mmol) were added. The mixture was then dissolved in 0.50 mL of NMP. The vial was tightly capped at and stirred at 80 °C for 24 h. After 24 h, 50 μL of pyrene (0.010 mmol, 0.20 M stock solution) as an internal standard was added. Deionized water and EtOAc were added, the organic layer was filtered through a silica pack (pipet column), and the solvent was removed by a rotary evaporator. The residue was dissolved in CDCl3 and characterized accordingly via 1H NMR. The yield was determined by integrating a product peak at 5.58 ppm against the pyrene peak at 8.06 ppm.
General Procedure for Robustness Screening
All reactions were performed at a 0.1 mmol scale and prepared in a glove box. A standard reaction was performed in the presence of one molar equivalent of an “additive”. After 24 h, 50 μL of pyrene (0.010 mmol, 0.20 M stock solution) as an internal standard was added. Deionized water and EtOAc were added, the organic layer was filtered through a silica pack (pipet column), and the solvent was removed by a rotary evaporator. The residue was dissolved in CDCl3 and characterized accordingly via 1H NMR. The yield was determined by integrating a product peak and starting material peak against the pyrene peak at 8.06 ppm.
General Procedure for Sensitivity Screening
All reactions were performed at a 0.1 mmol scale and prepared in a glove box. A standard reaction was performed in the presence of one molar equivalent of an “additive”. After 24 h, 50 μL of pyrene (0.010 mmol, 0.20 M stock solution) as an internal standard was added. Deionized water and EtOAc were added, the organic layer was filtered through a silica pack (pipet column), and the solvent was removed by a rotary evaporator. The residue was dissolved in CDCl3 and characterized accordingly via 1H NMR. The yield was determined by integrating a product peak 5.58 ppm against the pyrene peak at 8.06 ppm. For the catalyst and organozinc variations, 10% more or less than the control was used. For temperature, 20 °C in each direction from the standard was tested. For time, 12 h longer and shorter than the control was analyzed. The data were plotted into the radar diagram provided by Glorius and co-workers.16
Acknowledgments
We gratefully acknowledge the NIH NIGMS (R35GM133438) and The Pennsylvania State University (PSU) for their support of this work. We also thank Dr. Rishi Sapkota for the assistance with the isolation and characterization of compound 8. We thank Dr. Hemant Yennawar at the Pennsylvania State University for the X-ray crystallographic analysis of compound 2a. The X-ray instrument was funded by the NIH SIG S10 grants (1S10OD028589-01 and 1S10RR023439-01).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06417.
Experimental procedures and characterization data for all compounds; and copies of NMR spectra of all of the compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Dhungana R. K.; Shekhar K. C.; Basnet P.; Giri R. Transition Metal-Catalyzed Dicarbofunctionalization of Unactivated Olefins. Chem. Rec. 2018, 18, 1314–1340. 10.1002/tcr.201700098. [DOI] [PubMed] [Google Scholar]; b Wickham L. M.; Giri R. Transition Metal (Ni, Cu, Pd)-Catalyzed Alkene Dicarbofunctionalization Reactions. Acc. Chem. Res. 2021, 54, 3415–3437. 10.1021/acs.accounts.1c00329. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qi X. X.; Diao T. N. Nickel-Catalyzed Dicarbofunctionalization of Alkenes. ACS Catal. 2020, 10, 8542–8556. 10.1021/acscatal.0c02115. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Montgomery J. Nickel-catalyzed cyclizations, couplings, and cycloadditions involving three reactive components. Acc. Chem. Res. 2000, 33, 467–473. 10.1021/ar990095d. [DOI] [PubMed] [Google Scholar]; e Badir S. O.; Molander G. A. Developments in Photoredox/Nickel Dual-Catalyzed 1,2-Difunctionalizations. Chem 2020, 6, 1327–1339. 10.1016/j.chempr.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; A preprint of the current work was deposited on an archive. See:; f Wickham L. M.; Dhungana R. K.; Giri R. Ni-catalyzed Regioselective Reductive 1,3-Dialkenylation of Alkenes. ChemRxiv 2022, 10.26434/chemrxiv-2022-8jrzc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Derosa J.; Apolinar O.; Kang T.; Tran V. T.; Engle K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 2020, 11, 4287–4296. 10.1039/C9SC06006E. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang J. S.; Liu L.; Chen T.; Han L. B. Transition-Metal-Catalyzed Three-Component Difunctionalizations of Alkenes. Chem. – Asian J. 2018, 13, 2277–2291. 10.1002/asia.201800647. [DOI] [PubMed] [Google Scholar]
- a Xia T.; Xi Y.; Ding H.; Zhang Y.; Fang K.; Wu X.; Qu J.; Chen Y. Palladium(II)-catalyzed enantioselective intermolecular oxidative diarylation of internal enamides. Chem. Commun. 2022, 58, 9282–9285. 10.1039/D2CC03202C. [DOI] [PubMed] [Google Scholar]; b Ma Y. Y.; Zhang D. C.; Yan Z. Y.; Wang M. F.; Bian C. L.; Gao X. L.; Bunel E. E.; Lei A. W. Iron-Catalyzed Oxidative C-H/C-H Cross-Coupling between Electron-Rich Arenes and Alkenes. Org. Lett. 2015, 17, 2174–2177. 10.1021/acs.orglett.5b00775. [DOI] [PubMed] [Google Scholar]; c Werner E. W.; Sigman M. S. A Highly Selective and General Palladium Catalyst for the Oxidative Heck Reaction of Electronically Nonbiased Olefins. J. Am. Chem. Soc. 2010, 132, 13981–13983. 10.1021/ja1060998. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Urkalan K. B.; Sigman M. S. Palladium-Catalyzed Oxidative Intermolecular Difunctionalization of Terminal Alkenes with Organostannanes and Molecular Oxygen. Angew. Chem., Int. Ed. 2009, 48, 3146–3149. 10.1002/anie.200900218. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kalyani D.; Sanford M. S. Oxidatively intercepting Heck intermediates: Pd-catalyzed 1,2-and 1,1-arylhalogenation of alkenes. J. Am. Chem. Soc. 2008, 130, 2150–2151. 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]
- a García-Domínguez A.; Li Z.; Nevado C. Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 2017, 139, 6835–6838. 10.1021/jacs.7b03195. [DOI] [PubMed] [Google Scholar]; b Kuang Y. L.; Wang X. F.; Anthony D.; Diao T. N. Ni-catalyzed two-component reductive dicarbofunctionalization of alkenes via radical cyclization. Chem. Commun. 2018, 54, 2558–2561. 10.1039/C8CC00358K. [DOI] [PubMed] [Google Scholar]; c Jin Y.; Wang C. Ni-catalysed reductive arylalkylation of unactivated alkenes. Chem. Sci. 2019, 10, 1780–1785. 10.1039/C8SC04279A. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shu W.; Garcia-Dominguez A.; Quiros M. T.; Mondal R.; Cardenas D. J.; Nevado C. Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights. J. Am. Chem. Soc. 2019, 141, 13812–13821. 10.1021/jacs.9b02973. [DOI] [PubMed] [Google Scholar]; e Zhao L.; Meng X.; Zou Y. F.; Zhao J. S.; Wang L. L.; Zhang L. L.; Wang C. Directed Nickel-Catalyzed Diastereoselective Reductive Difunctionalization of Alkenyl Amines. Org. Lett. 2021, 23, 8516–8521. 10.1021/acs.orglett.1c03210. [DOI] [PubMed] [Google Scholar]; f Terao J.; Saito K.; Nii S.; Kambe N.; Sonoda N. Regioselective double alkylation of styrenes with alkyl halides using a titanocene catalyst. J. Am. Chem. Soc. 1998, 120, 11822–11823. 10.1021/ja982732l. [DOI] [Google Scholar]
- Giri R.; Shekhar K. C. Strategies toward Dicarbofunctionalization of Unactivated Olefins by Combined Heck Carbometalation and Cross-Coupling. J. Org. Chem. 2018, 83, 3013–3022. 10.1021/acs.joc.7b03128. [DOI] [PubMed] [Google Scholar]
- a Li W. F.; Boon J. K.; Zhao Y. Nickel-catalyzed difunctionalization of allyl moieties using organoboronic acids and halides with divergent regioselectivities. Chem. Sci. 2018, 9, 600–607. 10.1039/C7SC03149A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Derosa J.; Tran V. T.; Boulous M. N.; Chen J. S.; Engle K. M. Nickel-Catalyzed beta,gamma-Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Conjunctive Cross-Coupling. J. Am. Chem. Soc. 2017, 139, 10657–10660. 10.1021/jacs.7b06567. [DOI] [PubMed] [Google Scholar]; c Thapa S.; Dhungana R. K.; Magar R. T.; Shrestha B.; Shekhar K. C.; Giri R. Ni-catalysed regioselective 1,2-diarylation of unactivated olefins by stabilizing Heck intermediates as pyridylsilyl-coordinated transient metallacycles. Chem. Sci. 2018, 9, 904–909. 10.1039/C7SC04351A. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Basnet P.; Dhungana R. K.; Thapa S.; Shrestha B.; Shekhar K. C.; Sears J. M.; Giri R. Ni-Catalyzed Regioselective beta,delta-Diarylation of Unactivated Olefins in Ketimines via Ligand-Enabled Contraction of Transient Nickellacycles: Rapid Access to Remotely Diarylated Ketones. J. Am. Chem. Soc. 2018, 140, 7782–7786. 10.1021/jacs.8b03163. [DOI] [PubMed] [Google Scholar]; e Dhungana R. K.; Sapkota R. R.; Wickham L. M.; Niroula D.; Giri R. Ni-Catalyzed Regioselective 1,2-Dialkylation of Alkenes Enabled by the Formation of Two C(sp3)-C(sp3) Bonds. J. Am. Chem. Soc. 2020, 142, 20930–20936. 10.1021/jacs.0c09778. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Xu C.; Yang Z. F.; An L.; Zhang X. G. Nickel-Catalyzed Difluoroalkylation-Alkylation of Enamides. ACS Catal. 2019, 9, 8224–8229. 10.1021/acscatal.9b02488. [DOI] [Google Scholar]; g Gu J. W.; Min Q. Q.; Yu L. C.; Zhang X. G. Tandem Difluoroalkylation-Arylation of Enamides Catalyzed by Nickel. Angew. Chem., Int. Ed. 2016, 55, 12270–12274. 10.1002/anie.201606458. [DOI] [PubMed] [Google Scholar]; h Shrestha B.; Basnet P.; Dhungana R. K.; Shekhar K. C.; Thapa S.; Sears J. M.; Giri R. Ni-Catalyzed Regioselective 1,2-Dicarbofunctionalization of Olefins by Intercepting Heck Intermediates as Imine-Stabilized Transient Metallacycles. J. Am. Chem. Soc. 2017, 139, 10653–10656. 10.1021/jacs.7b06340. [DOI] [PubMed] [Google Scholar]; i Yang T.; Jiang Y.; Luo Y. X.; Lim J. J. H.; Lan Y.; Koh M. J. Chemoselective Union of Olefins, Organohalides, and Redox-Active Esters Enables Regioselective Alkene Dialkylation. J. Am. Chem. Soc. 2020, 142, 21410–21419. 10.1021/jacs.0c09922. [DOI] [PubMed] [Google Scholar]; j Yang T.; Chen X.; Rao W.; Koh M. J. Broadly Applicable Directed Catalytic Reductive Difunctionalization of Alkenyl Carbonyl Compounds. Chem 2020, 6, 738–751. 10.1016/j.chempr.2019.12.026. [DOI] [Google Scholar]
- a Yahiaoui S.; Fardost A.; Trejos A.; Larhed M. Chelation-Mediated Palladium(II)-Catalyzed Domino Heck-Mizoroki/Suzuki-Miyaura Reactions Using Arylboronic Acids: Increasing Scope and Mechanistic Understanding. J. Org. Chem. 2011, 76, 2433–2438. 10.1021/jo1018188. [DOI] [PubMed] [Google Scholar]; b Kusunuru A. K.; Jaladanki C. K.; Tatina M. B.; Bharatam P. V.; Mukherjee D. TEMPO-Promoted Domino Heck-Suzuki Arylation: Diastereoselective Cis-Diarylation of Glycals and Pseudoglycals. Org. Lett. 2015, 17, 3742–3745. 10.1021/acs.orglett.5b01722. [DOI] [PubMed] [Google Scholar]
- a Trzepizur D.; Brodzka A.; Koszelewski D.; Wilk M.; Ostaszewski R. Selective Palladium-Catalyzed α,β-Homodiarylation of Vinyl Esters in Aqueous Medium. Eur. J. Org. Chem. 2021, 2021, 6028–6036. 10.1002/ejoc.202101211. [DOI] [Google Scholar]; b Trejos A.; Fardost A.; Yahiaoui S.; Larhed M. Palladium(II)-catalyzed coupling reactions with a chelating vinyl ether and arylboronic acids: a new Heck/Suzuki domino diarylation reaction. Chem. Commun. 2009, 7587. 10.1039/b918358b. [DOI] [PubMed] [Google Scholar]
- a Weix D. J. Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res. 2015, 48, 1767–1775. 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Everson D. A.; Jones B. A.; Weix D. J. Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc. 2012, 134, 6146–6159. 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Anka-Lufford L. L.; Huihui K. M. M.; Gower N. J.; Ackerman L. K. G.; Weix D. J. Nickel-Catalyzed Cross-Electrophile Coupling with Organic Reductants in Non-Amide Solvents. Chem. – Eur. J. 2016, 22, 11564–11567. 10.1002/chem.201602668. [DOI] [PubMed] [Google Scholar]; d Shrestha R.; Dorn S. C. M.; Weix D. J. Nickel-Catalyzed Reductive Conjugate Addition to Enones via Allylnickel Intermediates. J. Am. Chem. Soc. 2013, 135, 751–762. 10.1021/ja309176h. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Dorn S. C. M.; Olsen A. K.; Kelemen R. E.; Shrestha R.; Weix D. J. Nickel-catalyzed reductive arylation of activated alkynes with aryl iodides. Tetrahedron Lett. 2015, 56, 3365–3367. 10.1016/j.tetlet.2015.02.120. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Huang L. B.; Olivares A. M.; Weix D. J. Reductive Decarboxylative Alkynylation of N-Hydroxyphthalimide Esters with Bromoalkynes. Angew. Chem., Int. Ed. 2017, 56, 11901–11905. 10.1002/anie.201706781. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wei X. F.; Shu W.; Garcia-Dominguez A.; Merino E.; Nevado C. Asymmetric Ni-Catalyzed Radical Relayed Reductive Coupling. J. Am. Chem. Soc. 2020, 142, 13515–13522. 10.1021/jacs.0c05254. [DOI] [PubMed] [Google Scholar]; h Williams C. M.; Johnson J. B.; Rovis T. Nickel-Catalyzed Reductive Carboxylation of Styrenes Using CO2. J. Am. Chem. Soc. 2008, 130, 14936. 10.1021/ja8062925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Anthony D.; Diao T. N. Asymmetric Reductive Dicarbofunctionalization of Alkenes via Nickel Catalysis. Synlett 2020, 31, 1443–1447. 10.1055/s-0040-1707900. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Anthony D.; Lin Q.; Baudet J.; Diao T.. Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes 2019, 58, 3198–3202, 10.1002/anie.201900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Tu H. Y.; Guo L.; Zhu S. Q.; Qing F. L.; Chu L. L. Intermolecular selective carboacylation of alkenes via nickel-catalyzed reductive radical relay. Nat. Commun. 2018, 9, 3488. 10.1038/s41467-018-05951-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt K. M.; Doyle A. G. Dialkyl Ether Formation by Nickel-Catalyzed Cross-Coupling of Acetals and Aryl Iodides. Angew. Chem., Int. Ed. 2015, 54, 9876–9880. 10.1002/anie.201503936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan R.; Shi C.; Zhang D. Q.; Tian Y.; Guo S. J.; Yao H. Q.; Lin A. J. Nickel-Catalyzed Reductive 1,2-Dialkynylation of Alkenes Bearing an 8-Aminoquinoline Directing Group. Org. Lett. 2019, 21, 8915–8920. 10.1021/acs.orglett.9b03147. [DOI] [PubMed] [Google Scholar]
- a Dhungana R. K.; Sapkota R. R.; Wickham L. M.; Niroula D.; Shrestha B.; Giri R. Ni-Catalyzed Arylbenzylation of Alkenylarenes: Kinetic Studies Reveal Autocatalysis by ZnX2. Angew. Chem., Int. Ed. 2021, 60, 22977–22982. 10.1002/anie.202110459. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dhungana R. K.; Shekhar K. C.; Basnet P.; Aryal V.; Chesley L. J.; Giri R. Ni(I)-Catalyzed β,δ-Vinylarylation of γ,δ-Alkenyl α-Cyanocarboxylic Esters via Contraction of Transient Nickellacycles. ACS Catal. 2019, 9, 10887–10893. 10.1021/acscatal.9b03574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Collins K. D.; Glorius F. Employing a robustness screen: rapid assessment of rhodium(III)-catalysed C-H activation reactions. Tetrahedron 2013, 69, 7817–7825. 10.1016/j.tet.2013.05.068. [DOI] [Google Scholar]; b Collins K. D.; Glorius F. A robustness screen for the rapid assessment of chemical reactions. Nat. Chem. 2013, 5, 597–601. 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]
- Pitzer L.; Schafers F.; Glorius F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem., Int. Ed. 2019, 58, 8572–8576. 10.1002/anie.201901935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.