

Abstract
Red blood cells (RBCs) are exposed to both external and internal sources of oxidants that challenge their integrity and compromise their physiological function and supply of oxygen to tissues. Autoxidation of oxyhemoglobin is the main source of endogenous RBC oxidant production, yielding superoxide radical and then hydrogen peroxide. In addition, potent oxidants from other blood cells and the surrounding endothelium can reach the RBCs. Abundant and efficient enzymatic systems and low molecular weight antioxidants prevent most of the damage to the RBCs and also position the RBCs as a sink of vascular oxidants that allow the body to maintain a healthy circulatory system. Among the antioxidant enzymes, the thiol-dependent peroxidase peroxiredoxin 2, highly abundant in RBCs, is essential to keep the redox balance. A great part of the RBC antioxidant activity is supported by an active glucose metabolism that provides reducing power in the form of NADPH via the pentose phosphate pathway. There are several RBC defects and situations that generate oxidative stress conditions where the defense mechanisms are overwhelmed, and these include glucose-6-phosphate dehydrogenase deficiencies (favism), hemoglobinopathies like sickle cell disease and thalassemia, as well as packed RBCs for transfusion that suffer from storage lesions. These oxidative stress-associated pathologies of the RBCs underline the relevance of redox balance in these anucleated cells that lack a mechanism of DNA-inducible antioxidant response and rely on a complex and robust network of antioxidant systems.
1. Introduction
Red blood cells (RBCs) are the most abundant cells in the blood. The average hematocrit of 40–45% correlates with a red blood cell count of 4.8–5.4 × 1012 cells per L, approximately 1000 times more than white blood cells and 20 times more than platelets. Their principal function is to transport oxygen to the tissues. To accomplish this function, RBCs have high intracellular concentration of hemoglobin (Hb), and binding of oxygen to Hb is highly regulated. Nevertheless, a side effect is the generation of superoxide radicals from autoxidation of Hb that must be controlled. In addition, reactive species produced in the vasculature (endothelium and other blood cells) can diffuse and reach the RBCs. To cope with this, RBCs are equipped with a battery of antioxidants, low molecular weight like glutathione (GSH), and enzymes like peroxiredoxin 2, which are described in detail below. The efficient decomposition of oxidant species along with repair mechanisms, the elimination through proteasomal degradation of altered proteins, and vesiculation of irreversibly damaged cellular structures1 keep the RBCs functional for 120 days in circulation. The redox status of the RBC is important not only to keep an adequate supply of oxygen to every tissue cell but also to keep a healthy circulatory system due to RBC interactions with other blood cells and the vascular endothelium.
RBCs lack organelles like the nucleus and mitochondria; thus, no new biosynthesis of proteins takes place in the mature erythrocyte, and energy relies on glycolysis. Glucose is the source of energy as ATP and also the source of reducing equivalents such as NADH (glycolysis) and NADPH (pentose phosphate pathway). ATP is mainly tasked with maintaining transmembrane ion gradients, membrane integrity, and the interaction with the cytoskeleton. This is crucial to maintain the human RBC biconcave shape that gives RBCs flexibility to circulate into capillaries as well as to prevent hemolysis that will release Hb into the intravascular space with deleterious consequences.
In this review we focus on the redox metabolism of the human RBC, describing oxidants and antioxidants involved in the maintenance of redox balance. Relevant pathologies associated with RBC oxidative stress like sickle cell disease and thalassemia are also described.
2. The Human RBC
Human RBCs are shaped as biconcave disks with 8 μm maximal diameter, 2 μm thickness, approximately 90 fL in volume, and 140 μm2 surface area.2,3 The biconcave shape results in a high surface area to volume ratio that not only favors gas exchange but also is essential for RBC deformation. Their particular shape and deformability allow RBCs to pass through narrow capillaries and the interstitial slits in the spleen.3 The ability of RBCs to deform depends on the properties and interactions of the RBC cytoskeleton, membrane components (proteins and lipids), and the cellular hydration state and viscosity.4 The plasma membrane of RBCs is composed of 43% lipids, 49% proteins, and 8% carbohydrates in mass.5 The lipid fraction is composed of cholesterol (45%, mol/mol) and phospholipids (55%, mol/mol). The most abundant phospholipids are phosphatidylethanolamine (PE), phosphatidylcholine (PC), and sphingomyelin (SM), with lower amounts of phosphatidylserine (PS), phosphatidylinositol (PI), and glycolipids.5,6 The lipids are distributed asymmetrically across the membrane, with negatively charged phospholipids (PS and PI) facing the cytosol and glycolipids facing the exterior. The exposure of PS in the outer face of the membrane is recognized by macrophages as a senescence signal, and these RBCs are subsequently phagocytosed and removed from circulation.7
The interaction between the cytoskeleton and the membrane proteins involves a large number of transmembrane proteins. The cytoskeleton gives RBCs their ability to deform and return to shape4 and is composed of long α- and β-spectrin heterotetramers that form coiled fibers anchored to the membrane by multiprotein nodal points that are bound to band 38. The most abundant transmembrane protein is the band 3 anion transport protein (SLC4A1), with over 106 copies per cell. It is an anion channel responsible for the bicarbonate/chloride exchange.9 Other transporter proteins are also abundant, such as the glucose transporter Glut1 (1.7 × 105 per cell) and aquaporin 1 (6 × 104 per cell), involved in water transport in response to osmotic gradients.9 ATPases in the membrane are less abundant but are responsible for maintaining ion gradients, and particularly relevant are the Na+/K+-ATPase and Ca2+-ATPase.9 Not as abundant but important in the membrane dynamics of RBCs is the mechano-sensitive PIEZO 1 calcium channel.10
The cytosol in RBCs is largely dominated by Hb (20 mM subunit concentration), followed by carbonic anhydrase (300 μM), Prx2 (250 μM), and then proteins involved in oxidant detoxification and the metabolism of glucose.9,11 Hb is a heterotetramer composed of two α-globin and two non-α-globin chains with one heme molecule bound to each globin. The α-globin locus, located on chromosome 16, contains the α1 and α2 genes, and the β-globin locus, on chromosome 11, contains the β, δ, and γ loci. According to the developmental stage, the major Hb tetramer changes from α2γ2 (HbF) in the fetal period to α2β2 (HbA) in adulthood. In mature RBCs, HbA is the major component, reaching a 5 mM concentration (20 mM heme). The redox state of the heme is very important since oxygen binds to the ferrous heme (FeII), forming oxyhemoglobin (oxyHb). The ferric form (FeIII) or methemoglobin (metHb) is unable to bind oxygen. MetHb forms spontaneously at a rate of 3% Hb per day, and its formation can be accelerated by drugs such as benzocaine and chemicals such as nitrite and can also have genetic causes.12 The RBC contains a metHb reductase enzyme system, which includes cytochrome b5 reductase and cytochrome b5, to catalyze the reduction of the heme iron at the expense of NADH.13
3. Reactive Species Derived from RBCs and Other Sources in the Vasculature
RBCs are exposed to both endogenous and exogenous reactive species. These reactive species, usually referred to in the literature as reactive oxygen species (ROS) or reactive nitrogen species (RNS), are small molecules, oxidants in general, some of which are radicals. It should be emphasized that ROS are not one chemical oxidant species but a wide group of molecules. We will briefly describe the main reactive species involved in oxidative damage to RBCs.
Superoxide
Superoxide (O2•–) is the one-electron reduction product of oxygen and can be produced in RBCs by oxyHb autoxidation that also leads to the production of metHb (Hb(FeIII)) (eq 1).
| 1 |
Autoxidation occurs at a slow rate (k = 4.5 × 10–7 s–1)14 and is accelerated in partially deoxygenated Hb.15 The reverse reaction (reduction of metHb by O2•–) is also possible with k = 4000 M–1 s–1.16 Furthermore, O2•– can be produced enzymatically by NADPH oxidases (Nox2, EC 1.6.3.1) or as a side product of mitochondrial respiration in endothelial cells and leukocytes.17 Also, the enzyme xanthine oxidase (EC 1.17.3.2) that binds to glycosaminoglycans in endothelial surfaces can also produce O2•–.17 Although O2•– can dismutate spontaneously to oxygen and hydrogen peroxide (H2O2) (eq 2), the enzyme superoxide dismutase (SOD1, EC 1.15.1.1), present in RBCs, accelerate this reaction several fold (see below).18
| 2 |
Superoxide per se is not very damaging and is not a strong oxidant; even more, it can act as a reductant sometimes. Reaction with lipid hydroperoxides has been proposed to yield alkoxyl radicals and promote lipid oxidation, eventually leading to RBC lysis,19 but the concentration of lipid hydroperoxides in fresh RBCs is very low. An alternative toxic pathway is the reaction with nitric oxide (NO•) to yield peroxynitrite (see below).
Hydrogen Peroxide
Most H2O2 derives from O2•–, but some oxidases can yield H2O2 directly (Figure 1).17 In addition to its microbicidal properties, H2O2 modulates different cell functions including endothelial cell proliferation and survival, platelet recruitment, insulin secretion, and cardiac remodeling induced by hypertension.20−25 H2O2 can diffuse across the RBC membrane very rapidly, without involving aquaporins.26 H2O2 is not a very oxidizing molecule but can yield the highly reactive hydroxyl radical (HO•) by reduction by metals (Fenton reaction) (eq 3) (Figure 1).
| 3 |
Under normal physiological conditions, the concentration of free or labile iron available for the Fenton reaction is kept very low by extracellular and intracellular proteins, preventing the deleterious reactions mediated by iron.27 However, different genetic disorders can led to iron over load.28 Such is the case of the ferrireductase Steap3, which reduces FeIII to FeII after DMT1 trans membrane transport.29 Mice lacking Steap3 are deficient in erythroid ferrireductase activity and suffer from an iron deficiency anemia.30 On the other hand, increased levels of Steap3 result in degradation of cellular membranes through lipid peroxidation, leading to a failure of RBC homeostasis and hemolysis/clearance of RBCs,31 likely because of Fenton-type reactions due to increased FeII.
Figure 1.
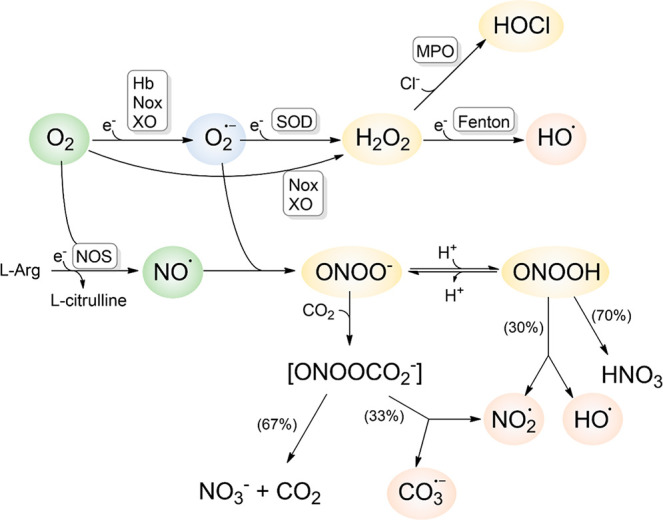
Reactive species produced in the vascular system relevant to RBCs. Oxygen and NO• are the two main ingredients required for the generation of reactive species that will lead to biomolecular damage. Details about each pathway are given in the text.
Two thirds of body iron is present in circulating RBCs as part of Hb.32 In the RBCs, the reaction of H2O2 with the heme of oxyHb yields the pro-oxidizing ferrylHb as an intermediate (eq 4), which can react with a second molecule of H2O2 to yield metHb (eq 5). The reaction of H2O2 with metHb yields ferrylHb with a protein radical that is readily detected by EPR (eq 6).33
| 4 |
| 5 |
| 6 |
To prevent this potentially harmful reactions, the RBCs are equipped with a robust system to reduce H2O2, including peroxiredoxin 2, catalase, and glutathione peroxidase, which will be discussed in detail below.34
Hydroxyl Radical
One of the most oxidizing radicals in biology is HO•.35 It can react with most biomolecules at diffusion-controlled rates, leading to protein, DNA, and lipid damage.36 It can be formed from the reduction of H2O2, mainly by reduced metal atoms, such as Cu+ and Fe2+ (Fenton reaction) (eq 3), and also from peroxynitrite homolysis (see below, Figure 1). In some conditions, excess labile iron in RBCs can contribute to oxidative damage by this mechanism (as in sickle cell disease, see below). Because of the extremely high rates of reaction of HO• with biomolecules, the best mechanism of defense in cells is to prevent its formation by consuming H2O2 very rapidly (see below).
Nitric Oxide
Despite being a radical molecule, NO• is formed in vivo as the product of specific enzymes called nitric oxide synthases, (NOS, EC 1.14.13.39).37 NO• is an autocrine and paracrine signaling molecule which promotes vascular relaxation, inhibits platelet aggregation, decreases inflammation, and modulates the neural activity.38 NOSs are complex enzymes that are constitutively expressed in endothelial cells (NOS3) and can be induced in leukocytes (NOS2).37 Recently an endogenous RBC NOS3 has been reported in low abundance and with functions still poorly understood.39 These enzymes catalyze the conversion of l-arginine to NO• and l-citrulline. The constitutive endothelial NOS3 responds to changes in Ca2+ concentration to increase the production of NO•. NO• produced by endothelial cells can diffuse through cell membranes virtually unhindered and reach underlying smooth muscle cells to cause relaxation through the activation of soluble guanylate cyclase.40,41 A large part of NO• will diffuse to the lumen of blood vessels, reacting mostly with oxyHb in RBCs to yield nitrate and metHb (k = 8.9 × 107 M–1 s–1, eq 7).42,43 The exact amount of NO• that will react with oxyHb depends mainly on the number of RBCs and the size of the RBC-free layer created near the vessel wall by the blood flow. The diffusion across this RBC-free layer will be the main barrier to NO consumption by RBCs.44
| 7 |
Another possible destiny of NO• is the reaction with O2•– to yield the highly oxidizing peroxynitrite (ONOO–) (Figure 1).
Peroxynitrite
The reaction between the two radicals, O2•– and NO•, occurs at diffusion-controlled rates to yield the peroxynitrite anion (ONOO–) (k = 4–16 × 109 M–1 s–1; eq 8) (Figure 1).45
| 8 |
Peroxynitrite is a powerful one- and two-electron oxidant.45 With a pKa of 6.8, at physiological pH peroxynitrite will be a mixture of ONOO– and the protonated peroxynitrous acid (ONOOH). ONOOH can decay relatively slowly to nitric acid plus a 30% fraction of HO• and nitrogen dioxide (NO2•) (eq 9) (Figure 1).
| 9 |
In RBCs, the main targets of peroxynitrite would be Prx2 (k = 1.4 × 107 M–1 s–1, pH 7.4 and 25 °C), oxyHb (k = 5.8 × 104 M–1 s–1), and CO2 (k = 5.8 × 104 M–1 s–1).46−49 Prx2 would detoxify peroxynitrite, but if Prx2 is oxidized the reaction with oxyHb occurs very rapidly with isomerization of peroxynitrite to nitrate, some production of superoxide anions, and finally oxidation of Hb to metHb47 (eq 10).
| 10 |
The reaction with CO2 yields the secondary radicals NO2• and the carbonate radical (CO3•–) in 33% yield (eq 11) (Figure 1).45
| 11 |
The carbonate radical is very reactive and will react rapidly with sulfur-containing molecules, aromatic residues in proteins, ascorbate, and urate (>107; 107–109; 109; 108 M–1 s–1, respectively).50
Nitrogen Dioxide
Sources of NO2• include peroxynitrite homolytic decay46 (Figure 1), the environment, as NO2• is one of the main components of smoke and smog,51 the autoxidation of NO• in lipid membranes,52 and the oxidation of nitrite by heme peroxidases.53 NO2• is a radical, and it is very reactive to a variety of biomolecules, including thiolate-containing molecules, ascorbate, and urate (k ∼ 108; 1.8–3.5 × 107; 2 × 107 M–1 s–1, respectively50), and it can also initiate lipid peroxidation.54 In RBCs, it is likely that both Hb thiols and GSH are the main targets of NO2•, whereas urate would be the main target in plasma.
Hypochlorous Acid
The leukocytes, neutrophils, and monocytes are recruited to sites of infection where they phagocytize and kill invading pathogens. Upon infection-related or inflammatory stimuli, these leukocytes generate large amounts of H2O2 via Nox2 and release myeloperoxidase (MPO).55 MPO is a hemeprotein that uses H2O2 to oxidize chloride to hypochlorous acid (HOCl) (Figure 1), which is highly cytotoxic.55 HOCl participates in both oxidation and chlorination reactions. Chlorination reactions are evident by the formation of chloramines and 3-chlorotyrosine. Reaction with sulfur-containing residues such as cysteine and methionine are expected to be the most important reactions in biology because of their abundance and reactivity (at pH 7.4, k = 3.6 × 108 and 3.4 × 107 M–1 s–1, respectively).56 HOCl and some chloramines can diffuse across the RBC membrane and react preferentially with cytosolic thiols.57 Unlike H2O2 and peroxynitrite, HOCl does not prefer Prx2 but will react with GSH and likely Hb thiols.57
Hydrogen Sulfide
H2S is not an oxidant but is a relevant redox reactive species with biological effects, including vasorelaxation, neurotransmission, and pro- or anti-inflammatory effects, depending on the pathology.58 Both endothelial cells and RBCs have enzymes that synthesize H2S, though unequally distributed. Endothelial cells produce H2S mainly through cystathionine γ-lyase (CSE) and mercaptopyruvate sulfur transferase (MST), whereas RBCs produce H2S via MST.59,60 H2S is slightly hydrophobic and can diffuse freely unhindered by plasma membranes, indicating that RBCs will also be exposed to H2S produced by the endothelium.61,62 H2S reacts with metHb in RBCs at moderate rates (kon = 3.2 × 103 M–1 s–1, eq 12) to form an intermediate iron-bound sulfidated Hb, which can slowly release the H2S (koff = 0.053 s–1).60 In the presence of O2, however, most H2S is rapidly oxidized to thiosulfate and iron-bound polysulfides through poorly characterized intermediates (eqs 13–15).60,63 Iron-bound polysulfides can further react with endogenous GSH to yield glutathione persulfide, which is more reactive than GSH and has been shown to be an important inhibitor of lipid peroxidation.63−65
| 12 |
| 13 |
| 14 |
| 15 |
H2S can also react with Hb to generate the green sulfhemoglobin. The most frequent formation of sulfhemoglobin in blood has been associated with misuse of sulfadrugs, rather than H2S poisoning.66 This green sulfhemoglobin results from the irreversible covalent reaction of sulfur to the pyrrole ring and leads to a 135-fold decrease in oxygen affinity.67 The mechanism of sulfhemoglobin formation is not clear, but it is postulated to involve reaction of H2S with an oxoferryl (Hb(FeIV=O) Por•+ or Hb(FeIV=O)) intermediate.68 The reactions of H2S with Hb are likely important in regulating the biological actions of H2S in the vascular system.
4. Antioxidant Systems in RBCs
The RBC contains several antioxidant systems that allow it to cope with extensive oxidative stress. These include low molecular weight systems involved in cytosolic protein and membrane lipid protection and enzymatic systems that can react and reduce mostly water-soluble oxidants. The following section gives a detailed description of the most relevant antioxidant systems in RBCs.
Superoxide dismutases (SODs, EC 1.15.1.1) are enzymes that catalyze the dismutation or disproportionation of two molecules of O2•– to O2 and H2O2 (eq 2).69 Interestingly, this reaction occurs spontaneously (k = 2 × 105 M–1 s–1), yet it is accelerated 4 orders of magnitude (2 × 109 M–1 s–1) by these enzymes. Considering the relatively high abundance of SOD and diffusion-limited reactivity, the enzyme-catalyzed reaction is the principal O2•– dismutation mechanism in aerobic organisms.70,71
SODs are metalloenzymes classified by the metal ions they bind. There are three forms in mammals: SOD1 and SOD3 that are copper−zinc superoxide dismutases (Cu/Zn SOD), and SOD2 that is a manganese SOD (MnSOD).71,72 RBCs contain Cu/Zn SOD1 which is accountable for at least 95% of the Cu content, and it is present at an approximate concentration of 4 μM.73,74 SOD1 is a homodimeric protein of 32 kDa in which each monomer contains a CuII and a ZnII ion in its structure.75 The catalytic metal is CuII, while ZnII has a rather indirect role in catalysis since it stabilizes the active site structure and adjusts its redox potential.76 During catalysis, the CuII ion is reversibly reduced and oxidized by two consecutive encounters with O2•– (eqs 16 and 17).
| 16 |
| 17 |
In the resting state, the oxidized CuII binds the oxygen atom of a water molecule and is connected to the ZnII ion by a histidine residue (His63). The first O2•– travels down a positively charged active site channel to bind the CuII and displaces the water molecule. Then, the bound O2•– reduces CuII to CuI which leads to O2 formation, the loss of the Cu–His61 coordination bond, and His63 protonation (eq 16). The second O2•– binds to the CuI and oxidizes it to CuII, while it receives two protons coming from the His63 and the solvent, which yield H2O2 (eq 17). As the H2O2 leaves the active site, the Cu–His63 coordination bond is reestablished, and the enzyme returns to the resting state.77,78
SOD1 activity is correlated with RBC physiology, protecting RBC proteins against O2•–-mediated damage. Studies on mice RBCs lacking SOD1 have shown an early increase in their size and a decreased life span (60% of that of control mice), ultimately leading to anemia. Furthermore, these RBCs exhibited high oxidant and metHb levels that had a concomitant increase with age. Moreover, SOD1-deficient RBCs exhibited bound immunoglobulins, and deposits of immune complexes were found in the glomeruli of these mice, which is a hallmark of autoimmune pathology.79 All these effects have been proven to be significantly reverted by the transgenic expression of SOD1.80 RBCs from SOD1 knockout mice also exhibited high levels of oxidized carbonic anhydrase 2 (CA2), which leads to its proteasomal degradation and RBC malfunction. Moreover, the accumulation of oxidized CA2 alters proteasomal activity and disturbs protein homeostasis in the RBC.81
Additionally, in human RBCs, SOD1 can be post-translationally phosphorylated and/or glutathionylated near the dimer interface. Specifically, the glutathionylation of Cys111 disrupts the dimer interface and yields SOD1 monomers that are significantly less active.82
Overall, SOD1 plays a main role in the RBC antioxidant system because it scavenges the O2•– that is continuously produced by oxyHb autoxidation. Although SOD1 activity prevents generation of more potent oxidants such as peroxynitrite (Figure 1), its reaction yields H2O2, which can further oxidize cellular components and/or produce HO•. Therefore, SOD1 must act concertedly with the H2O2 reduction systems in order to complete its antioxidant function.
Peroxiredoxins (Prx, EC 1.11.1.15) are peroxidases that reduce H2O2 and other hydroperoxides (ROOH), using highly reactive cysteine residues. They are ubiquitous, abundant, present in all cell compartments, and indispensable for aerobic life. There are three Prx isoforms in RBCs, namely, Prx1, Prx2, and Prx6.83 Among them, Prx2 is the most abundant in the RBC with a concentration of 240–410 μM.9,84,85
Prx2 belongs to the Prx1 class of the Prx family, also known as typical two-cysteine Prx. These Prx are homodimers (44 kDa) arranged in a head-to-tail fashion that further form toroid-shape homodecamers (220 kDa) (Figure 2). Dimerization is necessary for complete active site folding since each monomer has two halves of an active site: the amino-terminal (N-ter) head and the carboxi-terminal (C-ter) tail. During catalysis, the N-ter active site peroxidatic cysteine (CP) reacts with H2O2 to form a sulfenic acid on CP (CPSOH) (Figure 2). Then, CPSOH reacts with the C-term active site, resolving cysteine (CR) from an adjacent subunit (Figure 2), to form an intermolecular disulfide bond that is later reduced by the thioredoxin/thioredoxin reductase (Trx/TR) system at the expense of NADPH. Occasionally, the CPSOH can react further with H2O2 to form sulfinic (CPSO2) or sulfonic acid (CPSO3), which reversibly or irreversibly inactivates the enzyme.86
Figure 2.

Peroxiredoxin activity. The reduced Cys52 in Prx2 (CPSH) is oxidized by H2O2 and other oxidants to sulfenic acid (CP-SOH). This CPSOH reacts with the CRSH, forming an intermolecular disulfide bridge. The disulfide-oxidized Prx2 is predominantly a dimer and is reduced by Trx, TR, and NADPH. The oxidized CPSOH can alternatively react with a second oxidant molecule to yield the hyperoxidized sulfinic acid (CPSO2). The latter can either be repaired to the active enzyme by sulfiredoxin (Srx) or form stacked decamer high molecular weight structures. The structure of decameric Prx2 (5IJT) shows reactive cysteine residues in yellow, and each dimer is shown in green and blue, as sides of the pentagon.
The Prx2 CP can react with both H2O2 and peroxynitrite extremely fast, with rate constants of 1 × 108 and 1.4 × 107 M–1 s–1, respectively.45,49,88 Nevertheless, the catalytic cycle of Prx2 is significantly delayed by the CP--CR disulfide formation (0.3 s–1), which makes the enzyme prone to hyperoxidation, leading to the accumulation of CPSO2 and/or CPSO3. Prx2 switches its oligomeric form based on the redox state of CP and CR: the dithiol is a stable decamer, while the disulfide-bonded Prx2 is predominantly a covalent dimer.89,90 Prx2 in the RBCs is present mostly in the reduced state, and harsh oxidative insults are needed to accumulate oxidized Prx2.83 In some cases, the recycling of Prx2 is compromised by low NADPH availability, such as in G6PD deficiencies that will be discussed below.91
Prx2 is involved in different aspects of the RBC physiology. From an early stage, highly concentrated Prx2 is necessary in the erythropoiesis process, particularly in the erythroblast, acting as an antioxidant when Hb synthesis is at its peak and large amounts of heme and iron are handled.92 The absence of Prx2 at this stage has shown defective erythropoiesis and iron toxicity.
Furthermore, Prx2 specifically protects Hb against oxidation and is fundamental for the stabilization of its structure. Mice models lacking Prx2 show increased Hb oxidation, Heinz body precipitation, and hemolytic anemia,93 despite having intact catalase and GPx functionality.94,95 It has also been shown that the decameric state of Prx2 is needed to prevent H2O2-induced Hb aggregation.96
A small part of the RBC’s Prx2 pool is located in the cell membrane, where it has been associated with the cytoplasmic domain of a band 3 anion transport protein, spectrin, and the Gardos channel.97−99 Although the function of Prx2 in the RBC membrane is still elusive, the increase of membrane-bound Prx2 is a marker of RBC oxidation and stress.100,101 Interestingly, Prx2 also exhibits functions outside the RBC, as an enhancer of the cytotoxic activity of natural killer cells against tumor cells and as a proinflammatory cytokine that is excreted in exosomes.102,103
Thioredoxin is a small monomeric protein (12 kDa) which has a conserved active site sequence Cys-Gly-Pro-Cys. The N-terminal Cys attacks the target protein disulfide, generating a transient mixed disulfide (eq 18) that is then reduced by the C-terminal Cys in the active site of Trx to generate an intramolecular disulfide in Trx and the protein thiol (eq 19).104 RBCs contain Trx1 that participates in antioxidant defenses, acting as electron donors to several enzymes, including Prx2, and is reduced by Trx reductase (TR). Secreted Trx1 can mediate immune responses in association with Trx-interacting protein.8
| 18 |
| 19 |
Thioredoxin reductase (TR, EC 1.8.1.9) is a homodimeric selenocysteine-containing flavoprotein member of the pyridine nucleotide-disulfide oxidoreductase family. RBCs contain TR1, and each subunit contains FAD and NADPH binding domains. The dimer is accommodated head to tail. The electrons are transferred from NADPH to FAD (eq 20), then to the N-terminal redox-active dithiol (eq 21), then to the C-terminal selenylsulfide in the other subunit (eq 22), and finally to the disulfide substrate (eq 23).105
| 20 |
| 21 |
| 22 |
| 23 |
Substrates of TR include Trx, glutaredoxins, and others.106 Although a lower activity of TR has been measured in human RBCs compared to other cells, it is enough to keep Prx2 in the reduced state.83 The TR/Trx system appears to be connected to the GR/GSH system. For instance, when TR is downregulated, Trx can be reduced by Grx/GSH, and when GR is downregulated, TR can reduce GSSG to GSH.106
Catalases (EC 1.11.1.6) are ubiquitous enzymes that catalyze the decomposition of H2O2 into water and O2. They can be organized into four main groups: monofunctional catalases (typical catalases), bifunctional catalase-peroxidases, nonheme catalases, and miscellaneous proteins with minor catalytic activities.107 Human RBC catalase is found at a concentration of 11–12 μM (subunit concentration) and belongs to the group of monofunctional catalases. It is a tetrameric enzyme consisting of four identical subunits of 59.7 kDa, and each subunit contains a heme group, iron(III) porphyrin IX, and a tightly bound NADPH molecule.34,108
The decomposition of H2O2 occurs in two steps. In the first step, H2O2 oxidizes the heme iron (FeIII) to form the intermediate compound I, a π-porphyrin cation radical containing FeIV (catalase Fe•IV=O) (eq 24). The rate constant for this step per subunit is k = 0.6 × 107 M–1 s–1. One of the protons of the H2O2 molecule is transferred from one end to the other via a histidine residue in the active site, and this polarizes and breaks the O–O bond in the H2O2. In the next step (k = 1.7 × 107 M–1 s–1, per subunit), a second H2O2 molecule acts as a reductant, producing water and O2 and returning the enzyme to the FeIII resting state (eq 25).109,110 Unlike other enzymes, it is not possible to saturate catalase with H2O2, and kinetics follows a first-order reaction on H2O2 concentration.111
| 24 |
| 25 |
Despite being categorized as monofunctional, typical catalase can catalyze the oxidation of two-electron donors other than H2O2 from compound I. Compound I can also be reduced to inactive compound II by one-electron donors (eq 26), and this may be reduced to the native form by another one-electron reduction (eq 27). When compound II reacts with H2O2, inactive compound III is formed (eq 28).112,113
| 26 |
| 27 |
| 28 |
The oxidizing and reducing power during the normal catalytic cycle of catalase comes from H2O2 (eq 24 and eq 25), so NADPH does not appear to be essential for catalytic activity. Therefore, several hypotheses have been proposed about the role of NADPH in catalase, the main one being that it protects the enzyme from inactivation by H2O2 by preventing the formation of compound II.112,113
Glutathione peroxidase (EC 1.11.1.9) is part of the thiol-dependent antioxidant systems of RBCs. It catalyzes the reduction of hydroperoxides through a process that involves GSH, glutathione reductase (GR), and NADPH. Eight different glutathione peroxidases (GPx1–GPx8) are found in mammals, with GPx1 being the predominant one in RBCs at 1.5 μM.23,75 GPx4 is also present, although it is 20 times less abundant.9 Both isoforms are selenoenzymes, with an active site consisting of a catalytic tetrad formed by a selenocysteine residue, along with a glutamine, a tryptophan, and an asparagine.114−116 They differ, however, in their oligomeric state and their substrates. GPx1 is a homotetramer and reacts more rapidly with H2O2 and other small organic hydroperoxides, whereas GPx4 is monomeric and reacts more rapidly with larger and more complex molecules, such as phospholipid and cholesterol hydroperoxides, even when bound to the membrane surface.117−119
The catalytic cycle of GPx is based on a ping-pong mechanism and can be separated in two half-reactions, an oxidative and a reductive phase.120,121 During the oxidative phase, the hydroperoxide is reduced upon reaction with the active site of the enzyme, while its selenocysteine residue is oxidized to selenenic acid (k1 H2O2 = 4.1 × 107 M–1 s–1, k1t-BuOOH = 4.2 × 106 M–1 s–1) (eq 29).122,123 In the reductive half-reaction, the enzyme is regenerated in two subsequent steps. A first molecule of GSH binds to the enzyme through a selenosulfide bond (eq 30), which is then broken by a second GSH, yielding glutathione disulfide (GSSG) and the reduced form of the enzyme (eq 31).122,124,125
| 29 |
| 30 |
| 31 |
While GPx1 has a high specificity for GSH, this is not the case for all the GPx isoforms, as a gradual loss in substrate specificity has been observed. GPx4 can accept other protein thiols and even thiol groups in its own structure as electron donors.117,126
RBCs from GPx1 knockout mice showed increased cell susceptibility to lysis by organic peroxides, such as t-butyl hydroperoxide and cumene hydroperoxide.127,128 In these cases, the oxidative damage was often observed at the membrane level, with the appearance of newly oxidized thiols.129 It was also reported that GPx1 can translocate to the membrane of RBCs in conditions of oxidative stress, before Prx2 or catalase.130 In addition, a recent report indicates the anticorrelation of RBC hemolysis with functional Gpx4 (lyso-phospholipids).131 These results support the theory that, in RBCs, the main role of GPxs is to protect the lipid membrane from oxidative attack by hydroperoxides of a different nature.
Glutathione reductase (GR, EC 1.6.4.2) is a flavoenzyme that catalyzes the recycling of GSSG back to GSH at the expense of NADPH. With a homodimeric structure, both GR subunits are connected via a disulfide bond. Each one can be divided into four domains and present NADPH, FAD, and GSSG binding sites. The FAD domain holds a redox-active disulfide that takes part in the reduction of GSSG.132
GR presents a ping-pong mechanism of catalysis, with a cycle that can be separated in two, oxidative and reductive, half-reactions.121 In the beginning, NADPH binds to the enzyme and reduces the flavin (eq 32), which in turn establishes a charge-transfer complex with one of the cysteines of the active site (Cys63), breaking the previous disulfide bond (eq 33). NADP+ is released and replaced by a new molecule of NADPH. Next, a GSSG forms a mixed disulfide with the enzyme (eq 34), and after its reduction, two molecules of GSH are produced, along with the regeneration of the oxidized enzyme (eq 35).133
| 32 |
| 33 |
| 34 |
| 35 |
In RBCs, GR is found predominantly in its reduced form since the concentration of NADPH is five times larger than the Km for the enzyme.134 This allows GR to continuously maintain the levels of GSH in the millimolar range as well as preserve the balance of NADPH and NADP+ pools in the pentose phosphate pathway.135
Glutaredoxins (Grx, EC 1.20.4.1) are ubiquitous cysteine-dependent enzymes that catalyze both the formation and reduction of mixed disulfides between protein thiols and GSH.136 They can be classified into two groups: dithiolic and monothiolic. The first group are two-cysteine oxidoreductases that get their reduction equivalents from either GSH or TR. On the other hand, monothiolic Grx lacks oxidoreductase activity and has only one active site cysteine that it uses alongside GSH to assemble and transfer iron–sulfur clusters ([Fe–S]) to proteins.137,138
Two Grxs are found in the mature RBC, Grx1 and Grx3, and their concentration range is 4–8 μM and 0.6–0.8 μM, respectively.9 Grx1 is a two-cysteine Grx that can reduce both protein disulfides and protein–GSH mixed disulfides (deglutathionylation). In the first case, the amino-terminal (N-ter) active site cysteine attacks the protein disulfide to form a mixed disulfide between Grx and the protein (eq 36). Then, the C-terminal cysteine forms an intramolecular disulfide bond with the N-ter cysteine which yields the reduced protein (eq 37). Later, the oxidized Grx is reduced by two molecules of GSH to restore the dithiolic enzyme and form GSSG (eq 38), which ultimately will be reduced to GSH at the expense of GR and NADPH.
| 36 |
| 37 |
| 38 |
In the case of deglutathionylation, the enzyme exhibits classical ping-pong catalysis, where first the N-ter cysteine reacts with the glutathionylated protein or low molecular weight thiol, yielding glutathionylated Grx and reduced protein. Then, GSH reduces the Grx–GSH mixed disulfide, to form reduced Grx and GSSG, which is later reduced by GR and NADPH.139,140
Inside the RBCs, Grx1 has been found responsible for the deglutathionylation of Hb, phosphofructokinase, and the reduction of low molecular weight disulfides.141 Glutathionylation of Cys93 of the Hb β-chain is thought to be a protection mechanism against oxidation, yet it can disrupt the interaction between α and β subunits, affecting O2 and heme binding. Additionally, it has been shown that Grx1 reduces pyruvate kinase and restores thiol groups of proteins in the RBC membrane.142 Grx1 is also responsible for the reduction of dehydroascorbate to ascorbate (vitamin C), which acts as a reducing agent within RBCs.143
On a different note, Grx3 is a cytosolic, monothiolic, multidomain Grx, whose role in the mature RBC is still elusive. Nevertheless, studies on a zebrafish model propose that the function of Grx3 is important at the early erythroblast stage, specifically in the maturation of [Fe–S]-containing proteins, the heme synthesis pathway, and iron uptake and distribution.144
Glutathione (GSH), the tripeptide γ-glutamyl cysteinyl glycine (Figure 3), is considered the main intracellular low molecular weight antioxidant but is also a key determinant of redox signaling and xenobiotic metabolism and one of the most important ways of reducing power in the cell.145 In RBCs, the intracellular concentration of GSH is relatively high (0.4–3 mM146,147), and the normal physiological GSH/GSSG ratio is higher than ten.146 Biosynthesis of GSH occurs in the cytosol in a tightly regulated manner. Key determinants of GSH synthesis are the availability of the sulfur amino acid precursor, cysteine, and the activity of the rate-limiting enzyme, glutamate cysteine ligase (GCL, EC 6.3.2.2). GCL is a heterodimer composed of a catalytic (GCLC) and a modifier (GCLM) subunit.148 The holoenzyme is regulated by reversible protein phosphorylation and pyridine dinucleotide phosphate-dependent allostery.149 Glutathione synthetase (GS, EC 6.3.2.3), the second enzyme in GSH synthesis, catalyzes the condensation of γ-glutamylcysteine and glycine, to form GSH.146 Both enzymes depend on ATP production by glycolysis in the RBC.150
Figure 3.

Main low molecular weight antioxidants in RBCs are the water-soluble urate, glutathione, and ascorbate and the lipid-soluble α-tocopherol. These antioxidants can form stable radicals after one-electron oxidation that can be repaired by other antioxidants. The ultimate source of reducing power is given by NADPH, which can be used to yield GSH, which can be used to reduce DHA to ascorbate, which can reduce the urate radical and also the α-tocopheroxyl radical back to their reduced forms.
Cysteine for GSH synthesis is taken up from plasma by facilitated diffusion (L-transport system) and secondary active transport (alanine-serine-cysteine transporter, Asc-1) by RBCs.151,152 The liver and kidney play a fundamental role, providing GSH as a cysteine precursor for interorgan exchange.153 Hepatic GSH is transported into blood and rapidly degraded to cysteine in the circulation by plasma-membrane-bound enzymes γ-glutamyltranspeptidase (γ-GT) and dipeptidases of the kidney and other organs.154,155 γ-Glutamyl-cycle-encompassing GSH synthesis, transport, and catabolism coordinate the redox state of cells and tissues and thiol homeostasis.156 Transsulfuration and thiol/disulfide exchange reactions are additional sources of plasma cysteine.157,158
A significant percentage of GSH is produced de novo daily by RBCs to compensate the active export of GSSG and GSH conjugates.159,160 Carbon monoxide poisoning as well as oxidants like peroxynitrite promote the release of both GSH and GSSG from RBCs.160,161 The multidrug resistance-associated proteins (Mrp/Abcc) mediate GSH export and homeostasis in a variety of cells.162,163 Mrp/Abcc-1, -4, -5, and-10 were identified by mass spectrometry in RBC membranes.9 The Mrp proteins, in addition to mediating GSH efflux, also export GSSG, S-nitrosoglutathione, GSH–metal complexes, as well as other GSH S-conjugates. The ability to export both GSH and oxidized derivatives of GSH provides these transporters with the capacity to directly regulate the cellular thiol-redox status and, therefore, the ability to influence signaling and biochemical pathways.164 As mentioned above, GSH is regenerated from GSSG by the NADPH-dependent GR (Figure 3).
Ascorbate (vitamin C, AscH) cannot be synthesized by humans and must be obtained from the diet. At neutral pH, most of it is present in the anionic ascorbate form rather than as ascorbic acid. Ascorbate is an electron donor that can be oxidized by either one electron to the ascorbyl radical or two electrons to dehydroascorbate (DHA), but this is usually rapidly reduced back to ascorbate (Figure 3).165 Ascorbate can react with free radicals in the cytosol and also keep α-tocopherol (vitamin E) in the reduced state in the plasma membrane (Figure 3).166−168
Unlike other human cells, there is no active transport for ascorbate in RBCs.165 Instead, DHA is transported into the RBC by glucose transporters GluT, where it is reduced back to ascorbate by GSH and Grx.169 The concentration of DHA in plasma is estimated to be 1–2% that of ascorbate.165 The concentration of ascorbate in human RBCs is directly proportional and slightly lower than in plasma, amounting to 35 μM in average.170 At this concentration, ascorbate will likely not act as a direct free radical scavenger but may aid in keeping α-tocopherol reduced in the membrane and participate in enzymatic reactions. Human RBCs contain the duodenal cytochrome b561 isoform in the membrane that was shown to transport reducing equivalents from ascorbate in the cytosol to the exterior and may contribute to maintaining plasma ascorbate in the reduced state.171
RBCs from type 2 diabetes patients are mechanically more fragile than control RBCs.172 It was proposed that excess glucose competes with DHA transport and leads to lower intracellular concentration of ascorbate that then leads to increased membrane rigidity and cell fragility.173 The causes are not clear and may involve enzymatic reactions that use ascorbate as a substrate rather than antioxidant effects or a combination of both.173
The addition of either ascorbate or DHA to blood stored for transfusion only slightly decreased storage lesions.174,175 In contrast, the addition of ascorbate together with N-acetyl cysteine (NAC) to RBCs stored for transfusion resulted in overall improvement of RBC quality, in particular, GSH and α-tocopherol levels, leading to lower rates of lipid oxidation. The glycolytic flux was diminished, but ATP and NADH were higher than in the control, and NADPH increased only transiently. A decrease in hemolysis was observed only for 21 days of storage.176 Millimolar concentrations of ascorbate obtained by high dose intravenous infusion of vitamin C were found to increase metHb formation and Prx2 oxidation, suggesting that high concentrations may be detrimental to RBCs.177
Vitamin E encompasses eight fat-soluble compounds containing a chromane ring with a hydroxyl group at C-6 and a polyprenoid side chain, with three isopentyl units at the C-2 position. When the polyprenoid chain is saturated, the isomers are tocopherols, and when it is unsaturated, they are tocotrienols. According to the number and position of methyl groups in the aromatic ring, they are called α, β, δ, and γ tocopherol. These compounds are synthesized mainly by plants and cyanobacteria. Although γ-tocopherol is the most abundant in the diet, α-tocopherol shows greater bioavailability in plasma and human tissues (Figure 3).178,179 It is preferentially retained by the organism, thanks to the α-tocopherol transfer protein, which is expressed in the liver and presents greater selectivity for α-tocopherol than other compounds.180,181
Although there is some controversy about its main role, α-tocopherol is considered the most important antioxidant-protecting membrane lipid against oxidative damage.182,183 Lipid peroxidation mainly affects polyunsaturated fatty acids (PUFAs) and leads to the formation of lipid hydroperoxides, lipid alcohols, and aldehydes. Membrane lipid peroxidation begins with the attack of reactive species capable of removing a hydrogen atom from a PUFA. PUFAs are particularly susceptible to these oxidants since they possess easily oxidizable bisallylic hydrogens.184 Hydrogen abstraction leads to a carbon-centered radical that tends to stabilize by molecular rearrangement to produce a conjugated diene, which rapidly reacts with oxygen to give a lipoperoxyl radical. Lipoperoxyl radicals abstract hydrogen atoms from other lipid molecules, forming a lipid hydroperoxide and fueling the chain reaction of lipid peroxidation.185 The hydroxyl group in α-tocopherol competes with the unsaturated chains for the reaction with the lipoperoxyl radical, forming a less reactive tocopheroxyl radical, thus preventing the propagation of the chain reaction (Figure 3).35 This radical can be reduced to α-tocopherol by ascorbate through its oxidation to ascorbyl radical, two of which can dismutate to ascorbate and DHA.168,186 Alternatively, the tocopheroxyl radical can be further oxidized to the stable form, α-tocopherylquinone (Figure 3).
The concentration of α-tocopherol in RBCs has been determined to be 1.7–7.8 μM.187−193 This concentration is in agreement with kinetic predictions that indicate that the α-tocopherol/lipid ratio in membranes must be of the order of 1/1000 in order to be an effective chain-breaking antioxidant.35
Interestingly, it has been seen that intracellular parasites, including the RBC-infecting Plasmodium falciparum, are capable of synthesizing tocopherol as a defense mechanism against oxidative stress.194−197
Uric acid is the final product of the catabolism of purine bases. Its formation is catalyzed by the enzyme xanthine oxidase (XO, EC 1.17.3.2), which converts hypoxanthine to xanthine and xanthine to uric acid. This series of reactions uses O2 as an electron acceptor, thus yielding O2•– and H2O2 as secondary products. The antioxidant potential of uric acid resides in its ability to react with strong oxidants, such as peroxyl radicals and HO•. Urate can be oxidized by one-electron transfer to a urate radical anion, which can then be recycled in the presence of ascorbate (Figure 3). In a two-electron transfer reaction, urate is oxidized predominantly to allantoin (Figure 3) and also parabanic acid, oxaluric acid, and other compounds.198−200
In mammals, xanthine oxidase is mostly located in the liver, but it has also been found in epithelial cells of mammary glands and capillary endothelial cells in adipose, cardiac, and lung tissues.201 The RBC, on the other hand, does not show xanthine oxidase activity, so uric acid is not expected to be produced by the RBC.202 However, they are exposed to high concentrations of uric acid present in plasma (200–400 μM), where it represents one of the most important soluble antioxidants, accounting for 30–65% of the peroxyl-radical-scavenging capacity.203,204 Furthermore, it was revealed in earlier studies that RBCs are able to uptake uric acid, but it should be noted that there is not a consensus value reported for the intracellular concentration reached, and the mechanism is not fully understood. The main route was proposed to be an active transporter, with simple diffusion occurring in the absence of ATP.205−208
Regarding its possible antioxidant role, in vitro studies have shown that uric acid could help neutralize the oxidant species produced from Hb autoxidation. It was reported that it can limit the rate of formation of metHb, probably by reacting with intermediates like NO2• and ferrylHb.205,206 Experiments performed in RBCs from volunteers before and after physical exercise, where oxidative stress is increased, support this idea of uric acid as an antioxidant. The results exhibited an increase in uric acid concentrations inside RBCs, which decreased after an hour, following the appearance of allantoin.209 Other studies carried out in RBC ghosts have shown that uric acid added externally could protect RBC membranes from oxidative stress. Lipid peroxidation was observed to be diminished in ghosts exposed to t-butyl hydroperoxide or 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH) when treated with uric acid.205,210,211 Along the same lines, more recent observations made in RBCs stored for transfusion indicate there could be a beneficial effect in supplementing samples with uric acid.212 RBC concentrates that proceeded from donors with higher levels of uric acid in plasma manifested less deterioration during storage. Mainly, a lower percentage of echinocytes, band 3 proteolysis, and a diminished binding of calpain and Prx2 to the membrane were observed.213
5. Glucose Metabolism in RBCs and Generation of NADPH
RBCs do not contain mitochondria, and the glucose metabolism is dominated by glycolysis and the pentose phosphate pathway (PPP). Glycolysis provides ATP needed to maintain the ion gradients across the membrane and NADH for metHb reduction to oxyHb and pyruvate reduction to lactate. The PPP provides NADPH, one of the most important biological carriers of reducing equivalents, working usually as a coenzyme. This molecule is ubiquitously distributed in different cells, where it plays an essential role in redox homeostasis. In human RBCs, the concentration of NADPH has been reported to range from 16 to 44.9 μM.214−219
In RBCs, NADPH provides reducing power which is used by different antioxidant enzymes to fight against oxidative damage,220−223 and it is mainly produced by glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD), enzymes involved in the PPP. Glucose-6-phosphate dehydrogenase catalyzes the first step of the PPP, converting glucose-6-phosphate to 6-phosphogluconolactone, using NADP+ as the substrate and Mg2+ as the cofactor and producing NADPH. The Km for NADP+ has been reported to be 4.2 × 10–6 M at pH = 7.6.224 The third step of the PPP is catalyzed by 6-phosphogluconate dehydrogenase. This enzyme uses Mn2+ as a cofactor and NADP+ as a substrate to catalyze the decarboxylation of 6-phosphogluconate, producing ribulose-5-phosphate, CO2, and NADPH. A Km = 20 μM at pH = 8 has been reported.225
The fluctuations of O2 concentrations to which RBCs are subjected in the circulation regulates the flux of glucose to generate ATP by glycolysis or NADPH by the PPP.226 In the pulmonary capillaries, where O2 concentration is relatively high, the glycolytic enzymes are inhibited by their association to band 3, pushing glucose and the flux of electrons to the PPP. Conversely, in peripheral vascular beds with lower O2 tensions, deoxygenated Hb binds band 3, displacing the glycolytic enzymes which became active.227,228 Oxidative modification of GAPDH reroutes glycolysis to PPP to fuel NADPH biosynthesis.229,230
NADPH is used in RBCs as a substrate of two enzymes that are relevant to support the antioxidant defenses, GR and TR. On one hand, GR recycles GSSG to GSH, which is used by GPx and Grx. On the other hand, TR keeps Trx in the reduced state, which serves as an electron delivery system that is used by Prx2 and reduction of other cysteines due to its disulfide reductase activity.231 As will be discussed below, glucose-6-phosphate dehydrogenase deficiency is a common human enzymopathy that leads to several phenotypes ranging from neonatal jaundice to acute anemia, indicating how important NADPH is to prevent oxidative stress in RBCs.232
Recent studies have shown that the exposure of RBCs to different exogenous substances (exposome), including prescribed and over-the-counter drugs, smoking, as well as drinking coffee and taurine-rich beverages, can influence glucose use and NADPH production, impacting the capacity of the cells to cope with oxidants.220−223
6. Integrating the Oxidant Challenges and the Antioxidant Defenses of RBCs
RBCs are exposed to both endogenous and exogenous oxidants. In circulation and mainly in microcirculation, RBCs come in contact with oxidants generated by surrounding cells and tissues. Endothelial cells, and leukocytes in particular, express the enzymes devoted to the production of NO•, O2•–, and H2O2. In leukocytes, the inducible isoform of the NOS (NOS2, EC 1.14.13.39) and the NADPH-oxidase (Nox2, EC 1.6.3.1) generates NO• and O2•–.233,234 Both leukocytic enzymes are activated during inflammation, and in many cases MPO is released and produces HOCl. In blood-vessel-lining endothelial cells, NO• and H2O2 are produced by NOS3 and Nox1, 2, 4, and 5, contributing to maintain the normal blood flow.235 The imbalance in the relative production of endothelial-derived relaxation and contraction factors alters endothelial physiology and may lead to impairment of the normal blood flow.17 Moreover, uncontrolled production of oxidants has been involved in the development of chronic diseases, such as atherosclerosis and neurodegenerative diseases.236 As mentioned above, NO• and O2•– are weak oxidants, but they can give rise to more potent oxidant molecules like peroxynitrite, H2O2, and HO• (Figure 1).11,237,238
The oxidants generated both in the vasculature and by the surrounding tissues converge toward the blood. Due to their abundance and the high membrane permeability, RBCs are preferential targets for the oxidants reaching the bloodstream. In fact, uncharged NO•, H2O2, ONOOH, and HOCl freely diffuse through the lipid bilayer, while anion channels, like the exceedingly abundant band 3 anion transport protein, allows the permeation of O2•– and ONOO– through the RBC membrane.26,41,239 Once inside the RBC, the oxidants are rapidly decomposed by a complex and concerted antioxidant machinery, resulting in a marked concentration gradient across the membrane and promoting the entry and decomposition of more oxidant molecules, supporting the role of RBCs as an efficient sink of oxidants, protecting cells and tissues from uncontrolled oxidative stress.
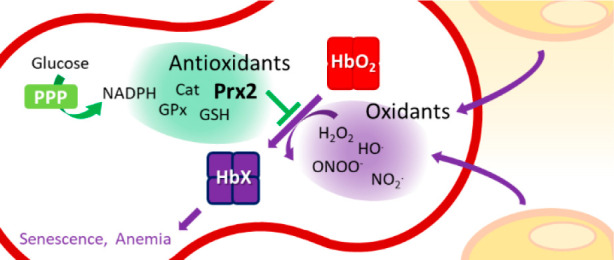
The antioxidant systems in RBCs act in concert to detoxify most of the reactive species produced by RBCs and their surroundings. OxyHb autoxidation generates O2•– that is rapidly converted to H2O2 by SOD1. There has been a long debate about the relative importance of the different antioxidant systems in RBCs responsible for H2O2 detoxification. Kinetics and further experimental confirmation indicate that the first line of defense of RBCs against H2O2 is Prx2, supported by its abundance and very high reaction rate (Table 1).34 Prx2 activity is mainly sustained by Trx/TR and ultimately NADPH from the PPP.34 When NADPH is depleted and Prx2 is completely oxidized, catalase acts as the second line of defense. In many experimental conditions Prx2 is completely oxidized, and catalase has been observed as the main antioxidant enzyme, resulting in a persistent confounding factor in the literature.34 GPx1 is expected to play a minor role in H2O2 detoxification mainly because of its lower abundance34,85,98 but seems to be more important in the reduction of lipid hydroperoxides.124
Table 1. Concentrations and Rate Constants of the RBC Components Responsible for H2O2 Removal.
| Reactant with H2O2 | Concentration (μM) | Second-order rate constant (M–1 s–1) | Pseudo-first-order rate constant (s–1) |
|---|---|---|---|
| Peroxiredoxin-2 | ∼30034,98,240 | 1 × 10845,49,88 | ∼30,000 |
| Catalase | 1134 | 6 × 106109 | 66 |
| Glutathione peroxidase 1 | ∼134,242 | 2 × 107243 | ∼20 |
| Hemoglobin, oxyHb | 20,000244 | 100244 | 2 |
| Glutathione, GSH | 1,500246 | 0.4245 | 6.3 × 10–4 |
The lipids in the membrane are protected against oxidation by α-tocopherol and ascorbate that act in concert to reduce lipid peroxyl radicals to lipid hydroperoxides. The α-tocopheroxyl radical is reduced by ascorbate, and the resulting DHA is reduced by Grx and GSH. The lipid hydroperoxides are then reduced by GPx and GSH to the corresponding alcohols to prevent the formation of the highly reactive alkoxyl radicals. Protein thiols can be protected from irreversible oxidation by glutathionylation,245 and this modification can be reversed by Grx1. The products of H2S reaction with metHb, in particular the polysulfides, could yield glutathione persulfide and other persulfides that could also protect against protein and lipid oxidation in RBCs, but this remains to be proven.
Peroxynitrite will also react preferentially with Prx2, while NO2•, HO•, and CO3•–, which are more reactive and less selective, will react with the most abundant target, Hb. It all indicates that the RBC is well-equipped to prevent the formation of these highly reactive species that will damage Hb, essential for the main RBC function. Most of these reactions are shown in Figure 4, intended to illustrate the complexity, redundancy, and robustness of RBC antioxidant systems.
Figure 4.

Antioxidant systems in RBCs are robust and redundant, allowing the detoxification of several oxidants (shown in red boxes) such as O2•–, H2O2, and ONOO– that could lead to more potent and less selective oxidants such as NO2• and HO•, which could result in hemoglobin damage, affecting RBC functionality. Oxidants can be generated endogenously or can be from other cells in the blood vessels. The RBC contains low molecular weight antioxidants, such as GSH, ascorbate, and α-tocopherol, and several enzymatic systems (in ovals of different colors). Reduction reactions are shown by green arrows. The reducing power for the antioxidant systems in RBCs is ultimately provided by NADPH from glucose-6-P and the pentose phosphate pathway. Details of the different pathways are given in the text.
7. Alterations in RBC Redox Homeostasis Led to Diseases
Oxidative stress was found in several hereditary RBC diseases, including the hemoglobinopathies (sickle cell disease and thalassemia) and glucose-6-phosphate dehydrogenase deficiency. Although oxidative stress is not the primary etiology of these diseases, oxidative damage to the RBC plays a crucial role in early removal of RBCs from circulation or provoking hemolysis; thus, anemia is a common feature to all these patients. A single mutation in the β globin gene of Hb, the substitution of the sixth glutamic acid for valine (Glu6Val, HbS), is the cause of sickle cell disease (SCD). Under hypoxic conditions, the deoxygenated state of HbS polymerizes as fibers in RBCs, which deform into sickle-shaped cells that occlude capillaries and cause intravascular hemolysis. Polymerized HbS reduces cell deformability and impairs rheology and survival of RBCs. In heterozygotes, the coexistence of HbA and HbS in RBCs prevents polymerization of HbS, but SCD manifests in homozygotes. These people display anemia, feeding disorders, splenomegaly, and recurrent infections. In addition, vascular occlusion can cause cerebral infarction. There was an early report indicating HbS can autooxidize twice as fast as HbA; however, comparable rates were shown later.246,247 Nevertheless, hemolysis releases Hb, iron, and increased HO• production via Fenton reaction, indicating oxidative stress is an important feature of SCD.248 Free Hb released from intravascular hemolysis of the sickle erythrocytes induces endothelial dysfunction by depleting endothelial NO•, heme-mediated inflammation, and iron overload.249 Desferoxamine as an iron chelator and a catalase mimetic were shown to decrease oxidative stress and inflammation in murine models of SCD.250,251 Hydroxyurea, approved by the FDA to treat SCD, increases fetal HbF with no β-chain and reduces the tendency for HbS to polymerize.252
Thalassemia is defined as a quantitative imbalance of α- and β-Hb chain production. α-Thalassemia develops when a gene deletion in the α-globin locus (α1 and α2) occurs. The excess of β-chains leads to the formation of Hb β4 that is stable but incapable of carrying oxygen. Conversely, β-thalassemia (or minor thalassemia) is mainly caused by a missense mutation (single amino acid substitution) in the β-globin gene. The excess of α-chains cannot form a tetramer; heme is easily released; oxidative stress in RBCs is installed; and anemia is observed. It was reported that an elevated content of GSH in RBCs significantly reduced the sensitivity of thalassemic RBCs to hemolysis and phagocytosis by macrophages.253
As mentioned before, glucose-6-phosphate dehydrogenase (EC 1.1.1.49, G6PD) participates in the generation of NADPH. G6PD deficiency is the most common human enzyme defect, an X-linked hereditary genetic defect due to mutations in the g6pd gene (about 140 mutations have been described, mostly single amino acid substitution). The clinical manifestation is hemolytic anemia triggered by exogenous agents like drugs and fava beans (so-called favism).232 The G6PD enzyme is critical to protect RBCs against oxidative stress, and subjects with deficiency in this enzyme are at risk of hemolysis under certain conditions. An increase in oxidative markers has been observed in these patients, and management of G6PD deficiency is to prevent hemolysis by avoiding oxidative stress. Successful treatment on favism-induced rats was reported with antioxidant β-carotene and also with a small-molecule activator AG1 that promotes the G6PD dimeric state on patient RBCs.254,255
8. RBC Aging in Circulation and in Packed RBC for Transfusion
Despite the effectivity of the RBC antioxidant systems, these hematic cells are constantly challenged. Moreover, in the capillaries, the increase of partially oxygenated Hb raises the rate of Hb autoxidation256 as well as exposure to the oxidants generated by other cells and tissues.257 This repeated exposure to oxidants deteriorates the capacity of RBCs to cope with them and to fulfill their physiological function. The increased metHb concentration impairs the capacity of RBCs to transport oxygen but also increases the adherence of Hb to the RBC membrane, impacting ATP synthesis and exposing its molecular components to the deleterious effects of superoxide and derived oxidants. The oxidative insult disrupts the interactions between membrane lipids and proteins and the cytoskeleton, compromising the ability of RBCs to squeeze and transverse narrow capillaries, affecting also the transport of ions and organic molecules like glucose through the membrane.257 The accumulation of altered proteins and lipids leads to vesiculation of the plasma membrane with the consequent release of extracellular vesicles in order to remove damaged and potentially toxic molecules.258−260 While vesiculation is an important homeostatic mechanism, excessive shedding of membrane parts inevitably leads to less deformable RBCs that are more prone to lysis.261 These aging-related changes are enhanced in RBCs affected by genetic disorders and also in RBCs exposed to oxidative stress.260,262
The lifespan of mature RBCs is ∼120 days. The removal of senescent or damaged RBCs involves tightly regulated molecular mechanisms, with participation of splenic and liver macrophages.263−265 The phagocytic response of these macrophages is triggered by the absence of CD47 and the exposure of phosphatidylserine in the outer membrane leaflet of senescent or damaged RBCs.266 The progressive loss of elasticity and increased rigidity are also responsible for the recognition of RBCs by the phagocytic cells.265,267 Another important mechanism promoting the engulfment of RBCs is triggered by the formation of antigens on the cells’ surface due to the interaction between denatured Hb and band 3, disrupting the membrane structure and leading to clustering of membrane proteins and the formation of protein aggregates. Band-3-centered protein aggregates become targets for opsonization by naturally occurring antibodies.267−269 Those changes also occur in some genetic disorders, such as sickle cell disease (SCD), spherocytosis, and thalassemia as well as acquired pathologies such as sepsis, malaria, or diabetes, shortening the life span of affected RBCs.270−272
Under storage of packed RBCs for transfusion, several aging RBC phenotypes are evident, dominated by the so-called “storage lesions”.273 This term brings together a set of progressive structural and metabolic changes in RBCs stored for transfusion. These lesions include the oxidation of lipids and proteins and affect RBC metabolism by compromising the activity of key enzymes, leading to a marked decrease in ATP and 2,3-diphosphoglycerate levels.274 The decrease in ATP deregulates cation homeostasis and alters membrane asymmetry, triggering the exposure of phosphatidylserine and phosphatidylethanolamine, normally confined to the inner bilayer.275 The structural disorganization of the membrane and the cytoskeleton, together with the lack of control in calcium homeostasis, leads to a progressive loss of the biconcave disc shape and the ability of the RBC to squeeze, properties that allow rapid gas exchange with the environment and traversing narrow capillary beds, characteristics of healthy RBCs.276
Additionally, functional and structural changes have been observed at the level of band 3.277 This protein, in addition to mediate the membrane Cl–/HCO3– exchange, is also a key regulator of the RBC cytoskeleton dynamics and cell metabolism.9,278 During storage, a disruption of the interaction of band 3 with cytoskeleton proteins and a progressive clustering of the protein has been observed.97,279 These changes, which also involve other proteins and lipids of the membrane, lead to changes in the shape of RBCs and give rise to extracellular vesicles containing altered cellular components.280 Cellular reducing power is also affected as a consequence of metabolic changes, with a decrease in GSH and NADPH,146,281 compromising the ability of RBCs to respond to oxidative stress, both in the transfusion bag and in the circulation after transfusion. Under normal conditions, band 3 acts as a nucleation center for various enzymes of the glycolytic pathway in RBCs, regulating the flow of glucose toward ATP generation or NADP reduction, depending on the oxygen level. This fine metabolic regulation mediated by band 3 is disrupted in aged cells by oxidation, fragmentation, and nonenzymatic glycation of the protein.278 These structural and functional changes are reproduced in RBCs carrying altered Hb and in RBCs lacking or carrying band 3 polymorphisms.282−285 Moreover, altered RBCs and the extracellular vesicles released by them can trigger an inflammatory response, evolving to major complications in the transfusion recipient.286 Although leukoreduction has improved several parameters, concerns about the safety of RBCs stored for longer periods and the patient outcomes still persist.287−289 At present, most of the efforts in blood storing for transfusion are directed to decrease these aging- and oxidative-related changes in order to ensure a safe therapy for the transfusion recipients. Furthermore, there are genetic and environmental differences between donors which make blood a nonstandardized therapeutic tool. A genome-wide association study (GWAS) identified G6PD polymorphism to impair RBC recovery after transfusion and modulate disease severity in hemolytic diseases.290−292
9. Conclusions
RBCs are exposed to oxidants of endogenous and exogenous origin but are well-equipped to cope with these oxidants. The antioxidant defense includes low molecular weight molecules such as GSH, ascorbate, urate, and α-tocopherol, enzymes such as SOD1 and catalase, and multienzymatic systems, such as Prx2/Trx/TR and Gpx/GSH/GR, that ultimately depend on the reducing power provided by the PPP in the form of NADPH. Many of these enzymes react very rapidly with mildly oxidizing reactive species, such as O2•–, H2O2, and ONOO–, suggesting that the main purpose of these antioxidant systems is to avoid the formation of the more potent and less selective oxidants HO•, NO2•, and CO3•– that will damage Hb and compromise RBC function. Even though RBCs are very robust to oxidant damage, congenital defects including hemoglobinopathies and G6PD deficiency result in less viable RBCs, related to rapid accumulation of damaged biomolecules in these RBCs. Furthermore, the RBCs that are stored for transfusion for medium to long periods of time also suffer from storage lesions related to the oxidant damage of biomolecules.
RBCs in circulation are exposed to constant oxidative stress, and maintenance of an adequate redox balance is essential to pursue their physiological function and to preserve Hb as an oxygen carrier as well as a flexible membrane for sustained microcirculation dynamics.
Acknowledgments
Financial support was provided by Universidad de la República (CSIC I+D grupos 46725 to A.D. and CSIC I+D 2020 to M.N.M.) and Agencia Nacional de Investigación e Innovación, ANII (FMV_1_2019_1_155597 to L.T.). M.N.M., F.O., S.F.V., A.C.L., N.S., L.T., and A.D. were partially supported by PEDECIBA, Uruguay. F.O. and A.C.L. were partially supported by scholarships from Comisión Académica de Posgrado, Universidad de la República. S.F.V., N.S., and M.D. were partially supported by scholarships from ANII.
Glossary
Abbreviations
- RBC
red blood cell
- Hb
hemoglobin
- GSH
glutathione
- GSSG
glutathione disulfide
- PE
phosphatidylethanolamine
- PC
phosphatidylcholine
- SM
sphingomyelin
- PS
phosphatidylserine
- PI
phosphatidylinositol
- band 3
band 3 anion transport protein (SLC4A1)
- Prx2
peroxiredoxin 2
- oxyHb
oxyhemoglobin
- metHb
methemoglobin
- O2•–
superoxide
- Nox2
NADPH oxidase 2
- SOD1
superoxide dismutase 1
- H2O2
hydrogen peroxide
- NO•
nitric oxide
- HO•
hydroxyl radical
- NOS
nitric oxide synthases
- ONOO–
peroxynitrite
- ONOOH
peroxynitrous acid
- NO2•
nitrogen dioxide
- CO3•–
carbonate radical
- MPO
myeloperoxidase
- HOCl
hypochlorous acid
- Trx
thioredoxin
- TR
thioredoxin reductase
- cat
catalase
- GPx
glutathione peroxidase
- GR
glutathione reductase
- Grx
glutaredoxin
- AscH
ascorbate
- DHA
dehydroascorbate
- XO
xanthine oxidase
- PPP
pentose phosphate pathway
- G6PD
glucose-6-phosphate dehydrogenase
- 6PGD
6-phosphogluconate dehydrogenase
- SCD
sickle cell disease
The authors declare no competing financial interest.
References
- D’Alessandro A.; Hansen K. C.; Eisenmesser E. Z.; Zimring J. C. Protect, Repair, Destroy or Sacrifice: A Role of Oxidative Stress Biology in Inter-Donor Variability of Blood Storage?. Blood Transfus 2019, 17 (4), 281–288. 10.2450/2019.0072-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren C. E.; Brittenham G. M.; Hasselblad V. Statistical and Graphical Evaluation of Erythrocyte Volume Distributions. Am. J. Physiol. 1987, 252, H857–H866. 10.1152/ajpheart.1987.252.4.H857. [DOI] [PubMed] [Google Scholar]
- Li H.; Lu L.; Li X.; Buffet P. A.; Dao M.; Karniadakis G. E.; Suresh S. Mechanics of Diseased Red Blood Cells in Human Spleen and Consequences for Hereditary Blood Disorders. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (38), 9574–9579. 10.1073/pnas.1806501115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisjes R.; Bogdanova A.; van Solinge W. W.; Schiffelers R. M.; Kaestner L.; van Wijk R. Squeezing for Life - Properties of Red Blood Cell Deformability. Front Physiol 2018, 9, 656. 10.3389/fphys.2018.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawata Y.The Red Blood Cell as a Model. In Cell Membrane; Wiley, 2006. [Google Scholar]
- Himbert S.; D’Alessandro A.; Qadri S. M.; Majcher M. J.; Hoare T.; Sheffield W. P.; Nagao M.; Nagle J. F.; Rheinstädter M. C. The Bending Rigidity of the Red Blood Cell Cytoplasmic Membrane. PLoS One 2022, 17 (8), e0269619 10.1371/journal.pone.0269619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabas M.; Coupland L. A.; Cromer D.; Winterberg M.; Teoh N. C.; D’Rozario J.; Kirk K.; Bröer S.; Parish C. R.; Enders A. Mice Deficient in the Putative Phospholipid Flippase ATP11C Exhibit Altered Erythrocyte Shape, Anemia, and Reduced Erythrocyte Life Span. J. Biol. Chem. 2014, 289 (28), 19531–19537. 10.1074/jbc.C114.570267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux S. E. Anatomy of the Red Cell Membrane Skeleton: Unanswered Questions. Blood 2016, 127 (2), 187–199. 10.1182/blood-2014-12-512772. [DOI] [PubMed] [Google Scholar]
- Bryk A. H.; Wiśniewski J. R. Quantitative Analysis of Human Red Blood Cell Proteome. J. Proteome Res. 2017, 16 (8), 2752–2761. 10.1021/acs.jproteome.7b00025. [DOI] [PubMed] [Google Scholar]
- Svetina S. Theoretical Bases for the Role of Red Blood Cell Shape in the Regulation of Its Volume. Frontiers in Physiology 2020, 10.3389/fphys.2020.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turell L.; Möller M. N.; Orrico F.; Randall L. M.; Steglich M.; Villar S.; Denicola A.; Thomson L.. Chapter 25 - Thiols in Blood. In Redox Chemistry and Biology of Thiols; Alvarez B., Comini M. A., Salinas G., Trujillo M., Eds.; Academic Press, 2022; pp 585–615. 10.1016/B978-0-323-90219-9.00025-X. [DOI] [Google Scholar]
- Skold A.; Cosco D. L.; Klein R. Methemoglobinemia: Pathogenesis, Diagnosis, and Management. South Med. J. 2011, 104 (11), 757–761. 10.1097/SMJ.0b013e318232139f. [DOI] [PubMed] [Google Scholar]
- Hultquist D. E.; Passon P. G. Catalysis of Methaemoglobin Reduction by Erythrocyte Cytochrome B5 and Cytochrome B5 Reductase. Nat. New Biol. 1971, 229 (8), 252–254. 10.1038/newbio229252a0. [DOI] [PubMed] [Google Scholar]
- Johnson R.; Goyette G.; Ravindranath Y.; Ho Y.-S. Hemoglobin Autoxidation and Regulation of Endogenous H2O 2 Levels in Erythrocytes. Free radical biology & medicine 2005, 39, 1407–1417. 10.1016/j.freeradbiomed.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Abugo O. O.; Rifkind J. M. Oxidation of Hemoglobin and the Enhancement Produced by Nitroblue Tetrazolium. J. Biol. Chem. 1994, 269 (40), 24845–24853. 10.1016/S0021-9258(17)31468-0. [DOI] [PubMed] [Google Scholar]
- Sutton H. C.; Roberts P. B.; Winterbourn C. C. The Rate of Reaction of Superoxide Radical Ion with Oxyhaemoglobin and Methaemoglobin. Biochem. J. 1976, 155 (3), 503–510. 10.1042/bj1550503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretón-Romero R.; Lamas S. Hydrogen Peroxide Signaling in Vascular Endothelial Cells. Redox Biol. 2014, 2, 529–534. 10.1016/j.redox.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich I. Superoxide Radical: An Endogenous Toxicant. Annu. Rev. Pharmacol Toxicol 1983, 23, 239–257. 10.1146/annurev.pa.23.040183.001323. [DOI] [PubMed] [Google Scholar]
- Lynch R. E.; Fridovich I. Permeation of the Erythrocyte Stroma by Superoxide Radical. J. Biol. Chem. 1978, 253 (13), 4697–4699. 10.1016/S0021-9258(17)30446-5. [DOI] [PubMed] [Google Scholar]
- Li J.; Stouffs M.; Serrander L.; Banfi B.; Bettiol E.; Charnay Y.; Steger K.; Krause K.-H.; Jaconi M. E. The NADPH Oxidase NOX4 Drives Cardiac Differentiation: Role in Regulating Cardiac Transcription Factors and MAP Kinase Activation. Mol. Biol. Cell 2006, 17 (9), 3978–3988. 10.1091/mbc.e05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijles D. N.; Cull J. J.; Markou T.; Cooper S. T. E.; Haines Z. H. R.; Fuller S. J.; O’Gara P.; Sheppard M. N.; Harding S. E.; Sugden P. H.; Clerk A. Redox Regulation of Cardiac ASK1 (Apoptosis Signal-Regulating Kinase 1) Controls P38-MAPK (Mitogen-Activated Protein Kinase) and Orchestrates Cardiac Remodeling to Hypertension. Hypertension 2020, 76 (4), 1208–1218. 10.1161/HYPERTENSIONAHA.119.14556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli P.; Carnevale R.; Di Santo S.; Bartimoccia S.; Sanguigni V.; Lenti L.; Finocchi A.; Mendolicchio L.; Soresina A. R.; Plebani A.; Violi F. Inherited Human Gp91phox Deficiency Is Associated with Impaired Isoprostane Formation and Platelet Dysfunction. Arterioscler Thromb Vasc Biol. 2011, 31 (2), 423–434. 10.1161/ATVBAHA.110.217885. [DOI] [PubMed] [Google Scholar]
- Plecitá-Hlavatá L.; Jabůrek M.; Holendová B.; Tauber J.; Pavluch V.; Berková Z.; Cahová M.; Schröder K.; Brandes R. P.; Siemen D.; Ježek P. Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes 2020, 69 (7), 1341–1354. 10.2337/db19-1130. [DOI] [PubMed] [Google Scholar]
- Van Buul J. D.; Fernandez-Borja M.; Anthony E. C.; Hordijk P. L. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxid Redox Signal 2005, 7, 308–317. 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- Violi F.; Carnevale R.; Loffredo L.; Pignatelli P.; Gallin J. I. NADPH Oxidase-2 and Atherothrombosis: Insight From Chronic Granulomatous Disease. Arterioscler Thromb Vasc Biol. 2017, 37 (2), 218–225. 10.1161/ATVBAHA.116.308351. [DOI] [PubMed] [Google Scholar]
- Orrico F.; Lopez A. C.; Saliwonczyk D.; Acosta C.; Rodriguez-Grecco I.; Mouro-Chanteloup I.; Ostuni M. A.; Denicola A.; Thomson L.; Möller M. N. The Permeability of Human Red Blood Cell Membranes to Hydrogen Peroxide Is Independent of Aquaporins. J. Biol. Chem. 2022, 298 (1), 101503. 10.1016/j.jbc.2021.101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Pantopoulos K. Regulation of Cellular Iron Metabolism. Biochem. J. 2011, 434 (3), 365–381. 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissot P.; Pietrangelo A.; Adams P. C.; de Graaff B.; McLaren C. E.; Loréal O. Haemochromatosis. Nat. Rev. Dis Primers 2018, 4, 18016. 10.1038/nrdp.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D. R.; Lane D. J. R.; Becker E. M.; Huang M. L.-H.; Whitnall M.; Suryo Rahmanto Y.; Sheftel A. D.; Ponka P. Mitochondrial Iron Trafficking and the Integration of Iron Metabolism between the Mitochondrion and Cytosol. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (24), 10775–10782. 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami R. S.; Campagna D. R.; Greer E. L.; Antiochos B.; McDonald A.; Chen J.; Sharp J. J.; Fujiwara Y.; Barker J. E.; Fleming M. D. Identification of a Ferrireductase Required for Efficient Transferrin-Dependent Iron Uptake in Erythroid Cells. Nat. Genet. 2005, 37 (11), 1264–1269. 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie H. L.; Hay A. M.; de Wolski K.; Waterman H.; Lebedev J.; Fu X.; Culp-Hill R.; D’Alessandro A.; Gorham J. D.; Ranson M. S.; Roback J. D.; Thomson P. C.; Zimring J. C. Differences in Steap3 Expression Are a Mechanism of Genetic Variation of RBC Storage and Oxidative Damage in Mice. Blood Advances 2019, 3 (15), 2272–2285. 10.1182/bloodadvances.2019000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbaspour N.; Hurrell R.; Kelishadi R. Review on Iron and Its Importance for Human Health. J. Res. Med. Sci. 2014, 19 (2), 164–174. [PMC free article] [PubMed] [Google Scholar]
- Nagababu E.; Rifkind J. M. Reaction of Hydrogen Peroxide with Ferrylhemoglobin: Superoxide Production and Heme Degradation. Biochemistry 2000, 39 (40), 12503–12511. 10.1021/bi992170y. [DOI] [PubMed] [Google Scholar]
- Orrico F.; Möller M. N.; Cassina A.; Denicola A.; Thomson L. Kinetic and Stoichiometric Constraints Determine the Pathway of H2O2 Consumption by Red Blood Cells. Free Radic Biol. Med. 2018, 121, 231–239. 10.1016/j.freeradbiomed.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Buettner G. R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, Alpha-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300 (2), 535–543. 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O– in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17 (2), 513–886. 10.1063/1.555805. [DOI] [Google Scholar]
- Stuehr D. J.; Haque M. M. Nitric Oxide Synthase Enzymology in the 20 Years after the Nobel Prize. Br. J. Pharmacol. 2019, 176 (2), 177–188. 10.1111/bph.14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran N.; Garcia T.; Aniqa M.; Ali S.; Ally A.; Nauli S. Endothelial Nitric Oxide Synthase (ENOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed Sci. Res. 2022, 15 (2), 155–179. 10.34297/AJBSR.2022.15.002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese-Krott M. M.; Kelm M. Endothelial Nitric Oxide Synthase in Red Blood Cells: Key to a New Erythrocrine Function?. Redox Biology 2014, 2, 251–258. 10.1016/j.redox.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denicola A.; Souza J. M.; Radi R.; Lissi E. Nitric Oxide Diffusion in Membranes Determined by Fluorescence Quenching. Arch. Biochem. Biophys. 1996, 328 (1), 208–212. 10.1006/abbi.1996.0162. [DOI] [PubMed] [Google Scholar]
- M?ller M.; Cuevasanta E.; Orrico F.; Lopez A. C.; Thomson L.; Denicola A.. Diffusion and Transport of Reactive Species Across Cell Membranes. In Advances in Experimental Medicine and Biology; Bioactive Lipids in Health and Disease; Springer Nature Switzerland AG, 2019. [DOI] [PubMed] [Google Scholar]
- Herold S.; Exner M.; Nauser T. Kinetic and Mechanistic Studies of the NO*-Mediated Oxidation of Oxymyoglobin and Oxyhemoglobin. Biochemistry 2001, 40 (11), 3385–3395. 10.1021/bi002407m. [DOI] [PubMed] [Google Scholar]
- Joshi M. S.; Ferguson T. B.; Han T. H.; Hyduke D. R.; Liao J. C.; Rassaf T.; Bryan N.; Feelisch M.; Lancaster J. R. Nitric Oxide Is Consumed, Rather than Conserved, by Reaction with Oxyhemoglobin under Physiological Conditions. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (16), 10341–10346. 10.1073/pnas.152149699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Samouilov A.; Lancaster J. R.; Zweier J. L. Nitric Oxide Uptake by Erythrocytes Is Primarily Limited by Extracellular Diffusion Not Membrane Resistance. J. Biol. Chem. 2002, 277 (29), 26194–26199. 10.1074/jbc.M201939200. [DOI] [PubMed] [Google Scholar]
- Portillo-Ledesma S.; Randall L. M.; Parsonage D.; Dalla Rizza J.; Karplus P. A.; Poole L. B.; Denicola A.; Ferrer-Sueta G. Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry 2018, 57 (24), 3416–3424. 10.1021/acs.biochem.8b00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Sueta G.; Radi R. Chemical Biology of Peroxynitrite: Kinetics, Diffusion, and Radicals. ACS Chem. Biol. 2009, 4 (3), 161–177. 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- Romero N.; Radi R.; Linares E.; Augusto O.; Detweiler C. D.; Mason R. P.; Denicola A. Reaction of Human Hemoglobin with Peroxynitrite: ISOMERIZATION TO NITRATE AND SECONDARY FORMATION OF PROTEIN RADICALS *. J. Biol. Chem. 2003, 278 (45), 44049–44057. 10.1074/jbc.M305895200. [DOI] [PubMed] [Google Scholar]
- Denicola A.; Freeman B. A.; Trujillo M.; Radi R. Peroxynitrite Reaction with Carbon Dioxide/Bicarbonate: Kinetics and Influence on Peroxynitrite-Mediated Oxidations. Arch. Biochem. Biophys. 1996, 333 (1), 49–58. 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- Manta B.; Hugo M.; Ortiz C.; Ferrer-Sueta G.; Trujillo M.; Denicola A. The Peroxidase and Peroxynitrite Reductase Activity of Human Erythrocyte Peroxiredoxin 2. Archives of biochemistry and biophysics 2009, 484 (2), 146–154. 10.1016/j.abb.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Möller M. N.; Rios N.; Trujillo M.; Radi R.; Denicola A.; Alvarez B. Detection and Quantification of Nitric Oxide-Derived Oxidants in Biological Systems. J. Biol. Chem. 2019, 294 (40), 14776–14802. 10.1074/jbc.REV119.006136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto O.; Bonini M. G.; Amanso A. M.; Linares E.; Santos C. C. X.; De Menezes S. L. Nitrogen Dioxide and Carbonate Radical Anion: Two Emerging Radicals in Biology. Free Radic Biol. Med. 2002, 32 (9), 841–859. 10.1016/S0891-5849(02)00786-4. [DOI] [PubMed] [Google Scholar]
- Möller M. N.; Li Q.; Lancaster J. R.; Denicola A. Acceleration of Nitric Oxide Autoxidation and Nitrosation by Membranes. IUBMB Life 2007, 59 (4–5), 243–248. 10.1080/15216540701311147. [DOI] [PubMed] [Google Scholar]
- van der Vliet A.; Janssen-Heininger Y. M. W. Hydrogen Peroxide as a Damage Signal in Tissue Injury and Inflammation: Murderer, Mediator, or Messenger?. J. Cell. Biochem. 2014, 115 (3), 427–435. 10.1002/jcb.24683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor W. A.; Lightsey J. W. Mechanisms of Nitrogen Dioxide Reactions: Initiation of Lipid Peroxidation and the Production of Nitrous Acid. Science 1981, 214 (4519), 435–437. 10.1126/science.214.4519.435. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C.; Kettle A. J.; Hampton M. B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. 10.1146/annurev-biochem-060815-014442. [DOI] [PubMed] [Google Scholar]
- Storkey C.; Davies M. J.; Pattison D. I. Reevaluation of the Rate Constants for the Reaction of Hypochlorous Acid (HOCl) with Cysteine, Methionine, and Peptide Derivatives Using a New Competition Kinetic Approach. Free Radical Biol. Med. 2014, 73, 60–66. 10.1016/j.freeradbiomed.2014.04.024. [DOI] [PubMed] [Google Scholar]
- Stacey M. M.; Peskin A. V.; Vissers M. C.; Winterbourn C. C. Chloramines and Hypochlorous Acid Oxidize Erythrocyte Peroxiredoxin 2. Free Radical Biol. Med. 2009, 47 (10), 1468–1476. 10.1016/j.freeradbiomed.2009.08.022. [DOI] [PubMed] [Google Scholar]
- Benchoam D.; Cuevasanta E.; Möller M. N.; Alvarez B. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxidants (Basel) 2019, 8 (2), 48. 10.3390/antiox8020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Szabo C.; Ichinose F.; Ahmed A.; Whiteman M.; Papapetropoulos A. The Role of H2S Bioavailability in Endothelial Dysfunction. Trends Pharmacol. Sci. 2015, 36 (9), 568–578. 10.1016/j.tips.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V.; Yadav P. K.; Kurthen A.; Banerjee R. Sulfide Oxidation by a Noncanonical Pathway in Red Blood Cells Generates Thiosulfate and Polysulfides. J. Biol. Chem. 2015, 290 (13), 8310–8320. 10.1074/jbc.M115.639831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevasanta E.; Denicola A.; Alvarez B.; Möller M. N. Solubility and Permeation of Hydrogen Sulfide in Lipid Membranes. PLoS One 2012, 7 (4), e34562 10.1371/journal.pone.0034562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai J. C.; Missner A.; Kügler P.; Saparov S. M.; Zeidel M. L.; Lee J. K.; Pohl P. No Facilitator Required for Membrane Transport of Hydrogen Sulfide. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (39), 16633–16638. 10.1073/pnas.0902952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V.; Yadav P. K.; An S.; Seravalli J.; Cho U.-S.; Banerjee R. Structural and Mechanistic Insights into Hemoglobin-Catalyzed Hydrogen Sulfide Oxidation and the Fate of Polysulfide Products. J. Biol. Chem. 2017, 292 (13), 5584–5592. 10.1074/jbc.M117.774943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchoam D.; Semelak J. A.; Cuevasanta E.; Mastrogiovanni M.; Grassano J. S.; Ferrer-Sueta G.; Zeida A.; Trujillo M.; Möller M. N.; Estrin D. A.; Alvarez B. Acidity and Nucleophilic Reactivity of Glutathione Persulfide. J. Biol. Chem. 2020, 295 (46), 15466–15481. 10.1074/jbc.RA120.014728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barayeu U.; Schilling D.; Eid M.; Xavier da Silva T. N.; Schlicker L.; Mitreska N.; Zapp C.; Gräter F.; Miller A. K.; Kappl R.; Schulze A.; Friedmann Angeli J. P.; Dick T. P. Hydropersulfides Inhibit Lipid Peroxidation and Ferroptosis by Scavenging Radicals. Nat. Chem. Biol. 2022, 10.1038/s41589-022-01145-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg R. O.; Smith H. L. Sulfhemoglobinemia: A Study of 62 Clinical Cases. American Heart Journal 1951, 42 (4), 582–588. 10.1016/0002-8703(51)90153-6. [DOI] [PubMed] [Google Scholar]
- Carrico R. J.; Blumberg W. E.; Peisach J. The Reversible Binding of Oxygen to Sulfhemoglobin. J. Biol. Chem. 1978, 253 (20), 7212–7215. 10.1016/S0021-9258(17)34486-1. [DOI] [PubMed] [Google Scholar]
- Ríos-González B. B.; Román-Morales E. M.; Pietri R.; López-Garriga J. Hydrogen Sulfide Activation in Hemeproteins: The Sulfheme Scenario. J. Inorg. Biochem 2014, 133, 78–86. 10.1016/j.jinorgbio.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord J. M.; Fridovich I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). J. Biol. Chem. 1969, 244 (22), 6049–6055. 10.1016/S0021-9258(18)63504-5. [DOI] [PubMed] [Google Scholar]
- Fridovich I. The Biology of Oxygen Radicals. Science 1978, 201 (4359), 875–880. 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide Radical and Superoxide Dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- Abreu I. A.; Cabelli D. E. Superoxide Dismutases-a Review of the Metal-Associated Mechanistic Variations. Biochim. Biophys. Acta 2010, 1804 (2), 263–274. 10.1016/j.bbapap.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Gärtner A.; Weser U. Erythrocuprein (Cu2Zn2 Superoxide Dismutase) Is the Major Copper Protein of the Red Blood Cell. FEBS Lett. 1983, 155 (1), 15–18. 10.1016/0014-5793(83)80199-9. [DOI] [PubMed] [Google Scholar]
- Gleitzmann J.; Raab A.; Schulze D.; Wätzig H.; Feldmann J.; Swart C. Accurate and Precise Quantification of Cu,Zn-SOD in Human Red Blood Cells Using Species-Specific Double and Triple IDMS. J. Anal. At. Spectrom. 2016, 31 (9), 1922–1928. 10.1039/C5JA00459D. [DOI] [Google Scholar]
- Eleutherio E. C. A.; Silva Magalhães R. S.; de Araújo Brasil A.; Monteiro Neto J. R.; de Holanda Paranhos L. SOD1, More than Just an Antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701. 10.1016/j.abb.2020.108701. [DOI] [PubMed] [Google Scholar]
- Nedd S.; Redler R. L.; Proctor E. A.; Dokholyan N. V.; Alexandrova A. N. Cu,Zn-Superoxide Dismutase without Zn Is Folded but Catalytically Inactive. J. Mol. Biol. 2014, 426 (24), 4112–4124. 10.1016/j.jmb.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tainer J. A.; Getzoff E. D.; Richardson J. S.; Richardson D. C. Structure and Mechanism of Copper, Zinc Superoxide Dismutase. Nature 1983, 306 (5940), 284–287. 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Cantini F.; D’Amelio N.; Gaggelli E. Human SOD1 before Harboring the Catalytic Metal: Solution Structure of Copper-Depleted, Disulfide-Reduced Form. J. Biol. Chem. 2006, 281 (4), 2333–2337. 10.1074/jbc.M506497200. [DOI] [PubMed] [Google Scholar]
- Iuchi Y.; Okada F.; Onuma K.; Onoda T.; Asao H.; Kobayashi M.; Fujii J. Elevated Oxidative Stress in Erythrocytes Due to a SOD1 Deficiency Causes Anaemia and Triggers Autoantibody Production. Biochem. J. 2007, 402 (2), 219–227. 10.1042/BJ20061386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuchi Y.; Okada F.; Takamiya R.; Kibe N.; Tsunoda S.; Nakajima O.; Toyoda K.; Nagae R.; Suematsu M.; Soga T.; Uchida K.; Fujii J. Rescue of Anaemia and Autoimmune Responses in SOD1-Deficient Mice by Transgenic Expression of Human SOD1 in Erythrocytes. Biochem. J. 2009, 422 (2), 313–320. 10.1042/BJ20090176. [DOI] [PubMed] [Google Scholar]
- Homma T.; Fujii J. Oxidative Stress Caused by an SOD1 Deficiency Triggers the Accumulation of Oxidatively Modified Carbonic Anhydrase II in Erythrocytes. Reactive Oxygen Species 2018, 6 (16), 289–298. 10.20455/ros.2018.847. [DOI] [Google Scholar]
- Wilcox K. C.; Zhou L.; Jordon J. K.; Huang Y.; Yu Y.; Redler R. L.; Chen X.; Caplow M.; Dokholyan N. V. Modifications of Superoxide Dismutase (SOD1) in Human Erythrocytes: A Possible Role in Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2009, 284 (20), 13940–13947. 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low F. M.; Hampton M. B.; Peskin A. V.; Winterbourn C. C. Peroxiredoxin 2 Functions as a Noncatalytic Scavenger of Low-Level Hydrogen Peroxide in the Erythrocyte. Blood 2007, 109 (6), 2611–2617. 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- Moore R. B.; Mankad M. V.; Shriver S. K.; Mankad V. N.; Plishker G. A. Reconstitution of Ca(2+)-Dependent K+ Transport in Erythrocyte Membrane Vesicles Requires a Cytoplasmic Protein. J. Biol. Chem. 1991, 266 (28), 18964–18968. 10.1016/S0021-9258(18)55157-7. [DOI] [PubMed] [Google Scholar]
- Cho C.-S.; Kato G.; Yang S.; Bae S.; Lee J.-S.; Gladwin M.; Rhee S. G. Hydroxyurea-Induced Expression of Glutathione Peroxidase 1 in Red Blood Cells of Individuals with Sickle Cell Anemia. Antioxidants & redox signaling 2010, 13, 1–11. 10.1089/ars.2009.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann B.; Hecht H.-J.; Flohé L. Peroxiredoxins. Biol. Chem. 2002, 383 (3–4), 347–364. 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- Dalla Rizza J.; Randall L. M.; Santos J.; Ferrer-Sueta G.; Denicola A. Differential Parameters between Cytosolic 2-Cys Peroxiredoxins, PRDX1 and PRDX2. Protein Sci. 2019, 28 (1), 191–201. 10.1002/pro.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood Z. A.; Poole L. B.; Hantgan R. R.; Karplus P. A. Dimers to Doughnuts: Redox-Sensitive Oligomerization of 2-Cysteine Peroxiredoxins. Biochemistry 2002, 41 (17), 5493. 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- Kristensen P.; Rasmussen D. E.; Kristensen B. I. Properties of Thiol-Specific Anti-Oxidant Protein or Calpromotin in Solution. Biochem. Biophys. Res. Commun. 1999, 262 (1), 127–131. 10.1006/bbrc.1999.1107. [DOI] [PubMed] [Google Scholar]
- Cheah F.-C.; Peskin A. V.; Wong F.-L.; Ithnin A.; Othman A.; Winterbourn C. C. Increased Basal Oxidation of Peroxiredoxin 2 and Limited Peroxiredoxin Recycling in Glucose-6-Phosphate Dehydrogenase-Deficient Erythrocytes from Newborn Infants. FASEB J. 2014, 28 (7), 3205–3210. 10.1096/fj.14-250050. [DOI] [PubMed] [Google Scholar]
- Rabilloud T.; Berthier R.; Vinçon M.; Ferbus D.; Goubin G.; Lawrence J. J. Early Events in Erythroid Differentiation: Accumulation of the Acidic Peroxidoxin (PRP/TSA/NKEF-B). Biochem. J. 1995, 312 (3), 699–705. 10.1042/bj3120699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.-H.; Kim S.-U.; Yu S.-L.; Kim S. H.; Park D. S.; Moon H.-B.; Dho S. H.; Kwon K.-S.; Kwon H. J.; Han Y.-H.; Jeong S.; Kang S. W.; Shin H.-S.; Lee K.-K.; Rhee S. G.; Yu D.-Y. Peroxiredoxin II Is Essential for Sustaining Life Span of Erythrocytes in Mice. Blood 2003, 101 (12), 5033–5038. 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- Johnson R. M.; Goyette G.; Ravindranath Y.; Ho Y. S. Red Cells from Glutathione Peroxidase-1-Deficient Mice Have Nearly Normal Defenses against Exogenous Peroxides. Blood 2000, 96 (5), 1985–1988. 10.1182/blood.V96.5.1985. [DOI] [PubMed] [Google Scholar]
- Ho Y.-S.; Xiong Y.; Ma W.; Spector A.; Ho D. S. Mice Lacking Catalase Develop Normally but Show Differential Sensitivity to Oxidant Tissue Injury. J. Biol. Chem. 2004, 279 (31), 32804–32812. 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- Han Y.-H.; Kim S.-U.; Kwon T.-H.; Lee D.-S.; Ha H.-L.; Park D.-S.; Woo E.-J.; Lee S.-H.; Kim J.-M.; Chae H.-B.; Lee S. Y.; Kim B. Y.; Yoon D. Y.; Rhee S. G.; Fibach E.; Yu D.-Y. Peroxiredoxin II Is Essential for Preventing Hemolytic Anemia from Oxidative Stress through Maintaining Hemoglobin Stability. Biochem. Biophys. Res. Commun. 2012, 426 (3), 427–432. 10.1016/j.bbrc.2012.08.113. [DOI] [PubMed] [Google Scholar]
- Matte A.; Bertoldi M.; Mohandas N.; An X.; Bugatti A.; Brunati A. M.; Rusnati M.; Tibaldi E.; Siciliano A.; Turrini F.; Perrotta S.; De Franceschi L. Membrane Association of Peroxiredoxin-2 in Red Cells Is Mediated by the N-Terminal Cytoplasmic Domain of Band 3. Free Radical Biol. Med. 2013, 55, 27–35. 10.1016/j.freeradbiomed.2012.10.543. [DOI] [PubMed] [Google Scholar]
- Moore R. B.; Mankad M. V.; Shriver S. K.; Mankad V. N.; Plishker G. A. Reconstitution of Ca (2+)-Dependent K+ Transport in Erythrocyte Membrane Vesicles Requires a Cytoplasmic Protein. J. Biol. Chem. 1991, 266 (28), 18964–18968. 10.1016/S0021-9258(18)55157-7. [DOI] [PubMed] [Google Scholar]
- Plishker G. A.; Chevalier D.; Seinsoth L.; Moore R. B. Calcium-Activated Potassium Transport and High Molecular Weight Forms of Calpromotin. J. Biol. Chem. 1992, 267 (30), 21839–21843. 10.1016/S0021-9258(19)36688-8. [DOI] [PubMed] [Google Scholar]
- Rinalducci S.; D’Amici G. M.; Blasi B.; Vaglio S.; Grazzini G.; Zolla L. Peroxiredoxin-2 as a Candidate Biomarker to Test Oxidative Stress Levels of Stored Red Blood Cells under Blood Bank Conditions. Transfusion 2011, 51 (7), 1439–1449. 10.1111/j.1537-2995.2010.03032.x. [DOI] [PubMed] [Google Scholar]
- Bayer S. B.; Maghzal G.; Stocker R.; Hampton M. B.; Winterbourn C. C. Neutrophil-Mediated Oxidation of Erythrocyte Peroxiredoxin 2 as a Potential Marker of Oxidative Stress in Inflammation. FASEB J. 2013, 27 (8), 3315–3322. 10.1096/fj.13-227298. [DOI] [PubMed] [Google Scholar]
- Shau H.; Gupta R. K.; Golub S. H. Identification of a Natural Killer Enhancing Factor (NKEF) from Human Erythroid Cells. Cell Immunol 1993, 147 (1), 1–11. 10.1006/cimm.1993.1043. [DOI] [PubMed] [Google Scholar]
- Mullen L.; Hanschmann E.-M.; Lillig C. H.; Herzenberg L. A.; Ghezzi P. Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion. Mol. Med. 2015, 21, 98–108. 10.2119/molmed.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnér E. S.; Holmgren A. Physiological Functions of Thioredoxin and Thioredoxin Reductase. Eur. J. Biochem. 2000, 267 (20), 6102–6109. 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Zhong L.; Arnér E. S. J.; Holmgren A. Structure and Mechanism of Mammalian Thioredoxin Reductase: The Active Site Is a Redox-Active Selenolthiol/Selenenylsulfide Formed from the Conserved Cysteine-Selenocysteine Sequence. Proc. Natl. Acad. Sci. U.S.A. 2000, 97 (11), 5854–5859. 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Holmgren A. The Thioredoxin Antioxidant System. Free Radical Biol. Med. 2014, 66, 75–87. 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- Nicholls P.; Fita I.; Loewen P. C.. Enzymology and Structure of Catalases. In Adv. Inorg. Chem.; Academic Press, 2000; Vol. 51, pp 51–106. 10.1016/S0898-8838(00)51001-0. [DOI] [Google Scholar]
- Kirkman H. N.; Gaetani G. F. Catalase: A Tetrameric Enzyme with Four Tightly Bound Molecules of NADPH. Proc. Natl. Acad. Sci. U. S. A. 1984, 81 (14), 4343–4347. 10.1073/pnas.81.14.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B.; Greenstein D. S.; Roughton F. J. W. The Mechanism of Catalase Action. I. Steady-State Analysis. Arch. Biochem. Biophys. 1952, 37 (2), 301–321. 10.1016/0003-9861(52)90194-X. [DOI] [PubMed] [Google Scholar]
- Goyal M. M.; Basak A. Human Catalase: Looking for Complete Identity. Protein Cell 2010, 1 (10), 888–897. 10.1007/s13238-010-0113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi H.[13] Catalase in Vitro. In Methods in Enzymology; Oxygen Radicals in Biological Systems; Academic Press, 1984; Vol. 105, pp 121–126. 10.1016/S0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- Kirkman H. N.; Galiano S.; Gaetani G. F. The Function of Catalase-Bound NADPH. J. Biol. Chem. 1987, 262 (2), 660–666. 10.1016/S0021-9258(19)75835-9. [DOI] [PubMed] [Google Scholar]
- Kirkman H. N.; Rolfo M.; Ferraris A. M.; Gaetani G. F. Mechanisms of Protection of Catalase by NADPH. Kinetics and Stoichiometry. J. Biol. Chem. 1999, 274 (20), 13908–13914. 10.1074/jbc.274.20.13908. [DOI] [PubMed] [Google Scholar]
- Flohe L.; Günzler W. A.; Schock H. H. Glutathione Peroxidase: A Selenoenzyme. FEBS Lett. 1973, 32 (1), 132–134. 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- Flohé L.; Toppo S.; Orian L. The Glutathione Peroxidase Family: Discoveries and Mechanism. Free Radic Biol. Med. 2022, 187, 113–122. 10.1016/j.freeradbiomed.2022.05.003. [DOI] [PubMed] [Google Scholar]
- Toppo S.; Vanin S.; Bosello V.; Tosatto S. C. E. Evolutionary and Structural Insights into the Multifaceted Glutathione Peroxidase (Gpx) Superfamily. Antioxid Redox Signal 2008, 10 (9), 1501–1514. 10.1089/ars.2008.2057. [DOI] [PubMed] [Google Scholar]
- Flohé L.; Toppo S.; Cozza G.; Ursini F. A Comparison of Thiol Peroxidase Mechanisms. Antioxid Redox Signal 2011, 15 (3), 763–780. 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- Weaver K.; Skouta R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10 (4), 891. 10.3390/biomedicines10040891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini F.; Maiorino M.; Gregolin C. The Selenoenzyme Phospholipid Hydroperoxide Glutathione Peroxidase. Biochim. Biophys. Acta 1985, 839 (1), 62–70. 10.1016/0304-4165(85)90182-5. [DOI] [PubMed] [Google Scholar]
- Flohé L.; Brand I. Kinetics of Glutathione Peroxidase. Biochim. Biophys. Acta 1969, 191 (3), 541–549. 10.1016/0005-2744(69)90347-7. [DOI] [PubMed] [Google Scholar]
- Deponte M. Glutathione Catalysis and the Reaction Mechanisms of Glutathione-Dependent Enzymes. Biochim. Biophys. Acta 2013, 1830 (5), 3217–3266. 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- Ursini F.; Maiorino M.; Brigelius-Flohé R.; Aumann K. D.; Roveri A.; Schomburg D.; Flohé L. Diversity of Glutathione Peroxidases. Methods Enzymol 1995, 252, 38–53. 10.1016/0076-6879(95)52007-4. [DOI] [PubMed] [Google Scholar]
- Takebe G.; Yarimizu J.; Saito Y.; Hayashi T.; Nakamura H.; Yodoi J.; Nagasawa S.; Takahashi K. A Comparative Study on the Hydroperoxide and Thiol Specificity of the Glutathione Peroxidase Family and Selenoprotein P. J. Biol. Chem. 2002, 277 (43), 41254–41258. 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- Flohé L.; Loschen G.; Günzler W. A.; Eichele E.; Glutathione Peroxidase V. The Kinetic Mechanism. Hoppe Seylers Z. Physiol Chem. 1972, 353 (6), 987–999. 10.1515/bchm2.1972.353.1.987. [DOI] [PubMed] [Google Scholar]
- Epp O.; Ladenstein R.; Wendel A. The Refined Structure of the Selenoenzyme Glutathione Peroxidase at 0.2-Nm Resolution. Eur. J. Biochem. 1983, 133 (1), 51–69. 10.1111/j.1432-1033.1983.tb07429.x. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohé R.; Maiorino M. Glutathione Peroxidases. Biochim. Biophys. Acta 2013, 1830 (5), 3289–3303. 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Johnson R. M.; Ho Y.-S.; Yu D.-Y.; Kuypers F. A.; Ravindranath Y.; Goyette G. W. The Effects of Disruption of Genes for Peroxiredoxin-2, Glutathione Peroxidase-1, and Catalase on Erythrocyte Oxidative Metabolism. Free Radic. Biol. Med. 2010, 48 (4), 519–525. 10.1016/j.freeradbiomed.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. M.; Goyette G.; Ravindranath Y.; Ho Y.-S. Oxidation of Glutathione Peroxidase-Deficient Red Cells by Organic Peroxides. Blood 2002, 100 (4), 1515–1516. 10.1182/blood-2002-04-1124. [DOI] [PubMed] [Google Scholar]
- Johnson R. M.; Ravindranath Y.; ElAlfy M. S.; El-Alfy M.; Goyette G. Oxidant Damage to Erythrocyte Membrane in Glucose-6-Phosphate Dehydrogenase Deficiency: Correlation with in Vivo Reduced Glutathione Concentration and Membrane Protein Oxidation. Blood 1994, 83 (4), 1117–1123. 10.1182/blood.V83.4.1117.bloodjournal8341117. [DOI] [PubMed] [Google Scholar]
- Rocha S.; Gomes D.; Lima M.; Bronze-da-Rocha E.; Santos-Silva A. Peroxiredoxin 2, Glutathione Peroxidase, and Catalase in the Cytosol and Membrane of Erythrocytes under H2O2-Induced Oxidative Stress. Free Radic. Res. 2015, 49 (8), 990–1003. 10.3109/10715762.2015.1028402. [DOI] [PubMed] [Google Scholar]
- Stolwijk J. M.; Stefely J. A.; Veling M. T.; van‘t Erve T. J.; Wagner B. A.; Raife T. J.; Buettner G. R. Red Blood Cells Contain Enzymatically Active GPx4 Whose Abundance Anticorrelates with Hemolysis during Blood Bank Storage. Redox Biol. 2021, 46, 102073. 10.1016/j.redox.2021.102073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieme R.; Pai E. F.; Schirmer R. H.; Schulz G. E. Three-Dimensional Structure of Glutathione Reductase at 2 A Resolution. J. Mol. Biol. 1981, 152 (4), 763–782. 10.1016/0022-2836(81)90126-1. [DOI] [PubMed] [Google Scholar]
- Berkholz D. S.; Faber H. R.; Savvides S. N.; Karplus P. A. Catalytic Cycle of Human Glutathione Reductase near 1 A Resolution. J. Mol. Biol. 2008, 382 (2), 371–384. 10.1016/j.jmb.2008.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington D. J.; Rosemeyer M. A. Glutathione Reductase from Human Erythrocytes. Catalytic Properties and Aggregation. Eur. J. Biochem. 1976, 67 (1), 231–238. 10.1111/j.1432-1033.1976.tb10654.x. [DOI] [PubMed] [Google Scholar]
- Chang J. C.; van der Hoeven L. H.; Haddox C. H. Glutathione Reductase in the Red Blood Cells. Ann. Clin Lab Sci. 1978, 8 (1), 23–29. [PubMed] [Google Scholar]
- Lillig C. H.; Berndt C.; Holmgren A. Glutaredoxin Systems. Biochim. Biophys. Acta 2008, 1780 (11), 1304–1317. 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Couturier J.; Jacquot J.-P.; Rouhier N. Evolution and Diversity of Glutaredoxins in Photosynthetic Organisms. Cell. Mol. Life Sci. 2009, 66 (15), 2539–2557. 10.1007/s00018-009-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero E.; de la Torre-Ruiz M. A. Monothiol Glutaredoxins: A Common Domain for Multiple Functions. Cell. Mol. Life Sci. 2007, 64 (12), 1518–1530. 10.1007/s00018-007-6554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallogly M. M.; Starke D. W.; Mieyal J. J. Mechanistic and Kinetic Details of Catalysis of Thiol-Disulfide Exchange by Glutaredoxins and Potential Mechanisms of Regulation. Antioxid Redox Signal 2009, 11 (5), 1059–1081. 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillig C. H.; Berndt C. Glutaredoxins in Thiol/Disulfide Exchange. Antioxid Redox Signal 2013, 18 (13), 1654–1665. 10.1089/ars.2012.5007. [DOI] [PubMed] [Google Scholar]
- Mieyal J. J.; Starke D. W.; Gravina S. A.; Dothey C.; Chung J. S. Thioltransferase in Human Red Blood Cells: Purification and Properties. Biochemistry 1991, 30 (25), 6088–6097. 10.1021/bi00239a002. [DOI] [PubMed] [Google Scholar]
- Terada T.; Oshida T.; Nishimura M.; Maeda H.; Hara T.; Hosomi S.; Mizoguchi T.; Nishihara T. Study on Human Erythrocyte Thioltransferase: Comparative Characterization with Bovine Enzyme and Its Physiological Role under Oxidative Stress. J. Biochem 1992, 111 (5), 688–692. 10.1093/oxfordjournals.jbchem.a123819. [DOI] [PubMed] [Google Scholar]
- van Zwieten R.; Verhoeven A. J.; Roos D. Inborn Defects in the Antioxidant Systems of Human Red Blood Cells. Free Radic Biol. Med. 2014, 67, 377–386. 10.1016/j.freeradbiomed.2013.11.022. [DOI] [PubMed] [Google Scholar]
- Haunhorst P.; Hanschmann E.-M.; Bräutigam L.; Stehling O.; Hoffmann B.; Mühlenhoff U.; Lill R.; Berndt C.; Lillig C. H. Crucial Function of Vertebrate Glutaredoxin 3 (PICOT) in Iron Homeostasis and Hemoglobin Maturation. Mol. Biol. Cell 2013, 24 (12), 1895–1903. 10.1091/mbc.e12-09-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. C. GLUTATHIONE SYNTHESIS. Biochim. Biophys. Acta 2013, 1830 (5), 3143–3153. 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amen F.; Machin A.; Touriño C.; Rodríguez I.; Denicola A.; Thomson L. N-Acetylcysteine Improves the Quality of Red Blood Cells Stored for Transfusion. Arch. Biochem. Biophys. 2017, 621, 31–37. 10.1016/j.abb.2017.02.012. [DOI] [PubMed] [Google Scholar]
- van ’t Erve T. J.; Wagner B. A.; Ryckman K. K.; Raife T. J.; Buettner G. R. The Concentration of Glutathione in Human Erythrocytes Is a Heritable Trait. Free Radic Biol. Med. 2013, 65, 742–749. 10.1016/j.freeradbiomed.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith O. W.; Mulcahy R. T. The Enzymes of Glutathione Synthesis: Gamma-Glutamylcysteine Synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 73, 209–267. 10.1002/9780470123195.ch7. [DOI] [PubMed] [Google Scholar]
- Toroser D.; Yarian C. S.; Orr W. C.; Sohal R. S. Mechanisms of γ-Glutamylcysteine Ligase Regulation. Biochim. Biophys. Acta 2006, 1760 (2), 233–244. 10.1016/j.bbagen.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Xiong Y.; Wang Y.; Wang Z.; Zhang A.; Zhao N.; Zhao D.; Yu Z.; Wang Z.; Yi J.; Luan X. Inhibition of Glutathione Synthesis via Decreased Glucose Metabolism in Stored RBCs. Cell Physiol Biochem 2018, 51 (5), 2172–2184. 10.1159/000495864. [DOI] [PubMed] [Google Scholar]
- Young J. D.; Wolowyk M. W.; Jones S. M.; Ellory J. C. Red-Cell Amino Acid Transport. Evidence for the Presence of System ASC in Mature Human Red Blood Cells. Biochem. J. 1983, 216 (2), 349–357. 10.1042/bj2160349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn B.; Dunstan R. H.; Macdonald M. M.; Borges N.; Roberts T. K. Evidence That Human and Equine Erythrocytes Could Have Significant Roles in the Transport and Delivery of Amino Acids to Organs and Tissues. Amino Acids 2020, 52 (5), 711–724. 10.1007/s00726-020-02845-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookhtens M.; Kaplowitz N. Role of the Liver in Interorgan Homeostasis of Glutathione and Cyst(e)Ine. Semin Liver Dis 1998, 18 (4), 313–329. 10.1055/s-2007-1007167. [DOI] [PubMed] [Google Scholar]
- Griffith O. W.; Meister A. Glutathione: Interorgan Translocation, Turnover, and Metabolism. Proc. Natl. Acad. Sci. U. S. A. 1979, 76 (11), 5606–5610. 10.1073/pnas.76.11.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith O. W. Glutathione Turnover in Human Erythrocytes. Inhibition by Buthionine Sulfoximine and Incorporation of Glycine by Exchange. J. Biol. Chem. 1981, 256 (10), 4900–4904. 10.1016/S0021-9258(19)69341-5. [DOI] [PubMed] [Google Scholar]
- Priora R.; Coppo L.; Margaritis A.; Di Giuseppe D.; Frosali S.; Summa D.; Heo J.; Di Simplicio P. The Control of S-Thiolation by Cysteine via Gamma-Glutamyltranspeptidase and Thiol Exchanges in Erythrocytes and Plasma of Diamide-Treated Rats. Toxicol. Appl. Pharmacol. 2010, 242 (3), 333–343. 10.1016/j.taap.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Finkelstein J. D. Pathways and Regulation of Homocysteine Metabolism in Mammals. Semin Thromb Hemost 2000, 26 (3), 219–225. 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- Jones D. P.; Go Y.-M.; Anderson C. L.; Ziegler T. R.; Kinkade J. M.; Kirlin W. G. Cysteine/Cystine Couple Is a Newly Recognized Node in the Circuitry for Biologic Redox Signaling and Control. FASEB J. 2004, 18 (11), 1246–1248. 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- Dass P. D.; Bermes E. W.; Holmes E. W. Renal and Hepatic Output of Glutathione in Plasma and Whole Blood. Biochim. Biophys. Acta 1992, 1156 (1), 99–102. 10.1016/0304-4165(92)90102-Z. [DOI] [PubMed] [Google Scholar]
- Rossi R.; Milzani A.; Dalle-Donne I.; Giannerini F.; Giustarini D.; Lusini L.; Colombo R.; Di Simplicio P. Different Metabolizing Ability of Thiol Reactants in Human and Rat Blood: Biochemical and Pharmacological Implications. J. Biol. Chem. 2001, 276 (10), 7004–7010. 10.1074/jbc.M005156200. [DOI] [PubMed] [Google Scholar]
- Thom S. R.; Kang M.; Fisher D.; Ischiropoulos H. Release of Glutathione from Erythrocytes and Other Markers of Oxidative Stress in Carbon Monoxide Poisoning. J. Appl. Physiol (1985) 1997, 82 (5), 1424–1432. 10.1152/jappl.1997.82.5.1424. [DOI] [PubMed] [Google Scholar]
- Sharma K. G.; Mason D. L.; Liu G.; Rea P. A.; Bachhawat A. K.; Michaelis S. Localization, Regulation, and Substrate Transport Properties of Bpt1p, a Saccharomyces Cerevisiae MRP-Type ABC Transporter. Eukaryot Cell 2002, 1 (3), 391–400. 10.1128/EC.1.3.391-400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N.; Hammond C. L.; Cunningham J. B.; Krance S. M.; Marchan R. Molecular Mechanisms of Reduced Glutathione Transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A Families of Membrane Proteins. Toxicol. Appl. Pharmacol. 2005, 204 (3), 238–255. 10.1016/j.taap.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Ballatori N.; Krance S. M.; Marchan R.; Hammond C. L. Plasma Membrane Glutathione Transporters and Their Roles in Cell Physiology and Pathophysiology. Mol. Aspects Med. 2009, 30 (1–2), 13–28. 10.1016/j.mam.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padayatty S. J.; Levine M. Vitamin C: The Known and the Unknown and Goldilocks. Oral Dis 2016, 22 (6), 463–493. 10.1111/odi.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu A.; Han D.; Packer L. Vitamin E Recycling in Human Erythrocyte Membranes. J. Biol. Chem. 1993, 268 (15), 10906–10913. 10.1016/S0021-9258(18)82071-3. [DOI] [PubMed] [Google Scholar]
- May J. M.; Qu Z. C.; Mendiratta S. Protection and Recycling of Alpha-Tocopherol in Human Erythrocytes by Intracellular Ascorbic Acid. Arch. Biochem. Biophys. 1998, 349 (2), 281–289. 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- Niki E.; Saito T.; Kawakami A.; Kamiya Y. Inhibition of Oxidation of Methyl Linoleate in Solution by Vitamin E and Vitamin C. J. Biol. Chem. 1984, 259 (7), 4177–4182. 10.1016/S0021-9258(17)43026-2. [DOI] [PubMed] [Google Scholar]
- Montel-Hagen A.; Kinet S.; Manel N.; Mongellaz C.; Prohaska R.; Battini J.-L.; Delaunay J.; Sitbon M.; Taylor N. Erythrocyte Glut1 Triggers Dehydroascorbic Acid Uptake in Mammals Unable to Synthesize Vitamin C. Cell 2008, 132 (6), 1039–1048. 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Li H.; Tu H.; Wang Y.; Levine M. Vitamin C in Mouse and Human Red Blood Cells: An HPLC Assay. Anal. Biochem. 2012, 426 (2), 109–117. 10.1016/j.ab.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D.; May J. M.; Koury M. J.; Asard H. Human Erythrocyte Membranes Contain a Cytochrome B561 That May Be Involved in Extracellular Ascorbate Recycling. J. Biol. Chem. 2006, 281 (52), 39852–39859. 10.1074/jbc.M606543200. [DOI] [PubMed] [Google Scholar]
- Lippi G.; Mercadanti M.; Aloe R.; Targher G. Erythrocyte Mechanical Fragility Is Increased in Patients with Type 2 Diabetes. Eur. J. Intern Med. 2012, 23 (2), 150–153. 10.1016/j.ejim.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Tu H.; Li H.; Wang Y.; Niyyati M.; Wang Y.; Leshin J.; Levine M. Low Red Blood Cell Vitamin C Concentrations Induce Red Blood Cell Fragility: A Link to Diabetes Via Glucose, Glucose Transporters, and Dehydroascorbic Acid. EBioMedicine 2015, 2 (11), 1735–1750. 10.1016/j.ebiom.2015.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval J. S.; Fontes J.; Banerjee U.; Yazer M. H.; Mank E.; Palmer A. F. Ascorbic Acid Improves Membrane Fragility and Decreases Haemolysis during Red Blood Cell Storage. Transfus Med. 2013, 23 (2), 87–93. 10.1111/tme.12013. [DOI] [PubMed] [Google Scholar]
- Sanford K.; Fisher B. J.; Fowler E.; Fowler A. A.; Natarajan R. Attenuation of Red Blood Cell Storage Lesions with Vitamin C. Antioxidants (Basel) 2017, 6 (3), 55 10.3390/antiox6030055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotta V.; Gevi F.; D’alessandro A.; Zolla L. Storing Red Blood Cells with Vitamin C and N-Acetylcysteine Prevents Oxidative Stress-Related Lesions: A Metabolomics Overview. Blood Transfus 2014, 12 (3), 376–387. 10.2450/2014.0266-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A. G.; Pullar J. M.; Cook J.; Spencer E. S.; Vissers M. C.; Carr A. C.; Hampton M. B. Peroxiredoxin 2 Oxidation Reveals Hydrogen Peroxide Generation within Erythrocytes during High-Dose Vitamin C Administration. Redox Biol. 2021, 43, 101980. 10.1016/j.redox.2021.101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traber M. G.; Kayden H. J. Preferential Incorporation of Alpha-Tocopherol vs Gamma-Tocopherol in Human Lipoproteins. Am. J. Clin. Nutr. 1989, 49 (3), 517–526. 10.1093/ajcn/49.3.517. [DOI] [PubMed] [Google Scholar]
- Podda M.; Weber C.; Traber M. G.; Packer L. Simultaneous Determination of Tissue Tocopherols, Tocotrienols, Ubiquinols, and Ubiquinones. J. Lipid Res. 1996, 37 (4), 893–901. 10.1016/S0022-2275(20)37587-8. [DOI] [PubMed] [Google Scholar]
- Arita M.; Sato Y.; Miyata A.; Tanabe T.; Takahashi E.; Kayden H. J.; Arai H.; Inoue K. Human Alpha-Tocopherol Transfer Protein: CDNA Cloning, Expression and Chromosomal Localization. Biochem. J. 1995, 306 (2), 437–443. 10.1042/bj3060437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosomi A.; Arita M.; Sato Y.; Kiyose C.; Ueda T.; Igarashi O.; Arai H.; Inoue K. Affinity for Alpha-Tocopherol Transfer Protein as a Determinant of the Biological Activities of Vitamin E Analogs. FEBS Lett. 1997, 409 (1), 105–108. 10.1016/S0014-5793(97)00499-7. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohé R.; Davies K. J. A. Is Vitamin E an Antioxidant, a Regulator of Signal Transduction and Gene Expression, or a “junk” Food? Comments on the Two Accompanying Papers: ‘Molecular Mechanism of Alpha-Tocopherol Action’ by A. Azzi and ‘Vitamin E, Antioxidant and Nothing More’ by M. Traber and J. Atkinson. Free Radic Biol. Med. 2007, 43 (1), 2–3. 10.1016/j.freeradbiomed.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Traber M. G.; Atkinson J. Vitamin E, Antioxidant and Nothing More. Free Radic Biol. Med. 2007, 43 (1), 4–15. 10.1016/j.freeradbiomed.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner B. A.; Buettner G. R.; Burns C. P. Free Radical-Mediated Lipid Peroxidation in Cells: Oxidizability Is a Function of Cell Lipid Bis-Allylic Hydrogen Content. Biochemistry 1994, 33 (15), 4449–4453. 10.1021/bi00181a003. [DOI] [PubMed] [Google Scholar]
- Halliwell B.; Gutteridge J. M. Oxygen Toxicity, Oxygen Radicals, Transition Metals and Disease. Biochem. J. 1984, 219 (1), 1–14. 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki E. Interaction of Ascorbate and Alpha-Tocopherol. Ann. N.Y. Acad. Sci. 1987, 498, 186–199. 10.1111/j.1749-6632.1987.tb23761.x. [DOI] [PubMed] [Google Scholar]
- Chow C. K. Distribution of Tocopherols in Human Plasma and Red Blood Cells. Am. J. Clin. Nutr. 1975, 28 (7), 756–760. 10.1093/ajcn/28.7.756. [DOI] [PubMed] [Google Scholar]
- Mino M.; Kitagawa M.; Nakagawa S. Red Blood Cell Tocopherol Concentrations in a Normal Population of Japanese Children and Premature Infants in Relation to the Assessment of Vitamin E Status. Am. J. Clin. Nutr. 1985, 41 (3), 631–638. 10.1093/ajcn/41.3.631. [DOI] [PubMed] [Google Scholar]
- Lehmann J.; Rao D. D.; Canary J. J.; Judd J. T. Vitamin E and Relationships among Tocopherols in Human Plasma, Platelets, Lymphocytes, and Red Blood Cells. Am. J. Clin. Nutr. 1988, 47 (3), 470–474. 10.1093/ajcn/47.3.470. [DOI] [PubMed] [Google Scholar]
- Vatassery G. T.; Smith W. E.; Quach H. T. A Liquid Chromatographic Method for the Simultaneous Determination of Alpha-Tocopherol and Tocopherolquinone in Human Red Blood Cells and Other Biological Samples Where Tocopherol Is Easily Oxidized during Sample Treatment. Anal. Biochem. 1993, 214 (2), 426–430. 10.1006/abio.1993.1518. [DOI] [PubMed] [Google Scholar]
- Peuchant E.; Carbonneau M. A.; Dubourg L.; Thomas M. J.; Perromat A.; Vallot C.; Clerc M. Lipoperoxidation in Plasma and Red Blood Cells of Patients Undergoing Haemodialysis: Vitamins A, E, and Iron Status. Free Radic Biol. Med. 1994, 16 (3), 339–346. 10.1016/0891-5849(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Burton G. W.; Joyce A.; Ingold K. U. Is Vitamin E the Only Lipid-Soluble, Chain-Breaking Antioxidant in Human Blood Plasma and Erythrocyte Membranes?. Arch. Biochem. Biophys. 1983, 221 (1), 281–290. 10.1016/0003-9861(83)90145-5. [DOI] [PubMed] [Google Scholar]
- Cuerq C.; Restier L.; Drai J.; Blond E.; Roux A.; Charriere S.; Michalski M.-C.; Di Filippo M.; Levy E.; Lachaux A.; Peretti N. Establishment of Reference Values of α-Tocopherol in Plasma, Red Blood Cells and Adipose Tissue in Healthy Children to Improve the Management of Chylomicron Retention Disease, a Rare Genetic Hypocholesterolemia. Orphanet J. Rare Dis 2016, 11 (1), 114. 10.1186/s13023-016-0498-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanco J. M. F.; Sussmann R. A. C.; Verdaguer I. B.; Gabriel H. B.; Kimura E. A.; Katzin A. M. Tocopherol Biosynthesis in Leishmania (L.) Amazonensis Promastigotes. FEBS Open Bio 2019, 9 (4), 743–754. 10.1002/2211-5463.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan S.; Tripathi T.; Saudagar P. Critical Insight into Plausible Acquired Tocopherol Pathway in Neglected Human Trypanosomatids. ACS Omega 2021, 6 (47), 31396–31403. 10.1021/acsomega.1c05046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussmann R. A. C.; Angeli C. B.; Peres V. J.; Kimura E. A.; Katzin A. M. Intraerythrocytic Stages of Plasmodium Falciparum Biosynthesize Vitamin E. FEBS Lett. 2011, 585 (24), 3985–3991. 10.1016/j.febslet.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Sussmann R. A. C.; Fotoran W. L.; Kimura E. A.; Katzin A. M. Plasmodium Falciparum Uses Vitamin E to Avoid Oxidative Stress. Parasit Vectors 2017, 10 (1), 461. 10.1186/s13071-017-2402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H.; Halliwell B. Action of Biologically-Relevant Oxidizing Species upon Uric Acid. Identification of Uric Acid Oxidation Products. Chem. Biol. Interact 1990, 73 (2–3), 235–247. 10.1016/0009-2797(90)90006-9. [DOI] [PubMed] [Google Scholar]
- Hicks M.; Wong L. S.; Day R. O. Identification of Products from Oxidation of Uric Acid Induced by Hydroxyl Radicals. Free Radic Res. Commun. 1993, 18 (6), 337–351. 10.3109/10715769309147501. [DOI] [PubMed] [Google Scholar]
- Becker B. F. Towards the Physiological Function of Uric Acid. Free Radic Biol. Med. 1993, 14 (6), 615–631. 10.1016/0891-5849(93)90143-I. [DOI] [PubMed] [Google Scholar]
- Bachmann K. Chapter 8 - Drug Metabolism. In Pharmacology; Hacker M., Messer W., Bachmann K., Eds.; Academic Press: San Diego, 2009; pp 131–173. 10.1016/B978-0-12-369521-5.00008-7. [DOI] [Google Scholar]
- Lewis S. E.; Rosencrance C. B.; De Vallance E.; Giromini A.; Williams X. M.; Oh J.-Y.; Schmidt H.; Straub A. C.; Chantler P. D.; Patel R. P.; Kelley E. E. Human and Rodent Red Blood Cells Do Not Demonstrate Xanthine Oxidase Activity or XO-Catalyzed Nitrite Reduction to NO. Free Radic Biol. Med. 2021, 174, 84–88. 10.1016/j.freeradbiomed.2021.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautin Y. Y.; Johnson R. J. Uric Acid: The Oxidant-Antioxidant Paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27 (6), 608–619. 10.1080/15257770802138558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner D. D.; Burton G. W.; Ingold K. U.; Barclay L. R.; Locke S. J. The Relative Contributions of Vitamin E, Urate, Ascorbate and Proteins to the Total Peroxyl Radical-Trapping Antioxidant Activity of Human Blood Plasma. Biochim. Biophys. Acta 1987, 924 (3), 408–419. 10.1016/0304-4165(87)90155-3. [DOI] [PubMed] [Google Scholar]
- Smith R. C.; Gore J. Z.; Doyle M. P. Degradation of Uric Acid during Autocatalytic Oxidation of Oxyhemoglobin Induced by Sodium Nitrite. Free Radic Biol. Med. 1991, 11 (4), 373–377. 10.1016/0891-5849(91)90153-T. [DOI] [PubMed] [Google Scholar]
- Arduini A.; Mancinelli G.; Radatti G. L.; Hochstein P.; Cadenas E. Possible Mechanism of Inhibition of Nitrite-Induced Oxidation of Oxyhemoglobin by Ergothioneine and Uric Acid. Arch. Biochem. Biophys. 1992, 294 (2), 398–402. 10.1016/0003-9861(92)90702-X. [DOI] [PubMed] [Google Scholar]
- Lassen U. V.; Overgaard-Hansen K. Hypoxanthine as Inhibitor of Uric Acid Transport in Human Erythrocytes. Biochim. Biophys. Acta 1962, 57, 111–117. 10.1016/0006-3002(62)91085-5. [DOI] [PubMed] [Google Scholar]
- Lucas-Heron B.; Fontenaille C. Urate Transport in Human Red Blood Cells. Activation by ATP. Biochim. Biophys. Acta 1979, 553 (2), 284–294. 10.1016/0005-2736(79)90232-3. [DOI] [PubMed] [Google Scholar]
- Kandár R.; Štramová X.; Drábková P.; Křenková J. A Monitoring of Allantoin, Uric Acid, and Malondialdehyde Levels in Plasma and Erythrocytes after Ten Minutes of Running Activity. Physiol Res. 2014, 63 (6), 753–762. 10.33549/physiolres.932696. [DOI] [PubMed] [Google Scholar]
- Ames B. N.; Cathcart R.; Schwiers E.; Hochstein P. Uric Acid Provides an Antioxidant Defense in Humans against Oxidant- and Radical-Caused Aging and Cancer: A Hypothesis. Proc. Natl. Acad. Sci. U. S. A. 1981, 78 (11), 6858–6862. 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki M.; Motoyama T.; Mino M. Synergistic Inhibition of Oxidation in RBC Ghosts by Vitamins C and E. Ann. N.Y. Acad. Sci. 1989, 570 (1), 474–477. 10.1111/j.1749-6632.1989.tb14961.x. [DOI] [Google Scholar]
- Bardyn M.; Chen J.; Dussiot M.; Crettaz D.; Schmid L.; Längst E.; Amireault P.; Tissot J.-D.; Jolicoeur M.; Prudent M. Restoration of Physiological Levels of Uric Acid and Ascorbic Acid Reroutes the Metabolism of Stored Red Blood Cells. Metabolites 2020, 10 (6), 226. 10.3390/metabo10060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounakas V. L.; Georgatzakou H. T.; Kriebardis A. G.; Papageorgiou E. G.; Stamoulis K. E.; Foudoulaki-Paparizos L. E.; Antonelou M. H.; Papassideri I. S. Uric Acid Variation among Regular Blood Donors Is Indicative of Red Blood Cell Susceptibility to Storage Lesion Markers: A New Hypothesis Tested. Transfusion 2015, 55 (11), 2659–2671. 10.1111/trf.13211. [DOI] [PubMed] [Google Scholar]
- Zerez C. R.; Lee S. J.; Tanaka K. R. Spectrophotometric Determination of Oxidized and Reduced Pyridine Nucleotides in Erythrocytes Using a Single Extraction Procedure. Anal. Biochem. 1987, 164 (2), 367–373. 10.1016/0003-2697(87)90506-9. [DOI] [PubMed] [Google Scholar]
- Wagner T. C.; Scott M. D. Single Extraction Method for the Spectrophotometric Quantification of Oxidized and Reduced Pyridine Nucleotides in Erythrocytes. Anal. Biochem. 1994, 222 (2), 417–426. 10.1006/abio.1994.1511. [DOI] [PubMed] [Google Scholar]
- Ogasawara Y.; Funakoshi M.; Ishii K. Determination of Reduced Nicotinamide Adenine Dinucleotide Phosphate Concentration Using High-Performance Liquid Chromatography with Fluorescence Detection: Ratio of the Reduced Form as a Biomarker of Oxidative Stress. Biol. Pharm. Bull. 2009, 32 (11), 1819–1823. 10.1248/bpb.32.1819. [DOI] [PubMed] [Google Scholar]
- Micheli V.; Simmonds H. A.; Bari M.; Pompucci G. HPLC Determination of Oxidized and Reduced Pyridine Coenzymes in Human Erythrocytes. Clin. Chim. Acta 1993, 220 (1), 1–17. 10.1016/0009-8981(93)90002-L. [DOI] [PubMed] [Google Scholar]
- Al-Ali A. K. Pyridine Nucleotide Redox Potential in Erythrocytes of Saudi Subjects with Sickle Cell Disease. Acta Haematol 2002, 108 (1), 19–22. 10.1159/000063062. [DOI] [PubMed] [Google Scholar]
- Stocchi V.; Cucchiarini L.; Magnani M.; Chiarantini L.; Palma P.; Crescentini G. Simultaneous Extraction and Reverse-Phase High-Performance Liquid Chromatographic Determination of Adenine and Pyridine Nucleotides in Human Red Blood Cells. Anal. Biochem. 1985, 146 (1), 118–124. 10.1016/0003-2697(85)90405-1. [DOI] [PubMed] [Google Scholar]
- Nemkov T.; Stefanoni D.; Bordbar A.; Issaian A.; Palsson B. O.; Dumont L. J.; Hay A.; Song A.; Xia Y.; Redzic J. S.; Eisenmesser E. Z.; Zimring J. C.; Kleinman S.; Hansen K. C.; Busch M. P.; D’Alessandro A. Blood Donor Exposome and Impact of Common Drugs on Red Blood Cell Metabolism. JCI Insight 2021, 10.1172/jci.insight.146175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanoni D.; Fu X.; Reisz J. A.; Kanias T.; Nemkov T.; Page G. P.; Dumont L.; Roubinian N.; Stone M.; Kleinman S.; Busch M.; Zimring J. C.; D’Alessandro A. for the Recipient Epidemiology and Donor Evaluation Study-III (REDS III). Nicotine Exposure Increases Markers of Oxidant Stress in Stored Red Blood Cells from Healthy Donor Volunteers. Transfusion 2020, 60 (6), 1160–1174. 10.1111/trf.15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro A.; Fu X.; Reisz J. A.; Kanias T.; Page G. P.; Stone M.; Kleinman S.; Zimring J. C.; Busch M. for the Recipient Epidemiology and Donor Evaluation Study-III (REDS III). Stored RBC Metabolism as a Function of Caffeine Levels. Transfusion 2020, 60 (6), 1197–1211. 10.1111/trf.15813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolone L.; Roy M. K.; Hay A. M.; Morrison E. J.; Stefanoni D.; Fu X.; Kanias T.; Kleinman S.; Dumont L. J.; Stone M.; Nemkov T.; Busch M. P.; Zimring J. C.; D’Alessandro A. Impact of Taurine on Red Blood Cell Metabolism and Implications for Blood Storage. Transfusion 2020, 60 (6), 1212–1226. 10.1111/trf.15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon R. G.[24] Glucose 6-Phosphate Dehydrogenase from Erythrocytes. In Methods in Enzymology; Carbohydrate Metabolism; Academic Press, 1966; Vol. 9, pp 126–131. 10.1016/0076-6879(66)09030-X. [DOI] [Google Scholar]
- Pearse B. M.; Rosemeyer M. A. 6-Phosphogluconate Dehydrogenase from Human Erythrocytes. Methods Enzymol 1975, 41, 220–226. 10.1016/S0076-6879(75)41051-5. [DOI] [PubMed] [Google Scholar]
- Lewis I. A.; Campanella M. E.; Markley J. L.; Low P. S. Role of Band 3 in Regulating Metabolic Flux of Red Blood Cells. Proc. Natl. Acad. Sci. U.S.A. 2009, 106 (44), 18515–18520. 10.1073/pnas.0905999106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa M. C.; Alinovi C. C.; Galtieri A.; Russo A.; Giardina B. Allosteric Properties of Hemoglobin and the Plasma Membrane of the Erythrocyte: New Insights in Gas Transport and Metabolic Modulation. IUBMB Life 2008, 60 (2), 87–93. 10.1002/iub.15. [DOI] [PubMed] [Google Scholar]
- Mohandas N.; Gallagher P. G. Red Cell Membrane: Past, Present, and Future. Blood 2008, 112 (10), 3939–3948. 10.1182/blood-2008-07-161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinalducci S.; Marrocco C.; Zolla L. Thiol-Based Regulation of Glyceraldehyde-3-Phosphate Dehydrogenase in Blood Bank–Stored Red Blood Cells: A Strategy to Counteract Oxidative Stress. Transfusion 2015, 55 (3), 499–506. 10.1111/trf.12855. [DOI] [PubMed] [Google Scholar]
- Reisz J. A.; Wither M. J.; Dzieciatkowska M.; Nemkov T.; Issaian A.; Yoshida T.; Dunham A. J.; Hill R. C.; Hansen K. C.; D’Alessandro A. Oxidative Modifications of Glyceraldehyde 3-Phosphate Dehydrogenase Regulate Metabolic Reprogramming of Stored Red Blood Cells. Blood 2016, 128 (12), e32–e42. 10.1182/blood-2016-05-714816. [DOI] [PubMed] [Google Scholar]
- Kuhn V.; Diederich L.; Keller T. C. S.; Kramer C. M.; Lückstädt W.; Panknin C.; Suvorava T.; Isakson B. E.; Kelm M.; Cortese-Krott M. M. Red Blood Cell Function and Dysfunction: Redox Regulation, Nitric Oxide Metabolism, Anemia. Antioxid Redox Signal 2017, 26 (13), 718–742. 10.1089/ars.2016.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellini M. D.; Fiorelli G. Glucose-6-Phosphate Dehydrogenase Deficiency. Lancet 2008, 371 (9606), 64–74. 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- Linley E.; Denyer S. P.; McDonnell G.; Simons C.; Maillard J.-Y. Use of Hydrogen Peroxide as a Biocide: New Consideration of Its Mechanisms of Biocidal Action. J. Antimicrob. Chemother. 2012, 67 (7), 1589–1596. 10.1093/jac/dks129. [DOI] [PubMed] [Google Scholar]
- Panday A.; Sahoo M. K.; Osorio D.; Batra S. NADPH Oxidases: An Overview from Structure to Innate Immunity-Associated Pathologies. Cell Mol. Immunol 2015, 12 (1), 5–23. 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (23), 5839–5848. 10.1073/pnas.1804932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum H.; Roebuck K. A. Oxidant Stress and Endothelial Cell Dysfunction. Am. J. Physiol., Cell Physiol. 2001, 280 (4), C719–741. 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- Forman H. J.; Bernardo A.; Davies K. J. A. What Is the Concentration of Hydrogen Peroxide in Blood and Plasma?. Arch. Biochem. Biophys. 2016, 603, 48–53. 10.1016/j.abb.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Flannagan R. S.; Cosío G.; Grinstein S. Antimicrobial Mechanisms of Phagocytes and Bacterial Evasion Strategies. Nat. Rev. Microbiol 2009, 7 (5), 355–366. 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- Denicola A.; Souza J. M.; Radi R. Diffusion of Peroxynitrite across Erythrocyte Membranes. Proc. Natl. Acad. Sci. U.S.A. 1998, 95 (7), 3566–3571. 10.1073/pnas.95.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C.-S.; Kato G. J.; Yang S. H.; Bae S. W.; Lee J. S.; Gladwin M. T.; Rhee S. G. Hydroxyurea-Induced Expression of Glutathione Peroxidase 1 in Red Blood Cells of Individuals with Sickle Cell Anemia. Antioxidants & redox signaling 2010, 13 (1), 1–11. 10.1089/ars.2009.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfeitas R.; Selvaggio G.; Antunes F.; Coelho P. M. B. M.; Salvador A. Hydrogen Peroxide Metabolism and Sensing in Human Erythrocytes: A Validated Kinetic Model and Reappraisal of the Role of Peroxiredoxin II. Free Radic Biol. Med. 2014, 74, 35–49. 10.1016/j.freeradbiomed.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Antunes F.; Salvador A.; Pinto R. E. PHGPx and Phospholipase A2/GPx: Comparative Importance on the Reduction of Hydroperoxides in Rat Liver Mitochondria. Free Radic. Biol. Med. 1995, 19 (5), 669–677. 10.1016/0891-5849(95)00040-5. [DOI] [PubMed] [Google Scholar]
- Giulivi C.; Davies K. J. A Novel Antioxidant Role for Hemoglobin. The Comproportionation of Ferrylhemoglobin with Oxyhemoglobin. J. Biol. Chem. 1990, 265 (32), 19453–19460. 10.1016/S0021-9258(17)45394-4. [DOI] [PubMed] [Google Scholar]
- Niwa T. Protein Glutathionylation and Oxidative Stress. J. Chromatogr B Analyt Technol. Biomed Life Sci. 2007, 855 (1), 59–65. 10.1016/j.jchromb.2006.12.029. [DOI] [PubMed] [Google Scholar]
- Sheng K.; Shariff M.; Hebbel R. P. Comparative Oxidation of Hemoglobins A and S. Blood 1998, 91 (9), 3467–3470. 10.1182/blood.V91.9.3467. [DOI] [PubMed] [Google Scholar]
- Hebbel R. P.; Morgan W. T.; Eaton J. W.; Hedlund B. E. Accelerated Autoxidation and Heme Loss Due to Instability of Sickle Hemoglobin. Proc. Natl. Acad. Sci. U. S. A. 1988, 85 (1), 237–241. 10.1073/pnas.85.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer J.; Ghoti H.; Rachmilewitz E.; Koren A.; Levin C.; Fibach E. Red Blood Cells, Platelets and Polymorphonuclear Neutrophils of Patients with Sickle Cell Disease Exhibit Oxidative Stress That Can Be Ameliorated by Antioxidants. Br. J. Hamaetol. 2006, 132 (1), 108–113. 10.1111/j.1365-2141.2005.05834.x. [DOI] [PubMed] [Google Scholar]
- Kato G. J.; Steinberg M. H.; Gladwin M. T. Intravascular Hemolysis and the Pathophysiology of Sickle Cell Disease. J. Clin Invest 2017, 127 (3), 750–760. 10.1172/JCI89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood K. C.; Hebbel R. P.; Granger D. N. Endothelial Cell NADPH Oxidase Mediates the Cerebral Microvascular Dysfunction in Sickle Cell Transgenic Mice. FASEB J. 2005, 19 (8), 989–991. 10.1096/fj.04-3218fje. [DOI] [PubMed] [Google Scholar]
- Mahaseth H.; Vercellotti G. M.; Welch T. E.; Bowlin P. R.; Sonbol K. M.; Hsia C. J.C.; Ma L.; Bischof J. C.; Hebbel R. P.; Belcher J. D. Polynitroxyl Albumin Inhibits Inflammation and Vasoocclusion in Transgenic Sickle Mice. J. Lab Clin Med. 2005, 145 (4), 204–211. 10.1016/j.lab.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Sundd P.; Gladwin M. T.; Novelli E. M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol 2019, 14, 263–292. 10.1146/annurev-pathmechdis-012418-012838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer J.; Atlas D.; Fibach E. N-Acetylcysteine Amide (AD4) Attenuates Oxidative Stress in Beta-Thalassemia Blood Cells. Biochim. Biophys. Acta 2008, 1780 (2), 249–255. 10.1016/j.bbagen.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Hwang S.; Mruk K.; Rahighi S.; Raub A. G.; Chen C.-H.; Dorn L. E.; Horikoshi N.; Wakatsuki S.; Chen J. K.; Mochly-Rosen D. Correcting Glucose-6-Phosphate Dehydrogenase Deficiency with a Small-Molecule Activator. Nat. Commun. 2018, 9 (1), 4045. 10.1038/s41467-018-06447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koriem K. M. M.; Arbid M. S. Evaluating of β-Carotene Role in Ameliorating of Favism-Induced Disturbances in Blood and Testis. J. Complement Integr Med. 2018, 10.1515/jcim-2017-0164. [DOI] [PubMed] [Google Scholar]
- Balagopalakrishna C.; Manoharan P. T.; Abugo O. O.; Rifkind J. M. Production of Superoxide from Hemoglobin-Bound Oxygen under Hypoxic Conditions. Biochemistry 1996, 35 (20), 6393–6398. 10.1021/bi952875+. [DOI] [PubMed] [Google Scholar]
- Mohanty J.; Nagababu E.; Rifkind J. M. Red Blood Cell Oxidative Stress Impairs Oxygen Delivery and Induces Red Blood Cell Aging. Front. Physiol. 2014, 10.3389/fphys.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriebardis A. G.; Antonelou M.; Stamoulis K.; Economou-Petersen E.; Margaritis L.; Papassideri I. RBC-Derived Vesicles during Storage: Ultrastructure and Lipid Raft Proteins Participation. Vox Sang 2008, 95, 190. [DOI] [PubMed] [Google Scholar]
- Ciana A.; Achilli C.; Gaur A.; Minetti G. Membrane Remodelling and Vesicle Formation During Ageing of Human Red Blood Cells. Cell Physiol Biochem 2017, 42 (3), 1127–1138. 10.1159/000478768. [DOI] [PubMed] [Google Scholar]
- Sudnitsyna J.; Skverchinskaya E.; Dobrylko I.; Nikitina E.; Gambaryan S.; Mindukshev I. Microvesicle Formation Induced by Oxidative Stress in Human Erythrocytes. Antioxidants (Basel) 2020, 9 (10), 929 10.3390/antiox9100929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safeukui I.; Buffet P. A.; Deplaine G.; Perrot S.; Brousse V.; Ndour A.; Nguyen M.; Mercereau-Puijalon O.; David P. H.; Milon G.; Mohandas N. Quantitative Assessment of Sensing and Sequestration of Spherocytic Erythrocytes by the Human Spleen. Blood 2012, 120 (2), 424–430. 10.1182/blood-2012-01-404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferru E.; Pantaleo A.; Carta F.; Mannu F.; Khadjavi A.; Gallo V.; Ronzoni L.; Graziadei G.; Cappellini M. D.; Turrini F. Thalassemic Erythrocytes Release Microparticles Loaded with Hemichromes by Redox Activation of P72Syk Kinase. Haematologica 2014, 99 (3), 570–578. 10.3324/haematol.2013.084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M.; Lutz R. J.; Cotter T. G.; O’Connor R. Erythrocyte Survival Is Promoted by Plasma and Suppressed by a Bak-Derived BH3 Peptide That Interacts with Membrane-Associated Bcl-X(L). Blood 2002, 99 (9), 3439–3448. 10.1182/blood.V99.9.3439. [DOI] [PubMed] [Google Scholar]
- Franco R. S. The Measurement and Importance of Red Cell Survival. Am. J. Hematol 2009, 84 (2), 109–114. 10.1002/ajh.21298. [DOI] [PubMed] [Google Scholar]
- Klei T. R. L.; Meinderts S. M.; van den Berg T. K.; van Bruggen R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Frontiers in Immunology 2017, 10.3389/fimmu.2017.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges M. D.; Sesti-Costa R. Macrophages: Key Players in Erythrocyte Turnover. Hematology, Transfusion and Cell Therapy 2022, 44, 574. 10.1016/j.htct.2022.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosale N. G.; Rouhiparkouhi T.; Bradshaw A. M.; Dimova R.; Lipowsky R.; Discher D. E. Cell Rigidity and Shape Override CD47’s “Self”-Signaling in Phagocytosis by Hyperactivating Myosin-II. Blood 2015, 125 (3), 542–552. 10.1182/blood-2014-06-585299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R.; Labotka R.; Low P. S. Isolation and Characterization of the Hemichrome-Stabilized Membrane Protein Aggregates from Sickle Erythrocytes. Major Site of Autologous Antibody Binding. J. Biol. Chem. 1988, 263 (27), 13766–13773. 10.1016/S0021-9258(18)68308-5. [DOI] [PubMed] [Google Scholar]
- Lutz H. U. Naturally Occurring Anti-Band 3 Antibodies in Clearance of Senescent and Oxidatively Stressed Human Red Blood Cells. Transfus Med. Hemother 2012, 39 (5), 321–327. 10.1159/000342171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarczyk P.; Mleczko-Sanecka K. The Multiple Facets of Iron Recycling. Genes 2021, 12 (9), 1364. 10.3390/genes12091364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuypers F. A.; Yuan J.; Lewis R. A.; Snyder L. M.; Kiefer C. R.; Bunyaratvej A.; Fucharoen S.; Ma L.; Styles L.; de Jong K.; Schrier S. L. Membrane Phospholipid Asymmetry in Human Thalassemia. Blood 1998, 91 (8), 3044–3051. 10.1182/blood.V91.8.3044.3044_3044_3051. [DOI] [PubMed] [Google Scholar]
- Wood B.; Gibson D.; Tait J. Increased Erythrocyte Phosphatidylserine Exposure in Sickle Cell Disease: Flow-Cytometric Measurement and Clinical Associations. Blood 1996, 88 (5), 1873–1880. 10.1182/blood.V88.5.1873.1873. [DOI] [PubMed] [Google Scholar]
- Kim-Shapiro D. B.; Lee J.; Gladwin M. T. Storage Lesion. Role of Red Cell Breakdown. Transfusion 2011, 51 (4), 844–851. 10.1111/j.1537-2995.2011.03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevi F.; D’Alessandro A.; Rinalducci S.; Zolla L. Alterations of Red Blood Cell Metabolome during Cold Liquid Storage of Erythrocyte Concentrates in CPD-SAGM. J. Proteomics 2012, 76, 168–180. 10.1016/j.jprot.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Bosman G. J. C. G. M.; Cluitmans J. C. A.; Groenen Y. A. M.; Werre J. M.; Willekens F. L. A.; Novotný V. M. J. Susceptibility to Hyperosmotic Stress-Induced Phosphatidylserine Exposure Increases during Red Blood Cell Storage. Transfusion 2011, 51 (5), 1072–1078. 10.1111/j.1537-2995.2010.02929.x. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Prudent M.; D’Alessandro A. Red Blood Cell Storage Lesion: Causes and Potential Clinical Consequences. Blood Transfus 2019, 17 (1), 27–52. 10.2450/2019.0217-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman G. J. C. G. M.; Stappers M.; Novotný V. M. J. Changes in Band 3 Structure as Determinants of Erythrocyte Integrity during Storage and Survival after Transfusion. Blood Transfus 2010, 8, s48–s52. 10.2450/2010.008S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro A.; Kriebardis A.; Rinalducci S.; Antonelou M.; Hansen K.; Papassideri I.; Zolla L. An Update on Red Blood Cell Storage Lesions, as Gleaned through Biochemistry and Omics Technologies. Transfusion 2015, 55, 205. 10.1111/trf.12804. [DOI] [PubMed] [Google Scholar]
- Karon B. S.; Hoyer J. D.; Stubbs J. R.; Thomas D. D. Changes in Band 3 Oligomeric State Precede Cell Membrane Phospholipid Loss during Blood Bank Storage of Red Blood Cells. Transfusion 2009, 49 (7), 1435–1442. 10.1111/j.1537-2995.2009.02133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.-L.; Williams K. J.; Werth V. P.. Chapter Four - Microvesicles in Autoimmune Diseases. In Advances in Clinical Chemistry; Makowski G. S., Ed.; Elsevier, 2016; Vol. 77, pp 125–175. 10.1016/bs.acc.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Peters A. L.; van Bruggen R.; de Korte D.; Van Noorden C. J. F.; Vlaar A. P. J. Glucose-6-Phosphate Dehydrogenase Activity Decreases during Storage of Leukoreduced Red Blood Cells. Transfusion 2016, 56 (2), 427–432. 10.1111/trf.13378. [DOI] [PubMed] [Google Scholar]
- Jana S.; Strader M. B.; Meng F.; Hicks W.; Kassa T.; Tarandovskiy I.; De Paoli S.; Simak J.; Heaven M. R.; Belcher J. D.; Vercellotti G. M.; Alayash A. I. Hemoglobin Oxidation-Dependent Reactions Promote Interactions with Band 3 and Oxidative Changes in Sickle Cell-Derived Microparticles. JCI Insight 2018, 3 (21), 120451. 10.1172/jci.insight.120451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo A.; Ferru E.; Pau M. C.; Khadjavi A.; Mandili G.; Mattè A.; Spano A.; De Franceschi L.; Pippia P.; Turrini F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p (72) Syk. Oxid Med. Cell Longev 2016, 2016, 6051093. 10.1155/2016/6051093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo A.; Giribaldi G.; Mannu F.; Arese P.; Turrini F. Naturally Occurring Anti-Band 3 Antibodies and Red Blood Cell Removal under Physiological and Pathological Conditions. Autoimmun Rev. 2008, 7 (6), 457–462. 10.1016/j.autrev.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Issaian A.; Hay A.; Dzieciatkowska M.; Roberti D.; Perrotta S.; Darula Z.; Redzic J.; Busch M. P.; Page G. P.; Rogers S. C.; Doctor A.; Hansen K. C.; Eisenmesser E. Z.; Zimring J. C.; D’Alessandro A. The Interactome of the N-Terminus of Band 3 Regulates Red Blood Cell Metabolism and Storage Quality. Haematologica 2021, 106 (11), 2971–2985. 10.3324/haematol.2020.278252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohé S.; Kobbe P.; Nast-Kolb D. Immunological Reactions Secondary to Blood Transfusion. Injury 2007, 38 (12), 1405–1408. 10.1016/j.injury.2007.09.028. [DOI] [PubMed] [Google Scholar]
- Shah A.; Brunskill S. J.; Desborough M. J.; Doree C.; Trivella M.; Stanworth S. J. Transfusion of Red Blood Cells Stored for Shorter versus Longer Duration for All Conditions. Cochrane Database Syst. Rev. 2018, 12, CD010801. 10.1002/14651858.CD010801.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruun-Rasmussen P.; Kragh Andersen P.; Banasik K.; Brunak S.; Johansson P. I. Intervening on the Storage Time of RBC Units and Its Effects on Adverse Recipient Outcomes Using Real-World Data. Blood 2022, 139 (25), 3647–3654. 10.1182/blood.2022015892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graw J. A.; Bünger V.; Materne L. A.; Krannich A.; Balzer F.; Francis R. C. E.; Pruß A.; Spies C. D.; Kuebler W. M.; Weber-Carstens S.; Menk M.; Hunsicker O. Age of Red Cells for Transfusion and Outcomes in Patients with ARDS. J. Clin Med. 2022, 11 (1), 245. 10.3390/jcm11010245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro A.; Fu X.; Kanias T.; Reisz J. A.; Culp-Hill R.; Guo Y.; Gladwin M. T.; Page G.; Kleinman S.; Lanteri M.; Stone M.; Busch M. P.; Zimring J. C. Recipient Epidemiology and Donor Evaluation Study-III (REDS III). Donor Sex, Age and Ethnicity Impact Stored Red Blood Cell Antioxidant Metabolism through Mechanisms in Part Explained by Glucose 6-Phosphate Dehydrogenase Levels and Activity. Haematologica 2021, 106 (5), 1290–1302. 10.3324/haematol.2020.246603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R. O.; D’Alessandro A.; Eisenberger A.; Soffing M.; Yeh R.; Coronel E.; Sheikh A.; Rapido F.; Carpia F. L.; Reisz J. A.; Gehrke S.; Nemkov T.; Thomas T.; Schwartz J.; Divgi C.; Kessler D.; Shaz B. H.; Ginzburg Y.; Zimring J. C.; Spitalnik S. L.; Hod E. A. Donor Glucose-6-Phosphate Dehydrogenase Deficiency Decreases Blood Quality for Transfusion. J. Clin Invest 2020, 130 (5), 2270–2285. 10.1172/JCI133530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page G. P.; Kanias T.; Guo Y. J.; Lanteri M. C.; Zhang X.; Mast A. E.; Cable R. G.; Spencer B. R.; Kiss J. E.; Fang F.; Endres-Dighe S. M.; Brambilla D.; Nouraie M.; Gordeuk V. R.; Kleinman S.; Busch M. P.; Gladwin M. T. Multiple-Ancestry Genome-Wide Association Study Identifies 27 Loci Associated with Measures of Hemolysis Following Blood Storage. J. Clin Invest 2021, 131 (13), x. 10.1172/JCI146077. [DOI] [PMC free article] [PubMed] [Google Scholar]