

Abstract
The ability to predict the intricate mechanistic behavior of ligands and associated structural determinants during protein–ligand (un)binding is of great practical importance in drug discovery. Ubiquitin specific protease‐7 (USP7) is a newly emerging attractive cancer therapeutic target with bound allosteric inhibitors. However, none of the inhibitors have reached clinical trials, allowing opportunities to examine every aspect of allosteric modulation. The crystallographic insights reveal that these inhibitors have common properties such as chemical scaffolds, binding site and interaction fingerprinting. However, they still possess a broader range of binding potencies, ranging from 22 nM to 1,300 nM. Hence, it becomes more critical to decipher the structural determinants guiding the enhanced binding potency of the inhibitors. In this regard, we elucidated the atomic‐level insights from both interacting partners, that is, protein–ligand perspective, and established the structure–activity link between USP7 inhibitors by using classical and advanced molecular dynamics simulations combined with linear interaction energy and molecular mechanics‐Poisson Boltzmann surface area. We revealed the inhibitor potency differences by examining the contributions of chemical moieties and USP7 residues, the involvement of water‐mediated interactions, and the thermodynamic landscape alterations. Additionally, the dissociation profiles aided in the establishment of a correlation between experimental potencies and structural determinants. Our study demonstrates the critical role of blocking loop 1 in allosteric inhibition and enhanced binding affinity. Comprehensively, our findings provide a constructive expansion of experimental outcomes and show the basis for varying binding potency using in‐silico approaches. We expect this atomistic approach to be useful for effective drug design.
Keywords: allosteric regulation, classical and steered MD simulation, conformational change, free energy landscape, LIE, MM‐PBSA
Abbreviations
- BL1 and BL2
blocking loop 1 and blocking loop 2
- cMD
classical molecular dynamics
- DUBs
deubiquitinating enzymes
- FEL
free energy landscape
- HB
hydrogen bonds
- LIE
linear interaction energy
- PCA
principal component analysis
- Rg
radius of gyration
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- SL
switching loop
- SMD
steered molecular dynamics
- Ubq
ubiquitin
- USP7
ubiquitin specific protease 7
- wHB
water‐mediated hydrogen bonds
1. INTRODUCTION
Ubiquitin specific protease‐7 (USP7) has been investigated as a drug target for oncology pathways. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 Beyond cancers, USP7 has also been implicated in immunotherapy, 10 glucose metabolism, 11 and viral infections 12 , 13 by modulating variety of proteins, including p53, MDM2, beta‐catenin, FOXP3, and ICPO. 14 , 15 , 16 , 17 Thus, USP7 is a promising therapeutic target, albeit one that is challenging to approach due to its complex regulatory mechanism. USP7 has a well‐defined multi‐domain conserved structure. At N‐terminal it has TRAF domain (~100 residues) that identifies substrates, followed by catalytic domain (~360 residues) and ends with C‐terminal region that contains five auxiliary domains as UBL12345 (ubiquitin‐like domains). 18 , 19 , 20 USP7 is inactive in its native state, but ubiquitin (Ubq) binding to its catalytic domain (CD) converts it to an active state, which acts as a switch‐on mechanism. 21 , 22 The activation mechanism occurs due to dynamical changes in blocking loop‐1 caused by Ubq at the allosteric checkpoint, and further its tail induces conformational changes in the switching loop, allowing USP7 to shift from a dormant to an active state. 21 , 23 Thus, BL1 and SL appear to be primary and secondary components, respectively, of USP7's biological activity, based on activation processes. These activation events facilitate USP7's ability to modulate substrates, and hence a thorough understanding of USP7's allosteric landscape will benefit oncology drug discovery.
To hinder this intricate activation mechanism of USP7, several crystals with allosteric inhibitors have been reported. We previously investigated the probable mechanism of inhibition of its reported allosteric inhibitors. 24 Studies have revealed that inhibitors hinder USP7 functional activity from a well‐characterized allosteric pocket (the Ubq binding site) located at the junction of the palm and finger sub‐domains of the USP7 catalytic domain. 22 , 24 , 25 , 26 Several co‐crystal bound inhibitors have exhibited anti‐cancer activity, although their binding potencies vary widely (from 22 nM to 1,300 nM), despite occupying the same binding site and reflecting similar binding mode, raising questions regarding their inhibitory mechanism and the causes of this variation. 22 , 26 , 27 Furthermore, these inhibitors also share the common core scaffold, which deepens the curiosity to explore further (Figure 1).
FIGURE 1.
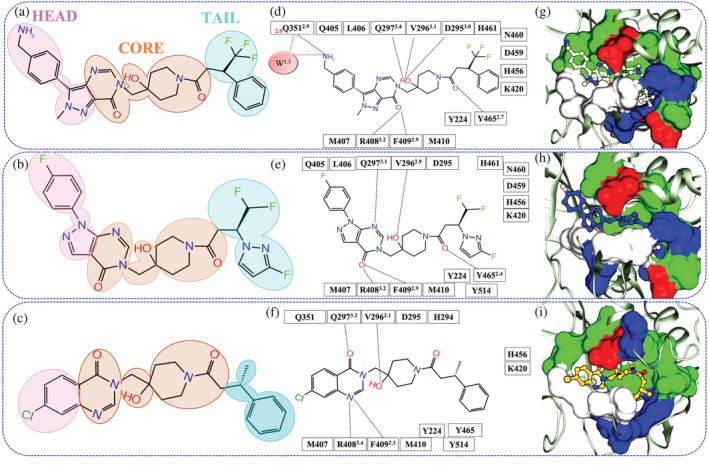
Schematic representation of the ligand interaction at crystal pose. (a‐c) The 2D images of ligand 8QQ, 8WK, and 9HS. The similar core of all ligands is shaded in orange while head and tail are shaded in pink and cyan. The panels (d‐f) represent the residue interaction with ligand within 3.5 Å of systems USP78QQ, USP78WK, and USP79HS, respectively, where hydrogen bonds (HB) and water‐mediated HB interactions (wHB) are shown in black and red dotted line. (g‐i) Chemical characteristics of the binding site in systems USP78QQ, USP78WK, and USP79HS respectively. The color‐coding of amino acids is as follows: green: polar; blue: basic; white: hydrophobic; and red: acidic. The notations in superscript and subscript are HB distances and wHB distances written in ‘black’ and ‘red’, respectively
Ligand–protein interactions transduce signals intracellularly that induce cellular responses. In this regard, the identification and quantification of protein and ligand determinants that are primarily responsible for enhanced biological activity constitute a fundamental and practical necessity for target‐specific drug discovery. Particularly, when ligands share the chemical moieties as well as bind to the same site using the common interactions with amino acids. The availability of co‐crystals of these compounds provides an opportunity for exploring the pivotal factors by comparing the protein's unbound‐to‐bound allosteric landscape at the atomic and energetic level. Furthermore, it is well‐recognized that one of the persistent factors for sustained drug efficacy and safety is not just its affinity but also the mean lifetime of the protein–ligand interaction, which needs to be underscored to identify the indispensable determinants. Hence, our goal is to study this landscape in USP7 by examining the binding pattern of dynamic motion, which alters their binding affinity allosterically, and reveals the causative structural determinants for higher or decreased binding potencies of inhibitors.
Deducing the mechanism of inhibition, thermodynamically favorable hot‐spots and structural determinants do hold a paramount relevance in the early stages of drug discovery 28 , 29 , 30 and are reliably addressed using classical molecular dynamics simulation (cMD) 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 along with thermodynamics calculation. 39 , 40 Also, advanced molecular modeling studies such as steered molecular dynamics (SMD) are known to characterize strong to weak binders, 41 conformational characterization of structural determinants, 42 , 43 the kinetics of protein‐ligand unbinding, 44 and predict unbinding pathways for the molecules. 45 , 46 Therefore, we have implemented both classical and advanced molecular dynamics simulations to investigate the possible source of the varying binding potencies of USP7 inhibitors. Starting from the crystallographic pose, the post‐processing cMD was used for thermodynamics and linear interaction energy (LIE) analysis, which provide significant insights at the atomic level. Here, we underscore the atom‐wise interactions of both interacting partners, that is, USP7 and its inhibitors. After that, the ligand dissociation study was performed to establish the link between experimental IC50 values and unbinding profiles and for the identification of persistent protein–ligand interactions. Overall, from microscopic information to conformational dynamicity, we have elucidated several persistent factors that have shed light on the variable spectrum of ligands. Our study provides an in‐depth quantitative analysis at the atomic level for improved and rationalized drug design.
2. RESULTS AND DISCUSSION
2.1. Crystal mining highlights variable interactions at allosteric checkpoint
Comparing the chemical architecture of ligands 8QQ, 8WK, and 9HS revealed that they share a core chemical moiety and can be separated into three regions: head, core, and tail (Figure 1a‐c). We observed the variation of chemical moieties at the head and tail regions of the respective ligands (Figure 1a‐c). The interaction fingerprinting of co‐crystals reveals that 11 residues Y224, D295, V296, Q297, M407, R408, F409, M410, K420, H456, and Y465 are shared by all three ligands (Figure 1d‐f). Furthermore, the ligands 8QQ and 8WK, whose binding modes are similar, share 15 residues from their core (Y224, D295, V296, Q297, Q405, L406, M407, R408, F409, M410, K420, H456, D459, N460, and Y465) (Figure 1d,e). However, their interaction pattern differs considerably with head and tail moieties. Considering the homogeneous nature of the pocket between both ligands, the residues Q351 and Y514 are unique with ligands 8QQ (head) and 8WK (tail), respectively (Figure 1d,e). On the other hand, the ligand 9HS is lined by unique residues Q351 and Y514 of 8QQ and 8WK, respectively (Figure 1f). However, it lacks three residues that surround the tail and are prevalent for 8QQ and 8WK: D459, N460, and H461 (Figure 1f).
In addition, the hydrogen bond (HB) network emphasizes the variable interactions because the ligand 8QQ forms seven HBs, one from the head (residue Q351) and six from the core (D295, V296, Q297, R408, F409, and Y465). The 8QQ also establishes an exclusive water‐mediated HB interaction (wHB) with residue Q351 (Figure 1d). The ligand 8WK's core shares five HBs with 8QQ, however, it lacks HB with residue D295 and wHB interaction with Q351. Finally, ligand 9HS forms four HBs and fails to interact with residues D295, Q351, and Y465 when compared to ligand 8QQ. Thus, the HB network clearly underlines a declining pattern from the strong ligand to the weak ligand: USP78QQ (7HBs) > USP78WK (5HBs) > USP79HS (4HBs). We observed that the binding pockets of three systems share a uniform blend of hydrophobic, polar, basic, and acidic interactions (Figure 1g‐i). However, we found a noticeable difference in the cavity formation in a static state. It was observed that 8QQ is buried in the pocket and residue Q351 supports its head moiety, whereas the other two ligands do not receive similar support (Figure 1g). The ligands 8WK and 9HS are solvent‐exposed, which is due to the difference in the backbone movement of the residue F409 when compared with system USP78QQ (Figure S1A). Consequently, this movement creates an additional gap between the side chains of the residues D295 and F409 in systems USP78WK and USP79HS, with distances 1.0 and 1.3 Å higher than USP78QQ, respectively (Figure S1B). Overall, this analysis revealed that ligand 8QQ would gain additional affinity due to (a) HB and wHB interactions with residue Q351 and (b) packing of the pocket due to the perpetuity of residues D295 and F409, making it less solvent exposed than the other two ligands.
2.2. cMD simulations assess the structural stability
Proteins are dynamic entities that populate conformational ensembles, and most functions of proteins depend on their dynamic nature. Hence, atomistic simulations were conducted to observe the dynamic behavior of the ligands. The structure stability analysis revealed that the backbone RMSD values of the systems' replicates are comparable (Figure S2), and hence, the most stable trajectories in each case were considered for the analysis. The RMSD analysis shows that systems, USP78WK and USP79HS fluctuate more than USP7APO; however, USP78QQ fluctuates less and attains stability after a 50 ns evolving period (Figure 2a). Unlike USP78QQ, the other two systems have attained convergence after 200 ns (Figure 2a). The RMSD distribution of all systems showed a unimodal distribution curve except for the system USP78WK (Figure 2b). It is intriguing to observe that systems, USP78WK and USP79HS have shown fluctuations from the interval of 125 to 200 ns during simulations. We tried to elucidate the reason behind this fluctuation, and upon manual inspection of the trajectories, we found significant fluctuations in the finger sub‐domain. However, a similar fluctuation is not captured in the system USP78QQ. Further, the ligand RMSD values reveal that 8QQ has better stability as compared to other ligands (Figure 2c,d). Additionally, the binding site (BS) RMSD shows that the flexibility of BS residues in the bound systems is reduced when compared with the USP7APO (Figure 2e) and ligand 8QQ significantly reduces the backbone deviations of the pocket (Figure 2e,f).
FIGURE 2.
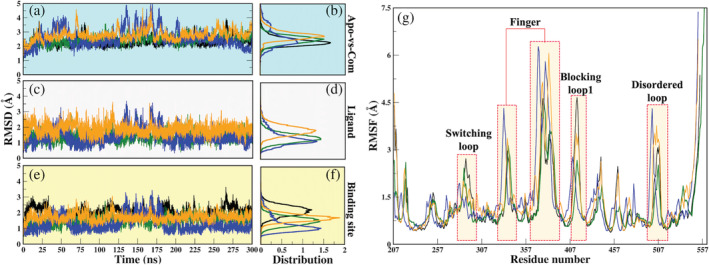
Dynamical characteristics of systems elucidated through classical MD simulations. (a, b) Root mean square deviation (RMSD) evolution through the course of 300 ns of protein in APO and Com systems are plotted over its backbone atoms and its distribution, respectively, (c, d) ligand RMSD and its distribution, (e, f) RMSD of the binding site. Color coding is followed as black: APO; green: USP78QQ; blue: USP78WK; and orange: USP79HS, (g) the Cɑ atomic fluctuation are represented by RMSF. The regions showing significant fluctuations are highlighted in transparent bars. MD, molecular dynamics
The RMSF profiles reveal that the three key regions: SL, finger sub‐domain, and BL1, have shown the highest Cɑ atomic fluctuations. We observed that the RMSF values of the replicated systems are also comparable (Figure S3). In comparison to USP7APO, 8QQ only reduces the protein's overall flexibility (Figure 2g). The other two ligands cause higher atomic fluctuations in the protein, notably at finger and BL1. The cMD simulation revealed a fluctuation from residues E495 to V507, which is a disordered loop. This loop is flexible in nature, and did not crystallize in USP7APO, USP78WK, and USP79HS crystals. However, it can be found in the crystal structure of USP78QQ. Despite the disordered tendency, it was found that the ligand 8QQ had only been able to reduce its flexibility. The HB calculations on the whole simulated trajectory revealed that USP78QQ was forming 9HBs as compared to systems USP78WK (6HBs) and USP79HS (3HBs) (Table S2). Further, the Rg values have predicted USP78QQ as the most compact system (Figure S4). Since the finger subdomain is a key determinant for USP7 protein as it accommodates Ubq and allows it to activate USP7, 47 , 48 the ligand 8QQ is able to constrain its flexibility along with other important regions, BL1 and the disordered loop.
2.3. Analysis of net calculated binding free energies
The MM‐PBSA method was applied to all clusters in each system, which revealed that cluster A is the lowest energy cluster in each case (Table S3) (detailed methodology in supporting information section 2.5.1). The results reveal that the system USP78QQ has shown the highest net binding free energy of ∆G bind − 45.04 kcal/mol, followed by USP78WK and USP79HS, with net ∆G bind of −34.20 and − 27.67 kcal/mol, respectively (Table S4). We observed that the electrostatic energy (∆E elec) is more favorable in USP78QQ than in other systems, but the van der Waals (∆E vdw) contribution is nearly the same in all the systems. (Table S4). The entropy of USP78QQ is lower as compared to USP78WK and USP79HS, which in turn does not affect its enthalpy, thereby achieving the highest binding affinity. Furthermore, the ligand desolvation energy results show that ligand 8QQ has a much higher desolvation energy than ligands 8WK and 9HS (Table S5, supporting result section 1.1). Thus, the net calculated total ∆G bind values followed the same trend as the experimental ∆G bind, where the ligand 8QQ has shown a better binding affinity, and a similar trend is also reflected in the desolvation energy of ligands (Tables S4‐5).
2.4. Deciphering residue‐wise energetic contributions
The consistency of the residual contribution was evaluated during dynamics using decomposition analysis and was further compared with static state interactions. The residues having energy contributions of ≤−1.0 kcal/mol are considered to have a more significant impact 49 ; however, we have used ≤−0.5 kcal/mol as a cut‐off to comprehend maximum contributors. The details of residue‐wise energy decomposition are mentioned in Figure S5A‐C. From the triplicates of each system, we observed that the key residues have a similar trend of energy contribution, showing a good convergence (Figure S6).
The residual contribution patterns in static and dynamic states were almost comparable (Figures 1 and S5). However, among different systems, we noticed a change in the energetic contribution (Figure S5). The comparison of the crystal‐versus‐MD state reveals that residue D295 was not involved in systems USP78QQ while the interaction was present in the crystal state. However, it gains interaction with residue N422 (Figures 1d and S5A). In system USP78WK, two residues, D295 and M410, lose interaction during dynamics, while no additional residues are observed (Figure S5B). USP79HS retains its crystallographic interaction with residue D295 and gains interaction with N460 and H461 (Figure S5C). Residue D295 loses its contribution in two systems, indicating that it is not a stable interaction; nonetheless, its contribution in the least potent system is also poor (−0.97 kcal/mol) (Figure S5C). Although crystal and MD pose interactions varied slightly, the fact that most residues are recurring and new residues are identified is encouraging. The residues identified with these inhibitors do correlate with inhibitor P5091. 24 Thereafter, we analyzed intra‐protein residue interaction patterns and observed substantial variances in energy contribution patterns. We found that the USP78QQ, USP78WK, and USP79HS have 17, 15, and 14 contributions, respectively, with 15 residues shared by USP78QQ and USP78WK, 13 residues by USP78QQ and USP79HS, and 12 residues by USP78WK and USP79HS (Figure S7). This reveals high conservity in binding patterns among these systems. Furthermore, a comparison of binding energies reveals that eight residues (Y224, V296, Q351, R408, F409, K420, N422, N460) contribute more in USP78QQ, four residues (Q207, M407, H461, and Y465) in USP78WK and five residues (Q405, L406, M410, H456, and Y514) in USP79HS (Figure S8). This spectrum of interaction energies shows that USP78QQhas a greater residue contribution than other systems, which could explain its better binding affinity. However, it is too early to link these values to varying potencies.
Overall, from this analysis, residue N422 for USP78QQ is a new finding that was missing in crystal interactions as well as other simulated systems. Also, it is now evident that despite the similar pattern of interaction, the spectrum of binding energies is varying, and among them, the ligand 8QQ is hoping for major contributions. Despite sharing the common core scaffold, ligand 8QQ exhibited dominant energy contribution, prompting us to explore the ligands atom‐wise assuming that the difference might be arising between them at atomic level.
2.5. Atom‐wise interaction profiling
We have performed LIE to identify the crucial moieties and the contribution of the ligand's atoms to understand their energy difference with the contributing residues. From MM‐PBSA analysis, we elucidated the most significant contributors that aid ligand stability (Figure S5), and hence these residues were chosen for LIE analysis to examine the variable spectrum in energy distribution. Since LIE considers vdW and electrostatic components, we have mapped the residues based on the vdW distance (6.0 Å), required for interaction between two atoms. Based on this distance, we have curated the atom‐to‐atom interaction between ligands and residues, which would reflect the distribution of residues across the dissection of ligands (head, core, and tail moieties). For each atom in the ligands, 8QQ, 8WK, and 9HS, the energy value was computed using LIE's linear equation. Atom‐wise, ligand 8QQ has C:29, N:6, O:3, and F:3 (Table S6), ligand 8WK has C:24, N:7, O:3, and F:4 (Table S7), and ligand 9HS has C:24, N:3, O:3, and Cl:1 atom (Table S8).
To better understand the mechanistic details of ligand action, we analyzed the head, core, and tail regions of ligands (Figure 1a‐c). It was found that the majority of the atom‐wise energy exchange between ligands and residues is observed from the core (common among all). In system USP78QQ, the head moiety interacts with 6 residues (Q297, Q351, M407, R408, F409, and M410), the core moiety with 11 residues (Y224, V296, Q297, Q405, L406, M407, R408, F409, H461, Y465 and Y514), and a tail moiety with 6 residues (Y224, K420, H456, N460, H461, and Y465). Among them, residues Q297, M407, R408, and F409 are shared between head and core moieties as well, while residues Y224, H461, and Y465 are shared by the core and tail moieties (Figure 3a). In the USP78WK system, 5 residues (Q297, Q351, L406, M407, and R408) interact with 8WK's head moiety, and except L406, the rest residues are common with 8QQ, and 11 residues (Y224, V296, Q297, Q405, L406, M407, R408, F409, H461, Y465, and Y514) connect with core moieties, which are also shared with 8QQ. The ligand 8WK's tail moiety takes up seven residues (F409, K420, H456, N460, H461, Y465, and Y514). The exceptional residues are L406 (head) and F409 (tail), rest are identical and establish contact with the tail moiety of 8QQ (Figure 3a,b). Within this system, four residues Q297, L406, M407, and R408 are shared by head and core moieties, and four residues F409, H461, Y465 and Y514 are shared between core and tail moieties (Figure 3b). Lastly, in system USP79HS, 9HS's head moiety interacts with 2 residues M407 and M410, the core moiety with 11 residues (Y224, D295, V296, Q405, M407, R408, F409, M410, N460, H461, and Y514) and a tail with seven residues (R408, F409, H456, N460, H461, Y465, and Y514) (Figure 3c). Both M407 and M410 are shared between the head and core moiety in this system, and five residues (R408, F409, N460, H461, and Y514) are shared between the core and tail moieties (Figure 3c).
FIGURE 3.
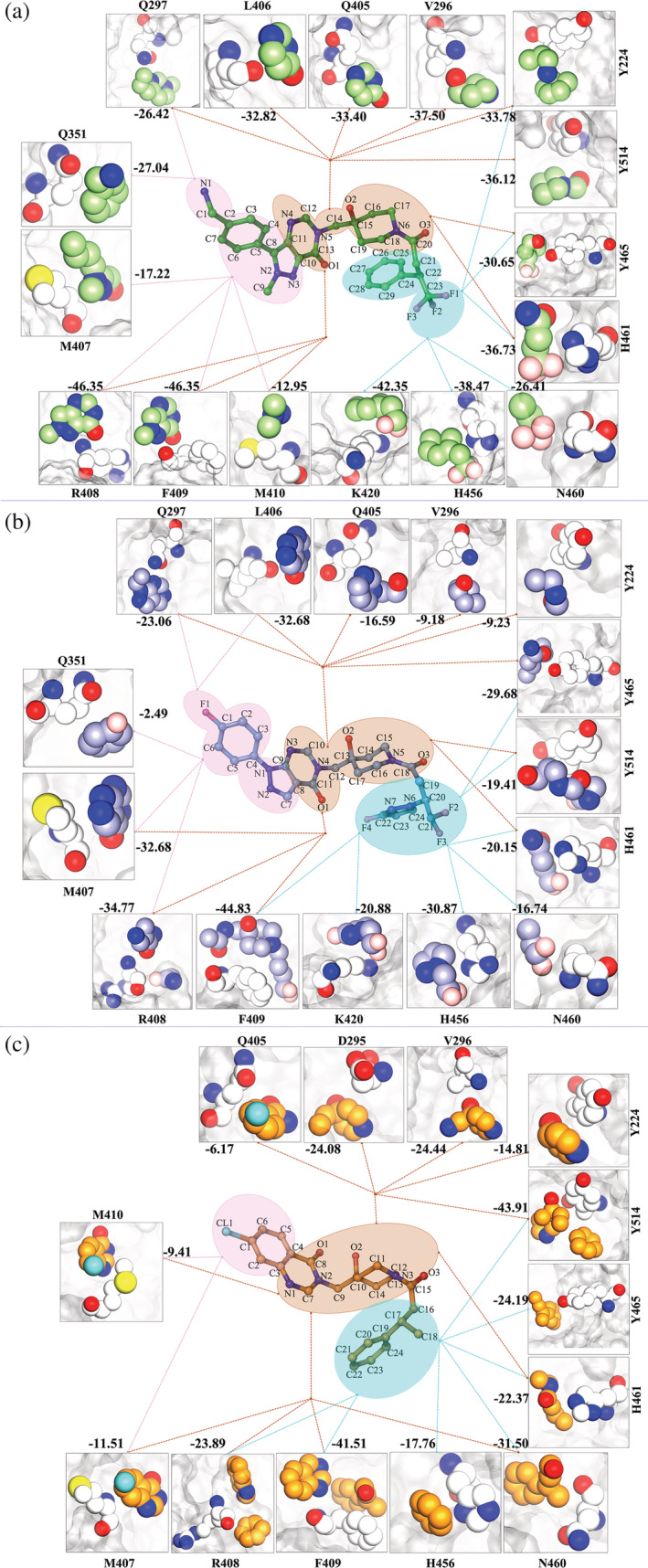
Atom‐wise energetic contributions of the ligand with respect to binding site residues as calculated through linear interaction energy. The residues and ligands are shown in VDW representation; Panel 1: USP78QQ; Panel 2: USP78WK; and Panel 3: USP79HS. The dotted lines represent the interaction from a specific moiety. All values are in kcal/mol. VDW, van der Waals
Overall, comparison of the ligand's head, core, and tail moieties reveals substantial changes in the interaction pattern. The LIE analysis shows that at the head region, the ligand 9HS has the least interaction, while ligands 8QQ and 8WK share five residues (Figure 3a‐c). Furthermore, the ligand 9HS interacts only with M407 and M410, which are also shared by the other two ligands (Figure 3a‐c). The core and tail moiety have majorly similar interactions in all the three ligands, except for D295 interacting with the core moiety of ligand 9HS. Hence, we have understood the differences in the distribution of the key residues around the head, core, and tail moieties in each system, which has demarcated the interaction pattern. Furthermore, we have now quantified them in the context of moiety‐wise energetic contribution.
2.6. LIE proposes atomic‐level modifications
Exploring the system at the atomic level may yield insights into similar‐pattern‐variable‐spectrum. The LIE analysis provided a platform to investigate the interaction energies of individual ligand atoms with residues in the neighborhood of vdW radii. It might provide a clear distinction between the energetic environment of the binding sites in each system. Our results indicate that the residues in USP78QQ receive more energy from the atoms of its ligand as compared to other systems (Figure 3a‐c and Figure S9). We observed that 13 residues (Y224, V296, Q297, Q351, Q405, L406, R408, F409, M410, K420, H456, H461, and Y465), which are common for all the three ligands, have shown dominant interaction energy with ligand 8QQ, while only 2 residues, N460 and Y514, have a high contribution to ligand 9HS (Figure S9). The energy value of the head moiety of 8WK and 9HS is significantly lower when compared with the ligand 8QQ. Head moiety in ligand 8QQ has an overall contribution of −38.09 kcal/mol, while 8WK and 9HS have −17.65 and 0.77 kcal/mol, respectively (Tables S6‐S8). Because of this, several analogous interactions (Q297, Q351, M407, and R408) between 8QQ and 8WK have variable energies (Figure 3a,b and S9). Further comparison of head moieties of ligands 8QQ and 9HS reveals that they are capturing two similar interactions M407 and M410 (Figure 3a), but despite this, the energy containment from the head moiety is entirely different. Residues M407 and M410 have cumulative energies of −30.17 and − 20.92 kcal/mol with ligands 8QQ and 9HS, respectively (Figure 3a,c). The analysis reveals that the head moiety of ligand 8QQ is providing a higher contribution to the similar residues than that of ligand 9HS. Also, the C2 atom of 9HS is providing an unfavorable contribution to the head moiety, which is reducing its total energy (Table S8). Similarly, the ligand 8QQ's C7 atom also provided an unfavorable energetic contribution but was nullified by two polar hydrogens, which have favorable electrostatic energy (Table S6). The H2 and H3 atoms are associated with N1, which facilitates the donor–acceptor interactions and hence this atom is critically important in the pocket as compared to the F1 atom in 8WK and the CL1 atom in 9HS. Furthermore, the presence of both NH2 and water is important for ligand stability, as demonstrated by MD simulation in which the water molecules were neglected, resulting in the instability of the head moiety (Figure S10).
The second region of ligands, the core, reveals a significant energy difference. Cumulatively, atoms of the core in 8QQ possess an energy of −68.29 kcal/mol, which is huge in comparison to the core of ligand 8WK, −19.88 kcal/mol, and 9HS, −37.80 kcal/mol (Tables S6‐S8). Because of this variation, residues in USP78QQ have superseded most interactions of the core (Figure S9). Despite the similar interactions (explained in the above section) with the core, the atoms of 8QQ provide a more favorable contribution to their respective residues than other ligands (Table S6‐S8). It is interesting to observe that despite the missing pyrazolo moiety in 9HS, the core has granted more energy to the residues as compared to the moieties of ligand 8WK. It happened because numerous atoms in ligand 8WK's core, C10, C11, C12, H1, C16, C17, and O3, have granted unfavorable energy penalties to the ligand, but atoms of the core were favorable for 9HS (Tables S7 and S8). Consequently, the residual comparison revealed that three residues Y224, D295, and V296 had received more energy from 9HS and two residues Q405 and M407 from the ligand 8WK, while two key residues R408 and F409 were nearly identical (Figures 3b,c and S9).
The last regions of ligands: tail, unveil the energy spectrum, where the tail of 8QQ contains the highest energy of −48.95 kcal/mol, followed by ligand 8WK at −30.24 kcal/mol and lastly ligand 9HS at −24.07 kcal/mol. In all ligands, the carbon atoms were mostly favorable (Tables S6–S8). However, the most potent ligand, 8QQ, gained additional energy from its F1 and F2 atoms (Tables S6–S8). At the tail, residue K420 was critical to both 8QQ and 8WK; however, there was a significant energy difference (Figure S9). Residues H456, H461, and Y465 that were shared with the core as well have obtained greater energy from the 8QQ atoms (Figure 3a‐c). Residue Y514, which is shared between the core and tail moiety, has superseded the interaction with ligand 9HS (Figure S9), which is also reflected in MM‐PBSA analysis (Figure S5).
Overall, LIE has nearly unraveled the same‐pattern‐variable‐spectrum landscape, which reveals the favorable and unfavorable atoms in respective ligands. Despite the comparable interaction, the mild and weak ligands have been unable to contribute to the interacting residues like the most potent ligand. The energy differences between the ligands are now clearly defined by the amino group (NH2) at the head and fluorine atoms at the tail. The scaffold of the core serves as the energy reservoir for all ligands; yet, despite their striking similarity, the atoms of 8QQ in the core moiety exhibit significant energy differences when compared to the other two ligands. Given that we now have clear evidence of the spectrum of atom‐by‐atom exchange between protein and ligands, the energy variations between the atoms of distinct ligands may account for their variable potencies. Lastly, LIE further adds crucial information for the ligand‐based design approach. It has underlined a key scaffold at the core, an electropositive amino group at the head, which might induce dual interactions, and electronegative atoms at the tail. Although the SAR of these molecules is well established in their respective works, the ligand's comparative study is required to explore the possible factors for increased potency, which is carried out in this study. Hence, energy quantification through LIE at atomic level will give significant direction to computational/medicinal chemist for designing of more potent and selective therapeutics leads from this atomistic approach.
2.7. The free energy landscape portrays USP78QQ as the most converged system
PCA was used on all of the simulated systems to capture the important conformational changes (methodology is mentioned in the supporting information section 2.6). Among all the PCs, the first two PCs of the system were considered significant for recording the largest variance of Cɑ displacements during MD simulations (Figures S11 and S12, supporting results section 1.2).
FEL's objectives were (1) to distinguish between APO and bound systems, and (2) to compare the three bound systems in order to determine the effect of ligands on the landscape of protein binding. To ascertain this, we examined the trajectory for all simulated systems connecting the minima and sub‐conformational spaces along PC1 and PC2 (Figure 4). The basins are numbered as per their appearance w.r.t. time during cMD simulations. The occurrence of multiple energy basins at distant places reflects certain conformational changes; on the other hand, one basin with a smooth and deeper energy basin reflected the dynamic solidity of the conformational state and indicated higher stability of the system. Our results indicated that USP79HS has displayed the most different pattern amongst all systems (Figure 4). USP7APO, USP78QQ, and USP78WK have captured two minima; however, USP79HS has shown three stable states (Figure 4a,e,i,m). Furthermore, minima's conformations indicate that the variations were mainly obtained by the structural determinants of USP7, while other regions are well‐aligned (Figure 4). The comparison of bound systems reveals that the finger sub‐domain has experienced the highest fluctuation in USP78WK followed by USP79HS, while its fluctuation was minimized in USP78QQ (Figure 4f,j,n). A similar trend was observed for BL1 as well (Figure 4f,j,n). The SL was found to be most fluctuating in USP7APO, while it was minimized in bound systems (Figure 4b,f,j,n). Excluding systems USP7APO and USP78QQ, the finger sub‐domain in the other two systems has shown bidirectional movement (Figure 4j,n). The directional shifts from minimaI to minimaII in the USP78WK system are outward to inward, but the opposite was seen in the USP79HS system. (Figure 4j,n). The other bi‐directional movement was observed in BL1 of all systems (Figure 4b,f,j,n). Lastly, the negligible movement was observed for SL in the minimas of respective systems (Figure 4b,f,j,n).
FIGURE 4.
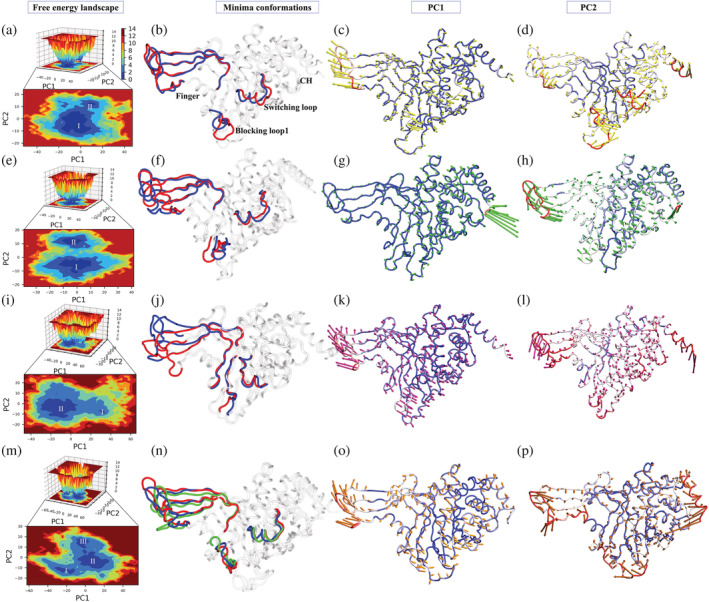
FEL analysis and porcupine plots of different systems in bound and unbound states. The panels (a, e, i, m) of 2D and 3D free energy landscape (FEL) plots between PC1 (x‐axis) and PC2 (y‐axis) for APO, USP78QQ, USP78WK, and USP79HS, respectively. The color bar represents the Gibbs free energies in the plot ranging from the lowest energy (blue) to highest energy (red) conformation states. The low energy minima states are shown in the 2D FEL plots. The minima extracted from FEL plots are superimposed for all simulated structures depicting the conformational changes in the panels (b, f, j, n). The blue, red, and green conformations represent the minima I–III respectively in the panels (b, f, j, n). Porcupine plots were generated using extreme PC1 (panels c, g, k, o) and PC2 projections (panels d, h, l, p) for all the four simulated systems. The direction of the green and red arrows (in PC1 and PC2 respectively) at each Cɑ shows its direction of motion and the length of the arrow depicts its strength. The protein is represented in tube form. The regions that are highly fluctuating are labeled
Furthermore, the porcupine plots of PC1 and PC2 show the highest atomic fluctuations in the above‐mentioned regions as well (Figure 4). The porcupine plot of the PC1 component suggests major fluctuations of the finger sub‐domain in systems USP7APO, USP78WK, and USP79HS showing anti‐correlated motion with the rest of the protein (Figure 4c,g,k,o). Surprisingly, the system USP78QQ shows a pretty different pattern, where finger sub‐domain fluctuations are reduced significantly along PC1 while PC2 captures it (Figure 4g). PC2 has also collected the other variations in the same regions as PC1 in all the systems (Figure 4d,h,l,p). The directionality of atomistic motions of USP7APO, USP78WK, and USP79HS varies when PC1 and PC2 are compared; nonetheless, the motions remain anti‐correlated for USP78WK, USP79HS, and USP78QQ (Figure 4d,h,l,p). Moreover, at the deepest minima state, ligand 8QQ attains the most favorable electrostatic complementarity in the binding pocket as compared to other ligands, 8WK and 9HS (Figure S13).
Overall, USP78QQ has displayed different behavior when compared with other systems. The major fluctuating regions (Finger, BL1, and SL) that are identified through PCA hold deep importance in the structure–function of USP7. The binding of 8QQ minimizes the atomistic fluctuations of the finger sub‐domain, which is not achieved in other systems, reflecting its tight packing and rigidifying the protein's internal wiring. A similar effect was observed in the RMSF profiles where the fluctuations at the finger sub‐domain were minimized, specifically in USP78QQ (Figure 2g). It is also evident from the analysis that the atomistic motions in USP78QQ, USP78WK, and USP79HS are suppressed, smooth, and not smooth, respectively. As a result of PCA and FEL analyses, the strong binding pattern of ligand 8QQ, whose effect is observed on the protein, has been highlighted, enlightening the spectrum of varying binding potency. It also unfolds similar‐pattern‐variable‐spectrum as, despite the similar binding pattern, the capability to minimize the atomistic fluctuation varies, which could be related to their binding potency.
2.8. PMFs and force profiles along the unbinding reaction coordinates
The SMD simulation was implemented to embrace the outcomes of cMD simulations. While both the approaches are unrelated, the unbinding events may actually highlight the probable cause of the variable potencies of ligands. The SMD simulation has been used to rank the inhibitors and quantitatively discriminate binders from non‐binders. 41 In our instance, all ligands were good binders, so this approach might also be used to analyze variability. Also, as per Tiwary et al., 44 drug efficacy is determined not only by binding affinity but also by its activity during unbinding, which may highlight rate‐limiting steps, crucial residues attempting to hold the ligands, and structural determinants/critical components of the protein involved during dissociation.
The analysis reveals that the PMF of ligand 8QQ is higher than the other two ligands (Figure 5a). Also, SMD simulations have provided encouraging results as the PMF profiles of the ligands are correlating well with the experimental IC50 values. The PMF profiles show a significant variation from the most potent (8QQ) to the least potent (9HS) ligands, indicating varied binding patterns which are responsible for ligand 9HS to dissociate early as compared to the other two potent ligands (Figure 5a). Since the binding potency of ligand 8WK (52 nM) is comparatively less than that of ligand 8QQ (22 nM), the same is reflected in the PMF profiles (Figure 5a). The force profiles suggest that ligands 8QQ and 8WK have attained the highest rupture force as compared to the least potent ligands (Figure 5b). The ligand 9HS begins to dissociate from the binding pocket at an early stage (0.5 ns), whereas ligands 8QQ and 8WK begin at 1.3 ns (Figure 5b). Following the first force peak, all three systems have minor peaks, showing that certain residues try to hold ligands during dissociation. (Figure 5b). We observed a distinguished HB pattern among the systems (Figure 5c). We observed the highest number of HBs for ligand 8QQ, followed by 8WK and 9HS (Figure 5c).
FIGURE 5.
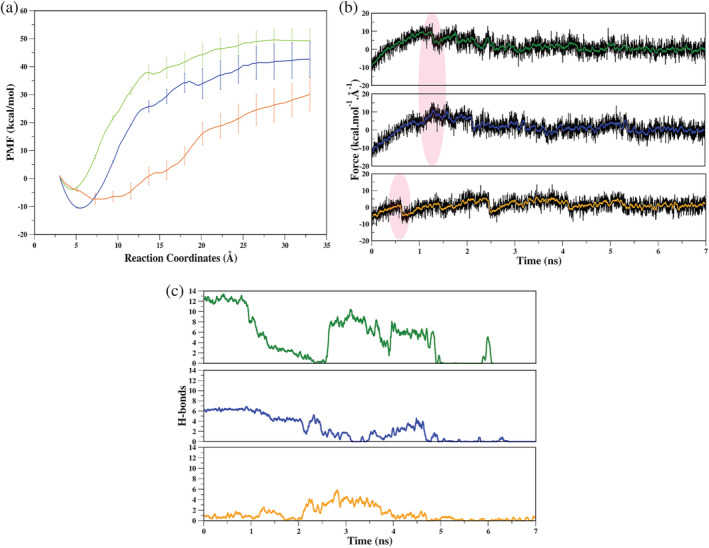
Quantitative analysis of SMD simulations. (a) Potential mean force (PMF) profiles with respective error bars of each ligand, (b) average force profile as a function of time for each system, (c) the number of HBs during dissociation. In all three plots, the color coding is as follows: green: USP78QQ; blue: USP78WK and orange: USP79HS. HBs, hydrogen bonds; SMD, steered molecular dynamics
Based on the quantitative estimation, it is apparent that the ligands have dissociated from the pocket in the order of their experimental binding potencies, which is an encouraging result and helps us establish the benchmark for distinguishing the weak and strong binders. These results support experimental results but do not clarify the ligand potency spectrum. We did notice a distinct pattern among ligands, which may be due to exit pathways or critical structural determinants. Hence, thermodynamically stable protein‐ligand interactions were quantified using SMD trajectories.
2.9. Analysis of the ligand's exit pathways
We observed that each ligand in the individual systems had chosen its own path (each distinct from the others), resulting in three separate exit routes from a common CV (Figure 6). The sampling of ligand representatives at each state confirms different exit routes (Figure 6a,d,g), and the last representative has been found fully solvated (Figure 6b,e,h). The dissociation via SMD simulation analysis claims three different paths from the binding site, path1 via BL1 and BL2, path2 between BL1 and SL, and path3 via SL (Figure 6c,f,i). The structural relevance of these determinants has been addressed in the preceding sections, and hence, ligands with varied determinants may provide substantial insights. It will be interesting to focus on path1 and path3 as both the most potent and least potent ligands have drawn clear demarcation between their exit routes by occupying two different critical determinants of USP7.
FIGURE 6.
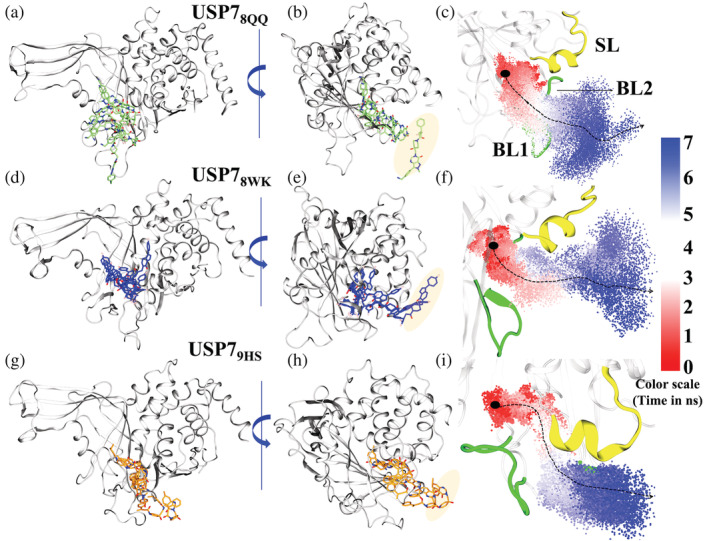
Ligand unbinding pathways. (a, d, g) The 8QQ, 8WK and, 9HS ligand states at every stage (0–7) of unbinding process. Ligands 8QQ, 8WK, and 9HS are rendered in Licorice and colored in lime, blue, and orange respectively while protein is rendered in New Cartoon and colored in white. (b, e, h) The rotated view of the protein which shows the ligand in complete solvent state. The coloring and representation are the same as previous notations. The shading over ligands represents its respective final solvated state. (c, f, i) The complete path of ligand exit from the pocket. Three main structural components that are critical for protein activity are highlighted, blocking loop1 (BL1), blocking loop2 (BL2) and switching loop (SL). BL1 and BL2 are colored in green and SL is colored in yellow. Red: white: blue shading are the transitions of ligands which represent the starting: intermediate: pre‐solvated states. The dotted arrow shows the direction preferred by ligands during exit
2.10. Energetically relevant states reveal new interactors
The dissociation pathways of ligands were found to be different from each other. Since the parameters (CV, force, and velocity) were the same for all the SMD simulated systems, their tendency to be drawn toward specific determinants makes them interesting to explore. We divided the unbinding process of ligands into three states: the starting state (S), a semi‐bound intermediate state (I) in which the ligand has exited the native pocket but is still forming interactions, and pre‐solvated state (P) which represents the last contact with protein. Collectively, these states further will be denoted as S‐I‐P, which are represented for each system (Figure 7a,e,f). The SMD trajectories of each system were subjected to MM‐PBSA, and further, the per‐residue decomposition energy cut‐off −0.5 kcal/mol was used to quantify the contributing residues.
FIGURE 7.
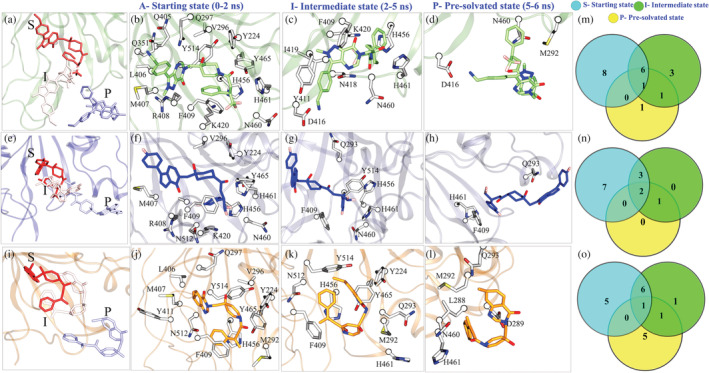
Ligand's exit highlights the energy‐relevant states of USP78QQ (a‐d), USP78WK (e‐h), and USP79HS (i‐l). Venn diagrams (m‐o) represent the common and unique number of interactions for each system respectively
2.10.1. Initial contacts
The initial residue contacts from t = 0‐2 ns of simulated systems reveal differences in each system with their crystallographic interaction patterns. After comparison of USP78QQ with two other systems, we find that after the force is exerted, the ligand 8QQ forms an interaction with 14 residues (Figure 7b), followed by ligands 8WK (Figure 7f) and 9HS (Figure 7j), each attaining 11 residues. Out of 14 residues in USP78QQ, 10 residues: Y224, V296, M407, R408, F409, K420, H456, N460, H461, and Y465 are common with USP78WK, and 9 residues: Y224, V296, Q297, L406. M407, F409, H456, Y465, and Y514 are common with USP79HS (Figure 7c,g,k,m). This pattern, with a loss of four residues each in 8WK and 9HS ligands, is possibly because they have started to dissociate early when compared with ligand 8QQ. Furthermore, in the comparison with the energy pattern of cMD simulations, most of the residues in USP78QQ were retained; nevertheless, it lost three residues, Q405, M410, and N422, which were certainly minor contributors (Figure S5) and lose their association once the ligand starts dissociating. USP78WK loses four residues Q297, Q351, L406, and Y514 and similarly USP79HS, R408, M410, K420, and N460 when compared with their cMD simulation pattern (Figure S5B,C). It is also observed that residue N512 is gained by both systems, USP78WK and USP79HS, and another residue M292 is also gained by USP79HS (Figure 7f,j). The S‐state interactions show that in the USP78QQ system, the majority of residues try to hold the ligand despite the force exerted. However, this was not similar for the other two systems, which lost native pocket at an early stage. Also, the appearance of residues M292 and N512 reveals different paths for ligand 8WK and 9HS from 8QQ.
2.10.2. Intermediate contacts
At I‐state from t = 2‐5 ns, four new residues Y411, D416, N418, and I419 were found to hold ligand 8QQ; however, only one new residue Q293 was observed for ligand 8WK and 9HS (Figure 7c,g,k). In the semi‐bound state, ligand 8QQ forms additional contacts while maintaining earlier interactions with residues F409, K420, H456, N460, and H461 (Figure 7c,m). 8WK also preserves five interactions with residues F409, H456, N460, H461, and Y514 (Figure 7g), but ligand 9HS has been able to hold the majority of previous state interactions (Y224, M292, F409, H456, H461, Y465, and Y514), which reflects that 9HS undergoes minimum change (Figure 7c,g,k,n). For system USP78QQ, residues D416, N418, and I419 are located on BL1, and for systems USP78WK and USP79HS, the occurrence of new residues M292 and Q293 is on SL. The interaction pattern during the exit confirms the exit paths of each ligand previously mentioned in the above section (Figure 6). We know the structural determinants occupied by the respective ligands from the exit path, and now we have been able to connect the relevance of ligand interaction with their new residues from the I‐state. It is also worth noting that the cMD hotspots are also strong contributors in SMD because residues Y224, F409, K420, H456, N460, H461, Y465, and Y514 attempt to hold ligands until 5 ns, implying their importance for ligand binding and affinity.
2.10.3. Pre‐solvated contacts
In the P‐state, ligand 8QQ interacts with M292 for a short time (described later), whereas ligand 8WK shows no change. However, two new residues, L288 and Q289, were found associated with ligand 9HS (Figure 7d,h,l,o). As we previously stated, ligand 8QQ solely interacts with BL1. Though the P‐state has revealed that ligand 8QQ has a modest connection with SL as well, the predominant interaction is still with BL1 (Figure 7). Because of the large size of ligand 8QQ and the protrusion of residue M292 toward the solvent‐exposed area during egress, an atomic contact occurred, resulting in M292 occurring before solvation. Based on the minimal interaction with one residue belonging to SL, it will be too naive to comment on the intensity of 8QQ interaction with SL. Alike cMD, a similar interaction pattern was observed in SMD simulations as well, as many residues were shared between ligands during their exit. Hence, to differentiate this spectrum, we have estimated the sustainability of these residues stage‐wise to understand their overall contribution to the respective ligands.
2.11. Sustained contacts
The quantification of the above pattern reveals that most of the native pocket residues have established their contact till 2 ns. However, our focus was to elucidate residues that have shown interaction at intermediate and pre‐solvated states to identify new interactors. From the heatmap analysis of stage‐wise residue contributions, we found the sustained contacts for each ligand during the SMD simulations (Figure 8a‐c). Sustained contacts, that is, residues that are retained with the dissociating ligand for the longer duration of the simulation. Not only duration, but we have also considered the cumulative energy of each residue that they have contributed to the dissociating ligands (Figure 8d,e). According to the findings, ligands 8QQ, 8WK, and 9HS had sustained interactions of nine (R408, F409, D416, N418, I419, K420, H456, N460, and H461), five (Q293, F409, H456, H461, and Y514), and seven (Y224, M292, F409, H456, H461, Y465, and Y514) residues, respectively, that were coupled for at least 4 ns in the initial or later states (Figure 8a‐c). Among them, residues F409, H456, and H461 were shared between all systems. Further, we have used −4.0 kcal/mol as the cut‐off to identify residues contributing cumulatively. The high threshold was chosen because we are now considering the cumulative energies of each residue rather than their individual energies. The USP78QQ system has nine residues that have crossed the threshold, followed by USP79HS with six residues and USP78WK with five residues (Figure 8d,f). Despite the similar interactors, there is a huge difference in the energy spectrum, for example, residue F409 is trying to hold ligands in each system. However, in system USP78QQ its cumulative contribution is −17.00 and around −12.00 kcal/mol in the other two systems. Furthermore, the HBs pattern revealed that only system USP78QQ has been able to form HBs with residue D416 in the P state, but no HBs are observed for the other two systems (Figure S14a‐c). Also, the HBs in USP78WK were found till 5 ns; however in the USP79HS had HBs till 4 ns (Figure S14b,c). From this analysis, we observed that USP79HS has the weakest interaction in its S state, whereas the USP78QQ and USP78WK have significant associations in the same time period, which might be the possible cause of 9HS's early dissociation (Figure S14a‐c).
FIGURE 8.
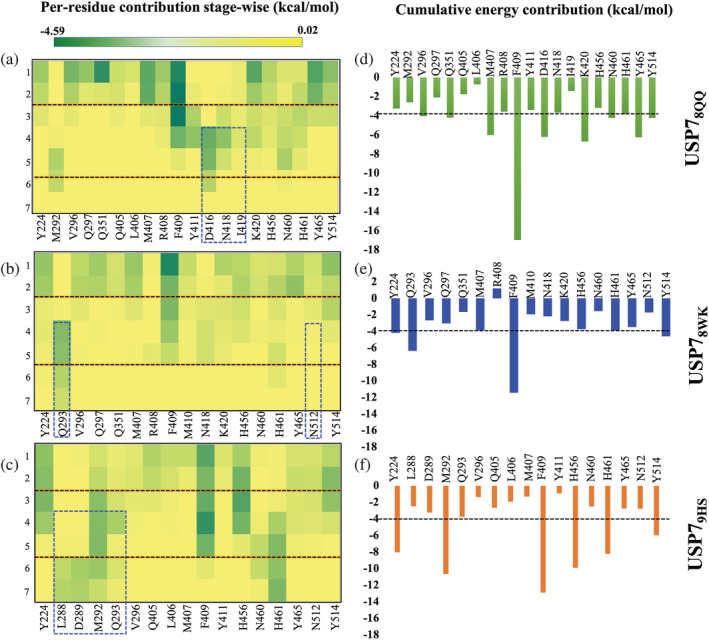
Heatmaps of the dissociation profiles of all three systems. (a‐c) Per‐residue energy contribution in each system USP78QQ, USP78WK, and USP79HS, respectively, was elucidated and is shown in the form of heatmap. The key residues at every stage were identified based on their energetic contribution toward the ligand (cut off −0.5 kcal/mol). (d‐f) The bar graph shows the cumulative energy of residues in respective systems throughout the dissociation time‐line as obtained from the heatmap. The residues highlighted in the box are unique interactors as identified by SMD simulation. SMD, steered molecular dynamics
2.12. Conformational changes demarcate the importance of structural determinants
We have now elucidated the unbinding pathways and the involvement of structural determinants during the dissociation process. It is now clear that BL1 is critical for ligand 8QQ, while SL is key for ligand 9HS, along with a modest impact from SL and BL2 for ligand 8WK. Finally, we attempted to observe conformational changes in BL1, BL2, and SL during unbinding and compared them to those observed in cMD simulations. As previously stated, these determinants are crucial for USP7 activity since they work in the following chronological order: (1) Attachment of ubiquitin (Ubq) leads to USP7 activation, 22 (2) to accommodate Ubq, BL1 should shift away from the Ubq binding site (bound allosteric site of inhibitors) 23 as found in its APO structure, (3) After accommodating Ubq, it must extend its C‐term tail toward the SL, causing a conformational shift 21 and allowing USP7 to enter into a transition from an inactive to an active state. From the chronology, it can be perceived that BL1 is the primary component of USP7 and SL is the secondary component, both of which are essential for its functionality. BL1 is Ubq's first facilitator; its tendency to shift away allows Ubq to sit and stretch its tail toward the catalytic core. If BL1's inherent nature is constrained, Ubq may not extend its tail to the catalytic center, altering determinant SL's conformation and activating USP7. Hence, in the context of Ubq binding, BL1 appears to be a more important determinant than SL.
Consequently, we are now using this information to explore the conformational changes induced during binding and unbinding events, which might shed light on the cause of varying potencies in ligands. The sampling of BL1 during cMD reveals that it is highly fluctuating in USP7APO, which relates well with the experimental statement 23 (Figure 9a.1). Further, the binding of Ubq reduced the flexibility of BL1, as did the binding of two other ligands, 8QQ and 8WK. However, the binding of 9HS had no significant impact on BL1, and instead increased its flexibility (Figure 9a.1‐4). The structural appearance in USP7Ubq, USP78QQ, and USP78WK highlights that BL1 has a stable β‐sheet architecture throughout cMD simulations, while USP79HS follows the trend of USP7APO (Figure 9a, right panel). This implies that potent inhibitors have not only reduced BL1's flexibility but have also preserved its secondary structure, whereas the least potent inhibitor did not reduce. Further, it was found that the BL2 conformations were minimized in all systems when compared with USP7APO (Figure 9b.1‐4). The attachment of Ubq has conformationally induced β‐sheet structure in BL2 (Figure 9b, right panel); however, in other systems it is in the form of loops, and despite this, the inhibitors have been able to restrict its flexibility (Figure 9b.1‐4). Further, we observed SL dynamicity during cMD in all systems (Figure 9c.1‐4). USP7APO state highlights SL in the form of a loop completely; however, upon the binding of Ubq, certain portions of this loop are converted into five residue helices, which justifies the conformational change in SL. Our results confirm that these conformations are further maintained during MD simulations (Figure 9, right panel). However, notable differences are observed in ligand‐bound cases, where these helices are more stabilized than USP7APO and even USP7Ubq (Figure 9c.1‐4). The deviation of SL is rigorously minimized by 8QQ and 8WK, since their deviation is below USP7APO, while the deviation of the least potent system, USP79HS, exceeds USP7APO RMSD (Figure 9c.2‐4). Based on these observations, we tried to undermine an explanation for the reduced flexibility of BL1 and SL in the cases of 8QQ and 8WK but not in 9HS. We found that residues V296 and Q297 act as anchor points for SL, which are protruding toward the binding pocket. Similarly, residues R408, F409, and K420 are acting as an anchor for BL1, now if we recall our MM‐PBSA and LIE results, these residues have shown high contribution for USP78QQ mainly (Figures 3, S5, S7 and S8). The most potent ligand, 8QQ, has attained a strong contribution, which has seized the internal wiring of these determinants and thus might be a justifiable cause for their high potency. The conformational change analysis has supported us well in understanding similar‐pattern‐variable‐spectrum landscapes. Moreover, conformational sampling of these determinants during SMD simulations has supplemented the outcomes of cMD simulations. In SMD simulations, we saw the opposite RMSD trend; since the system is under constant strain, contacts must be constantly forming and breaking, leading to the fluctuating RMSD for the ligands and determinants. Also, the determinants that are not captured by the respective ligands may have stable RMSD. As per SMD results, BL1 has shown high divergence in USP78QQ, followed by USP78WK, and least in USP79HS (Figure 9d). The sampling of BL1 also suggests more structural divergence from starting to solvate state in USP78QQ as compared to the other two systems (Figure 9d, right panel). For BL2, considerable divergence was attained in USP78QQ and USP79HS (Figure 9e), although the structural divergence of each system did not vary much (Figure 9e, right panel). The most distinguished results were obtained for SL, as the highest divergence was achieved in USP79HS, while for the other two systems it was almost similar (Figure 9f). SMD results confirm the exit paths of each system as BL1 was highly affected in USP78QQ and SL was mostly affected in USP79HS. In the context of USP7 activity, the exit pathways and preferred structural determinants by each ligand are now extremely important to elucidate the variable spectrum of ligands.
FIGURE 9.
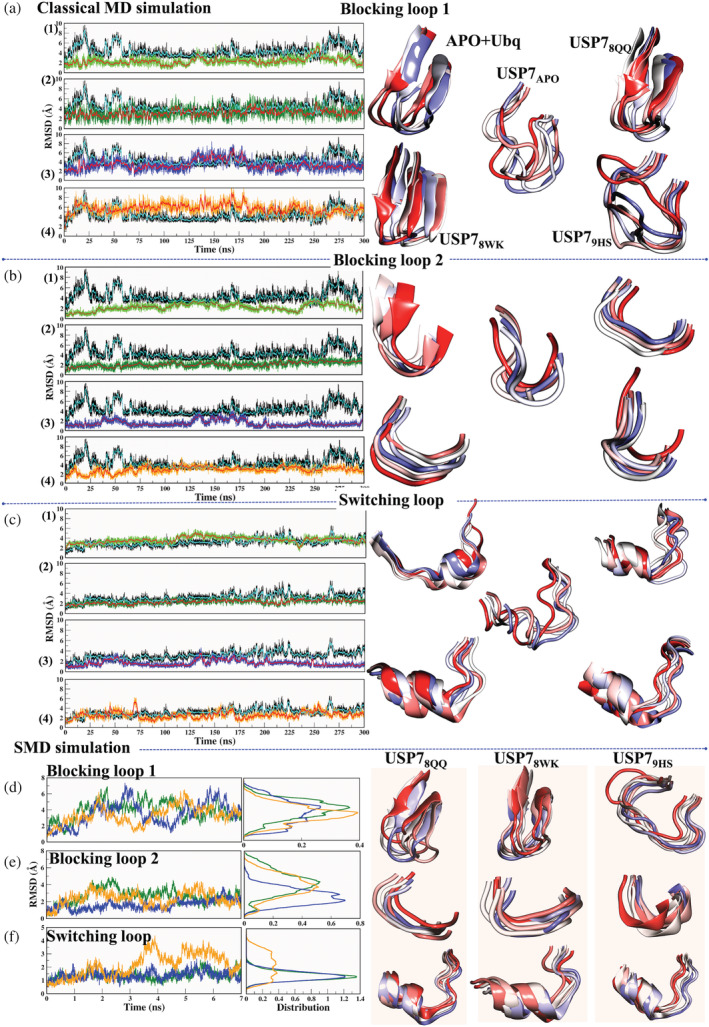
The time‐versus‐RMSD plot for (a,d) Blocking loop 1, (b,e) Blocking loop 2, and (c,f) switching loop during classical MD and SMD simulations, respectively. The panels within a, b, and c correspond to system APO‐versus‐(1) APO + UBQ, (2) USP78QQ, (3) USP78WK, and (4) USP79HS, respectively. The panels (d‐f) are for the systems USP78QQ (green), USP78WK (blue), and USP79HS (orange). The sampling of the loops during simulations (right panel) is represented in New Cartoon with the time‐step color method red (0 ns) to blue (300 ns) for classical MD simulation and red (0 ns) to blue (7 ns) for SMD simulation at equal time intervals. MD, molecular dynamics; RMSD, root mean square deviation; SMD, steered molecular dynamics
Finally, recalling the chronology of USP7 activation, the primary component is occupied by the most potent ligand, 8QQ, and according to SMD simulation, ligand 8QQ is attempting to maintain interaction with BL1 during dissociation, which may prevent Ubq from performing its biological activity. Ligand 9HS occupies secondary component SL but leaves the native allosteric pocket early, which may allow Ubq to extend the tail into the binding site. As a result, it can be perceived that the ligand 8QQ has a high potency because it occupies the primary component, whereas SL has a lower potency because it occupies the secondary component.
3. CONCLUSIONS
USP7 is a clinically validated target in the oncology field. Medicinal chemistry efforts aimed at identifying the inhibitors against USP7 have proved fruitful in the past 4–5 years, which are yet to be explored as none of them entered into clinical trials. In this regard, one of the challenges is to address the ligands that chemically have a common core scaffold, similar binding mode and interaction fingerprinting yet possess variable experimental potencies. It is fascinating to explore at the atomic level and to find out the pivotal determinants responsible for enhanced binding potency.
Herein, a detailed atomistic description was carried out using three molecules: 8QQ, 8WK, and 9HS from the reported co‐crystals, using a multifaceted technique. Starting from post‐processing cMD analysis that revealed USP78QQ is the most stable system as it significantly reduces the flexibility of key regions (finger sub‐domain and BL1). Further, the thermodynamics analysis justified the stability of USP78QQ as the free energy of this system was the highest and also the identified energy spectrum of other systems corroborates well with reported experimental values. LIE was also carried out to differentiate the energy spectrum of these ligands' atom‐wise, and it showed that the chemical moiety of 8QQ is more energetically favorable to establish the stable adduct than others. Understanding molecular interactions at an atomic level provides immense possibilities to enhance the structure–activity relationship (SAR). In this regard, the atomic exchange between binding site residues and functional groups of ligands and their feasibility toward betterment of binding affinity are key for drug designing. The FEL analysis revealed that the USP78QQ has the deepest minimas and also the atomistic fluctuations were significantly reduced compared to other systems, which justifies the tight packing of ligand 8QQ. We further performed the drug dissociation characterization using SMD to undermine most aspects of the binding pocket. It is well known that not only the binding affinity but also the average longevity of the protein–ligand complex is a crucial factor for drug efficacy; thus, the dissociation study seems crucial to elucidate the mechanistic behavior of ligands, and SMD has been demonstrated to be a useful technique in the field of structure‐based drug design. The SMD simulations provide two crucial pieces of information: (i) a force (or PMF) profile that can be associated qualitatively and possibly quantitatively with protein–ligand binding affinities; and (ii) a precise atomistic description of a ligand's unbinding mechanism. The SMD analysis revealed that PMF was the highest in the most potent system, USP78QQ, and thus correlates well with the experimental IC50 values. Further, two different exit paths of ligands from the binding site were identified, first via blocking loops (BL1 and BL2) and second via switching loop (SL). We identified that these loops play a crucial role during unbinding as they hold the exit of drugs by making durable interactions. As per the activation mechanism, these determinants are important for USP7's functional activity as they guide Ubq attachment to the misaligned catalytic triad. From co‐crystal analysis, we find for the Ubq attachment, the BL1 and SL appear as primary and secondary components. As a result, the greater affinity for 8QQ could be related to the fact that it occupies the primary component (exit via BL1), preventing Ubq from extending its tail toward the catalytic center, whereas 9HS exits the binding pocket early (exit via SL), leaving a vacancy for Ubq to fill in. Furthermore, the dissociation analysis highlighted new and critical residues Q293, H294, D416, and N418 located on these determinants, which were not previously reported. These residues are found to be responsible for holding these ligands during their exit. Overall, this atomistic approach provides molecular insights that are helpful for understanding the link between the varying potencies and their mechanisms of action. Such insights are essential for modifying the ligands for improved efficacy.
4. MATERIAL AND METHODS
The methodological in‐silico workflow is represented in Scheme 1 (Supporting Information).
4.1. System preparation
The protein structures of Apo and co‐crystal of USP7 were retrieved from the RCSB (Research Collaboratory for Structural Bioinformatics) protein data bank (PDB‐ID: 4M5W [USP7APO]: resolution 2.2 Å), 50 (PDB‐ID: 5N9T [USP78QQ]: resolution 1.7 Å), 25 (PDB‐ID: 5NGE [USP78WK]: resolution 2.3 Å), 22 and (PDB‐ID: 5VSK [USP79HS]: resolution 3.3 Å). 51 The inhibitor‐bound systems are collectively denoted as COMs in the study. We have chosen only these inhibitors' bound states because they reflect varied binding potency, as mentioned earlier (Introduction sub‐section). Furthermore, the rationale for selecting these inhibitor‐bound systems was the sharing of similar chemical scaffolds, moieties, and interaction maps (>80% of these inhibitors share the same residues). We have also included Ubq bound crystals (PDB ID: 5JTJ [USP7Ubq]: resolution 3.3 Å) 52 for understanding the structural changes induced upon Ubq binding as well as comparing them with the ligand‐bound crystals. The considered crystal structures have a well‐defined catalytic domain distinguished by their finger, palm, and thumb subdomains. However, it was observed that crystals USP78WK and USP79HS have missing loops at BL1 and BL2. USP78WK has a break at BL2 (D459‐H501), while USP79HS has missing residues at BL1 (M410‐N418) and BL2 (G500‐H509). The blocking loops are essential components of USP7 and hence, to maintain structural integrity during dynamics, the loop breaks were interpolated using the PLOP algorithm. 24 All systems were prepared using the Protein Preparation Wizard module of Maestro (Schrödinger Release 2020‐1). 53 , 54 The hydrogen and bond orders were added using PRIME. The hydrogen bond (HB) optimization and restrained minimization were also done for the systems using the OPLS3 force field model. 55 , 56 The Maestro and VMD software were used for plotting interaction maps.
4.2. Atomistic molecular dynamics simulation
4.2.1. Classical MD simulation
Proteins are dynamic entities that populate conformational ensembles, and most functions of proteins depend on their dynamic character; hence, detailed cMD simulations were conducted to observe the dynamics of ligands and their impact on respective systems. Systems USP7APO, USP78QQ, USP78WK, and USP79HS were subjected to a 300 ns simulation; however, two additional trajectories from the same seed, each of 300 ns, were simulated for each co‐crystal system to ensure reproducible and convincing results. The details of simulated systems are mentioned in Table S1. The simulations of all systems were conducted using the AMBER16 package. 57 , 58 , 59 The complete MD simulation protocol is described in Supporting information section 2.1. The post‐processing and clustering of trajectories were performed using the TCL script of VMD 60 (see supporting information section 2.2).
4.2.2. Steered MD simulation
The unbinding of ligands from proteins 41 , 61 was facilitated using a time‐dependent external force. The transition between the bound and unbound states is achieved by adding the standard Hamiltonian harmonic time‐dependent potential acting on a descriptor. The descriptor in our work is Cα atom of residue and an index atom of ligand. In this transition process, the exerted force and external work can be calculated. 41 , 61 SMD simulations were performed using the AMBER package 62 , 63 and following the complete ASMD tutorial 26 (http://ambermd.org/tutorials/advanced/tutorial26/). A constant pulling velocity of 0.02 Å/ps was used for SMD studies. According to the stiff spring approximation, 64 , 65 a sufficiently stiff spring constant of 10 kcal mol−1·A−2 was used. For each system, equil.rst of cMD simulation was used. The systems were set up for seven stages, and at each stage, 40 trajectories of 1 ns were simulated. Overall, for one system 280 ns of SMD simulation was performed. We chose residue Y514 as a reaction coordinate for SMD simulations because it is critical for ligand binding, acts as a gate‐keeper for ubiquitin binding, and undergoes conformational change. 24 It also forms strong energetic interactions. After completing the first stage, a python script, ASMD.py was executed to determine which of the ASMD simulations' work values are the closest to the Jarzynski Average. 66 The python script is available on the tutorial webpage. Hence, the trajectory closest to the Jarzynski average was considered to run for the next subsequent stage of SMD simulations. The same steps are repeated until seven stages. The transition of the complete unbinding process was measured by calculating force and constructing the PMF profiles (see supporting information sections 2.3 and 2.4). The trajectories were saved every 20 ps, and the calculated force and work were saved for every stage. All of these AMBER‐based SMD simulations were performed using the 40 processors and GTX 1080 GPU card, which took 24 hr to complete one stage comprising 40 trajectories each of 1 ns.
4.3. Linear interaction energy analysis
The LIE was employed to calculate the binding free energy from the change in electrostatic (ele) and van der Waals (vdW) interaction energies between the ligand and its environment upon complex formation. 67 , 68 In this study, we have calculated the atom‐wise energy of all the ligands. Since the architecture of these three ligands is almost similar, we were keen to understand the contribution of each moiety of different ligands toward the binding pocket. This may also shed light on variable ligand potencies and suggest crucial atoms for drug design.
For LIE analysis, we picked the lowest energy cluster of each system. The atom‐wise energies are determined by single‐point calculations with the sander package. Ligands 8QQ, 8WK, and 9HS have 41, 38, and 31 atoms, respectively. Every atom was considered an “index” for calculation. The non‐bonded interactions were computed between atoms of ligands and residues of protein at each frame of the lowest energy cluster.
The binding free energy is estimated by combining differences in the electrostatic and van der Waals interaction energies in a linear equation with the coefficients α and β and possibly a constant term γ 69
| (1) |
AUTHOR CONTRIBUTIONS
Mitul Srivastava: Conceptualization (equal); data curation (equal); formal analysis (lead); methodology (lead); validation (equal); visualization (equal); writing – original draft (lead); writing – review and editing (equal). Lovika Mittal: Data curation (supporting); formal analysis (supporting); methodology (supporting); validation (supporting); visualization (supporting); writing – original draft (supporting); writing – review and editing (supporting). Anita Kumari: Formal analysis (supporting); writing – original draft (supporting). Ashish Kumar Agrahari: Formal analysis (supporting). Mrityunjay Singh: Formal analysis (supporting); investigation (supporting); methodology (supporting); validation (supporting). Rajani Mathur: Supervision (supporting). Shailendra Asthana: Conceptualization (lead); formal analysis (equal); funding acquisition (lead); investigation (equal); project administration (lead); resources (lead); software (lead); supervision (lead); writing – original draft (equal); writing – review and editing (equal).
CONFLICTS OF INTEREST
The authors declare that they do not have conflict of interest.
Supporting information
Appendix S1 Supporting Information.
ACKNOWLEDGMENTS
The authors acknowledge the anonymous reviewers for valuable feedback and improving the content of our article. The authors would like to acknowledge DBT, THSTI, and DPSRU for providing support, research opportunities, and computational facilities.
Srivastava M, Mittal L, Kumari A, Agrahari AK, Singh M, Mathur R, et al. Characterizing (un)binding mechanism of USP7 inhibitors to unravel the cause of enhanced binding potencies at allosteric checkpoint. Protein Science. 2022;31(9):e4398. 10.1002/pro.4398
Review Editor: John Kuriyan
REFERENCES
- 1. Huang X, Dixit VM. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016;26:484–498. 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng Q, Huang T, Zhang L, et al. Dysregulation of ubiquitin‐proteasome system in neurodegenerative diseases. Front Aging Neurosci. 2016;8:303. 10.3389/fnagi.2016.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: New therapeutic opportunities. Cell Biochem Biophys. 2011;60:61–68. [DOI] [PubMed] [Google Scholar]
- 4. Song MS, Salmena L, Carracedo A, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP‐PML network. Nature. 2008;455:813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu H‐T, Kuo Y‐C, Hung J‐J, et al. K63‐polyubiquitinated HAUSP deubiquitinates HIF‐1α and dictates H3K56 acetylation promoting hypoxia‐induced tumour progression. Nat Commun. 2016;7:13644. 10.1038/ncomms13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tavana O, Li D, Dai C, et al. HAUSP deubiquitinates and stabilizes N‐Myc in neuroblastoma. Nat Med. 2016;22:1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Q, Ma S, Song N, et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J Clin Invest. 2016;126:2205–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou Z, Yao X, Li S, et al. Deubiquitination of Ci/Gli by Usp7/HAUSP regulates hedgehog signaling. Dev Cell. 2015;34:58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anon. Forkhead FOXO transcription factors in development and disease. current topics in developmental biology. 2018; Available from: 10.1016/s0070-2153(18)x0002-7 [DOI] [PubMed]
- 10. van Loosdregt J, Fleskens V, Fu J, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg‐cell‐suppressive capacity. Immunity. 2013;39:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall JA, Tabata M, Rodgers JT, Puigserver P. USP7 attenuates hepatic gluconeogenesis through modulation of FoxO1 gene promoter occupancy. Mol Endocrinol. 2014;28:912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meredith M, Orr A, Everett R. Herpes simplex virus type 1 immediate‐early protein Vmw110 binds strongly and specifically to a 135‐kDa cellular protein. Virology. 1994;200:457–469. [DOI] [PubMed] [Google Scholar]
- 13. Holowaty MN, Zeghouf M, Wu H, et al. Protein profiling with Epstein‐Barr nuclear antigen‐1 reveals an interaction with the herpesvirus‐associated ubiquitin‐specific protease HAUSP/USP7. J Biol Chem. 2003;278:29987–29994. [DOI] [PubMed] [Google Scholar]
- 14. Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. [DOI] [PubMed] [Google Scholar]
- 15. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma P, Yang X, Kong Q, et al. The ubiquitin ligase RNF220 enhances canonical Wnt signaling through USP7‐mediated deubiquitination of β‐catenin. Mol Cell Biol. 2014;34:4355–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma J, Martin JD, Xue Y, et al. C‐terminal region of USP7/HAUSP is critical for deubiquitination activity and contains a second mdm2/p53 binding site. Arch Biochem Biophys. 2010;503:207–212. [DOI] [PubMed] [Google Scholar]
- 19. Faesen AC, Dirac AMG, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. Mechanism of USP7/HAUSP activation by its C‐terminal ubiquitin‐like domain and allosteric regulation by GMP‐synthetase. Mol Cell. 2011;44:147–159. [DOI] [PubMed] [Google Scholar]
- 20. Holowaty MN, Sheng Y, Nguyen T, Arrowsmith C, Frappier L. Protein interaction domains of the ubiquitin‐specific protease, USP7/HAUSP. J Biol Chem. 2003;278:47753–47761. [DOI] [PubMed] [Google Scholar]
- 21. Hu M, Li P, Li M, et al. Crystal structure of a UBP‐family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. [DOI] [PubMed] [Google Scholar]
- 22. Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small‐molecule inhibitors. Nature. 2017;550:481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Molland K, Zhou Q, Mesecar AD. A 2.2 Å resolution structure of the USP7 catalytic domain in a new space group elaborates upon structural rearrangements resulting from ubiquitin binding. Acta Crystallogr F Struct Biol Commun. 2014;70:283–287. 10.1107/s2053230x14002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Srivastava M, Suri C, Singh M, Mathur R, Asthana S. Molecular dynamics simulation reveals the possible druggable of USP7. Oncotarget. 2018;9:34289–34305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gavory G, O'Dowd CR, Helm MD, et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol. 2018;14:118–125. [DOI] [PubMed] [Google Scholar]
- 26. O'Dowd CR, Helm MD, Shane Rountree JS, et al. Identification and structure‐guided development of pyrimidinone based USP7 inhibitors. ACS Med Chem Lett. 2018;9:238–243. 10.1021/acsmedchemlett.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamberto I, Liu X, Seo H‐S, et al. Structure‐guided development of a potent and selective non‐covalent active‐site inhibitor of USP7. Cell Chem Biol. 2017;24:1490–1500.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Copeland RA, Pompliano DL, Meek TD. Drug‐target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. [DOI] [PubMed] [Google Scholar]
- 29. Núñez S, Venhorst J, Kruse CG. Target‐drug interactions: First principles and their application to drug discovery. Drug Discov Today. 2012;17:10–22. [DOI] [PubMed] [Google Scholar]
- 30. Guo D, Heitman LH, IJzerman AP. The role of target binding kinetics in drug discovery. ChemMedChem. 2015;10:1793–1796. [DOI] [PubMed] [Google Scholar]
- 31. Mittal L, Tonk RK, Awasthi A, Asthana S. Targeting cryptic‐orthosteric site of PD‐L1 for inhibitor identification using structure‐guided approach. Arch Biochem Biophys. 2021;713:109059. [DOI] [PubMed] [Google Scholar]
- 32. Kumari A, Mittal L, Srivastava M, Pathak DP, Asthana S. Conformational characterization of the co‐activator binding site revealed the mechanism to achieve the bioactive state of FXR. Front Mol Biosci. 2021;8:658312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mittal L, Srivastava M, Kumari A, Tonk RK, Awasthi A, Asthana S. Interplay among structural stability, plasticity, and energetics determined by conformational attuning of flexible loops in PD‐1. J Chem Inf Model. 2021;61:358–384. [DOI] [PubMed] [Google Scholar]
- 34. Rajendran V, Purohit R, Sethumadhavan R. In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in Lamin A/C protein. Amino Acids. 2012;43:603–615. 10.1007/s00726-011-1108-7. [DOI] [PubMed] [Google Scholar]
- 35. Rajendran V, Sethumadhavan R. Drug resistance mechanism of PncA in Mycobacterium tuberculosis . J Biomol Struct Dyn. 2014;32:209–221. [DOI] [PubMed] [Google Scholar]
- 36. Kumar Bhardwaj V, Purohit R, Kumar S. Himalayan bioactive molecules as potential entry inhibitors for the human immunodeficiency virus. Food Chem. 2021;347:128932. [DOI] [PubMed] [Google Scholar]
- 37. Singh R, Bhardwaj VK, Sharma J, Das P, Purohit R. Identification of selective cyclin‐dependent kinase 2 inhibitor from the library of pyrrolone‐fused benzosuberene compounds: An in silico exploration. J Biomol Struct Dyn. 2021;1–9. 10.1080/07391102.2021.1900918. [DOI] [PubMed] [Google Scholar]
- 38. Rajendran V. Structural analysis of oncogenic mutation of isocitrate dehydrogenase 1. Mol Biosyst. 2016;12:2276–2287. [DOI] [PubMed] [Google Scholar]
- 39. Sharma J, Bhardwaj VK, Das P, Purohit R. Identification of naturally originated molecules as γ‐aminobutyric acid receptor antagonist. J Biomol Struct Dyn. 2021;39:911–922. [DOI] [PubMed] [Google Scholar]
- 40. Rajendran V, Gopalakrishnan C, Purohit R. Impact of point mutation P29S in RAC1 on tumorigenesis. Tumour Biol. 2016;37:15293–15304. [DOI] [PubMed] [Google Scholar]
- 41. Patel JS, Berteotti A, Ronsisvalle S, Rocchia W, Cavalli A. Steered molecular dynamics simulations for studying protein‐ligand interaction in cyclin‐dependent kinase 5. J Chem Inf Model. 2014;54:470–480. [DOI] [PubMed] [Google Scholar]
- 42. Mittal L, Srivastava M, Asthana S. Conformational characterization of linker revealed the mechanism of cavity formation by 227G in BVDV RDRP. J Phys Chem B. 2019;123:6150–6160. [DOI] [PubMed] [Google Scholar]
- 43. Rajendran V, Gopalakrishnan C, Sethumadhavan R. Pathological role of a point mutation (T315I) in BCR‐ABL1 protein—A computational insight. J Cell Biochem. 2018;119:918–925. [DOI] [PubMed] [Google Scholar]
- 44. Tiwary P, Limongelli V, Salvalaglio M, Parrinello M. Kinetics of protein–ligand unbinding: Predicting pathways, rates, and rate‐limiting steps. Proc Natl Acad Sci U S A. 2015;112:E386–E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Capelli AM, Costantino G. Unbinding pathways of VEGFR2 inhibitors revealed by steered molecular dynamics. J Chem Inform Model. 2014;54:3124–3136. 10.1021/ci500527j. [DOI] [PubMed] [Google Scholar]
- 46. Niu Y, Li S, Pan D, Liu H, Yao X. Computational study on the unbinding pathways of B‐RAF inhibitors and its implication for the difference of residence time: Insight from random acceleration and steered molecular dynamics simulations. Phys Chem Chem Phys. 2016;18:5622–5629. 10.1039/c5cp06257h. [DOI] [PubMed] [Google Scholar]
- 47. Valles GJ, Bezsonova I, Woodgate R, Ashton NW. USP7 is a master regulator of genome stability. Front Cell Dev Biol. 2020;8:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pozhidaeva A, Valles G, Wang F, et al. USP7‐specific inhibitors target and modify the Enzyme's active site via distinct chemical mechanisms. Cell Chem Biol. 2017;24:1501–1512.e5. [DOI] [PubMed] [Google Scholar]
- 49. Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. 1994;98:1978–1988. 10.1021/j100058a043. [DOI] [Google Scholar]
- 50. Molland K, Zhou Q, Mesecar AD. A 2.2 Å resolution structure of the USP7 catalytic domain in a new space group elaborates upon structural rearrangements resulting from ubiquitin binding. Acta Crystallographica Section F Structural Biology Communications [Internet] 2014;70:283–287. Available from: 10.1107/s2053230x14002519 [DOI] [PMC free article] [PubMed]
- 51. Lamberto I, Liu X, Seo H‐S, Schauer NJ, Iacob RE, Hu W, Das D, Mikhailova T, Weisberg EL, Engen JR, et al. Structure‐Guided Development of a Potent and Selective Non‐covalent Active‐Site Inhibitor of USP7. Cell Chem Biol. 2017;24:1490–1500.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rougé L, Bainbridge TW, Kwok M, et al. Molecular understanding of USP7 substrate recognition and C‐terminal activation. Structure. 2016;24:1335–1345. [DOI] [PubMed] [Google Scholar]
- 53. Anang S, Kaushik N, Hingane S, et al. Potent inhibition of hepatitis E virus release by a cyclic peptide inhibitor of the interaction between viral open Reading frame 3 protein and host tumor susceptibility gene 101. J Virol. 2018;92:e00684‐18. 10.1128/JVI.00684-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sastry GM, Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J Comput‐Aided Mol Des. 2013;27:221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 55. Jorgensen WL, Maxwell DS, Tirado‐Rives J. Development and testing of the OPLS all‐atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- 56. Harder E, Damm W, Maple J, et al. OPLS3: A force field providing broad coverage of drug‐like small molecules and proteins. J Chem Theory Comput. 2016;12:281–296. [DOI] [PubMed] [Google Scholar]
- 57. Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Suri C, Hendrickson TW, Joshi HC, Naik PK. Molecular insight into γ–γ tubulin lateral interactions within the γ‐tubulin ring complex (γ‐TuRC). J Comput‐Aided Mol Des. 2014;28:961–972. [DOI] [PubMed] [Google Scholar]
- 59. Khurana H, Srivastava M, Chaudhary D, et al. Identification of diphenyl furan derivatives via high throughput and computational studies as ArgA inhibitors of Mycobacterium tuberculosis . Int J Biol Macromol. 2021;193:1845–1858. [DOI] [PubMed] [Google Scholar]
- 60. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 61. Potterton A, Husseini FS, Southey MWY, et al. Ensemble‐based steered molecular dynamics predicts relative residence time of A2A receptor binders. J Chem Theory Comput. 2019;15:3316–3330. 10.1021/acs.jctc.8b01270. [DOI] [PubMed] [Google Scholar]
- 62. Ozer G, Quirk S, Hernandez R. Adaptive steered molecular dynamics: Validation of the selection criterion and benchmarking energetics in vacuum. J Chem Phys. 2012;136:215104. 10.1063/1.4725183. [DOI] [PubMed] [Google Scholar]
- 63. Ozer G, Valeev EF, Quirk S, Hernandez R. Adaptive steered molecular dynamics of the long‐distance unfolding of neuropeptide Y. J Chem Theory Comput. 2010;6:3026–3038. 10.1021/ct100320g. [DOI] [PubMed] [Google Scholar]
- 64. Park S, Khalili‐Araghi F, Tajkhorshid E, Schulten K. Free energy calculation from steered molecular dynamics simulations using Jarzynski's equality. J Chem Phys. 2003;119:3559–3566. 10.1063/1.1590311. [DOI] [Google Scholar]
- 65. Park S, Schulten K. Calculating potentials of mean force from steered molecular dynamics simulations. J Chem Phys. 2004;120:5946–5961. 10.1063/1.1651473. [DOI] [PubMed] [Google Scholar]
- 66. Jarzynski C. Nonequilibrium equality for free energy differences. Phys Rev Lett. 1997;78:2690–2693. [Google Scholar]
- 67. Åqvist J, Luzhkov VB, Brandsdal BO. Ligand binding affinities from MD simulations. Acc Chem Res. 2002;35:358–365. 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- 68. Brandsdal BO, Österberg F, Almlöf M, Feierberg I, Luzhkov VB, Åqvist J. Free energy calculations and ligand binding. Adv Protein Chem. 2003;66:123–158. [DOI] [PubMed] [Google Scholar]
- 69. Gutiérrez‐de‐Terán H, Åqvist J. Linear interaction energy: Method and applications in drug design. Methods Mol Biol. 2012;819:305–323. 10.1007/978-1-61779-465-0_20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information.