

Summary
A complete understanding of the genetic determinants underlying mammalian physiology and disease is limited by the capacity for high-throughput genetic dissection in the living organism. Genome-wide CRISPR screening is a powerful method for uncovering the genetic regulation of cellular processes, but the need to stably deliver single guide RNAs to millions of cells has largely restricted its implementation to ex vivo systems. There thus remains a need for accessible high-throughput functional genomics in vivo. Here, we establish genome-wide screening in the liver of a single mouse and use this approach to uncover regulation of hepatocyte fitness. We uncover pathways not identified in cell culture screens, underscoring the power of genetic dissection in the organism. The approach we developed is accessible, scalable, and adaptable to diverse phenotypes and applications. We have hereby established a foundation for high-throughput functional genomics in a living mammal, enabling comprehensive investigation of physiology and disease.
Keywords: functional genomics, genome-scale screen, CRISPR, liver
Graphical abstract
Highlights
-
•
Stable, high-coverage delivery of a genome-scale sgRNA library to the mouse liver
-
•
Screen for hepatocyte fitness uncovers significant hits in single mice
-
•
Screen reveals genes uniquely required for hepatocyte fitness in a living organism
-
•
Approach is accessible and adaptable to diverse phenotypes and CRISPR applications
Improved understanding of physiology and disease requires methods for high-throughput genetic dissection in the organism. Here, Keys and Knouse establish genome-scale screening in the mouse liver and apply this to uncover regulation of hepatocyte fitness. This approach offers an accessible and adaptable platform for high-throughput functional genomics in the organism.
Introduction
Our ability to understand and modulate mammalian physiology and disease requires the capacity to determine how all genes contribute to any given phenotype. In cell culture systems, genome-wide screening approaches have provided the power to identify all genes that positively or negatively regulate a cellular process in a single experiment. However, these ex vivo systems cannot reproduce all of the cellular processes that occur in vivo, and, even when they can, they cannot recapitulate the entirety of extracellular factors that influence these phenomena in vivo. Although these limitations warrant studying cellular processes directly in the organism, probing complex phenotypes in the organism has historically required sacrificing experimental tractability. When it comes to inferring causal relationships, one is largely restricted to analyzing a single gene at a time using knockout mice. This discordance between experimental tractability and physiologic relevance has long limited our ability to understand mammalian physiology and disease. There is therefore a pressing need to bring high-throughput functional genomics into the organism.
Among the methods for high-throughput functional genomics in cell culture, genome-wide screening using CRISPR-Cas9 has emerged as a powerful approach.1,2 This entails delivering a single guide RNA (sgRNA) library alongside Cas9 to cells, an intervening period for protein depletion and phenotypic selection, and ultimately deep sequencing to detect changes in sgRNA abundance and thus hits in the screen. Critical to the success of the screen is delivering sgRNAs in a manner that is stable and has high coverage, with any given sgRNA being delivered to at least a few hundred cells, so that changes in sgRNA abundance can be evaluated at the end of screening with the power to identify significantly enriched and depleted sgRNAs. Lentivirus is the preferred method for delivering sgRNAs into cells as it allows for stable, single-copy integration of an sgRNA within a cell. While lentivirus is effective at delivering genome-wide sgRNA libraries to tens of millions of cells in culture, it is less efficient at delivering sgRNAs to cells in vivo. Although groups have successfully used lentivirus to transduce keratinocytes and neurons in vivo, the number of cells transduced per mouse is limited, and previous approaches required over 50 mice to be pooled for a single genome-scale screen.3,4 As such, genome-wide CRISPR screening has largely been limited to cell culture systems or cellular transplantation models.5,6
Bringing genome-scale screening into the organism requires overcoming the barriers to stable, high-coverage sgRNA delivery into tissues. In this regard, the mouse liver is an appealing target organ. Comprised of tens of millions of hepatocytes, a single mouse liver offers cell numbers compatible with genome-scale screening. Moreover, given the liver’s diverse metabolic functions and impressive regenerative capacity, hepatocytes exhibit a broad range of phenotypes that are ripe for genetic dissection. Unfortunately, efforts to deliver lentivirus to the liver have suffered from poor transduction efficiency and immune-mediated clearance of transduced hepatocytes.7,8 Although groups have leveraged other delivery methods to perform genetic screens in the liver, these methods all have limitations that prevent genome-scale enrichment and depletion screening. For example, adeno-associated virus (AAV) can be used to deliver sgRNAs to the majority of hepatocytes, but because AAV vectors typically remain episomal, the sgRNAs will not persist in proliferating cells. Analysis of AAV-based screens therefore requires individually amplifying and sequencing the genes targeted by the sgRNAs, dramatically limiting the number of genes that can be screened in any given experiment.9 Hydrodynamic tail vein injection of plasmids encoding transposons can also introduce sgRNAs into hepatocytes and offers the benefit of integration into the host genome. However, hydrodynamic tail vein injection only transfects 10%–40% of hepatocytes in a non-uniform manner, again limiting the scale of the screen.10,11,12,13 As such, genetic screens in the liver have largely been restricted to small-scale screens on the order of tens to hundreds of genes.
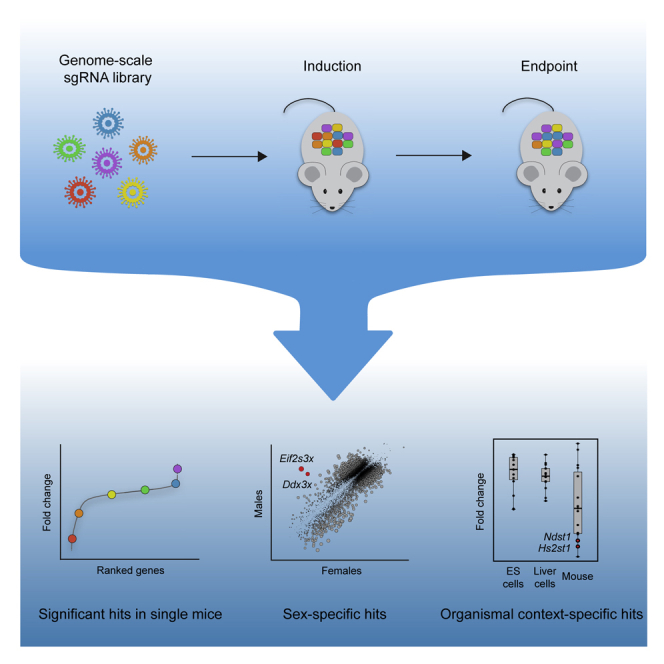
Here, we have established accessible, genome-scale, enrichment and depletion screening in the mouse liver. We developed an approach for stable, high-coverage delivery of a genome-scale sgRNA library into the liver of inducible Cas9 mice, allowing for Cas9 induction and phenotypic selection at any point in the animal’s lifetime. To validate this approach, we performed a screen for hepatocyte fitness in the neonatal liver. Our screen had the ability to uncover positive and negative regulators of hepatocyte fitness in individual mice with high reproducibility across mice. We discovered genes with sex-specific effects on hepatocyte fitness as well as genes that are uniquely required for hepatocyte fitness in a living organism. This approach is accessible and adaptable to diverse phenotypes and CRISPR methods and thereby provides a foundation for high-throughput genetic dissection in a living organism.
Results
Genome-scale sgRNA delivery in a single mouse liver
Given the advantages of lentiviral-mediated sgRNA delivery over other sgRNA delivery methods, we sought to establish efficient lentiviral sgRNA delivery to the liver. We hypothesized that intravenously injecting highly concentrated lentivirus into neonatal mice might avoid the poor transduction efficiency and immune clearance observed by others.14 To test this, we generated lentiviruses encoding a non-targeting sgRNA (sgAAVS1) alongside mCherry or mTurquoise2 (mTurq2) reporters and injected varying doses of an equal mixture of these two lentiviruses into postnatal day (PD) 1 mice (Figure 1A). We observed a dose-dependent increase in the percentage of transduced hepatocytes, with a dose of 5 × 107 transduction units (TU) transducing over 75% of hepatocytes (Figures 1B and 1C). Importantly, these transduced hepatocytes were distributed uniformly throughout the liver lobule and persisted into adulthood (Figures 1B, S1A, and S1B). Based on measurements of hepatocyte and liver volume and the transduction frequency of two fluorescent reporters, we estimated that a dose of 5 × 107 TU transduces approximately 10 million hepatocytes per PD1 liver with an average of just two integration events per cell (Figures 1C and S1C). This transduction efficiency would afford >200-fold coverage of a 100,000-feature sgRNA library in a single mouse. Our lentiviral approach therefore establishes an sgRNA delivery method that is wholly compatible with genome-scale screening in the mouse liver.
Figure 1.

Genome-scale sgRNA delivery in a single mouse liver
(A) Lentiviral vectors for U6-driven expression of an sgRNA and hepatocyte-specific expression of a fluorescent reporter (mCherry or mTurq2).
(B) Images of endogenous mCherry and mTurq2 fluorescence in livers from mice 4 days after injection with an equal mixture of sgAAVS1-mCherry and sgAAVS1-mTurq2 lentiviruses. Livers were counterstained with phalloidin (green) to label actin. Scale bars, 100 μm.
(C) Percentage of mCherry-, mTurq2-, and double-positive hepatocytes in livers from mice 4 days after injection with an equal mixture of sgAAVS1-mCherry and sgAAVS1-mTurq2 lentiviruses. Error bars indicate standard deviation. n = 3 mice per dose and 200 hepatocytes per mouse.
See also Figure S1.
We next asked whether we could use this sgRNA delivery approach as the basis for temporally controlled protein depletion in hepatocytes. We used commercially available loxP-stop-loxP-Cas9 (LSL-Cas9) mice, in which we could induce Cas9 in nearly all hepatocytes by injecting an AAV expressing Cre recombinase from the hepatocyte-specific Tbg promoter15 (AAV-Cre; Figures S2A and S2B). To evaluate the kinetics and efficiency of protein depletion, we selected two long-lived, non-essential proteins: the mitochondrial enzyme MAO-B (encoded by Maob) and the nuclear lamin Lamin B2 (encoded by Lmnb2).16,17 After delivering sgMaob-mCherry or sgLmnb2-mCherry lentivirus to PD1 mice, we injected PBS or AAV-Cre at PD5 and harvested livers at various time points to evaluate protein levels in individual hepatocytes (Figure 2A). By two weeks after Cas9 induction, MAO-B and Lamin B2 were depleted exclusively in mCherry-positive hepatocytes in mice injected with AAV-Cre (Figures 2B, 2C, S2C, and S2D). Importantly, this combination of lentiviral-mediated sgRNA delivery and AAV-Cre-mediated induction of Cas9 and the resulting gene targeting did not induce detectable hepatocyte damage or liver inflammation (Figures S2E–S2G). Hepatocyte turnover was also unaffected (Figures S2H and S2I). This approach therefore offers an effective platform for hepatocyte-specific protein depletion and genetic screening at any point in the animal’s lifetime without detectable confounding perturbation of the cells or tissue.
Figure 2.

Temporally controlled protein depletion in the mouse liver
(A) Scheme for inducing protein depletion in LSL-Cas9 mice.
(B) Images of livers from LSL-Cas9 mice injected with sgMaob-mCherry followed by PBS or AAV-Cre immunostained for mCherry (magenta), MAO-B (green), and actin (blue). Scale bars, 45 μm.
(C) Cytoplasmic MAO-B intensity per μm in mCherry-positive and mCherry-negative hepatocytes from LSL-Cas9 mice injected with sgMaob-mCherry followed by PBS or AAV-Cre. Closed and open circles represent values from male and female mice, respectively. n = 1 male and 1 female mouse per condition and 25 cells per mouse.
See also Figure S2.
A genome-scale screen in the liver
To enable genome-scale screening for diverse hepatocyte phenotypes, we sought to prepare an sgRNA library targeting all genes expressed in the developing, quiescent, and regenerating mouse liver. We performed RNA sequencing on livers at various time points during mouse development and after liver injury and determined that 13,266 protein-coding genes were expressed (FPKM > 0.3) at one or more time points (Figures 3A, S3A, and S3B; Table S1). We generated an sgRNA library targeting 13,189 of these genes (average of 5 sgRNAs per gene) alongside a previously published set of 6,500 control sgRNAs (∼2,000 non-targeting sgRNAs and ∼4,500 sgRNAs targeting exonic and intronic regions of control genes)18 for a total of 71,878 unique sgRNAs (Figure S3C; Table S2).
Figure 3.

A genome-scale screen for hepatocyte fitness in the neonatal mouse liver
(A) Number of protein-coding genes expressed in the liver as determined by RNA sequencing of livers at various time points.
(B) Scheme for performing a genome-scale screen for hepatocyte fitness in neonatal mice.
(C) Representation of sgRNAs in livers 4 days after injection with lentiviral library relative to the sgRNA representation in the plasmid library expressed as reads per million (RPM). n = 2 male and 2 female mice pooled into a single sequencing library. Pearson correlation r = 0.97.
(D) Pairwise comparisons of median fold change (log2) for each gene for each mouse at the endpoint of the screen.
(E) Genes ranked by median fold change across mice (log2) with significantly depleted genes denoted by red points and significantly enriched genes denoted by blue points (FDR < 0.05 by two-tailed Wilcoxon test). Core essential genes (red bars) are positioned below based on gene rank to demonstrate their significant depletion across mice. p < 2.2 × 10−16 by one-sided Kolmogorov-Smirnov test.
(F) Genes ranked by median fold change (log2) in each of the four mice. Highlighted are control gene sets consisting of tumor-suppressor genes in hepatocellular carcinoma (expected to enrich, blue) and genes required for hepatocyte viability (expected to deplete, red). Core essential genes (red bars) are positioned below based on gene rank to demonstrate their significant depletion in each mouse. Expected gene depletion p = 1 × 10−5, 1.6 × 10−4, 1.4 × 10−4, and 1 × 10−5 for male 1, female 1, male 2, and female 2, respectively, by one-sided Kolmogorov-Smirnov test, and expected gene enrichment p = 0.0032, 0.0052, 0.0053, and 0.0039 for male 1, female 1, male 2, and female 2, respectively, by one-sided Kolmogorov-Smirnov test. Core essential gene depletion p < 2.2 × 10−16 for each mouse by one-sided Kolmogorov-Smirnov test.
(G) Benjamini-Hochberg-adjusted Wilcoxon p-value (−log10) versus the median fold change across mice (log2) for each gene in the screen. Highlighted are control gene sets consisting of tumor-suppressor genes in hepatocellular carcinoma (expected to enrich, blue) and genes required for hepatocyte viability (expected to deplete, red). Expected depleted p = 3.5 × 10−6 by one-sided Kolmogorov-Smirnov test, and expected enriched p = 4.7 × 10−4 by one-sided Kolmogorov-Smirnov test.
See also Figure S3.
With this method in hand, we undertook a genome-wide screen for hepatocyte fitness (Figure 3B). To screen for the ability of hepatocytes to both persist and proliferate, we elected to screen over a three-week period in neonatal development when hepatocytes undergo approximately three population doublings to increase liver mass. We injected 5 × 107 TU of our lentiviral library into four female and four male LSL-Cas9 mice at PD1. At PD5, we harvested livers from two males and two females to evaluate the initial library representation. The sgRNA representation in these four livers correlated extremely well (Pearson r = 0.97) with the plasmid library, and we detected all sgRNAs, indicating that we can effectively deliver and recover a genome-scale sgRNA library from the neonatal mouse liver (Figure 3C; Table S3). In the remaining mice, we induced Cas9 at PD5 and harvested their livers at PD26 to evaluate the final library representation.
We used the model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK) algorithm19 to identify enriched and depleted genes based on statistical differences in their change in sgRNA abundance at PD26 relative to PD5 (Table S3). Using a false discovery rate cutoff of 0.05, we identified 6, 0, 0, and 2 significantly enriched and 364, 40, 386, and 297 significantly depleted genes in male 1, female 1, male 2, and female 2, respectively, indicating that our method can detect enriched and depleted genes in a single mouse. We also generated gene-level scores by calculating the median log2 fold change in the abundance of all sgRNAs targeting a given gene. We observed a strong correlation in gene scores across the four mice (Pearson r = 0.46 to 0.75; Figures 3D and S3D; Table S3). Indeed, the reproducibility we observed across mice is similar to that observed in cell culture screens in cases where replicates are performed (Pearson r = 0.59 to 0.65).20,21 We note that while female 1 was less well correlated with the rest of the mice, the correlation between female 1 and other mice was still significant (p < 2.2 × 10−16), and we had no technical reason to invalidate female 1’s data. We therefore considered all four mice as biological replicates without normalizing gene scores between mice for subsequent analyses.
To improve our power to identify significantly enriched and depleted genes, we combined the data from all four mice and calculated a unified gene score representing the median log2 fold change for each gene across mice (Table S3). Using a false discovery rate cutoff of 0.05, we identified 30 significantly enriched genes and 661 significantly depleted genes across all mice (Figure 3E; Table S3). Importantly, these gene scores were not positively correlated with gene expression or protein half-life,16 reaffirming that long-lived proteins were effectively depleted (Figures S3E and S3F). Collectively, these initial screen results establish the technical feasibility of genome-scale screening in the mouse liver. Importantly, while screening multiple mice in parallel increases the power to discover significant hits, a single mouse is sufficient to identify significantly enriched and depleted genes.
We next asked whether our screen reliably uncovered regulators of cell fitness. We first assessed whether genes established to be essential in cell culture22 were significantly enriched among the depleted genes. This set of core essential genes was indeed significantly depleted across all four mice and within each individual mouse (Figures 3E and 3F). To evaluate whether our screen could reliably reveal regulation specific to hepatocyte fitness in the organism, we assessed the gene scores for two sets of genes known to affect hepatocyte fitness in the organism: (1) a set of 13 genes established to act as tumor suppressors in hepatocellular carcinoma23 (expected to enrich), and (2) a set of seven genes required for hepatocyte viability24,25,26,27,28 (expected to deplete) (Table S3). Among the tumor suppressor genes, eight of the 13 genes were significantly enriched (false discovery rate [FDR] < 0.25; Figure 3G). Among the genes required for hepatocyte viability, all seven genes were significantly depleted (FDR < 0.25; Figure 3G). These two gene sets were also significantly enriched and depleted as expected within each individual mouse (Figure 3F). These results support our screen as a reliable platform for uncovering the genetic regulation of the phenotype in question.
Genome-scale screening in the organism affords unique insights
Genome-scale screening in the organism, by virtue of preserving the native state and context of the cell and phenotype under investigation, enables several biological insights not possible in cell culture. One such advantage is the ability to screen wild-type cells that do not carry pre-existing mutations. Most cell lines naturally harbor or inevitably acquire mutations that improve their viability and proliferation in culture,29 compromising the ability to query the function of these mutated genes in screens. As such, fitness screens in cell culture have an impaired ability to uncover tumor suppressor genes.30,31,32,33,34,35 In contrast, our screen readily recovered tumor suppressor genes. Over half of the 25 most enriched genes in our screen are established to act as tumor suppressor genes in at least one context36,37 (Table S3). Indeed, a set of the top 50 computationally predicted pan-cancer tumor suppressor genes36 was significantly enriched in our screen but not in fitness screens in mouse embryonic stem cell lines30,31 or human hepatocellular carcinoma cell lines32,33,34,35 (Figures 4A and S4A; Table S3). Our screen’s unique ability to, within only a few population doublings, reliably recover tumor suppressors highlights the power of being able to screen unmutated, wild-type cells and thereby probe all genes that may influence a phenotype.
Figure 4.

Genome-scale screening in the organism enhances discovery of tumor-suppressor genes and uncovers genes with sex-specific effects
(A) Cumulative fraction of tumor suppressor genes (cyan, blue) and other genes (black, gray) based on quantile-normalized median fold change (log2) of their gene scores across screens in mouse embryonic stem cells (ESCs) and our screen. ESCs p > 0.05 by one-sided Kolmogorov-Smirnov test, and our screen p = 6.5 × 10−3 by one-sided Kolmogorov-Smirnov test.
(B) Median fold change (log2) across males versus median fold change (log2) across females for each gene. Highlighted are genes uniquely enriched in females (blue), genes uniquely enriched in males (cyan), genes uniquely depleted in females (red), and genes uniquely depleted in males (pink). Point size is proportional to the absolute difference in median log2 fold change between females and males.
See also Figure S4.
A second advantage of screening directly in the organism is the opportunity to investigate how biological sex influences the relationship between genotype and phenotype. To determine whether biological sex affected hits in our screen, we compared the gene scores for all genes in males versus females. We identified three X-linked genes and nine autosomal genes with sex-specific effects on fitness (Figure 4B). The genes with the greatest difference between the sexes were the X-linked genes Ddx3x and Eif2s3x which were exclusively essential in females. Both Ddx3x and Eif2s3x facilitate protein synthesis. Both of these genes escape X inactivation and have paralogs on the Y chromosome, Ddx3y and Eif2s3y, with similar function.38,39 Thus, it is likely that disruption of Ddx3x and Eif2s3x causes a fitness defect in female hepatocytes, while male hepatocytes are functionally complemented by the Y chromosome paralogs. In this case, the sex-specific effect of these genes originates at a sex chromosome level. However, this approach is equally capable of identifying sex-specific effects that arise from hormonal or other differences between the sexes. Although uncovering sex-specific regulation of hepatocyte fitness was not the primary purpose of this study, these preliminary findings highlight the capacity to uncover such regulation by screening in the organism.
Major histocompatibility complex (MHC) class I and heparan sulfate biosynthesis are uniquely required for hepatocyte fitness in the organism
Perhaps the most valuable feature of genome-scale screening in the organism is the ability to investigate a phenotype in its native context. This captures all of the ways in which the cell interacts with the extracellular environment, many of which cannot be recapitulated in cell culture. To understand how hepatocyte fitness is regulated in the living organism, we first looked for patterns in the enriched and depleted genes by performing gene set enrichment analysis on the unified gene scores across the four mice. We did not identify any gene sets to be significantly enriched in our screen. However, we identified several gene sets that were significantly depleted in our screen (Figure 5A; Table S4). These gene sets included those previously established as essential for fitness in cell culture, including ribosome, proteasome, spliceosome, and RNA polymerase.1,2,17,22,32,33 However, we also identified several other gene sets not documented to be essential for cells in culture, including N-glycan biosynthesis, antigen processing/presentation, and glycosaminoglycan biosynthesis/heparan sulfate. Notably, these pathways all play major roles in the presentation or secretion of proteins at the cell surface,40,41 pointing to possible regulation of fitness from the extracellular environment that could only be identified by screening in the organismal context.
Figure 5.

Class I MHC and heparan sulfate biosynthesis are uniquely required for hepatocyte fitness in the organism
(A) KEGG gene sets exhibiting significant depletion (FDR q < 0.05) at the endpoint of the screen ranked by FDR q-value (−log10). Bars extending to the end of the plot indicate an FDR q-value of 0.
(B) KEGG gene sets exhibiting significant depletion (FDR q < 0.05) in our screen relative to screens in either mouse ESCs (dark gray bars) or human hepatocellular carcinoma (HCC) cell lines (light gray bars) ranked by FDR q-value (−log10) for our screen relative to mouse ESCs. Bars extending to the end of the plot indicate an FDR q-value of 0.
(C) Median fold change (log2) for genes in the KEGG gene set for antigen processing and presentation in quantile-normalized ESC screens, HCC cell line screens, and our screen. Genes uniquely depleted in our screen are highlighted in red. The bounds of the box indicate the first and third quartiles, and the whiskers extend to the furthest data point that is within 1.5 times the interquartile range.
(D) Median fold change (log2) for genes in the KEGG gene set for glycosaminoglycan biosynthesis and heparan sulfate in quantile-normalized ESC screens, HCC cell line screens, and our screen. Genes uniquely depleted in our screen are highlighted in red. The bounds of the box indicate the first and third quartiles, and the whiskers extend to the furthest data point that is within 1.5 times the interquartile range.
(E) Median fold change (log2) for genes in the heparan sulfate interactome in our screen. The bounds of the box indicate the first and third quartiles, and the whiskers extend to the furthest data point that is within 1.5 times the interquartile range.
(F) Scheme for determining effects of NDST1 knockdown on hepatocyte proliferation (top panel). Image of liver from postnatal day 15 mouse injected with 1 × 1010 genome copies (GC) of AAV-shNDST1 and AAV-shScramble on postnatal day 5 immunostained for Ki67 (white), GFP (green), and mCherry (magenta) and counterstained for Hoechst (blue) (left panel). Scale bar, 25 μm. Quantification of proliferation as inferred by Ki67 positivity in shNDST1 hepatocytes relative to shScramble hepatocytes (right panel). Bar and whiskers indicate mean and standard deviation across mice, respectively, and closed and open circles represent values from male and female mice, respectively. n = 2 male and 2 female mice and 200 cells per shRNA per mouse. ∗∗p = 0.0023 by two-tailed Fisher’s exact test.
See also Figure S5.
To determine whether any genes and pathways were indeed uniquely required for cell fitness in the organismal context, we compared our screen with fitness screens of mouse embryonic stem cells (ESCs) in culture30,31 and human hepatocellular carcinoma (HCC) cell lines in culture.32,33,34,35 We specifically chose ESCs and HCC cell lines in an effort to control for any species-specific and cell-lineage-specific requirements for cell fitness. We identified several gene sets that were significantly depleted in our screen relative to screens in either ESCs or HCC cell lines (Figure 5B; Table S4). We turned our focus to four gene sets that were significantly depleted in our screen relative to both the ESC screens and the HCC cell line screens: protein export, SNARE interactions in vesicular transport, antigen processing/presentation, and glycosaminoglycan biosynthesis/heparan sulfate. For the protein export and SNARE interactions gene sets, the most depleted genes in each set were depleted across all screens but to a greater extent in our screen (Figures S5A and S5B; Table S4). However, for the antigen processing/presentation and glycosaminoglycan biosynthesis/heparan sulfate gene sets, some genes were depleted exclusively in our screen, suggesting a unique requirement for aspects of these pathways in hepatocyte fitness in the organism (Figures 5C and 5D; Table S4).
Within the antigen processing/presentation gene set, Tap1 and B2m were uniquely and dramatically depleted in our screen, ranking as the 40th and 319th most depleted genes, respectively (Figure 5C). Both of these genes are involved in—and required for—presentation of antigens at the cell surface by the MHC class I pathway.41 Indeed, within the antigen processing/presentation gene set, eight of the 32 genes attributed to the MHC class I pathway were depleted in our screen, whereas none of the 13 genes attributed to the MHC class II pathway exhibited depletion (FDR < 0.25; Figure S5C). The MHC class I pathway presents intracellular antigens at the cell surface. At the cell surface, MHC class I can interact with both cytotoxic CD8 T cells and natural killer (NK) cells. The latter interaction can provide a pro-survival role by preventing NK cell cytotoxicity. However, loss of MHC class I alone should not be sufficient to induce NK cell cytotoxicity. Classically, NK cell activation requires both a loss of inhibitory signals, via loss of MHC class I, and presence of activating signals, expressed on the surface of transformed or infected cells.42 Although our screening approach involves viral infection of hepatocytes, any inflammation resulting from lentiviral and AAV vectors has been shown to resolve within 72 hours,43,44 and we indeed did not observe any liver inflammation in our system (Figures S2E–S2G). This result suggests that MHC class I may play an essential survival role even in untransformed and uninflamed cells in the organism.
Within the glycosaminoglycan biosynthesis/heparan sulfate gene set, Hs2st1 and Ndst1 were uniquely depleted in our screen (Figure 5D). These two genes encode enzymes involved in the biosynthesis of heparan sulfate, a glycosaminoglycan conjugated to plasma membrane or extracellular matrix proteins to form heparan sulfate proteoglycans (HSPGs).45 HSPGs can be classified into three groups based on their location: cell membrane (syndecans, glypicans), extracellular matrix (agrin, perlecan, type XVIII collagen), and secretory vesicles (serglycin). To determine whether the requirement for heparan sulfate reflected the requirement for a specific HSPG, we analyzed the performance of individual HSPGs in our screen. No single HSPG was essential, suggesting that the requirement for heparan sulfate arises from a redundant function performed by multiple HSPGs (Figure S5D).
There is indeed evidence that the syndecans can function redundantly.46 These transmembrane HSPGs play a variety of roles including facilitating attachment of cells to the extracellular matrix, protecting cytokines and growth factors from proteolysis, and serving as co-receptors for other transmembrane receptors. As such, these HSPGs interact with hundreds of other proteins in the extracellular matrix and at the cell surface. To gain insight into whether these interactions may be essential for hepatocyte fitness, we analyzed how proteins established to interact with HSPGs performed in our screen.47 We observed a handful of heparan-sulfate-interacting genes for which knockout caused significant fitness defects in hepatocytes (Figure 5E). This included B2m as well as the growth factor receptors Insr, Met, and Lgr4. These three growth factor receptors signal downstream of known hepatocyte mitogens.48,49 As such, the requirement for heparan sulfate biosynthesis in hepatocytes could be via HSPGs potentiating pro-survival or proliferative signals at the cell surface.
To examine the consequences of disrupted heparan sulfate biosynthesis by an alternative method, we used an orthologous AAV vector system in which we could deliver short hairpin RNA (shRNA) targeting NDST1 and thereby knock down NDST1 protein in the whole liver or individual hepatocytes (Figure S5E). To determine whether loss of NDST1 impaired hepatocyte proliferation, we injected neonatal mice with a low-dose mixture of AAV encoding shRNA targeting NDST1 alongside a GFP reporter (AAV-shNDST1) and AAV encoding a scramble shRNA alongside an mCherry reporter (AAV-shScramble) such that rare hepatocytes were expressing one of the shRNAs. We compared the proliferation of cells expressing shRNA targeting NDST1 with those expressing scramble shRNA within the same liver by immunostaining for the proliferation marker Ki67 alongside the fluorescent reporters. We observed a significant reduction in proliferation in the NDST1 knockdown hepatocytes compared with control knockdown hepatocytes, in agreement with our screen results (Figure 5F). We therefore find that an enzyme in the heparan sulfate biosynthesis pathway is required for hepatocyte proliferation in a cell-autonomous fashion. This finding, coupled to the above-mentioned observation that many growth factor receptors essential for hepatocyte fitness are established to interact with heparan sulfate, suggests that heparan sulfate may promote hepatocyte fitness by potentiating growth factor signaling. Further experimentation will be important to establish the specific mechanism by which heparan sulfate promotes hepatocyte fitness.
Discussion
Herein, we successfully developed a method for genome-scale CRISPR screening directly within a single mouse. Our approach involves lentiviral delivery of a genome-scale sgRNA library to a neonatal inducible Cas9 mouse followed by Cas9 induction and phenotypic selection at any point in the animal’s lifetime. We applied this approach to uncover regulators of hepatocyte fitness in the neonatal liver. Our screen reliably identified positive and negative regulation of fitness in a single mouse with high reproducibility across mice. Not surprisingly, we found that hepatocytes in the neonatal liver share many requirements for fitness with cells in culture. However, we also uncovered genes with sex-specific effects on hepatocyte fitness and genes that are uniquely required for hepatocyte fitness in the organism. Specifically, we found that hepatocytes in the liver, but not cells in culture, are dependent on the MHC class I and heparan sulfate biosynthesis pathways. We show that knockdown of the heparan sulfate biosynthesis enzyme NDST1 impairs hepatocyte proliferation in a cell-autonomous manner. The specific mechanism by which heparan sulfate promotes cell proliferation and if this holds true in other tissues will be important future directions. Notably, this finding provides an important consideration for the growing interest in antiviral therapies that interfere with HSPGs as a means of preventing entry of diverse viruses.50 Finally, we could not have discovered the sex-specific requirements nor the organism-specific requirements for hepatocyte fitness by screening a single cell line in culture. Our screen’s ability to uncover genetic regulation not identified in cell culture emphasizes the necessity and power of genome-scale screening in the living organism.
Our approach provides an adaptable and accessible method for the unbiased and comprehensive genetic dissection of diverse phenotypes within a living mouse. The stability of the sgRNA library in the liver and the inducible nature of Cas9 allow for phenotypic selection at any point in the animal’s lifetime. This selection can be performed, as in our screen, by evaluating changes in the bulk hepatocyte population over time. Alternatively, selection could be performed by isolating hepatocytes from the liver and enriching for a single-cell phenotype. This combined flexibility of Cas9 induction and phenotypic selection makes this method a powerful tool for screening myriad processes spanning universal cellular phenomena, development and aging, hepatocyte-specific functions, and liver disease. Moreover, given the extent to which biological sex impacts physiology and disease, this ability to investigate the interaction between biological sex and gene function lends even further value to this technology. Importantly, these diverse applications are all within reach as this versatile approach can be readily scaled yet minimally requires a single mouse and fewer reagents than a cell culture screen.
Genome-scale CRISPR-Cas9 screening in the liver alone has the power to offer novel insights into diverse aspects of mammalian physiology and disease, but the full potential of high-throughput functional genomics in the organism lies in expanding this technology to other organs and CRISPR applications. Introducing this technology into other tissues will similarly be predicated on the ability to achieve stable, high-coverage sgRNA delivery in these tissues. This will require developing methods for efficient lentiviral delivery to organs beyond the liver and achieving genome-scale coverage in organs with fewer cells. Once established in any organ, our overall approach can be readily adapted to incorporate other CRISPR-based techniques including CRISPR interference and activation. Our system therefore establishes the feasibility and foundation for genome-scale screening in a living organism. Building and expanding this platform will bring the experimental tractability once restricted to cell culture into the living organism, enabling unprecedented insight into mammalian physiology and disease.
Limitations of the study
In our approach, we evaluated the ability of lentivirus to infect hepatocytes by encoding a hepatocyte-specific promoter driving expression of a fluorescent reporter in the lentiviral genome. From this alone, we cannot determine whether our lentivirus is targeting other cell types in the liver. However, because we induce Cas9 with an AAV vector encoding Cre from a hepatocyte-specific promoter, gene editing and changes in sgRNA abundance should be specific to hepatocytes. We also note that while we can identify significantly enriched and depleted genes in a single mouse, we identify an even greater number of significantly enriched and depleted genes by screening multiple mice and combining data across all mice. As such, for experiments seeking to maximize the number of significant hits or to compare hits between males and females, it is advisable to screen and combine data from more than two males and two females. Finally, we note that our screen was carried out over a three-week period, which corresponds to approximately three hepatocyte population doublings. Extending this screen over a longer time period that encompasses a greater number of population doublings would likely uncover additional genes and pathways that regulate hepatocyte fitness specifically in the context of the whole organism.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Cas9 (Clone 7A9-3A3) | Abcam | Catalog #: ab191468 |
| Rabbit anti-Asialoglycoprotein receptor 1 (ASGR1, Clone 114) | Sino Biological | Catalog #: 50083-R114 |
| Rat anti-mCherry (Clone 16D7) | ThermoFisher Scientific | Catalog #: M11217 |
| Rabbit anti-Monoamine oxidase B (MAO-B, Polyclonal) | Novus Biologicals | Catalog #: NBP1-87493 |
| Rabbit anti-Lamin B2 (Clone EPR9701[B]) | Abcam | Catalog #: ab151735 |
| Mouse anti-Beta-actin (Clone AC-74) | Sigma-Aldrich | Catalog #: A2228 |
| Rabbit anti-CD45 (Polyclonal) | Abcam | Catalog #: ab10558 |
| Rabbit anti-Ki67 (Clone SP6) | Abcam | Catalog #: ab16667 |
| Rabbit anti-mCherry (Polyclonal) | Abcam | Catalog #: ab167453 |
| Chicken anti-GFP (Polyclonal) | Abcam | Catalog #: ab13970 |
| Rat anti-Ki67 (Clone SolA15) | ThermoFisher Scientific | Catalog #: 14-5698-80 |
| Rabbit anti-NDST1 (Polyclonal) | Sigma-Aldrich | Catalog #: SAB1307040 |
| Goat anti-Rabbit IgG H&L (HRP) | Abcam | Catalog #: ab205718 |
| Goat anti-Mouse IgG H&L (HRP) | Abcam | Catalog #: ab205719 |
| Bacterial and virus strains | ||
| Endura electrocompetent cells | Biosearch Technologies | Product Code: 60242-2 |
| AAV8-TBG-Cre | Addgene | Catalog #: 107787-AAV8 |
| AAV8-mCherry-U6-scrmb-shRNA | Vector Biolabs | |
| AAV8-GFP-U6-mNDST1-shRNA | Vector Biolabs | |
| Deposited data | ||
| Mouse liver RNA sequencing data | This paper | GEO: GSE215216 |
| CRISPR screen sequencing data | This paper | SRA: PRJNA887396 |
| Experimental models: Cell lines | ||
| Mouse: AML12 cells | American Type Culture Collection (ATCC) | ATCC #: CRL-2254, RRID: CVCL_0140 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain #: 000664, RRID: IMSR_JAX:000,664 |
| Mouse: LSL-Cas9+/+: B6J.129(B6N)-Gt(ROSA)26Sortm1(CAG-cas9∗,-EGFP)Fezh/J | The Jackson Laboratory | Strain #: 026175, RRID: IMSR_JAX:026175 |
| Oligonucleotides | ||
| Primer: sgRNA library cloning amplification primer forward: 5′-TTTCTTGGCTTTATATA TCTTGTGGAAAGGACGAAACACC-3′ |
This paper | |
| Primer: sgRNA library cloning amplification primer reverse: 5′-ATTTAAACTTGCTATGC TGTTTCCAGCATAGCTCTTAAAC-3′ |
This paper | |
| Primer: Sequencing library preparation downstream of sgRNA sequence in the reverse direction: 5′-AATGATAC GGCGACCACCGAGATCTACACCG ACTCGGTGCCACTTTT-3′ |
Wang et al.2 | |
| Primer: Sequencing library preparation upstream of sgRNA sequence in the forward direction: 5′-CAAGCAGAA GACGGCATACGAGATCnnnnnnTTTCT TGGGTAGTTTGCAGTTTT-3′ |
Wang et al.17 | |
| Primer: Sequencing read 1: 5′-GTTGATAACGGACTAG CCTTATTTAAACTTGCTAT GCTGTTTCCAGCATAGCT CTTAAAC-3′ |
This paper | |
| Primer: Sequencing index: 5′-TTTCAAGTTACGGTAAG CATATGATAGTCCATTTTAA AACATAATTTTAAAACTGCA AACTACCCAAGAAA-3′ |
Wang et al.2 | |
| Recombinant DNA | ||
| Plasmid: LentiCRISPRv2-Opti | Adelmann et al.51 | Addgene Catalog #: 163126 |
| Plasmid: pLentiCRISPRv2-Stuffer-HepmCherry | This paper | Addgene Catalog #: 192825 |
| Plasmid: pLentiCRISPRv2-Stuffer-HepmTurquoise2 | This paper | Addgene Catalog #: 192826 |
| Plasmid: pLentiCRISPRv2-sgAAVS1-HepmCherry | This paper | Addgene Catalog #: 192827 |
| Plasmid: pLentiCRISPRv2-sgAAVS1-HepmTurquoise2 | This paper | Addgene Catalog #: 192828 |
| Plasmid: pLentiCRISPRv2-sgMaob-HepmCherry | This paper | Addgene Catalog #: 192829 |
| Plasmid: pLentiCRISPRv2-sgLmnb2-HepmCherry | This paper | Addgene Catalog #: 192830 |
| Pooled sgRNA library: Mouse Liver CRISPR Knockout Library | This paper | Addgene Catalog #: 192824 |
| Plasmid: pCMV-VSV-G | Bob Weinberg | Addgene Catalog #: 8454 |
| Plasmid: psPAX2 | Didier Trono | Addgene Catalog #: 12260 |
| Software and algorithms | ||
| Volocity | Quorum Technologies | https://www.volocity4d.com/ |
| STAR 2.6.1a | Dobin et al.52 | https://github.com/alexdobin/STAR |
| featureCounts 1.6 | Liao et al.53 | https://www.rdocumentation.org/packages/Rsubread/versions/1.22.2/topics/featureCounts |
| DESeq2 1.22.2 | Love et al.54 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie 1.2.2 | Langmead et al.55 | https://bowtie-bio.sourceforge.net/index.shtml |
| MAGeCK-RRA 0.5.9.2 | Li et al.19 | https://sourceforge.net/p/mageck/wiki/Home/ |
| GSEA 4.1.0 | Subramanian et al.56 | https://www.gsea-msigdb.org/gsea/index.jsp |
| R 3.6.0 or 4.2.1 | https://www.r-project.org/ | |
| Prism 9.4.1 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| Data from CRISPR screens in mouse embryonic stem (ES) cells | Tzelepis et al.,30 Shohat et al.31 | |
| Data from CRISPR screens in human hepatocellular carcinoma (HCC) cell lines | Meyers et al.,32 Dempster et al.,33 Dempster et al.34 Pacini et al.35 |
https://figshare.com/articles/dataset/DepMap_22Q2_Public/19700056/2 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kristin A. Knouse (knouse@mit.edu).
Materials availability
Plasmids generated in this study, including the pooled sgRNA library, have been deposited at Addgene. Catalog numbers are listed in the key resources table.
Experimental model and subject details
Animals
C57BL/6J mice (strain 000664) and LSL-Cas9 mice (strain 026175) were purchased from the Jackson Laboratory. Mice were either singly- or group-housed with a 12-hour light-dark cycle (light from 7 AM to 7 PM, dark from 7 PM to 7 AM) in a specific-pathogen-free animal facility with unlimited access to food and water. Mating cages were supplemented with Eco Bedding (Pet Supplies Plus) to improve nest building and promote survival of neonatal mice. To deliver lentivirus, up to 100 μL of lentivirus in PBS was injected into the temporal vein of postnatal day one mice. For protein depletion tests, mice were injected with 1.25 × 107 transduction units (TU) of sgRNA-mCherry lentivirus. For the screen, mice were injected with 5 × 107 TU of sgRNA-mCherry lentiviral library. To deliver AAV-Cre, a stock solution of AAV8-TBG-Cre (Addgene 107787-AAV8) was diluted in PBS to a total volume of 20 μL and injected intraperitoneally into postnatal day five mice. For protein depletion tests and the screen, mice were injected with 2 × 1011 genome copies (GC) of AAV-TBG-Cre. To deliver AAV-shRNA, a stock solution of AAV8-mCherry-U6-scrmb-shRNA (AAV-shScramble, Vector Biolabs) and/or AAV8-GFP-U6-mNDST1-shRNA (AAV-shNDST1, Vector Biolabs) was diluted in PBS to a total volume of 20 μL and injected intraperitoneally into postnatal day five mice. To infect the entire liver to test protein depletion, mice were injected with 4 × 1011 GC of either AAV-shNDST1 or AAV-shScramble. To infect a subset of hepatocytes to compare proliferation, mice were injected with 1 × 1010 GC of both AAV-NDST1 and AAV-shScramble. All animal procedures were approved by the Massachusetts Institute of Technology Committee on Animal Care.
Cell lines
The male mouse hepatocyte cell line AML12 was purchased from the American Type Culture Collection (ATCC) and cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum, 10 μg/mL insulin, 5.5 μg/mL transferrin, 5 ng/mL selenium, and 40 ng/mL dexamethasone (ThermoFisher Scientific). HEK-293T cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin (ThermoFisher Scientific). All cell lines were cultured at 37°C with 5% CO2.
Method details
Vector construction
The pLentiCRISPRv2-Stuffer-HepmCherry and pLentiCRISPRv2-Stuffer-HepmTurquoise2 vectors were produced through the following steps: 1) removal of the EFS-NS promoter and Cas9 from the parental vector and insertion of a hepatocyte-specific promoter driving dsRed expression, 2) replacement of dsRed with mCherry or mTurquoise2, and 3) removal of the puromycin resistance cassette.
To produce pLentiCRISPRv2-Stuffer-HepdsRed-Puro, 100 ng of a synthetic gblock encoding the HS-CRM8-TTRmin module57 upstream of dsRed (Integrated DNA Technologies) and 1 μg of LentiCRISPRv2-Opti51 (gift from David Sabatini, Addgene plasmid #163126), a LentiCRISPRv2 derivative containing an optimized scaffold58 were digested sequentially with NheI and BamHI (New England Biolabs). The vector and fragment were purified using the QIAquick Gel Extraction Kit (Qiagen) and ligated with T4 DNA Ligase (New England Biolabs) in an 11 μL reaction to replace the EFS-NS promoter and Cas9 with the gblock fragment. 2.5 μL of the ligation was used to transform Stbl2 cells (Invitrogen) and DNA was isolated from ampicillin-resistant colonies with the QIAprep Spin Miniprep Kit (Qiagen). Clones were verified by Sanger sequencing (Quintara Biosciences) prior to retransformation and maxiprep using the ZymoPURE II Plasmid Maxiprep Kit (Zymo Research).
HS-CRM8-TTRmin-dsRed:
GAATTCGCTAGCACCGGCGCGCCGGGGGAGGCTGCTGGTGAATATTAACCAAGGTCACCCCAGTTATCGGAGGAGCAAACAGGGGCTAAGTCCACACGCGTGGTACCGTCTGTCTGCACATTTCGTAGAGCGAGTGTTCCGATACTCTAATCTCCCTAGGCAAGGTTCATATTTGTGTAGGTTACTTATTCTCCTTTTGTTGACTAAGTCAATAATCAGAATCAGCAGGTTTGGAGTCAGCTTGGCAGGGATCAGCAGCCTGGGTTGGAAGGAGGGGGTATAAAAGCCCCTTCACCAGGAGAAGCCGTCACACAGATCCACAAGCTCCTGACCGGTTCTAGAGCGCTGCCACCATGGTGCGCTCCTCCAAGAACGTCATCAAGGAGTTCATGCGCTTCAAGGTGCGCATGGAGGGCACCGTGAACGGCCACGAGTTCGAGATCGAGGGCGAGGGCGAGGGCCGCCCCTACGAGGGCCACAACACCGTGAAACTGAAGGTGACCAAGGGCGGCCCCCTGCCCTTCGCCTGGGACATCCTGTCCCCCCAGTTCCAGTACGGCTCCAAGGTGTACGTGAAGCACCCCGCCGACATCCCCGACTACAAGAAGCTGTCCTTCCCCGAGGGCTTCAAGTGGGAGCGCGTGATGAACTTCGAGGACGGCGGCGTGGTGACCGTGACCCAAGACTCCTCCCTGCAGGACGGCTGCTTCATCTACAAGGTGAAGTTCATCGGCGTGAACTTCCCCTCCGACGGCCCCGTAATGCAGAAGAAGACCATGGGCTGGGAGGCCTCCACCGAGCGCCTGTACCCCCGCGACGGCGTGCTGAAGGGCGAAATCCACAAGGCCCTGAAGCTGAAGGACGGCGGCCACTACCTGGTGGAGTTCAAGTCCATCTACATGGCCAAGAAGCCCGTGCAGCTGCCCGGCTACTACTACGTGGACTCCAAGCTGGACATCACCTCCCACAACGAGGACTACACCATCGTGGAGCAGTACGAGCGCACCGAAGGCCGCCACCACCTGTTCCTGGGATCCGGCGCAACAAACTTCTCTCTGCTGAAACAAGCCGGAGATGTCGAAGAGAATCCTGGACCGACCGAG
To construct pLentiCRISPRv2-Stuffer-HepmCherry-Puro and pLentiCRISPRv2-Stuffer-HepmTurquoise2-Puro, mCherry and mTurquoise2 were amplified from pKC027 and mTurquoise2-CMV (gifts from Iain Cheeseman), respectively, for 25 cycles with Q5 HotStart Polymerase (New England Biolabs) using the following primers:
pLC_EBFP2_F: 5′-GGTTCTAGAGCGCTGCCACCATGGTGAGCAAGGGCGAGGAG-3′
pLC_EBFP2_R: 5′-GCCGGATCCCTTGTACAGCTCGTCCATGCC-3′
Amplicons and pLentiCRISPRv2-Stuffer-HepdsRed-Puro were digested with XbaI and BamHI HF (New England Biolabs) and purified, ligated, transformed, and DNA was isolated and sequence verified as above.
To construct pLentiCRISPRv2-Stuffer-HepmCherry and pLentiCRISPRv2-Stuffer-HepmTurquoise2, a fragment encompassing the WPRE and 3′LTR was amplified from pLentiCRISPRv2-Stuffer-HepmCherry-Puro as above using the following primers:
Puro_removal_F: 5′-CGGCATGGACGAGCTGTACAAGTAATGAACGCGTTAAGTCGACAATCAACC-3′
Puro_removal_R: 5′-TCGAGGCTGATCAGCGGGTTTAAAC-3′
pLentiCRISPRv2-Stuffer-HepmCherry-Puro (or -HepmTurquoise2-Puro) was digested with BsrGI-HF and PmeI (New England Biolabs) and purified as above. NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) was used to assemble 25 ng each of vector and fragment in a 20 μL reaction for 15 min at 50°C. 50 μL of DH5-alpha cells were transformed with 2 μL assembly mix, and DNA was isolated and sequence verified as described above.
Individual sgRNAs were cloned as previously described,59 using the following oligonucleotides (Integrated DNA Technologies):
sgAAVS1_F: 5′-CACCGGGGCCACTAGGGACAGGAT-3′
sgAAVS1_R: 5′-AAACATCCTGTCCCTAGTGGCCCC-3′
sgMaob_F: 5′-CACCGACGGATAAAGGATATACTTG-3′
sgMaob_R: 5′-AAACCAAGTATATCCTTTATCCGTC-3′
sgLmnb2_F: 5′-CACCGAGGTACGGGAGACCCGACGG-3′
sgLmnb2_R: 5′-AAACCCGTCGGGTCTCCCGTACCTC-3′
Lentivirus preparation and concentration
HEK-293T cells were seeded at a density of 750,000 cells/mL in 20 mL viral production medium (IMDM, Thermo Fisher Scientific, supplemented with 20% heat-inactivated fetal bovine serum, GeminiBio) in T175 flasks. After 24 hours, media was changed to fresh viral production medium. At 32 hours post-seeding, cells were transfected with a mix containing 76.8 μL Xtremegene-9 transfection reagent (Thermo Fisher Scientific), 3.62 μg pCMV-VSV-G (gift from Bob Weinberg, Addgene plasmid #8454), 8.28 μg psPAX2 (gift from Didier Trono, Addgene plasmid #12260), and 20 μg sgRNA plasmid in Opti-MEM (Thermo Fisher Scientific) to a final volume of 1 mL. Media was changed 16 hours later to 55 mL of fresh viral production medium. At 48 hours after transfection, virus was collected and filtered through a 0.45 μm filter, aliquoted, and stored at −80°C until use.
To determine lentivirus titer, AML12 cells were transduced with a dilution series of lentivirus in the presence of 10 μg/mL polybrene for 16 hours. After four days, cells were harvested for flow cytometry analysis to determine percent of mTurquoise2-or mCherry-positive cells.
To concentrate lentivirus, lentiviral supernatant was ultracentrifuged at 23,000 RPM at 4°C for 2 hours in an SW 32 Ti swinging bucket rotor (Beckman Coulter). After centrifugation, media was decanted and pellets were air-dried at room temperature for 15 minutes. Pellets were then resuspended in PBS at room temperature for 30 minutes with gentle trituration. Concentrated lentivirus in PBS was stored for up to one week at 4°C prior to injection into mice.
Immunostaining
Livers were harvested and fixed in 4% paraformaldehyde in PBS at room temperature for 16–24 hours. Tissues were then washed with PBS and frozen in O.C.T. Compound (Tissue-Tek). Tissue sections of 12 to 30 μm thickness were prepared using a cryostat and adhered to Superfrost Plus Slides (Fisher Scientific). Slides were stored at −20°C until use. To visualize endogenous mCherry and mTurq2 fluorescence, slides were dried at room temperature for 15 minutes, rehydrated in PBS for 5 minutes, permeabilized with 1% Triton X-100 in PBS for 15 minutes, and counterstained with Alexa Fluor 488 Phalloidin (ThermoFisher Scientific) diluted 1:500 in blocking buffer (3% bovine serum albumin and 0.3% Triton X-100 in PBS). To immunostain for endogenous proteins, slides were dried at room temperature for 4–24 hours and rehydrated in PBS for 5 minutes. Antigen retrieval was then performed by pressure cooking slides in sodium citrate buffer (10 mM tri-sodium citrate dihydrate, 0.05% Tween 20, pH 6.0) for 20 minutes in an Instant Pot (Amazon). Slides were rinsed in PBS for 5 minutes, dried briefly, and sections outlined with an ImmEdge hydrophobic pen (Vector Laboratories). Sections were permeabilized with 1% Triton X-100 in PBS for 15 minutes and blocked with blocking buffer for one hour. Sections were then incubated in primary antibodies diluted in blocking buffer at room temperature for 12–24 hours. Sections were washed with blocking buffer three times for 10 minutes each. Sections were then incubated in AlexaFluor secondary antibodies (ThermoFisher Scientific) diluted 1:1,000 in blocking buffer at room temperature for 1–2 hours. In some cases, 5 μg/mL Hoechst 33342 (ThermoFisher Scientific) was added to the secondary antibody solution. Sections were washed with blocking buffer twice for 10 minutes each followed by one wash with PBS for 5 minutes. Slides were then mounted in Pro-Long Gold Antifade reagent (ThermoFisher Scientific).
The following primary antibodies were used: Cas9 (1:200, clone 7A9-3A3, Abcam ab191468), asialoglycoprotein receptor 1 (ASGR1) (1:500, clone 114, Sino Biological 50083-R114), mCherry (1:500, clone 16D7, ThermoFisher Scientific M11217), monoamine oxidase B (MAO-B) (1:1,000, Novus Biologicals NBP1-87493), lamin B2 (1:1,000, clone EPR9701(B), Abcam ab151735), actin (1:250, clone AC-74, Sigma Aldrich A2228), CD45 (1:500, Abcam ab10558), Ki67 (1:200, clone SP6, Abcam ab16667), mCherry (1:2,000, Abcam ab167453), GFP (1:1,000, Abcam ab13970), and Ki67 (1:200, clone SolA15, ThermoFisher Scientific 14-5698-80). The Cas9 antibody was directly conjugated to Alexa Fluor 647 using the Alexa Fluor 647 Antibody Labeling Kit (ThermoFisher Scientific). The actin antibody was directly conjugated to DyLight 405 using the DyLight 405 antibody labeling kit (ThermoFisher Scientific).
Image analysis
Images were acquired using either a CSU-22 spinning disc confocal head (Yokogawa) with Borealis modification (Andor) mounted on an Axiovert 200M microscope (Zeiss) with 10X or 40X objectives (Zeiss), an Orca-ER CCD camera (Hamamatsu), and MetaMorph acquisition software (Molecular Devices) or a McBain-Yokogawa spinning disk confocal head mounted on a Nikon Ti microscope with 20X objective (Nikon), a Clara CCD camera (Andor), and NIS Elements acquisition software (Nikon).
Images were analyzed using Volocity (Quorum Technologies). To determine the number of hepatocytes in the postnatal day one liver, liver volume was measured by volume displacement and hepatocyte volume was measured by immunostaining postnatal day one liver sections for the hepatocyte marker ASGR1 and actin. The proportion of a section occupied by ASGR1-positive cells was calculated to determine the proportion of liver volume comprised by hepatocytes and the hepatocyte volume was determined by measuring the x, y, and z dimensions of single hepatocytes, multiplying these three dimensions to calculate the volume of each hepatocyte, and averaging this volume across at least 29 cells per liver. To measure MAO-B and lamin B2 intensity, a single Z plane at the center of the cell was identified and the cytoplasm or nucleus was outlined to measure the signal intensity per μm. A similar procedure was done on sections stained only with secondary antibodies to calculate the average background intensity. This average background intensity was subtracted from each MAO-B and lamin B2 intensity measurement and the background-subtracted measurements were then normalized within a given sample (mCherry-positive or negative hepatocytes within a single liver). To measure proliferation in AAV-shRNA-infected livers, GFP-positive and mCherry-positive hepatocytes were first identified on the basis of GFP and mCherry signal alone. Once identified, these hepatocytes were analyzed for their Ki67 signal and scored as positive or negative for Ki67.
Hematoxylin and eosin staining
Livers were harvested and fixed in 4% paraformaldehyde in PBS at room temperature for 16–24 hours. Livers were embedded in Paraplast X-tra paraffin (Leica Biosystems). Tissue sections of 4 μm thickness were prepared using a microtome. Sections were stained with hematoxylin (3 minutes, Leica Biosystems) and eosin (10 seconds, Leica Biosystems) on a Tissue-Tek Prisma automated slide stainer (Sakura) and coverslipped on a Tissue Tek Glas g2 automated coverslipper (Sakura).
Immunoblotting
To prepare protein lysates from liver tissue, 50 mg of liver was homogenized in 1 mL of RIPA buffer (50 mM Tris pH 8.0, 150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing cOmplete protease inhibitors (Roche) using a Bio-Gen PRO200 handheld homogenizer (PRO Scientific). Homogenate was centrifuged at 18,000 G at 4°C for 20 minutes. Supernatant was combined with 5X sample buffer (250 mM Tris pH 6.8, 50% glycerol, 0.025% bromophenol blue, 5% sodium dodecyl sulfate, 5% beta-mercaptoethanol). Samples were separated on homemade polyacrylamide gels and transferred to Immobilon-FL membranes (Millipore) via wet transfer. Membranes were blocked in 5% milk in TBST (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween-20) for 1 hour at room temperature. Membranes were incubated in primary antibody diluted in blocking solution at 4 °C with rocking overnight and washed with TBST for 5 minutes five times. Membranes were incubated in HRP-conjugated secondary antibody diluted in blocking solution at room temperature with rocking for one hour and washed with TBST for five minutes five times. Membranes were incubated in ECL Prime Western Blotting Detection Reagent (GE Healthcare) for five minutes and imaged on an ImageQuant LAS 4000 imager (GE Healthcare).
The following primary antibodies were used: NDST1 (1:1,000, Sigma SAB1307040) and beta-actin (1:10,000, clone AC-74, Sigma A2228).
The following secondary antibodies were used: Rabbit (1:50,000, Abcam ab205718) and mouse (1:10,000, Abcam ab205719).
RNA sequencing
For surgical resection time points, partial hepatectomies were performed on 8 week-old mice as previously described.60 For toxic injury time points, 8 week-old mice were injected intraperitoneally with 2 μL/gram of 25% carbon tetrachloride diluted in corn oil (Sigma Aldrich). For all time points, livers from three male C57BL/6J mice were harvested, flushed with PBS, immediately immersed in RNAlater (Qiagen), incubated at room temperature for 24 hours, and stored at −20°C until future use. To isolate RNA, 30 mg of each tissue was removed from RNAlater and homogenized in 700 μL of QIAzol lysis reagent (Qiagen) using the TissueRuptor homogenizer (Qiagen). RNA was purified using the miRNeasy Kit (Qiagen) according to kit instructions and eluted in 30 μL of nuclease-free water. RNA sequencing libraries were prepared using KAPA mRNA Hyper-Prep Kit (KAPA Biosystems) according to manufacturer instructions. Briefly, 0.1–1 μg of total RNA was enriched for polyadenylated sequences using oligo-dT magnetic bead capture. The enriched mRNA fraction was then fragmented and first-strand cDNA generated using random primers. Strand specificity was achieved during second-strand cDNA synthesis by replacing dTTP with dUTP to quench the second strand during amplification. The resulting cDNA was A-tailed and ligated with indexed adapters. The library was amplified using a DNA polymerase that cannot incorporate past dUTPs to quench the second strand during PCR. The libraries were quantified using a KAPA qPCR Library Quantification Kit (KAPA Biosystems) as per manufacturer instructions. The samples were sequenced on a HiSeq 2500 (Illumina) based on qPCR concentrations. Base calls were performed by the instrument control software and further processed using the Offline Base Caller version 1.9.4 (Illumina). Samples were mapped with STAR version 2.6.1a52 to the mouse genome release mm10, using a gtf file from ENSEMBL version GRCm38.91, and setting the maximum intron length (“alignIntronMax”) parameter to 50000. featureCounts version 1.653 was run to assign reads to genes using the same gft file and setting “-s” parameter to 2. Gene counts were normalized with DESeq2 version 1.22.254. FPKMs were calculated using the function fpkm within the DESeq2 package. The FPKM values for the three replicates were averaged, and protein coding genes were selected based on the annotation in the gtf file.
sgRNA library preparation
Genes with an average FPKM >0.3 in any of the RNA sequencing time points were chosen to build a liver transcriptome-wide library. sgRNA sequences were designed using the Broad Institute GPP sgRNA Designer18,61 using the Azimuth 2.0 rule set. For genes which were not identified by the program, alternative gene names from ENSEMBL versions GRCm38.76 to 38.93 were attempted. A small number of designed sgRNAs targeted multiple genes; the sgRNA names and gene names were manually annotated to indicate all targeted genes for these cases. Non-targeting and control-gene-targeting sgRNAs18 were also included. sgRNA sequences from this control set that were identical to a sequence already in our library were annotated according to the targeted gene; those that did not overlap with sequences in our sgRNA library were annotated as control sgRNAs. The library contains 71,878 sgRNAs targeting 13,189 genes.
For sgRNAs beginning with a nucleotide other than G, a G was prepended. The following adapters were added to all sgRNA sequences:
Upstream: 5′-TATCTTGTGGAAAGGACGAAACACC-3′
Downstream: 5′-GTTTAAGAGCTATGCTGGAAACAGCATAGC-3′
Multiple rounds of cloning were combined to generate the final plasmid library. The oligonucleotide library (Agilent Technologies) was amplified for 16 cycles using Q5 HotStart Polymerase (New England Biolabs) using a gradient annealing temperature ranging from 50-62°C across 8, 50 μL reactions using the forward primer 5′-TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACC-3′ and the reverse primer 5′-ATTTAAACTTGCTATGCTGTTTCCAGCATAGCTCTTAAAC-3′ and the following program:
| 1 cycle | 98°C | 2 min |
| 16 cycles | 98°C | 10 sec |
| 50–62°C | 15 sec | |
| 72°C | 15 sec | |
| 1 cycle | 72°C | 2 min |
| 1 cycle | 10 °C | hold |
Reactions were pooled and purified by DNA Clean and Concentrator 5 (Zymo Research). pLentiCRISPRv2-Stuffer-HepmCherry was digested as described59 and either gel purified using a Zymoclean Gel DNA Recovery Kit (Zymo Research) followed by Ampure XP bead purification (Beckman Coulter) or DNA Clean and Concentrator 5. The library was assembled using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) in 4 × 20 μL reactions at 50°C for 1 h using 100 ng of vector per 5–10 ng of PCR amplicon. For each round of cloning, the reactions were combined and 2.5 μL of the assembly reaction or a control reaction without amplicon were used to transform NEB5-alpha cells (New England Biolabs) to measure background assembly. Subsequently, the assembly reactions were concentrated using Ampure XP beads, resuspended in 8 μL water, and used to electroporate 1–4 tubes of Endura electrocompetent cells (Biosearch Technologies) at 1.8 kV distributed over 2 cuvettes (0.1 cm gap width) per tube using a Micropulser Electroporator (Bio-Rad Laboratories). 10-fold serial dilutions of a 10 μL aliquot were plated on LB plates with ampicillin at 100 μg/mL to assess electroporation efficiency, and the remainder of each electroporation (2 cuvettes) was plated on LB agar supplemented with 100 μg/mL ampicillin in 4 × 245 mm square bioassay dishes (Corning). Plates were incubated overnight at 30°C and colonies were scraped the next morning. DNA was isolated using the ZymoPURE II Plasmid Maxiprep Kit (Zymo Research). Plasmid DNA from multiple rounds of assembly and electroporation were combined according to the measured electroporation efficiency to achieve 25-fold coverage of the library. sgRNA representation was measured by high-throughput sequencing as described below.
To improve coverage of some of the sgRNAs in the library, a second library containing ∼7,500 sgRNAs was synthesized and cloned as above, with the following modifications: assembly was performed using NEB Gibson Assembly mix (New England Biolabs) using a ratio of 200 ng vector: 10 ng sgRNA in each 20 μL reaction, and the final combined and concentrated reaction was used to electroporate a single tube of Endura cells.
Subsequent propagation of the plasmid library was performed using 50 ng plasmid library per single tube of Endura cells.
All steps were performed according to manufacturer’s instructions, except where noted.
Genomic DNA isolation
Livers were harvested from mice, separated into individual lobes, minced into 15 mg pieces using a razor blade, snap-frozen in liquid nitrogen, and stored at −80°C until use. Genomic DNA (gDNA) was isolated from livers using the illustra blood genomicPrep Mini Spin Kit (Cytiva) using one column for every 7.5 mg of tissue. The manufacturer’s protocol was used with the following modifications: 20 μL of 10 mg/mL Proteinase K (Millipore-Sigma) solution in water was added per 7.5 mg of tissue. Tissue was disrupted by thoroughly pipetting prior to adding lysis buffer, vortexing, and incubating at 56°C overnight. Elution was performed using 25 μL of water pre-heated to 70°C. Samples were combined by lobe and concentration was measured using the Qubit dsDNA HS Assay Kit (Invitrogen).
For the induction samples, equal amounts of gDNA from each lobe were combined within each mouse, and equal inputs from four mice were combined to prepare a single sequencing library. For the endpoint samples, gDNA from each lobe within a mouse was combined proportionally to the average lobe mass across mice measured at liver harvest. A sequencing library was prepared for each mouse individually using equal total gDNA input per mouse.
Sequencing library preparation and sequencing
All PCR reactions were performed in 50 μL reactions using ExTaq Polymerase (Takara Bio) with the following program:
| 1 cycle | 95°C | 5 min |
| 14 or 28 cycles | 95°C | 10 sec |
| 60°C | 15 sec | |
| 72°C | 45 sec | |
| 1 cycle | 72°C | 5 min |
| 1 cycle | 4°C | hold |
Using the following primers:
Forward: 5′-AATGATACGGCGACCACCGAGATCTACACCGACTCGGTGCCACTTTT-3′
Reverse: 5′-CAAGCAGAAGACGGCATACGAGATCnnnnnnTTTCTTGGGTAGTTTGCAGTTTT-3′
Where “nnnnnn” denotes the barcode used for multiplexing.
10 ng of plasmid DNA was amplified for 14 cycles in 4 × 50 μL reactions. 1, 3, or 6 μg of gDNA was initially amplified for 28 cycles in 50 μL test PCR reactions. Subsequently, 226 μg of gDNA (induction) was used in 38 reactions, or 75 μg of gDNA (endpoint) was used in 25 reactions per mouse. All reactions were cleaned and concentrated using Ampure XP beads prior to sequencing for 50 cycles on an Illumina Hiseq 2500 using the following primers:
Read 1 sequencing primer: 5′-GTTGATAACGGACTAGCCTTATTTAAACTTGCTATGCTGTTTCCAGCATAGCTCTTAAAC-3′
Index sequencing primer: 5′-TTTCAAGTTACGGTAAGCATATGATAGTCCATTTTAAAACATAATTTTAAAACTGCAAACTACCCAAGAAA-3′
Base calls were performed by the instrument control software and further processed using the Offline Base Caller (Illumina) v. 1.9.4.
Screen analysis
For initial measurement of sgRNA representation in the plasmid library or induction time point, sequencing reads were mapped to the library using Bowtie.55 First, a tab-delimited text file containing the sgRNA name in the first column and the sgRNA sequence in the second column was converted to fasta format using the following command: awk '{print ">"$1"∖n"$2}' <input tab delimited file> > <output fasta file>. Next, a Bowtie index of the sgRNA sequences was built using the following command: bowtie-build <input fasta file> <basename for bowtie index>. The fastq file reads were then aligned and a sam output file was generated using the following options: bowtie -3 30 -n 0 -l 20 -y -a -p 4 --nofw -S <BOWTIE INDEX> <input fastq file> <output sam filename>. Each sgRNA instance was counted and a tab-delimited summary file was generated using the following command: grep -v ˆ∖@ <sam alignment file> | awk -F"∖t" '{ if($2 = = "16" && $4 = 1 && $13 = = "MD:Z:20") print $3 }' | sort | uniq -c | awk '{ print $2"∖t"$1 }' > <out file with.count extension>. Raw counts were processed using MAGeCK for downstream analysis.19 The plasmid library and induction timepoint were used as control samples and to estimate variance, and each endpoint mouse was processed separately. For mouse 4, sgLmnb2_1 was removed prior to MAGeCK analysis, as the high representation of this sgRNA (an sgRNA used for development of the screening method) was likely due to contamination during sequencing library preparation. Counts data from screens in mouse ES cells from Tzelepis et al.30 (day 14) and Shohat et al.31 (day 18) were processed individually using MAGeCK. The corresponding plasmid libraries were used as control samples. For our screen, Shohat et al., and Tzelepis et al., the null distribution was generated using the matched control sgRNA set (Table S2). For Tzelepis et al., the three replicate day 14 samples were processed together to generate a single gene score, and those three samples were used to estimate variance. For all screens, the gene test FDR threshold was set to 0.05, the sgRNA p value was FDR-adjusted, and the gene score was calculated using the median. Twenty human hepatocellular carcinoma screens from the CRISPR (Chronos) Public 22Q2 release were downloaded from the Broad DepMap portal using “Liver” as a lineage filter and “Hepatocellular Carcinoma” as a lineage subtype filter32,33,34,35 (https://figshare.com/articles/dataset/DepMap_22Q2_Public/19700056/2).
All downstream analyses were performed in R version 3.6.0 or 4.2.1, and all plots were generated in either base R, using the R corrplot package, or in GraphPad Prism Version 9.4.1. Each sgRNA was given a pseudocount of 1 before calculating reads per million (RPM). For comparisons within our screen, the gene scores from individual mice were not normalized across mice, as each mouse serves as a replicate screen. The gene score for each gene across mice was tested against all gene scores using an unpaired two-sample Wilcoxon test. The p values from this test were adjusted using the Benjamini-Hochberg (FDR) procedure. The median log2 fold change across mice was used as input for pre-ranked gene set enrichment analysis (GSEA)56 using the c2.cp.kegg.v7.1.symbols.gmt gene sets. For comparisons between screens from different sources, all screens from all the sources in the specific comparison were quantile normalized to one another using the preprocessCore R package prior to calculating the median log2 fold change within the screens from each source. This normalized median log2 fold change was subtracted from the normalized median log2 fold change of our screens to generate a differential score used as input for pre-ranked GSEA using the c2.cp.kegg.v7.1.symbols.gmt gene sets. For converting mouse gene symbols to human gene symbols, the Mouse_Gene_Symbol_Remapping_to_Human_Orthologs_MSigDB.v7.1.chip was used (note that this excludes genes that have multiple annotations in either human or mouse).
Pearson correlation was used to compare gene effects between mice. Spearman correlation was used to compare gene effects with liver mRNA expression and protein half-life in hepatocytes.16 The TUSON dataset of predicted tumor suppressor genes was sorted by ascending FDR q value and the top 50 genes present in the compared datasets were used.36 Distribution differences were tested and p values were calculated using the Kolmogorov-Smirnov test. A one-sided test was used for gene sets for which a phenotype could be predicted (core essential genes, tumor suppressor genes, and control enriched and depleted genes); all other comparisons used a two-sided test.
For comparison of sex-specific fitness effects, only genes with an average of >2 sgRNAs detected across mice were considered. For sex-specific enriched genes, a median fold change (log2) > 0.5 across two mice of a given sex and an absolute median fold change (log2) difference of >0.25 compared to the other sex was required. To identify tumor-suppressor-like genes, a median fold change (log2) > −0.5 was required in mice of the other sex. For sex-specific depleted genes, a median fold change (log2) of < −0.5 across two mice of a given sex and an absolute median fold change (log2) difference of >0.75 compared to the other sex was required. To identify sex-specific essential genes, a median fold change (log2) > −0.5 was required in mice of the other sex.
Quantification and statistical analysis
Statistical analysis
The statistical details for any given experiment are provided in the corresponding figure legend. Additional information about statistical analysis can be found in the relevant Method Details sections.
Software
STAR version 2.6.1a
Conda version 4.9.2
Bowtie version 1.2.2
MAGeCK-RRA version 0.5.9.2
R version 3.6.0 or 4.2.1
featureCounts version 1.6
DESeq2 version 1.22.2
preprocessCore version 1.48.0
corrplot version 0.84
GSEA version 4.1.0
Mouse_Gene_Symbol_Remapping_to_Human_Orthologs_MSigDB.v7.1.chip
Human_Symbol_with_Remapping_MsigDB.v7.1.chip
c2.cp.kegg.v7.1.symbols.gmt
GraphPad Prism version 9.4.1
Acknowledgments
We thank Mehreen Khan and Keya Viswanathan of the Whitehead Institute Functional Genomics Platform for technical assistance; Amanda Chilaka and Sumeet Gupta of the Whitehead Institute Genome Technology Core for high-throughput sequencing; Inma Barrasa of the Whitehead Institute Bioinformatics and Research Computing Core for RNA sequencing analysis; the Whitehead Institute Flow Cytometry Core; the Whitehead Institute Keck Microscopy Facility; the Koch Institute Swanson Biotechnology Center Microscopy Facility; and the Koch Institute Swanson Biotechnology Center Histology Facility. We thank Agilent Technologies for their commitment to supporting academic research. We thank Iain Cheeseman, Jonathan Weissman, Peter Reddien, Raghu Chivukula, Sharon Grossman, Tobiloba Oni, and members of the Knouse laboratory for their comments on the manuscript. This work was supported by the National Institutes of Health NIH Director’s Early Independence Award (DP5-OD026369) to K.A.K. and the National Cancer Institute Koch Institute Support Grant (P30-CA14051). K.A.K. was also supported by the Scott Cook and Signe Ostby Fund.
Author contributions
H.R.K. and K.A.K. conceived the project, performed all experiments, analyzed the data, and wrote the manuscript.
Declaration of interests
H.R.K. and K.A.K. are co-inventors on a patent filed by the Whitehead Institute related to work in this manuscript.
Published: November 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100217.
Supplemental information
Average RNA expression (FPKM) for genes expressed in liver during development, adulthood, and regeneration. n = 3 male mice per time point.
Sequences for all sgRNAs used in our library and list of control sgRNAs used in our library, Shohat et al. 2019, and Tzelepis et al. 2016.
Count data for all sgRNAs in the plasmid library, induction library, and individual endpoint libraries, gene scores across mice with test for depletion, gene scores across mice with test for enrichment, list of control genes used to validate screen.
Depleted gene sets in our screen, depleted gene sets in our screen versus mouse embryonic stem (ES) cell screens, depleted gene sets in our screen versus human hepatocellular carcinoma (HCC) cell line screens, and gene scores for genes in indicated gene sets in our screen, mouse ES cell screens, and human HCC cell line screens.
Data and code availability
-
•
Sequencing data generated in this study have been deposited at GEO and SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beronja S., Janki P., Heller E., Lien W.-H., Keyes B.E., Oshimori N., Fuchs E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature. 2013;501:185–190. doi: 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wertz M.H., Mitchem M.R., Pineda S.S., Hachigian L.J., Lee H., Lau V., Powers A., Kulicke R., Madan G.K., Colic M., et al. Genome-wide in vivo CNS screening identifies genes that modify CNS neuronal survival and mHTT toxicity. Neuron. 2020;106:76–89.e8. doi: 10.1016/j.neuron.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S., Sanjana N.E., Zheng K., Shalem O., Lee K., Shi X., Scott D.A., Song J., Pan J.Q., Weissleder R., et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong M.B., Wang G., Chow R.D., Ye L., Zhu L., Dai X., Park J.J., Kim H.R., Errami Y., Guzman C.D., et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell. 2019;178:1189–1204.e23. doi: 10.1016/j.cell.2019.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfeifer A., Kessler T., Yang M., Baranov E., Kootstra N., Cheresh D.A., Hoffman R.M., Verma I.M. Transduction of liver cells by lentiviral vectors: analysis in living animals by fluorescence imaging. Mol. Ther. 2001;3:319–322. doi: 10.1006/mthe.2001.0276. [DOI] [PubMed] [Google Scholar]
- 8.Waddington S.N., Mitrophanous K.A., Ellard F.M., Buckley S.M.K., Nivsarkar M., Lawrence L., Cook H.T., Al-Allaf F., Bigger B., Kingsman S.M., et al. Long-term transgene expression by administration of a lentivirus-based vector to the fetal circulation of immuno-competent mice. Gene Ther. 2003;10:1234–1240. doi: 10.1038/sj.gt.3301991. [DOI] [PubMed] [Google Scholar]
- 9.Wang G., Chow R.D., Ye L., Guzman C.D., Dai X., Dong M.B., Zhang F., Sharp P.A., Platt R.J., Chen S. Mapping a functional cancer genome atlas of tumor suppressors in mouse liver using AAV-CRISPR–mediated direct in vivo screening. Sci. Adv. 2018;4:eaao5508. doi: 10.1126/sciadv.aao5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X., Calvisi D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014;184:912–923. doi: 10.1016/j.ajpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu C., Qi X., Du X., Zou H., Gao F., Feng T., Lu H., Li S., An X., Zhang L., et al. piggyBac mediates efficient in vivo CRISPR library screening for tumorigenesis in mice. Proc. Natl. Acad. Sci. USA. 2017;114:722–727. doi: 10.1073/pnas.1615735114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu M., Lu T., Jia Y., Luo X., Gopal P., Li L., Odewole M., Renteria V., Singal A.G., Jang Y., et al. Somatic mutations increase hepatic clonal fitness and regeneration in chronic liver disease. Cell. 2019;177:608–621.e12. doi: 10.1016/j.cell.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia Y., Li L., Lin Y.-H., Gopal P., Shen S., Zhou K., Yu X., Sharma T., Zhang Y., Siegwart D.J., et al. In vivo CRISPR screening identifies BAZ2 chromatin remodelers as druggable regulators of mammalian liver regeneration. Cell Stem Cell. 2022;29:372–385.e8. doi: 10.1016/j.stem.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen T.H., Bellodi-Privato M., Aubert D., Pichard V., Myara A., Trono D., Ferry N. Therapeutic lentivirus-mediated neonatal in vivo gene therapy in hyperbilirubinemic Gunn rats. Mol. Ther. 2005;12:852–859. doi: 10.1016/j.ymthe.2005.06.482. [DOI] [PubMed] [Google Scholar]
- 15.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M., et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathieson T., Franken H., Kosinski J., Kurzawa N., Zinn N., Sweetman G., Poeckel D., Ratnu V.S., Schramm M., Becher I., et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018;9:689. doi: 10.1038/s41467-018-03106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W., Köster J., Xu H., Chen C.-H., Xiao T., Liu J.S., Brown M., Liu X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015;16:281. doi: 10.1186/s13059-015-0843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viswanatha R., Li Z., Hu Y., Perrimon N. Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells. Elife. 2018;7 doi: 10.7554/elife.36333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hart T., Tong A.H.Y., Chan K., Leeuwen J.V., Seetharaman A., Aregger M., Chandrashekhar M., Hustedt N., Seth S., Noonan A., et al. Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3 (Bethesda) 2017;7:2719–2727. doi: 10.1534/g3.117.041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ally A., Balasundaram M., Carlsen R., Chuah E., Clarke A., Dhalla N., Holt R.A., Jones S.J.M., Lee D., Ma Y., et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grompe M., Al-Dhalimy M., Finegold M., Ou C.-N., Burlingame T., Kennaway N.G., Soriano P. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7:2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 25.Takehara T., Tatsumi T., Suzuki T., Rucker E.B., III, Hennighausen L., Jinushi M., Miyagi T., Kanazawa Y., Hayashi N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 26.Rimkunas V.M., Graham M.J., Crooke R.M., Liscum L. In vivo antisense oligonucleotide reduction of NPC1 expression as a novel mouse model for Niemann Pick type C– associated liver disease. Hepatology. 2008;47:1504–1512. doi: 10.1002/hep.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vick B., Weber A., Urbanik T., Maass T., Teufel A., Krammer P.H., Opferman J.T., Schuchmann M., Galle P.R., Schulze-Bergkamen H. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology. 2009;49:627–636. doi: 10.1002/hep.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cousin M.A., Conboy E., Wang J.-S., Lenz D., Schwab T.L., Williams M., Abraham R.S., Barnett S., El-Youssef M., Graham R.P., et al. RINT1 Bi-allelic variations cause infantile-onset recurrent acute liver failure and skeletal abnormalities. Am. J. Hum. Genet. 2019;105:108–121. doi: 10.1016/j.ajhg.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M., et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzelepis K., Koike-Yusa H., Braekeleer E.D., Li Y., Metzakopian E., Dovey O.M., Mupo A., Grinkevich V., Li M., Mazan M., et al. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016;17:1193–1205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shohat S., Shifman S. Genes essential for embryonic stem cells are associated with neurodevelopmental disorders. Genome Res. 2019;29:1910–1918. doi: 10.1101/gr.250019.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S., et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dempster J.M., Rossen J., Kazachkova M., Pan J., Kugener G., Root D.E., Tsherniak A. Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. bioRxiv. 2020;20:21–35. doi: 10.1101/720243. [DOI] [Google Scholar]
- 34.Dempster J.M., Boyle I., Vazquez F., Root D.E., Boehm J.S., Hahn W.C., Tsherniak A., McFarland J.M. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021;22:343. doi: 10.1186/s13059-021-02540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pacini C., Dempster J.M., Boyle I., Gonçalves E., Najgebauer H., Karakoc E., van der Meer D., Barthorpe A., Lightfoot H., Jaaks P., et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun. 2021;12:1661. doi: 10.1038/s41467-021-21898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davoli T., Xu A.W., Mengwasser K.E., Sack L.M., Yoon J.C., Park P.J., Elledge S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berletch J.B., Ma W., Yang F., Shendure J., Noble W.S., Disteche C.M., Deng X. Escape from X Inactivation varies in mouse tissues. PLoS Genet. 2015;11:10050799–e1005126. doi: 10.1371/journal.pgen.1005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bellott D.W., Hughes J.F., Skaletsky H., Brown L.G., Pyntikova T., Cho T.-J., Koutseva N., Zaghlul S., Graves T., Rock S., et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature. 2014;508:494–499. doi: 10.1038/nature13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cherepanova N., Shrimal S., Gilmore R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016;41:57–65. doi: 10.1016/j.ceb.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peaper D.R., Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu. Rev. Cell Dev. Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 42.Chan C.J., Smyth M.J., Martinet L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death Differ. 2014;21:5–14. doi: 10.1038/cdd.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Srivastava R.W.H.A. Innate immune responses to AAV Vectors. Front. Microbiol. 2011;2:194. doi: 10.3389/fmicb.2011.00194/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown B.D., Sitia G., Annoni A., Hauben E., Sergi L.S., Zingale A., Roncarolo M.G., Guidotti L.G., Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797–2805. doi: 10.1182/blood-2006-10-049312. [DOI] [PubMed] [Google Scholar]
- 45.Sarrazin S., Lamanna W.C., Esko J.D. Heparan sulfate proteoglycans. Csh Perspect Biol. 2011;3:a004952. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gopal S., Arokiasamy S., Pataki C., Whiteford J.R., Couchman J.R. Syndecan receptors: pericellular regulators in development and inflammatory disease. Open Biol. 2021;11 doi: 10.1098/rsob.200377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toledo A.G., Sorrentino J.T., Sandoval D.R., Malmström J., Lewis N.E., Esko J.D. A systems view of the heparan sulfate interactome. J. Histochem. Cytochem. 2020;69:105–119. doi: 10.1369/0022155420988661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huard J., Mueller S., Gilles E.D., Klingmüller U., Klamt S. An integrative model links multiple inputs and signaling pathways to the onset of DNA synthesis in hepatocytes. FEBS J. 2012;279:3290–3313. doi: 10.1111/j.1742-4658.2012.08572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan X., Yuan Y., Zeng G., Apte U., Thompson M.D., Cieply B., Stolz D.B., Michalopoulos G.K., Kaestner K.H., Monga S.P.S. β-Catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47:1667–1679. doi: 10.1002/hep.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cagno V., Tseligka E.D., Jones S.T., Tapparel C. Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses. 2019;11:596. doi: 10.3390/v11070596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adelmann C.H., Traunbauer A.K., Chen B., Condon K.J., Chan S.H., Kunchok T., Lewis C.A., Sabatini D.M. MFSD12 mediates the import of cysteine into melanosomes and lysosomes. Nature. 2020;588:699–704. doi: 10.1038/s41586-020-2937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 54.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;104:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chuah M.K., Petrus I., Bleser P.D., Guiner C.L., Gernoux G., Adjali O., Nair N., Willems J., Evens H., Rincon M.Y., et al. Liver-specific transcriptional modules identified by genome-wide in silico analysis enable efficient gene therapy in mice and non-human primates. Mol. Ther. 2014;22:1605–1613. doi: 10.1038/mt.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.-W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knouse K.A., Lopez K.E., Bachofner M., Amon A. Chromosome segregation fidelity in epithelia requires tissue architecture. Cell. 2018;175:200–211.e13. doi: 10.1016/j.cell.2018.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Average RNA expression (FPKM) for genes expressed in liver during development, adulthood, and regeneration. n = 3 male mice per time point.
Sequences for all sgRNAs used in our library and list of control sgRNAs used in our library, Shohat et al. 2019, and Tzelepis et al. 2016.
Count data for all sgRNAs in the plasmid library, induction library, and individual endpoint libraries, gene scores across mice with test for depletion, gene scores across mice with test for enrichment, list of control genes used to validate screen.
Depleted gene sets in our screen, depleted gene sets in our screen versus mouse embryonic stem (ES) cell screens, depleted gene sets in our screen versus human hepatocellular carcinoma (HCC) cell line screens, and gene scores for genes in indicated gene sets in our screen, mouse ES cell screens, and human HCC cell line screens.
Data Availability Statement
-
•
Sequencing data generated in this study have been deposited at GEO and SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.