

Abstract
Droplet-based microfluidic systems are ideally suited for the investigation of nucleation and crystallization processes. To best leverage the features of such platforms (including exquisite time resolution and high-throughput operation), sensitive and in situ detection schemes are needed to extract real-time chemical information about all species of interest. In this regard, the extension of conventional (UV, visible, and infrared) optical detection schemes to the X-ray region of the electromagnetic spectrum is of high current interest, as techniques such as X-ray absorption spectroscopy (XAS) provide for the element-specific investigation of the local chemical environment. Accordingly, herein, we report for the first time the integration of millisecond droplet-based microfluidics with XAS. Such a platform allows for the sensitive acquisition of X-ray absorption data from picoliter-volume droplets moving at high linear velocities. Significantly, the high-temporal resolution of the droplet-based microfluidic platform enables unprecedented access to the early stages of the reaction. Using such an approach, we demonstrate in situ monitoring of calcium carbonate precipitation by extracting XAS spectra at the early time points of the reaction with a dead time as low as 10 ms. We obtain insights into the kinetics of the formation of amorphous calcium carbonate (ACC) as a first species during the crystallization process by monitoring the proportion of calcium ions converted into ACC. Within the confined and homogeneous environment of picoliter-volume droplets, the ACC content reaches 60% over the first 130 ms. More generally, the presented method offers new opportunities for the real-time monitoring of fast chemical and biological processes.
Keywords: X-ray absorption, microfluidics, droplets, calcium carbonate
Introduction
In recent years, droplet-based microfluidic systems have emerged as powerful tools for the study of a variety of chemical and biological processes.1−5 In simple terms, droplet-based microfluidic systems generate, manipulate, and process discrete droplets contained within an immiscible carrier fluid. Such platforms enable the processing of monodisperse (femtoliter–nanoliter volume) droplets at rates in excess of tens of kilohertz and provide for precise control over each droplet in terms of its size, location, and chemical payload. The advantages of such platforms include low reagent consumption, efficient heat and mass transfer, and the ability to perform complex experiments in a high-throughput manner. Indeed, the ability to create and homogenize temperature or solute gradients within confined droplets makes them uniquely suited for the study of fast reactions.6,7 Moreover, mapping the position of each droplet as it moves downstream allows for the extraction of high-temporal resolution kinetic data, regardless of the acquisition time of the detection method being used. For example, in the case of reactive systems, it is possible to match volumetric flow rates and the locations where the system is interrogated in order to probe reaction times on a millisecond or sub-millisecond level.8 Indeed, it should be realized that droplet-based microfluidic systems are best exploited when combined with sensitive and high-throughput detection methods that are able to probe individual droplet payloads. To this end, a diversity of detection methods have been successfully used with droplet-based microfluidic platforms to allow for the real-time characterization of encapsulated species and reactions.9 That said, probing such small (normally picoliter) volumes is far from simple, due to reduced optical pathlengths and the fact each moving droplet can only be analyzed over a short time period.
As noted, in situ characterization tools are necessary for monitoring droplet contents in a rapid and efficient manner. In this context, a wide variety of optical tools, including those based on UV–visible absorption, fluorescence, photothermal, Raman, and IR spectroscopies, have been used to probe droplets in a sensitive and rapid manner. Of these, only Raman and IR spectroscopies are able to extract significant amounts of structural information in a direct manner. However, both approaches suffer from background issues that hamper their use in small volume environments. For example, (non-surface-enhanced) Raman signals can often be polluted by background fluorescence, and since water strongly absorbs in the infrared region of the electromagnetic spectrum, the application of IR-based tools in biological systems is frequently compromised.10,11 Accordingly, the utility of techniques based on X-ray absorption spectroscopy (XAS) is without doubt due to the fact that XAS is an element-selective detection method that allows chemical characterization via measurement of the binding energies of core electrons.12,13
In recent years, synchrotron-based detection techniques have been successfully interfaced with continuous-flow microfluidic platforms and used to study a variety of processes, including the nucleation and growth of nanomaterials, self-assembly processes, and protein unfolding.14−18 For example, XAS has been used to probe chemical species at laminar flow interfaces in a spatially resolved manner.19,20 Moreover, other X-ray-based analytical methods have been applied to droplet-based microfluidic platforms, although these have primarily been restricted to small-angle X-ray scattering (SAXS) studies.21−24 More recently, Levenstein et al. reported the coupling of X-ray diffraction (XRD) with droplet microfluidics for the identification of nucleating agents in calcium carbonate crystallization,25 determining that bioactive glasses are efficient nucleants for calcite. Finally, an X-ray-compatible cell for the study of static microliter-volume droplets was recently described by Xto and co-workers and applied to the investigation of calcium carbonate crystallization under controlled humidity.26 However, to the best of our knowledge, there has been no report of in situ reaction monitoring using XAS within a droplet-based microfluidic platform. XAS is a powerful method for probing chemical environments in a chemically specific manner. Importantly, and in contrast to diffraction-based methods, it can be used to study both crystalline and amorphous materials, revealing the oxidation state and electronic band structures of the atoms of interest. An XAS spectrum is obtained by measurement of the absorption coefficient as a function of the incident photon energy, either in a direct transmission mode or indirectly via emitted fluorescence photons. XAS measurements can be performed over large energy ranges; from “hard” (>5 keV) to “soft” (0.1–1 keV) X-rays. Tender X-rays exist between these two extremes (1–5 keV) and offer unique opportunities to study absorption edges that are of interest to the materials and life sciences communities, notably the K-edges of Na, Mg, Al, Si, P, S, Cl, K, and Ca.27 A key advantage when using droplet-based microfluidic tools to process samples for XAS analysis is the minimization of the sample exposure to the probe beam. Since droplets typically move at high linear velocities, their transit time through the optical probe volume is often not more than a few milliseconds, limiting the exposure of material in each droplet to the incoming X-rays. In this way, prolonged beam exposure that would otherwise alter or damage sensitive samples (such as highly valent transition metals) or could change chemical equilibria in aqueous solutions28 is eliminated. That said, there are challenges associated with the interrogation of droplet-based microfluidic tools with X-rays. Most notably, the microfluidic device must be robust enough to sustain beam exposure as well as strain from vacuum environments, while ensuring high transmission to X-rays.
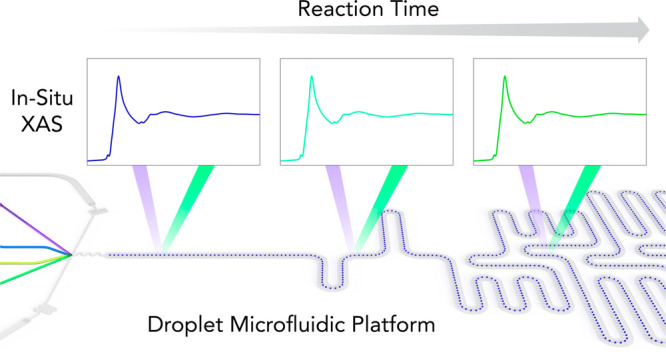
Herein, we present for the first time the combination of XAS with a high-throughput droplet-based microfluidic platform for in situ monitoring of fast processes and the extraction of kinetic data. We first describe the fabrication of an X-ray-compatible microfluidic device containing a microchannel network tailored for rapid kinetic studies. We then investigate the kinetics of amorphous CaCO3 formation by monitoring the evolution of the Ca K-edge XANES (X-ray absorption near-edge structure) spectra within picoliter-volume droplets at various positions along the length of the primary microfluidic channel. Understanding the detailed process of calcium carbonate precipitation from supersaturated salt solutions is of significant interest for many applications in geochemistry, biology, and industry.29−31 For example, numerous living organisms are able to precisely control calcium carbonate formation and its incorporation in various functional biomaterials.32 Moreover, calcium carbonate serves as a fundamental model system for nucleation and is the center of a yet unresolved controversy advocating either classical or nonclassical nucleation models.33−36 XAS measurements at the Ca K-edge enable the characterization of the local environment of calcium centers.37 XAS has also been used to analyze amorphous calcium carbonate intermediates and investigate their relevance for converting to crystalline phases.38 In another study, Politi et al. used X-ray absorption near-edge spectroscopy in the larval stages of the sea urchin spicules to reveal the initial formation of amorphous calcium carbonate (ACC) prior to its crystallization into calcite single crystals.39 Since confined reaction environments are useful in spatially controlling biomineral formation, droplet-based microfluidic platforms are particularly well-suited for studying both nucleation and crystallization processes in homogeneous environments. In this regard, it should be noted that droplet-based microfluidic systems have been successfully employed to controllably generate amorphous and crystalline CaCO3, with end-point characterization of samples (collected off-chip) achieved using Raman spectroscopy.40,41
Experimental Section
Design and Fabrication of the X-ray-Compatible Microfluidic Device
The X-ray-compatible microfluidic device comprises a single-layer elastomeric chip (fabricated using soft lithography) and an X-ray-transparent window (a Si3N4 membrane on a silicon frame) that accommodates measurements with tender X-rays (Figure 1a). For tender X-rays between 3090 and 4200 eV, the 450 nm thick Si3N4 window yields a transmission of 98%.42 XAS measurements along the observation channel are possible at locations were the Si3N4 window slit overlays the detection channel. Si3N4 windows were custom fabricated from Si3N4-coated Si wafers (Si3N4: 450 nm thick, Si: 525 μm thick, MicroChemicals GmbH, Ulm, Germany) via KOH etching following standard procedures.43 Photomasks were designed using AutoCAD 2018 (Autodesk, San Rafael, USA) and printed onto high-resolution photolithographic masks (Micro Lithography Services Ltd., Chelmsford, UK). The silicon layer is etched at a 54.7° angle; therefore, attention must be paid to mask design, such that desired feature dimensions are obtained. The polydimethylsiloxane (PDMS) part of the microfluidic device was fabricated following standard soft lithography procedures.44 Briefly, microfluidic channel patterns were designed using AutoCAD and printed onto high-resolution photolithographic masks. An SU-8 master mold, having a layer height of 40 μm, was fabricated via standard photolithography. After fabrication, the SU-8 mold was exposed to a chlorotrimethylsilane (Sigma-Aldrich, Buchs, Switzerland) vapor for at least 3 h in a desiccator at a pressure of 150 mbar to aid subsequent removal of cured PDMS. A 10:1 (w/w) mixture of PDMS base to curing agent (Elastosil RT 601 components A and B, Wacker Chemie AG, Basel, Switzerland) was poured onto the master mold, degassed in a desiccator for 15 min, and cured for 2 h at 70 °C. After the PDMS was peeled from the mold and devices were diced, inlets and outlets (0.76 μm diameter) were formed at desired locations using a hole puncher (Syneo, Florida, USA).
Figure 1.

(a) Schematic of the droplet-based microfluidic system for XAS measurements. A PDMS device is bonded to a Si3N4 membrane (450 nm thick) supported by a silicon frame (500 μm thick). The microfluidic device is then fixed to a copper holding plate for proper alignment inside the measurement chamber. (b) Schematic representation of the microfluidic device inside the measurement chamber. The X-ray beam enters the microfluidic channel at a 45° angle, with the detector being placed at a 90° angle with respect to the incoming beam. A light-emitting diode and lens coupled to a high-speed camera are used to visualize the droplets during analysis. (c) Schematic of the microfluidic channel network, showing the flow-focusing junction for droplet generation, the mixing region, and an elongated channel for kinetic measurements. The incoming X-ray beam is focused in the center of the channel for interrogation of the droplet stream.
Next, the structured PDMS substrates were aligned and bonded to the Si3N4 windows after a 20 s exposure to an air plasma (EMITECH K1000X, Quorum Technologies, East Sussex, United Kingdom). Two minutes after bonding, a 5% solution of tridecafluoro-1,1,2,2-tetrahydrooctyl)trichlorosilane (ABCR-chemicals, Karlsruhe, Germany) in HFE 7500 Novec oil was injected into all microchannels for a period of 5 min, followed by rinsing with pure HFE oil. This treatment renders the internal surfaces hydrophobic. The height of all microchannels in the fabricated device was 40 μm, and the width of the elongated channel for XAS measurements was 200 μm. Figure 1c depicts the microfluidic channel pattern used in this study. The device comprises five inlets, one for the continuous phase and four for the delivery of the aqueous reagents. In a first experiment, all aqueous inputs contained a CaCl2 solution to obtain a spectrum of free solvated calcium ions (Ca2+). Subsequently, in the kinetics experiment, the outer aqueous channels supplied the solution of CaCl2 on one side and of Na2CO3 on the other side, while the inner aqueous channels carried a dilute solution of HCl and a stream of water, creating a buffer layer to prevent the reagents from precipitating prematurely (see also Figure 3a). The design of the channel and its operation are described in more detail in the Results and Discussion section. Importantly, this channel design could be easily adapted and optimized for various applications, highlighting the versatility of the method.
Figure 3.

Kinetic analysis of ACC from CaCl2 and Na2CO3 solutions. (a) Schematic of the microfluidic device used for the kinetics measurements. The colored rectangles along the primary microchannel indicate the positions where XAS spectra were recorded and correspond to reaction times of 10, 130, 1100, and 2200 ms. (b) Ca K-edge XANES spectra of solvated calcium ions (Ca2+) (60 mM CaCl2) in droplets, Ca K-edge XANES spectra of impurity-free ACC under 80% humidity, and Ca K-edge XANES spectra measured at increasing reaction times. (c) Relative contribution from free calcium ions (Ca2+) and ACC as a function of time. Data were obtained from an LCF of each spectrum from the reference spectra.
Microfluidic Device Operation and XAS Measurements
Precision neMESYS syringe pumps (CETONI GmbH, Korbussen, Germany) were used to precisely control fluid flow rates. The flow rate of the continuous phase was typically set to 12 μL/min, and the total flow rate of the aqueous flows to 0.6 μL/min. At these flow rates, droplets were generated at a frequency of 630 Hz, with a droplet volume of approximately 15 pL. Gastight 10 mL and 250 μL syringes (Hamilton AG, Bonaduz, Switzerland) were used for the oil phase and for the aqueous phases, respectively. For the stated flow rates, syringes could be operated for up to 13 h without interruption or intervention. The tender X-ray energy range (between 3090 and 4200 eV) necessitates operation within a vacuum or helium environment to minimize attenuation of the incoming beam. Consequently, optimal sealing of all the fluidic connections is a necessity. Syringes were connected to polytetrafluoroethylene tubing (0.25 mm i.d. × 1/16 in. o.d., Fisher Scientific, Reinach, Switzerland) via a Luer-lock adapter, with the other ends connected to a short segment of Tygon tubing (TygonS-54-HL, i.d. 250 μm, Fisher Scientific, Reinach, Switzerland) via metal pins (TE Needle 30 GA 1/2 in. Lavender, Buchsteiner, Gingen, Germany). Tygon tubing was directly inserted into the inlets of the microfluidic device to create a tight fluidic connection able to withstand vacuum conditions without leakage.
XANES measurements at the Ca K-edge (E0 = 4038.5 eV) were carried out at the PHOENIX I Beamline, Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland). The microfluidic device was fixed onto a copper holder using double-sided tape and then mounted on an x-y-z stage for precise manipulation and alignment. The X-ray beam enters the microfluidic channel at an angle of 45° to the surface, with the solid-state fluorescence detector (Ketek GmbH, Germany) being placed at a 90° angle with respect to the incoming beam (Figure 1b). A light-emitting diode lamp was used for bright-field illumination of the device, with droplet formation being visualized using a zoom lens (Leica Z16 APO A) coupled to a high-speed camera (MotionBLITZ EoSens mini, MIKROTRON, Unterschleissheim, Germany). All measurements were carried out in a helium environment below atmospheric pressure. To generate this condition while avoiding significant stress on the Si3N4 membrane, the exhaust vial was connected via a 3-way valve to the inside of the chamber. In this way, the pressure at the outlet of the chip could be precisely controlled. Typically, measurements were performed as follows: with the valve open (exhaust vial equilibrated with the inside of the chamber), the chamber was pumped down to 100 mbar and then vented with He to 750 mbar. This cycle was repeated twice; the valve was then closed (yielding a pressure of 750 mbar He in the exhaust vial), and the chamber pumped to 600 mbar. Due to the low concentrations and small detection volume used here, long data acquisition times are needed to achieve appropriate signal-to-noise ratios. One scan typically took 8 min to acquire, with several scans (between 10 and 15) being averaged to obtain the final spectrum.
Materials and Reagents
Calcium chloride dihydrate, sodium carbonate, and hydrochloric acid 37% (Sigma-Aldrich, Buchs, Switzerland) were used as received. Purified water (Milli-Q, Merck Millipore, Burlington, USA) was used to prepare all solutions and used as the water stream on-chip. Aqueous solutions of 60 mM CaCl2, 80 mM Na2CO3, and 30 mM HCl were prepared and loaded into the syringes prior to each experiment. The continuous phase consisted of HFE-7500 oil (3M, Minnesota, USA) containing 1.25% (w/w) of fluorosurfactant (RAN Biotechnologies, Massachusetts, USA).
Results and Discussion
In Situ XAS Measurement in Picoliter Droplets
To assess the capability of our platform to acquire high-resolution XAS spectra, we first acquired a reference spectrum from solvated calcium ions (Ca2+) and compared this to a spectrum obtained from a bulk solution. All syringes were loaded with a 60 mM CaCl2 solution, and thus droplets contained only free solvated Ca2+. The X-ray beam was focused to a 50 × 75 μm area, which is slightly larger than the size of a droplet within the segmented flow. During initial experiments, we observed that exposure of the PDMS substrate to a focused X-ray beam yielded a distinct imprint on the surface within a few seconds (Figure S1). This imprint appears as a darkened area and is off-centered due to a parallax from the 45° angle of the incident beam. We attribute this to the appearance of colored centers in the PDMS bulk under beam exposure. Importantly, we have observed that the shape of the channel is not affected, and the stability of the droplets is not compromised. We leveraged this effect to estimate both the beam size and the beam position within the microfluidic channel. To precisely align the beam with the droplet train and thus maximize the signal, we first performed a cross-sectional scan across the channel with the incoming energy set to 4050 eV (Figure 2a). The Si XRF profile (originating from the PDMS substrate) clearly highlights the microchannel walls and the 200 μm wide microfluidic channel width, while the Ca XRF signal reports the centered position of the droplet train. It should be noted that Ca XRF also increases at the channel walls due to Si pileup counts overlapping with the Ca K-α line (see Figure S2 for a more detailed explanation). Subsequently, XAS spectra were acquired at the position giving the maximum Ca counts, which corresponds to the center of the droplet train. Figure 2b provides a comparison of Ca K-edge XANES spectra obtained from a 100 μm deep flow cell with a 10 mM CaCl2 solution (using a 50 μm X-ray beam) and from rapidly moving 15 pL droplets containing 60 mM CaCl2. The presented bulk spectrum was obtained by averaging five scans, while the droplet spectrum was obtained by averaging 14 scans from a 40 μm deep microchannel (one scan takes 8 min to acquire). As expected, signals from the microfluidic device were lower than those originating from the bulk flow cell, due to the reduced optical path length and the fact that droplets are separated by significant volumes of carrier oil. That said, the excellent correspondence between spectra originating from droplets and those from the flow cell confirms that high-resolution XAS spectra can be measured with excellent signal-to-noise within fast-flowing, picoliter-volume droplets.
Figure 2.

(a) Cross-sectional scans perpendicular to the microchannel axis with droplets containing 20 mM Ca2+, reporting variations in XRF (X-ray fluorescence) intensity for Si and Ca. The energy is maintained at 4050 eV. The variation in Si XRF (red) indicates the position of the channel walls, whereas Ca XRF (blue) reports droplets centered in the middle of the channel. The Ca XRF increases at the channel walls due to Si pileup overlapping with the Ca K-α line. The inset shows an image of the microfluidic device, with the corresponding cross section marked as a dotted white line. The scale bar is 100 μm. (b) Comparison of Ca K-edge XANES spectrum obtained from a bulk solution (CaCl2 10 mM) within the flow cell (black) and from picoliter-volume droplets (CaCl2 60 mM) (purple). Spectra have been vertically shifted to aid visualization.
Kinetics of Calcium Carbonate Precipitation in Droplets
Next, we employ our platform to monitor the early stage of CaCO3 precipitation from free solvated ions by mixing calcium chloride and sodium carbonate:
| 1 |
Time-resolved SAXS studies of ACC formation have previously reported that ACC is formed within the first second of mixing, highlighting the need to investigate the early times of the reaction on a millisecond time scale.45,46 These SAXS measurements, however, were limited by the minimum cluster size that can be detected, and thus observations of the first few milliseconds after mixing were precluded. The microfluidic channel network used in the current study was designed and optimized to measure kinetic data on a millisecond to second time scale. Indeed, we have recently used a similar design for the study of enzyme kinetics on sub-millisecond time scales.8 Briefly, the channel structure allows for the rapid acceleration of droplets immediately after droplet formation to decrease mixing times. This acceleration phase is followed by droplet deceleration to allow for efficient interrogation of droplets in the measurement channel. In the current work, the chip consists of four inlets to introduce aqueous reagents and one inlet to introduce the oil phase. The four inner channels unite at the flow-focusing junction where 15 pL volume droplets are generated at a rate of 630 per second. Immediately after formation, the droplets are accelerated and mixed via chaotic advection within a short winding section of the channel.47 An increase of the channel width in the main observation channel is used to slow droplets down and direct them toward the middle of the channel. XAS spectra are then acquired at various positions along the length of the channel (Figure 3a). Droplet velocity is extracted via analysis of bright-field movies and the reaction time corresponding to each measurement position calculated. The two side aqueous inlets are used to inject CaCl2 and Na2CO3, whereas the two central inlets are used to inject water and a HCl solution. The water and HCl streams act as a slightly acidic buffer layer to prevent precipitation at the interface between CaCl2 and Na2CO3 and ensure that mixing occurs after droplet formation. Moreover, such a configuration allows tuning of reagent concentrations and pH on-chip. In the current study, the flow rate of CaCl2 (60 mM) and Na2CO3 (80 mM) was 0.2 μL/min, whereas the flow rate of water and HCl (30 mM) was 0.1 μL/min. This yielded effective concentrations (in each droplet) of 20 mM for CaCl2, 26 mM for Na2CO3, and 5 mM for HCl. These concentrations yield a pH of 10.2 and supersaturation ratio of 2.022 for amorphous calcium carbonate, indicating that precipitation will occur after droplet generation (ACC will precipitate at a pH of 8.2 or greater).48 Using a slight excess of carbonate ensures that all the calcium ions will react with a carbonate radical, and thus there is no unreacted calcium that could be measured in the background.
CaCO3 exists as three anhydrous polymorphs (calcite, aragonite, and vaterite), as well as the highly unstable amorphous ACC phase. We measured in situ XANES spectra at four different positions along the microfluidic channel, corresponding to reaction times of 10, 130, 1100, and 2200 ms. The corresponding XANES spectra are presented in Figure 3b, along with reference spectra of free Ca2+ ions and ACC (measured in impurity-free ACC under 80% humidity, using an aerosol-based synthesis49). On the XANES spectra, the sharp intense peak from the rising absorption edge is referred to as the white line. The calcium K-edge XANES spectrum exhibits a distinct prepeak centered at 4039.0 eV as a result of the 1s–3d transition. The white line peak for solvated calcium ions has a signature energy centered at 4050.0 eV, whereas in the ACC phase, the white line of the Ca K-edge XANES shifts to 4049.0 eV. As the reaction time increases, the Ca white line continuously shifts to lower energies (∼1 eV) but does not show any shape change (inset of Figure 3a). This shift is correlated to the change in average oxygen coordination number (0.7 eV for each O atom)50 and tracks the movements of water molecules around the Ca ion. It is known that for a 6 M CaCl2 solution, the mean coordination number is 7.2 (±1.2) water molecules at an average Ca–O distance of 2.437 (±0.010 Å).51 Therefore, the 1 eV shift we observe points toward a decrease in the number of oxygen atoms toward six atoms around the Ca atom. For each time point, we determined the fraction of free Ca2+ ions and the fraction of ACC via a linear combination fitting of the reference spectra using the Athena software package.52 The resulting kinetic curve shown (Figure 3) consists of two kinetic phases: a steep increase of ACC fraction within the first 100 ms and a subsequent slower phase reporting a further increase of the ACC content. Such an observation confirms that ACC is the first intermediate species in the carbonate crystallization process.34 Importantly, for the concentrations employed here, ACC precipitation occurs quickly over the first few milliseconds of the reaction, with ACC formation reaching 60% after 130 ms. For the time scales investigated here, we do not observe any further crystallization of the ACC polymorph to calcite or vaterite phases within the confined volume of a droplet. These more stable crystalline polymorphs are know to form over longer time scales. Furthermore, our observation is in good agreement with previous results suggesting that confinement within small volumes drastically slows down the nucleation of crystalline phases of CaCO3.53,54 Finally, it can be seen that droplets offer increased control over the crystal polymorph, as previously reported,40 due to their homogeneous composition and the lack of contained impurities.
Conclusions
In summary, we have presented for the first time the combination of X-ray absorption spectroscopy with droplet-based microfluidics for in situ reaction monitoring with millisecond time resolution. Specifically, we have developed a versatile and X-ray-compatible microfluidic device that is easy to fabricate and can be used for a variety of applications. Importantly, we demonstrate that high-quality XAS spectra within picoliter-volume droplets moving at high velocity may be acquired in a reliable and robust fashion. To prove both the concept and utility of the approach, we show that monodisperse and impurity-free droplets provide well-defined environments in which to perform and probe CaCO3 crystallization. In this regard, we successfully monitored CaCO3 precipitation from free solvated ions and the formation of an amorphous phase on a millisecond to second time scale. We expect that the combination of XAS with droplet-based microfluidics will be of significant interest for the materials community at large and particularly for the investigation of nucleation and crystallization processes and the mechanisms of rapid reactive chemistries with an exceptional level of structural detail.
Acknowledgments
The authors acknowledge the Swiss Light Source at Paul Scherrer Institute for the award of beam time on the PHOENIX beamline. The authors would like to thank Dr. Tuncay Alan and Dr. Jason Brenker from Monash University for suggesting the use of an oxygen plasma to bond PDMS substrates to Si3N4 windows and for sharing the details for the bonding process prior to their own publication. The authors also thank Mr. Reto Wetter for technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmeasuresciau.1c00005.
Bright-field image of the microfluidic channel; cross-sectional measurement of Si and Ca XRF with corresponding XRF spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chiu D. T.; deMello A. J.; Di Carlo D.; Doyle P. S.; Hansen C.; Maceiczyk R. M.; Wootton R. C. R. Small but Perfectly Formed? Successes, Challenges, and Opportunities for Microfluidics in the Chemical and Biological Sciences. Chem. 2017, 2, 201–223. 10.1016/j.chempr.2017.01.009. [DOI] [Google Scholar]
- Suea-Ngam A.; Howes P. D.; Srisa-Art M.; Demello A. J. Droplet microfluidics: From proof-of-concept to real-world utility?. Chem. Commun. 2019, 55, 9895–9903. 10.1039/C9CC04750F. [DOI] [PubMed] [Google Scholar]
- Huebner A.; Sharma S.; Srisa-Art M.; Hollfelder F.; Edel J. B.; DeMello A. J. Microdroplets: A sea of applications?. Lab Chip 2008, 8, 1244–1254. 10.1039/b806405a. [DOI] [PubMed] [Google Scholar]
- Teh S. Y.; Lin R.; Hung L. H.; Lee A. P. Droplet microfluidics. Lab Chip 2008, 8, 198–220. 10.1039/b715524g. [DOI] [PubMed] [Google Scholar]
- Mashaghi S.; Abbaspourrad A.; Weitz D. A.; van Oijen A. M. Droplet microfluidics: A tool for biology, chemistry and nanotechnology. TrAC, Trends Anal. Chem. 2016, 82, 118–125. 10.1016/j.trac.2016.05.019. [DOI] [Google Scholar]
- Song H.; Ismagilov R. F. Millisecond Kinetics on a Microfluidic Chip Using Nanoliters of Reagents. J. Am. Chem. Soc. 2003, 125, 14613–14619. 10.1021/ja0354566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess D.; Rane A.; Demello A. J.; Stavrakis S. High-throughput, quantitative enzyme kinetic analysis in microdroplets using stroboscopic epifluorescence imaging. Anal. Chem. 2015, 87, 4965–4972. 10.1021/acs.analchem.5b00766. [DOI] [PubMed] [Google Scholar]
- Hess D.; Dockalova V.; Kokkonen P.; Bednar D.; Damborsky J.; deMello A.; Prokop Z.; Stavrakis S. Exploring mechanism of enzyme catalysis by on-chip transient kinetics coupled with global data analysis and molecular modeling. Chem. 2021, 7, 1066–1079. 10.1016/j.chempr.2021.02.011. [DOI] [Google Scholar]
- Liu W.-w.; Zhu Y. Development and application of analytical detection techniques for droplet-based microfluidics”-A review. Anal. Chim. Acta 2020, 1113, 66–84. 10.1016/j.aca.2020.03.011. [DOI] [PubMed] [Google Scholar]
- Cristobal G.; Arbouet L.; Sarrazin F.; Talaga D.; Bruneel J. L.; Joanicot M.; Servant L. On-line laser Raman spectroscopic probing of droplets engineered in microfluidic devices. Lab Chip 2006, 6, 1140–1146. 10.1039/b602702d. [DOI] [PubMed] [Google Scholar]
- Chan K. L. A.; Niu X.; Demello A. J.; Kazarian S. G. Generation of chemical movies: FT-IR spectroscopic imaging of segmented flows. Anal. Chem. 2011, 83, 3606–3609. 10.1021/ac200497a. [DOI] [PubMed] [Google Scholar]
- Koningsberger D. C.; Prins R.. X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS, and XANES; John Wiley & Sons: New York, 1988. [Google Scholar]
- Bordiga S.; Groppo E.; Agostini G.; Van Bokhoven J. A.; Lamberti C. Reactivity of surface species in heterogeneous catalysts probed by in situ x-ray absorption techniques. Chem. Rev. 2013, 113, 1736–1850. 10.1021/cr2000898. [DOI] [PubMed] [Google Scholar]
- Herbst M.; Hofmann E.; Förster S. Nucleation and Growth Kinetics of ZnO Nanoparticles Studied by in Situ Microfluidic SAXS/WAXS/UV-Vis Experiments. Langmuir 2019, 35, 11702–11709. 10.1021/acs.langmuir.9b01149. [DOI] [PubMed] [Google Scholar]
- Poulos A. S.; Nania M.; Lapham P.; Miller R. M.; Smith A. J.; Tantawy H.; Caragay J.; Gummel J.; Ces O.; Robles E. S. J.; et al. Microfluidic SAXS Study of Lamellar and Multilamellar Vesicle Phases of Linear Sodium Alkylbenzenesulfonate Surfactant with Intrinsic Isomeric Distribution. Langmuir 2016, 32, 5852–5861. 10.1021/acs.langmuir.6b01240. [DOI] [PubMed] [Google Scholar]
- Toft K. N.; Vestergaard B.; Nielsen S. S.; Snakenborg D.; Jeppesen M. G.; Jacobsen J. K.; Arleth L.; Kutter J. P. High-throughput small angle X-ray scattering from proteins in solution using a microfluidic front-end. Anal. Chem. 2008, 80, 3648–3654. 10.1021/ac800011y. [DOI] [PubMed] [Google Scholar]
- Kane A. S.; Hoffmann A.; Baumgärtel P.; Seckler R.; Reichardt G.; Horsley D. A.; Schuler B.; Bakajin O. Microfluidic mixers for the investigation of rapid protein folding kinetics using synchrotron radiation circular dichroism spectroscopy. Anal. Chem. 2008, 80, 9534–9541. 10.1021/ac801764r. [DOI] [PubMed] [Google Scholar]
- Ghazal A.; Lafleur J. P.; Mortensen K.; Kutter J. P.; Arleth L.; Jensen G. V. Recent advances in X-ray compatible microfluidics for applications in soft materials and life sciences. Lab Chip 2016, 16, 4263–4295. 10.1039/C6LC00888G. [DOI] [PubMed] [Google Scholar]
- Nagasaka M.; Yuzawa H.; Takada N.; Aoyama M.; Rühl E.; Kosugi N. Laminar flow in microfluidics investigated by spatially-resolved soft X-ray absorption and infrared spectroscopy. J. Chem. Phys. 2019, 151, 114201. 10.1063/1.5115191. [DOI] [PubMed] [Google Scholar]
- Chan E. M.; Marcus M. A.; Fakra S.; ElNaggar M.; Mathies R. A.; Alivisatos A. P. Millisecond kinetics of nanocrystal cation exchange using microfluidic X-ray absorption spectroscopy. J. Phys. Chem. A 2007, 111, 12210–12215. 10.1021/jp073474u. [DOI] [PubMed] [Google Scholar]
- Stehle R.; Goerigk G.; Wallacher D.; Ballauff M.; Seiffert S. Small-angle X-ray scattering in droplet-based microfluidics. Lab Chip 2013, 13, 1529–1537. 10.1039/c3lc41291a. [DOI] [PubMed] [Google Scholar]
- Lange T.; Charton S.; Bizien T.; Testard F.; Malloggi F. OSTE+ for: In situ SAXS analysis with droplet microfluidic devices. Lab Chip 2020, 20, 2990–3000. 10.1039/D0LC00454E. [DOI] [PubMed] [Google Scholar]
- Saldanha O.; Graceffa R.; Hémonnot C. Y. J.; Ranke C.; Brehm G.; Liebi M.; Marmiroli B.; Weinhausen B.; Burghammer M.; Köster S. Rapid Acquisition of X-Ray Scattering Data from Droplet-Encapsulated Protein Systems. ChemPhysChem 2017, 18, 1220–1223. 10.1002/cphc.201700221. [DOI] [PubMed] [Google Scholar]
- Pham N.; Radajewski D.; Round A.; Brennich M.; Pernot P.; Biscans B.; Bonneté F.; Teychené S. Coupling High Throughput Microfluidics and Small-Angle X-ray Scattering to Study Protein Crystallization from Solution. Anal. Chem. 2017, 89, 2282–2287. 10.1021/acs.analchem.6b03492. [DOI] [PubMed] [Google Scholar]
- Levenstein M. A.; Anduix-Canto C.; Kim Y.; Holden M. A.; González Niño C.; Green D. C.; Foster S. E.; Kulak A. N.; Govada L.; Chayen N. E.; et al. Droplet Microfluidics XRD Identifies Effective Nucleating Agents for Calcium Carbonate. Adv. Funct. Mater. 2019, 29, 1808172. 10.1002/adfm.201808172. [DOI] [Google Scholar]
- Xto J.; Wetter R.; Borca C. N.; Frieh C.; Van Bokhoven J. A.; Huthwelker T. Droplet-based in situ X-ray absorption spectroscopy cell for studying crystallization processes at the tender X-ray energy range. RSC Adv. 2019, 9, 34004–34010. 10.1039/C9RA06084G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrup P.; Leri A.; Tappero R. Applications of “Tender” Energy (1–5 keV) X-ray Absorption Spectroscopy in Life Sciences. Protein Pept. Lett. 2016, 23, 300–308. 10.2174/0929866523666160107114505. [DOI] [PubMed] [Google Scholar]
- Laanait N.; Callagon E. B. R.; Zhang Z.; Sturchio N. C.; Lee S. S.; Fenter P. X-ray-driven reaction front dynamics at calcite-water interfaces. Science (Washington, DC, U. S.) 2015, 349, 1330–1334. 10.1126/science.aab3272. [DOI] [PubMed] [Google Scholar]
- Beniash E.; Metzler R. A.; Lam R. S. K.; Gilbert P. U. P. A. Transient amorphous calcium phosphate in forming enamel. J. Struct. Biol. 2009, 166, 133–143. 10.1016/j.jsb.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chierici M.; Fransson A. Calcium carbonate saturation in the surface water of the Arctic Ocean: Undersaturation in freshwater influenced shelves. Biogeosciences 2009, 6, 2421–2432. 10.5194/bg-6-2421-2009. [DOI] [Google Scholar]
- Fathi A.; Mohamed T.; Claude G.; Maurin G.; Mohamed B. A. Effect of a magnetic water treatment on homogeneous and heterogeneous precipitation of calcium carbonate. Water Res. 2006, 40, 1941–1950. 10.1016/j.watres.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Mahamid J.; Aichmayer B.; Shimoni E.; Ziblat R.; Li C.; Siegel S.; Paris O.; Fratzl P.; Weiner S.; Addadi L. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6316–6321. 10.1073/pnas.0914218107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer D.; Völkel A.; Cölfen H. Stable prenucleation calcium carbonate clusters. Science (Washington, DC, U. S.) 2008, 322, 1819–1822. 10.1126/science.1164271. [DOI] [PubMed] [Google Scholar]
- Henzler K.; Fetisov E. O.; Galib M.; Baer M. D.; Legg B. A.; Borca C.; Xto J. M.; Pin S.; Fulton J. L.; Schenter G. K.; et al. Supersaturated calcium carbonate solutions are classical. Sci. Adv. 2018, 4, eaao6283. 10.1126/sciadv.aao6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget E. M.; Bomans P. H. H.; Goos J. A. C. M.; Frederik P. M.; De With G.; Sommerdijk N. A. J. M. The initial stages of template-controlled CaCO3 formation revealed by Cryo-TEM. Science (Washington, DC, U. S.) 2009, 323, 1455–1458. 10.1126/science.1169434. [DOI] [PubMed] [Google Scholar]
- Gebauer D.; Raiteri P.; Gale J. D.; Cölfen H. On classical and non-classical views on nucleation. Am. J. Sci. 2018, 318, 969–988. 10.2475/09.2018.05. [DOI] [Google Scholar]
- Martin-Diaconescu V.; Gennari M.; Gerey B.; Tsui E.; Kanady J.; Tran R.; Pécaut J.; Maganas D.; Krewald V.; Gouré E.; et al. Ca K-edge XAS as a probe of calcium centers in complex systems. Inorg. Chem. 2015, 54, 1283–1292. 10.1021/ic501991e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer D.; Gunawidjaja P. N.; Ko J. Y. P.; Bacsik Z.; Aziz B.; Liu L.; Hu Y.; Bergström L.; Tai C. W.; Sham T. K.; et al. Proto-calcite and proto-vaterite in amorphous calcium carbonates. Angew. Chem., Int. Ed. 2010, 49, 8889–8891. 10.1002/anie.201003220. [DOI] [PubMed] [Google Scholar]
- Politi Y.; Metzler R. A.; Abrecht M.; Gilbert B.; Wilt F. H.; Sagi I.; Addadi L.; Weiner S.; Gilbert P. Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17362–17366. 10.1073/pnas.0806604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashina A.; Meldrum F.; DeMello A. Calcium carbonate polymorph control using droplet-based microfluidics. Biomicrofluidics 2012, 6, 022001. 10.1063/1.3683162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Zeng M.; Gaule T.; McPherson M. J.; Meldrum F. C. Passive Picoinjection Enables Controlled Crystallization in a Droplet Microfluidic Device. Small 2017, 13, 1702154. 10.1002/smll.201702154. [DOI] [PubMed] [Google Scholar]
- Henke B. L.; Gullikson E. M.; Davis J. C. X-ray interactions: Photoabsorption, scattering, transmission, and reflection at E = 50–30, 000 eV, Z = 1–92. At. Data Nucl. Data Tables 1993, 54, 181–342. 10.1006/adnd.1993.1013. [DOI] [Google Scholar]
- Brugger J.; Beljakovic G.; Despont M.; De Rooij N. F.; Vettiger P. Silicon micro/nanomechanical device fabrication based on focused ion beam surface modification and KOH etching. Microelectron. Eng. 1997, 35, 401–404. 10.1016/S0167-9317(96)00210-9. [DOI] [Google Scholar]
- Zhao X.-M.; Xia Y.; Whitesides G. M. Soft lithographic methods for nano-fabrication. J. Mater. Chem. 1997, 7, 1069–1074. 10.1039/a700145b. [DOI] [Google Scholar]
- Bolze J.; Peng B.; Dingenouts N.; Panine P.; Narayanan T.; Ballauff M. Formation and growth of amorphous colloidal CaCO3 precursor particles as detected by time-resolved SAXS. Langmuir 2002, 18, 8364–8369. 10.1021/la025918d. [DOI] [Google Scholar]
- Liu J.; Pancera S.; Boyko V.; Shukla A.; Narayanan T.; Huber K. Evaluation of the particle growth of amorphous calcium carbonate in water by means of the Porod invariant from SAXS. Langmuir 2010, 26, 17405–17412. 10.1021/la101888c. [DOI] [PubMed] [Google Scholar]
- Song H.; Tice J. D.; Ismagilov R. F. A microfluidic system for controlling reaction networks in time. Angew. Chem., Int. Ed. 2003, 42, 768–772. 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- Allison J. D.; Brown D. S.; Novo-Gradac K. J.. Minteqa2/Prodefa2, a Geochemical Assessment Model for Environmental Systems, User’s Manual, version 3.0, 1991. [Google Scholar]
- Xto J. M.; Borca C. N.; Van Bokhoven J. A.; Huthwelker T. Aerosol-based synthesis of pure and stable amorphous calcium carbonate. Chem. Commun. 2019, 55, 10725–10728. 10.1039/C9CC03749G. [DOI] [PubMed] [Google Scholar]
- Sowrey F. E.; Skipper L. J.; Pickup D. M.; Drake K. O.; Lin Z.; Smith M. E.; Newport R. J. Systematic empirical analysis of calcium-oxygen coordination environment by calcium K-edge XANES. Phys. Chem. Chem. Phys. 2004, 6, 188–192. 10.1039/B311715D. [DOI] [Google Scholar]
- Fulton J. L.; Heald S. M.; Badyal Y. S.; Simonson J. M. Understanding the effects of concentration on the solvation structure of Ca2+ in aqueous solution. I: The perspective on local structure from EXAFS and XANES. J. Phys. Chem. A 2003, 107, 4688–4696. 10.1021/jp0272264. [DOI] [Google Scholar]
- Smith J. W.; Saykally R. J. Soft X-ray Absorption Spectroscopy of Liquids and Solutions. Chem. Rev. 2017, 117, 13909–13934. 10.1021/acs.chemrev.7b00213. [DOI] [PubMed] [Google Scholar]
- Stephens C. J.; Kim Y. Y.; Evans S. D.; Meldrum F. C.; Christenson H. K. Early stages of crystallization of calcium carbonate revealed in picoliter droplets. J. Am. Chem. Soc. 2011, 133, 5210–5213. 10.1021/ja200309m. [DOI] [PubMed] [Google Scholar]
- Tester C. C.; Wu C. H.; Weigand S.; Joester D. Precipitation of ACC in liposomes - A model for biomineralization in confined volumes. Faraday Discuss. 2012, 159, 345–356. 10.1039/c2fd20088k. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.