

Abstract
Rationale
Mucin homeostasis is fundamental to airway health. Upregulation of airway mucus glycoprotein MUC5B is observed in diverse common lung diseases and represents a potential therapeutic target. In mice, Muc5b is required for mucociliary clearance and for controlling inflammation after microbial exposure. The consequences of its loss in humans are unclear.
Objectives
The goal of this study was to identify and characterize a family with congenital absence of MUC5B protein.
Methods
We performed whole-genome sequencing in an adult proband with unexplained bronchiectasis, impaired pulmonary function, and repeated Staphylococcus aureus infection. Deep phenotyping over a 12-year period included assessments of pulmonary radioaerosol mucociliary clearance. Genotyping with reverse phenotyping was organized for eight family members. Extensive experiments, including immunofluorescence staining and mass spectrometry for mucins, were performed across accessible sample types.
Measurements and Main Results
The proband, and her symptomatic sibling who also had extensive sinus disease with nasal polyps, were homozygous for a novel splicing variant in the MUC5B gene (NM_002458.2: c.1938 + 1G>A). MUC5B was absent from saliva, sputum, and nasal samples. Mucociliary clearance was impaired in the proband, and large numbers of apoptotic macrophages were present in sputum. Three siblings heterozygous for the familial MUC5B variant were asymptomatic but had a shared pattern of mild lung function impairments.
Conclusions
Congenital absence of MUC5B defines a new category of genetic respiratory disease. The human phenotype is highly concordant with that of the Muc5b−/− murine model. Further study of individuals with decreased MUC5B production could provide unique mechanistic insights into airway mucus biology.
Keywords: MUC5B, mucociliary clearance, bronchiectasis
At A Glance Commentary
Scientific Knowledge on the Subject
Upregulation of the airway mucin glycoprotein MUC5B may be central to the pathogenesis of common (e.g., chronic obstructive pulmonary disease) and rare (e.g., idiopathic pulmonary fibrosis) lung diseases. The consequences of producing no MUC5B protein are unknown in humans but significant in mice, with impaired mucociliary clearance and chronic infection.
What This Study Adds to the Field
We performed a detailed characterization of a family with congenital absence of MUC5B, the chief component of human airway mucous. The two affected adult siblings had a variable phenotype of repeated Staphylococcus aureus infection, impairment of pulmonary function, bronchiectasis, and/or sinus disease with nasal polyps. Mucociliary clearance was impaired in the proband. These findings suggest that lessons learned from the Muc5b−/− murine model may be generalizable to humans. Hereditary mucin deficiency defines a new class of “hyposecretory” airway disease that is distinct from all other known genetic airway diseases, such as cystic fibrosis and primary ciliary dyskinesia.
Human airways are coated with a protective barrier of mucus that traps unwanted particulate matter and pathogens and eliminates them by way of mucociliary clearance (MCC) (1). The principal functioning components of this mucus are two secreted gel-forming mucin glycoproteins: MUC5B and MUC5AC (1). Mucin homeostasis is fundamental to airway health and disease (2). Individuals with mucoobstructive lung diseases (chronic obstructive pulmonary disease, cystic fibrosis [CF], primary ciliary dyskinesia [PCD], and non-CF bronchiectasis) consistently demonstrate elevated mucin concentrations (2). A common variant (rs35705950) in the promoter region of the MUC5B gene results in protein overexpression (3) and has been linked to several types of progressive lung fibrosis (4–6).
Inhibition of mucin production and secretion pathways, including MUC5B, has been suggested as a promising therapeutic target for treatment of progressive lung fibrosis (3). However, the clinical consequences of MUC5B deficiency are unclear. In mice, Muc5b is required for MCC and for controlling inflammation after microbial exposure (7). Muc5b−/− mice developed upper airway obstruction and spontaneous infection of upper and lower airways, followed by fatal bacteremia (7). It was hypothesized that these features are likely to be mimicked in humans (2) and that decreased mucin may contribute to age-related decline in MCC (8). A recent report provided preliminary evidence for an associated human phenotype, in its inclusion of two young siblings with severe early-onset respiratory disease who were homozygous for a nonsense variant in MUC5B (9). MUC5B is expressed during fetal development (10, 11), and it is unclear whether airways and parenchymal lung tissue would grow normally in its absence.
We describe here a family with congenital absence of MUC5B protein. The affected adult siblings have a variable phenotype of repeated Staphylococcus aureus infection, impairment of pulmonary function, bronchiectasis, and/or sinus disease with nasal polyps. The clinical and cellular phenotyping provides foundational support for an association between MUC5B deficiency and airway disease.
Preliminary data from this study were scheduled to be presented at the American Thoracic Society 2020 International Conference (May 15–20, 2020; Philadelphia, PA). That conference was canceled because of the global coronavirus disease (COVID-19) pandemic; however, the abstract was subsequently published (12).
Methods
Participant Recruitment
The proband was followed from ages 10 to 18 years in a specialized Bronchiectasis Clinic at The Hospital for Sick Children (Toronto, Canada) by author S.D.D. and is currently followed in a specialized Adult PCD and Related Disorders Clinic at St. Michael’s Hospital (Toronto, Canada) by author D.A.H. Medical history before age 10 years is based on parent report. Written informed consent was provided by each study participant, excepting that the 14-year-old sibling provided assent and her mother consented to the study on her behalf. All components of this research were approved by the local research ethics board (REB numbers 1000037726, 1000054690, and 1000009409).
Genetic Testing and Confirmatory Experiments
Whole-genome sequencing was performed for the proband at The Centre for Applied Genomics (Toronto, Canada), with DNA extracted from whole blood (see the Supplemental Methods in the online supplement) (13). We used an established variant annotation pipeline and filtering strategy that can facilitate the discovery of new gene–phenotype associations (14). Genotyping of the MUC5B variant (NM_002458.2: c.1938 + 1G>A) in the proband and all available family members was performed by Sanger sequencing using blood-derived DNA. Assessment of the impact of the DNA sequence variant on MUC5B mRNA splicing was done by targeted complementary DNA (cDNA) sequencing (Supplemental Methods). Saliva total RNAs were extracted for RT-PCR by standard procedures (Supplemental Methods).
Cell-based Experiments
Immunofluorescence staining was performed with MUC5B, MUC5AC, and anti–α-tubulin–fluorescein isothiocyanate antibodies on nasal airway cells differentiated in vitro from unrelated healthy control subjects and from the proband (Supplemental Methods). Label-free and labeled mass spectrometry–based detection and quantification of major mucins was performed on saliva, sputum, and/or nasal samples from family members and compared with control subjects (Supplemental Methods) (15–17). Sputum samples from the proband, two of her siblings, and an unrelated healthy control subject were assessed for macrophage number and morphology (Supplemental Methods). Hydration was measured by percent solids (Supplemental Methods). All samples were collected during nonexacerbation periods.
Pulmonary Radioaerosol MCC Measurements
Radioactive colloid aerosols (740 MBq of technetium-99m–labeled sulfur colloid; Pharmalucence) were administered by nebulizer into the airway of the proband over a 4-minute period. After a baseline static image, time-lapse imaging with a single-head γ camera (GE Millennium) was performed at 30 s/frame for 60 minutes with the patient in a sitting position. A static image and another 60 minutes of dynamic imaging were then obtained with the patient in a supine position. Static images were repeated in a sitting and a supine position at 24 hours after inhalation. Percent clearance was calculated using the baseline counts and the 24-hour decay-corrected counts, for the whole lung and by central and peripheral subregions.
Results
Clinical History
A 23-year-old female presented to our clinic at age 10 years with a chief complaint of recurrent pneumonia. She was born at term in North America after an uncomplicated pregnancy. Her birth weight was 2.7 kg. There was no neonatal respiratory distress. She was breastfed, and there was no household smoke exposure pre- or postnatally. She developed pneumonia at 1 month of life and required admission to the hospital. She went on to demonstrate recurrent chest infections, with exacerbations every 1–2 months. She had six to seven more hospitalizations for pneumonia in the first 5 years of life. Once she reached school age, her pneumonias were less severe, not requiring hospitalization; however, she has continued to require three to five courses of antibiotics per year. There was minimal cough and no sputum production between episodes of illness. X-rays were reportedly suggestive of bronchiectasis beginning around age 5 years. She never required ICU-level care or invasive ventilation. There was no history of recurrent ear infections after age 2 years, hearing loss, sinusitis, nasal polyps, congenital heart disease, laterality defects, dental problems, opportunistic infections, growth deficiency, reproductive health issues, or gastrointestinal symptoms.
An initial high-resolution thin-section chest computed tomography (CT) (HRCT) scan at age 10 years showed severe asymmetric cylindrical and saccular bronchiectasis (Figure 1A), most severe in the right upper and middle lobes but affecting all lobes of both lungs. Pulmonary function testing showed moderately impaired pulmonary function with a predicted FEV1 of 59% predicted (Table E1 in the online supplement). Lung volume measurement by plethysmography showed gas trapping and mild reduction in TLC, together suggesting mixed obstructive and restrictive defects typical of small airway disease (Table E1). HRCT imaging at age 12 years demonstrated radiographic progression of her lung disease, and at 22 years showed stable findings as compared with the HRCT scan at 12 years of age. Sputum cultures repeatedly grew methicillin-sensitive S. aureus, occasionally grew Haemophilus influenzae, and never grew Pseudomonas aeruginosa (Figure E1). She has been treated with an airway clearance regimen and antibiotics on an as-needed basis. Mucolytic agents were trialed and then discontinued because of a lack of clinical benefit. Pulmonary function testing at age 20 years showed lung volumes within normal limits (Table E1). Spirometry results from 33 separate assessments over a 12-year period suggest a gradual decline in pulmonary function (Figure E2). She now has a chronic nonproductive cough. Sinus CT at 22 years showed scattered mild mucosal thickening throughout the paranasal sinuses (Figure E3). Tympanometry and visual examination of the ear drums were normal. Extensive clinical and genetic investigations were negative, including normal cilia ultrastructure by transmission electron microscopy (Figure E4), normal cilia beating frequency by high-speed video microscopy (Figure E4), and a negative workup for primary immunodeficiencies (Supplemental History).
Figure 1.
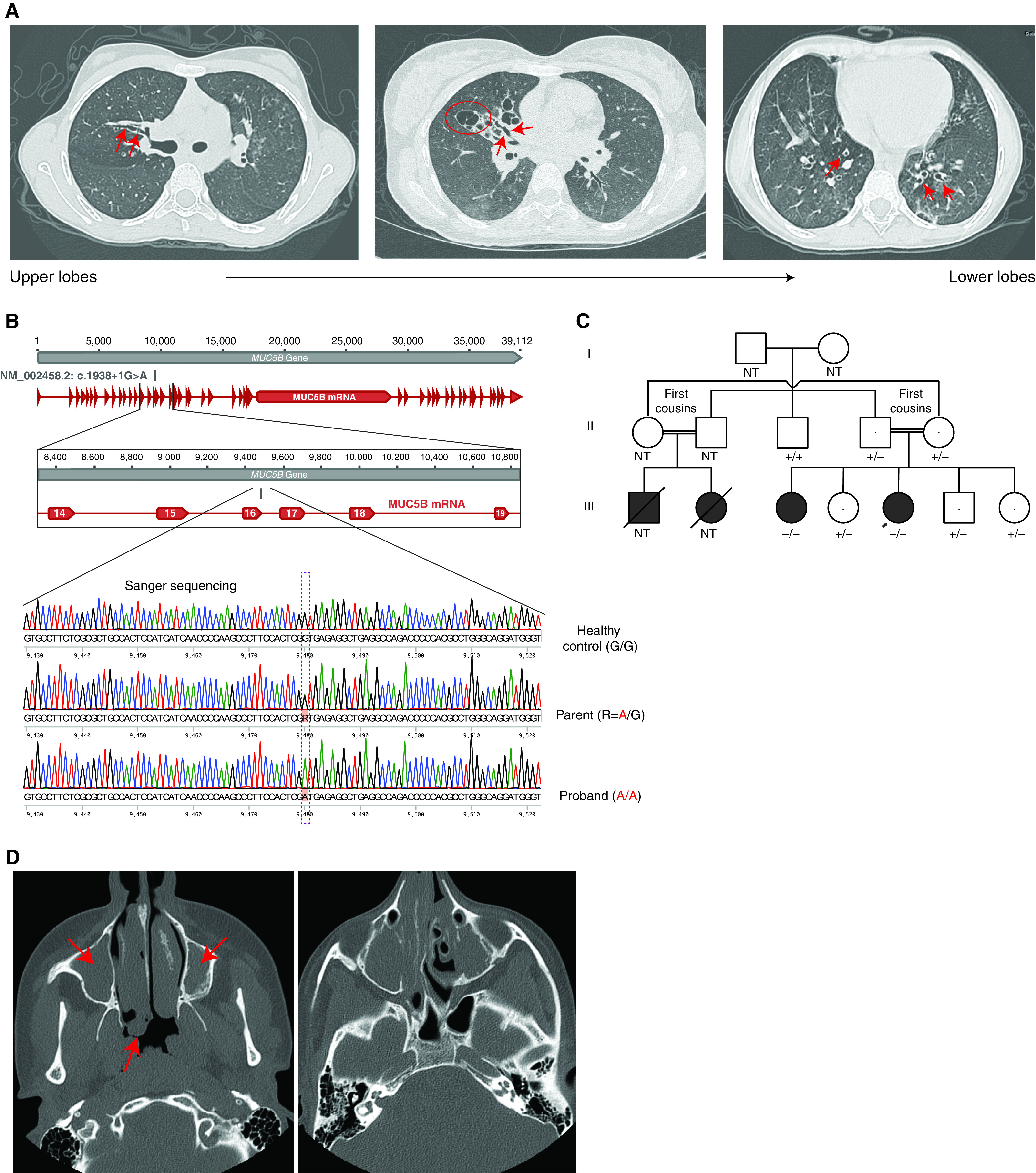
Phenotype and genotype information for the study participants. (A) High-resolution computed tomography (CT) images from the proband at age 10 years showing cylindrical (red arrows) and associated saccular bronchiectasis (e.g., red circle), most severe in the right upper and middle lobes. (B) Sanger sequencing chromatograms for an individual homozygous for the MUC5B reference allele (healthy control subject), an individual heterozygous for the MUC5B alternate allele (parent of proband), and an individual homozygous for the MUC5B alternate allele (proband). (C) Pedigree for the index family. −/− = homozygous for the alternate allele; +/− = heterozygous for the alternate allele; +/+ = homozygous for the reference allele. The arrow indicates the proband. (D) Volumetric axial sinus CT image from the proband’s affected sister at age 15 years showing extensive bilateral nasal polyposis (red arrows). NT = not tested.
Genome Sequencing and Targeted Familial Testing
Genome sequencing identified a novel homozygous variant in MUC5B (NM_002458.2: c.1938 + 1G>A) predicted to disrupt the consensus donor splice site at the exon 16/intron 16 boundary (Figure 1B). No alternative diagnosis was identified in the genome-wide sequence and copy number variation data, including with reannotation and reanalysis after a 3-year interval. There were no likely pathogenic or pathogenic variants in CFTR or genes associated with PCD and related phenotypes (Table E2) or with primary immunodeficiencies (Table E3), nor were there any rare, predicted damaging, homozygous coding variants in other biologically plausible candidate genes (Table E4).
The family history was significant for consanguinity, with her parents being first cousins of South Asian descent. Both parents were confirmed to be heterozygous for the familial MUC5B variant and were asymptomatic. Two cousins who died of respiratory disease in their early 20s had onset of symptoms in infancy and reportedly had recurrent pneumonias and bronchiectasis with no other known medical problems (Figure 1C). There was also a strong family history of asthma in second- and third-degree relatives in the maternal and paternal lineages.
Familial testing revealed that the proband’s 25-year-old sister is also homozygous for the familial MUC5B variant (Figure 1C). She reported a chronic nonproductive cough and a history of recurrent lung infections treated empirically with antibiotics. Pulmonary function testing revealed borderline to mild impairment in pulmonary function (Table E5), similar to her heterozygous siblings (Table E6). There was minor bronchial wall thickening without frank bronchiectasis on chest HRCT scan (Figure E5). However, she had a past history of bilateral nasal polyps requiring endoscopic nasal polypectomy at age 15 years. Testing for CF, including a sweat test (29 mmol/L) and CFTR recurrent mutation assay (Supplemental History), had been negative. The surgical pathology report noted that submucosal glands from the nasal polyps were normal and there was no increase in mucin (Figures E6 and E7). There was also extensive paranasal sinus disease on standard volumetric sinus CT scan (Figure 1D) without significant mastoid or middle ear disease. Similar to the proband, there was no history of recurrent ear infections after age 2 years, hearing loss, congenital heart disease, laterality defects, dental problems, opportunistic infections, growth deficiency, reproductive health issues, or gastrointestinal symptoms.
The proband’s other three siblings were heterozygous for the familial variant in MUC5B (Figure 1C). They were all asymptomatic; however, pulmonary function testing revealed a shared pattern of mild lung function impairments with a mixed obstructive/restrictive defect and gas trapping, suggestive of small airway disease (Table E6).
Molecular and Cellular Phenotyping
RT-PCR confirmed that the proband expressed a truncated MUC5B mRNA (Figure 2A). Gel extraction and Sanger sequencing of the truncated cDNA suggested that it lacked exon16 in the processed mature RNA (i.e., c.1844_1938del). This out-of-frame deletion would result in a frameshift [p.(Asn616Leufs*4)]. To confirm the consequences at a protein level, we performed immunofluorescence staining of the proband’s airway cells differentiated in vitro on an air–liquid interface, where airway cells form a pseudostratified cell layer mimicking in vivo condition. In contrast to healthy control subjects, MUC5B (but not MUC5AC) was absent in the patient’s cells (Figure 2B). To further validate these results in vivo, nasal wash, saliva, and induced sputum samples were subjected to mass spectrometry–based label-free proteomics and labeled mucin quantitation. MUC5B was not detected in any samples from the proband or her affected sister (Figure 2C). MUC5B was present in sputum, nasal wash, and saliva samples from the three unaffected heterozygous siblings but decreased compared with healthy control subjects (Figure 2C). This was confirmed by the labeled mass spectrometry–based absolute concentration analysis (Figure E8) (15).
Figure 2.
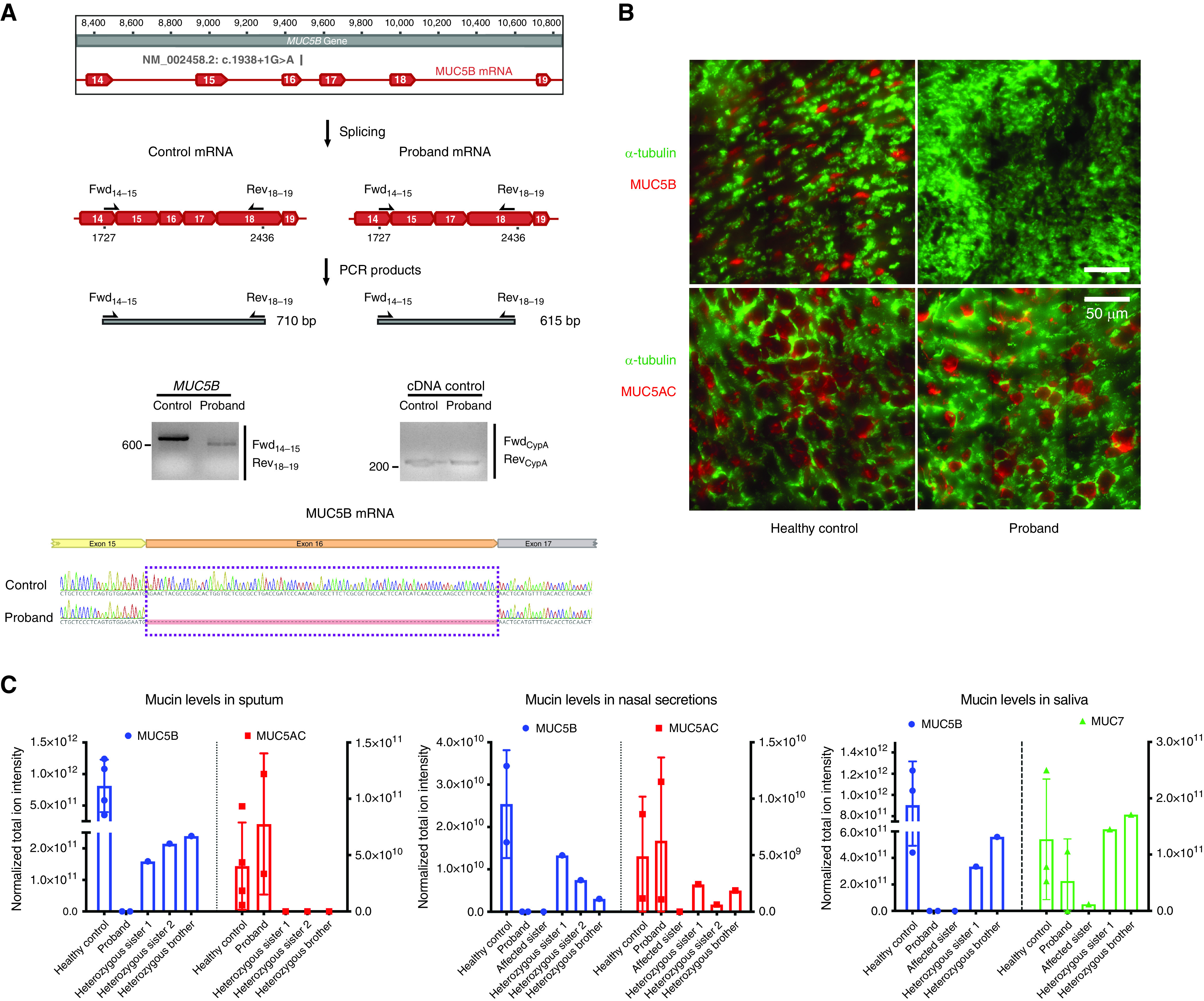
RNA- and protein-level consequences of the MUC5B genetic variant. (A) Functional analysis of the MUC5B c.1938 + 1G>A variant. Top: Schematic drawing of the MUC5B transcript (Ensembl ID: ENSG00000117983) and the primers for RT-PCR; middle: PCR amplification of MUC5B complementary DNA (cDNA) produced different-sized bands in the proband and in a healthy control subject. The left agarose gel compares a healthy control subject with a proband downstream of exon 15, revealing ∼95 bp of difference, reflective of the proband’s lack of exon 16. The expected fragment size for the healthy control subject is 710 bp. The right agarose gel is a loading control. The expected fragment size for the control CypA band is 296 bp. Images are from one experiment. Bottom: Amplified cDNA bands were gel purified and sequenced to validate the absence of exon 16 in the proband’s cells. Exon 15 is spliced with exon 17 with no additional or missing bases. (B) MUC5B and MUC5AC immunofluorescence staining of differentiated airway cells on an air–liquid interface from the proband and a healthy control subject. Images are representative results of more than three independent experiments with three biological replicates for healthy control subjects and three technical replicates for the proband. Scale bars, 50 μm. (C) Mass spectrometry quantification of major secreted mucins in saliva, sputum, and nasal samples from the available family members. Total ion intensity detected by mass spectrometry of all the detected mucin peptides are shown for quantitative comparison of the dominant mucins in sputum, nasal secretions, and saliva with comparison with the levels in healthy control subjects. No MUC5B peptide was detected in the samples from the proband or her affected sister. For mucins in sputum, n = 4 biological replicates for healthy control subjects, and n = 2, 1, 1, and 1 technical replicates for the proband and the other siblings, respectively. For mucins in nasal secretions, n = 2 biological replicates for healthy control subjects and n = 2, 1, 1, 1, and 1 technical replicates for the proband and the other siblings, respectively. For mucins in saliva, n = 3 biological replicates for healthy control subjects, and n = 2, 1, 1, and 1 technical replicates for the proband and the other siblings, respectively. Error bar stands for SEM. Fwd = forward; Rev = reverse.
Similar to the Muc5b−/− mice (7), large numbers of apoptotic macrophages and moderate numbers of neutrophils were present in sputum from the proband (Figure E9). The percent solids were approximately 2.5-fold higher in sputum samples from the proband (2.06% ± 0.6) compared with samples from her heterozygous siblings (0.77% ± 0.52) but lower than the previously reported values from cohorts with non-CF bronchiectasis (2.7% ± 2.0), CF (7.0% ± 2.7), and PCD (5.7% ± 1.7) (Figure E10) (17, 18).
Functional Assessment of Pulmonary MCC
The proband’s pulmonary radioaerosol MCC scan at age 23 years confirmed bolus formation and migration toward the buccal cavity, consistent with the prior scan at age 12 years that used an abbreviated pediatric protocol (Figure 3A; Supplemental History). The absence of an appreciable motile ciliary defect further distinguished the proband’s phenotype from PCD. Qualitatively, bolus motion was minimal in the sitting position and then marked in the supine position. Ventilation appeared impaired in the apex of the right lung. Quantitative analysis of imaging data obtained 24 hours after inhalation revealed decreased percent clearance relative to published norms (19), especially in the peripheral lung regions (54.0% of the mean control percent clearance; Figures 3B and 3C). This evidence of quantitatively impaired MCC, especially in the small airways, was consistent with the Muc5b−/− murine model (7).
Figure 3.
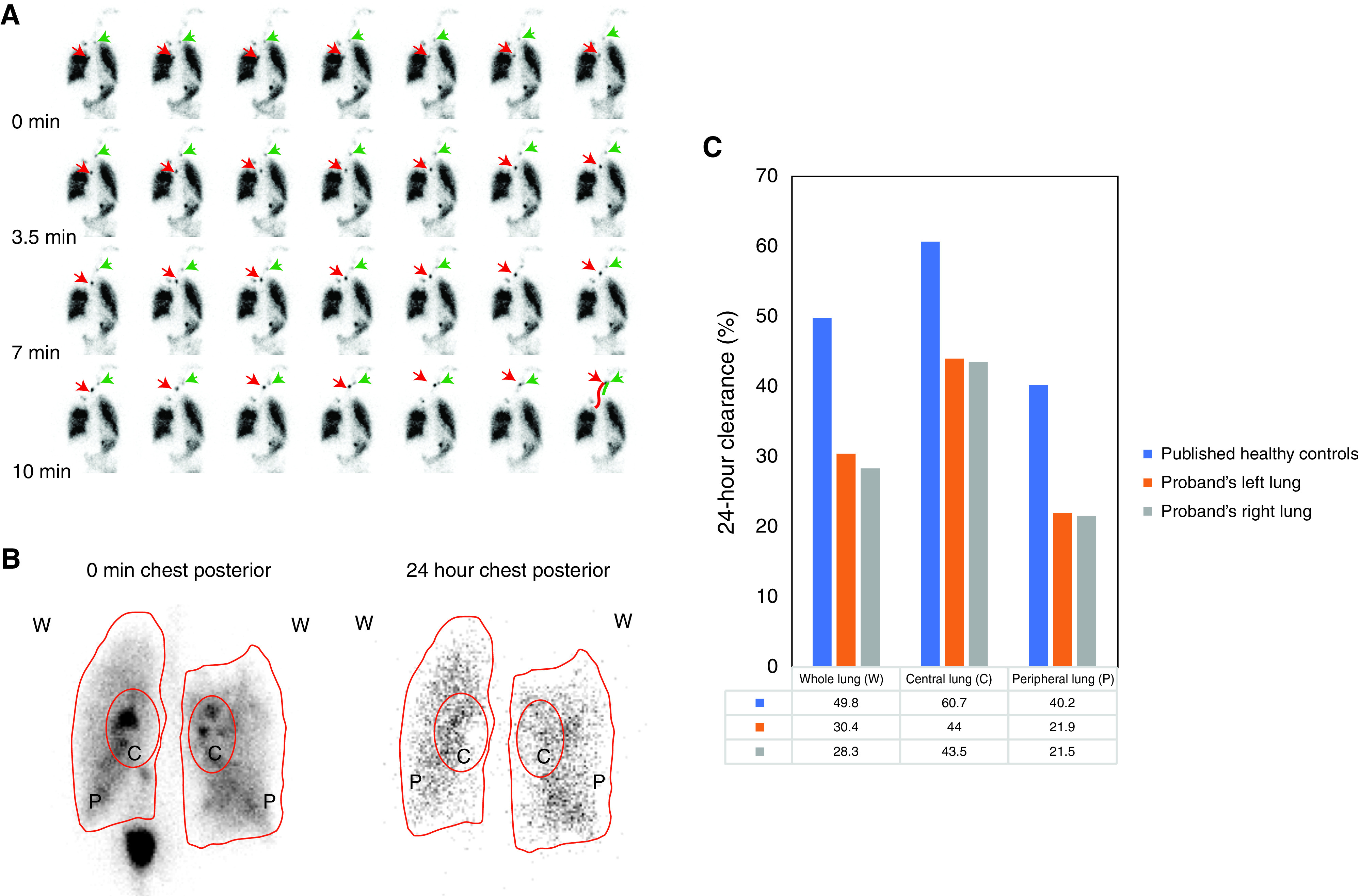
Assessment of mucociliary clearance. (A) Images from the pulmonary radioaerosol mucociliary clearance test performed in the proband. See text and Supplemental Methods for details. The red arrows and the green arrows represent the trajectory of two different boluses over time. (B) Radioactivity at 0 minutes and 24 hours for the chest posterior. (C) Quantitative radioactivity measurement shows impaired mucociliary clearance for the proband as compared with the reported healthy control subjects (18). The radioactivity is normalized by subtracting the background, and the 24-hour clearance is subject to decay correction. C = central lung; P = peripheral lung; W = whole lung.
Discussion
In this family, a biallelic loss-of-function variant in the MUC5B gene was associated with an airway phenotype. Adding to preliminary evidence recently published by another group (9), we demonstrated loss of protein in the airway mucus, fully characterized the associated respiratory phenotype, and advanced our still nascent understanding of the underlying disease mechanism. This report provides key evidence that MUC5B is crucial for maintenance of normal airway structure and function in humans. Our findings also suggest that lessons learned from the Muc5b−/− murine model (7) may be applicable to humans.
Pulmonary radioaerosol MCC was most impaired in the peripheral lung regions corresponding to the small airways. This pattern has been observed previously in individuals with CF (20). MUC5B is believed to be essential for both MCC and airway defense (1, 7, 8, 21), and the extent to which impaired MCC in the proband is a primary consequence of the MUC5B deficiency (vs. secondary, from infection and inflammation) is uncertain. As the main airway mucin (15), MUC5B provides infrastructure to the airway mucus gel to be transportable over cilia (7, 8). We speculate that lack of MUC5B in the lung surfaces will not only have an impact on the airway mucus properties and the barrier function but also adversely affect MCC. A limitation of this preliminary study is that assessment of MCC was not possible in the proband’s affected sibling; the degree to which bronchiectasis in the proband contributed to her impaired MCC remains unclear. Hereditary MUC5B deficiency may also compromise the innate lung defense against particular pathogens such as S. aureus in the trachea and the segmental bronchi, thereby allowing these pathogens to infiltrate more distally. The intrafamilial variability in expression could be explained in part by clinical and subclinical infections, with the pneumonias in infancy in the proband and her deceased cousins predisposing to more severe lung disease over time than was observed in the proband’s sister. The pattern of PFT abnormalities in the proband is like that of individuals with bronchiectasis of presumed infectious (nontuberculosis) etiology (22). We found no evidence in the genome sequencing data for an additional rare genetic modifier of expression in the proband, as was postulated as an explanation for phenotype discordance in the other reported family (9). The lower percent solids of the proband’s sputum, compared with individuals with CF, may account for the absence of P. aeruginosa that is known to require high percent mucus solids to make biofilms (23). An induced sputum sample from the affected sister was unavailable for study and would have been informative. Although nasal polyp biopsy was performed for one individual, another limitation of this study is that lung biopsy was not possible to facilitate morphologic analysis of the lung structure. In the prior report (9), lung biopsy at 18 months in a child with a homozygous nonsense variant in MUC5B revealed deficiency of alveolarization and interstitial fibrosis.
There are potential management implications arising from this diagnosis and its novel etiopathogenesis. Enhanced surveillance and aggressive treatment of intercurrent infections is warranted, as antibiotics during periods of exacerbation would be expected to be lifesaving. Susceptibility to S. aureus infection may be increased. An improved understanding of the natural history of hereditary mucin deficiency will facilitate prognostication and anticipatory care. A multisystem phenotype that extends beyond the airways cannot be ruled out. For example, although not observed to date in members of this index family, a negative impact on oral health and female reproduction is a concern given the apparent importance of MUC5B to saliva (24) and cervical mucus (25). The degree to which upregulation of other mucins can compensate for the absence of MUC5B in these complex antimicrobial environments is unclear and deserving of further study. Last, heterozygotes may have a subclinical phenotype, as was unexpectedly revealed by pulmonary function testing in the three asymptomatic siblings. Consistent with this hypothesis are two observations: 1) MUC5B is significantly constrained against monoallelic loss-of-function variation in the general population (26), and 2) Muc5b+/− mice have reduced MCC compared with wild-type littermates (8). Assessment of additional family members was not possible and would have been informative.
Upregulation of MUC5B is an integral feature of many common respiratory disorders and represents a promising therapeutic target. Sinopulmonary disease can also be associated with mucin deficiency. After excluding CF, PCD, and immunodeficiency, the diagnosis should be considered for individuals with cryptogenic bronchiectasis and/or recurrent pneumonias. Further study of humans with mono- or biallelic null alleles in MUC5B promises to provide unique mechanistic insights into airway mucus biology.
Acknowledgments
Acknowledgment
The authors thank the family whose participation made this study possible, the many healthcare providers involved in their care, Matthew Markovetz in the Hill Laboratory for assisting with experiments and analyses, Tayyaba Khan and Michael Sawras for coordinating patient visits and sample collection, and staff at The Centre for Applied Genomics.
Footnotes
Supported by NHLBI grant U54HL096458 (S.D.D., M. Knowles, and M.A.Z.); NIH/National Institute of Diabetes and Digestive and Kidney Diseases grant R01HL071798 (M. Knowles and M.A.Z.); R01HL110906 and R01HL103940 (M. Kesimer); NIH/National Institute of Diabetes and Digestive and Kidney Diseases grant P30 DK065988 and CF Foundation grant HILL20Y2-OUT (D.B.H.); and a grant from the Hospital for Sick Children Research Institute (N.P.). Genome sequencing was funded by the Centre for Genetic Medicine and the University of Toronto McLaughlin Centre. Airway cell in vitro measurements and experiments were funded by grants from the Ontario Thoracic Society and SickKids Foundation. The Genetic Disorders of Mucociliary Clearance Consortium (U54HL096458) is part of the National Center for Advancing Translational Sciences Rare Diseases Clinical Research Network (RDCRN) and supported by the RDCRN Data Management and Coordinating Center (U2CTR002818). RDCRN is an initiative of the Office of Rare Diseases Research funded through a collaboration between the National Center for Advancing Translational Studies and the NHLBI.
Author Contributions: S.D.D. conceived the study. V.M., C.R.M., and S.D.D. planned the initial experiments. G.C., Z.L., A.N.G., A.A., S.W., B.N., R.V., M. Khan, N.P., D.B.H, D.A.H., G.R., and S.D.D. carried out the experiments and acquired the data. G.C., Z.L., D.M., N.P., V.M., M. Kesimer, M. Knowles, M.A.Z., and S.D.D. analyzed and interpreted the data. G.C., Z.L., and S.D.D. drafted the manuscript, and the remaining authors revised it critically for important intellectual content. All authors give final approval of the version to be submitted.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202106-1456OC on January 13, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med . 2010;363:2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boucher RC. Muco-obstructive lung diseases. N Engl J Med . 2019;380:1941–1953. doi: 10.1056/NEJMra1813799. [DOI] [PubMed] [Google Scholar]
- 3. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med . 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hobbs BD, Putman RK, Araki T, Nishino M, Gudmundsson G, Gudnason V, et al. Overlap of genetic risk between interstitial lung abnormalities and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2019;200:1402–1413. doi: 10.1164/rccm.201903-0511OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med . 2013;368:2192–2200. doi: 10.1056/NEJMoa1216076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med . 2018;379:2209–2219. doi: 10.1056/NEJMoa1801562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature . 2014;505:412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grubb BR, Livraghi-Butrico A, Rogers TD, Yin W, Button B, Ostrowski LE. Reduced mucociliary clearance in old mice is associated with a decrease in Muc5b mucin. Am J Physiol Lung Cell Mol Physiol . 2016;310:L860–L867. doi: 10.1152/ajplung.00015.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alsamri MT, Alabdouli A, Alkalbani AM, Iram D, Tawil MI, Antony P, et al. Genetic variants of small airways and interstitial pulmonary disease in children. Sci Rep . 2021;11:2715. doi: 10.1038/s41598-021-81280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reid CJ, Gould S, Harris A. Developmental expression of mucin genes in the human respiratory tract. Am J Respir Cell Mol Biol . 1997;17:592–598. doi: 10.1165/ajrcmb.17.5.2798. [DOI] [PubMed] [Google Scholar]
- 11. Buisine MP, Devisme L, Copin MC, Durand-Réville M, Gosselin B, Aubert JP, et al. Developmental mucin gene expression in the human respiratory tract. Am J Respir Cell Mol Biol . 1999;20:209–218. doi: 10.1165/ajrcmb.20.2.3259. [DOI] [PubMed] [Google Scholar]
- 12. Liu Z, Costain G, Goczi A, Albulescu A, Walker S, Ngan B, et al. Hereditary mucin deficiency caused by biallelic loss-of-function of MUC5B defines a novel category of lung disease. Am J Respir Crit Care Med . 2020;201:A5353. doi: 10.1164/rccm.202106-1456OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med . 2018;20:435–443. doi: 10.1038/gim.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Costain G, Walker S, Marano M, Veenma D, Snell M, Curtis M, et al. Genome sequencing as a diagnostic test in children with unexplained medical complexity. JAMA Netw Open . 2020;3:e2018109. doi: 10.1001/jamanetworkopen.2020.18109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, Davis CW, et al. Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med . 2017;377:911–922. doi: 10.1056/NEJMoa1701632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reidel B, Radicioni G, Clapp PW, Ford AA, Abdelwahab S, Rebuli ME, et al. E-cigarette use causes a unique innate immune response in the lung, involving increased neutrophilic activation and altered mucin secretion. Am J Respir Crit Care Med . 2018;197:492–501. doi: 10.1164/rccm.201708-1590OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramsey KA, Chen ACH, Radicioni G, Lourie R, Martin M, Broomfield A, et al. Airway mucus hyperconcentration in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med . 2020;201:661–670. doi: 10.1164/rccm.201906-1219OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bush A, Payne D, Pike S, Jenkins G, Henke MO, Rubin BK. Mucus properties in children with primary ciliary dyskinesia: comparison with cystic fibrosis. Chest . 2006;129:118–123. doi: 10.1378/chest.129.1.118. [DOI] [PubMed] [Google Scholar]
- 19. Bennett WD, Wu J, Fuller F, Balcazar JR, Zeman KL, Duckworth H, et al. Duration of action of hypertonic saline on mucociliary clearance in the normal lung. J Appl Physiol (1985) . 2015;118:1483–1490. doi: 10.1152/japplphysiol.00404.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med . 2006;354:241–250. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 21. Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T, et al. Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med . 2019;199:715–727. doi: 10.1164/rccm.201804-0734OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopes AJ, Camilo GB, de Menezes SL, Guimarães FS. Impact of different etiologies of bronchiectasis on the pulmonary function tests. Clin Med Res . 2015;13:12–19. doi: 10.3121/cmr.2014.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA . 2006;103:18131–18136. doi: 10.1073/pnas.0606428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frenkel ES, Ribbeck K. Salivary mucins protect surfaces from colonization by cariogenic bacteria. Appl Environ Microbiol . 2015;81:332–338. doi: 10.1128/AEM.02573-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gipson IK, Moccia R, Spurr-Michaud S, Argüeso P, Gargiulo AR, Hill JA, III, et al. The amount of MUC5B mucin in cervical mucus peaks at midcycle. J Clin Endocrinol Metab . 2001;86:594–600. doi: 10.1210/jcem.86.2.7174. [DOI] [PubMed] [Google Scholar]
- 26. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome Aggregation Consortium Analysis of protein- coding genetic variation in 60,706 humans. Nature . 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]