

Abstract
Cerebral ischemia is the leading cause for long‐term disability and mortality in adults due to massive neuronal death. Currently, there is no pharmacological treatment available to limit progressive neuronal death after stroke. A major mechanism causing ischemia‐induced neuronal death is the excessive release of glutamate and the associated overexcitation of neurons (excitotoxicity). Normally, GABAB receptors control neuronal excitability in the brain via prolonged inhibition. However, excitotoxic conditions rapidly downregulate GABAB receptors via a CaMKII‐mediated mechanism and thereby diminish adequate inhibition that could counteract neuronal overexcitation and neuronal death. To prevent the deleterious downregulation of GABAB receptors, we developed a cell‐penetrating synthetic peptide (R1‐Pep) that inhibits the interaction of GABAB receptors with CaMKII. Administration of this peptide to cultured cortical neurons exposed to excitotoxic conditions restored cell surface expression and function of GABAB receptors. R1‐Pep did not affect CaMKII expression or activity but prevented its T286 autophosphorylation that renders it autonomously and persistently active. Moreover, R1‐Pep counteracted the aberrant downregulation of G protein‐coupled inwardly rectifying K+ channels and the upregulation of N‐type voltage‐gated Ca2+ channels, the main effectors of GABAB receptors. The restoration of GABAB receptors activated the Akt survival pathway and inhibited excitotoxic neuronal death with a wide time window in cultured neurons. Restoration of GABAB receptors and neuroprotective activity of R1‐Pep was verified by using brain slices prepared from mice after middle cerebral artery occlusion (MCAO). Treatment with R1‐Pep restored normal GABAB receptor expression and GABA receptor‐mediated K+ channel currents. This reduced MCAO‐induced neuronal excitability and inhibited neuronal death. These results support the hypothesis that restoration of GABAB receptor expression under excitatory conditions provides neuroprotection and might be the basis for the development of a selective intervention to inhibit progressive neuronal death after ischemic stroke.
Keywords: CaMKII, cerebral ischemia, GABAB receptor, interfering peptide, neuroprotection
There is no pharmacological treatment available to limit progressing neuronal death after cerebral ischemia. Here we show that targeting the interaction of GABAB receptors with CaMKII using a synthetic interfering peptide after an excitotoxic insult in cultured neurons or in brain slices from MCAO‐treated mice restored downregulated GABAB receptor expression and function, normalized aberrant expression of their major effectors, reduced neuronal excitability, activated the Akt survival pathway, and inhibited progressing neuronal death.

1. INTRODUCTION
Ischemic stroke is associated with high mortality and survivors often suffer from disabilities such as paralysis or disturbance of speech, which cause tremendous health and socio‐economic cost [1]. Disruption of blood supply to or within the brain deprives neurons of oxygen and energy. Currently, the only available pharmacological treatment for acute stroke is the application of thrombolytic agents to restore blood circulation early after the ischemic insult [2, 3]. If reperfusion is not rapidly initiated, this leads to metabolic derailment and death of neurons in the region surrounding the lesion core. The development of treatments to diminish progressive, delayed ischemic neuronal death remains a major challenge and calls for novel therapeutic strategies [4, 5, 6]. One promising strategy is the targeting of protein–protein interactions with small interfering peptides to modify signaling pathways that are altered in disease states. In fact, there is a variety of in vitro and in vivo preclinical examples advocating the power of this approach and some interfering peptides are currently in clinical trials [7, 8, 9, 10].
Excitotoxicity, caused by massive release of glutamate and over‐stimulation of glutamate receptors, is thought to be a major mechanism for progressive and delayed ischemia‐induced neuronal death [11, 12, 13, 14, 15]. Under physiological conditions, the activity of glutamate receptors and neuronal excitability is controlled by GABAB receptors [16]. GABAB receptors are heterodimeric G protein‐coupled receptors, comprising GABAB1 and GABAB2 subunits. They are expressed by virtually all neurons at pre‐ and postsynaptic locations where they convey prolonged inhibition [16]. In principle, ischemic over‐excitation of neurons should increase the activity of GABAB receptors, counteract over‐stimulation of glutamate receptors and limit progressing excitotoxic neuronal death. Unfortunately, this is not the case because excitotoxic conditions rapidly downregulate GABAB receptors [17, 18, 19, 20, 21, 22, 23]. Studies on cultured neurons demonstrated that sustained activation of glutamate receptors increased CaMKII‐dependent phosphorylation of serine 867 in the GABAB1 subunit [17]. CaMKIIβ‐mediated phosphorylation of internalized GABAB receptors serves as a signal for K63‐linked ubiquitination of GABAB1 at multiple sites by the E3‐ligase Mind‐bomb 2 and sorting of the receptors to lysosomal degradation [24, 25].
Since phosphorylation of GABAB1(S867) by CaMKIIβ plays a key role in the aberrant decreased abundance of GABAB receptors under excitotoxic conditions, we hypothesized that preventing the interaction of CaMKIIβ with GABAB receptors, and thereby inhibiting phosphorylation of GABAB1, would block the aberrant degradation of the receptors. This should restore normal cell surface expression of the receptors, reduce neuronal excitability and limit excitotoxic neuronal death. To this end, we developed an interfering peptide that inhibits the interaction of CaMKIIβ with GABAB receptors and tested its effects on cultured neurons exposed to excitotoxic conditions and in the middle cerebral artery occlusion (MCAO) mouse model of cerebral ischemia.
2. METHODS AND MATERIALS
2.1. Animals
In keeping with the 3R principles, we first thoroughly evaluated the interfering peptide in a series of in vitro experiments using cultured cortical neurons prepared from Wistar rat E18 embryos. We then tested the activity of the interfering peptide in the MCAO model of cerebral ischemia using 8‐ to 12‐week‐old male C57BL/6J mice. Mice were housed up to five per cage with a standard 12/12‐h light/dark cycle and food and water available ad libitum. The mice were randomly (random number generator) assigned to MCAO or sham‐operation. All animal experiments were approved by the Zurich cantonal veterinary office, Zurich, Switzerland (licence ZH152/16, ZH011/19, and ZH031/16).
2.2. Antibodies
Mouse Akt (1:250 for immunofluorescence [IF], Cell Signaling Technology #2920), rabbit pAkt‐S473 (1:1000 for IF, Cell Signaling Technology #4060), rabbit CaMKII (1:1000 for IF, 1:100 for PLA; Abcam #ab52476), mouse CaMKIIβ (1:1500 for Western blotting [WB], Thermo Fisher Scientific #13‐9800), rabbit phospho‐CaMKII(Thr286) (1:500 for WB, Cell Signaling Technology #12716), rabbit CaV2.2 (1:1000 for IF and 1:200 for Western blotting; Alomone Labs #ACC‐002), mouse GABAB1 (1:250 for IF and Western blotting, 1:100 for PLA; Abcam #ab55051), rabbit GABAB1b directed against the N‐terminus of GABAB1b (affinity‐purified, 1:100 for IF; custom made by GenScript) [26], rabbit GABAB2 directed against the N‐terminus of GABAB2 (affinity‐purified, used for cell surface staining, 1:25 for IF; custom made by GenScript) [27], rabbit GABAB2 (1:500 for IF, 1:100 for PLA, 1:800 for Western blotting; Abcam #ab75838), mouse GFP antibody (for immunoprecipitation, TaKaRa #632381), rabbit Kir3.2 (1:250 for IF and Western blotting, Alomone Labs #APC‐006), mouse GSK3β (1:250 for IF, Abcam #ab93926), rabbit pGSK3β‐S9 (1:1500 for IF, Abcam #ab131097), rabbit pGSK3β‐Y216 (1:1500 for IF, Abcam #ab75745), rabbit ubiquitin K63‐specific (clone Apu3, 1:50 for PLA; Millipore #05‐1308), rabbit NeuN (1:400 for IF; Millipore #ABN78), mouse phospho‐serine (1:150 for PLA, Sigma‐Aldrich #P5747), Alexa Fluor 647 and 488‐conjugated streptavidin (1:100; Jackson ImmunoResearch Laboratories #016‐600‐084 and #016‐540‐084). For immunofluorescence staining, secondary antibodies used were labeled with Alexa Fluor 488, 555, and 647 (1:2000), Cy‐3 (1:500) or Cy‐5 (1:300) (Jackson ImmunoResearch Laboratories) and for Western blotting antibodies were conjugated to IRDye 700CW or IRDye 800CW (LI‐COR Biosciences).
2.3. Interfering peptide
The interfering peptide was identified by screening a small library of synthetic peptides (15–25 amino acids long) comprising all intracellularly located amino acid sequences of GABAB1 for their ability to prevent the decreased abundance of GABAB receptors after stressing cultured cortical neurons for 1 h with the 50 μM glutamate. The interfering peptide (R1‐Pep) consists of GABAB1 amino acids 867–888 (SETQDTMKTGSSTNNNEEEKSR, rat sequence). A peptide (Ctrl‐Pep) containing the same amino acids, but in a randomly scrambled order was used as a negative control (TESRKNMKSSSTNEETGNDETQ). To render them cell‐permeable, both peptides were tagged at the N‐terminus with a peptide sequence derived from the Rabies virus glycoprotein (YTIWMPENPRPGTPCDIFTNSRGKRASNGGGG) [28] followed by nine arginine residues. Both peptides were custom‐synthetized by Pepmic Co., Ltd. Unless otherwise stated, the peptides were used at a concentration of 10 μg/ml.
2.4. Middle cerebral artery occlusion
MCAO was performed as described [29]. In brief, mice were anesthetized with isoflurane (Provet AG) and analgesia was provided by buprenorphine (0.1 mg/kg, s.c., Indivior Schweiz AG) and lidocaine (5 mg/kg, s.c., at the site of incision, Steuli Pharma). For transient MCAO, the left common carotid artery was exposed and the middle cerebral artery was occluded by inserting a 7‐0 silicone rubber‐coated monofilament (Catalog no.: 701956PK5Re, Doccol Corp.). After 60 min, the filament was withdrawn to permit reperfusion. After surgery, the mice were transferred to a 37°C warm recovery chamber for exactly 60 min and then sacrificed for electrophysiological or immunostaining experiments. Peptides were applied to brain slices 75–80 min after starting reperfusion. Electrophysiological recordings were performed 3–5 h after peptide application and immunohistochemical stainings were done 6 h after peptide treatment. For sham‐operated mice, the filament was inserted up to the left middle cerebral artery and immediately withdrawn to allow instant reperfusion.
2.5. Primary neuronal cultures
Primary neurons co‐cultured with glia were obtained from the cerebral cortex of Wistar rat E18 embryos. The cortices were dissected on ice in phosphate‐buffered saline (PBS) containing 5.5 mM glucose (Sigma‐Aldrich) and antibiotic‐antimycotic solution (1:100, Invitrogen), cut into small pieces, and digested in papain solution (0.5 mg/ml papain [Sigma‐Aldrich], 1 mg/ml bovine serum albumin [BSA, Sigma‐Aldrich], 10 μg/ml DNaseI [Roche Diagnostics], 10 mM glucose) for 15 min at 37°C. After two washes in DMEM (Gibco Life Technologies) containing 10% fetal calf serum (FCS, Gibco Life Technologies) and 1:100 antibiotic‐antimycotic solution (complete DMEM), the tissue was gently triturated. 60,000 cells were plated onto poly‐L‐lysine coated (50 μg/ml in PBS, Sigma‐Aldrich) coverslips (18 mm, Epredia, Gerhard Menzel GmbH) and placed in 12‐well cell culture plates (Sarstedt) and incubated in 2 ml complete DMEM overnight at 37°C/5% CO2. Then, the DMEM was exchanged with 2 ml NU‐medium (MEM [Gibco Life Technologies] with 15% NU serum [Corning], 2% B27 supplement [Gibco Life Technologies], 15 mM HEPES, 0.45% glucose, 1 mM sodium pyruvate, 2 mM GlutaMAX [Gibco Life Technologies]). Neuron/glia cultures were used after 11–18 days in vitro.
2.6. Glutamate and peptide treatment of cultured neurons
For inducing glutamate stress, 1 ml of the culture medium from each coverslip‐containing well was transferred to a fresh 12‐well plate and placed in the incubator. Then cultures were treated with glutamate (50 μM; Sigma‐Aldrich) and incubated for 1 h at 37°C and 5% CO2. The coverslips were briefly washed with PBS and transferred to the fresh 12‐well plate containing the saved original conditioned culture medium supplemented with R1‐Pep (10 μg/ml) or with Ctrl‐Pep (10 μg/ml) and incubated for 12–16 h before testing. For all experiments, except for the survival assay, the cultures were treated with peptides immediately after the glutamate incubation.
2.7. Culture and transfection of HEK 293 cells
HEK 293 cells (human embryonic kidney, ATCC) were cultured in DMEM (Gibco Life Technologies) containing 10% fetal bovine serum (Gibco Life Technologies) and penicillin/streptomycin (Gibco Life Technologies). HEK 293 cells were transfected with plasmids using the polyethyleneimine method according to the jet‐PEI protocol (Polyplus Transfection).
2.8. CaMKII assay
The effect of P1‐Pep on CaMKII activity was determined using the CycLex CaM kinase II Assay Kit (MBL; Cat# CY‐1173) according to the manufactures instructions using recombinant CaMKIIβ (Abcam, ab268377) and recombinant calmodulin (Abcam, ab94219) protein. The kinase reactions containing increasing concentrations of R1‐Pep or KN93 (10 μM; Sigma‐Aldrich) were run for 30 min at 30°C.
2.9. Western blotting
For Western blot analysis, neuron/glia co‐cultures were grown for 12 days on 6 cm culture dishes plated with 500,000 cells. Cultures were washed two times with ice‐cold PBS, harvested, and homogenized by sonication. The samples were incubated with Laemmli sample buffer (Bio‐Rad) for 1 h at 37°C and aliquots containing 25 μg protein were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) using mini‐gels (Mini Protean 3; Bio‐Rad). Proteins were transferred onto nitrocellulose membranes in a semi‐dry transfer cell (Trans‐Blot SD; Bio‐Rad) at 15 V for 75 min. After blotting, the transferred total proteins were stained with REVERT 700 Total Protein Stain (LI‐COR Biosciences) and detected by the ODYSSEY CLx scanner (LI‐COR Biosciences). After destaining, the blots were blocked for 1 h in PBS containing 5% nonfat dry milk at room temperature, followed by incubation with primary antibody overnight at 4°C in PBS containing 5% nonfat dry milk. The blots were then washed five times for 5 min with TBST and incubated with secondary antibodies for 1 h at room temperature. The blot was washed again with TBST and immunoreactivity was detected by the ODYSSEY CLx scanner (LI‐COR Biosciences). Immunoreactivity was quantified with the Image Studio software (LI‐COR Biosciences) and normalized to total protein in the corresponding lanes.
2.10. Immunoprecipitation
To test the interaction of R1‐Pep with CaMKIIβ, EGFP‐tagged CaMKIIβ was expressed in HEK 293 cells and incubated with/without biotin‐labeled R1‐Pep for 3 h. Cells were then harvested, homogenized in 5 mM Tris pH 7.4 containing protease inhibitors (Complete Mini, Roche Diagnostics), and centrifuged for 60 min at 60,000g. Aliquots of the supernatant were used for immunoprecipitation of CaMKII using GFP antibodies coupled to magnetic beads (Pierce Direct Magnetic IP/Co‐IP Kit; Thermo Fisher Scientific). Aliquots of the supernatant were incubated with 25 μl GFP‐coupled magnetic beads (5 μg antibody/25 μl beads) at 4°C overnight. Subsequently, the beads were extensively washed with buffer and precipitated proteins were eluted with Laemmli sample buffer. Eluted proteins were then subjected to electrophoresis and analyzed for the presence of CaMKII and R1‐Pep by Western blotting using CaMKII antibodies and Alexa Fluor 647‐conjugated streptavidin (Jackson ImmunoResearch Laboratories), respectively.
2.11. Immunofluorescence staining
2.11.1. Cultured neurons
To monitor cell surface expression of GABAB receptors, cultured neurons were washed with buffer A (25 mM HEPES pH 7.4, 119 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose) and incubated on ice with the antibodies directed against the extracellularly located N‐terminal amino acid sequence of GABAB2 diluted in buffer A, 10% normal donkey serum (NDS; Merk Millipore), for 2 h. After washing with ice‐cold buffer A, the cultures were incubated with secondary antibody for 1 h on ice, washed again, and fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences) containing 4% sucrose for 15 min at room temperature. The coverslips were either mounted on glass slides with DAKO fluorescence mounting medium (Agilent Technologies) or processed for staining of intracellular proteins.
To stain intracellular proteins, cultures were fixed with 4% PFA containing 4% sucrose for 15 min and permeabilized for 10 min with 0.2% Triton X‐100 in PBS. The cells were then incubated with primary antibody diluted in PBS containing 10% NGS overnight at 4°C, washed, and processed for staining with secondary antibodies as described above.
2.11.2. Brain slices
Following MCAO or sham surgery, mice were euthanized under isoflurane anesthesia and brains were removed and sliced into 90‐μm‐thick coronal sections using a vibratome (HM 650; Microm). Slices were then incubated in a modified 12‐well culture plate containing oxygenated artificial cerebrospinal fluid (ACSF pH 7.4, 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 1 mM MgCl2, 2 mM CaCl2, and 25 mM glucose). R1‐Pep or Ctrl‐Pep was added (10 μg/ml) to all slices simultaneously and incubated at room temperature for 6 h with constant oxygenation (95% O2 and 5% CO2). Brain slices were fixed with 4% PFA for 30 min at room temperature and processed for immunostaining as free‐floating sections. Brain slices were incubated with primary antibody prepared in 50 mM Tris pH 7.4, 150 mM NaCl containing 0.2% Triton X‐100 (Tris‐Triton), and 2% NDS overnight at 4°C. After series of intensive washing with Tris‐Triton, secondary antibody diluted in Tris‐Triton, 2% NDS was added for 1 h at room temperature. Following the final wash, brains were mounted onto glass slides (SuperFrost Plus; Thermo Fisher Scientific), dried, and coverslipped with DAKO fluorescence mounting medium.
2.11.3. Fluoro‐Jade C staining
Brains from mice that underwent sham or MCAO surgery were extracted and cut into 1 mm thick coronal slices using a brain matrix (Zivic Instruments). The slices were incubated with R1‐Pep or Ctrl‐Pep for 6 h (as described above), followed by incubation in 4% PFA overnight at 4°C. Slices were then cut using a cryostat into 14 μm sections and mounted onto glass slides (SuperFrost Plus; Thermo Fisher Scientific). For Fluoro‐Jade C (Sigma‐Aldrich) staining, slides containing the somatosensory cortex were first immersed in a basic alcohol solution consisting of 1% sodium hydroxide in 80% ethanol for 5 min and then rinsed for 2 min in 70% ethanol. After washing in distilled water for 2 min, slides were incubated in 0.06% potassium permanganate solution for 10 min and rinsed in water for 1 min. Slides were transferred for 10 min to a 0.0001% w/v solution of Fluoro‐Jade C dissolved in 0.1% acetic acid then rinsed three times in distilled water for 1 min and left to dry at room temperature. Finally, slides were cleared with xylene for 1 min and then coverslipped with DPX mounting media (Sigma‐Aldrich).
2.12. In‐situ proximity ligation assay
The in‐situ proximity ligation assay (PLA) is an extremely sensitive assay for the detection and quantification of protein–protein interactions and post‐translational modifications [30, 31]. Here, we used this technique for the analysis of GABAB receptor interaction with CaMKII, for studying K63‐linked ubiquitination, and phosphorylation of GABAB receptors as described previously [32]. Cells were fixed with 4% PFA for 20 min at room temperature, permeabilized with 0.2% Triton X‐100 in PBS, and then incubated overnight at 4°C with the appropriate pair of primary antibodies (diluted in PBS containing 5% BSA). Thereafter, in situ PLA was performed using the Duolink kit (Sigma‐Aldrich) according to the manufacturer's instructions.
Neurons were imaged for PLA signals using a confocal laser scanning microscope (see below). Quantitative analysis of the acquired images was performed using ImageJ. The soma of each neuron was defined and the number of fluorescent spots inside this area was counted using “Find maxima,” with the noise tolerance kept constant between different conditions within the same experiment. The PLA signals of neurons were normalized to the expression levels of GABAB1 and to the area analyzed.
2.13. Confocal laser scanning microscopy
2.13.1. Cultured neurons
Images from the immunocytochemistry experiments were acquired using the Zeiss LSM 700 or LSM 710 confocal microscope using a 40× plan‐apochromat oil differential interference contrast objective, 1.3 NA (Carl Zeiss AG) at an image resolution of 1024 × 1024 pixels. Laser intensities and detector gains were kept within the dynamic range of the fluorescent signal to avoid pixel saturation. An image stack of 5‐optical sections at 0.3 μm z‐spacing was recorded and fluorescence intensities were analyzed in ImageJ as detailed previously [21]. For quantification of cell surface staining, the outer and inner borders of the cell surface were carefully marked. The mean fluorescence intensity value of the inner border was subtracted from the mean intensity value of the outer border, so that only the fluorescence intensity from the cell surface remained. For quantification of the total staining, the outer border of the neuron was outlined and the mean fluorescence intensity was measured.
2.13.2. Brain slices
Images were acquired using an LSM 800 confocal microscope (Carl Zeiss) using Plan‐Apochromat 10×/0.45 and Plan‐Neofluar 25/0.8 objectives. Image stacks of nine optical sections spaced at 1 μm z‐spacing were taken from each brain slice. For quantification of GABAB receptor expression, an area of interest was defined and mean fluorescence values were determined using ImageJ. NeuN positive neurons were automatically counted in the selected area using ImageJ.
2.13.3. Image processing for display
The images used for quantification were quite dim to avoid saturation of signals and were not suitable for display in their original form. Therefore, the staining intensities of the images were linearly enhanced by the identical degree in all images belonging to one experiment.
2.14. Induction of excitotoxicity and analysis of neuronal death in culture
For induction of excitotoxicity, cells were supplemented with glutamate (50 μM; Sigma‐Aldrich) and incubated for 1 h at 37°C and 5% CO2. The coverslips were then briefly rinsed with PBS and further incubated in a conditioned culture medium supplemented with the peptides for 12–16 h before testing. For analysis of neuronal death, R1‐Pep or Ctrl‐Pep was added at various time intervals (0–24 h) after glutamate stress and cultures were analyzed after 48 h.
To quantify neuronal death, neurons were stained with NeuN antibodies and the total number of cells (neurons plus glia) was determined by staining with DAPI (included in the mounting medium, Fluoroshield; Sigma Aldrich). The ratio of NeuN‐positive neurons and total cells was used for the evaluation.
2.15. Electrophysiology
2.15.1. Cultured neurons
Whole‐cell patch‐clamp recordings were taken from neurons either treated with glutamate (50 μM) alone or glutamate and R1‐Pep (10 μg/ml) for 1 h before recording. Recording electrodes were filled with an internal solution containing (in mM) potassium gluconate (130), NaCl (5), EGTA (1), HEPES (10), Mg‐ATP (5), and Na‐GTP (0.5), pH 7.35–7.4 and osmolarity 290–300 mOsm. Neurons were superfused with an external solution containing (in mM) NaCl (120), NaHCO3 (26), NaH2PO4 (1.25), KCl (2.5), HEPES (5), Glucose (14.6), CaCl2 (2), and MgCl2 (1), pH 7.35–7.4 and osmolarity 310–320 mOsm at a flow rate between 2 and 3 ml/min.
Whole‐cell patch‐clamp recordings were acquired using a HEKA EPC10 amplifier and Patchmaster software. Neurons were voltage‐clamped at a holding potential of −50 mV. Baclofen was bath applied at 20 μM. Changes in the holding current were measured between the baselines before drug application to the peak current following drug application. The baseline current was defined as the average current within a 1 min epoch directly before addition of drugs, and the peak current was averaged over 1 min during the greatest increase in holding current following drug application. Series resistance was monitored before and after the experiment, and data were excluded if the series resistance changed by >20% during the recording.
Spontaneous postsynaptic currents were recorded before, during, and after the bath application of 10 μM CGP 56999 at a holding potential of −70 mV. Recordings were performed at 34°C using a Multiclamp 700B amplifier controlled by the Clampex acquisition software (Molecular Devices). Spontaneous activity was analyzed using the Mini Analysis Program (Synaptosoft) using the peak detection algorithm and the detected events were verified visually. The amplitude threshold was set two times the baseline rms noise level (5–10 pA).
2.15.2. Brain slices
After MCAO surgery (see above), 300 μm coronal brain sections containing somatosensory cortical areas were taken using a vibratome (HM 650; Microm), incubated in oxygenated artificial cerebrospinal fluid (ACSF, pH 7.4, osmolarity 315 mOsm) containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 2 CaCl2, and 25 glucose at 34°C for 30 min and then at room temperature until used.
Recordings were taken from pyramidal cells, which were identified by their relatively large soma and shape, a low input resistance, and according to their firing pattern induced by depolarizing current injection. Recordings were performed at 34 °C with continuous superfusion of oxygenated ACSF at 1–2 ml/min using a Multiclamp 700B amplifier controlled by the Clampex acquisition software (Molecular Devices). For all recordings, borosilicate glass patch pipettes (3.5–5 MΩ) were filled with intracellular solution containing (in mM): 135 mM potassium gluconate, 2 mM NaCl, 4 mM KCl, 4 mM EGTA, 10 mM HEPES, 4 mM Mg‐ATP, and 0.3 mM Na3GTP (osmolarity 290 mOsm, pH 7.3 with KOH).
To measure GABAB receptor‐mediated currents, neurons were held at −55 mV and current changes were measured in response to bath application of 100 μM baclofen. GABAB receptor‐mediated GIRK currents were confirmed by application of the GABAB receptor antagonist CGP 56999 (10 μM).
To determine neuronal excitability, current‐clamp experiments were performed using a series of 50 pA current increment steps of 250 ms duration. Neurons were maintained at their original resting membrane potential throughout the experiment. The resting membrane potential (V m) was measured immediately after acquiring the whole‐cell patch mode. Input resistance was calculated by injection of a 150 pA hyperpolarizing current step, and the firing threshold (V th) was calculated by injection of 5 pA depolarizing current steps until the first action potential was generated. Recordings were excluded from analysis if the series resistance (Rs) varied by 25%. The liquid junction potential was ~7 mV and not corrected.
2.16. Statistics
The experimental data were analyzed using GraphPad Prism (version 8.4.3) and are expressed as means ± standard deviation (SD). Data were analyzed by ordinary one‐way, Brown–Forsythe and Welch one‐way ANOVA or two‐way ANOVA followed by appropriate post hoc analysis (Tukey's or Dunnet T3 multiple comparison tests). All data sets displayed normal or lognormal distributions as analyzed by the D'Agostino‐Pearson test and QQ‐plots. Homogeneity of variance was tested using the Brown–Forsythe test. In case of significant deviation from homoscedasticity, Welch and Brown–Forsythe variations of ANOVA were used. Small to moderate deviations from equal variance in two‐way ANOVA were accepted as this test is fairly robust against the assumption of equal variances if the sample sizes are equal [33]. Sample size and the number of experiments performed are detailed in the respective figure legends. Differences between conditions were considered statistically significant when p < 0.05.
3. RESULTS
3.1. A small interfering peptide (R1‐Pep) inhibits the interaction of CaMKII with GABAB receptors
Under excitotoxic conditions, GABAB receptors are rapidly downregulated due to increased phosphorylation by CaMKIIβ [17], which serves as a signal for their lysosomal degradation [24]. This unfortunate cascade provokes excitotoxicity by reducing GABAB receptor‐mediated neuronal inhibition. To restore GABAB receptor‐mediated inhibition under excitotoxic conditions, which is assumed to provide neuroprotection, we identified a small interfering peptide that prevents the decreased abundance of GABAB receptors in cultured cortical neurons after glutamate stress. This peptide (designated R1‐Pep) comprises a sequence within the intracellular‐located C‐terminal domain of GABAB1 adjacent to the CaMKII phosphorylation site (Figure S1A, Supporting Information). To render it cell‐permeable, R1‐Pep was tagged at the N‐terminus with a peptide sequence derived from the Rabies virus glycoprotein [28], which delivers cargo to the brain and specifically targets neurons via a receptor‐mediated uptake mechanism [34]. In fact, cellular R1‐Pep uptake required expression of a GABAB receptor subunit at the cell surface (Figure S1B, Supporting Information). In all main experiments, the specificity of R1‐Pep was verified against a control peptide (Ctrl‐Pep), consisting of the same amino acids as R1‐Pep but in a random sequence (Figure S1A, Supporting Information).
As expected, the R1‐Pep inhibited the interaction of GABAB receptors with CaMKII under normal physiological conditions as well as the increased interaction after glutamate stress, as tested by the in situ PLA on cultured cortical neurons (Figure 1A). The control peptide (Ctrl‐Pep) neither affected the interaction of GABAB receptors with CaMKII under basal conditions nor in glutamate‐treated neurons.
FIGURE 1.
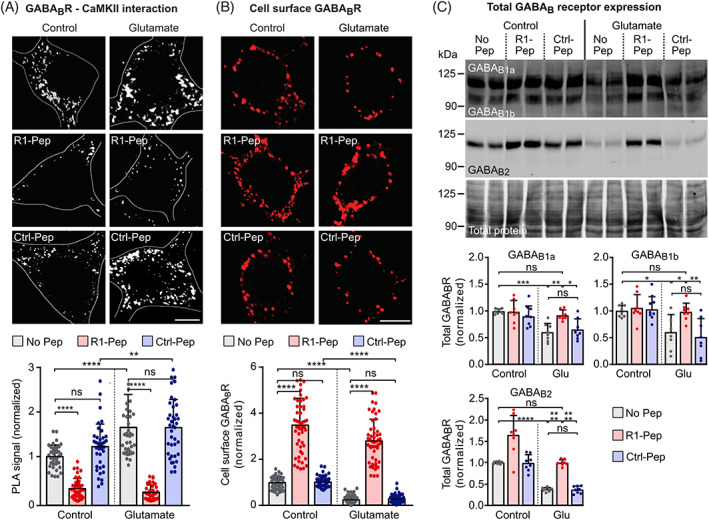
R1‐Pep blocked the interaction between CaMKII and GABAB1 and increased cell surface GABAB receptor expression. Cultures were stressed for 1 h with 50 μM glutamate and thereafter treated with R1‐Pep (10 μg/ml) or Ctrl‐Pep (10 μg/ml). The neurons were then incubated for 16 h with the peptides and analyzed. (A) R1‐Pep prevented the interaction of CAMKII with GABAB receptors as tested by in situ PLA (interaction is represented by white dots, scale bar: 5 μm). Bottom, quantification of in situ PLA signals. Signals were normalized to no Pep control. N = 39 neurons per condition from three independent experiments. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; **, p < 0.005;****, p < 0.0001). (B) R1‐Pep increased the cell surface expression of GABAB receptors (scale bar: 5 μm). Bottom, quantification of fluorescence intensities. Signals were normalized to no Pep control. N = 52 neurons per condition from three independent experiments. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; ****, p < 0.0001). (C) R1‐Pep prevented the decreased abundance of total GABAB receptors in glutamate stressed neurons as tested by Western blotting. N = 8 cultures per condition from four independent neuron preparations. Signals were normalized to no Pep control. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.0005; ****, p < 0.0001). PLA, proximity ligation assay.
3.2. R1‐Pep restores GABAB receptor expression and function after glutamate stress
Next, we tested whether preventing the interaction between CaMKII and GABAB receptors promotes cell surface expression of GABAB receptors. Application of R1‐Pep to cultured neurons after glutamate stress considerably increased cell surface expression levels of GABAB receptors as tested with an antibody directed against the N‐terminus of GABAB2 (Figure 1B). Both, under normal conditions and following glutamate stress, R1‐Pep treatment enhanced cell surface GABAB receptor expression levels that exceeded the normal cell surface expression in the absence of peptide. This indicates that R1‐Pep inhibits the degradation of GABAB receptors also under physiological conditions. Treatment of cultures with the control peptide (Ctrl‐Pep) neither affected basal cell surface expression of GABAB receptors nor their expression levels after glutamate stress (Figure 1B). Western blot experiments revealed the restoration of total expression of GABAB1a, GABAB1b, and GABAB2 subunits to normal control levels after glutamate stress (Figure 1C). However, an increased expression after R1‐Pep treatment was only observed under control conditions for the GABAB2 subunit.
R1‐Pep completely inhibited CaMKII‐mediated decreased abundance of GABAB receptors, as additional blocking of CaMKII activity with KN93 did not further increase cell surface expression of GABAB receptors (Figure S2, Supporting Information).
R1‐Pep dose‐dependently increased cell surface GABAB receptor expression levels in glutamate‐stressed neurons as determined with antibodies directed against GABAB2 (Figure 2A). Low dosage of R1‐Pep (0.5 and 1.0 μg/ml) reversed glutamate‐induced decreased abundance of cell surface receptors to control levels. With peptide dosage starting from 2.5 μg/ml and higher, we observed a significant increase in cell surface receptor expression. A concentration of 10 μg/ml R1‐Pep was used in the following experiments as this dosage provided a stable and consistent recovery in cell surface expression of GABAB receptors after glutamate stress.
FIGURE 2.
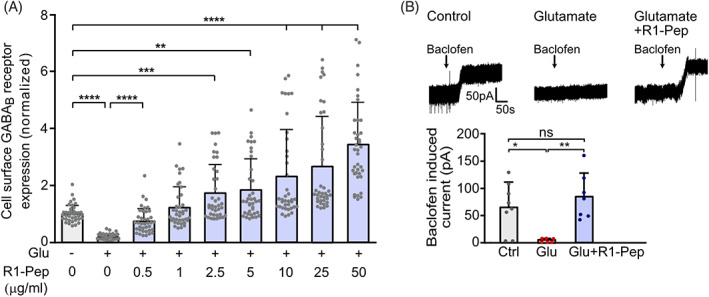
R1‐Pep dose‐dependently increased cell surface GABAB receptors after glutamate treatment and restored normal GABAB receptor function. Cultures were stressed for 1 h with 50 μM glutamate and thereafter treated with R1‐Pep. The neurons were then incubated for 16 h with the peptide and analyzed. (A) Neurons were incubated overnight with increasing concentrations of R1‐Pep and stained for surface expression of GABAB receptors using GABAB2 antibodies. Signals were normalized to no Pep/no Glu control. N = 38 neurons per condition from three independent experiments. One‐way ANOVA, Dunnett's T3 multiple comparison test (**, p < 0.01; ***, p < 0.001; ****, p < 0.0001). (B) R1‐Pep (10 μg/ml) restored normal GABAB receptor function. Baclofen‐induced currents were measured in glutamate‐stressed (Glu) and unstressed (Ctrl) neurons using whole‐cell patch‐clamp recordings. N = 8 (Ctrl), 6 (Glu), and 7 (Glu + R1‐Pep) neurons. One‐way ANOVA, Tukey's multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01).
Next, we tested the ability of R1‐Pep to restore GABAB receptor function following glutamate stress using whole‐cell patch‐clamp recordings from primary cultured neurons (Figure 2B). In line with the decreased abundance of the receptors, glutamate stress strongly reduced the currents induced by the GABAB receptor agonist baclofen when compared to control neurons. However, application of R1‐Pep prevented the reduction in baclofen‐induced currents (Figure 2B).
Overall, these findings indicate that inhibiting the CaMKII/GABAB receptor interaction with R1‐Pep restores GABAB receptor expression and function after glutamate stress.
3.3. R1‐Pep prevents CaMKII‐mediated phosphorylation and K63‐linked ubiquitination of GABAB receptors
Glutamate stress‐induced decreased abundance of GABAB receptors is triggered by CaMKII‐mediated phosphorylation of S867 of GABAB1 [17]. We therefore tested whether R1‐Pep prevents GABAB1(S867) phosphorylation on GABAB receptors expressed in HEK 293 cells co‐transfected with or without CaMKIIβ by in situ PLA (Figure 3). Co‐expression of CaMKIIβ significantly increased phosphorylation of GABAB receptors, which was reduced to control levels upon treatment with R1‐Pep. The increased PLA signals observed upon co‐expression with CaMKIIβ were due to phosphorylation of GABAB1 at S867 as expression of mutant receptor with inactivated CaMKII phosphorylation site (GABAB1(S867A)) did not show elevated phosphorylation upon co‐expression with CaMKIIβ (Figure 3).
FIGURE 3.
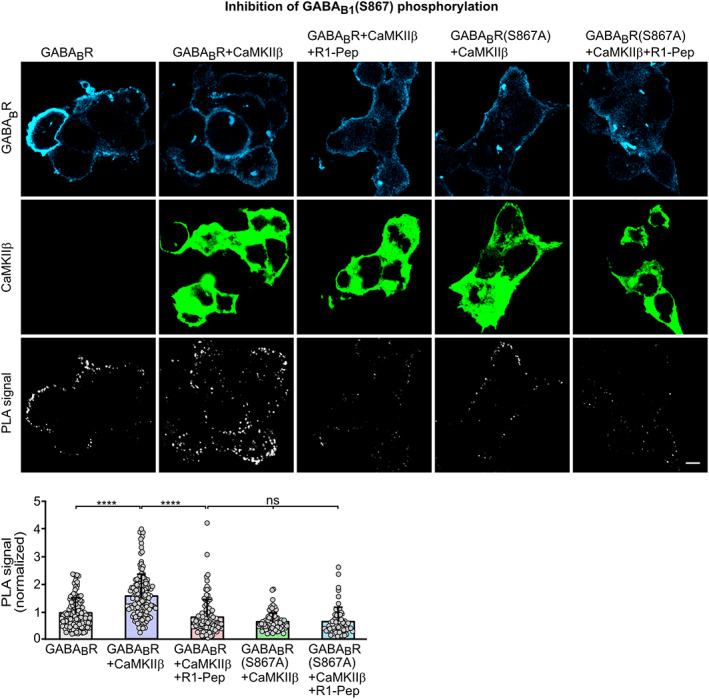
R1‐Pep prevented CaMKII‐mediated phosphorylation of S867 in GABAB1. HEK 293 cells were transfected with the indicated plasmids and tested for GABAB receptor phosphorylation by in situ PLA. Top: Representative images depicting staining for GABAB receptors, CaMKII and PLA signals (phosphorylation is represented by white dots; scale bar: 10 μm). Bottom, quantification of PLA signals. Signals were normalized to GABABR control. N = 62–157 cells from four independent preparations. One‐way ANOVA, Dunnett's T3 multiple comparison test (ns, p > 0.05; ****, p < 0.0001). PLA, proximity ligation assay.
CaMKII‐induced lysosomal degradation of GABAB1 requires K63‐linked ubiquitination of GABAB1 at multiple sites [24, 25]. Consistent with this observation, preventing the interaction of CaMKII with GABAB receptors using R1‐Pep inhibited K63‐linked ubiquitination of the receptors in glutamate treated as well as in untreated neurons as tested by in situ PLA (Figure 4).
FIGURE 4.
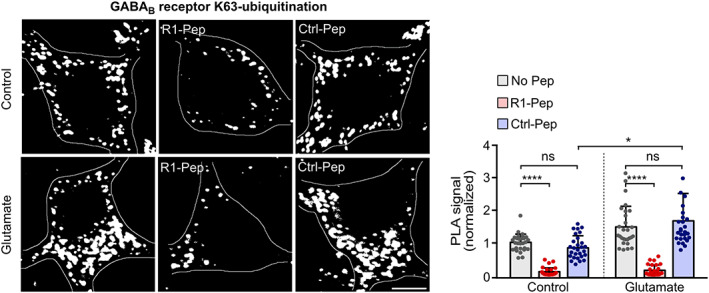
R1‐Pep treatment blocked K63‐linked ubiquitination of GABAB receptors. Cultures were stressed for 1 h with 50 μM glutamate and thereafter treated with R1‐Pep (10 μg/ml). The neurons were then incubated for 16 h with the peptide and tested for K63‐linked ubiquitination of GABAB receptors by in situ PLA (interaction is represented by white dots; scale bar: 5 μm). Bottom, quantification of in situ PLA signals. Signals were normalized to no pep control. N = 28 neurons per condition from two independent experiments. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; ****, p < 0.0001). PLA, proximity ligation assay.
These results indicate that R1‐Pep prevents CaMKII‐mediated phosphorylation of GABAB1 and thereby K63‐linked ubiquitination of the receptor, which is a prerequisite for their lysosomal degradation.
3.4. R1‐Pep treatment does not directly affect CaMKII function and expression
R1‐Pep interacted with CaMKII as shown by in situ PLA on cultured neurons (Figure 5A) and by co‐immunoprecipitation with CaMKII transiently expressed in HEK293 cells (Figure 5B). In addition, R1‐Pep did not directly affect the function of CaMKII as tested in an in vitro phosphorylation assay using recombinant CaMKIIβ protein (Figure 5C).
FIGURE 5.
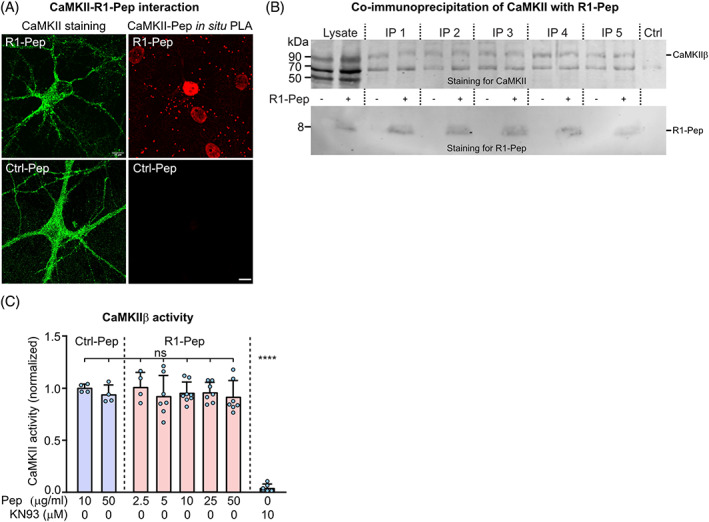
R1‐Pep interacted with CaMKII after glutamate stress but did not directly affect CaMKII activity. (A, B) CaMKII interacted with GABAB receptors. Cultured neurons were incubated overnight with R1‐Pep (10 μg/ml) or Ctrl‐Pep (10 μg/ml) and analyzed for interaction by in situ PLA (scale bar: 10 μm) (A) or co‐immunoprecipitation (B). For co‐immunoprecipitation, HEK 293 cells expressing EGFP‐tagged CaMKIIβ were incubated with or without biotin‐labeled R1‐Pep, lysed, and immunoprecipitated using GFP antibodies coupled to magnetic beads. Top, staining for CaMKII using CaMKII antibodies. Bottom, staining for R1‐Pep using biotin antibodies. The blot shows five independent immunoprecipitations. Ctrl, no lysate, and GFP‐magnetic beads were added to buffer. (C) P1‐Pep did not affect CaMKIIβ activity. Recombinant CaMKIIβ was incubated with R1‐Pep, Ctrl‐Pep, or the CaMKII inhibitor KN93 (10 μM) and tested for activity after 30 min. N = 4–8 from two (Ctrl‐Pep, 2.5 μg/ml R1‐Pep) and four experiments (R1‐Pep, 5–50 μg/ml). One‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05).
CaMKII regulates numerous intracellular processes and is critical for normal physiological functioning of neurons [35, 36]. Therefore, we tested whether R1‐Pep affects the expression level of CaMKII under normal physiological conditions, which in principle could impair numerous intracellular pathways. R1‐Pep did not affect the expression level of CaMKII, as tested with a pan antibody for CaMKII (Figure 6A) or an antibody recognizing specifically CaMKIIβ (Figure 6B). However, we found that R1‐Pep inhibited glutamate stress‐induced T286 autophosphorylation of CaMKII (Figure 6C), which renders CaMKII autonomously and persistently active [37, 38]. This required GABAB receptor activity since co‐application of the GABAB receptor antagonist CGP 56999 prevented the R1‐Pep effect. The inhibition of the autonomous activity of CaMKII could represent another beneficial effect of R1‐Pep, as upregulated CaMKII activity after cerebral ischemia has been reported to foster neuronal death [39, 40].
FIGURE 6.
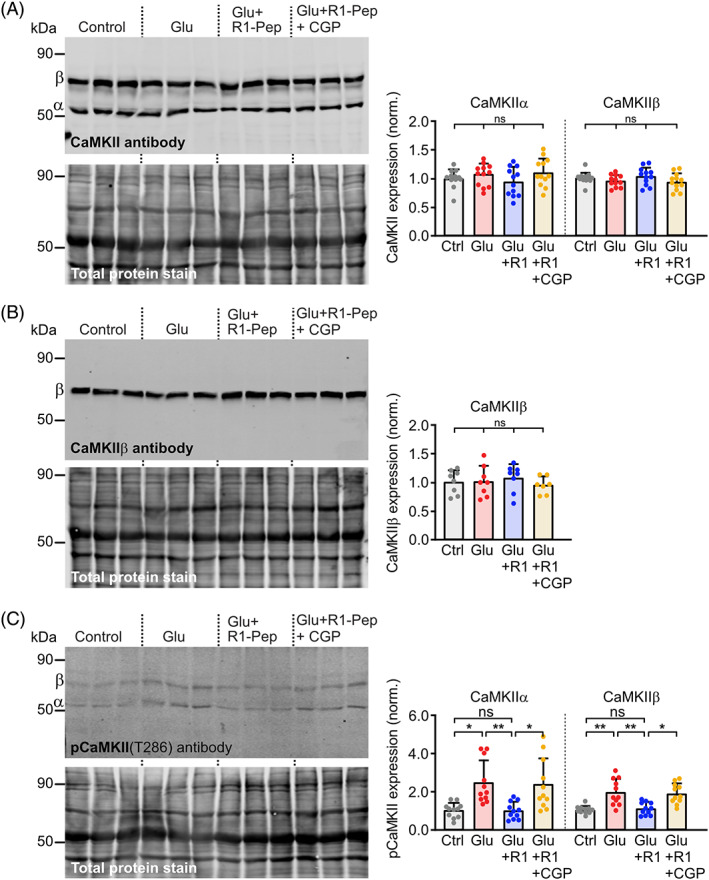
R1‐Pep did not affect CaMKII expression but stress‐induced CaMKII(T286) autophosphorylation. (A, B) Glutamate‐stressed neurons were treated for 16 h with R1‐Pep (10 μg/ml) or R1‐Pep plus the GABAB receptor antagonist CGP 56999 (10 μM) and then analyzed by Western blotting using antibodies directed against α, β, γ, and δ subtypes of CaMKII (A) or specifically to CaMKIIβ (B). Signals were normalized to control. N = 12 (A) or 8 (B) cultures per condition from four (A) and three (B) independent preparations, respectively. One‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05). (C) R1‐Pep inhibited glutamate stress‐induced T286 autophosphorylation of CaMKII. Glutamate‐stressed neurons were treated for 16 h with R1‐Pep (10 μg/ml) or R1‐Pep plus the GABAB receptor antagonist CGP 56999 and then analyzed by Western blotting using antibodies directed against pCaMKII(T286). Signals were normalized to control. N = 11 cultures per condition from four independent preparations. Two‐way ANOVA with Dunnett's T3 multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01).
The finding that R1‐Pep mediated inhibition of CaMKII(T286) autophosphorylation requires GABAB receptor activity implies the presence of endogenous receptor activity in the cultures. Indeed, electrophysiological experiments confirmed that our neuron/glia cultures display spontaneous GABAB receptor activity (Figure S3, Supporting Information).
3.5. R1‐Pep counteracts aberrant regulation of GABAB receptor downstream effectors
G protein‐coupled inwardly rectifying K+ channels (GIRK, Kir) [16, 41, 42] and voltage‐gated Ca2+ channels (VGCCs) [16, 43, 44, 45, 46] are the main downstream effectors conveying GABAB receptor‐mediated neuronal inhibition. Because GABAB receptors form signaling complexes with their downstream effectors [47], it is very likely that they are co‐regulated with GABAB receptors under excitotoxic conditions. Indeed, Kir3.2 channel expression was reduced in glutamate‐stressed neurons and R1‐Pep considerably enhanced Kir3.2 expression after glutamate stress (Figure 7A,B), in agreement with co‐regulation of Kir3.2 with GABAB receptors. Regulation of Kir3.2 channels by R1‐Pep required GABAB receptor activity since co‐application of the GABAB receptor antagonist CGP 56999 prevented R1‐Pep mediated normalization of Kir3.2 expression in glutamate stressed neurons (Figure 7B). Treatment of unstressed neurons with R1‐Pep did not alter Kir3.2 expression (Figure 7A). This finding implies that signal transduction via Kir3.2 channels induced by G protein‐coupled receptors other than GABAB receptors is not compromised in healthy neurons in the presence of R1‐Pep.
FIGURE 7.
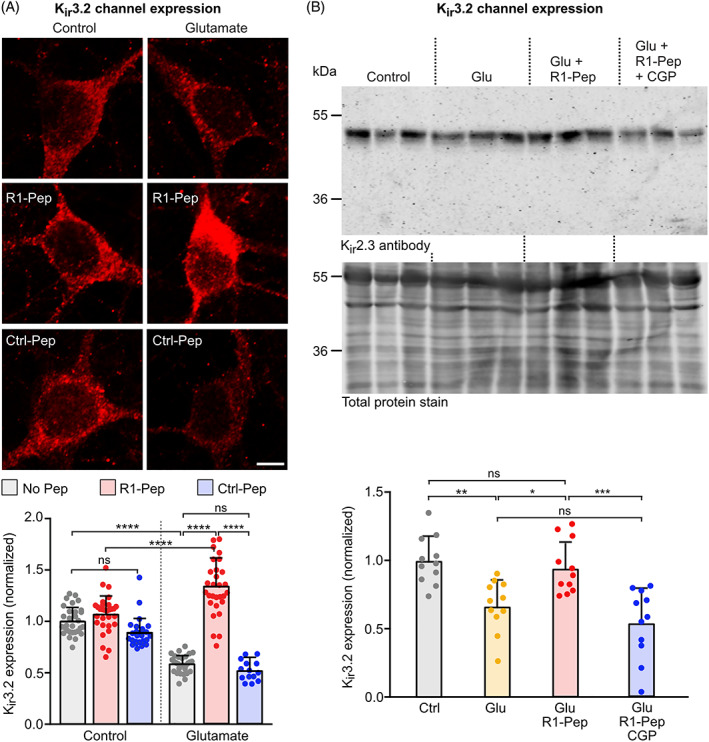
R1‐Pep treatment reduced aberrant expression of Kir3.2 channels after glutamate stress. Glutamate‐stressed neurons were treated for 16 h with R1‐Pep (10 μg/ml) or Ctrl‐Pep (10 μg/ml) and then stained with Kir3.2 antibodies. (A) Immunofluorescence staining: top, representative images (scale bar: 5 μm); bottom, quantification of fluorescence intensities. N = 30 neurons per condition from two independent experiments. Signals were normalized to no Pep control. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; ****, p < 0.0001). (B) Western blotting: top panel, staining for Kir3.2 channels; bottom panel, staining for total protein used for normalization. Control, untreated cultures; Glu, glutamate‐stressed cultures; Glu + R1‐Pep, glutamate‐stressed cultures treated with R1‐Pep; Glu + R1‐Pep + CGP, glutamate‐stressed cultures treated with R1‐Pep and the GABAB receptor antagonist CGP 56999. Signals were normalized to control. N = 11 cultures per condition from four independent neuron preparations. One‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01 ***, p < 0.0005).
In contrast to the decreased abundance of Kir 3.2 channels, we observed increased expression of N‐type VGCC in glutamate‐stressed neurons (Figure 8A,B). However, R1‐Pep treatment attenuated the increased expression of N‐type. As observed for Kir3.2 channels, regulation by R1‐Pep after glutamate stress required GABAB receptor activity as co‐application of CGP 56999 blocked the R1‐Pep effect (Figure 8B). The attenuation of the enhanced expression of N‐type VGCC expression upon R1‐Pep treatment presumably reduces Ca2+ overload in glutamate stressed neurons and reduces transmitter release.
FIGURE 8.
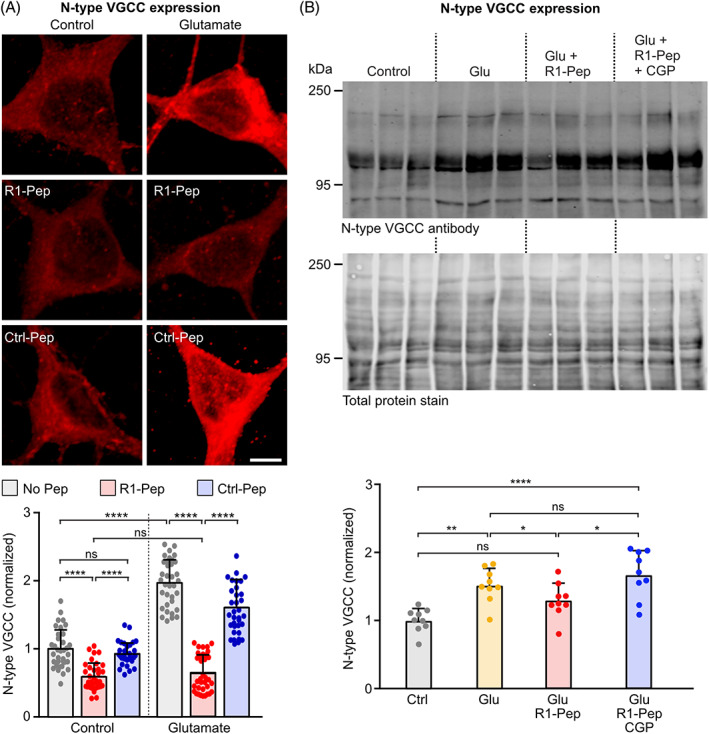
R1‐Pep treatment reduced aberrant expression of N‐type VGCCs. Glutamate‐stressed neurons were treated for 16 h with R1‐Pep (10 μg/ml) or Ctrl‐Pep (10 μg/ml) and then stained with antibodies against N‐type VGCCs. (A) Immunofluorescence staining: top, representative images (scale bar: 5 μm); bottom, quantification of fluorescence intensities. Signals were normalized to no Pep control. N = 35 neurons per condition from two independent experiments. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; ****, p < 0.0001). (B) Western blotting: top panel, staining for N‐type VGCC channels; bottom panel, staining for total protein used for normalization. Control, untreated cultures; Glu, glutamate‐stressed cultures; Glu + R1‐Pep, glutamate‐stressed cultures treated with R1‐Pep; Glu + R1‐Pep + CGP, glutamate‐stressed cultures treated with R1‐Pep and the GABAB receptor antagonist CGP 56999. Signals were normalized to control. N = 9 cultures per condition from three independent neuron preparation. One‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01 ****, p < 0.0001). VGCC, voltage‐gated Ca2+ channel.
3.6. R1‐Pep‐mediated restoration of GABAB receptors inhibits glutamate‐induced neuronal death
We then tested if application of R1‐Pep indeed limits excitotoxic neuronal death. Cortical neuron cultures stressed for 1 h with glutamate were treated with R1‐Pep at different time intervals thereafter (0, 3, 6, 12, and 24 h) and surviving neurons were counted after 48 h. As expected, R1‐Pep treatment following glutamate stress inhibited progressive neuronal death at all time points tested, even when added 24 h after the excitotoxic insult (Figure 9A). Treatment of cultures with the control peptide (Ctrl‐Pep) failed to show any effect. This finding verified our hypothesis that preventing the decreased abundance of GABAB receptors under excitotoxic conditions limits progressive neuronal death and revealed a very promising wide time window for the neuroprotective activity of R1‐Pep. These results are supported by the findings that persistent activation of GABAB receptors by baclofen mediates neuroprotection via activation of the phosphatidylinositol‐4,5‐bisphosphate 3 kinase (PI3K)/Akt (protein kinase B) pathway, which inhibits apoptosis [48, 49, 50]. Indeed, we found that treatment of glutamate‐stressed neurons with R1‐Pep activated the Akt survival cascade (Figure S4, Supporting Information).
FIGURE 9.
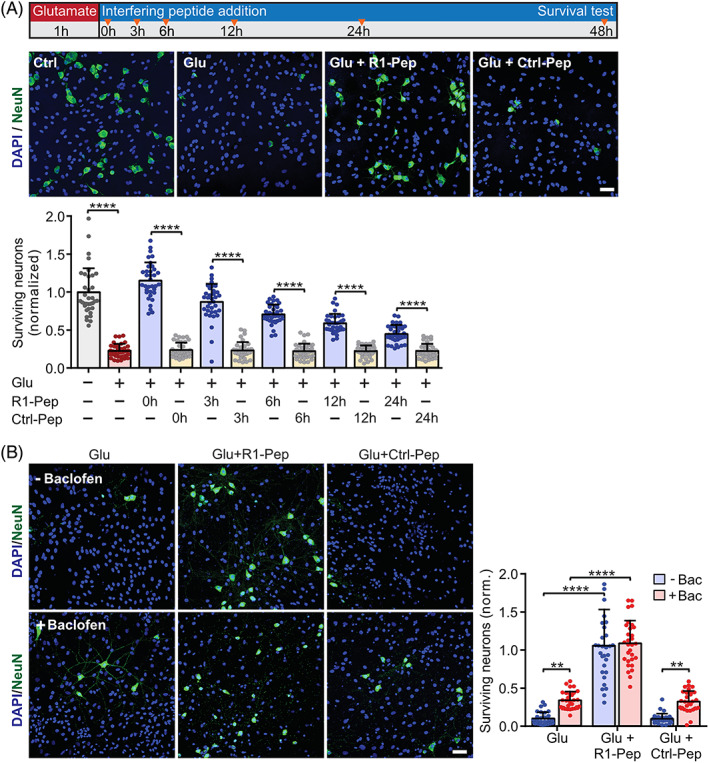
R1‐Pep provided neuroprotection within a wide time window and surpassed the neuroprotective activity of baclofen. (A) R1‐Pep treatment inhibited progressive glutamate‐induced neuronal death. Neurons were stressed with glutamate and treated with no peptide, R1‐Pep (10 μg/ml) or Ctrl‐Pep (10 μg/ml) for 0, 3, 6, 12, and 24 h thereafter. After 48 h, the cultures were stained with DAPI for total cells (glia plus neurons) and with an antibody directed against NeuN for neurons. Top, schematic representation of the experimental design; below, the representative images (scale bar: 25 μm) depict the condition where the peptides were immediately administered after the glutamate stress (time 0 h in the scheme). Signals were normalized to untreated control neurons. N = 35 frames (fields of view) per experimental condition from three independent experiments. Two‐way ANOVA with Tukey's multiple comparison (****, p < 0.0001). (B) The GABAB receptor agonist baclofen exhibited considerably less neuroprotection than R1‐Pep. Neurons were stressed with glutamate and immediately treated with no peptide, R1‐Pep (10 μg/ml) or Ctrl‐Pep (10 μg/ml) thereafter. After 48 h, the cultures were stained for surviving neurons as in (A; scale bar: 25 μm). Signals were normalized to untreated control neurons. N = 30 frames (fields of view) per experimental condition from three independent experiments. Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; **, p < 0.01; ****, p < 0.0001).
The GABAB receptor agonist baclofen provides some neuroprotective activity under ischemic conditions [49, 51, 52, 53, 54, 55, 56, 57, 58, 59]. However, this protection may be limited due to the decreased abundance of GABAB receptor expression following the excitotoxic insult. Therefore, we compared the neuroprotection between R1‐Pep and baclofen treatments. R1‐Pep fully protected the neurons if applied immediately after glutamate stress whereas baclofen exhibited only a partial neuroprotective effect (Figure 9B).
3.7. Application of R1‐Pep restores GABAB receptor function and is neuroprotective in a mouse model of focal cerebral ischemia
To test whether R1‐Pep is also effective in a mouse model of focal cerebral ischemia, we subjected the mice to MCAO for 1 h and subsequently tested the effect of R1‐Pep ex‐vivo on brain slices containing the somatosensory cortex. We first confirmed that R1‐Pep was taken up by neurons in brain slices (Figure S5, Supporting Information). In sections of the cerebral cortex of MCAO‐treated mice, we observed reduced expression of GABAB receptors, which was restored to normal levels after incubation of the slices with R1‐Pep for 6 h (Figure 10A,A′). This time interval was chosen for the analysis since we previously observed a significantly decreased abundance of GABAB receptors and a small, but consistent, loss of neurons in the somatosensory cortex at 6 h after the ischemic insult [54].
FIGURE 10.
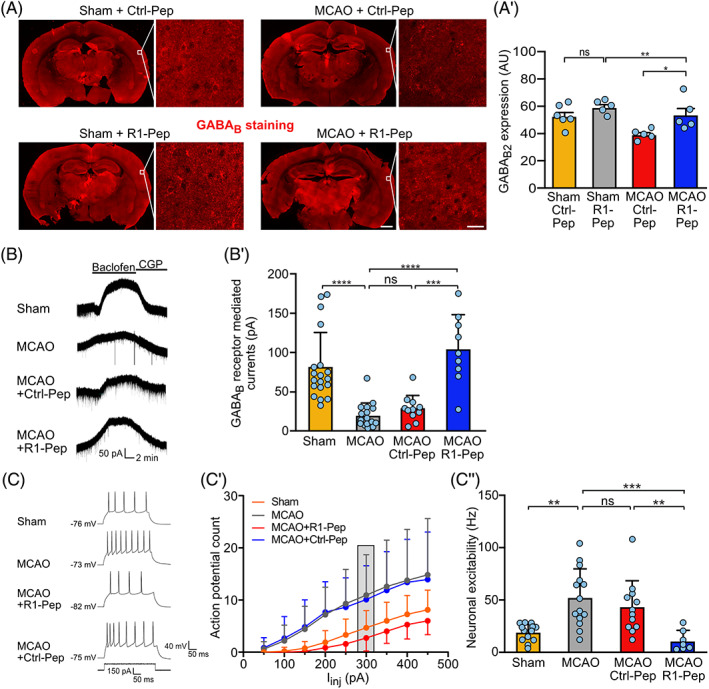
R1‐Pep normalized receptor expression and GABAB receptor‐mediated inhibition after MCAO‐induced cerebral ischemia. (A) Administration of R1‐Pep restored GABAB receptor expression after MCAO. Brain slices containing the somatosensory cortex prepared from sham and MCAO treated mice were incubated for 6 h with R1‐Pep (10 μg/ml) and then stained for GABAB receptor expression using GABAB2 antibodies. Representative images show overviews (scale bar: 1 mm) and higher magnifications (scale bars: 20 μm). (A′) Quantification of signal intensities shown in (A). The data represent the average of multiple frames (fields of view) per mouse (n = 5–6 mice per condition). Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05, *, p < 0.05; **, p < 0.005). (B, C) R1‐Pep restored GABAB receptor‐mediated currents and reduced neuronal excitability. Brain slices prepared from the somatosensory cortex of sham‐operated and MCAO mice were analyzed using whole‐cell patch‐clamp recordings 3–5 h after treatment with peptide or saline. (B) Representative baclofen‐evoked current traces. (B′) Quantification of baclofen‐evoked current traces. N = 18 (Sham), 16 (MCAO), 9 (MCAO+R1‐Pep), and 11 (MCAO+Ctrl‐Pep) neurons. One‐way ANOVA, Tukey's multiple comparison test (ns, p > 0.05; ***, p ≤ 0.001; ****, p ≤ 0.0001). (C‐C″) Neuronal excitability was determined by injecting increasing current steps. (C) Representative current traces. (C′) Quantification of action potential firing evoked by increasing current injections. (C″) Bar graph and statistical evaluation of data depicted in the gray box in (C′). N = 13 (Sham), 14 (MCAO), 7 (MCAO+R1‐Pep), and 12 (MCAO+Ctrl‐Pep) neurons. One‐way ANOVA, Dunnett's T3 multiple comparison test (ns, p > 0.05; **, p = 0.01 ***, p = 0.001). MCAO, middle cerebral artery occlusion.
Next, we examined how GABAB receptor‐mediated currents were altered after cerebral ischemia. Using whole‐cell patch‐clamp recordings, we found that 1 h MCAO strongly reduced GABAB receptor‐mediated currents (Figure 10B,B′), in line with the decreased abundance of these receptors (Figure 10A) and our previous data [54]. Administration of R1‐Pep restored GABAB receptor‐mediated currents to levels comparable to sham‐operated mice, while treatment with Ctrl‐Pep had no effect (Figure 10B,B′).
We then examined whether the reduced GABAB receptor‐mediated inhibition is accompanied with increased neuronal excitability. We detected enhanced neuronal firing in MCAO‐treated mice when compared to sham‐operated controls (Figure 10C,C′,C″). This was accompanied with an increased input resistance (Figure S6A and Table S1, Supporting Information) and depolarized resting membrane potential (Figure S6B and Table S1, Supporting Information). Incubation of the slices with R1‐Pep hyperpolarized the resting membrane potential and lowered the input resistance, resulting in a reduced neuronal firing rate (Figure S6A,B and Table S1, Supporting Information). No change in the action potential threshold was observed in all conditions tested (Figure S6C and Table S1, Supporting Information). Thus, blocking the interaction of CaMKII with GABAB receptors after cerebral ischemia restored normal GABAB receptor‐mediated inhibition and reduced neuronal exitability.
Finally, we tested for a potential neuroprotective activity of R1‐Pep. In somatosensory cortices derived from MCAO‐treated mice, the number of neurons was significantly reduced (89 ± 2.2%, p < 0.05) compared to sham‐operated mice (100 ± 2.3%) 6 h after MCAO (Figure 11A,A′). In line with our in vitro data, administration of R1‐Pep about 1 h after MCAO for 6 h completely prevented the loss of neurons (101 ± 1.7%, p < 0.05). We confirmed this finding using Fluoro‐Jade C, which specifically stains degenerating neurons. R1‐Pep significantly reduced the number of degenerating neurons in the somatosensory cortex of MCAO‐treated mice (Figure 11B,B′).
FIGURE 11.
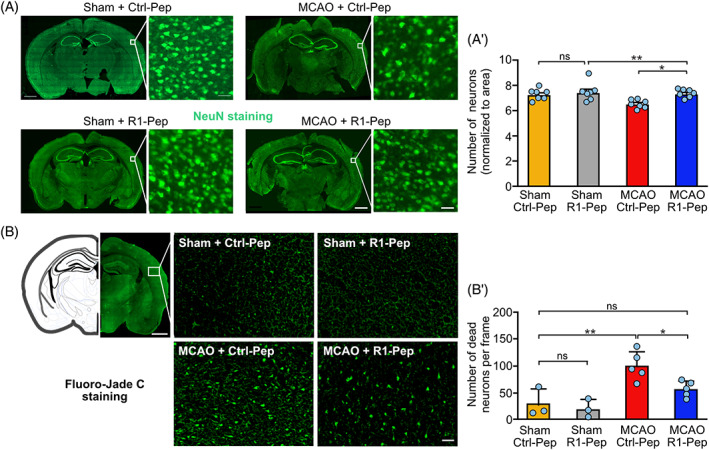
R1‐Pep reduced neuronal death after MCAO. Brain slices prepared from the somatosensory cortex of sham and MCAO treated mice were incubated for 6 h with R1‐Pep and then stained for the presence of neurons using NeuN antibodies (A) or for degenerating neurons using Fluoro‐Jade C staining (B) (scale bars: Overviews, 1 mm; higher magnifications, 50 μm). (A′, B′) Quantification of neurons. The data represent the average of multiple frames (fields of view) per mouse (A′: N = 7 mice per condition, B′: N = 3 for sham and n = 5 for MCAO). Two‐way ANOVA with Tukey's multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01). MCAO, middle cerebral artery occlusion.
The data indicate that restoration of normal GABAB receptor expression and function after cerebral ischemia reduced neuronal excitability and increased neuronal survival.
4. DISCUSSION
In ischemic stroke, a massive increase in intracellular Ca2+ by sustained activation of glutamate receptors is the main cause for progressive excitotoxicity‐induced neuronal death in the ischemic penumbra (an area surrounding the ischemic core that is metabolically and functionally compromised, but still viable and is considered amenable to neuroprotective treatments [11, 12, 13, 14, 15]). GABAB receptors normally control the activity of glutamate receptors and the excitability of neurons [16, 60, 61], but cannot provide adequate inhibition to counteract ischemia‐induced neuronal hyperexcitability because of its decreased abundance induced by CaMKII activation during ischemic insult [17, 18, 19, 20, 21, 22, 23]. In this study, we developed an interfering peptide that inhibited the interaction of GABAB receptors with CaMKII. Targeting the activity of CaMKII directly using specific inhibitors is not a preferred option as this kinase is involved in numerous important brain functions and can promote survival as well as apoptotic pathways [40, 62]. In contrast, specifically preventing the interaction of GABAB receptors with CaMKII under excitotoxic conditions would be restricted to pathways controlled by GABAB receptors. Our findings on cultured neurons indicate that administration of R1‐Pep after glutamate stress indeed restored expression of GABAB receptors and inhibited progressive neuronal death. Mechanistically, cell surface expression of GABAB receptors depends on a precise regulation and balance of recycling, degradation, and delivery of newly synthetized receptors [63]. GABAB receptors are constitutively internalized at a high rate and are then recycled to the cell surface or are degraded in lysosomes [64]. Phosphorylation of GABAB1 by CaMKII recruits the E3 ubiquitin ligase MIB2 resulting in the K63‐linked ubiquitination of the receptor at multiple sites, which tags the receptors for lysosomal degradation [24, 25]. Blocking recycling results in fast degradation of the receptors, whereas inhibition of lysosomal degradation favors recycling and increases cell surface expression [65]. R1‐Pep prevents phosphorylation of GABAB receptors by CaMKII and thereby degradation of the receptors. This reconstitutes recycling of the receptors and, together with delivery of newly synthetized receptors, normalizes cell surface GABAB receptor expression levels.
Normal expression levels and activity of CaMKII are fundamental to neuronal physiology. CaMKII regulates a variety of intracellular processes important for neuronal homeostasis and plays a key role in controlling synaptic plasticity [35, 66]. Therefore, it was important that administration of R1‐Pep did not affect CaMKII expression under physiological conditions to avoid undesired side effects. An effect on CaMKII expression was not an unlikely scenario, as binding of R1‐Pep to CaMKII might trigger proteasomal degradation of the complex. Fortunately, R1‐Pep did not affect CaMKII expression levels and hence R1‐Pep should not affect CaMKII‐mediated pathways in healthy neurons. However, glutamate stress increased autophosphorylation of CaMKII, which renders it persistently active. Administration of R1‐Pep inhibited CaMKII(T268) autophosphorylation in a GABAB receptor activity‐dependent manner, which most likely reduces the detrimental effects of elevated CaMKII activity under ischemic conditions and thereby contributes to the neuroprotective activity of R1‐Pep.
R1‐Pep also displayed a normalizing effect on the expression levels of the main GABAB receptor effectors conveying neuronal inhibition in glutamate‐stressed neurons. While Kir3.2 channels were concomitantly downregulated with GABAB receptors, N‐type VGCC was upregulated. This aberrant regulation of GABAB receptor effectors fosters the detrimental effects of glutamate receptor overstimulation because it further compromises GABAB receptor‐mediated inhibition and increases Ca2+ overload in neurons, respectively. Restoration of GABAB receptor expression with R1‐Pep also largely normalized the aberrant expression levels of Kir3.2 channels and N‐type and N‐type VGCC, which all are under the direct control of GABAB receptors. The mechanism(s) involved in this regulation requires GABAB receptor activity but are otherwise unclear. Because of the co‐regulation of Kir 3.2 channels and GABAB receptors, it is likely that they are regulated, at least partially, as a signaling complex via the CaMKII pathway delineated for GABAB receptors [17, 21, 25]. The expression of N‐type VGCCs is potentially regulated via the cAMP‐PKA and/or ERK 1/2‐CREB pathways, which are controlled by GABAB receptors [67].
In addition to controlling neuronal excitability, GABAB receptor activity promotes neuronal survival via the PI3K/Akt‐GSK3β pathway [50], which is a major neuronal survival cascade [68]. Accordingly, restoration of GABAB receptor expression after glutamate stress increased the activation of Akt and blocked the pro‐apoptotic activity of GSK3β. It is very likely that the combined effects of restored GABAB receptor activity via different pathways (reduction of neuronal activity, normalization of CaMKII activity, normalization of VGCC and Kir 3.2 channel expression, and activation of Akt) contributed to the efficient inhibition of progressing excitotoxic neuronal death. We also found that R1‐Pep inhibited ongoing neuronal death even after 24 h following glutamate stress, which demonstrates that R1‐Pep has a wide time window for neuroprotection.
For verifying the activity of R1‐Pep in vivo, we selected the MCAO mouse model of cerebral ischemia because this model mimics the pathophysiological changes found in most human strokes [69]. In line with the in vitro experiments, application of R1‐Pep about 1 h after termination of MCAO for 3–6 h on brain slices ex vivo restored normal GABAB receptor expression and GABAB receptor‐mediated inhibition, reduced neuronal excitability, and prevented neuronal death in the somatosensory cortex, which belongs to the ischemic penumbra that is accessible for neuroprotection [69].
A limitation of our study is that R1‐Pep exhibited no solid reproducible effect upon systemically application of R1‐Pep via i.v. injection, most likely due to rapid enzymatic degradation of the peptide in the circulation (data not shown). This made it impossible to analyze its neuroprotective activity in the MCAO mouse model in vivo. As acute brain slices are viable and usable for experiments for about 6 h, this restricted the time span for our ex vivo analysis. One general technical issue associated with peptide therapeutics is their susceptibility to proteolytic degradation and potential fast renal excretion after systemic injection [70]. In addition, for targeting the brain, another problem represents the blood–brain barrier, which may limit peptide delivery to an insufficient amount into neurons. Therefore, future experimentation needs to focus on the optimization of R1‐Pep for systemic administration. An effective way to increase the proteolytic stability of peptides is the partial substitution of naturally occurring l‐amino acids with d‐amino acids in positions not essential for peptide activity [71, 72, 73]. Alanine scanning for identification of the essential amino acid residues required for activity in the R1‐Pep and the replacement of non‐essential amino acid by their D isomers should be performed to optimize its stability. In addition, intranasal application of R1‐Pep might be a promising strategy as this route circumvents the circulation and its associated problems and has been shown to efficiently deliver high concentrations of peptides or proteins into the brain [74, 75]. Another promising strategy is the delivery of R1‐Pep via nanoparticles. Nanoparticles have been shown to protect cargo from degradation, slow down renal excretion and cross the blood–brain barrier [76, 77, 78]. Two recently published nanoparticle‐based approaches successfully delivered neuroprotective drugs/peptides into the brain after cerebral ischemia in mice and rats. The first approach is based on wheat germ agglutinin modified polyethylene glycol‐PLGA nanoparticles. These nanoparticles very efficiently delivered an interfering peptide targeting an NMDA receptor interaction into the brain via intranasal application [75]. The second approach is based on betulinic acid nanoparticles [79]. Betulinic acid itself exhibits antioxidant activity and has thus the added benefit of intrinsic neuroprotective activity.
R1‐Pep optimized for stability, formulation, and application route for systemic application will permit testing for the wide time window for neuroprotection we observed in our in vitro experiments and for investigating a potentially favorable side effect profile.
The only approved pharmacological treatment of stroke is the thrombolytic drug tissue plasminogen activator (tPA). Unfortunately, the time window for restoring blood flow to the affected tissue is rather narrow (3–4.5 h after stroke) and only suitable for a limited number of patients [2, 3]. Potential neuroprotective agents have been tested in clinical trials but with limited success [4, 5, 6]. An obvious target for a neuroprotective strategy is the NMDA receptor, which triggers apoptosis under ischemic conditions via activation of neuronal nitric oxide (NO) synthase [80]. However, because NMDA receptors are involved in many fundamental brain functions, including triggering survival as well as death pathways, global inhibition of NMDA receptors induces severe side effects [12, 81, 82]. A specific strategy to inhibit NMDA receptor‐mediated NO production by using an interfering peptide to block the interaction of these receptors with PSD95 provided neuroprotection in animal models of stroke [80, 83] and showed some neuroprotective activity in clinical trials [84, 85]. However, the limitation of this approach is that it very specifically disrupts one downstream event contributing to apoptotic cell death (NMDA receptor‐mediated NO production) but did not affect other stroke relevant factors contributing to neuronal death (e.g., Ca2+ overload and downstream effects). In view of the complex detrimental mechanisms contributing to neuronal death in cerebral ischemia, it is rather unlikely that targeting a single downstream pathway is sufficient to efficiently limit progressive neuronal death. Hence, the restoration of GABAB receptor expression following cerebral ischemia is a promising approach, as it interferes with multiple critical elements of the neurotoxic cascade, including deleterious neuronal hyperexcitability, exaggerated glutamate release, autonomous activity of CaMKII, and increased expression of N‐type VGCCs, while supporting the activation of neuronal survival pathway.
AUTHOR CONTRIBUTIONS
Karthik Balakrishnan, Mohammad Hleihil, Musadiq A. Bhat, Robert P. Ganley, Markus Vaas, and Dietmar Benke performed experiments, collected, and analyzed data. Jan Klohs and Hanns Ulrich Zeilhofer provided methodological support and interpreted data. Dietmar Benke, Karthik Balakrishnan, and Mohammad Hleihil designed the study and wrote the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
The authors are grateful to Giovanna Bosshard for dissecting the E18 rat cortex and Thomas Grampp for excellent technical assistance. This study was supported by the Swiss National Science Foundation (grants 31003A_156648 and 31003A_182325 to D.B.) and the Foundation for Research in Science and the Humanities at the University of Zurich (grant STWF‐18‐005 to D.B.).
Balakrishnan K, Hleihil M, Bhat MA, Ganley RP, Vaas M, Klohs J, et al. Targeting the interaction of GABAB receptors with CaMKII with an interfering peptide restores receptor expression after cerebral ischemia and inhibits progressive neuronal death in mouse brain cells and slices. Brain Pathology. 2023;33(1):e13099. 10.1111/bpa.13099
Karthik Balakrishnan and Mohammad Hleihil are co‐first authors and contributed equally to this study.
Funding information Foundation for Research in Science and the Humanities at the University of Zurich, Grant/Award Number: STWF‐18‐005; Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung, Grant/Award Numbers: 31003A_156648, 31003A_182325
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available from the corresponding author on request.
REFERENCES
- 1. Feigin VL, Norrving B, Mensah GA. Global burden of stroke. Circ Res. 2017;120(3):439–48. [DOI] [PubMed] [Google Scholar]
- 2. Siket MS, Cadena R. Novel treatments for transient ischemic attack and acute ischemic stroke. Emerg Med Clin N Am. 2021;39(1):227–42. [DOI] [PubMed] [Google Scholar]
- 3. Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: an updated systematic review and meta‐analysis. Lancet. 2012;379(9834):2364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55(3):363–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matei N, Camara J, Zhang JH. The next step in the treatment of stroke. Front Neurol. 2020;11:582605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59(3):467–77. [DOI] [PubMed] [Google Scholar]
- 7. Bruzzoni‐Giovanelli H, Alezra V, Wolff N, Dong CZ, Tuffery P, Rebollo A. Interfering peptides targeting protein‐protein interactions: the next generation of drugs? Drug Discov Today. 2018;23(2):272–85. [DOI] [PubMed] [Google Scholar]
- 8. Ellert‐Miklaszewska A, Poleszak K, Kaminska B. Short peptides interfering with signaling pathways as new therapeutic tools for cancer treatment. Future Med Chem. 2017;9(2):199–221. [DOI] [PubMed] [Google Scholar]
- 9. Lee AC, Harris JL, Khanna KK, Hong JH. A comprehensive review on current advances in peptide drug development and design. Int J Mol Sci. 2019;20(10):2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenbaum MI, Clemmensen LS, Bredt DS, Bettler B, Strømgaard K. Targeting receptor complexes: a new dimension in drug discovery. Nat Rev Drug Discov. 2020;19(12):884–901. [DOI] [PubMed] [Google Scholar]
- 11. Bano D, Ankarcrona M. Beyond the critical point: an overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci Lett. 2018;663:79–85. [DOI] [PubMed] [Google Scholar]
- 12. Choi DW. Excitotoxicity: still hammering the ischemic brain in 2020. Front Neurosci. 2020;14:579953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kostandy BB. The role of glutamate in neuronal ischemic injury: the role of spark in fire. Neurol Sci. 2012;33(2):223–37. [DOI] [PubMed] [Google Scholar]
- 14. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–568. [DOI] [PubMed] [Google Scholar]
- 15. Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47(2):122–9. [DOI] [PubMed] [Google Scholar]
- 16. Chalifoux JR, Carter AG. GABAB receptor modulation of synaptic function. Curr Opin Neurobiol. 2011;21:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guetg N, Aziz SA, Holbro N, Turecek R, Rose T, Seddik R, et al. NMDA receptor‐dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB1 . Proc Natl Acad Sci U S A. 2010;107(31):13924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang L, Li Q, Wen R, Yu Z, Li N, Ma L, et al. Rho‐kinase inhibitor prevents acute injury against transient focal cerebral ischemia by enhancing the expression and function of GABA receptors in rats. Eur J Pharmacol. 2017;797:134–42. [DOI] [PubMed] [Google Scholar]
- 19. Kantamneni S, Gonzalez‐Gonzalez IM, Luo J, Cimarosti H, Jacobs SC, Jaafari N, et al. Differential regulation of GABAB receptor trafficking by different modes of N‐methyl‐D‐aspartate (NMDA) receptor signaling. J Biol Chem. 2014;289(10):6681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JY, Kim N, Yenari MA, Chang W. Mild hypothermia suppresses calcium‐sensing receptor (CaSR) induction following forebrain ischemia while increasing GABAB receptor 1 (GABAB‐R1) expression. Transl Stroke Res. 2011;2(2):195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maier PJ, Marin I, Grampp T, Sommer A, Benke D. Sustained glutamate receptor activation down‐regulates GABAB receptors by shifting the balance from recycling to lysosomal degradation. J Biol Chem. 2010;285(46):35606–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terunuma M, Vargas KJ, Wilkins ME, Ramirez OA, Jaureguiberry‐Bravo M, Pangalos MN, et al. Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc Natl Acad Sci U S A. 2010;107(31):13918–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu X, Hu H, Li Z, Lin R, Mao J, Chen L. Gua Lou Gui Zhi decoction attenuates poststroke spasticity via the modulation of GABAB receptors. Mol Med Rep. 2015;12(4):5957–62. [DOI] [PubMed] [Google Scholar]
- 24. Zemoura K, Balakrishnan K, Grampp T, Benke D. Ca2+/Calmodulin‐dependent protein kinase II (CaMKII) β‐dependent phosphorylation of GABAB1 triggers lysosomal degradation of GABAB receptors via mind bomb‐2 (MIB2)‐mediated Lys‐63‐linked ubiquitination. Mol Neurobiol. 2019;56(2):1293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zemoura K, Trumpler C, Benke D. Lys‐63‐linked ubiquitination of γ‐aminobutyric acid (GABA), type B1, at multiple sites by the E3 ligase mind Bomb‐2 targets GABAB receptors to lysosomal degradation. J Biol Chem. 2016;291(41):21682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benke D, Honer M, Michel C, Bettler B, Mohler H. γ‐Aminobutyric acid type B receptor splice variant proteins GBR1a and GBR1b are both associated with GBR2 in situ and display differential regional and subcellular distribution. J Biol Chem. 1999;274(38):27323–30. [DOI] [PubMed] [Google Scholar]
- 27. Benke D, Michel C, Mohler H. Structure of GABAB receptors in rat retina. J Recept Signal Transduc. 2002;22(1–4):253–66. [DOI] [PubMed] [Google Scholar]
- 28. Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448(7149):39–43. [DOI] [PubMed] [Google Scholar]
- 29. Vaas M, Enzmann G, Perinat T, Siler U, Reichenbach J, Licha K, et al. Non‐invasive near‐infrared fluorescence imaging of the neutrophil response in a mouse model of transient cerebral ischaemia. J Cereb Blood Flow Metab. 2017;37(8):2833–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leuchowius KJ, Jarvius M, Wickstrom M, Rickardson L, Landegren U, Larsson R, et al. High content screening for inhibitors of protein interactions and post‐translational modifications in primary cells by proximity ligation. Mol Cell Proteomics. 2010;9(1):178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995–1000. [DOI] [PubMed] [Google Scholar]
- 32. Maier PJ, Zemoura K, Acuna MA, Yevenes GE, Zeilhofer HU, Benke D. Ischemia‐like oxygen and glucose deprivation mediates down‐regulation of cell surface γ‐aminobutyric acidB receptors via the endoplasmic reticulum (ER) stress‐induced transcription factor CCAAT/enhancer‐binding protein (C/EBP)‐homologous protein (CHOP). J Biol Chem. 2014;289(18):12896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu L‐W, Yang F‐Q, Ae A, Qin S. A parametric bootstrap approach for two‐way ANOVA in presence of possible interactions with unequal variances. J Multivar Anal. 2013;115:172–80. [Google Scholar]
- 34. Fu A, Zhao Z, Gao F, Zhang M. Cellular uptake mechanism and therapeutic utility of a novel peptide in targeted‐delivery of proteins into neuronal cells. Pharm Res. 2013;30(8):2108–17. [DOI] [PubMed] [Google Scholar]
- 35. Shioda N, Fukunaga K. Physiological and pathological roles of CaMKII‐PP1 signaling in the brain. Int J Mol Sci. 2018;19(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takemoto‐Kimura S, Suzuki K, Horigane SI, Kamijo S, Inoue M, Sakamoto M, et al. Calmodulin kinases: essential regulators in health and disease. J Neurochem. 2017;141(6):808–18. [DOI] [PubMed] [Google Scholar]
- 37. Chin D, Means AR. Mechanisms for regulation of calmodulin kinase IIα by Ca2+/calmodulin and autophosphorylation of threonine 286. Biochemistry. 2002;41(47):14001–9. [DOI] [PubMed] [Google Scholar]
- 38. Lucić V, Greif GJ, Kennedy MB. Detailed state model of CaMKII activation and autophosphorylation. Eur Biophys J. 2008;38(1):83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Araki S, Osuka K, Takata T, Tsuchiya Y, Watanabe Y. Coordination between calcium/calmodulin‐dependent protein kinase II and neuronal nitric oxide synthase in neurons. Int J Mol Sci. 2020;21(21):7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU. CaMKII in cerebral ischemia. Acta Pharmacol Sin. 2011;32(7):861–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gähwiler BH, Brown DA. GABAB‐receptor‐activated K+ current in voltage‐clamped CA3 pyramidal cells in hippocampal cultures. Proc Natl Acad Sci U S A. 1985;82(5):1558–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein‐coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19(3):687–95. [DOI] [PubMed] [Google Scholar]
- 43. Bussieres N, El Manira A. GABAB receptor activation inhibits N‐ and P/Q‐type calcium channels in cultured lamprey sensory neurons. Brain Res. 1999;847(2):175–85. [DOI] [PubMed] [Google Scholar]
- 44. Chen G, van den Pol AN. Presynaptic GABAB autoreceptor modulation of P/Q‐type calcium channels and GABA release in rat suprachiasmatic nucleus neurons. J Neurosci. 1998;18(5):1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mintz IM, Bean BP. GABAB receptor inhibition of P‐type Ca2+ channels in central neurons. Neuron. 1993;10(5):889–98. [DOI] [PubMed] [Google Scholar]
- 46. Santos AE, Carvalho CM, Macedo TA, Carvalho AP. Regulation of intracellular [Ca2+] and GABA release by presynaptic GABAB receptors in rat cerebrocortical synaptosomes. Neurochem Int. 1995;27(4–5):397–406. [DOI] [PubMed] [Google Scholar]
- 47. Schwenk J, Perez‐Garci E, Schneider A, Kollewe A, Gauthier‐Kemper A, Fritzius T, et al. Modular composition and dynamics of native GABAB receptors identified by high‐resolution proteomics. Nat Neurosci. 2015;19(2):233–42. [DOI] [PubMed] [Google Scholar]
- 48. Keegan BM, Beveridge TJ, Pezor JJ, Xiao R, Sexton T, Childers SR, et al. Chronic baclofen desensitizes GABAB‐mediated G‐protein activation and stimulates phosphorylation of kinases in mesocorticolimbic rat brain. Neuropharmacology. 2015;95:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu L, Li CJ, Lu Y, Zong XG, Luo C, Sun J, et al. Baclofen mediates neuroprotection on hippocampal CA1 pyramidal cells through the regulation of autophagy under chronic cerebral hypoperfusion. Sci Rep. 2015;5:14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tu H, Xu C, Zhang W, Liu Q, Rondard P, Pin JP, et al. GABAB receptor activation protects neurons from apoptosis via IGF‐1 receptor transactivation. J Neurosci. 2010;30(2):749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Babcock AM, Everingham A, Paden CM, Kimura M. Baclofen is neuroprotective and prevents loss of calcium/calmodulin‐dependent protein kinase II immunoreactivity in the ischemic gerbil hippocampus. J Neurosci Res. 2002;67(6):804–11. [DOI] [PubMed] [Google Scholar]
- 52. Fu P, Wu Q, Hu J, Li T, Gao F. Baclofen protects primary rat retinal ganglion cells from chemical hypoxia‐induced apoptosis through the Akt and PERK pathways. Front Cell Neurosci. 2016;10:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han D, Zhang QG, Yong L, Li C, Zong YY, Yu CZ, et al. Co‐activation of GABA receptors inhibits the JNK3 apoptotic pathway via the disassembly of the GluR6‐PSD95‐MLK3 signaling module in cerebral ischemic‐reperfusion. FEBS Lett. 2008;582(9):1298–306. [DOI] [PubMed] [Google Scholar]
- 54. Hleihil M, Vaas M, Bhat MA, Balakrishnan K, Benke D. Sustained baclofen‐induced activation of GABAB receptors after cerebral ischemia restores receptor expression and function and limits progressing loss of neurons. Front Mol Neurosci. 2021;14:726133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lal S, Shuaib A, Ijaz S. Baclofen is cytoprotective to cerebral ischemia in gerbils. Neurochem Res. 1995;20(2):115–9. [DOI] [PubMed] [Google Scholar]
- 56. Lu Y, Li CJ, Chen C, Luo P, Zhou M, Li C, et al. Activation of GABAB2 subunits alleviates chronic cerebral hypoperfusion‐induced anxiety‐like behaviours: a role for BDNF signalling and Kir3 channels. Neuropharmacology. 2016;110(Pt A):308–21. [DOI] [PubMed] [Google Scholar]
- 57. Xu J, Li C, Yin XH, Zhang GY. Additive neuroprotection of GABAA and GABAB receptor agonists in cerebral ischemic injury via PI‐3K/Akt pathway inhibiting the ASK1‐JNK cascade. Neuropharmacology. 2008;54(7):1029–40. [DOI] [PubMed] [Google Scholar]
- 58. Zhang F, Li C, Wang R, Han D, Zhang QG, Zhou C, et al. Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neuroscience. 2007;150(4):938–49. [DOI] [PubMed] [Google Scholar]
- 59. Zhou C, Li C, Yu HM, Zhang F, Han D, Zhang GY. Neuroprotection of γ‐aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J Neurosci Res. 2008;86(13):2973–83. [DOI] [PubMed] [Google Scholar]
- 60. Chalifoux JR, Carter AG. GABAB receptor modulation of voltage‐sensitive calcium channels in spines and dendrites. J Neurosci. 2011;31(11):4221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ouyang C, Guo L, Lu Q, Xu X, Wang H. Enhanced activity of GABA receptors inhibits glutamate release induced by focal cerebral ischemia in rat striatum. Neurosci Lett. 2007;420(2):174–8. [DOI] [PubMed] [Google Scholar]
- 62. Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, et al. Calcium/calmodulin‐dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J Biol Chem. 2012;287(11):8495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Benke D. Mechanisms of GABAB receptor exocytosis, endocytosis, and degradation. Adv Pharmacol. 2010;58:93–111. [DOI] [PubMed] [Google Scholar]
- 64. Grampp T, Sauter K, Markovic B, Benke D. γ‐Aminobutyric acid type B receptors are constitutively internalized via the clathrin‐dependent pathway and targeted to lysosomes for degradation. J Biol Chem. 2007;282:24157–65. [DOI] [PubMed] [Google Scholar]
- 65. Grampp T, Notz V, Broll I, Fischer N, Benke D. Constitutive, agonist‐accelerated, recycling and lysosomal degradation of GABAB receptors in cortical neurons. Mol Cell Neurosci. 2008;39(4):628–37. [DOI] [PubMed] [Google Scholar]
- 66. Bayer KU, Schulman H. CaM kinase: still inspiring at 40. Neuron. 2019;103(3):380–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tu H, Rondard P, Xu C, Bertaso F, Cao F, Zhang X, et al. Dominant role of GABAB2 and Gβγ for GABAB receptor‐mediated‐ERK1/2/CREB pathway in cerebellar neurons. Cell Signal. 2007;19(9):1996–2002. [DOI] [PubMed] [Google Scholar]
- 68. Rai SN, Dilnashin H, Birla H, Singh SS, Zahra W, Rathore AS, et al. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. 2019;35(3):775–95. [DOI] [PubMed] [Google Scholar]
- 69. Sicard KM, Fisher M. Animal models of focal brain ischemia. Exp Transl Stroke Med. 2009;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kalafatovic D, Giralt E. Cell‐penetrating peptides: design strategies beyond primary structure and Amphipathicity. Molecules. 2017;22(11):E1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu M, Li C, Pazgier M, Mao Y, Lv Y, Gu B, et al. D‐peptide inhibitors of the p53‐MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc Natl Acad Sci U S A. 2010;107(32):14321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Partial D‐amino acid substitution: improved enzymatic stability and preserved ab recognition of a MUC2 epitope peptide. Proc Natl Acad Sci U S A. 2005;102(2):413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D‐peptide inhibitors of HIV‐1 entry. Proc Natl Acad Sci U S A. 2007;104(43):16828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dufes C, Olivier JC, Gaillard F, Gaillard A, Couet W, Muller JM. Brain delivery of vasoactive intestinal peptide (VIP) following nasal administration to rats. Int J Pharm. 2003;255(1–2):87–97. [DOI] [PubMed] [Google Scholar]
- 75. Li R, Huang Y, Chen L, Zhou H, Zhang M, Chang L, et al. Targeted delivery of intranasally administered nanoparticles‐mediated neuroprotective peptide NR2B9c to brain and neuron for treatment of ischemic stroke. Nanomedicine. 2019;18:380–90. [DOI] [PubMed] [Google Scholar]
- 76. Ding S, Khan AI, Cai X, Song Y, Lyu Z, Du D, et al. Overcoming blood‐brain barrier transport: advances in nanoparticle‐based drug delivery strategies. Mater Today. 2020;37:112–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kim J, Ahn SI, Kim Y. Nanotherapeutics engineered to cross the blood‐brain barrier for advanced drug delivery to the central nervous system. J Ind Eng Chem. 2019;73:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li X, Tsibouklis J, Weng T, Zhang B, Yin G, Feng G, et al. Nano carriers for drug transport across the blood‐brain barrier. J Drug Target. 2017;25(1):17–28. [DOI] [PubMed] [Google Scholar]
- 79. Deng G, Ma C, Zhao H, Zhang S, Liu J, Liu F, et al. Anti‐edema and antioxidant combination therapy for ischemic stroke via glyburide‐loaded betulinic acid nanoparticles. Theranostics. 2019;9(23):6991–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor‐ PSD‐95 protein interactions. Science. 2002;298(5594):846–50. [DOI] [PubMed] [Google Scholar]
- 81. Ge Y, Chen W, Axerio‐Cilies P, Wang YT. NMDARs in cell survival and death: implications in stroke pathogenesis and treatment. Trends Mol Med. 2020;26(6):533–51. [DOI] [PubMed] [Google Scholar]
- 82. Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med. 2004;4(2):131–6. [DOI] [PubMed] [Google Scholar]
- 83. Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD‐95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483(7388):213–7. [DOI] [PubMed] [Google Scholar]
- 84. Hill MD, Goyal M, Menon BK, Nogueira RG, McTaggart RA, Demchuk AM, et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE‐NA1): a multicentre, double‐blind, randomised controlled trial. Lancet. 2020;395(10227):878–87. [DOI] [PubMed] [Google Scholar]
- 85. Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, et al. Safety and efficacy of NA‐1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2012;11(11):942–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author on request.