

Summary
Condensins are evolutionarily conserved molecular motors that translocate along DNA and form loops. To address how DNA topology affects condensin translocation, we applied auxin-inducible degradation of topoisomerases I and II and analyzed the binding and function of an interphase condensin that mediates X chromosome dosage compensation in C. elegans. TOP-2 depletion reduced long-range spreading of condensin DC from its recruitment sites and shortened 3D DNA contacts measured by Hi-C. TOP-1 depletion did not affect long-range spreading but resulted in condensin DC accumulation within expressed gene bodies. Both TOP-1 and TOP-2 depletion resulted in X chromosome derepression indicating that condensin DC translocation at both scales is required for its function. Together, the distinct effects of TOP-1 and TOP-2 suggest two distinct modes of condensin DC association with chromatin: long-range DNA loop extrusion that requires decatenation/unknotting of DNA and short-range translocation across genes that requires resolution of transcription-induced supercoiling.
Graphic Abstract
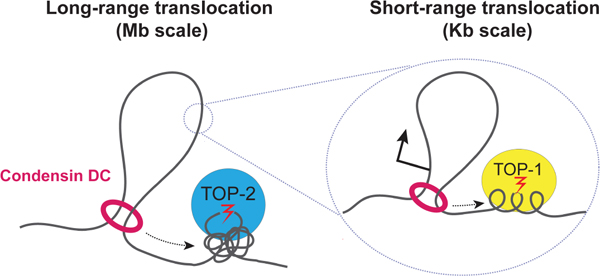
eTOC Blurb
Condensins are molecular motors that translocate along DNA forming loops. In vivo, DNA supercoiling and knots may affect condensin loop extrusion. Morao et al. show that topoisomerase II is required for long-range processivity of loop extrusion by condensin DC whereas topoisomerase I is required for short-range translocation across transcribed genes.
Introduction
Eukaryotic genome compaction is dynamically regulated throughout the cell cycle to accommodate essential functions including gene expression during interphase and chromosome segregation during cell division. A key mechanism that achieves high levels of genome compaction is the formation of DNA loops. In addition to genome packaging, DNA looping modulates gene expression by bringing together regulatory sequences that are far apart in linear distance and by organizing the genome into functional domains that impact gene regulation (Merkenschlager and Nora, 2016). DNA looping is performed by an evolutionary conserved family of ring-shaped motor proteins known as structural maintenance of chromosomes (SMC) complexes, which have been proposed to pull DNA through their opening in a process named “loop extrusion” (Banigan and Mirny, 2020; Uhlmann, 2016). This model is supported by single molecule experiments allowing the direct imaging of DNA loop extrusion by SMC complexes in vitro (Ganji et al., 2018; Golfier et al., 2020; Kong et al., 2020; Pradhan et al., 2022).
We reason that SMC complexes may be challenged by DNA supercoiling and entanglements in vivo. Transcription and replication machineries open and translocate along the double helix, forcing DNA to rotate around its axis. This generates negative and positive supercoiling behind and ahead the translocating machineries, respectively (Liu and Wang, 1987). In addition to supercoiling, DNA molecules can be entangled, especially after replication where positive supercoiling at the replication fork causes the intertwining of the newly replicated duplexes leading to their catenation (Nitiss, 2009). These topological stresses are resolved by topoisomerases, a family of enzymes with DNA cleavage and re-ligation activity. Topoisomerases are classified based on whether they cleave one or two DNA strands in type I (e.g. topoisomerase I) and type II (e.g. topoisomerase II) (Baranello et al., 2013). While both topoisomerase I and II can resolve positive and negative supercoils, TOP-1 has been described to have a major role in the relaxation of transcription-induced supercoiling (Durand-Dubief et al., 2010; Teves and Henikoff, 2014), and TOP-2 is uniquely capable of resolving DNA catenations that require its double-stranded DNA cutting and passage activity (Baranello et al., 2013).
Inactivation of topoisomerase II and condensin leads to two main phenotypes: impaired chromosome compaction and failure to resolve sister chromatids (Piskadlo and Oliveira, 2017; Uhlmann, 2016). Computational simulations suggest that these functions stem from the interplay between loop extrusion and DNA-duplex passage (Goloborodko et al., 2016). It was proposed that topological entanglements were pushed and spatially localized by loop extrusion, promoting unknotting by topoisomerase II (Orlandini et al., 2019; Racko et al., 2018). Supporting this model, evidence from different organisms indicates that in the absence of condensin, topoisomerase II-induced DNA entanglements increase (Baxter et al., 2011; Charbin et al., 2014; Dyson et al., 2020; Piskadlo et al., 2017; Sen et al., 2016), thus suggesting that condensin can control the activity of topoisomerase II to decatenate sister chromatids. An important open question is how DNA entanglements and supercoiling that are constantly being generated across the cell cycle, in turn affect loop extrusion by condensin and other SMC complexes.
To address how condensin translocation is modulated by DNA topology in vivo, a system that allows measurement of the direction and the extent of translocation is needed. Such a unique eukaryotic system exists for a specialized condensin DC that forms the core of the X chromosome Dosage Compensation complex in C. elegans (Ercan, 2015; Meyer, 2005) (Figure 1A). Condensin DC is exclusively recruited to the two X chromosomes in hermaphrodites by SDC-2, SDC-3 and DPY-30, which are enriched at a small number of sequence specific recruitment elements on the X (rex sites) (Albritton et al., 2017; Ercan et al., 2007; Jans et al., 2009). Condensin DC spreads linearly along the X chromosome and concentrates at active promoters and other accessible gene regulatory elements (Ercan et al., 2009; Jans et al., 2009; Street et al., 2019). The activity of condensin DC regulates topologically associating domains (TADs), the compaction of X chromosomes (Anderson et al., 2019; Crane et al., 2015; Jimenez et al., 2021; Lau et al., 2014) and halves transcription initiation of both X chromosomes in hermaphrodites to equalize it to that of the single X in males (Kramer et al., 2015; Kruesi et al., 2013). The X-specific recruitment and linear spreading of condensin DC, along with the strong rex sites acting as TAD boundaries allows the analysis of condensin DC translocation and loop formation. Furthermore, this system provides a framework to study the interplay between condensin and topoisomerase II outside of mitosis.
Figure 1. TOP-1 and TOP-2 overlap with condensin DC on the X chromosome and bind to active promoters across the genome.
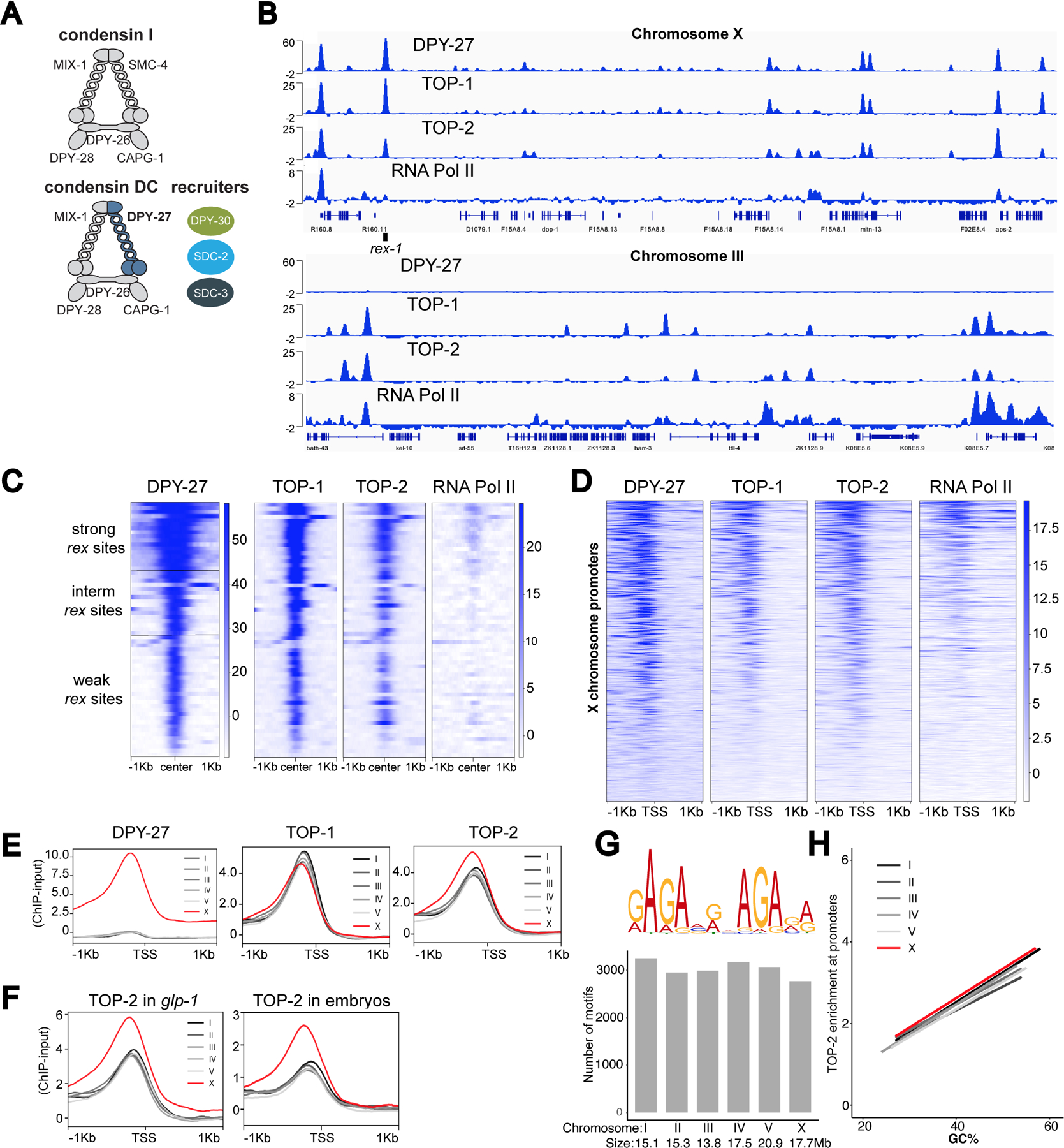
A) Schematic representation of the C. elegans condensin DC, which differs from condensin I by one subunit, the SMC-4 variant, DPY-27. Binding of condensin DC to the X chromosomes requires the recruiter proteins: SDC-2, SDC-3 and DPY-30.
B) DPY-27, TOP-1, TOP-2 and RNA Pol-II ChIP-seq profiles at representative regions on chromosomes X (top) and III (bottom). Normalized ChIP minus input coverage is shown. Genes are displayed and the location of rex-1 is indicated. ChIPs were performed in L2/L3 worms.
C) Heatmap showing ChIP-seq signal for DPY-27, TOP-1, TOP-2 and RNA Pol-II across a 2 Kb window centered around 64 rex sites classified into three strength categories (weak, intermediate and strong) described in (Albritton et al., 2017).
D) Heatmap showing DPY-27, TOP-1, TOP-2 and RNA Pol-II ChIP-seq signals across X chromosome transcription start sites (TSS) defined by (Kruesi et al., 2013) using GRO-seq.
E) Average DPY-27, TOP-1 and TOP-2 ChIP-seq scores are plotted across a 2 Kb window centered around TSSs on X and autosomes. Unlike TOP-1, TOP-2 is enriched on the X chromosomes.
F) Average TOP-2 ChIP-seq scores are plotted across TSSs on X and autosomes in glp-1(q224) adults lacking a germline and WT embryos. TOP-2 ChIP signal is higher on the X chromosomal promoters compared to autosomes in somatic cells.
G) Top: MEME was used to perform a motif search across 150 bp regions centered on TOP-2 ChIP-seq summits. Top scoring motif is shown. Bottom: The C. elegans genome was scanned for instances of the motif using PWMScan. The number of hits (p-value cutoff 1e-5) obtained per chromosome are shown.
H) The GC content of 250 bp windows centered on TSSs was plotted on the X axis and TOP-2 ChIP-seq scores across the same window were plotted on the Y axis. Data was fitted to a linear model.
Here we analyzed condensin DC binding and function upon auxin-inducible depletion of TOP-1 and TOP-2 using ChIP-seq, mRNA-seq and Hi-C. TOP-2 depletion hindered the long-range spreading of condensin DC, resulting in its accumulation around the strong rex sites and in the shortening of 3D DNA contacts on the X, suggesting that TOP-2 is required for the processivity of condensin DC loop extrusion. TOP-1 depletion resulted in gene-body accumulation of condensin DC and RNA Pol II but did not reduce the range of condensin DC spreading from the strong rex sites, thus revealing two distinct modes of condensin DC translocation. Translocation across long distances that necessitates TOP-2, and short-range translocation across gene bodies that requires TOP-1. X chromosomes were derepressed upon both TOP-1 and TOP-2 depletion, thus condensin DC mediated transcriptional repression involves both long-range and local translocation.
Results
TOP-1 and TOP-2 localize to active promoters across the genome and overlap with condensin DC on the X
We first reasoned that if topoisomerases regulate condensin DC translocation, they may co-localize on chromatin. To test this, we analyzed binding of the two major somatic topoisomerases in C. elegans, TOP-1 (M01E5.5) and TOP-2 (K12D12.1), in L2/L3 larvae by ChIP-seq (Jaramillo-Lambert et al., 2016; Lee et al., 1998). For TOP-2, we used a published strain, in which the endogenous top-2 gene is tagged with GFP (Ladouceur et al., 2017). C. elegans TOP-1 has two isoforms, TOP-1α, which contains five exons and TOP-1β, which lacks the second exon (Lee et al., 1998). The functional differences between the two isoforms have not been explored in detail but the second exon, which is missing in TOP-1β, was reported to be dispensable for catalytic activity (Kim et al., 1996). Therefore, both isoforms are expected to regulate supercoiling. To tag both isoforms, we introduced a degron-GFP tag at the end of exon five using CRISPR/Cas9.
The ChIP-seq profile of DPY-27, the SMC-4 variant in condensin DC, shows strong enrichment at recruitment sites (rex), differing levels of moderate enrichment at promoters and a baseline signal across the X chromosome (Ercan et al., 2007; Jans et al., 2009; Street et al., 2019) (Figure 1B). Both TOP-1 and TOP-2 were also enriched at the rex sites (Figure 1C), and bound to X chromosome promoters, in a manner largely proportional to condensin DC (Figure 1D).
Unlike condensin DC, TOP-1 and TOP-2 also bind to the autosomal promoters (Figure 1B lower panel). As in other organisms (Durand-Dubief et al., 2010; Heldrich et al., 2020; Uusküla-Reimand et al., 2016), our data shows that in C. elegans TOP-1 and TOP-2 binding correlate with transcriptional activity (Figure 1D).
TOP-2 binding is enriched at X chromosome promoters
Strikingly, unlike TOP-1, TOP-2 binding was higher on the X chromosome compared to autosomes (Figure 1E and S1A). TOP-2 and condensin are key regulators of mitotic chromosome structure (Kinoshita and Hirano, 2017). Here, the specific association between TOP-2 and an X-specific condensin highlights the relevance of our system to conserved mechanisms of eukaryotic chromosome compaction.
In C. elegans, condensin DC is only expressed in somatic cells, therefore if X enrichment of TOP-2 is related to condensin DC function, it should happen in the soma (Strome et al., 2014). Indeed, TOP-2 ChIP-seq showed X enrichment in glp-1 mutant adults (Figure 1F), which lack the germline when grown at the restrictive temperature (Figure S1B). X-enrichment of TOP-2 was also observed in mixed-stage embryos, which contain only two germ cells, indicating that like condensin DC, TOP-2 is enriched on X chromosomes in somatic cells throughout development (Figure 1F).
To address if there are X-specific sequence features that recruit TOP-2, we performed a motif search under TOP-2 binding peaks across the genome. This analysis yielded one prominent motif, which was not enriched on the X (Figure 1G). The next sequence feature we considered is GC content, as C. elegans X chromosome promoters have higher GC content than autosomes (Ercan et al., 2011). While TOP-2 binding across all chromosomes correlates positively with GC content (Figure 1H), for sites with the same GC content, X chromosomal binding was higher than that of autosomes. Thus, both sequence features failed to explain TOP-2 enrichment on the X (Figure 1H).
Recruitment elements on the X (rex) sites recruit TOP-2
In bacteria, the SMC complex MukBEF recruits topoisomerase IV to ori regions (Nicolas et al., 2014) and in mammalian cells, cohesin interacts with TOP-2 (Uusküla-Reimand et al., 2016) and is required for TOP-2 binding at loop anchors (Canela et al., 2019). To address if condensin DC is required for TOP-2 binding on the X, we introduced a degron-GFP tag at the C-terminus of DPY-27 and expressed the TIR-1 component of the degradation machinery exclusively in somatic cells (Zhang et al., 2015) (Figure 2A). Insertion of the tag in the presence of TIR-1 already impaired DPY-27 resulting in reduced binding and X upregulation without auxin (Figure S2A, S2B and S2C). Auxin treatment further reduced DPY-27, eliminating binding (Figure 2B, S2A and S2B). In both partial and complete DPY-27 depletion conditions, TOP-2 enrichment on the X chromosome promoters was maintained (Figure 2B and 2C). However, since TOP-2 binding correlates with transcription, we cannot exclude that the X derepression resulting from DPY-27 depletion may counter the potential reduction of TOP-2 levels (Figure S2C).
Figure 2. Recruitment elements on the X (rex) sites recruit TOP-2.
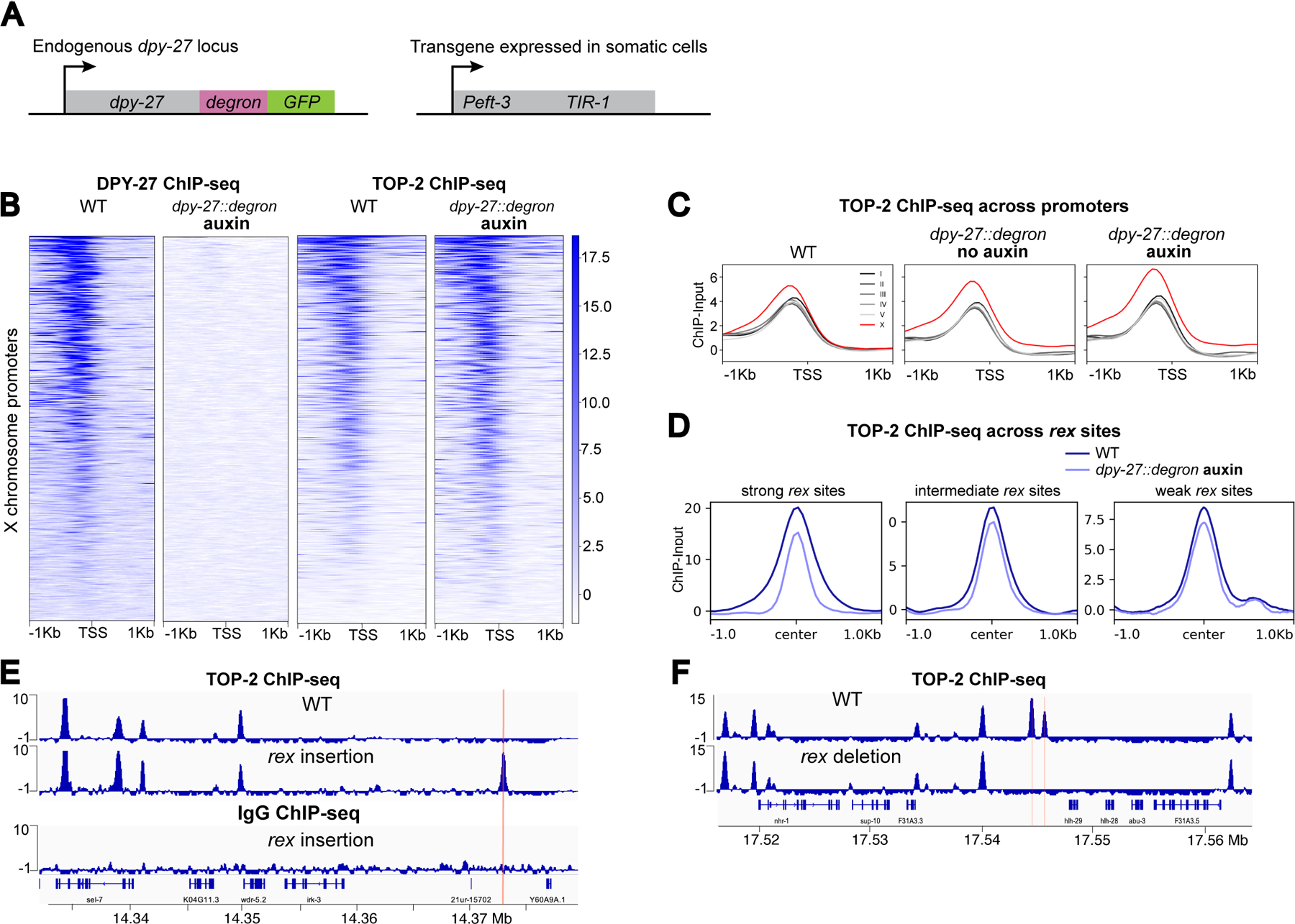
A) Schematic representation of components of the Auxin Inducible Degradation system used for DPY-27 depletion.
B) Heatmap showing DPY-27 and TOP-2 ChIP-seq signals around X chromosome TSSs in WT and DPY-27 depletion conditions. dpy-27::degron L2/L3 worms were treated with auxin for 60 min.
C) Average TOP-2 ChIP-seq scores in WT, DPY-27 partial (no-auxin) and complete (auxin) depletion conditions are plotted across a 2 Kb window centered around TSSs on X and autosomes.
D) Average TOP-2 ChIP-seq scores in WT and DPY-27 depletion conditions are plotted across a 2 Kb window centered at rex sites. TOP-2 signal reduces at the strong rex sites in the absence of condensin DC.
E) TOP-2 and IgG only control ChIP-seq in wild-type and in a strain carrying an ectopic rex-8 insertion on the X chromosome. ChIPs were performed in L2/L3 worms. rex insertion was sufficient to recruit TOP-2 to the ectopic location, indicated by a red line.
F) TOP-2 ChIP-seq in wild-type and in a strain in which ~100 bp containing the two recruiting motifs of the endogenous rex-41 have been deleted. Deletion of rex-41 eliminated TOP-2 binding at the site of deletions marked by a red line.
Interestingly, despite the maintenance of TOP-2 binding at promoters, its binding over strong rex sites was reduced in the absence of condensin DC, whereas enrichment at intermediate and weak rex sites was less affected (Figure 2D). Strong recruitment sites consist of clusters of a 12 bp motif that are important for condensin DC recruitment (Ercan et al., 2007; Jans et al., 2009). To test if strong rex sites recruit TOP-2, we used insertion of a strong rex at an ectopic location that normally lacks TOP-2 (Jimenez et al., 2021). Ectopically inserted rex-8 (358 bp) recruited condensin DC and TOP-2, indicating sufficiency (Figure 2E). IgG and RNA Pol-II ChIP-seq showed background levels at the insertion site validating specificity (Figure S2D). We also performed TOP-2 ChIP-seq in a strain in which ~100 bp containing the two recruiting motifs of the endogenous rex-41 have been deleted (Albritton et al., 2017). Here, the TOP-2 binding domain spanning around 2.5 Kb disappeared (Figure 2F). Thus, rex sequences are both necessary and sufficient for TOP2 binding to the rex sites.
TOP-1 and TOP-2 depletion results in X chromosome transcriptional upregulation
To determine the functional impact of topoisomerases on condensin DC mediated chromosome-wide transcriptional repression, we performed mRNA-seq in TOP-1 and TOP-2 depleted conditions. For this, we endogenously tagged top-1 and top-2 with degron-GFP using CRISPR/Cas9 in the strain expressing TIR-1 in somatic cells (Figure 3A). The resulting strains were healthy and produced normal levels of progeny (Figure S3A and S3B). To avoid defects stemming from chromosome segregation, we performed short-term depletion in L2/L3 larvae where most somatic cells are no longer dividing (Sulston and Horvitz, 1977). For both TOP-1 and TOP-2, nuclear GFP signal could no longer be detected in somatic cells after one hour of auxin treatment (Figure 3B) and ChIP-seq experiments showed strong and uniform depletion across the genome (Figure 3C and 3D). We performed mRNA-seq after thirty minutes, one hour and two hours of auxin-mediated TOP-1 or TOP-2 degradation. As controls, we used top-1 or top-2 degron-tagged worms that were not treated with auxin (no-auxin) (Figure 3A), as well as a strain that lacks the degron-GFP tag but expresses TIR-1, and was treated with auxin (no-tag auxin) (Figure 3A).
Figure 3. TOP-1 and TOP-2 depletion results in X chromosome transcriptional upregulation).
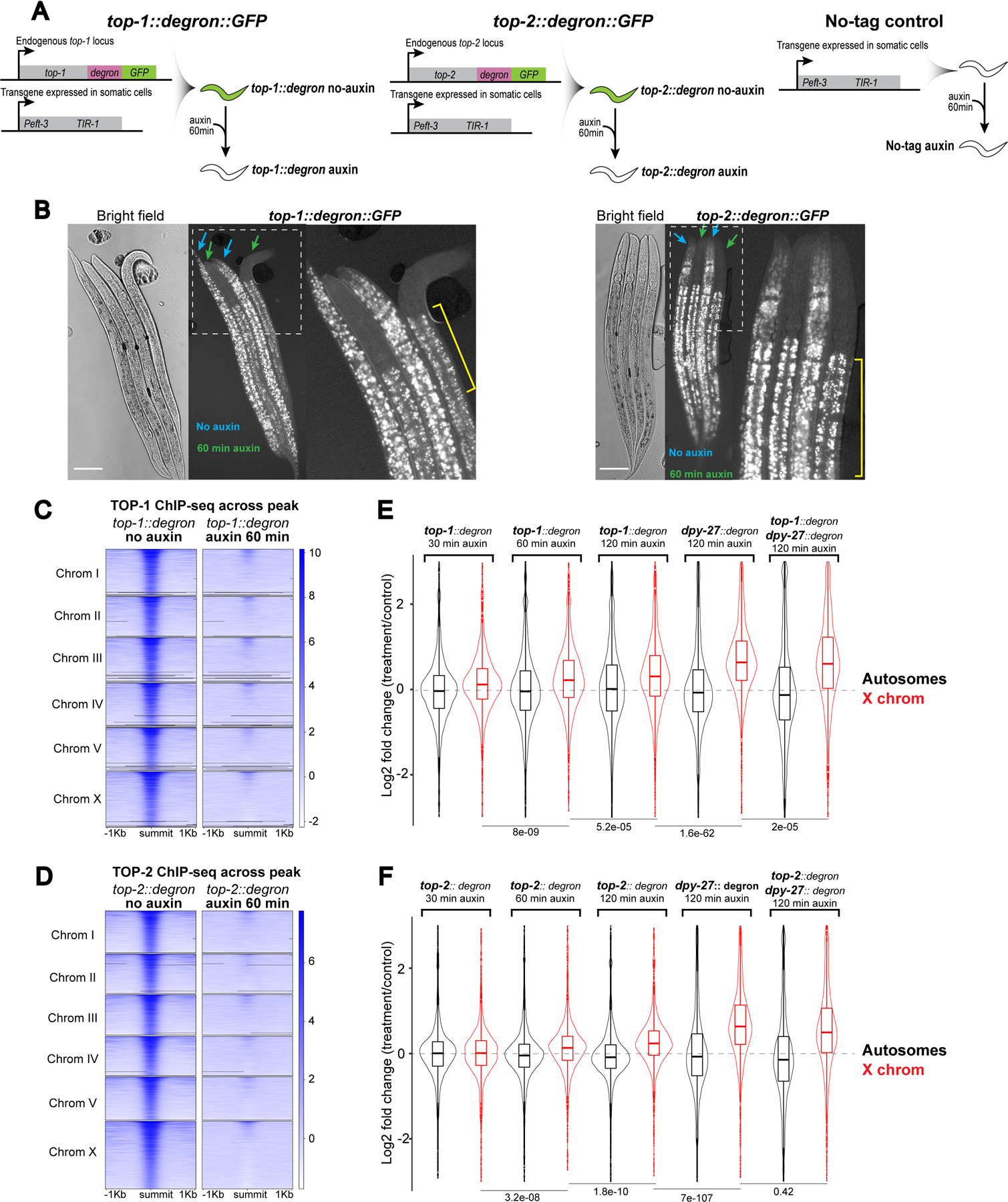
A) Schematic representation of the Auxin Inducible Degradation system used for TOP-1 and TOP-2 depletion. As controls, we used top-1 or top-2 degron-tagged worms that were not treated with auxin (no-auxin), as well as a strain lacking the degron-GFP tag but containing the TIR-1 transgene, that was treated with auxin (no-tag auxin). Depletion experiments were performed by incubating L2/L3 worms on plates containing 1 mM auxin.
B) top-1::degron::GFP (left) and top-2::degron::GFP (right) L2/L3 worms were incubated in auxin (green arrows) and no-auxin (blue arrows) plates for 60 min, followed by imaging. Rightmost images show higher magnification views of the outlined regions. Yellow brackets indicate autofluorescent gut granules (Hermann et al., 2005). Scale bars: 64.5 μm.
C-D) Heatmap showing TOP-1 (C) and TOP-2 (D) ChIP-seq signal in no-auxin and auxin conditions across TOP-1 and TOP-2 peaks, respectively.
E) mRNA-seq was performed after 30, 60 and 120 min of TOP-1 auxin-depletion and after 120 min of DPY-27 and TOP-1;DPY-27 depletion. For TOP-1 depletion, the distribution of log2 fold changes between the auxin treated and no-auxin conditions are shown for autosomes and X chromosomes. For DPY-27 and TOP-1;DPY-27 depletion the distribution of log2 fold changes between the degron-tagged auxin and no-tag auxin conditions are shown. Two-tailed independent two-sample t-test p-values are indicated. TOP-1 depletion does not enhance the level of X chromosome derepression observed in DPY-27 depletion.
F) mRNA-seq in TOP-2 depleted condition as in (E). Similar to TOP-1, TOP-2 depletion did not enhance the X derepression observed in DPY-27 depletion, suggesting that both TOP-1 and TOP-2 repress X chromosomes within the condensin-DC mediated dosage compensation pathway.
In both TOP-1 and TOP-2 depleted conditions, average X chromosomal gene expression increased progressively with longer depletion (Figure 3E, 3F and S3C). To test if the repression mediated by the topoisomerases is part of the dosage compensation pathway, we performed double depletion of condensin DC along with topoisomerase I or II. The additional depletion of topoisomerases did not enhance the derepression observed in the single condensin DC depletion (Figure 3E and 3F), suggesting that topoisomerases contribute to X chromosome repression through the regulation of condensin DC.
TOP-2 depletion reduces the size of condensin DC mediated DNA contacts on the X chromosomes
We reasoned that TOP-2 may regulate the processivity of loop extrusion by condensin DC through the resolution of DNA catenations and knots. To test this idea, we performed Hi-C in larvae depleted of TOP-2 by one hour auxin treatment. Upon TOP-2 knockdown, the TADs on the X appeared weaker, as seen by decreased insulation at TAD boundaries along the chromosome (Figure 4A and S4B). There was no strong effect on autosomes, indicating that TOP-2 is especially required for X chromosome 3D organization (Figure S4A).
Figure 4. TOP-2 depletion leads to shorter DNA interactions on the X chromosomes).
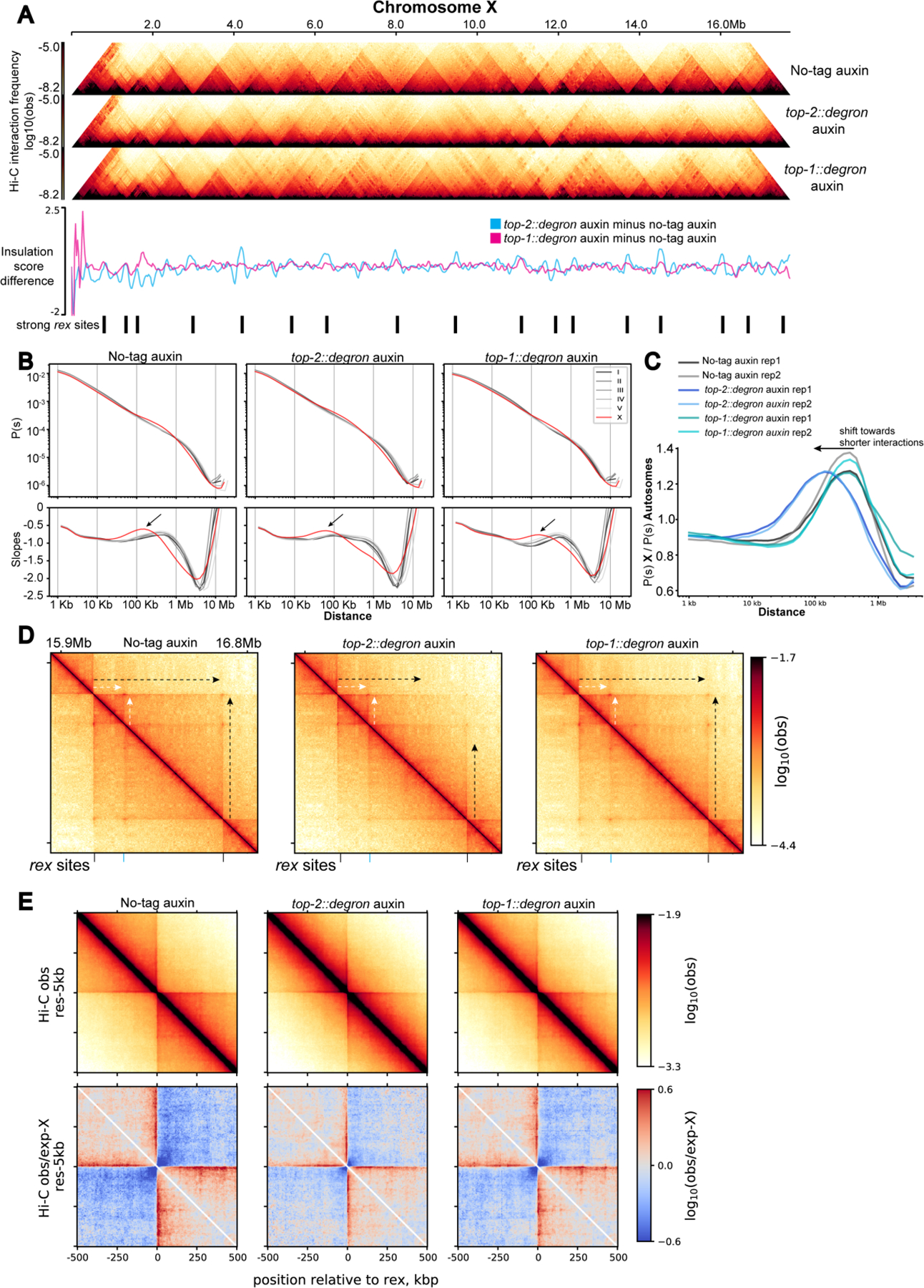
A) Hi-C heatmap of chromosome X in no-tag auxin, top-2::degron auxin, and top-1::degron L2/L3 worms treated with auxin for 60 min. The difference in the insulation scores (subtraction) between the depletion condition and the control are shown below. The positions of the 17 strong rex sites described in (Albritton et al., 2017) are indicated.
B) P(s) and its log-derivative showing the average contact probability and its slopes as a function of genomic distance. Black arrows indicate the local maxima of the slope which has been interpreted as the average loop size (Gassler et al., 2017). Interactions that are enriched on the X chromosome fall within the 100Kb-1Mb distance range in the control and TOP-1 depleted conditions and get shorter in the TOP-2 depleted condition.
C) X-enriched chromosomal contacts are visualized by autosome normalized distance decay curves. For the same genomic distance, contact probability of the X-chromosome, P(s) X, is divided by that of the autosomes, P(s) Autosomes.
D) Hi-C snapshot of 1MB region on X-chromosome. Black arrows indicate long stripes emanating from strong rex sites. White arrows indicate short stripes of nested TAD. Strong (black line) and intermediate (blue line) rex sites are indicated at the bottom.
E) Top: On-diagonal pile-up analysis showing the average Hi-C map across +/− 500 Kb regions surrounding the 17 strong rex sites. 500kb is chosen based on the mean genomic distance between strong rex sites being approximately 1MB. Bottom: for the observed-expected matrix, the expected values are computed from the X-chromosomes (expected-X), since the X-chromosomes have distinct P(s) compared to the autosomes.
Within the frame of the loop extrusion hypothesis, if TOP-2 is required for condensin-DC processivity, 3D DNA contacts on the X chromosome should shorten upon TOP-2 depletion. Indeed, Hi-C contact probabilities plotted as a function of genomic distance, showed reduced distance-range of interactions on the X. In control larvae, autosome curves align together reflecting a similar range of DNA contacts, while X displays increased interactions in the distance range of TADs (~100 Kb - 1Mb) producing a characteristic hump shape (Figure 4B left panel). TOP-2 degradation led to a leftward shift of the hump, indicating a shift towards shorter-range interactions (Figure 4B middle panel). Dividing the distance decay curve of the X chromosome by that of autosomes highlighted the X-enrichment of 3D DNA contacts centering approximately at ~300 Kb in wild type (Figure 4C) and ~100 Kb in TOP-2 depletion.
A second prediction of reduced condensin DC processivity is that Hi-C ‘stripes’ should get shorter upon TOP-2 depletion. Stripes are produced by the asymmetric reeling-in of DNA through the SMC ring, which results in a single locus (anchor) forming interactions with progressively more distant loci (Fudenberg et al., 2017; Vian et al., 2018). On the X, stripes are anchored at strong rex sites that form TAD boundaries (Anderson et al., 2019; Crane et al., 2015; Jimenez et al., 2021). In line with reduced processivity, stripes of around 600 Kb originating from two strong rex sites are reduced (Figure 4D, black arrows), whereas stripes flanking a smaller nested TAD are not affected (Figure 4D, white arrows). A metaplot analysis of interactions around the 17 strong rex sites also shows the overall shortening of stripes emanating from strong rex sites (Figure 4E). Since the strong rex sites are stripe anchors, they are brought together in 3D space. Indeed, rex-rex interactions are among the most frequent long-range interactions on the X chromosome (Crane et al., 2015). Consistent with shorter stripes, upon TOP-2 depletion, rex-rex interactions were lost (Figure S4C). Thus, both predictions, shorter X-specific 3D DNA contacts and shorter stripes, are met. Therefore, we conclude that TOP-2 is required for the processivity of condensin DC loop extrusion.
TOP-2 degradation reduces the spreading of condensin DC along the X chromosome
To directly test the idea that TOP-2 is required for condensin DC processivity, we analyzed condensin DC binding by ChIP-seq upon one hour of TOP-2 depletion. DPY-27 ChIP-seq in the no-tag auxin and no-auxin controls shows strong and narrow enrichment at rex sites and moderate peaks evenly distributed across the X (Figure 5A, Figure S5A). Upon TOP-2 degradation, DPY-27 binding concentrated around the strong rex sites and progressively decreased in a distance dependent manner creating “mountains” of condensin DC centered around strong rex sites (Figure 5A). Subtracting DPY-27 ChIP-seq scores (top-2 depletion minus control) highlights the increased binding nearby strong rex sites and decreased binding across regions located far away from rex sites (Figure S5B). Consistent with an effect on processivity, DPY-27 peaks broadened around recruitment sites and binding at promoters located near rex sites increased (Figure 5B and 5C), whereas binding at promoters located far away from rex sites was reduced (Figure 5C).
Figure 5. Spreading of condensin DC is hindered in the absence of TOP-2).
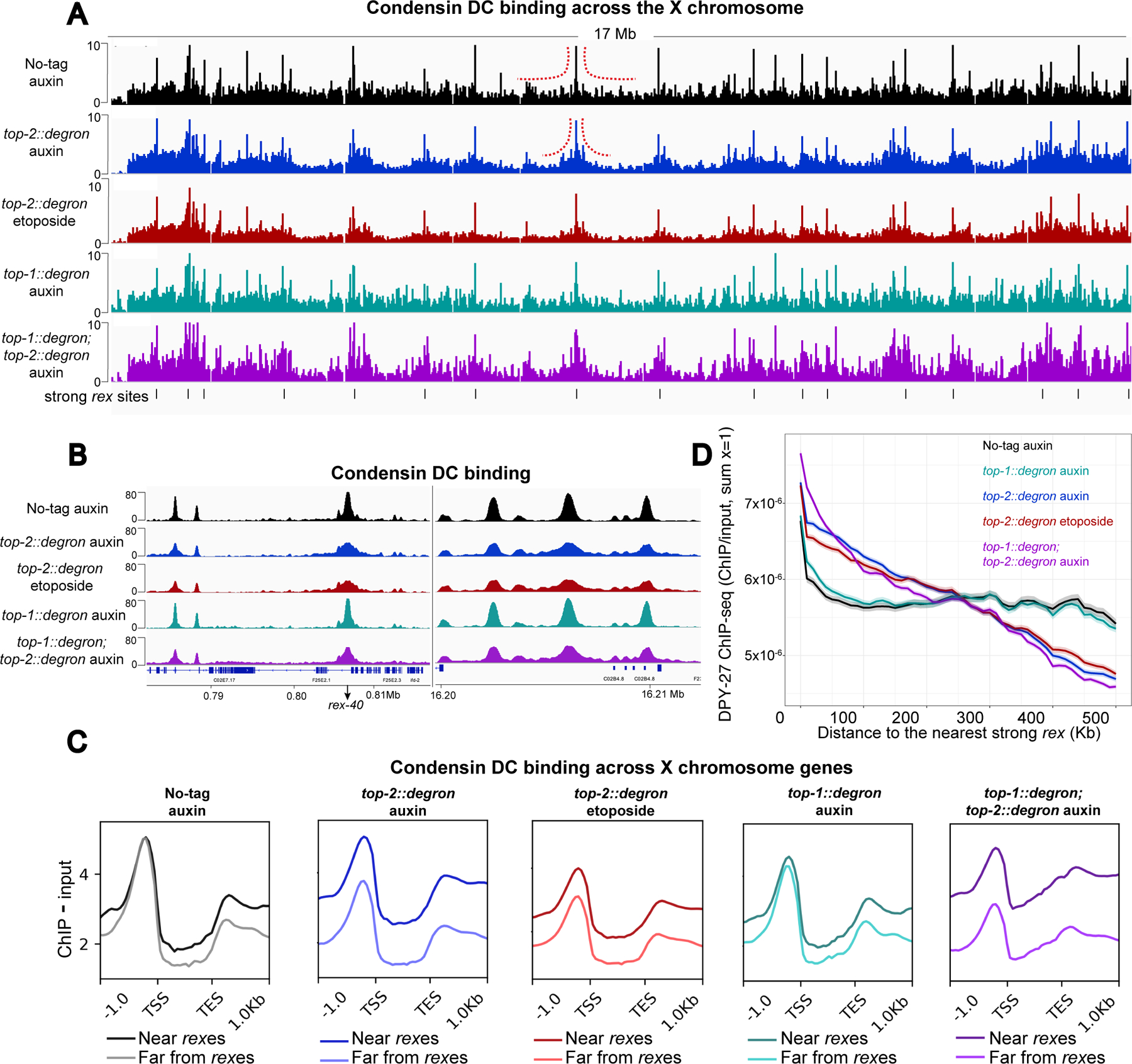
A) X chromosome view of DPY-27 ChIP-seq profile in control (no-tag auxin), TOP-1, TOP-2, double TOP-1/TOP-2 depleted and etoposide treated top-2::degron worms. L2/L3 worms were treated with 1mM auxin for 60 minutes. For etoposide treatment, top-2::degron L2/L3 worms were incubated with 12.5–50 μM etoposide for 60 minutes. Black lines at the bottom indicate the location of strong rex sites. Normalized ChIP minus Input coverages are shown. Red dashed lines indicate changes in DPY-27 binding profile around rex sites between the control and TOP-2 depleted condition. DPY-27 signal accumulates around the strong rex sites upon TOP-2 depletion and etoposide treatment.
B) DPY-27 ChIP-seq profile in control, TOP-1, TOP-2, double TOP-1/TOP-2 depleted and etoposide treated worms at two representative regions of the X chromosome.
C) Genes on the X chromosome were classified according to their distance to a strong rex site into two categories: near (within 400Kb of a strong rex site) and far (more than 400Kb away from a strong rex site). Average DPY-27 ChIP-seq scores are plotted across X chromosome genes belonging to the two categories in control, TOP-1, TOP-2, double TOP-1/TOP-2 depleted and etoposide treated worms. 1 Kb upstream of the TSS and downstream of the TES are included. TOP-2 depletion reduced DPY-27 binding at genes away from the strong rex sites.
D) Metaplot of normalized DPY-27 ChIP-seq within a sliding window of 100 Kb and step size of 10 Kb moving away from strong rex sites. The main line indicates the average and the range indicates 95% confidence interval of each window. In wild type and TOP-1 depletion, DPY-27 spreads from the rex sites and shows an even distribution. Upon TOP-2 depletion, DPY-27 signal is higher near the rex sites and reduced with distance from the rex.
We quantified the progressive decrease of DPY-27 spreading by plotting DPY-27 ChIP-seq scores as a function of distance from the closest strong rex site using a sliding window (Figure 5D). While condensin DC in no-tag auxin control larvae is evenly distributed, upon TOP-2 depletion, DPY-27 ChIP-seq signal progressively declines with increased distance from the rex sites (Figure 5D). This binding pattern is consistent with decreased spreading of condensin DC complexes and along with the Hi-C data presented above, supports the conclusion that in the absence of TOP-2, condensin DC is less processive.
The requirement for TOP-2 could be related to its decatenating activity or a non-catalytic function. To differentiate between these possibilities, instead of auxin depletion, we treated top-2::degron worms with etoposide, which blocks TOP-2 during double-strand cleavage (Montecucco et al., 2015). One hour of etoposide treatment resulted in a similar accumulation of condensin DC around strong rex sites as one hour of auxin treatment (Figure 5 and S5C). Etoposide treatment did not affect the ChIP-seq profile of TOP-2, thus the effect of etoposide on condensin DC is not due to a reduction in TOP-2 binding (Figure S5D and S5E). These results support the conclusion that the effect of TOP-2 on condensin DC translocation is linked to its catalytic activity, unless etoposide also affects a novel non-catalytic function of TOP-2 by altering its dynamic turnover on DNA.
TOP-1 degradation results in accumulation of condensin DC within gene bodies
Next, we wondered how TOP-1 contributes to condensin DC-mediated X chromosome repression (Figure 3C). Single-strand cleavage activity of TOP-1 is efficient to remove supercoiling but is not able to resolve catenations and knots (Pommier et al., 2016). Unlike TOP-2, TOP-1 depletion did not show any effect on condensin DC’s long-range spreading (Figure 5). Furthermore, double depletion of TOP-1 and TOP-2 did not enhance the accumulation of condensin DC around strong rex sites that was observed in the single TOP-2 depletion (Figure 5D). Consistent with the lack of effect on the long-range translocation of condensin DC, TOP-1 depletion did not shorten the range of X-enriched 3D DNA contacts (Figure 4 and S4). Thus, catenations and knots resolved by TOP-2 specifically reduce the long-range processivity of condensin DC.
Instead, TOP-1 depletion resulted in increased DPY-27 ChIP-seq signal within gene-bodies and around transcription end sites (Figure 6A). We reasoned that accumulation of transcription induced supercoiling in the absence of TOP-1 may cause the accumulation of DPY-27. Consistent with this, DPY-27 binding within gene bodies was higher for genes with higher expression (Figure 6B). Furthermore, DPY-27 ChIP-seq enrichment within gene bodies increased with gene length, which correlates with stronger accumulation of supercoiling due to RNA Polymerase II elongating over longer distances (Liu and Wang, 1987) (Figure 6C).
Figure 6. TOP-1 degradation results in accumulation of condensin DC within gene bodies).
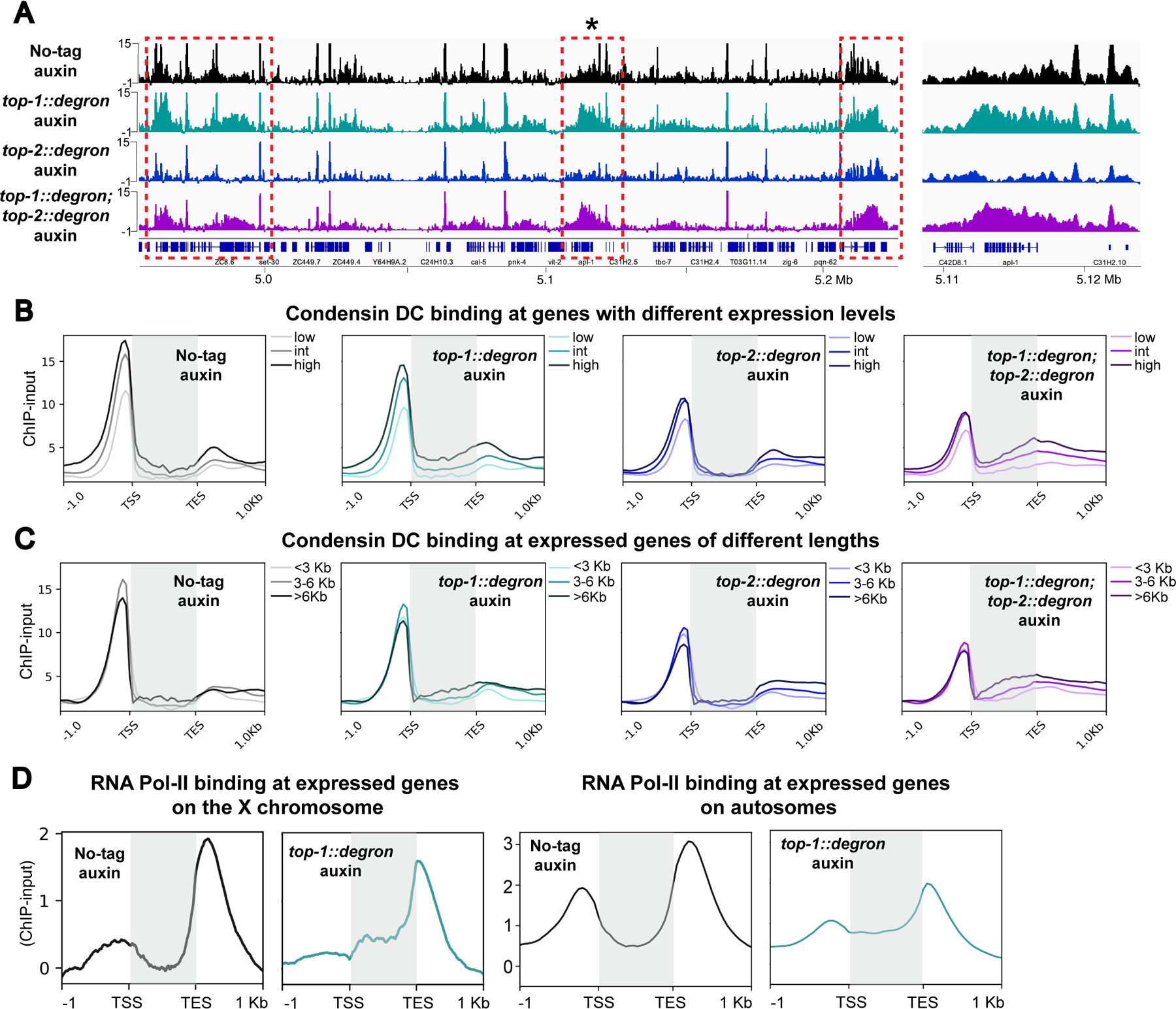
A) Left: DPY-27 ChIP-seq profile in control (no-tag auxin), TOP-1, TOP-2 and double TOP-1/TOP-2 depletions at a representative region of chromosome X. Upon TOP-1 depletion, DPY-27 signal increases within gene bodies (dashed red boxes). Right: Zoomed-in view of region indicated with an asterisk on the left. Normalized ChIP-input coverages are shown.
B) Average DPY-27 ChIP-seq scores in control (no-tag auxin), TOP-1, TOP-2 and double TOP-1/TOP-2 depletion are plotted across X genes, grouped based on their expression level. Gene bodies are highlighted by gray boxes. DPY-27 signal increases within gene bodies upon TOP-1 depletion and this accumulation is enhanced when both topoisomerases are depleted.
C) Average DPY-27 ChIP-seq scores in the same conditions as in panel B are plotted across expressed genes on the X chromosome grouped based on their length into three categories: <3 Kb, 3–6 Kb and >6 Kb. Gene bodies are highlighted by gray boxes.
D) Average RNA Pol-II ChIP-seq scores in control (no-tag auxin), and TOP-1 depletion conditions across expressed genes suggest that elongating polymerases stall upon transcription-induced supercoiling accumulation on both X chromosomal and autosomal gene bodies. Gene bodies are highlighted by gray boxes.
If TOP-1 depletion results in the accumulation of transcription-induced supercoiling, it should also hinder progression of RNA Pol II (Teves and Henikoff, 2014). Indeed, RNA Pol II ChIP-seq after one hour of TOP-1 depletion showed reduced signal around transcription start sites and increased signal within gene bodies on the X and autosomes (Figure 6D). Together these results suggest that the resolution of transcription-generated supercoiling by TOP-1 is required for condensin DC translocation across gene bodies and transcriptional repression of the X chromosomes.
Distinct effects of TOP-1 and TOP-2 in condensin translocation across the X chromosomes
Both topoisomerases I and II are involved in the relaxation of supercoiling that originates from transcription (Pommier et al., 2016). In this context, topoisomerase I and II act in a redundant manner, although exceptions have been described for long and highly expressed genes (Durand-Dubief et al., 2010; Joshi et al., 2012; King et al., 2013; Kouzine et al., 2013; Sperling et al., 2011). Consistent with some redundancy, double depletion of TOP-1 and TOP-2 exacerbated accumulation of condensin DC between TSS-TES (Figure 6B and 6C). Thus, TOP-2 also contributes, but TOP-1 has the major role in the resolution of transcription-induced supercoiling.
Importantly, the distinct effects of TOP-1 and TOP-2 on chromosome-wide distribution of condensin DC suggests that long-range translocation, which is sensitive to TOP-2, is not affected by gene-body accumulation of condensin DC upon TOP-1 depletion. In other words, condensin DC translocation across transcribed genes was hindered in the absence of TOP-1, but this blockage did not affect spreading from recruitment sites (Figure 5A and 5D). Furthermore, stronger gene-body accumulation of condensin DC in the double TOP-1;TOP-2 depletion did not result in a further reduction of spreading from recruitment sites, like one would expect if long-range spreading required the complex to translocate across transcribed genes (Figure 5D). This suggests that condensin DC complexes spreading across long distances from recruitment sites can bypass transcription-induced supercoiling whereas condensin DC complexes translocating within gene bodies are susceptible to such topological constraints.
Discussion
In this study we provide evidence for a role of topoisomerases in the regulation of condensin DC translocation in vivo. Our data indicate that topoisomerases I and II regulate condensin DC translocation at different genomic scales. TOP-1 is required for the local translocation of condensin DC through genes which necessitates the resolution of transcription-generated supercoiling. TOP-2 is required for long-range translocation of condensin DC that generates 3D DNA contacts across distances of hundreds of Kb. We propose that TOP-2 contributes to condensin DC processivity by the resolution of DNA knots and catenates, although a novel non-catalytic function remains possible and requires further investigation.
TOP-2 promotes condensin DC long-range translocation and loop formation
The enrichment of 3D DNA contacts and TADs on the X chromosomes requires the recruitment of condensin DC (Crane et al., 2015). Strong rex sites are necessary and sufficient to increase condensin DC binding (Albritton et al., 2017; Jans et al., 2009; McDonel et al., 2006) and to create loop-anchored TADs (Anderson et al., 2019; Jimenez et al., 2021), suggesting that they function to both load and block condensin DC-mediated DNA loop extrusion. Whether rex sites create Hi-C stripes by being preferential entry points for condensin DC or by blocking loop extrusion is unclear. Regardless, the specific requirement for TOP-2 to create Hi-C stripes and increase the distance range of 3D contacts on the X chromosome strongly suggests that TOP-2 is required for the processivity of condensin DC loop extrusion. One simulation study showed that reducing TOP-2 mediated DNA chain passing with fixed loop extrusion parameters enhances TAD structures (Nuebler et al., 2018). Our work suggests that future simulations should consider the possibility that the properties of loop extrusion depend on the frequency of chain passing in order to understand the contribution of DNA topology and loop extrusion on 3D genome folding features.
The strong rex sites act as TAD boundaries and recruit TOP-2
Modeling experiments proposed that as SMC complexes extrude, they could push and localize catenations and knots, thus promoting their relaxation by recruiting TOP-2 to highly entangled regions (Orlandini et al., 2019; Racko et al., 2018). This model is supported by the observations that mammalian TOP2B colocalizes with CTCF/cohesin (Canela et al., 2017; Manville et al., 2015; Uusküla-Reimand et al., 2016) and induces double strand breaks (DSBs) at DNA loop anchors (Canela et al., 2019, 2017; Gothe et al., 2019). Our results are also supportive of this model, since TOP-2 binding at the TAD boundaries created by strong rex sites is reduced in the absence of condensin DC, and strong rex sites recruit TOP-2. Therefore, it is possible that condensin DC induces topological configurations that recruit TOP-2 to TAD boundaries.
Continuous addition and removal of DNA knots by TOP-2 may regulate condensin processivity
In vivo there seems to be a limit in the capacity of condensins to push through intertwined chromatin. The fact that TOP-2 is required for condensin DC translocation in larvae suggests that topological constraints are generated even in somatic cells that have largely completed their divisions. In addition, only one hour of TOP-2 depletion or etoposide treatment reduced condensin DC processivity. Thus, we propose that loop extrusion by condensins continually requires TOP-2 strand passage activity that resolves DNA catenates/knots during interphase. These topological constraints may be short-lived entanglements that were proposed to be formed and resolved by TOP-2 due to transcription induced positive supercoiling (Valdés et al., 2019). The balance between the rate of generation and resolution of such entanglements would be maintained in the presence of TOP-2 but its acute depletion may result in unresolved entanglements that have the potential to stall condensin translocation.
TOP-1 is required for condensin DC translocation across transcribed gene bodies
Here we show that in C. elegans, TOP-1 is the main relaxer of transcription-generated supercoiling across the genome and propose that on the X, this relaxation activity is important for the translocation of condensin DC across genes. The relaxation of transcription-induced supercoiling by topoisomerases is required for transcription elongation (Durand-Dubief et al., 2010; Pommier et al., 2016). Therefore, accumulation of condensin DC at gene bodies could be the result of stalled RNA Pol II complexes. On one hand, high concentrations of transcribing RNA Pol II may act as permeable barriers that slow condensin loop extrusion (Brandão et al., 2019; Tran et al., 2017). On the other hand, RNA Pol II may directly push condensin or move it through the energy originated from transcription-induced supercoiling (Busslinger et al., 2017; Davidson et al., 2016; Rusková and Račko, 2021).
TOP-1 and TOP-2 contribute to condensin-DC mediated transcriptional repression
The fact that depletion of both TOP-1 and TOP-2 caused X chromosome upregulation suggests that condensin DC mediated chromosome-wide repression requires both long and short-range translocation. We speculate that the long-range translocation of condensin DC, which requires TOP-2, may be important to spread the complex along the entire chromosome to achieve the chromosome-wide dosage compensation. The link between condensin DC binding and repression was established in X-autosome fusion chromosomes in which condensin DC spreading into the autosome portion decreases linearly with distance from the X and the level of condensin DC spreading correlates with the level of repression (Street et al., 2019). Once condensin DC reaches gene promoters, short-range translocation through the gene body, which requires TOP-1, may underlie the actual repression by interfering with transcription initiation. In yeast, condensin was reported to bind active gene bodies during mitosis to prevent transcription induced chromosome segregation defects (Sutani et al., 2015). Mechanism of transcription repression by condensin DC may be related to such local binding to genes and co-opted from a mitotic function by condensins.
A model for DNA catenation and transcription-induced supercoiling regulating in vivo condensin translocation
Here we present a model in which condensin DC’s linear spreading over long distances requires resolution of DNA entanglements by TOP-2 and translocation over genes requires the resolution of DNA supercoiling by TOP-1 (Figure 7). In wild type cells, as condensin moves along chromatin, chances of encountering yet-unresolved DNA catenations increase, resulting in reduced binding with increased distance from the rex sites. This would explain the gradual decrease in condensin DC spreading into the autosomal region of X-autosome fusion chromosomes (Ercan et al., 2009). Even distribution of rex sites (~1 Mb apart across the X) would accomplish the chromosome-wide distribution of condensin DC. At the local scale, another pool of condensin DC may be translocating over genes in a manner sensitive to transcription-induced supercoiling and thus require TOP-1.
Figure 7. Model for regulation of condensin DC translocation by topoisomerases I and II.
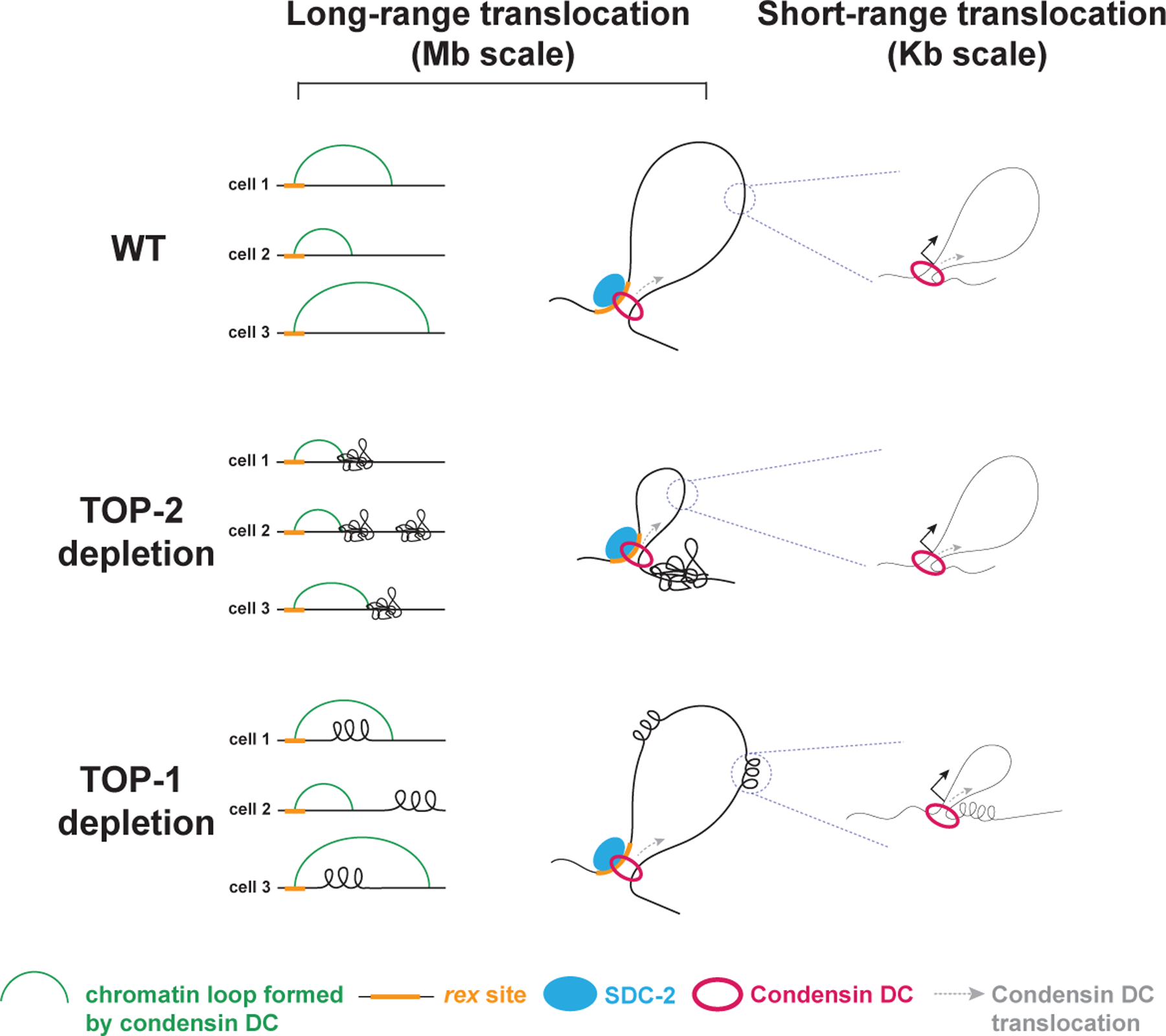
Condensin DC translocation along the X chromosomes occurs at two different distance scales that are distinctly regulated by the two topoisomerases. The long-range spreading mediates ~100kb-1Mb 3D X-enriched DNA contacts and may be due to loop extrusion. This spreading likely requires the resolution of topological constraints such as DNA catenates and knots by TOP-2 but is capable of bypassing transcription-induced supercoiling that accumulates in the absence of TOP-1. The short-range translocation occurs across gene bodies and requires TOP-1, but does not affect the long-range translocation and the 3D DNA contacts. At rex sites SDC-2 may promote condensin DC processivity enabling it to spread over Mb-scale distances, thus distributing the complex to accomplish chromosome-wide dosage compensation. Some condensin DC complexes could dissociate from SDC-2 at promoters and engage in a shorter scale translocation within gene-bodies that cannot overcome transcription-induced supercoiling, and may interfere with RNA Polymerase II to reduce transcription initiation.
What determines the different behaviors of condensin DC? One distinguishing factor between complexes found at recruitment sites versus gene promoters is the level of the main condensin DC recruiter protein, SDC-2, which shows high binding at rex sites and limited spreading across the X chromosome (Ercan et al., 2009). Similar to the cohesin loader, NIPBL, SDC-2 could associate with extruding condensin DC complexes and act as a processivity factor allowing condensin to overcome obstacles. Condensin DC extruding complexes that dissociate from SDC-2 could engage in a different mode of DNA translocation that involves movement through the gene in a transcription-coupled manner and is sensitive to DNA supercoiling.
Our model opens a new line of research to understand the mechanism by which condensin and TOP-2 function together. While the role of condensin regulating TOP-2 is better understood, here we show that TOP-2 in turn, promotes condensin processivity. Thus upon TOP-2 depletion or inhibition, blocked condensin complexes may contribute to previously reported condensation defects (Piskadlo and Oliveira, 2017). Our work also contrasts condensin to cohesin, another SMC complex whose loop extrusion activity was proposed to be equally sensitive to TOP-1 and TOP-2 inhibition (Neguembor et al., 2021). It is possible that unlike condensin which has two distinct modes of translocation including one that “skips over” genes, cohesin may have to move through transcribed regions and is thus additionally sensitive to TOP-1 depletion. In summary, our study sheds light into how different types of topological constraints impact condensin translocation and function. Deciphering how loop extrusion by SMCs is impacted by factors inherent to the chromatin fiber will contribute to the better understanding of the mechanisms by which the genome is dynamically organized throughout the cell cycle.
Limitations of the study
Although we interpret our results using DNA loop extrusion as a framework, it is possible that long-range condensin DC translocation along chromatin in vivo uses additional mechanisms. Since condensin DC spreads linearly over Mb distances in X-autosome fusion chromosomes (Ercan et al., 2009), this non-extrusion mechanism should also be linear. In this case, in the absence of TOP-2, X chromosome structure should change in a way that affects long-range 3D DNA contacts, and the translocation of condensin DC, concentrating it around the strong recruitment sites to produce the ChIP-seq profiles we observe (Figure 5A).
Here we should also note that by the nature of the acute depletion set up, our conclusions are limited to the maintenance of condensin-DC mediated X chromosome structure and repression. For example, whether condensin DC is required for the establishment, rather than maintenance of higher TOP-2 binding on the X chromosome is unclear. In addition, given that baseline reduction of DPY-27 in the dpy-27::degron strain, we cannot exclude the possibility of secondary effects associated with the derepression of the X chromosome that could affect TOP-2 binding in unpredictable ways.
STAR methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sevinc Ercan (se71@nyu.edu).
Materials availability
Worm strains generated in this study are available upon request to lead contact.
Data and code availability
Genomic data is available at Gene Expression Omnibus (GEO) series numbers GSE188851. Accession numbers of the data sets generated in this study are listed in Supplemental table 2,3 and 4.
All original code is available at https://doi.org/10.5281/zenodo.7125333 and in the supplementary information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Worm strains and growth
Worms were grown and maintained at 20–22°C on Nematode Growth Medium (NGM) plates containing E. coli strains OP50–1 and/or HB101. Mixed developmental stage embryos were obtained by bleaching gravid adults in 0.5 M NaOH and 1.2% bleach. To isolate synchronized L2/L3 worms, gravid adults were bleached, and embryos were hatched overnight in M9 buffer (22 mM KH2PO4 monobasic, 42.3 mM Na2HPO4, 85.6 mM NaCl, 1mM MgSO4). The resulting starved L1s were grown for 24 hours at 22°C. For glp-1 adult worms, gravid adults grown at the permissive temperature (15°C) were bleached and starved L1s were grown at the restrictive temperature (25°C) for three days.
Generation of top-1::degron::GFP:
The degron-GFP tag was inserted at the 3’ end of top-1 (M01E5.5) using the CRISPR/Cas9 system. A 20 bp crRNA (AM56) was designed to target the end of the last top-1 exon. Repair templates consisting of a 15 bp flexible linker (GlyGlyGlyGlySer) and the degron-GFP tag flanked by 120 bp single stranded overhangs complementary to top-1 were generated by PCR using AM57F&R and AM04&AM20 primers, and pLZ29 as a template. The injection mix containing S.pyogenes Cas9 3NLS (10 mg/ml, IDT), crRNA (2 nmol, IDT), tracrRNA (5 nmol, IDT), dsDNA donors, and pCFJ90 (pharynx mCherry marker,(Frøkjær-Jensen et al., 2008)) was prepared as described in (Dokshin et al., 2018). Around 30 F1s that were positive for the mCherry co-injection marker were transferred to individual plates. F2 worms were screened by PCR using primers AM58F&R. PCR products of the expected size were sequenced to confirm in-frame insertion. Sanger sequencing results are provided in supplemental table 5. A list of primers is provided in supplementary table 1.
Generation of top-2::degron::GFP:
Same as for top-1::degron::GFP with the following reagents:
crRNA: AM30
Primers to generate repair template: AM22F&R
Genotyping primers: SS02F&R
Sanger sequencing results are provided in supplemental table 6.
Generation of dpy-27::degron::GFP:
Same as for top-1::degron::GFP with the following reagents:
crRNA: LS37
Primers to generate repair template: AM21F&R
Genotyping primers: LS40F&R
Sanger sequencing results are provided in supplemental table 7.
METHOD DETAILS
Auxin treatment:
Auxin (indole-3-acetic-acid, Fisher 87–51-4) was resuspended in 100% ethanol to a concentration of 400 mM. Auxin plates were prepared by adding resuspended auxin to NGM media before pouring the plates, to a concentration of 1 mM.
Synchronized L2/L3 worms were washed three times with M9 and split into two aliquots. Half of the worms were transferred to NGM 10 cm plates containing 1mM of auxin. The other half were placed in normal NGM 10 cm plates (no-auxin control) for the indicated time. A maximum of 300 μL of settled worms was placed in one 10 cm plate. Worms were then washed one time with M9 and processed accordingly to future application. For ChIP and Hi-C, worms were crosslinked in 2% formaldehyde for 30 minutes, followed by quenching in 125mM glycine for 5 minutes, one wash with M9 and two washes with PBS, PMSF and protease inhibitors. For RNA-seq, worms were stored in Trizol.
Etoposide treatment:
Synchronized L2/L3 worms were washed three times with M9 and incubated for one hour in etoposide (Sigma-Aldrich, E1383–100MG) diluted at the desired concentration in 5mL of M9. Similar results were obtained with 12.5 μM, 25 μM and 50 μM of etoposide. Worms were then washed twice time with M9 followed by crosslinking performed as indicated in the auxin treatment section.
ChIP-seq
Two biological replicates with matching input samples were performed for each experiment (Supplemental table 2). Around 100 μL of pelleted embryos, L2/L3 larvae and adult worms were dounce-homogenized with 30 strokes in FA buffer (50 mM HEPES/KOH pH 7.5, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 150 mM NaCl) supplemented with PMSF and protease inhibitors (Calbiochem, 539131). Dounced worms were sonicated in 0.1% sarkosyl for 15 minutes using a Bioruptor-pico to obtain chromatin fragments between 200 and 800 bp. Protein concentration was determined using Bradford assay (Biorad 500–0006). 2 mg of protein extract was used per ChIP and 5% was taken out to use as input DNA. The remaining of the protein extract was incubated with 3 to 10 ug of antibody at 4°C rotating overnight in a volume of 440µL. 40µL of Protein A and/or G Sepharose beads that were previously washed 3 times with FA buffer, were added to the immunoprecipitation and incubated rotating at 4°C for 2 hours. Beads were washed with 1mL of each of the following buffers: 2 times with FA buffer, 1 time with FA-1mM NaCl buffer, 1 time with FA-500mM NaCl buffer, 1 time with TEL buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0) and 2 times with TE buffer.
Immunoprecipitated chromatin was eluted from beads by incubating in ChIP elution buffer (1% SDS, 250 mM NaCl, 10 mM Tris pH 8.0, 1 mM EDTA) at 65°C for 30 minutes, treated with Proteinase K and reverse crosslinked at 65°C overnight. Half of the ChIP DNA and 30 ng of input DNA were used for library preparation. End repair was performed in T4 ligase reaction buffer (New England Biolabs, NEB), 0.4mM of dNTPs, 20 U of T4 Polynucleotide kinase (NEB), 3.5 U of Large (Klenow) fragment (NEB) and 6 U of T4 DNA polymerase for 30 minutes at 20°C in a total volume of 33 μL. Reaction was cleaned using Qiagen MinElute PCR purification kit. A-tailing reaction was performed in NEB buffer 2, 0.2 mM of dATPs and 7.5 U of Klenow fragment-exo (NEB) at 37°C for 60 minutes in 25 μL. Reaction was cleaned using Qiagen MinElute PCR purification kit. Illumina TruSeq adapters were ligated to DNA fragments in a reaction containing 2X Quick ligation buffer (NEB), 0.25 μM of adapter and 2 μL of Quick ligase (NEB) for 20 minutes at 23°C in 40 μL. Reaction was cleaned using Agencourt AMPure XP beads and the eluted DNA was PCR amplified in 50 μL using Phusion Polymerase and TruSeq primers, forward: AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC G*A, reverse: CAA GCA GAA GAC GGC ATA CGA GA*T, where * represents a phosphorothioate bond. PCR reactions were cleaned using Qiagen MinElute PCR purification kit. The eluted DNA was run on a 1.5% agarose gel and fragments between 250–600 bp were gel extracted using Qiagen gel extraction kit. Library concentration was determined using KAPA Library quantification kit. Single-end 75 bp sequencing was performed using the Illumina NextSeq 500 at the New York University Center for Genomics and Systems Biology, New York, NY.
mRNA-seq
L2/L3 larvae were collected for two biological replicates per condition and stored in Trizol (Invitrogen) at −70°C (Supplemental table 3). Total RNA was purified following manufacturer’s instructions after freeze-cracking samples five times. RNA was cleaned up using Qiagen RNeasy MinElute Cleanup kit and quantified using Nanodrop. 1 μg of RNA was run on 1% agarose gel complemented with bleach to assess quality (Aranda et al., 2012). mRNAs were purified using Sera-Mag Oligo (dT) beads (Thermo Scientific) from 10 μg of total RNA, followed by fragmentation using kit from Ambion (AM8740) and cleaning using Qiagen RNeasy minElute kit. First strand cDNA synthesis was performed using Superscript III system (Invitrogen, 18080–051). dNTPs were removed by ethanol precipitation. Second strand synthesis was performed using DNA polymerase I (Invitrogen) in the presence of dATPs, dCTPs, dGTPs, dUTPs and RNase H (Invitrogen). cDNA was purified using Qiagen RNeasy MinElute Cleanup kit. Stranded Illumina libraries were prepared as indicated for ChIP with the following modification: before PCR amplification, uridine digestion was performed at 37°C for 15 minutes in 50 μL with 1 U of Uracil-N-Glycosylase (ThermoScientific EN0361) (Parkhomchuk et al., 2009). Single-end 75 bp sequencing was performed using the Illumina NextSeq 500 at the New York University Center for Genomics and Systems Biology, New York, NY.
Hi-C
Two biological replicates were performed for each experiment (Supplemental table 4). Crosslinked L2/L3 worms collected as described above were resuspended in 20 ul of PBS per 50ul of worm pellet then dripped into liquid nitrogen containing mortar. The worms were grounded with pestle until fine powder. Grounded worms were crosslinked again in 2% formaldehyde using the TC buffer as described by the Arima Hi-C kit, which uses two 4-base cutters, DpnII and HinfI. For one of the Hi-C samples, JK93, Arima Hi-C high-coverage kit was used which includes two additional enzymes DdeI and MseI. The Arima manufacturer’s protocol was followed including the recommended method of the library preparation using KAPA Hyper Prep Kit. Paired-end 150 bp sequencing was performed using the Illumina Novaseq 6000 at the New York University Center for Genomics and Systems Biology, New York, NY.
Microscopy
For direct comparison, top-1::degron::GFP and top-2::degron::GFP worms incubated with and without auxin were aligned side by side prior to imaging. For this, 5 μL of 100 mM sodium azide were added to a 2% agar pad positioned on top of a coverslip and 4–6 L2/L3 larvae were transferred to the liquid and aligned using a hair pick (Figure 3B). For glp-1 mutants (Figure S1B), adult worms grown at the permissive (16°C) and restrictive (25°C) temperatures were immobilized with 100 mM sodium azide immediately before imaging. Images were acquired with a Zeiss Axio Imager A2 microscope using a 10x and 20x objective and the AxioVision Rel.4.8 software.
Brood size and embryonic lethality analysis
For brood size, single late L4 worms were placed in individual plates and transferred to a new plate every 24 hours for a total of 4 days. Viable progeny was counted after 48 hours of each transfer.
The progeny of 8 worms of each genotype was analyzed.
For embryonic lethality, late L4 worms of each genotype were transferred to plates for 24 hours. Gravid adults were then transferred to a new 35mm plate and removed after 2 hours. The eggs laid on each plate were counted immediately after removing the parent and the viable progeny was quantified 48 hours later. Embryonic lethality represents the percentage of unhatched eggs. The progeny of 8 worms of each genotype was analyzed.
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-seq data processing and analysis:
Bowtie2 version 2.4.2 was used to align 75 bp single-end reads to WS220 with default parameters (Langmead and Salzberg, 2012). Bam sorting and indexing was performed using samtools version 1.11 (Danecek et al., 2021). BamCompare tool in Deeptools version 3.5.0 was used to normalize for the sequencing depth using CPM and create ChIP-Input coverage and ChIP/Input ratios with a bin size of 10 bp and 200 bp read extension (Ramírez et al., 2016). Only reads with a minimum mapping quality of 20 were used, and mitochondrial DNA, PCR duplicates, and blacklisted regions were removed (Amemiya et al., 2019). The average coverage data was generated by averaging ChIP-Input enrichment scores per 10 bp bins across the genome. Heatmaps and average-profile plots of ChIP-seq scores across different annotations were produced using Deeptools in Galaxy (doi:10.1093/nar/gkw343). The transcription start site annotations used are from GRO-seq experiments (Kruesi et al., 2013). Average score over the regions was calculated using 50 bp non-overlapping bins. In figure 5C, 6B, C and D, genes were scaled to 1000 bp.
For alignments and sliding window analysis: Input subtracted ChIP signal across X-chromosome was binned into 100 bp resolution. The signal across the X-chromosome is normalized to unity. Each bin on X-chromosome was assigned to the closest strong rex site defined in (Albritton et al., 2017). Then, the normalized signal was plotted as a function of linear distance away from rex sites with sliding window size of 100 kb and step size of 10 kb, where the main line indicates the average and the wider range indicates the 95% confidence interval of the window. The code is available at: https://github.com/ercanlab/2022_Morao_et_al
For motif analysis: MEME (version 5.3.3) (Machanick and Bailey, 2011) was used to find DNA sequence motifs enriched at TOP-2 binding sites. 150 bp sequences centered on TOP-2 ChIP summits were analyzed using the parameters: minimum motif size 6 bp; maximum motif size 12 bp; expected motif occurrence set to any number of occurrences and other parameters set to default. The position weight matrix (pwm) of the top enriched motif was saved from the MEME output and used in PWMScan (Ambrosini et al., 2018) to scan the C. elegans ce10 genome for genome wide occurrences of the motif. The p-value cut-off was set to 1e-5, and other parameters were set as default. The hits obtained with PWMScan were used in R to get the sequence motif chromosomal distribution.
For GC content analysis: The GC content of 250-bp sequences centered around the TSS was obtained using bedtools (version 2.27.1) NucBed function with the C. elegans ce10 genome and default parameters (Quinlan and Hall, 2010). The TOP-2 ChIP binding for each region was obtained using deeptools (version 3.3.1) multiBigWigSummary function with the ‘BED-file’ option and other parameters as default.
RNA-seq data processing and analysis:
We aligned reads to the WS220 genome version using HISAT2 version 2.2.1 (Kim et al., 2019) with the parameter --rna-strandness R. Count data was calculated using HTSeq version 0.13.5 (Anders et al., 2015). The raw counts were normalized FPKM using cufflinks version 2.2.1 (Roberts et al., 2011), and then FPKM was converted to TPM. The raw counts were used for the R package DESeq2 version 1.30.0 (Love et al., 2014). Violin and box plots were produced in R using ggplot2 (http://ggplot2.org). Outliers were not plotted.
HiC data processing and analysis:
Hi-C data analysis: The Hi-C data was mapped to ce10 (WS220) reference genome using default parameters of the Juicer pipeline version 1.5.7 (Durand et al., 2016). The biological replicates were combined using juicer’s mega.sh script. The mapping statistics from the inter_30.txt output file are provided in Supplementary Table 4. The inter_30.hic outputs were converted to cool format using the hicConvertFormat of HiCExplorer version 3.6 (Ramírez et al., 2018; Wolff et al., 2020, 2018) in two steps using the following parameters: 1) --inputFormat hic, --outputFormat cool, 2) -- inputFormat cool --outputFormat cool --load_raw_values. The cool file was balanced using cooler version 0.8.11 using the parameters: --max-iters 500, --mad-max 5, --ignore-diags 2 (Abdennur and Mirny, 2020). The balanced cool file was used for all downstream analysis. For computing insulation score, hicexplorer hicFindTADs function was used with the following parameters: -m 10kb-binned.cool, --correctForMultipleTesting fdr, --minDepth 80000, --maxDepth 200000, --step 40000. The output bedgraph file was converted to a bigwig file using bedGraphToBigwig (Kent et al., 2010), which was used for chromosome-wide visualization using hicPlotTADs and later for on-diagonal pile-up analysis. For off-diagonal rex-rex meta-dot plot, hicexplorer hicAggregateContacts function was used with parameters: -m 5kb-binned cool, --range 100000:3000000, --operationType mean, -- mode intra-chr, --transform obs/exp, --plotType 3d, --vMin 0.5 --vMax 3, --chromosomes X. For computing log-binned P(s), its log-derivative, and on-diagonal pile-up analysis at rex sites, cooltools version 0.4.0 (https://github.com/open2c/cooltools) was used. For visualizing ChIP-seq data with Hi-C data in python, pyBigwig version 0.3.18 (https://github.com/deeptools/pyBigWig) was used. For both on and off diagonal pile-up analysis, the 17 strong rex sites defined in (Albritton et al., 2017) were used. The jupyter notebook for computational workflow is publicly available https://github.com/ercanlab/2022_Morao_et_al
Supplementary Material
Table S1. List of the primers used in this study, related to STAR methods section: worm strains and growth
Table S2. ChIP-seq data generated in this study, related to STAR method section: ChIP-seq
Table S3. mRNA-seq data generated in this study, related to STAR method section: mRNA-seq
Table S4. Hi-C data generated in this study, related to STAR method section: Hi-C
Table S5. Sanger sequencing results for strain ERC84 (top-1::degron::GFP), related to STAR methods section: worm strains and growth
Table S6. Sanger sequencing results for strain ERC83 (top-2::degron::GFP), related to STAR methods section: worm strains and growth
Table S7. Sanger sequencing results for strain ERC82 (dpy-27::degron::GFP), related to STAR methods section: worm strains and growth
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP, rabbit polyclonal | Abcam | Cat# ab290 |
| Anti-RNA Pol II, mouse monoclonal | Abcam | Cat# ab817 |
| Anti-RNA Pol II, mouse monoclonal | Millipore | Cat# 05–952-I-100UG |
| Anti-TOP-2, rabbit polyclonal | Ladouceur et al, 2017 | N/A |
| Anti-IgG, rabbit polyclonal | Abcam | Cat# ab46540 |
| Anti-DPY-27, rabbit polyclonal | Covance Research Products Inc | Cat# JL00001_DPY27, RRID: AB_2616039 |
| Bacterial and virus strains | ||
| E. coli OP50 | Caenorhabditis Genetics Center | OP50 |
| E. coli HB101 | Caenorhabditis Genetics Center | HB101 |
| Chemicals, peptides, and recombinant proteins | ||
| Etoposide | Sigma Aldrich | Cat# E1383 |
| Protein A Sepharose beads | Thermo Fisher Scientific | Cat# 17–5280-01 |
| Protein G Sepharose beads | Thermo Fisher Scientific | Cat# 45–000-116 |
| Sera-Mag* Oligo(dT) Magnetic Particles | Thermo Fisher Scientific | Cat# 3815–2103-010150 |
| 10x Fragmentation Buffer & stop solution | Ambion | Cat# AM8740 |
| Superscript III First strand synthesis system | Invitrogen | Cat# 18080–051 |
| Alt-R S.p. Cas9 Nuclease 3NLS | IDT | Cat# 93007249 |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | Cat# 93007209 |
| Alt-R CRISPR-Cas9 crRNA | IDT | Cat# 93014091 |
| indole-3-acetic-acid | Thermo Fisher Scientific | Cat# Thermo Fisher Scientific |
| protease inhibitors | Calbiochem | Cat# 539131 |
| Bio-Rad Protein Assay Dye Reagent Concentrate | Biorad | Cat# 500–0006 |
| Trizol | Life technologies | Cat# 15596018 |
| Formaldehyde | Sigma | Cat# 252549 |
| Critical commercial assays | ||
| Arima Hi-C kit | Arima genomics | N/A |
| Deposited data | ||
| ChIP-seq, RNA-seq and Hi-C raw and analyzed data. Accession numbers can be found in supplementary tables 2, 3 and 4 | This paper | GSE188851 |
| Original code | This paper | https://doi.org/10.5281/zenodo.7125333 |
| Imaging data | This paper | https://data.mendeley.com/datasets/bjgtrsp2t9/1 |
| Experimental models: Organisms/strains | ||
| C.elegans ERC82 ers54[dpy-27::degron::GFP] III; ieSi57 [eft-3p::TIR1::mRuby::unc-54 3’UTR + Cbr-unc-119(+)] II | This paper | AM01 dpy-27::degron ::GFP |
| C.elegans ERC83 ers55[top-2::degron::GFP] II; ieSi57 [eft-3p::TIR1::mRuby::unc-54 3’UTR + Cbr-unc-119(+)] II | This paper | SS01A top-2::degron ::GFP |
| C.elegans ERC84 ers56[top-1::degron::GFP] I; ieSi57 [eft-3p::TIR1::mRuby::unc-54 3’UTR + Cbr-unc-119(+)] II | This paper | AM05 top-1::degron ::GFP |
| C.elegans ERC69 ersIs33[X:11093924–11094281[rex-8], X:14373128] | Jimenez et al, 2021 | LS05 rex- 8 insertion |
| C.elegans ERC38 ers30[delX:17544437–17544484, delX:17545624–17545624] | Albritton et al, 2017 | SEA04 rex-41 depletion |
| C. elegans JK1107 glp-1(q224) III | Caenorhabditis Genetics Center | glp-1 mutant |
| C. elegans MDX53 top-2::sfGFP-3xFLAG; mCherry::H2B | Paul Maddox lab | MDX53 |
| C.elegans N2: WT strain from Bristol | Caenorhabditis Genetics Center | N2 |
| C. elegans CA1200 ieSi57 [eft-3p::TIR1::mRuby::unc-54 3’UTR + Cbr-unc-119(+)] II | Caenorhabditis Genetics Center | CA1200 |
| Oligonucleotides | ||
| See Supplementary table 1 | This paper | N/A |
| Recombinant DNA | ||
| pLZ29 | Dernburg lab | RRID:Addgene_71719 |
| pCFJ90 | Frøkjær-Jensen et al, 2008 | RRID:Addgene_19327 |
| Software and algorithms | ||
| bowtie2 (2.4.2) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| samtools (1.11) | Danecek et al., 2021 | http://www.htslib.org/ |
| deeptools (3.5.0) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/index.html |
| MEME (5.3.3) | Machanick and Bailey, 2011 | https://meme-suite.org/meme/ |
| PWMScan | Ambrosini et al., 2018 | https://ccg.epfl.ch/pwmtools/pwmscan.php |
| bedtools (2.27.1) | Quinlan et al., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| HISAT2 (2.2.1) | Kim et al., 2019 | http://daehwankimlab.github.io/hisat2/ |
| Htseq (0.13.5) | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| cufflinks (2.2.1) | Roberts et al., 2011 | http://cole-trapnell-lab.github.io/cufflinks/ |
| DEseq2 (1.30.0) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| juicer (1.5.7) | Durand et al., 2016 | https://github.com/aidenlab/juicer |
| HiCexplorer (3.6) | Ramírez et al., 2018; Wolff et al., 2020, 2018 | https://hicexplorer.readthedocs.io/en/latest/index.html |
| bedGraphToBigwig | Kent et al., 2010 | https://www.encodeproject.org/software/bedgraphtobigwig/ |
| cooler (0.8.11) | Abdennur, N., and Mirny, L., 2020 | https://github.com/open2c/cooler |
| cooltools (0.4.0) | https://doi.org/10.5281/zenodo.6324229 | https://cooltools.readthedocs.io/en/latest/ |
| pyBigwig (0.3.18) | https://doi.org/10.5281/zenodo.45238 | https://github.com/deeptools/pyBigWig |
| Other | ||
Highlights.
Auxin inducible degradation of topoisomerases I and II in C. elegans
TOP-2 is required for long-range processivity of condensin DC loop extrusion
TOP-1 is required for translocation of condensin DC across transcribed genes
TOP-1 and TOP-2 contribute to condensin DC mediated gene repression
Acknowledgements
AKM, SE, DO and research in this manuscript were supported by NIGMS of the National Institutes of Health under award numbers R01 GM107293 and R35 GM130311. Jun Kim was partially supported by NIGMS Predoctoral Fellowship T32HD007520. We thank Paul Maddox for the TOP-2 antibody and the top-2::sfGFP strain. We thank Gencore at the NYU Center for Genomics and Systems Biology for sequencing and raw data processing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
References:
- Abdennur N, Mirny LA, 2020. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316. 10.1093/bioinformatics/btz540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albritton SE, Kranz A-L, Winterkorn LH, Street LA, Ercan S, 2017. Cooperation between a hierarchical set of recruitment sites targets the X chromosome for dosage compensation. Elife 6, e23645. 10.7554/eLife.23645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Groux R, Bucher P, 2018. PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics 34, 2483–2484. 10.1093/bioinformatics/bty127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, Boyle AP, 2019. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. 10.1038/s41598-019-45839-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W, 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EC, Frankino PA, Higuchi-Sanabria R, Yang Q, Bian Q, Podshivalova K, Shin A, Kenyon C, Dillin A, Meyer BJ, 2019. X Chromosome Domain Architecture Regulates Caenorhabditis elegans Lifespan but Not Dosage Compensation. Dev Cell 51, 192–207.e6. 10.1016/j.devcel.2019.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda PS, LaJoie DM, Jorcyk CL, 2012. Bleach Gel: A Simple Agarose Gel for Analyzing RNA Quality. Electrophoresis 33, 366–369. 10.1002/elps.201100335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banigan EJ, Mirny LA, 2020. Loop extrusion: theory meets single-molecule experiments. Current Opinion in Cell Biology, Cell Nucleus 64, 124–138. 10.1016/j.ceb.2020.04.011 [DOI] [PubMed] [Google Scholar]
- Baranello L, Kouzine F, Levens D, 2013. DNA Topoisomerases. Transcription 4, 232–237. 10.4161/trns.26598 [DOI] [PubMed] [Google Scholar]
- Baxter J, Sen N, Martínez VL, Carandini MEMD, Schvartzman JB, Diffley JFX, Aragón L, 2011. Positive Supercoiling of Mitotic DNA Drives Decatenation by Topoisomerase II in Eukaryotes. Science 331, 1328–1332. 10.1126/science.1201538 [DOI] [PubMed] [Google Scholar]
- Brandão HB, Paul P, Berg A.A. van den Rudner DZ, Wang X, Mirny LA, 2019. RNA polymerases as moving barriers to condensin loop extrusion. PNAS 116, 20489–20499. 10.1073/pnas.1907009116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, Peters JM, 2017. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507. 10.1038/nature22063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Maman Y, Huang SN, Wutz G, Tang W, Zagnoli-Vieira G, Callen E, Wong N, Day A, Peters J-M, Caldecott KW, Pommier Y, Nussenzweig A, 2019. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Molecular Cell 75, 252–266.e8. 10.1016/j.molcel.2019.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon K-R, Pekowska A, Zhang H, Rao SSP, Huang S-C, Mckinnon PJ, Aplan PD, Pommier Y, Aiden EL, Casellas R, Nussenzweig A, 2017. Genome Organization Drives Chromosome Fragility. Cell 170, 507–521.e18. 10.1016/j.cell.2017.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbin A, Bouchoux C, Uhlmann F, 2014. Condensin aids sister chromatid decatenation by topoisomerase II. Nucleic Acids Research 42, 340–348. 10.1093/nar/gkt882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ, 2015. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. 10.1038/nature14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H, 2021. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed]
- Davidson IF, Goetz D, Zaczek MP, Molodtsov MI, Huis In ‘t Veld PJ, Weissmann F, Litos G, Cisneros DA, Ocampo-Hafalla M, Ladurner R, Uhlmann F, Vaziri A, Peters J-M, 2016. Rapid movement and transcriptional re-localization of human cohesin on DNA. EMBO J 35, 2671–2685. 10.15252/embj.201695402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokshin GA, Ghanta KS, Piscopo KM, Mello CC, 2018. Robust Genome Editing with Short Single-Stranded and Long, Partially Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics 210, 781–787. 10.1534/genetics.118.301532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL, 2016. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98. 10.1016/j.cels.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Dubief M, Persson J, Norman U, Hartsuiker E, Ekwall K, 2010. Topoisomerase I regulates open chromatin and controls gene expression in vivo. The EMBO Journal 29, 2126–2134. 10.1038/emboj.2010.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson S, Segura J, Martínez-García B, Valdés A, Roca J, 2020. Condensin minimizes topoisomerase II-mediated entanglements of DNA in vivo. The EMBO Journal n/a, e105393. 10.15252/embj.2020105393 [DOI] [PMC free article] [PubMed]
- Ercan S, 2015. Mechanisms of x chromosome dosage compensation. J Genomics 3, 1–19. 10.7150/jgen.10404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan S, Dick LL, Lieb JD, 2009. The C. elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Curr Biol 19, 1777–1787. 10.1016/j.cub.2009.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, Lieb JD, 2007. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nat Genet 39, 403–408. 10.1038/ng1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan S, Lubling Y, Segal E, Lieb JD, 2011. High nucleosome occupancy is encoded at X-linked gene promoters in C. elegans. Genome Res 21, 237–244. 10.1101/gr.115931.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Wayne Davis M, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, Jorgensen EM, 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40, 1375–1383. 10.1038/ng.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, Mirny LA, 2017. Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb Symp Quant Biol 82, 45–55. 10.1101/sqb.2017.82.034710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, Dekker C, 2018. Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105. 10.1126/science.aar7831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassler J, Brandão HB, Imakaev M, Flyamer IM, Ladstätter S, Bickmore WA, Peters J, Mirny LA, Tachibana K, 2017. A mechanism of cohesin‐dependent loop extrusion organizes zygotic genome architecture. EMBO J 36, 3600–3618. 10.15252/embj.201798083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golfier S, Quail T, Kimura H, Brugués J, 2020. Cohesin and condensin extrude DNA loops in a cell cycle-dependent manner. eLife 9, e53885. 10.7554/eLife.53885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloborodko A, Imakaev MV, Marko JF, Mirny L, 2016. Compaction and segregation of sister chromatids via active loop extrusion. Elife 5, e14864. 10.7554/eLife.14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothe HJ, Bouwman BAM, Gusmao EG, Piccinno R, Petrosino G, Sayols S, Drechsel O, Minneker V, Josipovic N, Mizi A, Nielsen CF, Wagner E-M, Takeda S, Sasanuma H, Hudson DF, Kindler T, Baranello L, Papantonis A, Crosetto N, Roukos V, 2019. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Molecular Cell 75, 267–283.e12. 10.1016/j.molcel.2019.05.015 [DOI] [PubMed] [Google Scholar]
- Heldrich J, Sun X, Vale-Silva LA, Markowitz TE, Hochwagen A, 2020. Topoisomerases Modulate the Timing of Meiotic DNA Breakage and Chromosome Morphogenesis in Saccharomyces cerevisiae. Genetics 215, 59–73. 10.1534/genetics.120.303060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, Fonarev P, Grant BD, Priess JR, 2005. Genetic Analysis of Lysosomal Trafficking in Caenorhabditis elegans. MBoC 16, 3273–3288. 10.1091/mbc.e05-01-0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jans J, Gladden JM, Ralston EJ, Pickle CS, Michel AH, Pferdehirt RR, Eisen MB, Meyer BJ, 2009. A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes Dev 23, 602–618. 10.1101/gad.1751109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Lambert A, Fabritius AS, Hansen TJ, Smith HE, Golden A, 2016. The Identification of a Novel Mutant Allele of topoisomerase II in Caenorhabditis elegans Reveals a Unique Role in Chromosome Segregation During Spermatogenesis. Genetics 204, 1407–1422. 10.1534/genetics.116.195099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez DS, Kim J, Ragipani B, Zhang B, Street LA, Kramer M, Albritton SE, Winterkorn L, Ercan S, 2021. Condensin DC spreads linearly and bidirectionally from recruitment sites to create loop-anchored TADs in C. elegans 10.1101/2021.03.23.436694 [DOI] [PMC free article] [PubMed]
- Joshi RS, Piña B, Roca J, 2012. Topoisomerase II is required for the production of long Pol II gene transcripts in yeast. Nucleic Acids Res 40, 7907–7915. 10.1093/nar/gks626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D, 2010. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. 10.1093/bioinformatics/btq351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL, 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim Y-C, Lee JH, Jang YJ, Chung IK, Koo H-S, 1996. cDNA Cloning, Expression, and Chromosomal Localization of Caenorhabditis elegans DNA Topoisomerase I. European Journal of Biochemistry 237, 367–372. 10.1111/j.1432-1033.1996.00367.x [DOI] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang H-S, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, Chamberlain SJ, Philpot BD, Zylka MJ, 2013. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62. 10.1038/nature12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita K, Hirano T, 2017. Dynamic organization of mitotic chromosomes. Current Opinion in Cell Biology, Cell Nucleus 46, 46–53. 10.1016/j.ceb.2017.01.006 [DOI] [PubMed] [Google Scholar]
- Kong M, Cutts EE, Pan D, Beuron F, Kaliyappan T, Xue C, Morris EP, Musacchio A, Vannini A, Greene EC, 2020. Human Condensin I and II Drive Extensive ATP-Dependent Compaction of Nucleosome-Bound DNA. Molecular Cell 79, 99–114.e9. 10.1016/j.molcel.2020.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Gupta A, Baranello L, Wojtowicz D, Ben-Aissa K, Liu J, Przytycka TM, Levens D, 2013. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat Struct Mol Biol 20, 396–403. 10.1038/nsmb.2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer M, Kranz A-L, Su A, Winterkorn LH, Albritton SE, Ercan S, 2015. Developmental Dynamics of X-Chromosome Dosage Compensation by the DCC and H4K20me1 in C. elegans. PLOS Genetics 11, e1005698. 10.1371/journal.pgen.1005698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ, 2013. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife 2, e00808. 10.7554/eLife.00808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladouceur A-M, Ranjan R, Smith L, Fadero T, Heppert J, Goldstein B, Maddox AS, Maddox PS, 2017. CENP-A and topoisomerase-II antagonistically affect chromosome length. Journal of Cell Biology 216, 2645–2655. 10.1083/jcb.201608084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AC, Nabeshima K, Csankovszki G, 2014. The C. elegans dosage compensation complex mediates interphase X chromosome compaction. Epigenetics Chromatin 7, 31. 10.1186/1756-8935-7-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Jang YJ, Koo H-S, 1998. Alternative splicing in the Caenorhabditis elegans DNA topoisomerase I gene. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression 1396, 207–214. 10.1016/S0167-4781(97)00209-1 [DOI] [PubMed] [Google Scholar]
- Liu LF, Wang JC, 1987. Supercoiling of the DNA template during transcription. PNAS 84, 7024–7027. 10.1073/pnas.84.20.7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL, 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville CM, Smith K, Sondka Z, Rance H, Cockell S, Cowell IG, Lee KC, Morris NJ, Padget K, Jackson GH, Austin CA, 2015. Genome-wide ChIP-seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol Open 4, 1436–1447. 10.1242/bio.014308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonel P, Jans J, Peterson BK, Meyer BJ, 2006. Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature 444, 614–618. 10.1038/nature05338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, Nora EP, 2016. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genom. Hum. Genet 17, 17–43. 10.1146/annurev-genom-083115-022339 [DOI] [PubMed] [Google Scholar]
- Meyer BJ, 2005. X-Chromosome dosage compensation. WormBook 1–14. 10.1895/wormbook.1.8.1 [DOI] [PMC free article] [PubMed]
- Montecucco A, Zanetta F, Biamonti G, 2015. Molecular mechanisms of etoposide. EXCLI J 14, 95–108. 10.17179/excli2015-561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neguembor MV, Martin L, Castells-García Á, Gómez-García PA, Vicario C, Carnevali D, AlHaj Abed J, Granados A, Sebastian-Perez R, Sottile F, Solon J, Wu C, Lakadamyali M, Cosma MP, 2021. Transcription-mediated supercoiling regulates genome folding and loop formation. Molecular Cell 10.1016/j.molcel.2021.06.009 [DOI] [PMC free article] [PubMed]
- Nicolas E, Upton AL, Uphoff S, Henry O, Badrinarayanan A, Sherratt D, 2014. The SMC Complex MukBEF Recruits Topoisomerase IV to the Origin of Replication Region in Live Escherichia coli. mBio 5. 10.1128/mBio.01001-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL, 2009. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9, 327–337. 10.1038/nrc2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, Mirny LA, 2018. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc Natl Acad Sci U S A 115, E6697–E6706. 10.1073/pnas.1717730115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandini E, Marenduzzo D, Michieletto D, 2019. Synergy of topoisomerase and structural-maintenance-of-chromosomes proteins creates a universal pathway to simplify genome topology. Proc Natl Acad Sci U S A 116, 8149–8154. 10.1073/pnas.1815394116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A, 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37, e123. 10.1093/nar/gkp596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskadlo E, Oliveira RA, 2017. A Topology-Centric View on Mitotic Chromosome Architecture. International Journal of Molecular Sciences 18, 2751. 10.3390/ijms18122751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskadlo E, Tavares A, Oliveira RA, 2017. Metaphase chromosome structure is dynamically maintained by condensin I-directed DNA (de)catenation. eLife 6, e26120. 10.7554/eLife.26120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, Nitiss JL, 2016. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17, 703–721. 10.1038/nrm.2016.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan B, Kanno T, Igarashi MU, Baaske MD, Wong JSK, Jeppsson K, Björkegren C, Kim E, 2022. The Smc5/6 complex is a DNA loop extruding motor 10.1101/2022.05.13.491800 [DOI] [PMC free article] [PubMed]
- Quinlan AR, Hall IM, 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racko D, Benedetti F, Goundaroulis D, Stasiak A, 2018. Chromatin Loop Extrusion and Chromatin Unknotting. Polymers (Basel) 10. 10.3390/polym10101126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T, 2018. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9, 189. 10.1038/s41467-017-02525-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T, 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L, 2011. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol 12, R22. 10.1186/gb-2011-12-3-r22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusková R, Račko D, 2021. Entropic Competition between Supercoiled and Torsionally Relaxed Chromatin Fibers Drives Loop Extrusion through Pseudo-Topologically Bound Cohesin. Biology 10, 130. 10.3390/biology10020130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Leonard J, Torres R, Garcia-Luis J, Palou-Marin G, Aragón L, 2016. Physical Proximity of Sister Chromatids Promotes Top2-Dependent Intertwining. Molecular Cell 64, 134–147. 10.1016/j.molcel.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling AS, Jeong KS, Kitada T, Grunstein M, 2011. Topoisomerase II binds nucleosome-free DNA and acts redundantly with topoisomerase I to enhance recruitment of RNA Pol II in budding yeast. Proc Natl Acad Sci U S A 108, 12693–12698. 10.1073/pnas.1106834108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street LA, Morao AK, Winterkorn LH, Jiao C-Y, Albritton SE, Sadic M, Kramer M, Ercan S, 2019. Binding of an X-Specific Condensin Correlates with a Reduction in Active Histone Modifications at Gene Regulatory Elements. Genetics 212, 729–742. 10.1534/genetics.119.302254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Kelly WG, Ercan S, Lieb JD, 2014. Regulation of the X Chromosomes in Caenorhabditis elegans. Cold Spring Harb Perspect Biol 6, a018366. 10.1101/cshperspect.a018366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR, 1977. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Developmental Biology 56, 110–156. 10.1016/0012-1606(77)90158-0 [DOI] [PubMed] [Google Scholar]
- Sutani T, Sakata T, Nakato R, Masuda K, Ishibashi M, Yamashita D, Suzuki Y, Hirano T, Bando M, Shirahige K, 2015. Condensin targets and reduces unwound DNA structures associated with transcription in mitotic chromosome condensation. Nat Commun 6, 7815. 10.1038/ncomms8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, Henikoff S, 2014. Transcription-generated torsional stress destabilizes nucleosomes. Nat Struct Mol Biol 21, 88–94. 10.1038/nsmb.2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NT, Laub MT, Le TBK, 2017. SMC Progressively Aligns Chromosomal Arms in Caulobacter crescentus but Is Antagonized by Convergent Transcription. Cell Reports 20, 2057–2071. 10.1016/j.celrep.2017.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F, 2016. SMC complexes: from DNA to chromosomes. Nature Reviews Molecular Cell Biology 17, 399–412. 10.1038/nrm.2016.30 [DOI] [PubMed] [Google Scholar]
- Uusküla-Reimand L, Hou H, Samavarchi-Tehrani P, Rudan MV, Liang M, Medina-Rivera A, Mohammed H, Schmidt D, Schwalie P, Young EJ, Reimand J, Hadjur S, Gingras A-C, Wilson MD, 2016. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol 17, 182. 10.1186/s13059-016-1043-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés A, Coronel L, Martínez-García B, Segura J, Dyson S, Díaz-Ingelmo O, Micheletti C, Roca J, 2019. Transcriptional supercoiling boosts topoisomerase II-mediated knotting of intracellular DNA. Nucleic Acids Research 47, 6946–6955. 10.1093/nar/gkz491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Pękowska A, Rao SSP, Kieffer-Kwon K-R, Jung S, Baranello L, Huang S-C, El Khattabi L, Dose M, Pruett N, Sanborn AL, Canela A, Maman Y, Oksanen A, Resch W, Li X, Lee B, Kovalchuk AL, Tang Z, Nelson S, Di Pierro M, Cheng RR, Machol I, St Hilaire BG, Durand NC, Shamim MS, Stamenova EK, Onuchic JN, Ruan Y, Nussenzweig A, Levens D, Aiden EL, Casellas R, 2018. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178.e20. 10.1016/j.cell.2018.03.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J, Bhardwaj V, Nothjunge S, Richard G, Renschler G, Gilsbach R, Manke T, Backofen R, Ramírez F, Grüning BA, 2018. Galaxy HiCExplorer: a web server for reproducible Hi-C data analysis, quality control and visualization. Nucleic Acids Res 46, W11–W16. 10.1093/nar/gky504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J, Rabbani L, Gilsbach R, Richard G, Manke T, Backofen R, Grüning BA, 2020. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res 48, W177–W184. 10.1093/nar/gkaa220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ward JD, Cheng Z, Dernburg AF, 2015. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 142, 4374–4384. 10.1242/dev.129635 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the primers used in this study, related to STAR methods section: worm strains and growth
Table S2. ChIP-seq data generated in this study, related to STAR method section: ChIP-seq
Table S3. mRNA-seq data generated in this study, related to STAR method section: mRNA-seq
Table S4. Hi-C data generated in this study, related to STAR method section: Hi-C
Table S5. Sanger sequencing results for strain ERC84 (top-1::degron::GFP), related to STAR methods section: worm strains and growth
Table S6. Sanger sequencing results for strain ERC83 (top-2::degron::GFP), related to STAR methods section: worm strains and growth
Table S7. Sanger sequencing results for strain ERC82 (dpy-27::degron::GFP), related to STAR methods section: worm strains and growth
Data Availability Statement
Genomic data is available at Gene Expression Omnibus (GEO) series numbers GSE188851. Accession numbers of the data sets generated in this study are listed in Supplemental table 2,3 and 4.
All original code is available at https://doi.org/10.5281/zenodo.7125333 and in the supplementary information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.