

Abstract
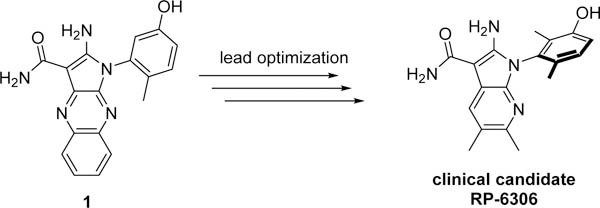
PKMYT1 is a regulator of CDK1 phosphorylation and is a compelling therapeutic target for the treatment of certain types of DNA damage response cancers due to its established synthetic lethal relationship with CCNE1 amplification. To date, no selective inhibitors have been reported for this kinase that would allow for investigation of the pharmacological role of PKMYT1. To address this need compound 1 was identified as a weak PKMYT1 inhibitor. Introduction of a dimethylphenol increased potency on PKMYT1. These dimethylphenol analogs were found to exist as atropisomers that could be separated and profiled as single enantiomers. Structure-based drug design enabled optimization of cell-based potency. Parallel optimization of ADME properties led to the identification of potent and selective inhibitors of PKMYT1. RP-6306 inhibits CCNE1-amplified tumor cell growth in several preclinical xenograft models. The first-in-class clinical candidate RP-6306 is currently being evaluated in Phase 1 clinical trials for treatment of various solid tumors.
Keywords: PKMYT1, CCNE1 amplification, RP-6306, Atropisomer
Graphical Abstract
INTRODUCTION
Cyclins are primary regulators of cellular growth and interact with cyclin-dependent kinases (CDKs) to initiate events required for cell cycle progression. The CCNE1 locus encodes the protein cyclin E1, which complexes with cyclin-dependent kinase 2 (CDK2) and drives cells from G1 phase to S phase.1 In cancer, amplification of the CCNE1 gene and/or deregulation of cyclin E1 frequently occurs at an early stage of tumorigenesis and forces cancer cells into S phase prematurely. Excessive replication, origin firing, and inadequate pools of nucleotides cause replication fork stalling, leading to replication stress and DNA damage.2 In the absence of functional p53, this causes genomic instability as cells move into mitosis with damaged DNA. Protein Kinase, Membrane Associated Tyrosine/Threonine 1 (PKMYT1), a member of the WEE family of serine/threonine-kinases, phosphorylates threonine 14 (Thr14) of cyclin-dependent Kinase 1 (CDK1), which inhibits its ability (when complexed with cyclin B) to trigger mitosis.3 In contrast, WEE1 phosphorylates tyrosine 15 (Tyr15) of CDK1 and is implicated in the regulation of both CDK1 and CDK2 activity.4 PKMYT1 function does not appear to be critical in the unperturbed cell cycle whereas WEE1 function is essential for cell cycle progression of cells.5 However, the absence of functional PKMYT1 in a genetically-vulnerable tumor, such as with CCNE1-amplification, causes cells to lose major checkpoint regulation leading to hyperactive CDK1, unscheduled mitosis and catastrophic DNA damage, ultimately resulting in cell death.6 No selective inhibitors have been previously reported for PKMYT1 that would allow for the investigation of the pharmacological activity. In this manuscript we report the discovery of the first potent, selective, and orally bioavailable PKMYT1 inhibitor RP-6306.
RESULTS AND DISCUSSION
COMPOUND STRUCTURE-ACTIVITY-RELATIONSHIPS AND OPTIMIZATION
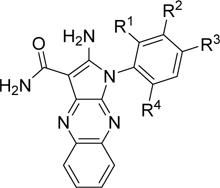
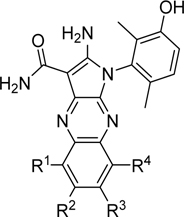
To identify a chemical starting point for a selective PKMYT1 inhibitor, we chose to conduct a focused screen of 560 known kinase inhibitors7, 8, 9 using a fluorescence polarization based competitive displacement assay with Tracer 178 binding probe (Thermofisher PV5593). Among a list of non-selective SRC/ABL inhibitors (dasatinib,11 bosutinib12 and PD-17395513) that scored as potent hits, we were particularly attracted by compound 1 which has previously been disclosed as a non-specific ephrin inhibitor.14,15 This appeared to be an excellent lead structure, particularly in light of the 50-fold selectivity observed over the highly homologous enzyme WEE1 (Table 1). We then began modifications of this structure to understand which elements were important to PKMYT1 inhibition, beginning with the structure-activity relationship (SAR) of the phenol ring. It was quickly determined that the 3-phenol of compound 1 was essential for PKMYT1 inhibition, as both the 6-tolyl derivative 214 and the 4-phenol analog 3 were found to be inactive in our biochemical adenosine diphosphate (ADP) release enzymatic assay (Table 1). Moving the methyl on the phenol ring from the 6-position in compound 1 to the 2-position in analog 4 provided improved potency while the unsubstituted phenol 514 was considerably less potent, suggesting a torsion angle requirement between the phenol ring and the tricyclic system. Addition of a second methyl substituent to compound 1 to give the 2,6-dimethyl phenol 6 further improved the potency suggesting that enforcing a large dihedral angle with the tricyclic ring system was beneficial. Replacement of the phenol of compound 6 with substituents such as methoxy in compound 7 and chloro in compound 8 deleteriously affected potency, highlighting the requirement for the hydrogen bond donor aspect of the phenol. Replacing the phenol by an aniline also yielded an inactive analog 9. These findings inspired a search for a phenol isostere. Indazole16 10 retained some potency, while other hydrogen bond donor analogs such as the indole 11, benzotriazole 12, benzimidazole 13, and difluoromethyl 14 were all inactive. The introduction of a methyl at the para position relative to the hydrogen bond donor of the indazole to give analog 15 provided only a small improvement in potency and confirmed our preference for the phenol. Substitution of each methyl of analog 6 by chloro yielded the three analogs 16, 17, and 18 with similar potency in the enzymatic assay compared to compound 6.
Table 1.
Compound 1 phenol SAR
| ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | PKMYT1 Enzymatic1 IC50 (μM) | WEE1 Enzymatic IC50 (μM) | |
| 1 | H | OH | H | Me | 0.068 | 3.7 |
| 2 | H | H | H | Me | >20 | - |
| 3 | H | H | OH | Me | 8.4 | - |
| 4 | Me | OH | H | H | 0.010 | 2.2 |
| 5 | H | OH | H | H | 0.37 | 19.6 |
| 6 | Me | OH | H | Me | 0.008 | 0.58 |
| 7 | Me | OMe | H | Me | 5.3 | - |
| 8 | Me | Cl | H | Me | >20 | - |
| 9 | H | NH2 | H | Me | >20 | - |
| 10 |
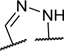
|
H | H | 0.182 | 5.2 | |
| 11 |
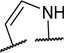
|
H | H | >20 | - | |
| 12 |
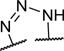
|
H | H | >20 | - | |
| 13 |
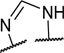
|
H | H | >20 | - | |
| 14 | H | CHF2 | H | Me | >20 | - |
| 15 |
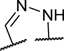
|
H | Me | 0.053 | 3.2 | |
| 16 | Cl | OH | H | Me | 0.005 | 0.94 |
| 17 | Me | OH | H | Cl | 0.010 | 0.23 |
| 18 | Cl | OH | H | Cl | 0.008 | 0.22 |
The enzymatic PKMYT1 assay is a luminescent ADP detection assay where the activity of PKMYT1 is measured by quantifying the amount of ADP produced during an enzymatic reaction in the presence of ATP.10
With the phenol SAR well-defined, we turned our attention to the carboxamide and aminopyrrole groups. These groups are known to form important hydrogen bonds to the hinge region of Ephrin A314 and our attempts at modification suggested that these interactions were similarly important to PKMYT1 (vide infra). Alkyl substitution of the carboxamide NH2 of compound 1 yielded analogs 19–21 with a severe loss in potency (Table 2). In contrast, replacement of the pyrrole NH2 with hydrogen (compound 22) was reasonably well tolerated. Replacement of the pyrrole NH2 with a chlorine (compound 23) resulted in a dramatic loss in potency. Extending our exploratory SAR to the tricyclic ring, each nitrogen of the pyrazine of compound 1 was individually replaced by CH to yield analogs 24 and 25, the latter being around five-fold more potent than the analogous pyrazine analog 1 in the enzymatic assay (Table 2).
Table 2.
Compound 1 carboxamide, pyrrolo-NH2, and pyrazine nitrogens SAR
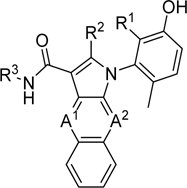
| ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | A1 | A2 | PKMYT1 enzymatic IC50 (μM) | |
| 19 | H | NH2 | Me | N | N | 0.69 |
| 20 | H | NH2 | Et | N | N | 4.1 |
| 21 | H | NH2 | CH2CH2OH | N | N | 1.2 |
| 22 | Me | H | H | N | N | 0.011 |
| 23 | Me | Cl | H | N | N | 2.4 |
| 24 | H | NH2 | H | N | CH | 0.029 |
| 25 | H | NH2 | H | CH | N | 0.012 |
To better evaluate our most potent compounds, a cell-based activity assay was developed to monitor phosphorylation of CDK1 (Thr14), a direct substrate of PKMYT1. This assay confirmed the potency boost associated with the 2,6-dimethyl phenol motif of analog 6.compare to both mono methyl analogs 1 and 4. Interestingly, the chloro analogs 17 and 18 without a methyl at R4 that showed comparable potency in the enzymatic assay (Table 1) were significantly less potent than analog 16 in the cell-based assay despite good cell permeability (analog 17 Caco2 Papp A-B=14.1 x 10−6 cm/s, efflux ratio (ER) = 1.2; analog 18 Caco2 Papp A-B=18.8 x 10−6 cm/s, ER=0.4). 2-Chloro phenol analog 16 showed favorable cell potency compared to 2,6-dimethyl phenol analog 6 but such chloro substitution resulted in notable increase in metabolism in a human hepatocyte assay. Increasing the acidity of a phenol by adding electron withdrawing groups can result in an increase in the glucuronidation rate.17 Despite the attractive enzymatic potency of des-amino analog 22 and reduced TPSA, the cellular activity was markedly reduced relative to the parent compound 6, discouraging us from further pursuit of this chemotype. Consistent with its increased potency in the enzymatic assay, analog 25 was also more potent than the parent compound 1 in the cell-based assay.
Starting from non-specific ephrin inhibitor compound 114,15 we deemed necessary to interrogate the selectivity of key compounds over a representative member of the ephrin family. Promega NanoBRET™ cell-based assays19 were developed for both PKMYT1 and EPHB3 in HEK293T cells. As expected, the initial non-specific ephrin inhibitor 1 showed higher affinity for EPHB3 compared to PKMYT1 (Table 3). We were delighted to observe that this initial selectivity profile was reversed for the 2,6-dimethyl phenol analog 6. We were also pleased to find that removing the nitrogen on the carboxamide side of the tricyclic scaffold had a favorable impact on the PKMYT1 selectivity over EPHB3 (analog 25). The rationales behind the PKMYT1 potency boost observed for the 2,6-dimethyl phenol motif and the removal of the carboxamide-side pyrazine’s nitrogen, which drives the enhanced PKMYT1 selectivity ratios over EPHB4, are discussed in the co-crystal structures section.
Table 3.
Cell-based potency and selectivity of initial analogs.
| Compound | PKMYT1 enzymatic IC50 (μM) | PKMYT1 cell assay2 IC50 (μM) | PKMYT1 nanoBRET IC50 (μM) | EPHB3 nanoBRET IC50 (μM) | EPHB3 selectivity ratio |
|---|---|---|---|---|---|
| 1 | 0.068 | >3 | 0.085 | 0.007 | 0.08 |
| 4 | 0.010 | >3 | - | - | - |
| 6 | 0.008 | 0.13 | 0.001 | 0.018 | 18 |
| 16 | 0.005 | 0.07 | 0.001 | 0.007 | 7 |
| 17 | 0.010 | 0.38 | - | - | - |
| 18 | 0.008 | 0.92 | - | - | - |
| 22 | 0.011 | 0.56 | - | - | - |
| 25 | 0.012 | 0.36 | 0.005 | 0.007 | 1.4 |
The PKMYT1 cell-based activity assay was developed to monitor pCDK1 (Thr14), a direct substrate of PKMYT1, based on the Amplified Luminescent Proximity Homogeneous Assay (Alpha) technology.18 This assay measures the phosphorylation status of the CDK1 Thr14 residue in FUOV1 cells, a high grade ovarian serous adenocarcinoma CCNE1-amplified cell line, which we established having a high level of endogenous pCDK1 (Thr14) that we can exploit for screening purposes.
To explore the SAR of the phenyl ring in the tricyclic scaffold, all four possible bromo-phenyl regioisomers of analog 6 were prepared (compounds 26–29) to enable further diversification of each position by transition metal-mediated transformations. Although all four bromo analogs (26–29) displayed low nanomolar potencies in the enzymatic assay, compound 29 was found to have superior potency in the cell-based activity assay (Table 4). The three most promising bromo regioisomers (26, 28, and 29) were each derivatized to provide representative nitrile, pyrazole and cyclopentene analogs 30–38. All these analogs were very potent in the enzymatic assay, but the differences observed in the cell-based assay suggested that the lower limit of the enzymatic assay had been reached (Table 4).
Table 4.
Potency of substituted phenyl ring analogs.
| ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | PKMYT1 Enzymatic IC50 (μM) | PKMYT1 cell assayIC50 (μM) | |
| 26 | H | H | H | Br | <0.003 | 0.19 |
| 27 | H | H | Br | H | 0.005 | 1.37 |
| 28 | H | Br | H | H | <0.003 | 0.36 |
| 29 | Br | H | H | H | <0.003 | 0.021 |
| 30 | H | H | H | CN | 0.004 | 0.036 |
| 31 | H | H | H |
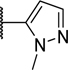
|
<0.003 | 0.12 |
| 32 | H | H | H |
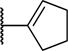
|
<0.003 | 0.55 |
| 33 | H | CN | H | H | 0.005 | 0.20 |
| 34 | H |
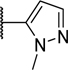
|
H | H | 0.004 | 0.17 |
| 35 | H |
|
H | H | 0.005 | 0.30 |
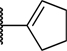
| 36 | CN | H | H | H | 0.005 | 0.034 |
| 37 |
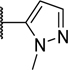
|
H | H | H | 0.006 | 0.32 |
| 38 |
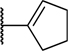
|
H | H | H | 0.008 | 1.05 |
Interestingly, upon supercritical fluid chromatographic (SFC) purification of compound 29 with a chiral stationary phase, two compounds were isolated in equal amounts. It was determined that the single bromo regioisomer 29 existed as two stable atropisomers (Class III),20 the eutomer 39 and the distomer 40 (Figure 1 and Table 6). The stereochemistry of the eutomer 39 was confirmed by X-ray crystallography of this compound bound to PKMYT1 (Figure 3). The thermal stability of compound 39 was investigated by heating aliquots of a DMSO solution at various temperatures for 1 hour, followed by chiral chromatography analysis to detect the potential presence of compound 40 (Table 5). Interconversion of 39 to 40 was not detected when the solution was heated at up to 150 °C indicating a highly stable atropisomer. In contrast, when the mono methyl compound 1 was separated into eutomer 41 and distomer 42, the interconversion of 41 to 42 took place between 50 °C and 70 °C, and a racemic mixture was observed after 1 hour at 120 °C. A similar thermal stability profile was observed for the des-amino analog 22 where the eutomer 43 and distomer 44 were found to fully racemize after 1 hour at 150 °C. The rotational stability of the dimethyl phenol linked to this aminopyrrole ring system allowed for the isolation and further characterization of each atropisomer as distinct compounds. Each atropisomer were isolated from SFC and the subsequent characterization was conducted on the pure enantiomers that showed inhibition of PKMYT1.
Figure 1.
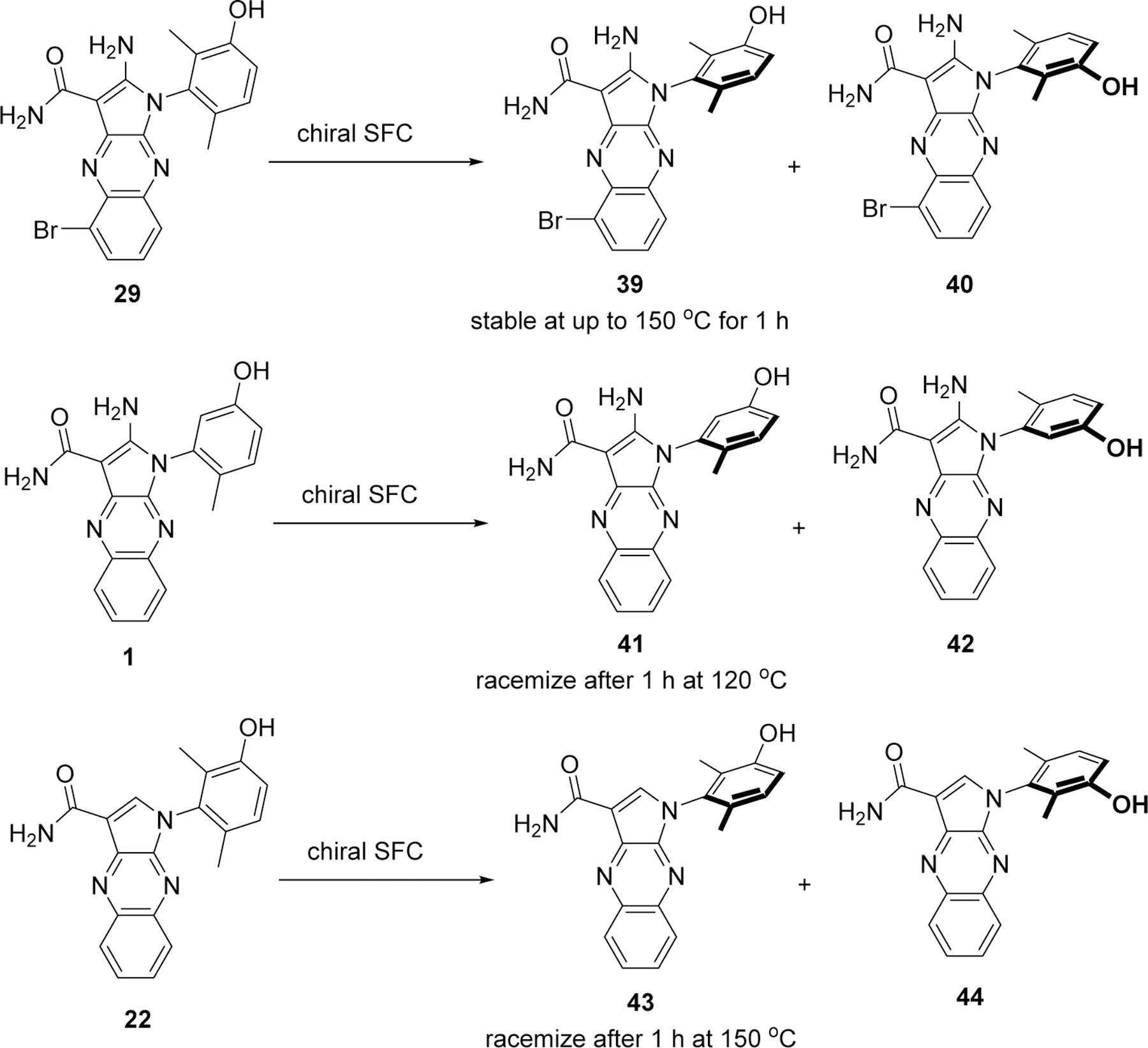
Separation of representative atropisomers.
Table 6.
Potency and selectivity of bicyclic pyrrolopyrazine analogs
| |||||
|---|---|---|---|---|---|
| R1 | R2 | PKMYT1 cell assay IC50 (μM) | PKMYT1 nanoBRET IC50 (μM) | EPHB3 selectivity ratio | |
| 39 | n.a. | n.a. | 0.020 | 0.009 | 3.3 |
| 45 | H | H | 0.626 | 0.012 | 168 |
| 46 | Me | Me | 0.073 | 0.005 | 31 |
| 47 | H | Me | 0.488 | - | - |
| 48 | Me | H | 0.108 | - | - |
| 49 | cPr | H | 0.042 | 0.001 | 15 |
| 50 | tBuOH | H | 0.026 | 0.023 | 2.2 |
| 51 |
|
H | 0.025 | - | - |
| 52 |
|
H | 0.050 | - | - |
| 53 |
|
H | 0.034 | - | - |
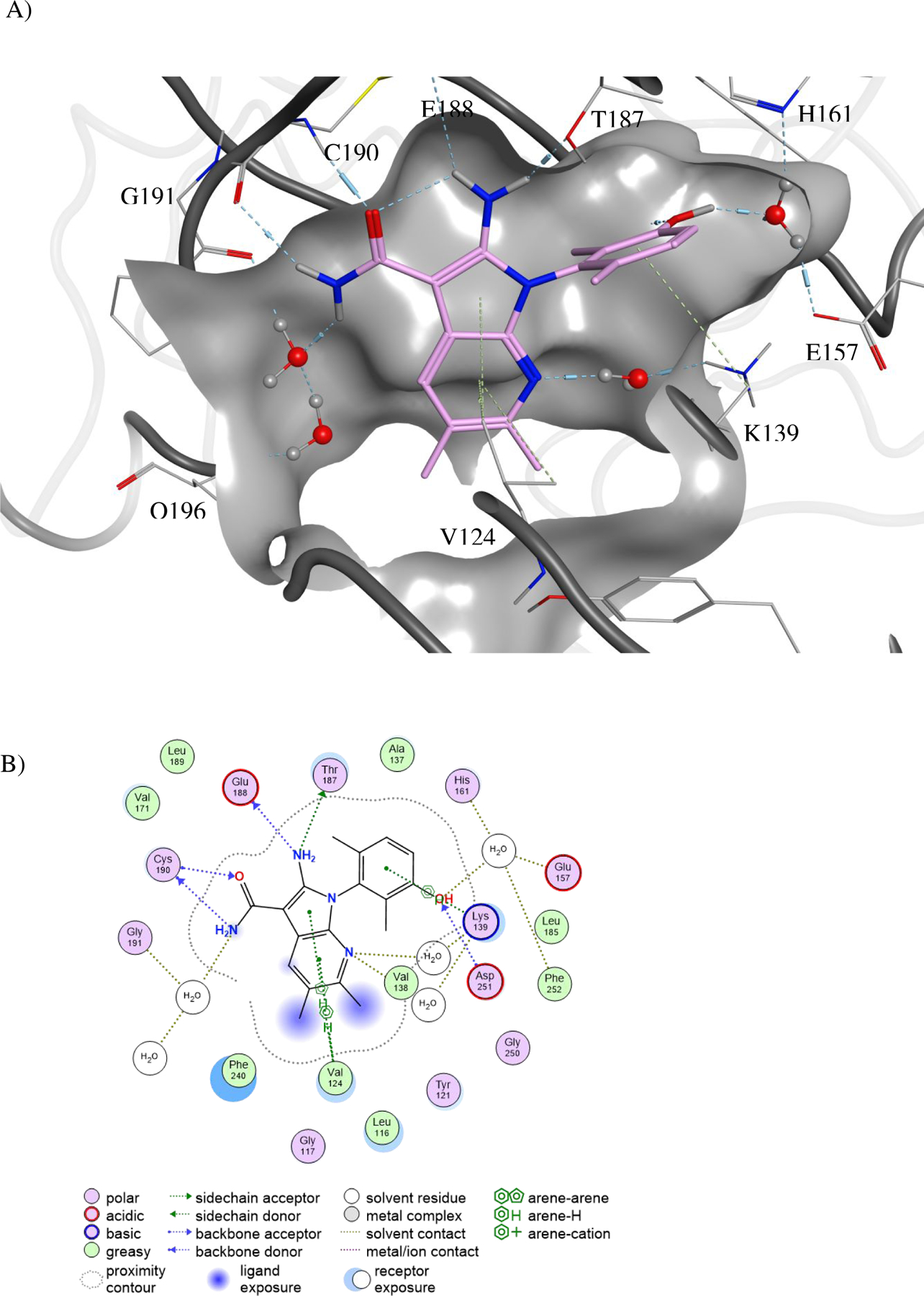
Figure 3.
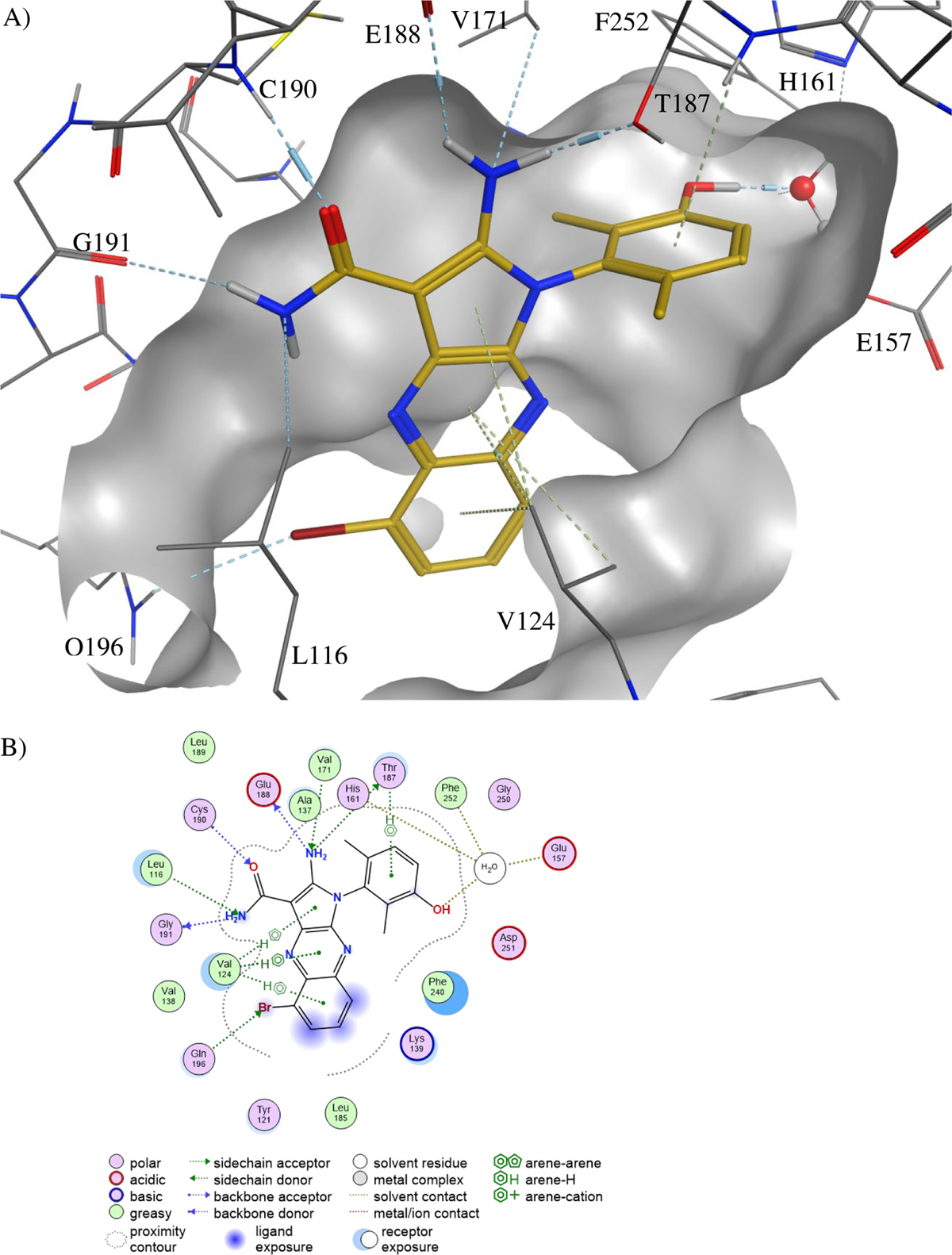
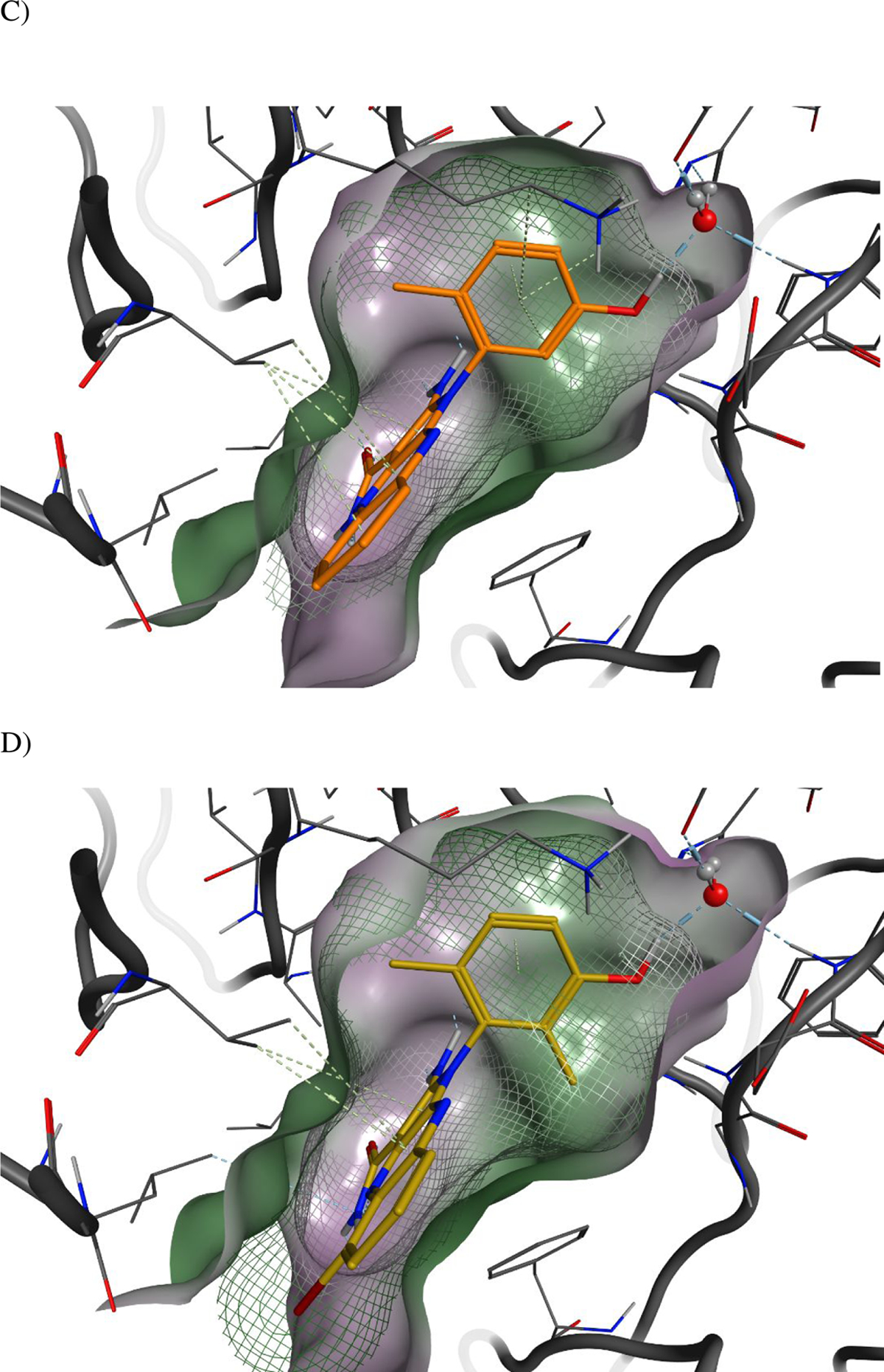
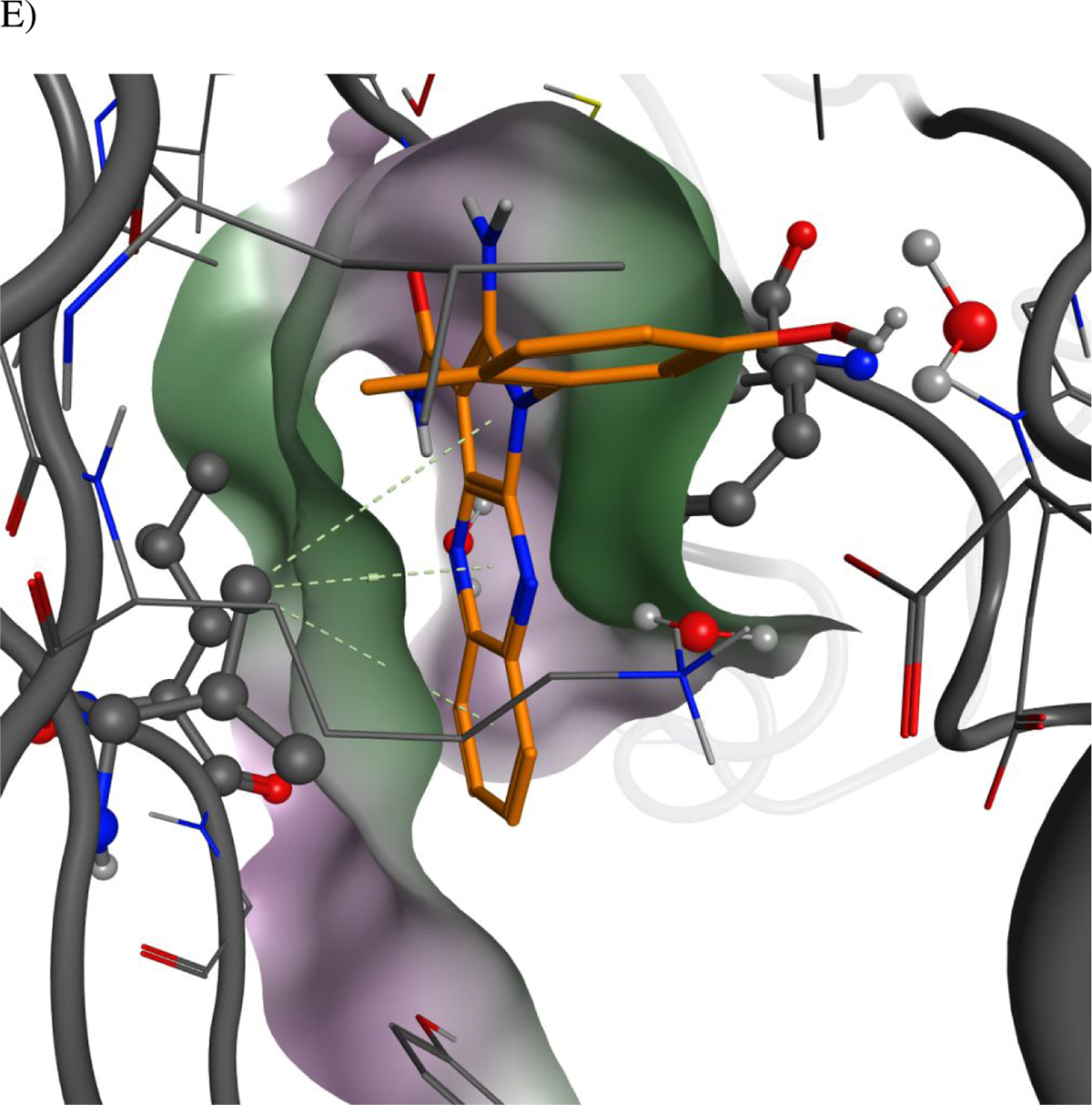
The binding mode of 39 to PKMYT1.
A) Ribbons representation of 39 bound to PKMYT1 (crystal structure PDB ID 8D6D). 39 is shown as sticks with golden carbon atoms, and part of the solvent-accessible surface of the binding pocket is shown in light gray. Oxygen is rendered in red, nitrogen in blue, carbon in yellow, bromine in brown, and polar hydrogens in white. Favorable interactions are highlighted with dashed lines with stronger hydrogen bonds additionally highlighted with cylinders (as calculated by the Molecular Operating Environment, MOE, from the Chemical Computing Group, Inc.)22. A bridging water molecule is shown in ball-and-stick representation.
B) 2D plot of the interactions that compound 39 forms with PKMYT1, as calculated by MOE.
C) Surface-surface complementarity of the 6-monomethyl phenol of analog 41 bound to PKMYT1 (crystal structure PDB ID 8D6F). The solvent-accessible surface of the protein is shown as a solid surface, with polar regions in purple and hydrophobic patches in green. The solvent-accessible surface of the ligand is displayed as a mesh.
D) Surface-surface complementarity of the 2,6-dimethyl phenol motif of analog 39 bound to PKMYT1. The solvent-accessible surfaces of the protein and ligand are represented, using the same convention as in panel C.
E) The carboxamide-side pyrazine’s nitrogen is sandwiched between the side-chains of hydrophobic residues. The solvent-accessible surfaces of the protein and ligand are represented, using the same convention as in panel C.
Table 5.
Thermal stability of atropisomers.
| Temperature1 | %ee2 | ||
|---|---|---|---|
| °C | 39 | 413 | 43 |
| 22 | 100 | 96 | 100 |
| 50 | 100 | 96 | 100 |
| 70 | 100 | 74 | 100 |
| 90 | 100 | 44 | 100 |
| 120 | 100 | 0 | 52 |
| 150 | 100 | 0 | 4 |
Temperature at which a 1 mg/mL DMSO solution was heated for 1 h.
Determined by chiral SFC or HPLC analysis (absorption at 254–270 nm).
41 used in this experiment was isolated from chiral SFC purification with a %ee of 96%.
Despite the encouraging potency of these substituted tricyclic derivatives, they suffered from unfavorable ADME (absorption, distribution, metabolism, and excretion) and physicochemical (for example: solubility and lipophilicity) properties. Truncating the fused aryl ring from tricyclic compounds such as 39 provided bicyclic pyrrolopyrazine analog 45 with generally more desirable physicochemical and ADME profile albeit with a significant loss in potency (Table 6 and Table 7). Analog 45 demonstrated an improvement in the in vitro clearance in human hepatocytes, in vivo unbound clearance and bioavailability in rat, Caco2 permeability, reversible CYP3A4, 2D6 and 2C9 (cytochrome P450) inhibition and time-dependent CYP3A4 inhibition.
Table 7.
ADME profile of selected bicyclic analogs.
| Rat %PPB1 | Rat %F | Rat IV CLunb (mL/min/k g)2 | CYP inhibition (3A4, 2D6, 2C9) IC50 (μM) | Caco2 Papp A to B x10−6 cm/s (ER) | Human hepatocyte CLint (μL/min/106 cells) | CYP3A4 TDI (IC50 shift) | |
|---|---|---|---|---|---|---|---|
| 39 | 99.7 | 12 | 7430 | 9, 19, 3 | 7.5 (1.1) | 33 | 5.4 |
| 45 | 81.0 | 56 | 81 | >30,>30,>30 | 19 (1.1) | <3 | 1 |
| 46 | 89.2 | 20 | 505 | >30,>30, >30 | 19.8 (1.1) | <3 | 1 |
| 49 | 95.1 | 25 | 939 | 13, 8.2, 4.6 | 11.8 (0.6) | 9.7 | >2.3 |
| 50 | 48.2 | 21 | 41 | >30,>30, >30 | 1.7 (4.1) | 4.5 | 1 |
| 51 | 62.8 | 18 | 151 | >30,>30, >30 | 5.8 (4.7) | <3 | 1 |
| 52 | 92.8 | 4.5 | 1250 | >30,>30, 8 | 10.8 (2.0) | 24 | 1 |
| 53 | 95.7 | 4.4 | 1721 | 23,>30, 9 | 10.1 (3.1) | 36 | 1 |
Rat plasma protein binding at 1 μM.
CLunb = (Total CL/fraction unbound)
The potency loss resulting from the ring truncation of compound 39 to yield the unsubstituted bicyclic pyrrolopyrazine analog 45 was almost completely recovered by introducing two methyls to the truncated scaffold to afford analog 46 with promising cell-based potency (Table 6). Monomethyl substituents at either at R2 (47) or R1 (48) were also well tolerated, although the increased cell-based potency at R1 prompted us to prioritize exploration of this vector. This work yielded a number of potent analogs such as 49–53 (Table 6). We were pleased to see that the beneficial impact of ring truncation extended beyond ADME properties. Indeed, the PKMYT1 over EPHB3 selectivity window for the pyrrolopyrazine analog 45 was greatly improved over the tricyclic analogs 6 and 39 (Table 3 and Table 6). Although this selectivity advantage was lost when the pyrrolopyrazine was substituted with a polar group such as tBuOH (50), the addition of small, non-polar substituents preserved much of this selectivity as seen for the bismethyl analog 46 and the cyclopropyl analog 49 (Table 6). Such small, non-polar substituents maintained favorable potency. Our focus was thus directed towards the enantiopure bicyclic analogs with small, non-polar substituents, capitalizing on these substantial ADME and selectivity advantages.
Analogs 46 and 49-53 were profiled to evaluate their potential for drug-drug interactions, metabolic stability, and oral bioavailability (Table 7). As seen with the pyrrolopyrazine analog 45 described above, time-dependent CYP3A4 inhibition was not detected, and minimal reversible CYP inhibition was observed for the majority of these bicyclic analogs with the exception of the cyclopropyl analog 49. Pharmacokinetic studies in the rat showed that analogs with lower unbound clearance (<1000 mL/min/kg) had moderately oral bioavailability with %F ranging from 18% - 25% (compound 46-51, Table 7). When R1 was substituted with heteroaryls such as compound 52 and 53, a negative impact was observed on the rat unbound clearance. In general, the bicyclic analogs were highly permeable in Caco2 cells, but an impact on permeability was noticed when polar groups were introduced as with compounds 50 and 51 (Table 7).
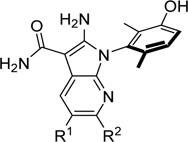
Taking advantage of the earlier observation that removing a nitrogen from the tricyclic ring system improved potency (see analogs 24 and 25 in Table 2), we next removed the nitrogen on the carboxamide side of the bicyclic pyrrolopyrazine scaffold. This afforded the 7-azaindole analogs 54-57 and RP-6306 which showed improved cell-based potency and increased selectivity over EPHB3. Several of these 7-azaindole analogs showed <25 nM potency in the cell-based PKMYT1 assay and >100-fold selectivity in the nanoBRET assays. The azaindoles also had favorable PK properties as shown by the reduced unbound clearance and improved oral bioavailability in the rat (Table 8). Consistent with the knowledge acquired with the pyrrolopyrazine analogs 46 and 49, a favorable selectivity profile over EPHB3 was obtained for the 7-azaindole analogs substituted with small, non-polar groups as exemplified by the cPr analog 56, and especially the methyl (54), the chloro (55), and the bismethyl (RP-6306) analogs (Table 8).
Table 8.
Potency, rat clearance and selectivity of 7-azaindole analogs.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | PKMYT1 cell assay IC50 (μM) | Rat %F | Rat %PPB1 | Rat IV CLunb (mL/mi n/kg)2 | PKMYT1 nanoBRET IC50 (μM) | EPHB3 Selectivity ratio | |
| 54 | Me | H | 0.024 | 62 | 73.8 | 76.0 | 0.001 | 372 |
| 55 | Cl | H | 0.017 | 37 | 88.1 | 215 | 0.001 | 186 |
| 56 | cPr | H | 0.005 | 28 | 80.3 | 170 | 0.003 | 30 |
| 57 | Cl | Me | 0.007 | 24 | 93.1 | 731 | - | - |
| RP-6306 | Me | Me | 0.014 | 48 | 87.7 | 198 | 0.002 | 131 |
Rat plasma protein binding at 1 μM.
CLunb = (Total CL/fraction unbound)
Most importantly, the preferred 7-azaindole analogs 55 and RP-6306 displayed advantageous ADME profiles, i.e. good stability in human hepatocytes, high permeability and minimal efflux in Caco-2 cells, a favorable pharmacokinetic profile in rodents, and no detectable reversible or time-dependent CYP inhibition (Table 9). As a result, these compounds were further characterized to show low human hepatocyte induction, favorable non-rodent PK (dog and monkey), and no hERG inhibition (Table 9). Despite overall similar profiles, RP-6306 was selected for development based on the overall superior PK characteristics in the four preclinical species studied. Additionally, RP-6306 showed a reduced propensity for CYP3A4 induction in a single donor hepatocyte study compare to compound 54 and 55. The atropisomeric stability of RP-6306 was evaluated and no interconversion to the distomer was detected when a DMSO solution of RP-6306 was heated at up to 150 °C.
Table 9.
Advanced ADME profile of promising azaindole analogs.
| 55 | RP-6306 | |
|---|---|---|
| Human hepatocytes Clint (μL/min/million cells) | 4.5 | 3.2 |
| CD-1 mouse PK (%F, CL) | 21%, 59.2 | 37%, 30.4 |
| CD rat PK (%F, CL) | 37%, 25.6 | 48%, 24.4 |
| Beagle dog PK (%F, CL) | 33%, 21.3 | 75%, 13.1 |
| Cyno monkey PK (%F, CL) | 32%, 19.4 | 29%, 23.8 |
| % PPB (R/H)* | 88.1%, 89.6% | 87.7%, 84.2% |
| Papp A to B x10−6 cm/s (ER) Caco2 | 18.3 (2.4) | 15.2 (2.1) |
| CYP 3A4, 2D6, 2C9 inhibition IC50 (μM) | >30, >30, 26 | >30, >30, >30 |
| CYP3A4 TDI (IC50 shift) | 1 | 1 |
| 3A4 hepatocyte induction at 3 μM** | 26.7% | 12.5% |
| hERG patch clamp IC50 | nd | >100 μM |
Rat and human plasma protein binding at 1 μM
*, % of positive control 10 μM rifampicin in single donor of human hepatocytes
RP-6306 KINASE SELECTIVITY PROFILE
RP-6306 was tested in a Kinativ™ Colo-205 cell lysate kinase binding assay21 at 1.2 μM (85x its cellular IC50) to identify binding to off-target kinases. At this high concentration, RP-6306 bound to only 6 of the 274 kinases detected, mostly within the ephrin family (Figure 2). Because PKMYT1 could not be detected in the Kinativ™ Colo-20 cell lysate analysis, it was not possible to determine the selectivity ratio for RP-6306 against these six kinases using this technique. Therefore, our panel of NanoBRET™ cell-based assays was expanded to include five ephrins (A1, A2, B2, B3, and B4), FRK, the promiscuous c-SRC, and the related WEE1. RP-6306 showed a high degree of selectivity (29x to 4000x) over these kinases in these cellular binding assays (Table 10).
Figure 2.
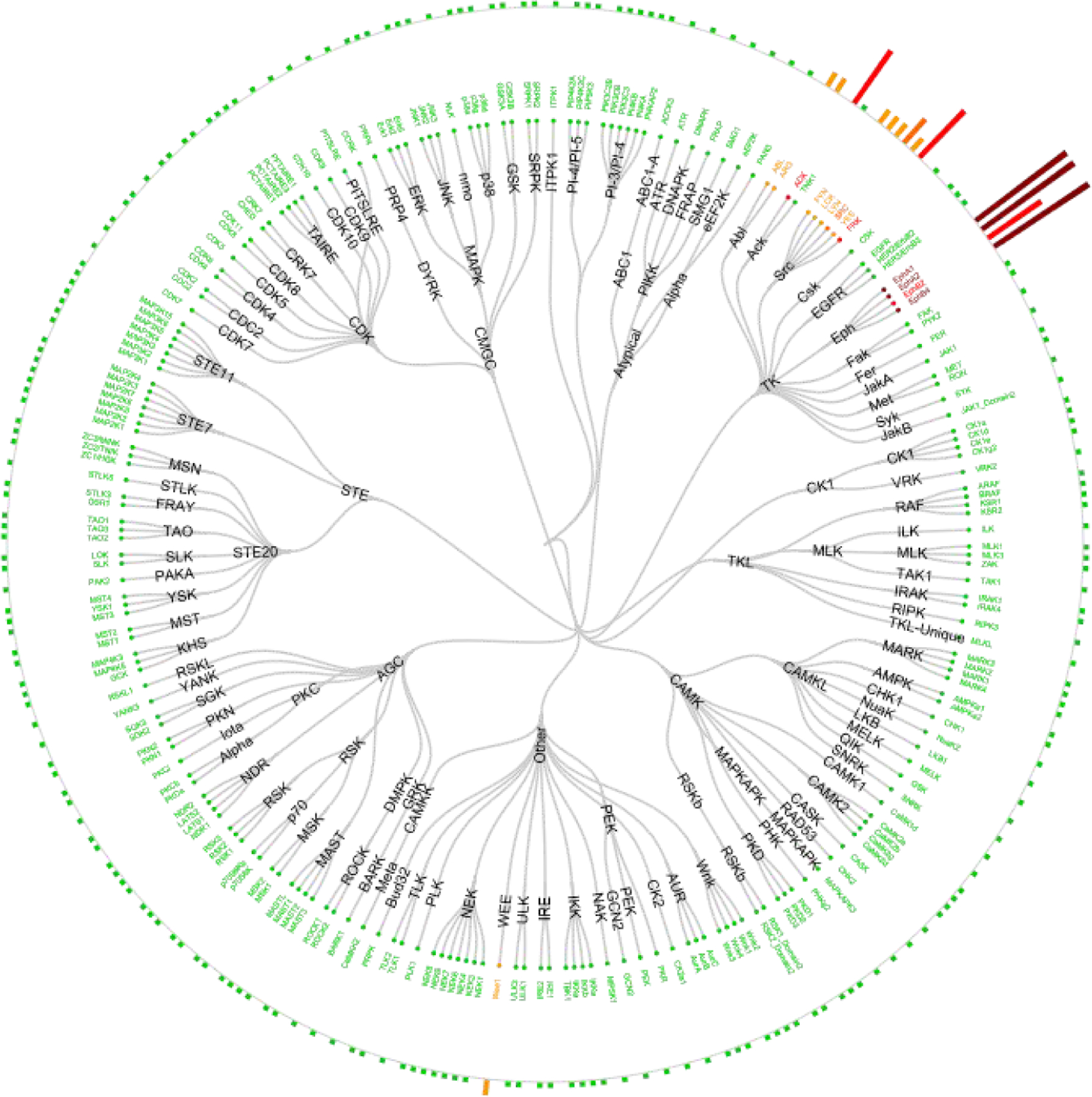
Kinativ™ Colo-20 cell lysate kinase binding profile of RP-6306 at 1.2 μM.
Table 10.
RP-6306 NanoBRET PKMYT1 selectivity against Kinativ™-flagged kinases.
| PKMYT1 IC50 (uM) | EPHA1 | EPHA2 | EPHB2 | EPHB3 | EPHB4 | FRK | SRC | WEE1 | |
|---|---|---|---|---|---|---|---|---|---|
| RP-6306 | 0.002 | 29x | 69x | 189x | 131x | 138x | 570x | >4150x | 2050x |
CO-CRYSTAL STRUCTURES OF INHIBITORS BOUND TO PKMYT1
Co-crystal structures of four PKMYT1 inhibitors bound to the kinase domain of PKMYT1 were solved at 2.15 to 2.58 Å resolution by molecular replacement (see Table S1 in methods section for X-ray data collection and refinement statistics). In the crystal structure of 39 bound to PKMYT1 (Figure 3), five hydrogen bonds were apparent that explained the importance of the pharmacophore. The primary amino group forms hydrogen bonds with both the hydroxyl of the gatekeeper residue Thr187 and the backbone carbonyl of Glu188 in the hinge region. The amino group of the carboxamide forms a hydrogen bond with the backbone carbonyl of Gly191 in the hinge, while the carbonyl of the carboxamide forms a hydrogen bond with the backbone amino of Cys190 in the hinge. The hydroxyl of the phenol forms a water-mediated hydrogen bond with the carboxylate of Glu157, the backbone amino of Phe252, and the imidazole of His161. The phenyl ring of the phenol displays favorable hydrophobic packing with the methyl of Thr187. The tricyclic aromatic rings have favorable hydrophobic packing with the side chain of Val124. In addition, the bromine displays a weak but favorable electrostatic interaction with the amino group in the side chain of Gln196. Similar interactions were observed in the co-crystal structures of 28 and 41 bound to PKMYT1. Images of these structures and detailed 2D plots of the interactions are available in the Supporting Information. Inspection of the surface-surface complementarity of the 6-monomethyl phenol of analog 41 revealed some unoccupied hydrophobic space between the phenol and the protein (Figure 3C), within which a water molecule cannot fit. With the 2,6-dimethyl phenol motif of analog 39, the surface-surface complementarity is optimal (Figure 3D). The carboxamide-side pyrazine’s nitrogen is sandwiched between the side-chains of hydrophobic residues Val124 and Leu116 on one side and Phe240 on the other, suggesting a preference for a carbon at this position (Figure 3E).
The crystal structure of RP-6306 bound to PKMYT1 is shown in Figure 4. Overall, the key interactions between this inhibitor and the kinase domain of PKMYT1 are very similar to 39. However, the amino of the carboxamide of RP-6306 forms an additional water-mediated hydrogen bond with the backbone carbonyl of Gly191, and the pyridine nitrogen forms a water-mediated hydrogen bond with the amino in the sidechain of Lys139. In addition, the oxygen of the phenol of RP-6306 forms a hydrogen bond with the backbone amino of Asp251 (which is obscured by the surface). As observed in the co-crystal structure of 39, the primary amino group of RP-6306 forms a hydrogen bond with the hydroxyl of the gatekeeper residue Thr187. This interaction with Thr187 is likely responsible for the selectivity against the highly homologous kinase WEE1 as this residue is the single residue difference (Thr vs Asn376) in the active site compared to PKMYT1.
Figure 4.
The binding mode of RP-6306 to PKMYT1.
A) RP-6306 (crystal structure PDB ID 8D6E) is shown as sticks with pink carbon atoms, and part of the solvent-accessible surface of the binding pocket is rendered in light gray. Oxygen is rendered in red, nitrogen in blue, carbon in pink, bromine in brown, and polar hydrogens in white. Favorable interactions are highlighted with dashed lines with stronger hydrogen bounds additionally highlighted with cylinders (as calculated by MOE). Water molecules are shown as balls-and-sticks.
B) 2D plot of the interactions that compound RP-6306 forms with PKMYT1, as calculated by MOE.
INHIBITION OF PKMYT1 INHIBITS GROWTH OF CCNE1-AMPLIFIED XENOGRAFT TUMORS
The synthetic lethal relationship between CCNE1 amplification and the absence of PKMYT1 activity was confirmed both genetically and chemically (with RP-6306) through comparison of the growth sensitivity of isogenic fallopian tube cells (FT282 cells) engineered to overexpress CCNE1 relative to wild type cells.6 Furthermore, multiple cancer cell lines and xenograft models with amplified CCNE1 or cyclin E over-expression showed greater growth inhibition compared to normal counterparts.6 Using a very sensitive and robust CCNE1-amplified ovarian xenograft model (OVCAR3) we sought to understand the relationship between target inhibition, RP-6306 exposure and efficacy. Oral dosing of RP-6306 formulated in chow at 15, 50 and 300 ppm (equivalent to approximately 3, 10 and 60 mg/kg/day) resulted in a statistically significant and dose-dependent reduction in OVCAR3 tumor growth (Figure 5A). Although there was slight body weight loss at the highest dose of 300 ppm initially, RP-6306, formulated in chow did not cause a decrease in food consumption and was well tolerated over a 21-day treatment period (Figure 5B and C). At day 2, 5 and 22, the steady state free plasma levels of RP-6306 were measured in chow-fed animals in the early morning and late afternoon to capture an average exposure per day. The compound exposure was stable over the course of 22 days but was less than dose proportional at doses above 150 ppm (Figure 5D). In a parallel study, when RP-6306 was administered PO twice daily (BID), a dose-dependent increase in anti-tumor efficacy in the OVCAR3 model was observed up to the maximum tolerated dose of 20 mg/kg.6 The efficacy and compound pharmacokinetic (PK) parameters are summarized in Table 11.
Figure 5. RP-6306 free plasma exposure and in vivo efficacy in the OVCAR3 CCNE1 amplified xenograft model.
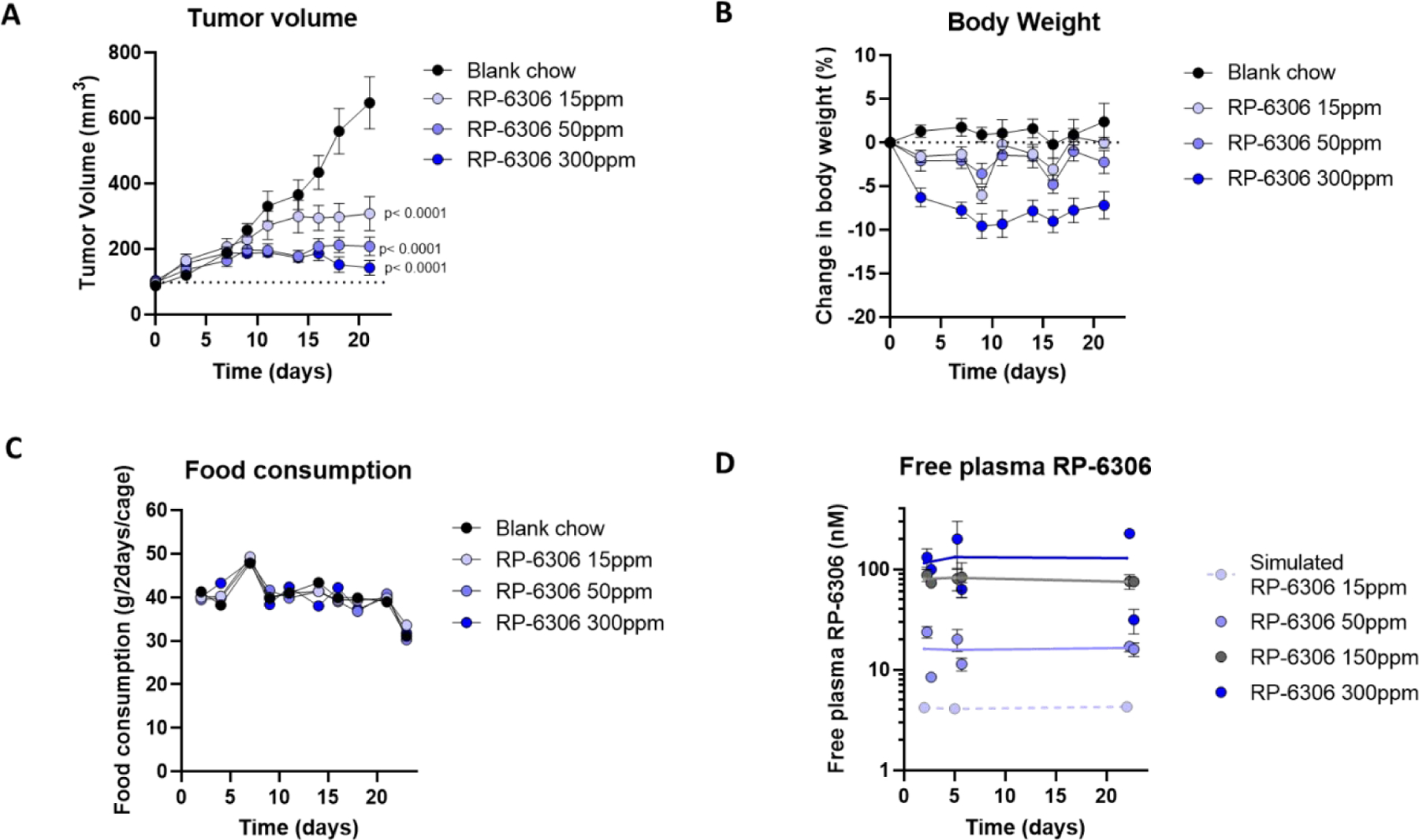
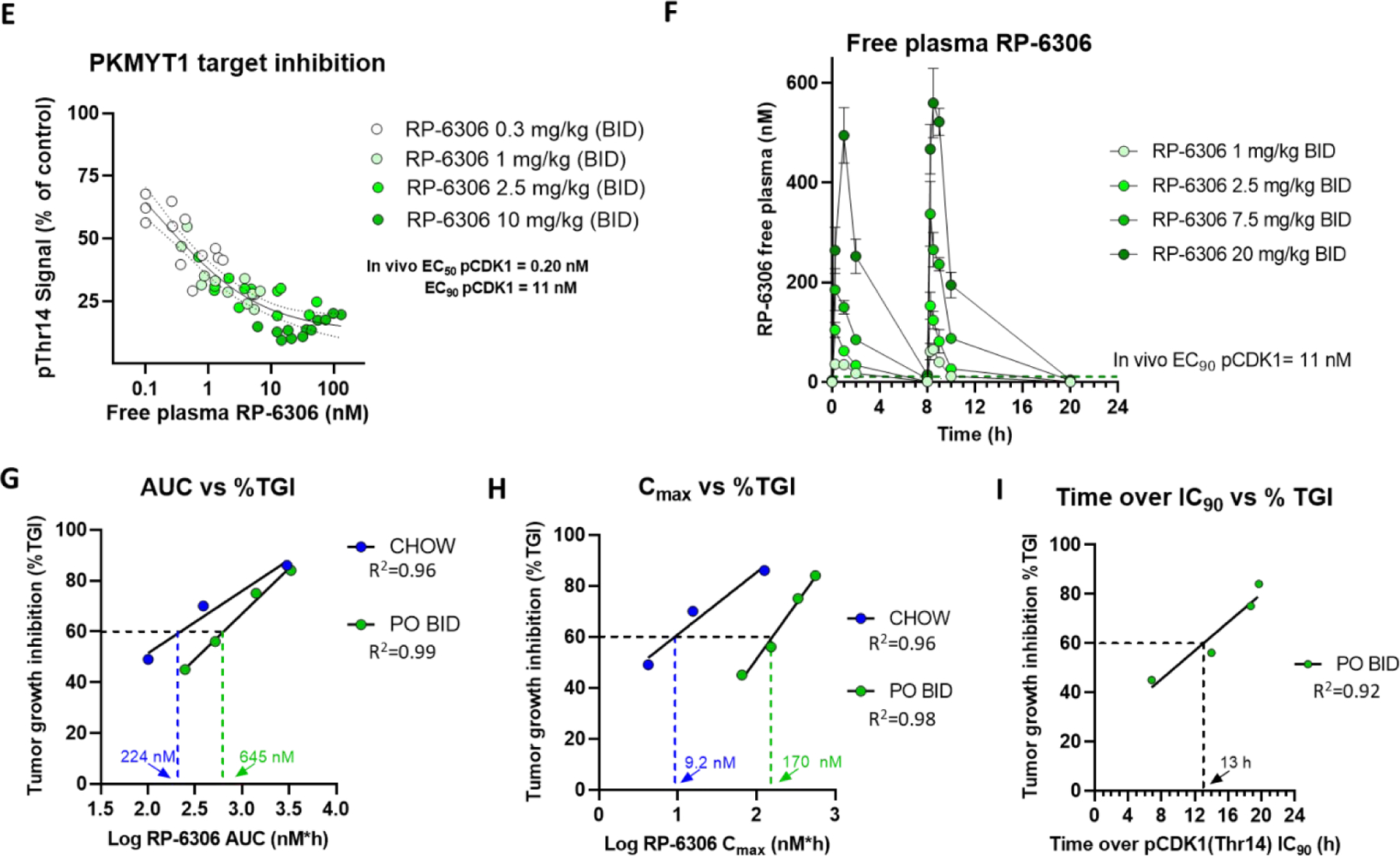
A) Tumor xenograft volume and B) change in body weight in OVCAR3-bearing mice treated with RP-6306 formulated in chow for 21 days. Results are expressed as mean tumor volume ± SEM, N=8 mice / group. Statistical significance relative to vehicle control was established by One-Way ANOVA followed by Fisher’s LSD test (GraphPad Prism v8). C) The two-day mean chow consumption in mice receiving blank chow or chow mixed with RP-6306 at the indicated concentrations D) Measured free plasma levels of RP-6306 in chow formulation at the indicated doses measured at 6:30am and 4:30pm on Days 2, 5 and 22. The 15 ppm dose was simulated, assuming linearity from the 50 ppm dose. E) The proportion of OVCAR3 tumor pCDKThr14 signal relative to vehicle treated mice for each dose at 2, 6 and 10 h post PO dosing; mean ± SEM (N=4/group/time point). The tumor pCDK1(Thr14) EC50 was determined by a non-linear dose-response model (GraphPad Prism v9.30). F) Pharmacokinetics of RP-6306 administered PO BID at the indicated doses. G-I) The relationship between measured tumor growth inhibition (TGI) and free plasma RP-6306 exposure (AUC) G), Cmax H) or time over pCDK1(Thr14) EC90 I) at each chow (A, D) and BID dose (F and Gallo et al.6) evaluated in efficacy studies.
Table 11.
Summary of RP-6306 PK parameters and efficacy in the OVCAR3 xenograft model.
| Dose PO | Mean Free AUC0-∞ (nM*h) | Mean Free Cmax (nM) | Time over EC90 (h) | Mean TGI (%) |
|---|---|---|---|---|
| 15 ppm chow* | 101 | 4.2 | 0 | 49 |
| 50 ppm chow | 388 | 15.5 | 24 | 70 |
| 300 ppm chow | 3013 | 126 | 24 | 86 |
| 1 mg/kg BID | 216 | 65.4 | 6.84 | 45 |
| 2.5 mg/kg BID | 414 | 153 | 14.0 | 56 |
| 7.5 mg/kg BID | 1410 | 337 | 18.7 | 75 |
| 20 mg/kg BID | 3320 | 560 | 19.7 | 84 |
simulated pharmacokinetics, AUC= area under the concentration vs time curve
To further investigate the pharmacokinetic/pharmacodynamic (PK/PD) relationship of RP-6306 with PKMYT1 target engagement, phosphorylation of the PKMYT1 substrate CDK1(Thr14) in OVCAR3 tumors was evaluated at 2, 6 and 10 h post PO BID dosing by ELISA from tumor homogenates. The effective free plasma concentration (EC50) to inhibit pCDK1(Thr14) by 50 % was calculated as 0.20 nM and EC90 as 11 nM. The results demonstrate potent in vivo PKMYT1 target inhibition and a direct relationship between RP-6306 free plasma levels and tumor pCDK1(Thr14) inhibition (Figure 5E). The pharmacokinetics of RP-6306 administered PO BID are shown in Figure 5F and illustrate the rapid clearance of RP-6306 in mouse plasma, yet substantial target coverage at dose as low as 2.5 mg/kg. The PK/efficacy relationship demonstrates a strong correlation between efficacy and free RP-6306 plasma exposure (area under the concentration vs time curve (AUC) and maximal concentration (Cmax) (Table 11). Interestingly, better efficacy is observed with lower, more sustained levels of RP-6306 provided in the chow formulation compared to the high peak-to-trough ratio of BID formulation (Figure 5G, H). For example, to achieve a 60% TGI, a sustained RP-6306 exposure of 224 nM*h is required compared to greater than twice that exposure with BID dosing. A sustained Cmax of 9.2 nM (just under 90% pCDK1(Thr14) target inhibition), for 24 h generates a 60% TGI (Figure 5H) compared to a Cmax of 170 nM generating the same efficacy on a BID schedule. Our analysis suggests that in either dosing scenario, RP-6306 levels maintained above EC90 for at least 13 h are required to generate efficacy (Figure 5I). These results illustrate the value of utilizing chow formulations in addition to PO dosing by gavage as tools to evaluate pre-clinical PK/efficacy relationships. Together, our data suggests that in vivo, prolonged PKMYT1 target inhibition is required for efficacy which may provide guidance to maximize efficacy in the ongoing RP-6306 clinical trials (NCT04855656).
SYNTHETIC CHEMISTRY
All the analogs in Table 1 were prepared as depicted in Scheme 1 using an approach adapted from the reported synthesis of compounds 1, 2, and 5.14 One chloro of 2,3-dichloroquinoxaline (58) was substituted with malononitrile to afford 59,14 and the remaining chloro was subsequently displaced with an arylamine to afford the aminopyrroles 62–73. Alternatively, the aminopyrroles 74 and 75 were obtained by inverting the sequence, where one chloro of 58 was initially displaced with an arylamine, and the remaining chloro was subsequently substituted with malononitrile. Hydrolysis of the nitrile to the carboxamide upon treatment with sulfuric acid, followed by a final deprotection of the aryl group when required, yielded analogs 1–18.
Scheme 1.
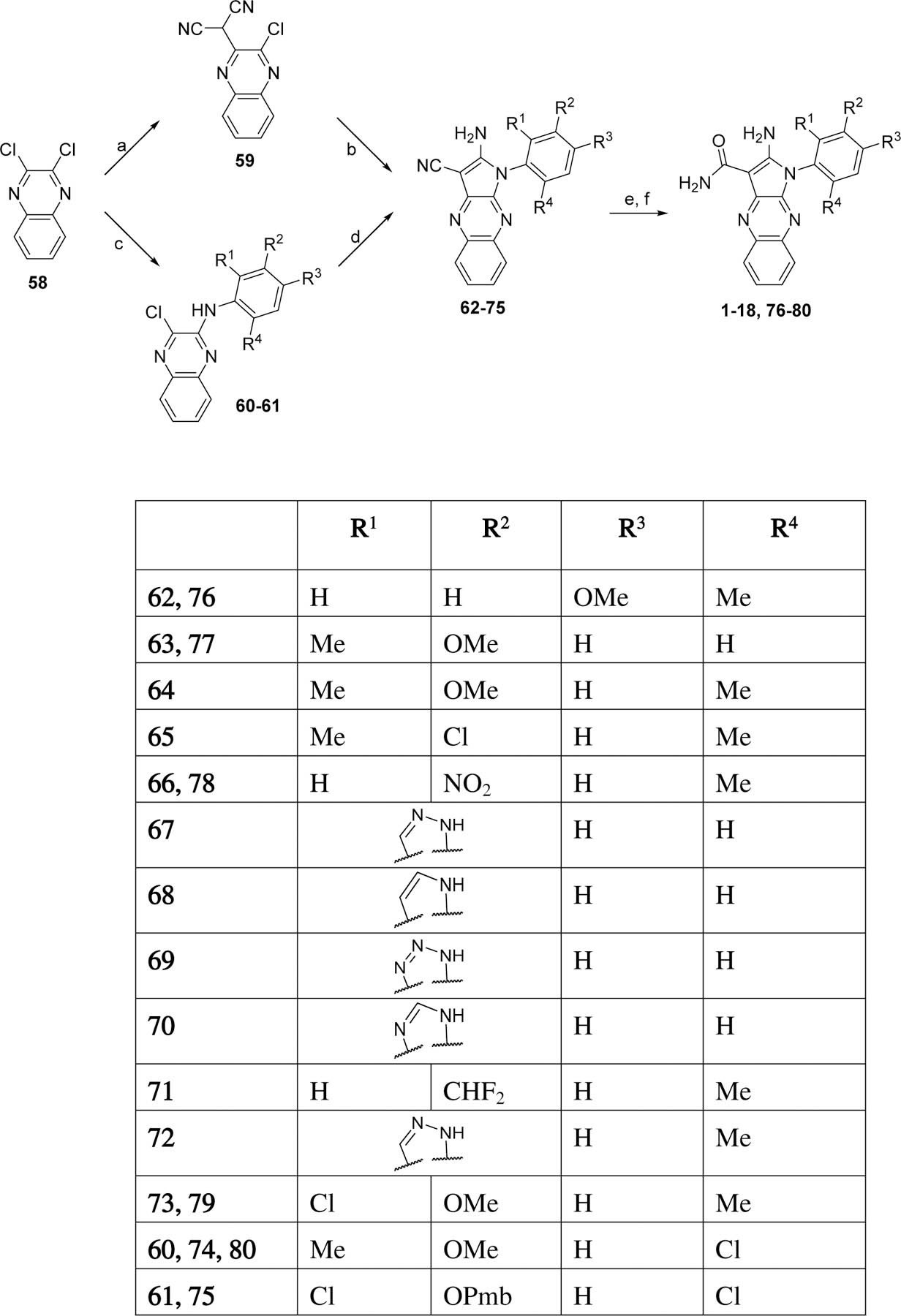
Preparation of analogs for phenol ring SARs
Reagents and conditions. a. malononitrile, NaH, DME; b. ArNH2, NMP; c. ArNH2, KOtBu, THF; d. malononitrile, NaH, Pd(PPh3)4, dioxane; e. H2SO4; f. BBr3 for methoxy deprotection or H2, Pd/C for NO2 reduction.
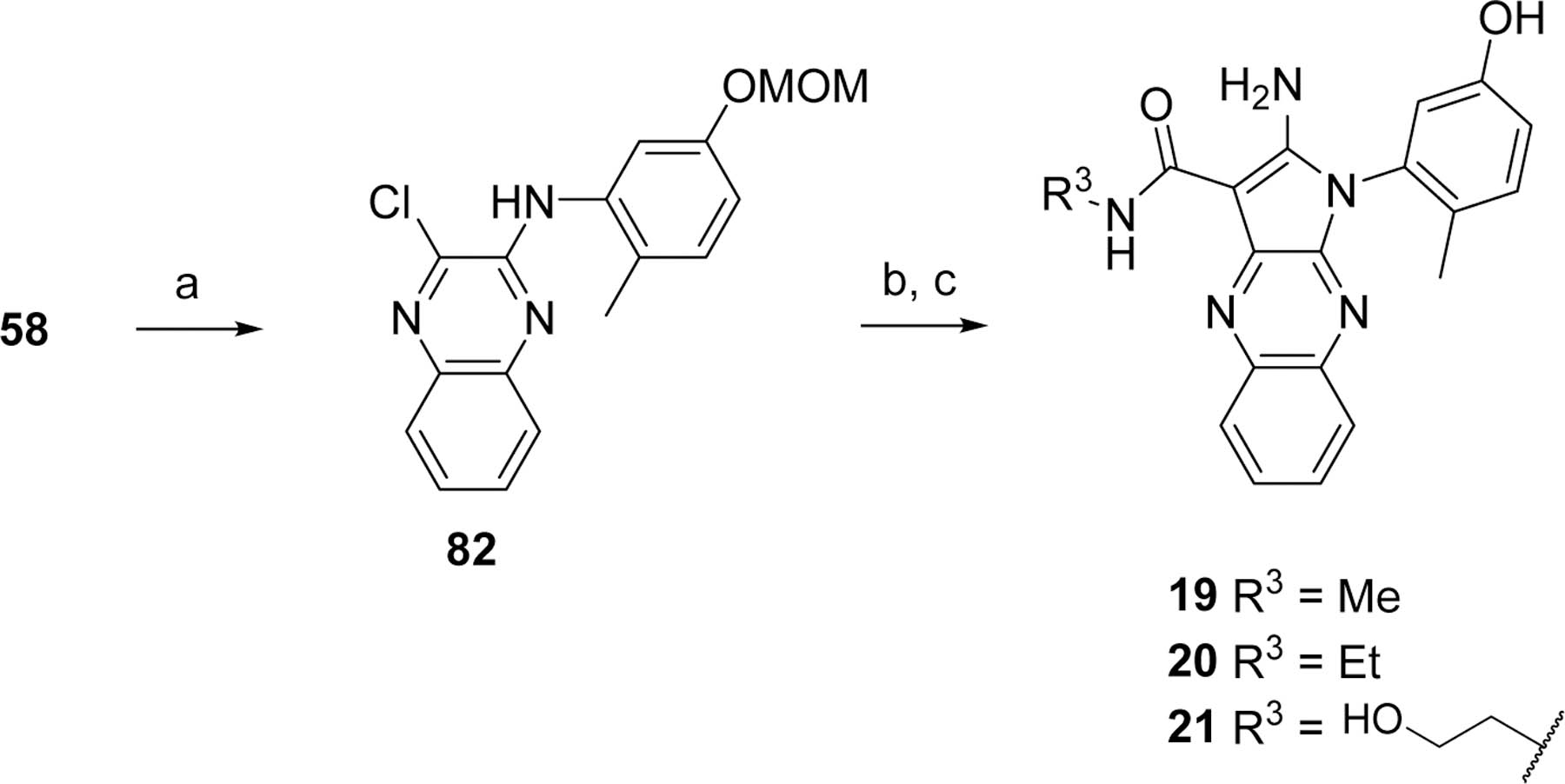
Amides 19–21 described in Table 2 were prepared as depicted in Scheme 2. One chloro of 58 was substituted with 5-(methoxymethoxy)-2-methyl-aniline (81)23, 24 under palladium-catalyzed C-N coupling conditions25 to afford compound 82. Substitution of the remaining chloro with selected 2-cyano-acetamides followed by O-MOM deprotection yielded analogs 19–21.
Scheme 2.
Preparation of substituted carboxamide analogs.
Reagents and conditions. a. 81 NaOtBu, Pd2(dba)3, XantPhos, toluene; b. R3NH(CO)CH2CN, KOtBu, THF or Cs2CO3, DMF; c. HCl, dioxane.
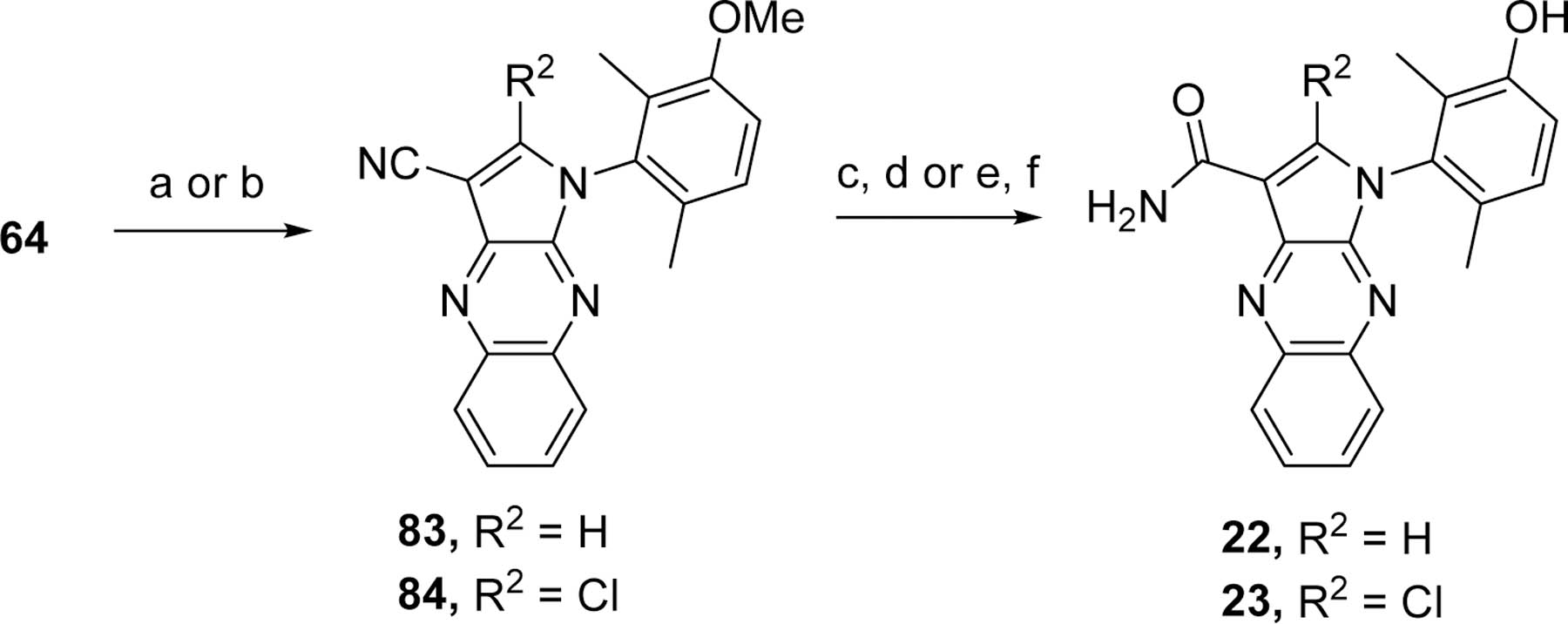
Des-amino pyrrole analogs 22 and 23 described in Table 2 were obtained upon treatment of the amino nitrile intermediate 64 with tBuONO, and the resulting diazonium was either protonolyzed26 to yield intermediate 83 or treated with CuCl27 to obtain chlorinated compound 84, respectively, as depicted in Scheme 3. Hydrolysis of the nitrile of intermediate 83 to the carboxamide upon treatment with sulfuric acid, followed by methoxy deprotection upon treatment with BBr3, yielded analogs 22. The methoxy deprotection of the less stable chloro intermediate 84 was achieved by a treatment with boron trichloride and tetrabutyl ammonium iodide (TBAI).28 The nitrile hydrolysis to the carboxamide was then completed using the Ghaffar-Parkins catalyst29 to afford analog 23.
Scheme 3.
Preparation of analogs with pyrrole NH2 replacements.
Reagents and conditions. a. tBuONO, THF; b. tBuONO, CuCl, ACN; c. H2SO4; d. BBr3, CH2Cl2; e. BCl3, TBAI, CH2Cl2; f. Ghaffar-Parkins catalyst, EtOH, H2O.
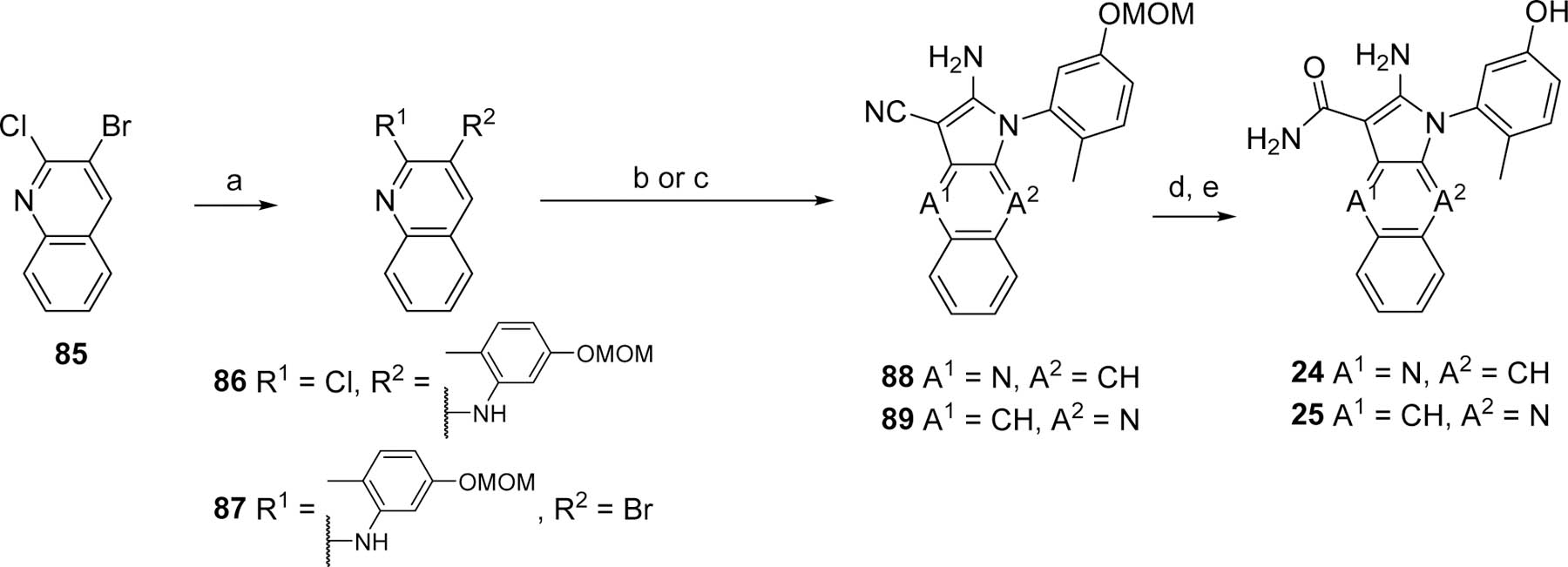
The preparation of analogs 24 and 25 described in Table 2 is depicted in Scheme 4. Both syntheses were initiated with 3-bromo-2-chloroquinoline (85). The initial C-N coupling reaction with 81 is not specific and yielded intermediates 86 and 87, that afforded aminopyrroles 88 and 89, respectively, upon substitution of the remaining halogen with malononitrile. In the case of the 2-chloropyridine 86, the substitution with malononitrile was achieved under SNAR conditions.30 For the 3-bromopyridine 87 the substitution with malononitrile was done under palladium-catalyzed conditions.31 Analogs 24 and 25 were obtained after O-MOM deprotection with HCl, followed by nitrile hydrolysis to the carboxamide with H2SO4.
Scheme 4.
Preparation of N-regioisomers 24 and 25.
Reagents and conditions. a. 81, NaOtBu, Pd2(dba)3, Xantphos, toluene; b. malononitrile, KOtBu, DME; c. malononitrile, NaH, Pd(PPh3)4; d. HCl, dioxane; e. H2SO4.
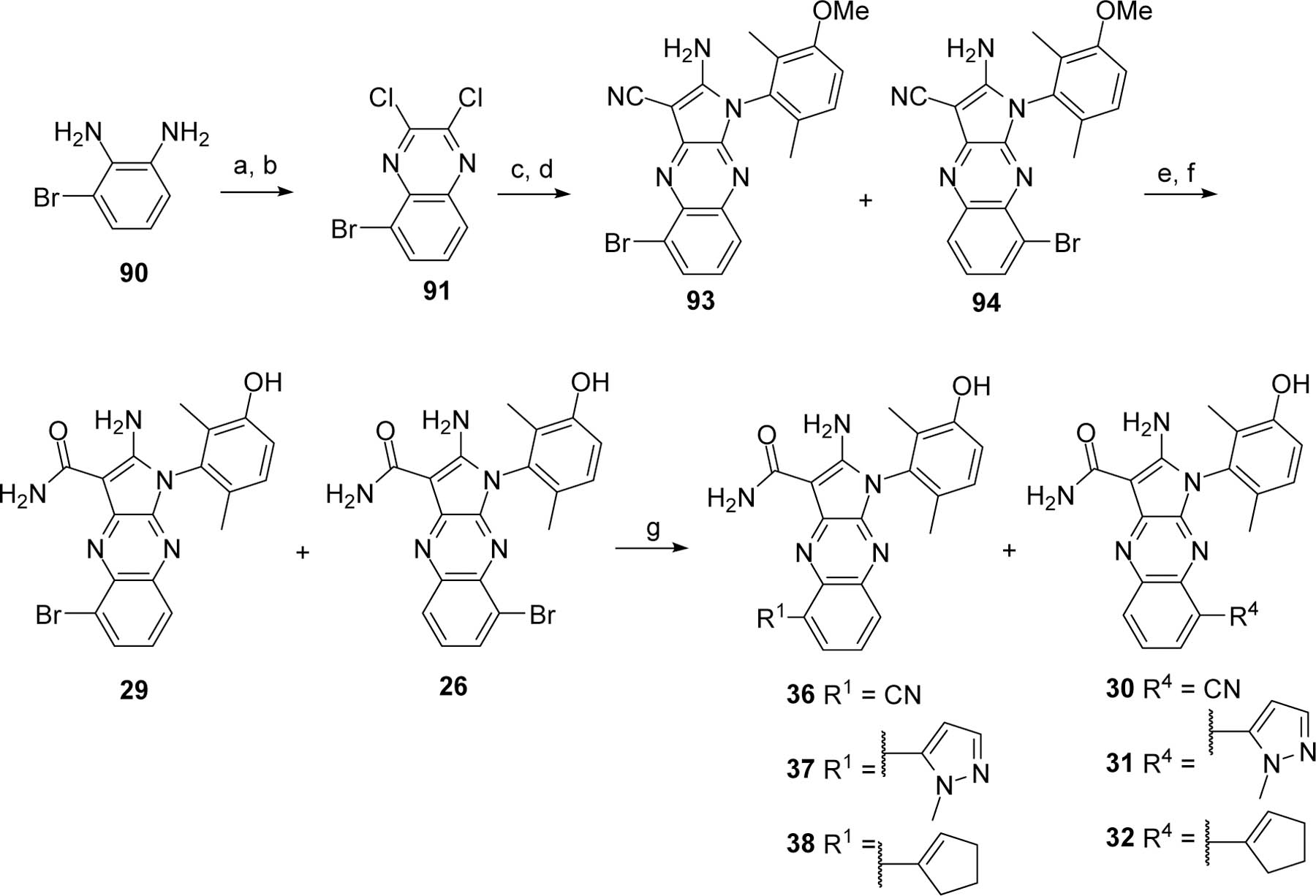
The syntheses of the four bromo regioisomers 26–29 were initiated with the bromobenzene-1,2-diamine 90 or 95 that provided the corresponding bromo 2,3-dichloroquinoxaline 91 or 96, respectively, in two steps upon treatment with diethyl oxalate and then thionyl chloride according to a known procedure32 (Scheme 5 and 6). One chloro of the 2,3-dichloroquinoxaline 91 or 96 was substituted with malononitrile, and the remaining chloro was displaced with 3-methoxy-2,6-dimethyl-aniline (92)33 under SNAR conditions to yield a mixture of the aminopyrroles 93 and 94 (Scheme 5) or 97 and 98 (Scheme 6). Hydrolysis of the nitrile and methoxy deprotection yielded pure analogs 26 and 29 or 27 and 28 after chromatographic separations. The position of the bromines were confirmed by X-ray structures of analogs 28 and 29 (structure discussed in Figure 3 and Supporting Information). The brominated analogs 26, 28, and 29 were derivatized by transition metal-mediated transformations34, 35 to provide analogs 30–38 (Scheme 5 and 6).
Scheme 5.
Preparation and derivatization of the bromo regioisomers 26 and 29
Reagents and conditions. a. diethyl oxalate; b. SOCl2, DMF cat; c. malononitrile, NaH, DME; d. 92, NMP; e. H2SO4; f. BBr3, CH2Cl2; g. RB(OR)2, Pd(dppf)Cl2.CH2Cl2, K2CO3, DMF or CuCN, DMF.
Scheme 6.
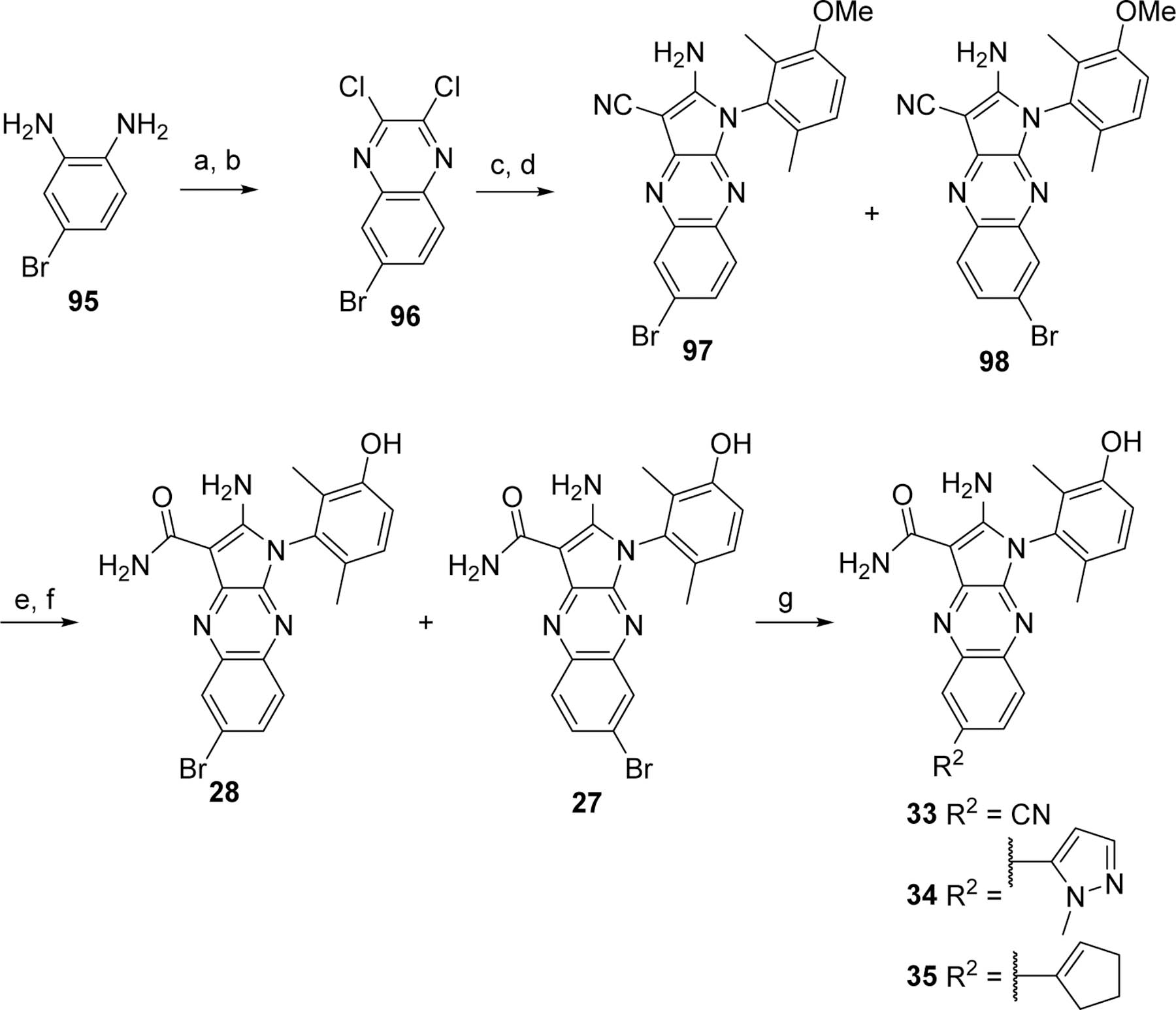
Preparation and derivatization of the bromo regioisomers 27 and 28.
Reagents and conditions. a. diethyl oxalate, reflux; b. SOCl2, DMF cat.; c. Malononitrile, NaH, DME; d. 92, NMP; e. H2SO4; f. BBr3, CH2Cl2; g. RB(OR)2, Pd(dppf)Cl2.CH2Cl2, K2CO3, DMF or CuCN, DMF.
The synthesis of the bicyclic pyrrolopyrazine analogs 45–47 was initiated by substituting one chloro of a symmetrical 2,3-dichloro-pyrazine, such as 99 or 100, with malononitrile under SNAr conditions to yield 101 and 102. The remaining chloro was then substituted with 92 to afford the aminopyrroles 103 or 104 (Scheme 7). Intermediate 103 was brominated with NBS to afford compound 105. The nitriles of 103, 104, and 105 were hydrolyzed to the carboxamides upon treatment with sulfuric acid and cleavage of the methoxy protecting group with boron tribromide provided the compounds 106 and 107, as well as the bromo intermediate 108 which was methylated to provide compound 109. Racemates 106, 107 and 109 were purified by chiral SFC to yield the analogs 45–47.
Scheme 7.
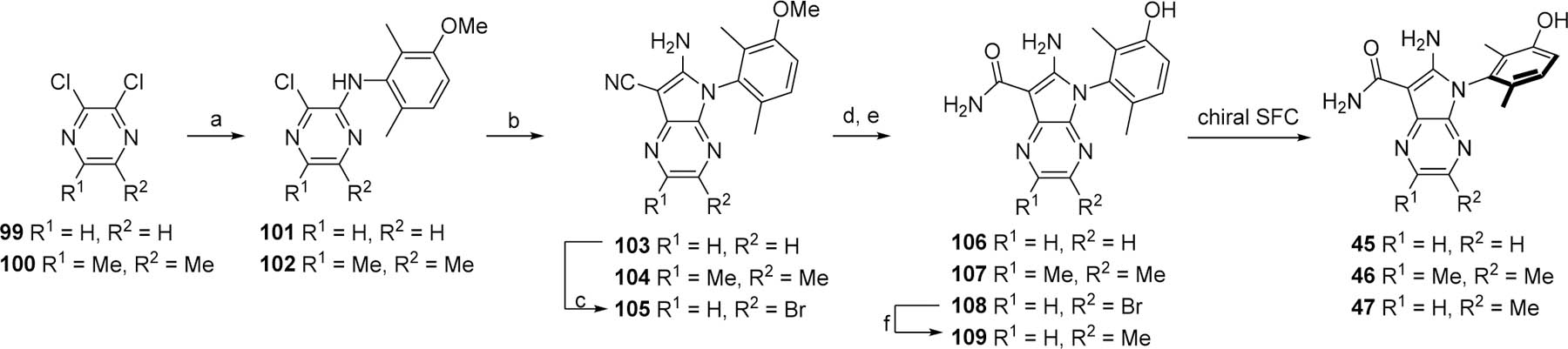
Preparation of bicyclic pyrrolopyrazine analogs 45–47.
Reagents and conditions. a. Malononitrile, NaH, THF; b. 92, NMP or 92 KOtBu, Pd-PEPPSI™-SIPr, NMP; c. NBS, DMF; d. H2SO4; e. BBr3, CH2Cl2; f. MeMgBr, ZnCl2, THF, then Pd(PPh3)4, THF.
The synthesis of the R1-substituted pyrrolopyrazine analogs 48–53 was initiated by converting the amino group of the 5-bromo-6-chloropyrazin-2-amine (110) to a hydroxyl (111) that was further benzylated to generate 112 (Scheme 8). The bromo was then substituted by 92 under C-N coupling conditions to afford 113. The chloro was displaced by malononitrile to afford the aminopyrrole 114. The nitrile was hydrolyzed to the carboxamide upon treatment with sulfuric acid with concomitant cleavage of the benzyl group to provide 115. The resulting hydroxyl was converted to the triflate 116. The methoxy protecting group was cleaved to generate the versatile intermediate 117. This intermediate was derivatized to provide compounds 118–124 as racemates which were purified by chiral SFC to yield the analogs 48–53.
Scheme 8.
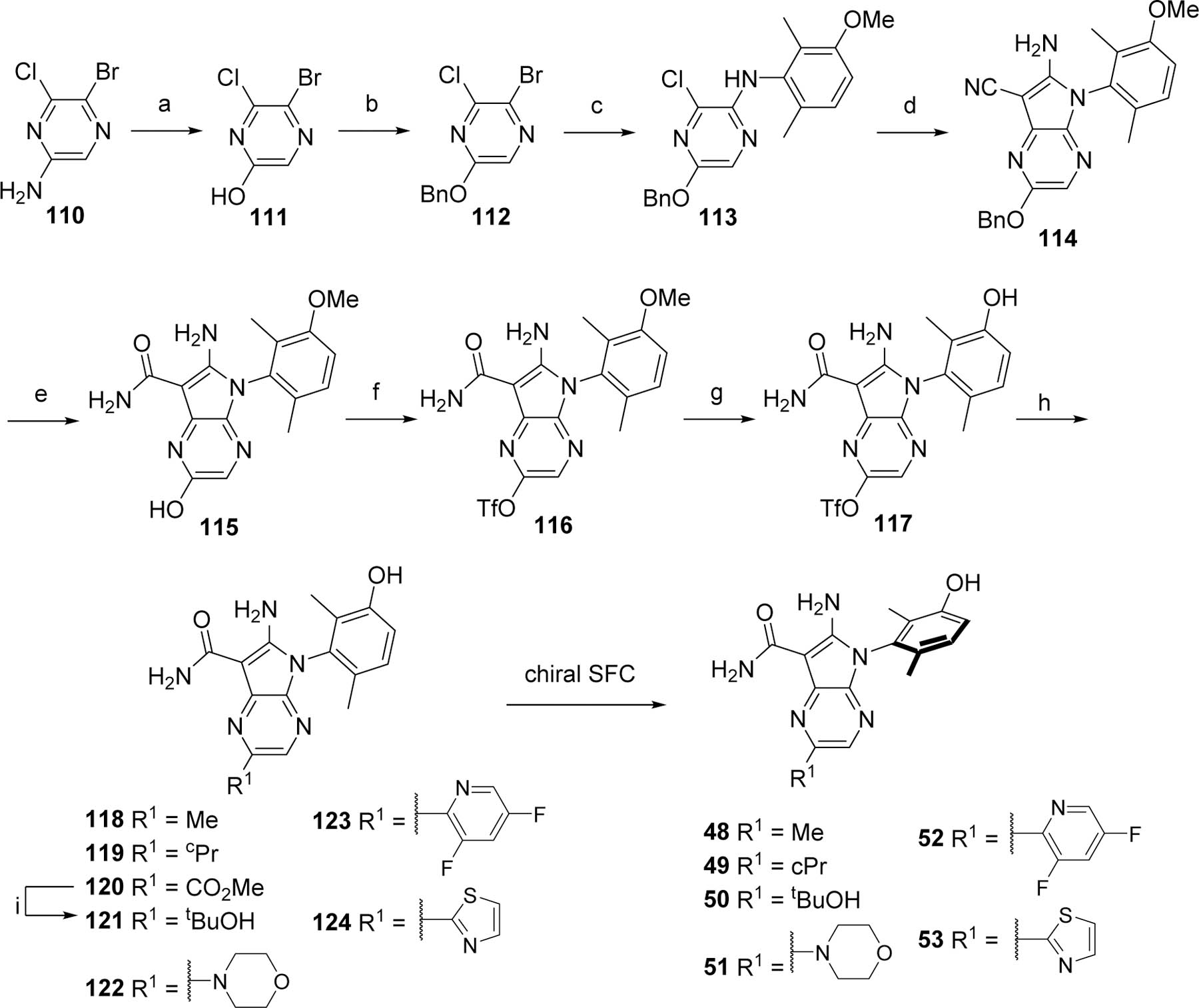
Preparation of bicyclic pyrrolopyrazine analogs 49–53.
Reagents and conditions. a. NaNO2, H2SO4; b. BnBr, Ag2CO3, toluene; c. 92, Pd2(dba)3, Xantphos, toluene; d. Malononitrile, NaH, Pd(PPh3)4, DME; e. H2SO4; f. PhNTf2, Cs2CO3, DMF; g. BBr3, CH2Cl2; h. OTf derivatization; i. MeMgCl, THF.
The syntheses of 7-azaindole analogs 54, and 56–57 were initiated by substituting the most electrophilic bromo of 2,3-dibromo-5-nitropyridine (125) or the chloro of 3-bromo-2-chloro-6-methyl-5-nitropyridine (126) by 92 under SNAr conditions to afford 127 or 128 (Scheme 9). The remaining bromo was substituted by malononitrile under metal-mediated conditions to afford the aminoazaindole 129 or 130. The resulting amino group was protected with a Boc group to yield compound 131 or 132. The nitro was reduced to the NH2 133 or 134, and the resulting NH2 was converted to the halogenated derivatives 135 or 136 under Sandmeyer conditions.36 The NHBoc-protecting group was cleaved thermally or by treatment with hydrochloric acid, and the nitrile was hydrolyzed to the carboxamide to provide intermediates 137 and 138. Cleavage of the methoxy protecting group with boron tribromide provided the compounds 139 and 142. Compound 142 was purified by chiral SFC to provide analogs 57. The bromo of compound 139 was substituted by a methyl or a cyclopropyl to give compounds 140 or 141 respectively which were purified by chiral SFC to yield the analogs 54 and 56.
Scheme 9.
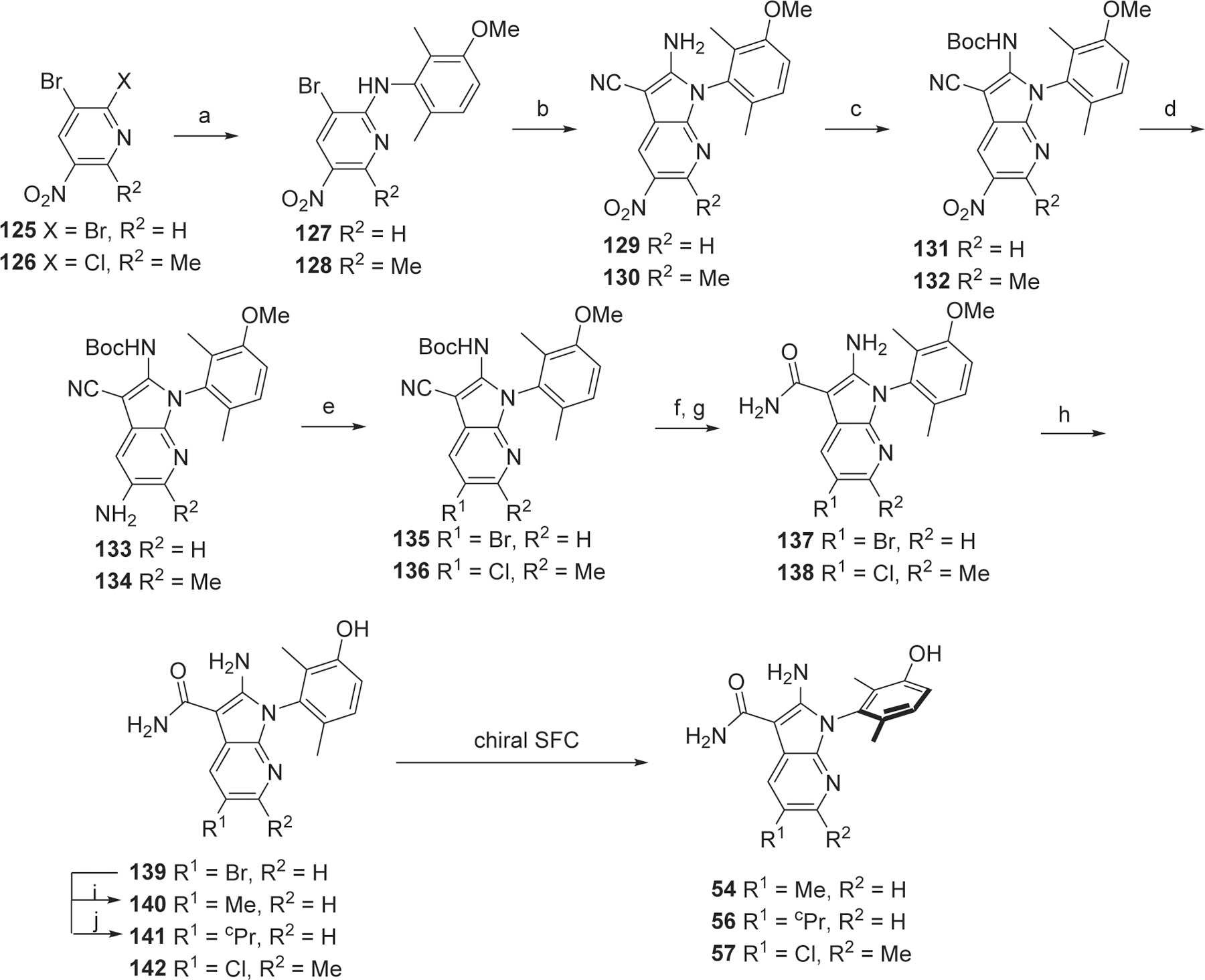
Preparation of 7-azaindole analogs 54 and 56–57.
Reagents and conditions. a. 92, 2,6-lutidine, NMP; b. Malononitrile, NaH, Pd(dppf)Cl2·CH2Cl2, DME; c. Boc2O, Et3N, 4-DMAP, THF, then ethylenediamine; d. H2, Pd/C, DCM, MeOH; e. tBuONO, CuX2, ACN, DMF; f. HCl, EtOH or 160 °C, NMP; g. LiOH, H2O2, EtOH, H2O; h. BBr3, DCM; i. Me2Zn, Pd(PtBu3)2, THF; j. cPrZnBr, Pd(PtBu3)2, THF.
Although the preferred 7-azaindole analogs 55 and RP-6306 were initially obtained from the route depicted in Scheme 9, alternative syntheses were developed to better accommodate the gram-scale requirements for advanced profiling of these analogs. The specific synthesis for analog 55 is depicted in Scheme 10. Substitution of the fluoro of 3-bromo-5-chloro-2-fluoropyridine (143) by 92 afforded 144. The bromo was then substituted by malononitrile to afford the azaindole 145. The nitrile can be hydrolyzed to the carboxamide under basic or acidic conditions to yield 146. Analog 55 was obtained after methoxy deprotection using boron tribromide and chiral SFC purification.
Scheme 10.
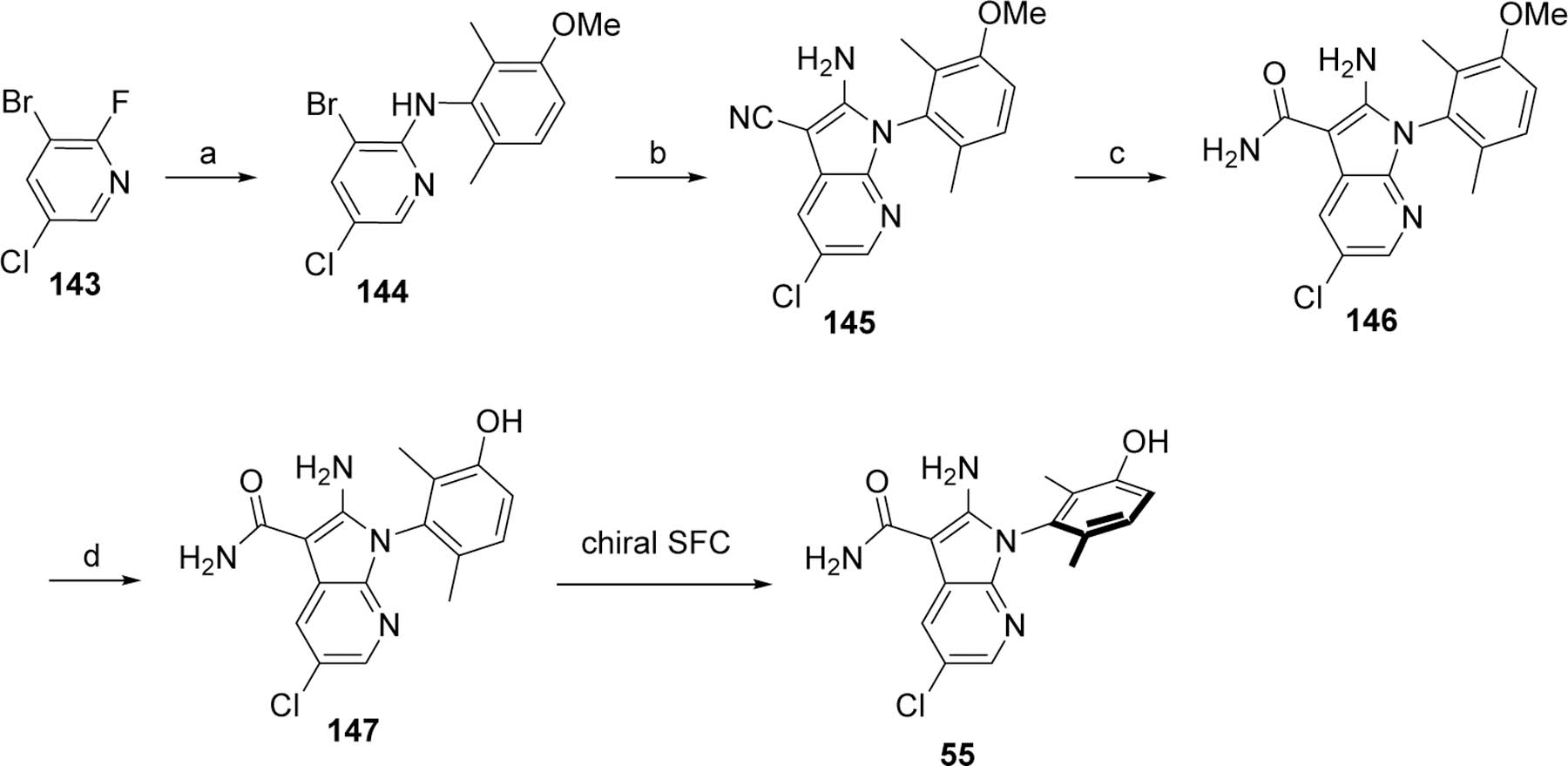
Preparation of analog 55.
Reagents and conditions. a. 92, LiHMDS, THF; b. Malononitrile, NaH, Pd(dppf)Cl2·CH2Cl2, DME; c. LiOH, H2O2, H2O, EtOH or H2SO4. d. BBr3, DCM.
The specific route for RP-6306 is depicted in Scheme 11. Conversion of the amino group of 3-bromo-5,6-dimethylpyridin-2-amine (148) into a hydroxyl 149, followed by a treatment with phosphorus oxybromide, yielded the dibromo 150. Substitution of the most active bromo by 92 under C-N coupling conditions afforded 151. The remaining bromo was substituted by malononitrile to afford the azaindole 152. The nitrile was hydrolyzed to the carboxamide upon treatment with sulfuric and methanesulfonic acids and the methoxy deprotection was accomplished by adding DL-methionine in a one-pot sequence to provide the racemate 153 that yielded RP-6306 after chiral SFC purification.
Scheme 11.
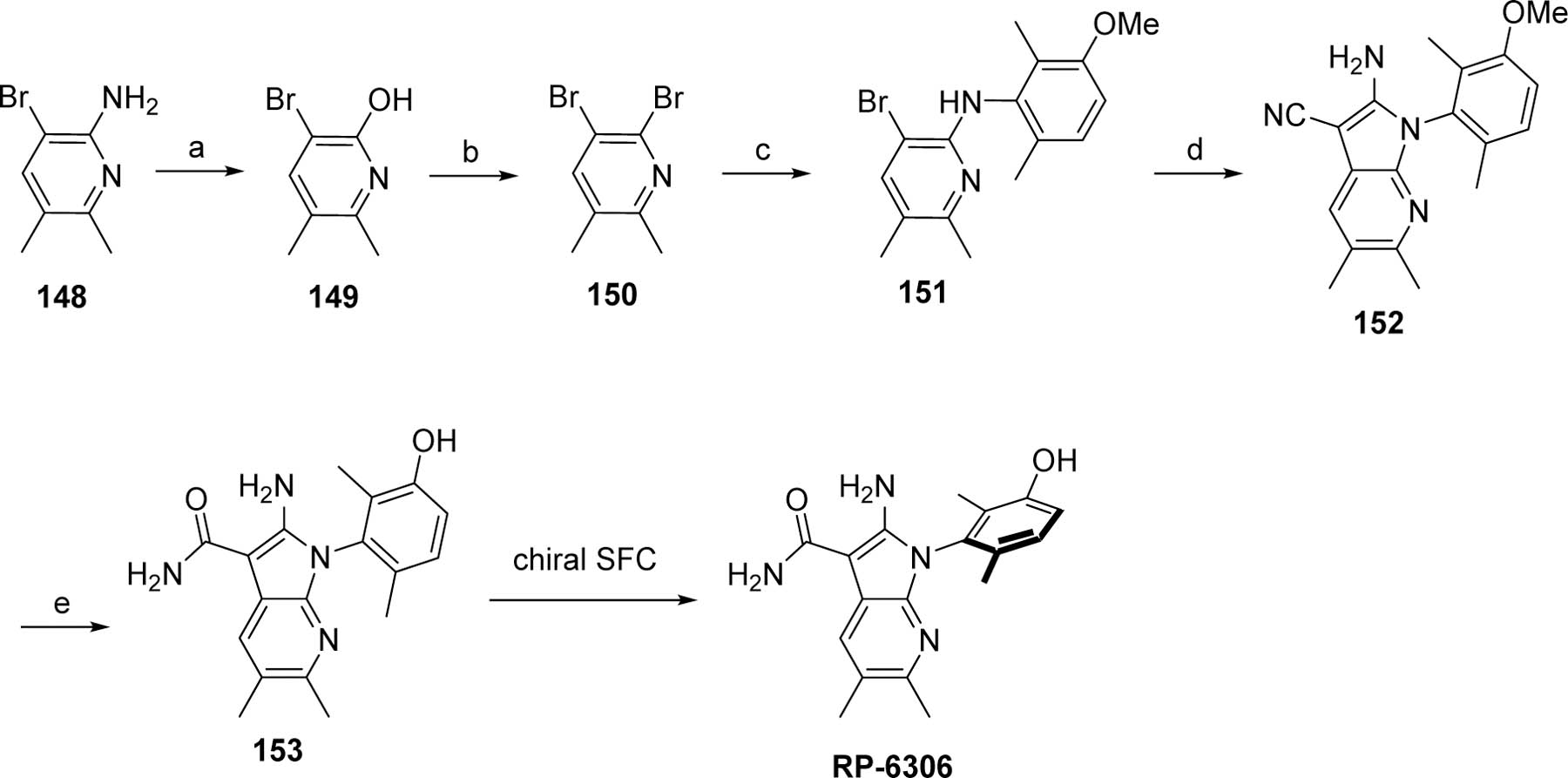
Alternative preparation of RP-6306.
Reagents and conditions. a. NaNO2, H2SO4, H2O; b. POBr3, DMF, toluene; c. 92, Pd2(dba)3, Xantphos, Cs2CO3; d. Malononitrile, NaOtBu, Pd(dppf)Cl2·CH2Cl2, DME; e. H2SO4, MeSO3H, H2O, then DL-methionine.
CONCLUSION
PKMYT1 is an important regulator of CDK1 phosphorylation and is a compelling target for treatment of certain types of DNA damage response cancers due to its established synthetic lethal relationship with CCNE1 amplification. Starting from the non-specific ephrin inhibitor 1 and supported by multiple co-crystal structures, the optimization of key properties including PKMYT1 cell-based potency, kinase selectivity and ADME properties yielded the orally bioavailable and selective PKMYT1 inhibitor RP-6306. This compound showed a favorable pharmacokinetic profile in preclinical species and was efficacious in a mouse xenograft model. The first-in-class clinical candidate RP-6306 is currently being evaluated in Phase 1 clinical trials (NCT04855656 and NCT05147272) to investigate the pharmacological role of PKMYT1 in the treatment of genetically-selected solid tumors.
EXPERIMENTAL SECTION
Molecular modeling
The crystal structures of the inhibitor:PKMYT1 complexes were analyzed with MOE (Molecular Operating Environment, Chemical Computing Group, Inc., Montreal, QC, Canada), beginning with the 2017 version and continuing through MOE 2020. MOE’s “QuickPrep” tool was used with the default settings to calculate the pKa shifts of titratable residues, add the hydrogen atoms, and flip His, Asn, or Gln residues to optimize the hydrogen bonding network. A restrained energy minimization was also performed on the ligand and the residues near it, as part of the default QuickPrep process. All molecular images were made using MOE.22
Mouse and Rat Pharmacokinetic Studies.
Female CD1 mice (20−30 g) or male Sprague−Dawley rats (200−300 g) were administered with the test compound at a dose of 1 mg/kg intravenously using a solution formulation. Oral bioavailability was determined at doses 2.5 or 5 mg/kg using the same formulation. In the mouse, RP-6306 and compound 55 were dosed as a suspension in 0.5% methyl cellulose. The blood samples for the IV experiment were collected at pre-dose, 5, 15, 30 min, 1, 2, 4, 8 and 24 h time points. The blood samples for the PO experiment were collected at pre-dose, 15, 30 min, 1, 2, 4, 6, 8 and 24 h time points. EDTA-plasma was obtained for the rat pharmacokinetic determinations while micro-sampled whole blood was collected for the mouse pharmacokinetic determinations using a previously described method.1 Mouse efficacy pharmacokinetic parameters were determined following doses of 2.5, 7.5 and 20 mg/kg PO from SCID-beige mice with micro-sampling at 30min, 2, 8, 8.5, 10 and 24 h time points. All samples were quantified using a reversed-phase liquid chromatography gradient coupled to electrospray mass spectrometry operated in positive mode. PK parameters were calculated using non-compartmental analysis.
Dog and Monkey Pharmacokinetic Study.
Male Beagle dogs (~10 kg) or Cynomolgus monkeys (~3 kg) were administered with the test compound at a dose of 0.5 mg/kg intravenously using a solution formulation. Oral bioavailability was determined at a dose of 1 mg/kg using the same formulation. The blood samples for the IV experiment were collected at pre-dose, 5, 15, 30 min, 1, 2, 4, 8 and 24 h time points. The blood samples for the PO experiment were collected at pre-dose, 15, 30 min, 1, 2, 4, 6, 8 and 24 h time points. EDTA-plasma was obtained for the pharmacokinetic determinations. All samples were quantified using a reversed-phase liquid chromatography gradient coupled to electrospray mass spectrometry operated in positive mode. PK parameters were calculated using non-compartmental analysis.
PKMYT1 enzymatic assay (ADP GLO)
To determine the IC50 of PKMYT1 inhibitor compounds, the ADP-GLO assay (Promega Corp.) was used. First, human recombinant PKMYT1 enzyme (Thermo Fisher # A33387) was diluted in the Enzyme Assay Buffer (70 mM Hepes, 3 mM MgCl2, 3 mM MnCl2, 50 μg/mL PEG20000, 3 μM Na-Orthovanadate (added fresh), 1.2 mM Dithiothreitol (added fresh) in a 5 μL volume and plated in white 384-well plates. Then, 5 μL of inhibitor or DMSO control was diluted in Enzyme Assay Buffer and added to the plate. The enzyme/compound mix was then incubated at room temperature for 15 minutes. Finally, the enzymatic reaction was started by the addition of 5 μL of ATP (diluted in Enzyme Assay Buffer) so that the final ATP concentration is 10 μM and the final PKMYT1 enzyme concentration was 18.5 nM. The enzymatic reaction was then incubated in a 30C incubator for 1 hour. At the end of the incubation period, 15 μL of ADP-GLO Reagent was added and the plate was incubated at room temperature for 40 minutes. Finally, 30 μL of the Kinase Detection Reagent was added and the plate was incubated at room temperature for 30 minutes after which the luminescence was measured using the Envision Plate reader (Perkin-Elmer). The IC50 was then determined for each compound screened in the assay. Reported IC50 in this manuscript are the geometrical mean of at least n=3 replicates.
PKMYT1/Kinases NanoBret Assay
To determine the affinity of compounds in the NanoBRET target engagement assay, HEK293 T cells were transfected with NanoLuc fusion vector DNA (PKMYT1 NanoLuc Fusion Vector or other kinases of interest all purchased from Promega Corp.) and Transfection carrier DNA using the Fugene HD Transfection reagent (Promega Corp.) in Opti-MEM No Phenol Red buffer. After an overnight incubation in a 37 °C/5% CO2 incubator, the transfected HEK293 T cells were trypsinized, counted and resuspended in Opti-MEM No Phenol Red buffer at a concentration of 200000 cells/mL. White 96-well plates were then plated with 85 μL of cells (17000 cells/well) to which 5 μL of the 20X tracer solution diluted in tracer dilution buffer (Promega Corp.) was added. Finally, 10 μL of the 10X compounds diluted in Opti-MEM No Phenol Red buffer was added and the plates were then incubated in a 37 °C/5% CO2 incubator for 2 hours. After this incubation, a 50 μL 3X solution of the Substrate/Inhibitor mix was added to the cells. The plate was then transferred to the Perkin Elmer EnVision Multimode plate reader where the Acceptor emission (610 nm) and the Donor emission (450 nm) are measured. Reported IC50 in this manuscript are the geometrical mean of at least n=3 replicates.
PKMYT1 cell-based activity assay (CDK1 pThr14 AlphaLISA)
To determine compound IC50, FUOV1 cells were plated into a 96-well TC-treated culture plate at 50000 cells/well in a final volume of 100 μL of media. The plates were then allowed to equilibrate in a biological safety cabinet for 30 minutes before being placed in a humidified incubator at 37 °C and 5% CO2 overnight. The next day, 2 μL of PKMYT1 inhibitors or DMSO were diluted in 400 μL of warmed culture media in a 96-well block using a Biomek FX liquid handler. Compounds were mixed in media and then 25 μL was dispensed into each well of the 96-well cell plate. Plates were centrifuged at 300 g for 10 seconds and then placed in the incubator for 2 hours. After the 2-hour incubation with compound, media was removed via aspiration using a multichannel pipette. 30 μL of 1X AlphaLISA lysis buffer (Perkin Elmer) supplemented with protease and phosphatase inhibitors as well as 1 mM PMSF, was added to each well. Plates were rotated at 500g for 20 minutes to facilitate lysis. Plates were then sealed with aluminum foil and frozen at −80 °C for at least 1 hour. Lysates were thawed at 37 °C for 10 minutes, then 10 μL of each lysate was transferred in duplicate to a white 384-well assay plate. Antibody mixture was prepared in 1X AlphaLISA assay buffer (Perkin Elmer) containing antibodies (5 nM final concentration for rabbit pThr14-CDK1 from Abcam #ab58509 and mouse total CDK1 from ThermoFisher Scientific #33–1800). 5 μL of antibody mixture was added to each well of the assay plate. Assay plate was sealed and stored at 4 °C overnight. The next day, AlphaLISA bead mixture (Perkin Elmer) was prepared in 1X AlphaLISA assay buffer. Anti-rabbit IgG Acceptor (Perkin Elmer #AL104C) and anti-mouse IgG Donor beads (Perkin Elmer #AS104D) were prepared to a concentration of 80 μg/ml in assay buffer. 5 μL of bead mixture was added to each well of the assay plate (20 μg/ml final concentration for each bead). The plate was protected from light and incubated for 2 hours at room temperature. After a 2 h incubation with beads, the plate was read using the Perkin Elmer EnVision Multimode plate reader with excitation at 680 nm and emission at 615 nm. Reported IC50 in this manuscript are the geometrical mean of at least n=3 replicates.
Kinase binding selectivity
Kinase binding selectivity was performed at ActivX Biosciences using the Kinativ kinase profiling platform. Unstimulated Colo205 cells were lysed in a lysis buffer of HEPES pH 7.8, 150 mM NaCl, 20 mM MnCl2, and 0.1 % Triton X-1000 and lysate was gel-filtered to remove endogenous nucleotides including ATP. The standard cell lysate preparations were labeled at a concentration of 5–10 mg/mL and were incubated with 1.2 μM of RP-6306 to establish binding to native kinases. An ADP-chemical probe with an acyl group off the β-phosphate was used to compete RP-6306 from the kinases. Once cell lysates were labelled with irreversible competitor probes, the probe bound proteins were purified using streptavidin beads and quantitation of enriched kinases was achieved through liquid chromatography-tandem mass spectrometry using ActivX’s proprietary data analysis pipeline 21. Each assay was performed in duplicate. The percentage changes in mass spectrometry signals reported were statistically significant (Student T-test score <0.04).
Chow formulation
A suspension of RP-6306 (600 mg) in acetone (80 mL) was sonicated for 30 sec, then heated to 60 °C for 5 min. The beige solution was slowly added to 1999 g of untreated feed (Purina Mills Rodent Meal 5002C) while mixing at speed 2 with the Kitchen Aid Stand Mixer 6QT 575Watt using the paddle attachment and the cover. The flask containing the solution was rinsed with 10 mL of acetone to ensure a complete transfer. The feed was mixed at speed 2 for 1.5 h. The treated feed was transferred into a clean, labeled bottle and vacuum was applied for 18 h. The 300 ppm formulated chow was transferred into a sterile bag.
128 g of RP-6306–300 ppm treated feed and 642 g of untreated feed were transferred in a clean mixing bowl. After mixing at speed 3 for 1.5 h, the mixing was halted, the mixture was stirred manually with a spatula with special attention to the surface of the bowl (i.e. bottom and sides), then the mixture was mixed for another 1.5 h at speed 3. The 50 ppm treated feed was transferred into a clean, sterile bag.
25 g of RP-6306–300 ppm treated feed and 475 g of untreated feed were transferred in a clean mixing bowl. After mixing at speed 3 for 1.5 h, the mixing was halted, the mixture was stirred manually with a spatula with special attention to the surface of the bowl (i.e. bottom and sides), then the mixture was mixed for another 1.5 h at speed 3. It was inspected for any large clumps, none were found. The 15 ppm treated feed was transferred into a clean, sterile bag.
The content and uniformity were verified prior to use using replicate 1 g samples of the formulated feed. Samples were extracted by mixing and sonication with 10 volumes of 1:1:1 methanol:acetonitrile:water. The extracts were centrifuged and the supernatant was diluted further with 1:1:1 methanol:acetonitrile:water prior to HPLC-UV analysis using a reversed-phase chromatography method. A 5-point calibration curve bracketing the concentration range of samples was used for quantitation.
Cell line-derived xenografts
OVCAR3 cells were implanted at 5×106 cells per mouse into the right flanks of female SCID-beige mice (5–7 weeks old; Charles River), in 1:1 matrigel: media (Matrigel Corning, cat# CB35248). When tumors reached the target size of 100–150 mm3, (n=8) mice were randomized to treatment groups and treatment with RP-6306 was initiated. In vivo studies involving cell-derived xenografts were performed at Repare Therapeutics, in a CCAC (Canadian Council on Animal Care)-accredited vivarium with an Institutional Animal Care Committee-approved protocol. RP-6306 was formulated in chow at 15–300 ppm or in 0.5% methylcellulose and orally administered twice daily (BID, 0–8 h) for a maximum of 21 days. Chow treated mice were acclimatized to blank chow prior to drug-formulated chow for 3–5 days. Tumor volume was measured using a digital caliper and calculated using the formula 0.52×L×W2. TGI was defined as the formula: % TGI= ((TVvehicle/last – TVvehicle/day0) - (TVtreated/last – TVtreated/day0)) / (TVvehicle/last – TVvehicle/day0) x100 calculated based on the means of the treatment groups at day 0 and last day of measurement. Change in body weight (BW) was calculated using the formula: % BW change = (BWlast-BWday0/ BWday0) x100. BW change was calculated based on individual body weight changes relative to day 0. Statistical significance relative to vehicle control or other test groups was established by one-way ANOVA followed by Fisher’s LSD test for multiple groups and unpaired t-test for two group comparisons (GraphPad Prism v9.0).
Blood and tumor tissue collection
To determine PKMYT1 target inhibition in vivo, whole blood was collected by cardiac puncture from OVCAR3 tumor bearing mice (n=4 per dose level and time point) under isoflurane anesthesia and transferred to tubes containing 0.1 M citric acid (3:1 citric acid:blood) and stored at −20 °C for LC-MS/MS analysis. Tumors were removed from mouse flanks (n=4) and cleared of surrounding mouse stroma. RP-6306 Quantitation by LC-MS-MS The extraction of whole blood samples was performed by protein precipitation using four volumes of acetonitrile. The sample extracts were analyzed using a Transcend LX2 / Ultimate 3000 liquid chromatography system coupled to a Thermo Altis triple quadrupole electrospray mass spectrometer (Thermo Fisher Scientific) operated in positive mode. Separations were performed using a 2 x 50mm, 2.8μm Pursuit XRS C8 HPLC column (Agilent). A reversed-phase linear gradient of water + 0.1% formic acid and 1:1 acetonitrile:MeOH was used to elute RP-6306 and the internal standard. Samples were quantified against a 10-point linear standard curve and 3 levels of quality control samples. Whole blood concentrations of RP-6306 were converted to free unbound plasma concentrations using an in vitro derived blood / plasma ratio = 1.2 and fraction unbound (fu) plasma = 0.185 from the CD-1 mouse strain. To simulate the free RP-6306 plasma levels at 15ppm, the mean Cmax/dose values were calculated from the 50, 150 and 300 ppm doses and then through linear regression (Excel), extrapolated to the 15 ppm dose. AUC was calculated using WinNonLin.
ELISA assay Tumor samples were homogenized in MSD Tris lysis buffer (Meso Scale Discovery, #R60TX-2) supplemented with 1X Halt Protease (Thermo Fisher Scientific, #78429) and phosphatase inhibitors (Thermo Fisher Scientific, #78426) using a Beadruptor tissue homogenizer (OMNI International) and clarified by centrifugation. ELISA was performed using an anti-CDK1 capture antibody Thermo Fisher Scientific #33–1800) and an anti- CDK1-pT14 detector antibody (Abcam #ab58509) with a secondary anti-rabbit HRP conjugate (Jackson Immunoresearch #111–035-144). The absorbance was measured in 96-well plate format on an EnVision2105 at 450 nm. Samples were quantified relative to a standard protein extract and an MSD lysis buffer used as a blank to control for inter-day variability.
Protein expression and purification
A plasmid expressing PKMYT1 (residues 76 to 362) fused to a TEV cleavable N-terminal 6XHis tag was a gift from Nicola Burgess-Brown at the Structural Genomics Consortia at Oxford (Addgene plasmid #39061; http//nt2.net/addgene:39061; RRID:addgene_39061). PKMYT1 protein was expressed in the BL21 DE3 RIL strain of E.coli. In brief, ten ml of overnight culture grown at 37 °C in Lysogeny Broth containing 50 mg/ml kanamycin was added to 1 L of Terrific Broth containing 50 mg/ml kanamycin. The diluted culture was grown at 37 °C with shaking at 180 rpm until an optical density of 0.8. Then the temperature was lowered to 16 °C and IPTG was added to the culture to a final concentration of 0.5 mM once the incubator temperature reached 20 degrees Celsius. The culture was incubated overnight, and the bacterial pellet was harvested by centrifugation at 6000 g for 15 minutes. Harvested bacterial pellet was lysed in buffer containing 50 mM HEPES pH 7.5, 500 mM NaCl, 5 mM imidazole, 5% glycerol, 0.5 mM TCEP, and 2 mM PMSF. Lysate was clarified by centrifugation, passed over a 5 mL HiTrap Ni-chelation column (GE LifeScience Inc.) and bound protein was eluted using a gradient from 5 mM to 500 mM imidazole. Fractions containing PKMYT1, as detected by SDS-PAGE, were pooled, concentrated, and loaded onto a Superdex S200 sizing column equilibrated in buffer containing 25 mM HEPES pH 7.5, 500 mM NaCl, and 0.5 mM TCEP for final polishing and buffer exchange.
Crystallography
Apo crystals of 6xHis-TEV-PKMYT176–362 were grown in hanging drop 24-well plates by mixing 2 μL of a solution containing 6.25 mg/ml protein with 1 μL of well solution (5.6 – 6.6% PEG3350, 0.2 M Na2SO4, 0.1 M Tris-HCl; pH 8.25 and 10% EG) at 4 °C for 1 day. The crystallization plate was then transferred to 20°C and crystals appeared in approximately 7 days. Inhibitor soaks were performed by transfer of apo crystals into a cryo-stabilization solution containing 12% PEG3350, 0.2 M Na2SO4, 0.1 M Tris-HCl pH 8.25, 25% ethylene glycol and 0.25 mM inhibitor, for 2 h. Crystals were then harvested and plunge frozen in liquid nitrogen.
Diffraction data was collected from single frozen crystals at 0.97918 Å wavelength on beamline NE-CAT beamline 24ID-C (APS, Chicago, Il) and processed with XDS.38 Molecular replacement was performed using Phaser with the structure of the PKMYT1 kinase domain (PDB: 3P1A) used as a search model.39 Refinement was performed using PHENIX with TLS parameters and torsion-angle NCS activated.40 Model building was performed using Coot.41 Software used in this project was curated by SBGrid.42 X-ray data collection and refinement statistics are summarized in Table S1.
Synthesis
Solvents and reagents were obtained from commercial suppliers and were used without further purification. UPLCMS analyses for reaction monitoring were performed on a Waters Acquity-H UPLC Class system using an Acquity UPLC HSS C18 2.1x30 mm column eluting with a gradient (1.86 min) of acetonitrile (15% to 98%) in water (both containing 0.1% formic acid) using electrospray ionization (ESI). Prep-HPLC separations were performed on a Teledyne Isco Combi Flash EZ Prep system using either Phenomenex Gemini® 5 μm NX-C18 110Å 150 x 21.2 mm column at a flow of 40 mL/min over 12 min (<100 mg or multiple injections of <100 mg) unless otherwise specified or HP C18 RediSep Rf gold column (>100 mg) eluting with a gradient of acetonitrile in water (both containing 0.1% formic acid) unless otherwise specified. Purifications by silica gel chromatography were performed on a Teledyne Isco Combi Flash Rf system using RediSep Rf silica gel columns. All compounds are >95% pure by HPLC analysis. Purity of final compounds was assessed by injection of a small aliquot on a Waters Acquity-H UPLC® Class system using an Acquity® UPLC BEH C18 2.1x50 mm column eluting with a gradient (7 min) of acetonitrile (2% to 98%) in water (both containing 0.1% formic acid). Magnetic resonance (NMR) spectra were obtained on a Varian 400 MHz NMR spectrometer with an Oxford NMR AS400 magnet and are referenced in ppm relative to the residual solvent peak in the indicated solvent. For 1H NMR spectra, multiplicities, coupling constants in hertz, and numbers of protons are indicated parenthetically. HRMS Samples were chromatographed using a Waters Acquity H-class UPLC system by employing a 4-minute aqueous gradient from 15 to 90% acetonitrile with 0.1% formic acid. High resolution mass spectra were collected using a Waters Xevo G2 Q-tof mass spectrometer operated in positive mode. A lockspray solution containing leucine enkephalin was used to maintain mass accuracy during analysis. Calibration was performed according to the manufacturer’s guidelines and the mass accuracy was determined within 5 ppm of the theoretical exact mass. The enantiomeric excess values were calculated based on the UV absorption (at 254 nm) areas for the two enantiomers. Structural assignments of the separated atropisomers were confirmed by biological activity where the eutomer was assigned to have the (S) configuration, which was confirmed by X-ray crystallography of some analogs including 39 and RP-6306.
2-(3-chloroquinoxalin-2-yl)propanedinitrile (59).
Adapting a known procedure,43 malononitrile (6.83 g, 104 mmol) was carefully added by portions to a vigorously stirring suspension of sodium hydride (60% dispersion in mineral oil, 4.07 g, 106 mmol) in DME (200 mL). After the addition, the stirring was continued for 20 min at RT and then 58 (10.16 g, 51.1 mmol) was added. The mixture was stirred at RT for 10 min and then heated to reflux for 1 h. The volatiles were evaporated under reduced pressure and cold aqueous 1M hydrochloric acid was added to the resulting deep brown residue to give a yellow precipitate that was filtered, washed with cold water and a minimum of cold ethanol to afford 59 (6.70 g, 57% yield) as a yellow solid. ESI MS m/z 227.0 [M - H]−. 1H NMR (400 MHz, DMSO-d6) δ 7.74 – 7.67 (m, 2H), 7.63 – 7.57 (m, 1H), 7.43 – 7.37 (m, 1H).
2-amino-1-(4-methoxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (62).
A microwave vial charged with 59 (250 mg, 1.1 mmol) and 4-methoxy-2-methyl-aniline (471 mg, 3.4 mmol) in NMP (2.5 mL) was capped and heated to 130 °C for 1 h. The mixture was cooled to RT, and a saturated aqueous NaHCO3 solution was added dropwise to give a precipitate. The solid was collected by filtration, washed with water then dried under vacuum to provide provided 62 (281 mg, quantitative yield) as a tan solid. ESI MS m/z 330.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (br s, 2H), 7.93 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.75 (ddd, J = 8.2, 1.5, 0.5 Hz, 1H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.47 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.36 (d, J = 8.7 Hz, 1H), 7.11 – 7.05 (m, 1H), 6.98 (ddd, J = 8.7, 3.0, 0.6 Hz, 1H), 3.86 (s, 3H), 1.97 (s, 3H).
2-amino-1-(4-methoxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (76).
A solution of 62 (281 mg, 0.853 mmol) in sulfuric acid (2 mL) was stirred at RT for 3 h. The mixture was then slowly poured into cold water with vigorous stirring and then made slightly alkaline with the addition of concentrated aqueous NH4OH. The solid formed was collected by filtration, washed with water twice, then dried under vacuum to provide 76 (200 mg, 67% yield) as a purple solid. ESI MS m/z 348.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (br s, 2H), 7.96 – 7.91 (m, 1H), 7.80 – 7.68 (m, 2H), 7.57 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.46 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.34 (br s, 1H), 7.10 (dd, J = 2.9, 0.8 Hz, 1H), 7.00 (ddd, J = 8.7, 2.9, 0.6 Hz, 1H), 3.86 (s, 3H), 1.99 (s, 3H).
2-amino-1-(4-hydroxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (3).
To a solution of 76 (100 mg, 0.288 mmol) in DCM (3 mL) was added BBr3 (1M in DCM, 0.87 mL, 0.87 mmol). After stirring for 2 h at RT, the volatiles were evaporated under reduced pressure. The residue was dissolved in a solution of 10% MeOH in DCM (5 mL) and the volatiles were evaporated under reduced pressure. The residue was dissolved in MeOH (5 mL) and the volatiles were evaporated under reduced pressure. The residue was taken in MeOH (5 mL), made basic with Et3N and the volatiles were evaporated under reduced pressure. The residue was purified by prep HPLC (35–65% MeCN in water, 0.1% formic acid modifier). The appropriate fractions were combined and lyophilized providing 3 (21 mg, 22% yield) as a lavender solid. ESI MS m/z 334.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.93 (dd, J = 8.3, 1.5 Hz, 1H), 7.76 (dd, J = 8.3, 1.5 Hz, 1H), 7.56 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.45 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.33 (br s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 2.7 Hz, 1H), 6.80 (dd, J = 8.5, 2.8 Hz, 1H), 1.92 (s, 3H).
2-amino-1-(3-methoxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (63).
Following the procedure used to prepare 62, using 59 (250 mg, 1.1 mmol) and 3-methoxy-2-methyl-aniline (459 mg, 3.35 mmol), and running the reaction at 130 °C for 1 h. The solid recovered by filtration was purified by silica gel chromatography eluting with a gradient of 20 to 60% EtOAc in heptane to yield 63 (86 mg, 23% yield) as a brown solid. ESI MS m/z 330.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (br s, 2H), 7.94 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.75 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.58 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.48 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.41 (ddd, J = 8.5, 7.9, 0.7 Hz, 1H), 7.23 (dd, J = 8.4, 1.0 Hz, 1H), 7.03 (dd, J = 8.0, 1.1 Hz, 1H), 3.91 (s, 3H), 1.83 (s, 3H).
2-amino-1-(3-methoxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (77).
Following the procedure used to prepare 76, a solution of 63 (1.02g, 3.11 mmol) was stirred in sulfuric acid (10 mL) for 1 h to provide 77 (990 mg, 92% yield) as an orange solid. ESI MS m/z 348.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.94 (dd, J = 8.3, 1.4 Hz, 1H), 7.80 – 7.73 (m, 2H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.49 – 7.40 (m, 2H), 7.35 (br s, 1H), 7.24 (dd, J = 8.5, 1.1 Hz, 1H), 7.05 (dd, J = 8.0, 1.0 Hz, 1H), 3.91 (s, 3H), 1.85 (s, 3H).
2-amino-1-(3-hydroxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (4).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 11 mL, 11 mmol) was added to a solution of 77 (990 mg, 2.85 mmol) in DCM (11 mL) and the mixture was stirred for 90 min at RT. The residue was purified by silica gel chromatography eluting with gradient of 0 to 10% MeOH in DCM to provide 4 (335 mg, 34% yield). ESI MS m/z 334.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 7.90 (dd, J = 8.3, 1.5 Hz, 2H), 7.78 – 7.67 (m, 2H), 7.53 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.42 (ddd, J = 8.3, 7.0, 1.5 Hz, 1H), 7.33 (s, 1H), 7.21 (t, J = 7.9 Hz, 1H), 7.03 (dd, J = 8.2, 1.2 Hz, 1H), 6.85 (dd, J = 7.8, 1.1 Hz, 1H), 1.76 (s, 3H).
2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (64).
Following the procedure used to prepare 62, using 59 (4.43 g, 19.4 mmol) and 3-methoxy-2,6-dimethyl-aniline (92)33 (9.03 g, 3.08 mmol), running the reaction at 150 °C in NMP (40 mL) for 75 min. In this instance a precipitate could not be obtained after the addition of saturated aqueous NaHCO3 solution. Therefore, the aqueous mixture was extracted with EtOAc twice, the combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated under reduced pressure to yield a residue that was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane to provide 64 (3.23 g, 48% yield) as a dark orange solid. ESI MS m/z 344.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (br s, 2H), 7.95 (dd, J = 8.3, 1.4 Hz, 1H), 7.76 (dd, J = 8.4, 1.4 Hz, 1H), 7.59 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.48 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.5 Hz, 1H), 3.87 (s, 3H), 1.86 (s, 3H), 1.78 (s, 3H).
2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (6).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 0.88 mL, 0.88 mmol) was added to a solution of 7 (106 mg, 0.293 mmol) in DCM (0.9 mL) and the mixture was stirred for 2 h at RT. The residue was purified by prep HPLC (35–65% MeCN in water, 0.1% formic acid modifier). The appropriate fractions were combined and lyophilized providing 6 (55 mg, 54% yield) as a bright orange solid. ESI MS m/z 348.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (br s, 1H), 8.01 (br s, 2H), 7.95 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.81 – 7.71 (m, 2H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.46 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.38 (br s, 1H), 7.12 (dt, J = 8.3, 0.7 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 1.83 (s, 3H), 1.76 (s, 3H).
2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (7).
A solution of 64 (130 mg, 0.379 mmol) in sulfuric acid (0,5 mL) was stirred at RT for 70 min. The mixture was then slowly poured into cold water with vigorous stirring and then made slightly alkaline with the addition of concentrated aqueous NH4OH. The solid formed was collected by filtration, washed with water twice, dried under reduced pressure, and purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in hexanes to provide 7 (94 mg, 70% yield) as an ochre solid. ESI MS m/z 362.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (br s, 2H), 8.57 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 8.44 – 8.32 (m, 2H), 8.20 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 8.08 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 8.01 (br s, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.78 (d, J = 8.5 Hz, 1H), 4.50 (s, 3H), 2.50 (s, 3H), 2.42 (s, 3H).
2-amino-1-(3-chloro-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (65).
Following the procedure used to prepare 62, using 59 (250 mg, 1.1 mmol) and 3-chloro-2,6-dimethyl-aniline (515 mg, 3.31 mmol), running the reaction at 130 °C for 3 h provided 65 (70 mg, 18% yield) as an orange solid. ESI MS m/z 348.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (br s, 2H), 7.96 (dd, J = 8.6, 1.4 Hz, 1H), 7.78 (dd, J = 8.3, 1.5 Hz, 1H), 7.65 – 7.57 (m, 2H), 7.50 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.41 – 7.34 (m, 1H), 1.99 (s, 3H), 1.92 (s, 3H).
2-amino-1-(3-chloro-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (8).
Following the procedure used to prepare 7, a solution of 65 (100 mg, 0.288 mmol) in sulfuric acid (2 mL) was stirred at RT for 30 min. The recovered solid from the filtration was purified by prep HPLC (20–80% MeCN in water, 0.1% formic acid modifier). Appropriate fractions were combined and lyophilized to provide 8 (3.9 mg, 4% yield). ESI MS m/z 366.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (s, 2H), 7.92 (dd, J = 8.3, 1.4 Hz, 1H), 7.80 – 7.66 (m, 2H), 7.65 – 7.51 (m, 2H), 7.43 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.37 (d, J = 4.7 Hz, 1H), 1.95 (s, 3H), 1.89 (s, 3H).
2-amino-1-(2-methyl-5-nitro-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (66).
Following the procedure used to prepare 63, using 59 (250 mg, 1.1 mmol) and 2-methyl-5-nitro-aniline (520 mg, 3.42 mmol), running the reaction at 130 °C for 2 h, and eluting with a gradient of 20 to 60% EtOAc in heptane provided 66 (242 mg, 63% yield) as a brown solid. ESI MS m/z 345.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.52 (d, J = 2.5 Hz, 1H), 8.40 (br s + dd, J = 8.5, 2.5 Hz, 3H), 7.96 (dd, J = 8.3, 1.4 Hz, 1H), 7.82 (dd, J = 8.6, 0.9 Hz, 1H), 7.75 (dd, J = 8.3, 1.4 Hz, 1H), 7.60 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.49 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 2.16 (s, 3H).
2-amino-1-(2-methyl-5-nitro-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (78).
Following the procedure used to prepare 76, a solution of 66 (625 mg, 1.82 mmol) in sulfuric acid (8 mL) for 1 h provided 78 (500 mg, 76% yield) as an orange solid. ESI MS m/z 363.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (d, J = 2.4 Hz, 1H), 8.41 (dd, J = 8.5, 2.5 Hz, 1H), 7.96 (dd, J = 8.3, 1.4 Hz, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.78 (br s, 1H), 7.76 (dd, J = 8.2, 1.4 Hz, 1H), 7.59 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.46 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.38 (br s, 1H), 2.17 (s, 3H).
2-amino-1-(5-amino-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (9).
A suspension of 78 (500 mg, 1.38 mmol) and palladium on carbon (10% w/w, 250 mg, 0.235 mmol) in DCM (10 mL) and MeOH (10 mL) was stirred under hydrogen atmosphere (balloon) at RT for 90 min after which the suspension was filtered on Celite. The filter cake was washed with DCM. Silica gel (5 g) was added to the filtrate that was evaporated under reduced pressure and purified by silica gel chromatography eluting with a gradient of 0 to 10% MeOH in DCM to afford 9 (240 mg, 52% yield). ESI MS m/z 333.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (dd, J = 8.3, 1.4 Hz, 2H), 7.75 (dd, J = 8.2, 1.5 Hz, 1H), 7.71 (d, J = 3.3 Hz, 1H), 7.53 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.43 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.33 (s, 1H), 7.15 – 7.07 (m, 1H), 6.70 (dd, J = 8.2, 2.4 Hz, 1H), 6.53 (d, J = 2.4 Hz, 1H), 5.23 (s, 2H), 1.78 (s, 3H).
2-amino-1-(1H-indazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (67).
Following the procedure used to prepare 63, using 59 (250 mg, 1.1 mmol) and 1H-indazol-4-amine (431 mg, 3.24 mmol)), running the reaction at 150 °C for 2 h, and eluting with a gradient of 0 to 20% MeOH in DCM provided 67 (140 mg, 38% yield) as a tan solid. ESI MS m/z 326.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 13.44 (s, 1H), 8.27 (br s, 2H), 8.01 – 7.93 (m, 1H), 7.88 (s, 1H), 7.80 (dt, J = 8.4, 0.8 Hz, 1H), 7.72 – 7.63 (m, 1H), 7.58 (ddd, J = 8.6, 7.0, 1.7 Hz, 2H), 7.46 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.33 (dd, J = 7.3, 0.7 Hz, 1H).
2-amino-1-(1H-indazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (10).
Following the procedure used to prepare 7, a solution of 67 (124 mg, 0.381 mmol) was stirred in sulfuric acid (1 mL) for 1h. The solid recovered from the filtration was purified by prep HPLC (30–60% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 10 (74 mg, 56% yield) as a yellow solid. ESI MS m/z 344.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 13.45 (s, 1H), 8.07 (br s, 2H), 7.96 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.89 – 7.75 (m, 3H), 7.69 (ddd, J = 8.2, 1.5, 0.5 Hz, 1H), 7.63 – 7.51 (m, 2H), 7.44 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.42 – 7.38 (m, 1H), 7.35 (dd, J = 7.3, 0.7 Hz, 1H).
2-amino-1-(1H-indol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (68).
Following the procedure used to prepare 63, using 59 (250 mg, 1.1 mmol) and 1H-indol-4-amine (422 mg, 3.19 mmol), running the reaction at 150 °C for 1 h, and eluting with a gradient of 20 to 100% EtOAc in heptane followed by reverse phase flash chromatography on C18 cartridge with a gradient of 10 to 100% MeCN in water, 0.1% formic acid modifier) provided 68 (57 mg, 38% yield) as an olive brown solid. ESI MS m/z 325.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 8.11 (br s, 2H), 7.96 – 7.90 (m, 1H), 7.68 – 7.62 (m, 2H), 7.56 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.46 – 7.40 (m, 2H), 7.32 (dd, J = 8.1, 7.5 Hz, 1H), 7.19 (dd, J = 7.4, 0.9 Hz, 1H), 6.10 – 6.02 (m, 1H).
2-amino-1-(1H-indol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (11).
Following the procedure used to prepare 7, a solution of 68 (57 mg, 0.176 mmol) was stirred in sulfuric acid (2 mL) for 2h. The solid recovered from the filtration was purified by prep HPLC (35–65% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 11 (20 mg, 33% yield) as a yellow fluffy solid. ESI MS m/z 343.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.52 (br s, 1H), 7.95 (dd, J = 8.3, 1.4 Hz, 1H), 7.81 (d, J = 3.3 Hz, 1H), 7.71 – 7.64 (m, 2H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.47 – 7.40 (m, 2H), 7.38 (br d, J = 3.2 Hz, 1H), 7.34 (dd, J = 8.2, 7.4 Hz, 1H), 7.21 (dd, J = 7.5, 0.9 Hz, 1H), 6.07 (ddd, J = 3.0, 2.0, 0.9 Hz, 1H).
2-amino-1-(1H-benzotriazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (69).
Following the procedure used to prepare 62, using 59 (250 mg, 1.1 mmol) and 1H-benzotriazol-4-amine (427 mg, 3.18 mmol, running the reaction at 150 °C for 1 h provided 69 (275 mg, quantitative yield) as a dark purple solid. ESI MS m/z 327.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.94 (dd, J = 8.5, 1.4 Hz, 1H), 7.86 (dd, J = 7.9, 1.2 Hz, 1H), 7.72 – 7.66 (m, 1H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.45 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.18 – 7.09 (m, 2H).
2-amino-1-(1H-benzotriazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (12).
Following the procedure used to prepare 7, a solution of 69 (97 mg, 0.297 mmol) in sulfuric acid (1 mL) for 1.5h. The solid recovered from the filtration was purified by prep HPLC (30–60% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 12 (2.6 mg, 3% yield) as a yellow fluffy solid. ESI MS m/z 345.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.25 – 8.05 (m, 4H), 7.97 (dd, J = 8.4, 1.3 Hz, 1H), 7.83 (br d, J = 3.2 Hz, 1H), 7.73 – 7.64 (m, 2H), 7.64 – 7.61 (m, 1H), 7.61 – 7.56 (m, 1H), 7.48 – 7.38 (m, 2H).
2-amino-1-(1H-benzimidazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (70).
Following the procedure used to prepare 63, using 59 (250 mg, 1.1 mmol) and 1H-benzimidazol-4-amine (454 mg, 3.41 mmol), running the reaction at 130 °C for 2 h, and purification by reverse phase chromatography on C18 cartridge (10–100% MeCN in WATER, 0.1% formic acid modifier) provided 70 (33 mg, 22% yield) as a brown solid. ESI MS m/z 326.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H), 7.95 (dd, J = 8.4, 1.4 Hz, 1H), 7.84 (br s, 1H), 7.67 (dd, J = 8.3, 1.5 Hz, 1H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.50 – 7.34 (m, 3H).
2-amino-1-(1H-benzimidazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (13).
Following the procedure used to prepare 7, a solution of 70 (33 mg, 0.101 mmol) was stirred in sulfuric acid (1 mL) for 45 min. The solid recovered from filtration was purified by prep HPLC (25–55% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 13 (8 mg, 23% yield) as a yellow fluffy solid. ESI MS m/z 344.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H), 7.96 (dd, J = 8.4, 1.4 Hz, 1H), 7.81 (s, 2H), 7.72 – 7.64 (m, 1H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.48 – 7.39 (m, 3H), 7.37 (br s, 1H).
2-amino-1-[5-(difluoromethyl)-2-methyl-phenyl]pyrrolo[3,2-b]quinoxaline-3-carbonitrile (71).
Following the procedure used to prepare 62, using 59 (50 mg, 0.22 mmol) and 5-(difluoromethyl)-2-methyl-aniline (103 mg, 0.66 mmol), running the reaction at 130 °C for 3 h provided 71 (160 mg, 70% yield) as a yellow solid. ESI MS m/z 350.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (s, 2H), 7.96 – 7.86 (m, 1H), 7.76 – 7.60 (m, 3H), 7.54 (dddd, J = 8.4, 6.9, 2.8, 1.5 Hz, 1H), 7.48 – 7.40 (m, 1H), 7.39 – 7.20 (m, 1H), 7.05 (t, J = 55.7 Hz, 1H), 2.07 – 1.88 (m, 3H).
2-amino-1-[5-(difluoromethyl)-2-methyl-phenyl]pyrrolo[3,2-b]quinoxaline-3-carboxamide (14).
Following the procedure used to prepare 7, a solution of 71 (150 mg, 0.429 mmol) was stirred in sulfuric acid (2 mL) for 30 min. The solid recovered from filtration was purified by prep HPLC (20–80% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 14 (46 mg, 29% yield). ESI MS m/z 368.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 2H), 7.95 (m, 1H), 7.76 (m, 3H), 7.73 – 7.67 (m, 2H), 7.62 – 7.53 (m, 1H), 7.46 (m, 1H), 7.41 – 7.35 (m, 1H), 7.10 (t, J = 55.7 Hz, 1H), 2.09 (s, 3H).
2-amino-1-(5-methyl-1H-indazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (72).
Following the procedure used to prepare 63, using 59 (250 mg, 1.1 mmol) and 5-methyl-1H-indazol-4-amine (495 mg, 3.36 mmol), running the reaction at 130 °C for 80 min. The solid was purified by preparative HPLC (Phenomenex Gemini) eluting with a gradient of CH3CN (30 to 60%) in water both containing 0.1% formic acid provided 72 (37 mg, 10% yield) as a dark brown solid. ESI MS m/z 340.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 13.32 (s, 1H), 8.25 (br s, 2H), 7.96 (dd, J = 8.4, 1.4 Hz, 1H), 7.75 (s, 1H), 7.71 (d, J = 8.7 Hz, 1H), 7.67 (dd, J = 8.3, 1.4 Hz, 1H), 7.58 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.49 – 7.43 (m, 2H), 2.10 (s, 3H).
2-amino-1-(5-methyl-1H-indazol-4-yl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (15).
Following the procedure used to prepare 7, a solution of 72 (35 mg, 0.10 mmol) was stirred in sulfuric acid (1 mL) for 1.5 h. The solid recovered by filtration was purified by prep HPLC (30–60% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 15 (15 mg, 41% yield) as a light beige fluffy solid. ESI MS m/z 358.2 [M + H]+. 1H NMR (500 MHz, DMSO-d6) δ 13.33 (s, 1H), 8.03 (br s, 2H), 7.96 (dd, J = 8.4, 1.4 Hz, 1H), 7.81 (br d, J = 3.3 Hz, 1H), 7.76 – 7.71 (m, 2H), 7.68 (dd, J = 8.3, 1.4 Hz, 1H), 7.57 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.43 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.39 (br s, 1H), 2.12 (s, 3H).
2-amino-1-(2-chloro-3-methoxy-6-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (73).
Following the procedure used to prepare 64, using 59 (250 mg, 1.1 mmol) and 2-chloro-3-methoxy-6-methyl-aniline44 (586 mg, 3.41 mmol), running the reaction at 130 °C for 7 h and eluting with a gradient of 20 to 100% EtOAc in heptane provided 73 (120 mg, 30% yield) as a brown orange solid. ESI MS m/z 364.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (br s, 2H), 7.95 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.77 (ddd, J = 8.2, 1.5, 0.5 Hz, 1H), 7.60 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.53 – 7.43 (m, 2H), 7.37 (d, J = 8.7 Hz, 1H), 3.94 (s, 3H), 1.99 (s, 3H).
2-amino-1-(2-chloro-3-methoxy-6-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (79).
Following the procedure used to prepare 76, a solution of 73 (49 mg, 0.135 mmol) was stirred in sulfuric acid (1 mL) for 1.5 h provided 79 (48 mg, 93% yield) as a light yellow solid. ESI MS m/z 382.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (br s, 2H), 7.95 (dd, J = 8.5, 1.4 Hz, 1H), 7.79 – 7.71 (m, 2H), 7.59 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.50 – 7.43 (m, 2H), 7.37 (br s + d, J = 8.7 Hz, 2H), 3.94 (s, 3H), 1.99 (s, 3H).
2-amino-1-(2-chloro-3-hydroxy-6-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (16).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 0.4 mL, 0.4 mmol) was added to a solution of 79 (48 mg, 0.126 mmol) in DCM (1 mL) and the mixture was stirred for 3 h at RT. Then more BBr3 (1M in DCM, 0.4 mL, 0.4 mmol) was added and the mixture was stirred for an additional 3 h. The residue was purified by prep HPLC (30–60% MeCN in water, 0.1% formic acid modifier) provided 16 (17 mg, 37% yield) as a light orange fluffy solid. ESI MS m/z 368.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (br s, 1H), 8.17 (br s, 2H), 7.95 (ddd, J = 8.3, 1.5, 0.5 Hz, 1H), 7.78 (ddd, J = 8.2, 1.5, 0.5 Hz, 1H), 7.75 (br d, J = 3.1 Hz, 1H), 7.58 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.47 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.39 (br s, 1H), 7.28 (dd, J = 8.5, 0.8 Hz, 1H), 7.16 (d, J = 8.5 Hz, 1H), 1.94 (s, 3H).
3-chloro-N-(6-chloro-3-methoxy-2-methyl-phenyl)quinoxalin-2-amine (60).
To a cold (0 °C) mixture of 58 (303 mg, 1.52 mmol) and 6-chloro-3-methoxy-2-methyl-aniline44 (507 mg, 2.95 mmol) in THF (10 mL) was added slowly a solution of potassium tert-butoxide in THF (1 M, 4.6 mL). After stirring for 1 h at 0°C, the mixture was quenched with saturated aqueous NH4Cl and diluted with EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc twice. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 30% EtOAc in hexanes to provide 60 (133 mg, 26% yield) as an off-white solid. ESI MS m/z 334.2 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.86 (ddd, J = 8.3, 1.5, 0.6 Hz, 1H), 7.65 (ddd, J = 8.4, 1.5, 0.6 Hz, 1H), 7.55 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.45 (ddd, J = 8.3, 7.0, 1.5 Hz, 1H), 7.31 (dd, J = 8.9, 0.7 Hz, 1H), 7.13 (br s, 1H), 6.82 (d, J = 8.8 Hz, 1H), 3.89 (s, 3H), 2.14 (s, 3H).
2-amino-1-(6-chloro-3-methoxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (74).
To a suspension of sodium hydride (60% dispersion in mineral oil, 54 mg, 1.41 mmol) in dioxane (2 mL), was added a solution of malononitrile (58 mg, 0.878 mmol) in dioxane (0.5 mL). After 20 min, 60 (133 mg, 0.398 mmol) in dioxane (2 mL) and Pd(PPh3)4 (48 mg, 0.041 mmol) were added. The mixture was flushed with nitrogen and heated to 100 °C for 1h under a nitrogen atmosphere. The mixture was cooled to RT, quenched with saturated aqueous NH4Cl, extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in hexanes to provide 74 (130 mg, 90% yield) as a yellow solid. ESI MS m/z 364.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (br s, 2H), 7.95 (dd, J = 8.4, 1.5 Hz, 1H), 7.77 (dd, J = 8.3, 1.4 Hz, 1H), 7.63 – 7.55 (m, 2H), 7.49 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.28 (d, J = 9.0 Hz, 1H), 3.92 (s, 3H), 1.90 (s, 3H).
2-amino-1-(6-chloro-3-methoxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (80).
Following the procedure used to prepare 76, a solution of 74 (130 mg, 0.357 mmol) in sulfuric acid (1 mL) for 2.5 h provided 80 (136 mg, 98% yield) as a light yellow solid. ESI MS m/z 382.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.99 – 7.92 (m, 1H), 7.81 – 7.76 (m, 1H), 7.75 (br s, 1H), 7.62 – 7.54 (m, 2H), 7.47 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.39 (br s, 1H), 7.28 (d, J = 9.0 Hz, 1H), 3.92 (s, 3H), 1.90 (s, 3H).
2-amino-1-(6-chloro-3-hydroxy-2-methyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (17).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 1 mL, 1 mmol) was added to a solution of 80 (70 mg, 0.183 mmol) in DCM (1 mL) and the mixture was stirred for 2 at RT. The residue was purified by prep HPLC (35–65% MeCN in water, 0.1% formic acid modifier) to provide 17 (36 mg, 53% yield) as a pale yellow fluffy solid. ESI MS m/z 368.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (br s, 1H), 8.18 (br s, 2H), 7.95 (dd, J = 8.5, 1.4 Hz, 1H), 7.82 – 7.76 (m, 1H), 7.75 (br d, J = 2.7 Hz, 1H), 7.58 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.47 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H), 7.42 – 7.34 (m, 2H), 7.09 (d, J = 8.8 Hz, 1H), 1.86 (s, 3H).
3-chloro-N-[2,6-dichloro-3-[(4-methoxyphenyl)methoxy]phenyl]quinoxalin-2-amine (61).
To a cold (0 °C) mixture of 58 (98 mg, 0.492 mmol) and 2,6-dichloro-3-[(4-methoxyphenyl)methoxy]aniline44 (292 mg, 0.979 mmol) in THF (4 mL) was added slowly a solution of potassium tert-butoxide in THF (1 M, 1.50 mL). After stirring for 1 h at 0°C, the mixture was quenched with saturated NH4Cl and diluted with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc twice. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified twice by silica gel chromatography eluting with a gradient of 0 to 50% EtOAc in hexanes to provide 61 (109 mg, 48% yield) as a sticky off-white foam. ESI MS m/z 462.0 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.88 (ddd, J = 8.3, 1.5, 0.6 Hz, 1H), 7.67 (ddd, J = 8.4, 1.5, 0.7 Hz, 1H), 7.58 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.49 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.43 – 7.38 (m, 2H), 7.36 (d, J = 9.0 Hz, 1H), 7.12 (s, 1H), 6.99 – 6.90 (m, 3H), 5.14 (s, 2H), 3.82 (s, 3H).
2-amino-1-[2,6-dichloro-3-[(4-methoxyphenyl)methoxy]phenyl]pyrrolo[3,2-b]quinoxaline-3-carbonitrile (75).
To a vial containing sodium hydride (60% dispersion in mineral oil, 15 mg, 0.391 mmol) in dioxane (2 mL), was added a solution of malononitrile (17 mg, 0.257 mmol) in dioxane (0.5 mL). After 20 min, 61 (59 mg, 0.128 mmol) in dioxane (1 mL) and Pd(PPh3)4 (15 mg, 0.013 mmol) were added, the mixture was flushed with nitrogen and heated to 100 °C for 1h under nitrogen atmosphere. The mixture was cooled to RT, quenched with saturated NH4Cl, extracted with EtOAc twice. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified twice by silica gel chromatography eluting with a gradient of 0 to 50% EtOAc in hexanes to provide 75 (40 mg, 63% yield) as a yellow solid. ESI MS m/z 490.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (br s, 2H), 7.96 (dd, J = 8.4, 1.5 Hz, 1H), 7.79 (dd, J = 8.3, 1.5 Hz, 1H), 7.75 (d, J = 9.1 Hz, 1H), 7.65 – 7.57 (m, 2H), 7.51 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.49 – 7.41 (m, 2H), 7.06 – 6.88 (m, 2H), 5.25 (dd, J = 19.2, 11.4 Hz, 2H), 3.77 (s, 3H).
2-amino-1-(2,6-dichloro-3-hydroxy-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (18).
Following the procedure used to prepare 7, a solution of 73 (71 mg, 0.145 mmol) was stirred in sulfuric acid (1 mL) for 1 h. The solid recovered from the filtration was purified by prep HPLC (35–65% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 18 (29 mg, 52% yield) as a fluffy yellow solid. ESI MS m/z 388.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.33 (br s, 2H), 7.95 (dd, J = 8.3, 1.4 Hz, 1H), 7.79 (dd, J = 8.3, 1.4 Hz, 1H), 7.74 (br s, J = 3.1 Hz, 1H), 7.60 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.56 (d, J = 9.0 Hz, 1H), 7.48 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.41 (br d, J = 3.1 Hz, 1H), 7.26 (d, J = 9.0 Hz, 1H).
3-chloro-N-[5-(methoxymethoxy)-2-methyl-phenyl]quinoxalin-2-amine (82).
In a 20 mL microwave vial, 58 (1 g, 5.0 mmol) was dissolved in toluene (10 mL). 5-(methoxymethoxy)-2-methyl-aniline (840 mg, 5.0 mmol) and sodium tert-butoxide (580 mg, 6.0 mmol), Pd2dba3 (230 mg, 0.251 mmol), and XantPhos (350 mg, 0.605 mmol) were added. The vial was purged with nitrogen, capped, and the mixture was heated to 110 °C for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% EtOAc in heptane to provide 83 (1.1 g, 66% yield). ESI MS m/z 330.24 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 7.77 (dd, J = 8.2, 1.4 Hz, 1H), 7.65 – 7.48 (m, 2H), 7.43 (m, 1H), 7.26 (d, J = 2.6 Hz, 1H), 7.17 (dd, J = 8.3, 0.8 Hz, 1H), 6.83 (dd, J = 8.3, 2.6 Hz, 1H), 5.14 (s, 2H), 3.34 (s, 3H), 2.08 (s, 3H).
2-amino-1-(5-hydroxy-2-methylphenyl)-N-methyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (19).
In a microwave vial, 82 (0.21 g, 0.637 mmol) was dissolved in THF (1.2 mL) at RT, followed by the addition of 2-cyano-N-methylacetamide (0.094 g, 0.955 mmol) and potassium tert-butoxide (0.427 g, 3.82 mmol). The mixture was capped and stirred at 80 °C for 16 h. Water (30 mL) was added to the mixture and solid precipitates was filtered, washed with water, dried under a flow of air and triturated with diethyl ether to yield crude 2-amino-1-(5-(methoxymethoxy)-2-methylphenyl)-N-methyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (0.2 g, 80%, LC-MS (ESI) m/z: 392.56 [M + H]+) that was dissolved in MeOH (5 mL) at RT. 4M HCl in Dioxane (2 mL) was added and the mixture was stirred for 2 h at RT. The volatiles were evaporated under reduced pressure, and the residue was purified by prep HPLC to afford 19 (48 mg, 28% yield) as a white solid. ESI MS m/z 348.46 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.75 (s, 1H), 8.11 – 7.96 (m, 3H), 7.79 (d, J = 8.4 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.50–7.56 (m, 1H), 7.32 (d, J = 8.0 Hz, 1H), 6.96 (dd, J = 8.4, 2.4 Hz, 1H), 6.82 (d, J = 2.4 Hz, 1H), 2.97 (d, J = 4.4 Hz, 3H), 1.89 (s, 3H).
2-amino-N-ethyl-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (20).
To a solution of 82 (0.22 g, 0.67 mmol) in DMF (3 mL), 2-cyano-N-ethylacetamide (0.089 g, 0.80 mmol) and cesium carbonate (1.09 g, 3.34 mmol) were added and the mixture was heated at 100 °C for 1 h. After cooling to RT, the mixture was slowly poured in icy water (10 mL) and extracted with EtOAc (3 X 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 30% EtOAc in hexanes to yield crude 2-amino-N-ethyl-1-(5-(methoxymethoxy)-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (0.08 g, 30%, ESI MS m/z 406.3 [M + H]+) that was dissolved in 4M HCl in 1,4-dioxane (1 mL). The mixture was stirred at for 1 h at RT. The volatiles were evaporated under reduced pressure and the residue was taken in EtOAc and washed with a saturated aqueous NaHCO3 solution. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 3% MeOH in DCM. The desired fractions were combined, evaporated under reduced pressure, and triturated with ether to afford 20 (22 mg, 35% yield) as a white solid. ESI MS m/z 362.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.20 (d, J = 5.6 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.58 (t, J = 7.2 Hz, 1H), 7.46 (t, J = 7.2 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 8.4, 2.4 Hz, 1H), 6.81 (d, J = 2.4 Hz, 1H), 3.48 – 3.40 (m, 2H), 1.88 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H).
2-amino-1-(5-hydroxy-2-methylphenyl)-N-(2-hydroxyethyl)-1H-pyrrolo [2,3-b]quinoxaline-3-carboxamide (21).
To a solution of 82 (0.12 g, 0.36 mmol) in DMF (1.2 mL) at RT, 2-cyano-N-(2-hydroxyethyl)acetamide (0.069 g, 0.55 mmol) and Cs2CO3 (0.59 g, 1.81 mmol) were added and the mixture was heated at 100 °C for 1 h. After cooling to RT, the mixture was slowly poured in icy water (30 mL) and extracted with EtOAc (3 X 10 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure to afford crude 2-amino-N-(2-hydroxyethyl)-1-(5-(methoxymethoxy)-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (150 mg, 98% yield, ESI MS m/z 422.34 [M + H]+) that was dissolved in 1,4-dioxane (1 mL). 4M HCl in 1,4-dioxane was added and the mixture was stirred for 2 h at RT. The volatiles were evaporated under reduced pressure, and the residue was purified by prep HPLC to afford 21 (19 mg, 14% yield) as a white solid. ESI MS m/z 378.26 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.40 (d, J = 5.2 Hz, 1H), 8.04 – 7.92 (m, 3H), 7.79 (d, J = 8.0 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.83 (s, 1H), 4.91 (t, J = 4.4 Hz, 1H), 3.62 (t, J = 6.0 Hz, 2H), 3.52 (t, J = 5.2 Hz, 2H), 1.89 (s, 3H).
1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (83).
To a solution of 64 (400 mg, 1.16 mmol) in THF (10 mL) was added tert-butyl nitrite (599 mg, 5.81 mmol). The mixture was stirred at RT for 30 min then refluxed for 4.75 h, cooled to RT and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in hexanes providing 83 (367 mg, 96% yield) as a light orange solid. ESI MS m/z 329.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.37 – 8.25 (m, 1H), 8.14 – 8.04 (m, 1H), 7.94 – 7.75 (m, 2H), 7.32 (dt, J = 8.5, 0.7 Hz, 1H), 7.18 (d, J = 8.5 Hz, 1H), 3.88 (s, 3H), 1.88 (s, 3H), 1.76 (s, 3H).
1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (22).
Following the procedure used to prepare 76, a solution of 83 (367 mg, 1.12 mmol) in sulfuric acid (2 mL) for 4.5 h provided 1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (356 mg, 92% yield). ESI MS m/z 347.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.35 – 8.27 (m, 1H), 8.14 – 7.99 (m, 2H), 7.88 – 7.73 (m, 3H), 7.29 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 8.5 Hz, 1H), 3.88 (s, 3H), 1.86 (s, 3H), 1.75 (s, 3H). Following the procedure used to prepare 3, BBr3 (1M in DCM, 1 mL, 1 mmol) was added to a solution of 1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (356 mg, 1.03 mmol) in DCM (3 mL) and the mixture was stirred for 1 h at RT. The residue was purified by reverse phase flash chromatography on C18 cartridge (10–100% MeCN in water, 0.1% formic acid modifier), appropriate fractions were combined and lyophilized to provide 22 (189 mg, 55% yield) as a beige fluffy solid. ESI MS m/z 333.4 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.82 (s, 1H), 8.37 – 8.24 (m, 1H), 8.11 – 8.04 (m, 2H), 7.91 – 7.73 (m, 3H), 7.11 (dt, J = 8.3, 0.7 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 1.81 (s, 3H), 1.71 (s, 3H).
2-chloro-1-(3-methoxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carbonitrile (84).
To a solution of tert-Butyl nitrite 90% (0.46 mL, 3.49 mmol) and Copper(I) chloride (576 mg, 5.82 mmol) in ACN (15 mL) at RT was added dropwise a solution of 64 (1.0 g, 2.91 mmol) in ACN (15 mL). The mixture was stirred at RT for 0.5 h and then heated at 65 °C for 2 h. The volatiles were removed under reduced pressure at RT. The residue was diluted with EtOAc and washed with water (3x50 mL) and brine. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure at RT to yield a brown oil that was adsorbed on 4 g of silica gel and purified by silica gel chromatography eluting with a gradient of 0 to 50% EtOAc in heptane to afford 84 (84.0 mg, 8% yield) as an orange solid. ESI MS m/z 363.0 [M + H]+. 1H NMR (400 MHz, CDCl3) δ ppm 1.82 (s, 3 H) 1.88 (s, 3 H) 6.14 (s, 1 H) 6.90 (d, J = 8.31 Hz, 1 H) 7.11 (d, J = 8.31 Hz, 1 H) 7.82 (dddd, J = 18.49, 8.34, 6.79, 1.59 Hz, 2 H) 8.12 (dd, J = 8.31, 1.47 Hz, 1 H) 8.38 (dd, J = 8.44, 1.34 Hz, 1 H)
2-chloro-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (23).
To a solution of 84 (83.0 mg, 0.23 mmol) and tetrabutylammonium iodide (93.0 mg, 0.25 mmol) in DCM (2 mL) at −78 °C in a sealed vial under nitrogen, was added dropwise boron trichloride 1M in DCM (0.6 mL, 0.60 mmol). The reaction was brought back to RT and stirred for 4 h. DCM was removed with a stream of nitrogen and the mixture was diluted with EtOAc. Sat. NaHCO3 was added, and the mixture left to stir 10 min. Phases were separated and the organic layer was washed once with sat. NaHCO3, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was adsorbed on silica gel (1 g) and purified by silica gel chromatography eluting with a gradient of 0 to 60% EtOAc in heptane to afford 2-chloro-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carbonitrile (30 mg, 34% yield) as a light orange solid. ESI MS m/z 349.0 [M + H]+. 1H NMR (400 MHz, CDCl3) δ ppm 1.82 (s, 3 H) 1.88 (s, 3 H) 6.14 (s, 1 H) 6.90 (d, J = 8.31 Hz, 1 H) 7.11 (d, J = 8.31 Hz, 1 H) 7.82 (dddd, J = 18.49, 8.34, 6.79, 1.59 Hz, 2 H) 8.12 (dd, J = 8.31, 1.47 Hz, 1 H) 8.38 (dd, J = 8.44, 1.34 Hz, 1 H). To a solution of 2-chloro-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carbonitrile (30 mg, 0.08 mmol) in ethanol (2.28 mL, 39 mmol) and water (0.23 mL, 12 mmol) was added Ghaffar-Parkins catalyst (3.4 mg, 0.008 mmol). The mixture was stirred 2.5 h at 80 °C. The solvent was reduced to a minimum using a stream of nitrogen and the crude was loaded (liquid deposit in DMSO) on a 30 g C18 column and purified using a gradient of MeCN/water from 0% to 100% MeCN. The pure fractions were combined and concentrated under reduced pressure to remove a maximum of the organic solvent. The remaining was lyophilized to afford 23 (30.0 mg, 62% yield) as an off-white solid. ESI MS m/z 367.2, 369.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.68 (s, 3 H), 1.78 (s, 3 H), 7.02 (d, J = 8.3 Hz, 1 H), 7.15 (d, J = 8.3 Hz, 1 H), 7.78 – 7.88 (m, 2 H), 7.94 (d, J = 2.0 Hz, 1 H), 8.06 (dd, J = 8.2, 1.6 Hz, 1 H), 8.30 (dd, J = 8.4, 1.3 Hz, 1 H), 8.36 (d, J = 2.2 Hz, 1 H), 9.75 (s, 1 H).
3-bromo-N-[5-(methoxymethoxy)-2-methyl-phenyl]quinolin-2-amine (87) and 2-chloro-N-(5-(methoxymethoxy)-2-methylphenyl)quinolin-3-amine (86).
In a 20 mL microwave vial, 85 (1 g, 4.12 mmol) and 5-(methoxymethoxy)-2-methyl-aniline (830 mg, 4.96 mmol) were dissolved in toluene (10 mL) at RT followed by the addition of sodium tert-butoxide (475 mg, 4.94 mmol), XantPhos (290 mg, 0.501 mmol), and Pd(OAc)2 (115 mg, 0.512mmol). The mixture was purged with nitrogen gas for 5 min and the vial was capped. The mixture was heated at 85 °C for 16 h. The volatiles were evaporated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 0 to 5% EtOAc in heptane to yield 87 (465 mg, 30% yield). ESI MS m/z 373.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.55 (s, 1H), 7.94 (s, 1H), 7.78 – 7.68 (m, 2H), 7.59 – 7.48 (m, 2H), 7.28 (m, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.73 (dd, J = 8.3, 2.6 Hz, 1H), 5.15 (s, 2H), 3.35 (s, 3H), 2.13 (s, 3H). Then the silica gel purification was continued with a gradient was 5% to 20% EtOAc in heptane to yield 86 (1.1 g, 66% yield). ESI MS m/z 329.2 [M + H]+. 1H NMR (400 MHz, DMSO) δ 7.83 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.64 (s, 1H), 7.54–7.46 (m, 3H), 7.25 (d, J = 8.4 Hz, 1H), 7.18 (s, 1H), 6.91 (s, 1H), 6.86 (d, J = 8.4 Hz, 1H), 5.17 (s, 2H), 3.38 (s, 3H), 2.08 (s, 3H).
2-amino-1-(5-(methoxymethoxy)-2-methylphenyl)-1H-pyrrolo[3,2-b]quinoline-3-carbonitrile (88).
In a microwave vial, malononitrile (0.054 g 0.82 mmol) was dissolved in DME (10 mL) at 0 °C followed by the addition of potassium tert-butoxide (0.36 g, 3.29 mmol). The mixture was stirred at 0 °C for 30 min and 86 (0.18 g, 0.55 mmol) was added. The vial was capped and heated at 150 °C for 3 h. After cooling to RT, water (30 mL) was added, and the mixture was extracted with EtOAc (3 X 30 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 70% EtOAc in hexanes to afford 88 (0.05 g, 25% yield). ESI MS m/z 359.5 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.42 (dd, J = 7.8, 1.2 Hz, 1H), 7.31 (s, 1H), 7.18 (m, 3H), 7.05 (m, 1H), 7.01 (d, J = 2.5 Hz, 1H), 6.87 (s, 1H), 6.68 (dd, J = 8.3, 2.5 Hz, 1H), 5.13 (s, 2H), 3.34 (s, 3H), 2.10 (s, 3H).
2-amino-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[3,2-b]quinoline-3-carboxamide (24).
4M HCl in dioxane (2 mL) was added to 88 (50 mg, 0.14 mmol) and the mixture was stirred for 3 h at RT. The volatiles were evaporated under reduced pressure and the residue was triturated with n-pentane to obtain impure 2-amino-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[3,2-b]quinoline-3-carbonitrile (50 mg, ESI MS m/z 315.2 [M+H]+) that was dissolved in sulfuric acid (2 mL) and mixture was stirred for 20 min. The mixture was slowly poured in icy water (30 mL) and the mixture was extracted with EtOAc (3 X 30 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by prep HPLC carried out using SUNFIRE C18 (250 X 19mm) 5 μm column and 0.1% TFA in water and 100% ACN as mobile phase. The combined pure fractions were lyophilized to afford 24 (3 mg, 6% yield). ESI MS m/z 333.3 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.90 (s, 1H), 8.32 (s, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8 Hz, 1H), 7.56–7.54 (m, 3H), 7.37–7.29 (m, 3H), 7.23 (s, 1H), 6.98 (d, J = 8 Hz, 2H), 6.81 (s, 1H), 1.88 (s, 3H).
2-amino-1-(5-(methoxymethoxy)-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoline-3-carbonitrile (89).
In a microwave vial, malononitrile (14 mg, 0.21 mmol) was dissolved in THF (3 mL) and mixture was cooled to 0 °C followed by addition of NaH (60% dispersion in oil, 17 mg, 0.42 mmol). The mixture was stirred at 0 °C for 30 min and 87 (40 mg, 0.10 mmol) and Pd(PPh3)4 (12 mg, 0.010 mmol) were added. The vial was capped and heated at 80 °C for 4 h, cooled to RT, diluted with water (3 mL) and extracted with EtOAc (10 mL x 3). The combined organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 20 to 50% EtOAc in hexanes to afford 89 (39 mg, 25% yield). ESI MS m/z 359.2 [M + H]+. 1H NMR (400 MHz, DMSO) δ 8.04 (s, 1H), 7.96 (d, J = 8 Hz, 1H), 7.74 (d, J = 8 Hz, 1H), 7.63 – 7.57 (m, 3H), 7.47–7.42 (m, 2H), 7.20 (d, J = 8.4 Hz, 1H), 7.06 (s, 1H), 5.24 (dd, J = 20.4, 6.8 Hz, 2H), 3.42 (s, 3H), 1.90 (s, 3H).
2-amino-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoline-3-carboxamide (25).
3M HCl in MeOH (2 mL) was added to 89 (38 mg, 0.10 mmol) and mixture was stirred for 3 h at RT. The volatiles were evaporated under reduced pressure and the residue was triturated with pentane to afford 2-amino-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoline-3-carbonitrile (32 mg, 90% yield, LCMS: m/z 315.2 [M+H]+) that was dissolved in sulfuric acid (2 mL) and stirred for 2 h. Saturated aqueous K2CO3 solution was added until slightly basic pH and the mixture was extracted with EtOAc (5 mL x 3). The combined organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 2 to 50% EtOAc in hexanes to provide a residue that was further purified by prep HPLC carried out using X-select Phenyl Hexyl (250 X 19 mm) 5 μm column and 0.1% FA in water and 100% ACN as mobile phase. The combined pure fractions were lyophilized to afford 25 (6 mg, 17% yield). ESI MS m/z 333.2 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.70 (s, 1H), 8.46 (s, 1H), 7.87 (d, J = 8 Hz, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.48–7.39 (m, 4H), 7.31 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 2.4 Hz, 2.4Hz, 2H), 6.89 (s, 1H), 6.75 (d, J = 2.4 Hz, 1H), 1.85 (s, 3H).
5-bromo-2,3-dichloro-quinoxaline (91).
Adapting a known procedure,32 a solution of 90 (37.50 g, 200.5 mmol) in diethyl oxalate (205 g, 1.40 mol, 190 mL) was refluxed for 4 h. The mixture was cooled to RT and EtOAc (500 mL) was added. The precipitate was filtered, washed with EtOAc three times, and dried under vacuum to give as a brown powder that was suspended in thionyl chloride (623g, 5.24 mol, 380 mL). A catalytic amount of DMF (2.36 g, 32.3 mmol, 2.5 mL) was added at RT and the mixture was refluxed for 4 h. The volatiles were evaporated under reduced pressure and the thick residue was poured slowly into icy water (500 mL) with vigorous stirring. The precipitate was filtered, dissolved in EtOAc (750 mL), dried with Na2SO4, and filtered. 40 g of silica gel was added to the filtrate, and the suspension was evaporated under reduced pressure to afford a brown residue that was purified by silica gel chromatography eluting with a gradient of 0 to 20% EtOAc in hexanes to yield 91 (35.4 g, 64% yield) as a white solid. ESI MS molecular ion not observed. 1H NMR (400 MHz, DMSO-d6) δ 8.24 – 8.19 (m, 1H), 8.05 – 8.00 (m, 1H), 7.81 – 7.74 (m, 1H).
2-amino-8-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carbonitrile (94) and 2-amino-5-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carbonitrile (93).
Adapting a known procedure,43 malononitrile (4.2 g, 63.5 mmol) was added portionwise to a vigorously stirred suspension of sodium hydride (60% dispersion in mineral oil, 2.6 g, 67 mmol) in DME (150 mL). After the addition, the stirring was continued for 30 min and then 91 (8.85 g, 32 mmol) was added. The mixture was stirred at RT for 15 min and then refluxed for 4 h. The volatiles were evaporated under reduced pressure and the resulting residue was poured by portion in cold aqueous 1M HCl to give a yellow precipitate that was filtered and washed with water to afford a mixture of 2-(5-bromo-3-chloro-quinoxalin-2-yl)malononitrile and 2-(8-bromo-3-chloro-quinoxalin-2-yl)malononitrile (9.0 g, 92% yield) (in about 1:1 ratio estimated by UPLCMS, LC−MS (ESI) m/z: 306.9 [M - H]−) as a yellow solid that was dissolved in in NMP (50 mL). 92 (13.3 g, 88 mmol) was added and the mixture was heated to 130 °C for 6 h, cooled to RT, and poured into vigorously stirring aqueous NaHCO3 sat. The precipitate was collected by filtration, washed with water, and residual water was removed by azeotropic evaporation with toluene under reduced pressure twice. The brown residue was taken in 550 mL of 15% MeOH in DCM and 25 g of silica gel was added. The mixture was evaporated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 20 to 60% EtOAc in hexanes to provide a mixture of 93 and 94 (8.9g, 72% yield) as an orange solid. ESI MS m/z 424.1 [M + H]+.
2-amino-5-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (29) and 2-amino-8-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (26).
Following the procedure used to prepare 76, a solution of 93 and 94 (5.0g, 11.8 mmol) in sulfuric acid (50 mL) was stirred for 1 h at RT to provide a mixture of 2-amino-8-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide and 2-amino-5-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (5.2 g, quantitative yield, ESI MS m/z 442.0 [M + H]+) as a yellow solid. Following the procedure used to prepare 3, BBr3 (1M in DCM, 36 mL, 36 mmol) was added to a solution of the yellow solid in DCM (36 mL) and the mixture was stirred for 2 h at RT. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 10% MeOH in DCM to obtain 3.75 g of a mixture of the two bromo regiomers that were separated by SFC (Column: ZymorSPHER HA-Dipyridyl, 30 x 150 mm, 5 μm; Conditions: Isocratic at 50% MeOH + 0.1% Formic Acid with 50% CO2; Flow Rate: 70 mL/min; outlet pressure 100 bar) providing 29 (RT 4.33 min, 1.14 g, 46% yield) as a white solid. ESI MS m/z 428.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 8.12 (m, 2H), 7.89 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (d, J = 3.3 Hz, 1H), 7.77 (dd, J = 8.3, 1.3 Hz, 1H), 7.48 (d, J = 3.3 Hz, 1H), 7.33 (dd, J = 8.3, 7.6 Hz, 1H), 7.08 (dt, J = 8.3, 0.8 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 1.79 (s, 3H), 1.72 (s, 3H); and 26 (RT 4.99 min, 520 mg, 22% yield) as a white solid. ESI MS m/z 428.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.68 (s, 1H), 8.12 (s, 2H), 7.96 (dd, J = 8.3, 1.3 Hz, 1H), 7.80 (dd, J = 7.6, 1.3 Hz, 1H), 7.70 (d, J = 3.1 Hz, 1H), 7.49 (dd, J = 8.3, 7.6 Hz, 1H), 7.44 (d, J = 3.2 Hz, 1H), 7.13 (dt, J = 8.3, 0.8 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 1.86 (d, J = 0.7 Hz, 3H), 1.78 (s, 3H). The position of the bromine in 29 was confirmed by Xray structure of 39.
6-bromo-2,3-dichloro-quinoxaline (96).
Adapting a known procedure,32 a solution of 95 (12.5 g, 66.8 mmol) in diethyl oxalate (70.2 g, 480 mmol, 65 mL) was refluxed for 16 h. The mixture was cooled to RT and EtOAc (175 mL) was added. The precipitate was filtered, washed with EtOAc three times, and dried under vacuum to give a brown powder that was suspended in thionyl chloride (81.5 g, 685 mmol, 50 mL). A catalytic amount of DMF (472 mg, 6.46 mmol, 0.5 mL) was added at RT and the mixture was refluxed for 8 h. The volatiles were evaporated under reduced pressure and the thick residue was poured slowly into icy water (200 mL) with vigorous stirring. The precipitate was filtered and dried under vacuum to yield 96 (35.4 g, 64% yield) as an orange solid. ESI MS molecular ion not observed. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (dd, J = 2.1, 0.5 Hz, 1H), 8.00 (d, J = 2.1 Hz, 1H), 7.97 (d, J = 0.5 Hz, 1H).
2-amino-6-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carbonitrile (97) and 2-amino-7-bromo-1-(3-methoxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carbonitrile (98).
Adapting a known procedure,43 malononitrile (475 mg, 7.19 mmol) was added portionwise to a vigorously stirred suspension of sodium hydride (60% dispersion in mineral oil, 285 mg, 7.44 mmol) in DME (30 mL). After the addition, the stirring was continued for 30 min and then 96 (1.0 g, 3.60 mmol) was added. The mixture was stirred at RT for 15 min and then refluxed for 2 h. The volatiles were evaporated under reduced pressure and the resulting residue was poured by portions in cold aqueous 1M HCl to give a brown mixture that was extracted twice with EtOAc, the combined organic layer was dried over Na2SO4 and evaporated under reduced pressure. The brown residue was dissolved in 10% MeOH in DCM and 4 g of silica gel was added. The suspension was evaporated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 0 to 10% MeOH in DCM to provide a mixture of 97 and 98 (750 mg, 68% yield, ESI MS m/z 306.9 [M - H]−) as one major and one minor regiomer (estimated by UPLCMS) as a yellow solid that was dissolved in NMP (5 mL). 92 (1.1 g, 7.3 mmol) was added and the mixture was heated to 130 °C for 3 h, cooled to RT, and poured into vigorously stirring aqueous NaHCO3 sat. The precipitate was collected by filtration, washed with water, and residual water was removed by azeotropic evaporation with toluene under reduced pressure twice. The brown residue was taken in 250 mL of 15% MeOH in DCM and 5 g of silica gel was added. The suspension was evaporated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 20 to 60% EtOAc in hexanes to provide a mixture of 97 and 98 (675 mg, 72% yield) as one major and one minor regiomer (estimated by UPLCMS) as an orange solid. ESI MS m/z 424.1 [M + H]+.
2-amino-7-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (27) and 2-amino-6-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (28).
Following the procedure used to prepare 76, a solution of a mixture of 93 and 94 (450 mg, 1.07 mmol) in sulfuric acid (5 mL) was stirred for 1 h at RT providing a mixture of 2-amino-6-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide and 2-amino-7-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (450 mg, 96% yield, ESI MS m/z 442.1 [M + H]+) as one major and one minor regiomer (estimated by UPLCMS) as a yellow solid. Following the procedure used to prepare 3, BBr3 (1M in DCM, 3.4 mL, 3.4 mmol) was added to a solution of the yellow solid in DCM (3.4 mL) and the mixture was stirred for 2 h at RT. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 10% MeOH in DCM to obtain 450 mg of a mixture of the bromo regiomers as one major and one minor regiomer (estimated by UPLCMS) that were separated by SFC (Column: Chiral Technologies OJ 10 x 250 mm, 5um; Conditions: Isocratic 40% ACN+EtOH 1:1, 10 mL/min 100 Bar) providing 27 (RT 3.30 min, 45 mg, 10% yield) as a white solid. ESI MS m/z 426.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.13 (d, J = 23.0 Hz, 2H), 7.97 (d, J = 2.3 Hz, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.74 – 7.61 (m, 2H), 7.41 (d, J = 3.3 Hz, 1H), 7.11 (dt, J = 8.3, 0.8 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 1.85 – 1.78 (m, 3H), 1.73 (s, 3H); and 28 (RT 6.56 min, 250 mg, 56% yield) as a white solid. ESI MS m/z 426.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.09 (d, J = 2.3 Hz, 3H), 7.66 (d, J = 8.8 Hz, 1H), 7.63 (dq, J = 4.9, 2.3 Hz, 1H), 7.50 (dd, J = 8.8, 2.3 Hz, 1H), 7.43 – 7.32 (m, 1H), 7.06 (dt, J = 8.2, 0.8 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 1.81 – 1.73 (m, 3H), 1.69 (s, 3H). The position of the bromine in 28 was confirmed by Xray structure.
2-amino-8-cyano-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (30).
Copper (I) cyanide (32 mg, 0.36 mmol) was added to a solution of 26 (50 mg, 0.12 mmol) in DMF (1 mL) and the suspension was stirred at 80 °C for 8 h. The mixture was filtered through a 0.45 micron PTFE filter and purified by preparative HPLC (30–80% MeCN in water, 0.1% formic acid modifier). The recovered tubes were combined and lyophilized to provide 26 (16 mg, 36% yield). ESI MS m/z 373.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.44 – 8.03 (m, 3H), 7.96 (dd, J = 7.3, 1.3 Hz, 1H), 7.79 – 7.32 (m, 3H), 7.10 (d, J = 8.3 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 1.82 (s, 3H), 1.74 (s, 3H).
2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-8-(2-methylpyrazol-3-yl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (31).
Pd(dppf)Cl2•CH2Cl2 (6 mg, 0.007 mmol) was added to 26 (30 mg, 0.070 mmol) and 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (21 mg, 0.105 mmol) in DMF (1 mL) and aqueous K2CO3 solution (2M, 0.21 mL, 0.21 mmol). After stirring at 80 °C for 2 h, the mixture was cooled to RT, filtered through a 0.45 micron PTFE filter and purified by prep HPLC (gradient of 20 to 80% MeCN in water, 0.1% formic acid modifier). The recovered tubes were combined and lyophilized to provide 31 (16 mg, 53% yield) as a pale-yellow solid. ESI MS m/z 428.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 1H), 8.02 (dd, J = 8.3, 1.5 Hz, 3H), 7.71 (d, J = 3.1 Hz, 1H), 7.61 (dd, J = 8.3, 7.2 Hz, 1H), 7.44 (dd, J = 7.2, 1.5 Hz, 1H), 7.41 – 7.35 (m, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.06 – 6.99 (m, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.23 (d, J = 1.8 Hz, 1H), 3.40 (s, 3H), 1.75 (d, J = 0.7 Hz, 3H), 1.68 (s, 3H).
2-amino-8-(cyclopenten-1-yl)-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (32).
Following the procedure used to prepare 31, using 26 and 2-(cyclopenten-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane afforded 32 (21 mg, 70% yield) as a pale-yellow solid. ESI MS m/z 414.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 7.98 (s, 2H), 7.79 (dd, J = 8.3, 1.4 Hz, 1H), 7.71 – 7.59 (m, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.38 (dd, J = 7.4, 1.4 Hz, 1H), 7.35 – 7.27 (m, 1H), 7.09 (d, J = 8.3 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.47 (t, J = 2.2 Hz, 1H), 2.71 (m, 2H), 2.36 – 2.23 (m, 2H), 1.82 (s, 3H), 1.75 (m, 5H).
2-amino-6-cyano-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (33).
Following the procedure used to prepare 30, cyanation of 28 yielded 33 (16 mg, 36% yield) as a pale-yellow solid. ESI MS m/z 373.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.42 (d, J = 1.8 Hz, 1H), 8.39 – 8.12 (m, 2H), 7.87 (d, J = 8.5 Hz, 1H), 7.71 (dd, J = 8.5, 1.9 Hz, 1H), 7.56 (s, 2H), 7.08 (d, J = 8.3 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 1.79 (s, 3H), 1.71 (s, 3H).
2-amino-7-(3,6-dihydro-2H-pyran-4-yl)-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (34).
Following the procedure used to prepare 31, using 28 and (2-methylpyrazol-3-yl)boronic acid afforded 34 (6 mg, 6% yield) as a pale yellow solid. ESI MS m/z 428.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.18 – 7.93 (m, 3H), 7.83 (dd, J = 8.5, 0.5 Hz, 1H), 7.72 (d, J = 4.9 Hz, 1H), 7.57 (dd, J = 8.6, 2.1 Hz, 1H), 7.47 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 3.0 Hz, 1H), 7.09 (dt, J = 8.3, 0.7 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 6.50 (d, J = 1.9 Hz, 1H), 3.92 (s, 3H), 1.81 (d, J = 0.7 Hz, 3H), 1.73 (s, 3H).
2-amino-7-(cyclopenten-1-yl)-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[3,2-b]quinoxaline-3-carboxamide (35).
Following the procedure used to prepare 31, using 28 and 2-(cyclopenten-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane afforded 35 (41 mg, 38% yield) as a pale-yellow solid. ESI MS m/z 414.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.11 – 7.84 (m, 2H), 7.81 (m, 1H), 7.75 – 7.68 (m, 1H), 7.66 (m, 2H), 7.33 (d, J = 3.3 Hz, 1H), 7.08 (d, J = 8.3 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.43 (m, 1H), 2.82 – 2.72 (m, 2H), 2.56 – 2.48 (m, 2H), 1.97 (p, J = 7.6 Hz, 2H), 1.79 (s, 3H), 1.71 (s, 3H).
2-amino-5-cyano-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (36).
Following the procedure used to prepare 30, cyanation of 29 yielded 36 (12 mg, 26% yield) as a pale-yellow solid. ESI MS m/z 373.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.31 (s, 2H), 8.18 – 8.02 (m, 2H), 7.69 (d, J = 3.1 Hz, 1H), 7.55 (d, J = 6.7 Hz, 1H), 7.51 (dd, J = 8.4, 7.3 Hz, 1H), 7.08 (d, J = 8.3 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 1.79 (s, 3H), 1.72 (s, 3H).
2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5-(2-methylpyrazol-3-yl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (37).
Following the procedure used to prepare 31, using 29 and (2-methylpyrazol-3-yl)boronic acid afforded 37 (10 mg, 20% yield) as a pale yellow solid. ESI MS m/z 428.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 7.95 (s, 2H), 7.76 (dd, J = 8.2, 1.6 Hz, 1H), 7.73 – 7.67 (m, 2H), 7.64 – 7.31 (m, 6H), 7.19 (d, J = 3.4 Hz, 1H), 7.09 (d, J = 8.3 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 1.81 (s, 3H), 1.73 (s, 3H).
2-amino-5-(cyclopenten-1-yl)-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (38).
Following the procedure used to prepare 31, using 29 and 2-(cyclopenten-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane afforded 38 (25 mg, 51% yield) as a pale yellow solid. ESI MS m/z 414.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 7.95 (s, 2H), 7.73 – 7.57 (m, 2H), 7.47 (dd, J = 7.3, 1.6 Hz, 1H), 7.43 – 7.32 (m, 2H), 7.08 (d, J = 8.3 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.48 (m, 1H), 2.91 (m, 2H), 2.54 (m, 2H), 1.98 (m, 2H), 1.79 (s, 3H), 1.72 (s, 3H).
(S)-2-amino-5-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (39) and (R)-2-amino-5-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]quinoxaline-3-carboxamide (40).
SFC separation of 29 (150 mg) using Mettler Toledo Minigram instrument equipped with a Chiral Technologies IC, 10 x 250 mm, 5 μm column eluting with 35% MeOH containing 10 mM ammonium formate with a flow rate of 10 mL/min yielded 40 (RT 6.00 min, 68 mg, 45% yield) as a white solid. ESI MS m/z 427.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 7.95 (s, 2H), 7.73 – 7.57 (m, 2H), 7.47 (dd, J = 7.3, 1.6 Hz, 1H), 7.43 – 7.32 (m, 2H), 7.08 (d, J = 8.3 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.48 (m, 1H), 2.91 (m, 2H), 2.54 (m, 2H), 1.98 (m, 2H), 1.79 (s, 3H), 1.72 (s, 3H), 100%ee, [α]23.4 −74.0 (c 0.1, MeOH); and 39 (RT 8.31 min, 60 mg, 40% yield) as a white solid. ESI MS m/z 427.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 7.97 (s, 2H), 7.73 – 7.56 (m, 2H), 7.48 (dd, J = 7.3, 1.6 Hz, 1H), 7.44 – 7.26 (m, 2H), 7.09 (d, J = 8.3 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 6.50 (m, 1H), 2.92 (m, 2H), 2.53 (m, 2H), 1.99 (m, 2H), 1.79 (s, 3H), 1.72 (s, 3H). 100%ee, [α]23.4D +80.0 (c 0.1, MeOH). The position of the bromine in 39 was confirmed by Xray structure.
(S)-2-amino-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (41) and (R)-2-amino-1-(5-hydroxy-2-methylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (42).
SFC separation of 1 (205 mg) using Mettler Toledo Minigram instrument equipped with a Chiral Technologies IC, 10 x 250 mm, 5 μm column eluting with 45% MeOH with a flow rate of 10 mL/min yielded 42 (RT 3.98 min, 72 mg, 35% yield) as a white solid. MS m/z 334.3 [M + H]+, 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 7.98 (s, 3H), 7.96 – 7.87 (m, 1H), 7.80 – 7.67 (m, 2H), 7.56 (m, 1H), 7.45 (m, 1H), 7.37 (s, 1H), 7.11 (dt, J = 8.3, 0.8 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 1.82 (s, 3H), 1.75 (s, 3H), 99%ee, [α]24.9D −90.0 (c 0.1, MeOH); and 41 (RT 6.81 min, 71 mg, 35% yield) as a white solid. MS m/z 334.3 [M + H]+, 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 7.97 (br s, 3H), 7.97 – 7.88 (m, 1H), 7.81 – 7.64 (m, 2H), 7.58 (m, 1H), 7.46 (m, 1H), 7.35 (br s, 1H), 7.11 (dt, J = 8.3, 0.8 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 1.84 (s, 3H), 1.74 (s, 3H), 96%ee, [α]25.1D +100.0 (c 0.1, MeOH).
(S)-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (43) and (R)-1-(3-hydroxy-2,6-dimethylphenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxamide (44).
SFC separation of 22 (180 mg) using Mettler Toledo Minigram instrument equipped with a Chiral Technologies IA, 10 x 250 mm, 5 μm column eluting with 40% isopropanol containing 10 mM ammonium formate with a flow rate of 10 mL/min yielded 44 (RT 3.06 min, 70 mg, 39% yield) as a white solid. MS m/z 333.4 [M + H]+, 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.82 (s, 1H), 8.37 – 8.24 (m, 1H), 8.11 – 8.04 (m, 2H), 7.91 – 7.73 (m, 3H), 7.11 (dt, J = 8.3, 0.7 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 1.81 (s, 3H), 1.71 (s, 3H), 100%ee, [α]25.3 −92.0 (c 0.1, MeOH); and 43 (RT 5.38 min, 70 mg, 39% yield) as a white solid. MS m/z 333.4 [M + H]+, 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.82 (s, 1H), 8.37 – 8.24 (m, 1H), 8.11 – 8.04 (m, 2H), 7.91 – 7.73 (m, 3H), 7.11 (dt, J = 8.3, 0.7 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 1.81 (s, 3H), 1.71 (s, 3H). 100%ee, [α]25.5D +99.0 (c 0.1, MeOH).
2-(3-chloropyrazin-2-yl)propanedinitrile (101).
Malononitrile (26.6 g, 403 mmol) was added dropwise with vigorous stirring to a suspension of NaH (60% dispersion in mineral oil, 16 g, 418 mmol) in DME (600 mL). The mixture was stirred for 30 min and then 99 (30 g, 201 mmol) was added. The mixture was stirred for 30 min and then heated to reflux for 1 h. The volatiles were evaporated under reduced pressure and the residue was treated with cold aqueous HCl 1 M to give a yellow solid that was recovered by filtration, washed with water and a minimum of cold ethanol to afford 101 (34.2 g, 95% yield) as a yellow solid. ESI MS m/z 177.0 [M - H]−. 1H NMR (500 MHz, DMSO-d6) δ 7.85 (d, J = 3.2 Hz, 1H), 7.56 (d, J = 3.2 Hz, 1H).
6-amino-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carbonitrile (103).
A microwave vial containing 101 (1.00 g, 5.60 mmol), 92 (2.54 g, 16.8 mmol) and NMP (10 mL) was capped, stirred at 150 °C for 1 h then at 200 °C for 8 h. The mixture was cooled to RT, poured into saturated aqueous NaHCO3 and diluted with water and EtOAc. The mixture was filtered through a pad of Celite and the layers were separated. The organic layer was dried over Na2SO4, filtered, adsorbed on silica gel and purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in hexanes. The appropriate fractions were combined, concentrated under reduced pressure. The residue was purified again by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM to provide 103 (346 mg, 21% yield) as a beige solid. ESI MS m/z 294.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 3.0 Hz, 1H), 7.78 (d, J = 3.0 Hz, 1H), 7.71 (s, 2H), 7.30 – 7.21 (m, 1H), 7.12 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 1.80 (d, J = 0.7 Hz, 3H), 1.71 (s, 3H).
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (106).
Following the procedure used to prepare 76, a solution of 103 (346 mg, 1.18 mmol) in sulfuric acid (4 mL) was stirred at RT for 1 h providing 6-amino-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (295 mg, 80% yield, ESI MS m/z 312.3 [M + H]+) as a beige solid. Following the procedure used to prepare 3, BBr3 (1M in DCM, 2.8 mL, 2.8 mmol) was added to a solution of the beige solid in DCM (2.8 mL) and the mixture was stirred for 2 h at RT. The residue was triturated with saturated NaHCO3. The solid was collected by filtration on Buchner, washed with water, air-dried, affording 106 (214 mg, 76% yield) as a light-yellow solid. ESI MS m/z 299.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.12 (d, J = 3.1 Hz, 1H), 7.75 (d, J = 3.1 Hz, 1H), 7.40 (br m, 2H), 7.21 (br m, 1H), 7.08 (d, J = 8.3 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 1.77 (s, 3H), 1.69 (s, 3H).
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (45).
SFC separation of 106 (214 mg) using Mettler Toledo Minigram instrument equipped with a Phenomenex Lux Cellulose-2, 10 x 250 mm, 5 μm column eluting with 55% 1:1 ACN:EtOH with a flow rate of 10 mL/min yielded 45 (RT 3.93 min, 76 mg, 36% yield) as a light beige fluffy solid. ESI MS m/z 299.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.12 (d, J = 3.1 Hz, 1H), 7.75 (d, J = 3.1 Hz, 1H), 7.40 (br m, 2H), 7.21 (br m, 1H), 7.08 (d, J = 8.3 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 1.77 (s, 3H), 1.69 (s, 3H), 100%ee, [α]26.5D +35.0 (c 0.1, MeOH). The distomer (RT 4.52 min) was not characterized.
2-(3-chloro-5,6-dimethyl-pyrazin-2-yl)propanedinitrile (102).
To a suspension of NaH (60% dispersion in mineral oil, 3.54 g, 92 mmol,) in THF (100 mL) at 0 °C was added malononitrile (3.99 g, 60.4 mmol) in THF (30 mL) dropwise via an addition funnel. The cold bath was removed at the end of the addition and the resulting mixture was allowed to stir for 45 min at RT. 100 (5.49 g, 31.0 mmol) and Pd(PPh3)4 (1.76 g, 1.52 mmol) were added, and the mixture was refluxed for 3.25 h. After cooling to RT, it was poured into 200 mL of cold 1N HCl and extracted with DCM (3x). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was adsorbed on silica using DCM/THF and purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in hexanes. Mix fractions were re-purified by a second silica gel chromatography using same conditions. Clean fractions from both columns were combined, concentrated under reduced pressure, and dried under vacuum to afford 102 (5.0 g, 78% yield) as an orange solid. ESI MS m/z 205.0 [M - H]−. 1H NMR (400 MHz, Chloroform-d) δ 5.44 (s, 1H), 2.62 (s, 3H), 2.61 (s, 3H).
6-amino-5-(3-methoxy-2,6-dimethyl-phenyl)-2,3-dimethyl-pyrrolo[2,3-b]pyrazine-7-carbonitrile (104).
A microwave vial was charged with 102 (1.01 g, 4.9 mmol), 92 (2.2 g, 14.6 mmol), potassium tert-butoxide (1.1 g, 9.8 mmol) and Pd-PEPPSI™-SIPr catalyst (171 mg, 0.25 mmol), flushed with nitrogen three times, then dry NMP (10 mL) was added, flushed again with nitrogen, capped and submitted to microwave irradiation (100 °C) for 30 min. The vial was diluted with EtOAc, saturated aqueous NH4Cl and water. The layers were separated, and the aqueous layer was extracted twice with EtOAc. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane to provide 104 (0.85 g, 54% yield) as a yellow solid. ESI MS m/z 322.3 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.20 (dt, J = 8.5, 0.8 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 4.96 (br s, 2H), 3.87 (s, 3H), 2.57 (s, 3H), 2.41 (s, 3H), 1.91 (s, 3H), 1.84 (s, 3H).
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2,3-dimethyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (107).
Following the procedure used to prepare 76, a solution of 104 (7.11 g, 22.1 mmol) in sulfuric acid (70 mL) was stirred at RT for 45 min providing 6-amino-5-(3-methoxy-2,6-dimethyl-phenyl)-2,3-dimethyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (7.5 g, quantitative yield) as a yellow solid. ESI MS m/z 341.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.45 (br s, 1H), 7.25 (d, J = 8.4 Hz, 1H), 7.18 – 7.04 (m, 4H), 3.85 (s, 3H), 2.47 (s, 3H), 2.31 (s, 3H), 1.81 (s, 3H), 1.71 (s, 3H). Following the procedure used to prepare 3, BBr3 (6.4 mL, 66.3 mmol) was added to a solution of the yellow solid in DCM (132 mL) and the mixture was stirred for 70 min at RT. The residue was triturated with saturated NaHCO3 (100 mL). The solid was collected by filtration, washed with water, air-dried, then adsorbed on silica gel and purified by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM. Mixed fractions were combined and re-purified by silica gel chromatography using the same conditions. The clean material from both columns was combined, concentrated then dried under vacuum, affording affording 107 (5.3 g, 74% yield). ESI MS m/z 327.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 7.45 (br s, 1H), 7.18 – 7.02 (m, 4H), 6.93 (d, J = 8.3 Hz, 1H), 2.48 – 2.45 (m, 3H), 2.35 – 2.24 (m, 3H), 1.81 – 1.73 (m, 3H), 1.68 (s, 3H).
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2,3-dimethyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (46).
SFC separation of 107 (5.40 g) using Waters Prep 100 SFC-MS instrument equipped with a Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm column eluting with 55% 1:1 ACN:EtOH with a flow rate of 70 mL/min yielded 46 (RT 4.07 min, 1.31 g, 20% yield) as a light beige fluffy solid. ESI MS m/z 327.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 7.45 (br s, 1H), 7.12 (br s, 1H), 7.09 (br s, 2H), 7.06 (d, J = 8.8 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 2.47 (s, 3H), 2.31 (s, 3H), 1.76 (s, 3H), 1.68 (s, 3H). 100%ee, [α]26.4D +54.0 (c 0.1, MeOH). The distomer (RT 4.81 min) was not characterized.
6-amino-3-bromo-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carbonitrile (105).
To a solution of 103 (600 mg, 2.05 mmol) in DMF (10 mL) was added NBS (436 mg, 2.45 mmol). The mixture was stirred for 10 min, diluted with water, stirred for 20 min then filtered. The solid was washed with water, dried under vacuum and purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in hexanes to provide 105 (350 mg, 46% yield). ESI MS m/z 374.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.86 (s, 2H), 7.23 (d, J = 8.5 Hz, 1H), 7.09 (d, J = 8.5 Hz, 1H), 3.81 (s, 3H), 1.77 (s, 3H), 1.69 (s, 3H).
6-amino-3-bromo-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (108).
Following the procedure used to prepare 7, a solution of 105 (350 mg, 0.94 mmol) in sulfuric acid (1 mL) was stirred at RT for 1 h. The solid recovered by filtration was purified by silica gel purification eluting with a gradient of 0 to 20% MeOH in DCM to provide 6-amino-2-bromo-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (300 mg, 82% yield) as a yellow solid. ESI MS m/z 392.0 [M + H]+. Following the procedure used to prepare 3, BBr3 (1M in DCM, 2.3 mL, 2.3 mmol) was added to a solution of the yellow solid in DCM (2.3 mL) and the mixture was stirred for 2 h at RT. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM affording 108 (263 mg, 91% yield). ESI MS m/z 378.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (s, 1H), 7.55 (s, 2H), 7.31 (s, 1H), 7.21 (s, 1H), 7.13 – 7.06 (m, 1H), 6.96 (d, J = 8.3 Hz, 1H), 1.78 (s, 3H), 1.70 (s, 3H).
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-3-methyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (109).
To a solution of methyl magnesium chloride (3 M, 0.3 mL, 0.9 mmol) in THF (1.5 mL) in a MW vial was added ZnCl2 (0.5 M in THF, 1.8 mL, 0.9 mmol) dropwise at RT. After addition, the resulting white suspension was stirred for 35 min at RT. 108 (69 mg, 0.180 mmol) in THF (1 mL) and Pd(PPh3)4 (25 mg, 0.02 mmol) were added. The vial was flushed with nitrogen, capped, transferred to a preheated (70 °C) heat block and stirred at this temperature for 18 h. The mixture was cooled to RT, diluted with 0.5N HCl (3 mL), and extracted twice with EtOAc. Combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by preparative HPLC using a gradient of 25 to 55 % ACN in water (both containing 0.1% formic acid) over 12 min at a flow of 40 mL/min on a Phenomenex Gemini® 5μm NX-C18 110Å 150 x 21.2 mm column. The recovered tubes were combined and lyophilized to yield 109 (60 mg, 93% yield) as a white fluffy solid. ESI MS m/z 312.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (br s, 1H), 8.02 (d, J = 0.7 Hz, 1H), 7.35 (br s, 1H), 7.23 (br s, 2H), 7.16 (br s, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 2.33 (d, J = 0.7 Hz, 3H), 1.77 (s, 3H), 1.68 (s, 3H).
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-3-methyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (47).
SFC separation of 109 (42 mg) using Mettler Toledo Minigram instrument equipped with a Phenomenex Lux Cellulose-2, 10 x 250 mm, 5 μm column eluting with 50% isopropanol with a flow rate of 10 mL/min yielded 47 (RT 3.81 min, 8,9 mg, 21% yield) as a white fluffy solid. ESI MS m/z 312.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (br s, 1H), 8.02 (d, J = 0.7 Hz, 1H), 7.35 (br s, 1H), 7.23 (br s, 2H), 7.16 (br s, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 2.33 (d, J = 0.7 Hz, 3H), 1.77 (s, 3H), 1.68 (s, 3H). 99%ee, [α]26.3D +55.0 (c 0.1, MeOH). The distomer (RT 4.38 min) was not characterized.
5-bromo-6-chloro-pyrazin-2-ol (111). Sodium nitrite (40 g, 580 mmol) was added portion wise to a solution of 110
(110 g, 528 mmol) in sulfuric acid (770 mL) at 0 °C under mechanical stirring. The resulting thick mixture was stirred at 0°C for 1 h and was then slowly poured in 6 L of cold water containing crushed ice maintaining temperature below 30 °C. The resulting precipitate was collected by filtration, washed with water then dried by azeotropic evaporation with toluene under reduced pressure twice to give 111 (104.6 g, 95% yield) as a pale beige solid. ESI MS m/z 208.9 [M - H]−. 1H NMR (400 MHz, DMSO-d6) δ 12.76 (br s, 1H), 7.97 (s, 1H).
5-benzyloxy-2-bromo-3-chloro-pyrazine (112).
Benzyl bromide (48 mL, 404 mmol) was added dropwise to a suspension of 111 (80 g, 382 mmol) and silver carbonate (216 g, 778 mmol) in toluene (2 L). After stirring for 3 h, the suspension was filtered on Celite. The filtrate was evaporated under reduced pressure to provide a yellow oil that was dissolved in warm EtOH. After slow addition of water under sonication, the precipitate was collected by filtration to provide 112 (85.2 g, 75% yield) as a beige solid. ESI MS molecular ion not observed. 1H NMR (400 MHz, DMSO-d6) δ 8.27 (s, 1H), 7.51 – 7.46 (m, 2H), 7.44 – 7.33 (m, 3H), 5.36 (s, 2H).
5-benzyloxy-3-chloro-N-(3-methoxy-2,6-dimethyl-phenyl)pyrazin-2-amine (113).
To a solution of 112 (90 g, 300 mmol) in toluene (1350 mL) were added potassium tert-butoxide (45.0 g, 401 mmol), 92 (48 g, 318 mmol), Pd2(dba)3 (14.4 g, 15.7 mmol) and Xantphos (18.0 g, 31 mmol). The mixture was degassed under vacuum and back filled with nitrogen. The resulting mixture was stirred at 80 °C for 45 min and then concentrated under reduced pressure. The residue was dissolved in DCM (500 mL), 200 g of silica gel was added, and the suspension was evaporated to under reduced pressure. The residue was purified on a pad of silica gel (1 kg of silica gel) eluting with a gradient of 0 to 15% EtOAc in hexanes to provide 113 (108.4 g, 98% yield) as a pale beige solid. ESI MS m/z 370.2 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.22 – 7.17 (m, 1H), 6.96 (d, J = 8.5 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 4.95 (s, 2H), 3.87 (s, 3H), 1.87 (s, 3H), 1.79 (s, 3H).
6-amino-2-benzyloxy-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carbonitrile (114).
To a solution of malononitrile (42.1 g, 637 mmol) in DME (1.8 L) was added portion wise NaH (60% dispersion in mineral oil, 25.0 g, 628 mmol). The resulting mixture was stirred for 30 min, then 113 (115 g, 311 mmol) in DME (500 mL) and Pd(PPh3)4 (17.7 g, 15.3 mmol) were added. The resulting mixture was stirred at reflux for 2 h, and then concentrated under reduced pressure to ~1 L. Water (1 L) was added slowly and the resulting biphasic mixture was stirred for 18 h with a mechanical stirrer. The resulting solid was recovered by filtration, washed with water, and dried under vacuum. Trituration in DCM provided the first batch of the desired material as a beige solid isolated by filtration. The mother liquor was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with a gradient of 10 to 60% EtOAc in hexanes to provide a second batch of the desired material. The two batches were combined to provide 114 (103.1 g, 83% yield) as a beige solid. ESI MS m/z 400.4 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.60 (s, 1H), 7.53 – 7.47 (m, 2H), 7.42 – 7.34 (m, 2H), 7.33 – 7.27 (m, 1H), 7.21 – 7.15 (m, 1H), 6.94 (d, J = 8.5 Hz, 1H), 5.45 (s, 2H), 4.91 (s, 2H), 3.84 (s, 3H), 1.90 (d, J = 0.7 Hz, 3H), 1.83 (s, 3H).
6-amino-2-hydroxy-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (115).
A solution of 114 (83 g, 208 mmol) in sulfuric acid (550 mL) was stirred with a mechanical stirrer for 18 h. The thick brown mixture was poured slowly in icy water (2 L) in an ice bath maintaining internal temperature below 20 °C while stirred with a mechanical stirrer. A pale-yellow solid precipitated out. The resulting suspension in an ice bath was slowly neutralized to basic pH with aqueous ammonium hydroxide (28% solution; 850 mL) while maintaining the internal temperature below 40 °C. The precipitate was collected by filtration, washed with water, and dried under vacuum to provide 115 (65.1 g, 96% yield) as a pale beige solid. ESI MS m/z 328.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (s, 1H), 7.24 (s, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.13 (m, 4H), 7.05 (d, J = 8.5 Hz, 1H), 3.80 (s, 3H), 1.90 – 1.75 (s, 3H), 1.69 (s, 3H).
[6-amino-7-carbamoyl-5-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazin-2-yl] trifluoromethanesulfonate (116).
To a solution of 115 (30.5 g, 93.2 mmol) and Cs2CO3 (34.9 g, 107 mmol) in DMF (300 mL) was added 1,1,1-trifluoro-N-phenyl-N-(trifluoromethylsulfonyl)methanesulfonamide (36.6 g, 103 mmol). The mixture was stirred for 1h, diluted with water (900 mL) and extracted with EtOAc (3x300 mL). The combined organic extracts were washed with water, brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 20 to 100% EtOAc in hexanes to provide 116 (28 g, 65% yield) as an off-white solid. ESI MS m/z 460.4 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.75 (s, 1H), 7.22 m, 2H), 6.97 (d, J = 8.5 Hz, 1H), 6.37 (s, 2H), 5.49 (s, 1H), 3.86 (s, 3H), 1.91 (s, 3H), 1.84 (s, 3H).
[6-amino-7-carbamoyl-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazin-2-yl] trifluoromethanesulfonate (117).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 9.3 mL, 9.3 mmol) was added to a solution of 116 (1.07 g, 2.33 mmol) in DCM (11 mL) and the mixture was stirred for 2 h at RT. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM to provide 117 (744 mg, 72% yield) as an off-white solid. ESI MS m/z 446.2 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (s, 1H), 7.39 – 7.26 (m, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.94 (s, 1H), 6.82 (d, J = 8.3 Hz, 1H), 5.62 (s, 1H), 1.90 (s, 3H), 1.86 (s, 3H).
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-methyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (118).
To a solution of methyl magnesium chloride (3 M, 1.08 mL, 3.24 mmol) in THF (9 mL) in a MW vial was added ZnCl2 (0.5 M in THF, 6.60 mL, 3,3 mmol) dropwise at RT. After addition, the resulting white suspension was stirred at RT for 50 min. Then to the zincate solution was added 117 (300 mg, 0.65 mmol) and Pd(PPh3)4 (74 mg, 0.064 mmol). The vial was flushed with nitrogen, capped, and transferred to a preheated (70 °C) heat block and stirred for 18 h. The mixture was cooled to RT, diluted with 0.5N HCl (25 mL), extracted with EtOAc (3 x 25 mL). Combined organic extracts washed with brine (25 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane to provide 118 (96 mg, 45% yield) as an amber solid. The solid was further purified by preparative HPLC using a gradient of 25 to 55 % ACN in water (both containing 0.1% formic acid) over 12 min at a flow of 40 mL/min on a Phenomenex Gemini® 5μm NX-C18 110Å 150 x 21.2 mm column to provide 118 (24 mg, 12% yield) as a white solid. ESI MS m/z 312.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 7.62 (t, J = 0.6 Hz, 1H), 7.48 (br s, 1H), 7.29 (br s, 2H), 7.18 (br s, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 2.46 (s, 3H), 1.76 (s, 3H), 1.68 (s, 3H).
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-methyl-pyrrolo[2,3-b]pyrazine-7-carboxamide (48).
SFC separation of 118 (24 mg) using Mettler Toledo Minigram instrument equipped with a Phenomenex Lux Cellulose-2, 10 x 250 mm, 5 μm column eluting with 50% isopropanol with a flow rate of 10 mL/min yielded 48 (RT 3.76 min, 6 mg, 25% yield) as a white fluffy solid. ESI MS m/z 312.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 7.62 (d, J = 0.7 Hz, 1H), 7.48 (br s, 1H), 7.30 (br s, 2H), 7.19 (br s, 1H), 7.07 (d, J = 8.3 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 2.46 (s, 3H), 1.76 (s, 3H), 1.68 (s, 3H). 99%ee, [α]26.5D +48.0 (c 0.1, MeOH). The distomer (RT 4.43 min) was not characterized.
6-amino-2-cyclopropyl-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (119).
To a solution of 117 (222 mg, 0.50 mmol) in DMF (5 mL) were added lithium chloride (48 mg, 1.1 mmol), and tributyl(cyclopropyl)stannane (330 mg, 1 mmol). The mixture was stirred at 120 °C for 18 h. The mixture was cooled to RT, filtered, and the filtrate was purified by preparative HPLC eluting with a gradient of CH3CN (25 to 60%) in water both containing 0.1% formic acid to afford 119 (36 mg, 21% yield) as an off-white solid. ESI MS m/z 338.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 7.66 (s, 1H), 7.29 (s, 1H), 7.24 (s, 2H), 7.10 (s, 1H), 7.03 (d, J = 8.3 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 2.13 – 2.05 (m, 1H), 1.72 (s, 3H), 1.64 (s, 3H), 0.97 – 0.84 (m, 4H).
(S)-6-amino-2-cyclopropyl-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (49).
SFC separation of 119 (36 mg) using Mettler Toledo Minigram instrument equipped with a Phenomenex Lux Cellulose-2, 10 x 250 mm, 5 μm column eluting with 50% isopropanol with a flow rate of 10 mL/min yielded 49 (RT 5.34 min, 6 mg, 25% yield) as a white fluffy solid. ESI MS m/z 338.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 7.66 (s, 1H), 7.43 (s, 2H), 7.29 (d, J = 8.7 Hz, 2H), 7.12 (s, 1H), 6.99 (d, J = 8.8 Hz, 1H), 2.07 (m, H), 1.73 (s, 3H), 1.11 – 0.75 (m, 4H). 99%ee, [a]25.2D +45.0 (c 0.1, MeOH). The distomer (RT 4.58 min) was not characterized.
methyl 6-amino-7-carbamoyl-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-2-carboxylate (120).
A solution of 117 (744 mg, 1.67 mmol), PdCl2(PPh3)2 (117 mg, 0.167 mmol) in a mixture of DMF (8 mL) and MeOH (8 mL) and Et3N (1.40 mL, 10.0 mmol) was heated at 70 °C under an atmosphere of carbon monoxide (balloon). The apparatus was previously flushed with carbon monoxide once. After 2 h, more PdCl2(PPh3)2 (117 mg, 0.167 mmol) was added, and the reaction was continued for 18 h. The mixture was cooled to RT, filtered through Celite, rinsing with MeOH and the filtrate was concentrated. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% of MeOH in DCM to provide a dark green sticky solid. The solid was dissolved in EtOAc and filtered through a pad of silica gel. The pad was washed with 5% MeOH in EtOAc and the volatiles were evaporated under reduced pressure to provide 120 (418 mg, 70% yield) as a light brown sticky solid. ESI MS m/z 356.1 [M + H]+.
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-(1-hydroxy-1-methyl-ethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (121).
A solution of 120 (322 mg, 0.906 mmol) in THF (12 mL) was cooled to −40 °C and MeMgCl solution in THF (3 M, 4.53 mL, 13.1 mmol) was added dropwise. The mixture was left to warm to RT for 18 h, quenched with saturated aqueous NH4Cl (25 mL), the pH was adjusted to 7–8 with 1N HCl and the mixture was extracted with DCM (3x). The combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM to provide 121 (103 mg, 32% yield) as a light tan solid. ESI MS m/z 338.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 7.99 (s, 1H), 7.50 (br s, 1H), 7.42 (br s, 2H), 7.23 (br s, 1H), 7.08 (d, J = 8.5 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 5.95 – 5.81 (m, 1H), 5.30 – 5.17 (m, 1H), 2.19 (s, 3H), 1.78 (s, 3H), 1.70 (s, 3H).
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-(1-hydroxy-1-methyl-ethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (50).
SFC separation of 121 (39 mg) using Mettler Toledo Minigram instrument equipped with a Phenomenex Lux Cellulose-2, 10 x 250 mm, 5 μm column eluting with 40% isopropanol containing 10 mM ammonium formate with a flow rate of 10 mL/min yielded 50 (RT 3.83 min, 10 mg, 26% yield) as an off-white solid. ESI MS m/z 356.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 1H), 7.99 (s, 1H), 7.51 (d, J = 3.2 Hz, 1H), 7.33 (s, 2H), 7.20 (s, 1H), 7.08 (d, J = 8.3 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 5.25 (s, 1H), 1.77 (s, 3H), 1.69 (s, 3H), 1.51 (s, 6H). 99%ee, [α]26.3D +44.0 (c 0.1, MeOH). The distomer (RT 4.07) was not characterized.
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-morpholino-pyrrolo[2,3-b]pyrazine-7-carboxamide (122).
To a solution of 117 (500 mg, 1.09 mmol) in NMP (4 mL) was added morpholine (598 mg, 6.86 mmol, 0.60 mL) and the mixture was stirred at 130 °C for 18 h. The mixture was then purified using prep-HPLC C18 column eluting with ACN/water/0.1% formic acid to provide 122 (80 mg, 19% yield) as an off-white solid. ESI MS m/z 383.2 [M + H]+.
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-morpholino-pyrrolo[2,3-b]pyrazine-7-carboxamide (51).
SFC separation of 122 (80 mg) using Mettler Toledo Minigram instrument equipped with a Phenomenex Lux Cellulose-2, 10 x 250 mm, 5 μm column eluting with 55% MeOH containing 10 mM ammonium formate with a flow rate of 10 mL/min yielded 51 (RT 5.22 min, 21 mg, 26% yield) as an off-white solid. ESI MS m/z 383.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 7.33 (s, 1H), 7.25 (s, 1H), 7.09 (s, 2H), 7.06 (s, 1H), 7.01 (d, J = 8.3 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 3.70 (m, 4H), 3.35 (m, 4H), 1.73 (s, 3H), 1.65 (s, 3H). 100%ee, [α]25.1D +47.0 (c 0.1, MeOH). The distomer (RT 5.89 min) was not characterized.
6-amino-2-(3,5-difluoro-2-pyridyl)-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (123).
A microwave vial was loaded with 117 (497 mg, 1.1 mmol), copper(I) iodide (33 mg, 0.17 mmol), lithium chloride (100 mg, 2.36 mmol), Pd(dppf)Cl2•CH2Cl2 (85 mg, 0.11 mmol), DMF (8 mL) and tributyl-(3,5-difluoro-2-pyridyl)stannane (888 mg, 2.20 mmol). The vial was purged with nitrogen, capped, and transferred to a preheated (110 °C) heat block for 2.25h. The reaction mixture was cooled to RT, concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 0 to 10% MeOH in DCM to provide 123 (659 mg, 61% yield) as a tan solid. ESI MS m/z 411.3 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.65 (d, J = 2.4 Hz, 1H), 8.36 (d, J = 0.8 Hz, 1H), 8.09 (ddd, J = 11.3, 9.0, 2.4 Hz, 1H), 7.54 (br s, 3H), 7.33 (br s, 1H), 7.10 (dt, J = 8.3, 0.7 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 1.81 (s, 3H), 1.73 (s, 3H).
(S)-6-amino-2-(3,5-difluoro-2-pyridyl)-5-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyrazine-7-carboxamide (52).
SFC separation of 123 (270 mg) using Mettler Toledo Minigram instrument equipped with a Chiral Technologies ID, 10 x 250 mm, 5 μm column eluting with 40% isopropanol containing 10 mM ammonium formate with a flow rate of 10 mL/min yielded 52 (RT 7.87 min, 62 mg, 23% yield) as an off-white solid. ESI MS m/z 411.4 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.65 (d, J = 2.4 Hz, 1H), 8.36 (d, J = 0.8 Hz, 1H), 8.09 (ddd, J = 11.3, 9.0, 2.4 Hz, 1H), 7.54 (br s, 3H), 7.33 (br s, 1H), 7.10 (dt, J = 8.3, 0.7 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 1.81 (s, 3H), 1.73 (s, 3H). 100%ee, [α]26.4D +62.0 (c 0.1, MeOH). The distomer (RT 6.02 min) was not characterized.
6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-thiazol-2-yl-pyrrolo[2,3-b]pyrazine-7-carboxamide (124).
The solution of 117 (6.0 g, 13.1 mmol), copper(I) iodide (400 mg, 2.10 mmol), lithium chloride (1.32 g, 31.1 mmol), Pd(dppf)Cl2•CH2Cl2 (950 mg, 1.21 mmol) and tributyl(thiazol-2-yl)stannane (9.52 g, 25.4 mmol) in DMF (100 mL) was degassed under vacuum and backfilled with nitrogen. The mixture was stirred at 110 °C for 8 h. The volatiles were removed under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 20 to 100% EtOAc in hexanes to provide 124 (2.74 g, 52% yield) as an off-white solid. ESI MS m/z 381.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.49 (s, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.78 (d, J = 3.2 Hz, 1H), 7.61 (s, 2H), 7.40 (s, 1H), 7.29 (s, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 1.77 (s, 3H), 1.69 (s, 3H).
(S)-6-amino-5-(3-hydroxy-2,6-dimethyl-phenyl)-2-thiazol-2-yl-pyrrolo[2,3-b]pyrazine-7-carboxamide (53).
SFC separation of 124 (2.74 g) using Waters Prep 15 SFC-MS instrument equipped with a Chiral Technologies ID, 10 x 250 mm, 5 μm column eluting with 40% isopropanol containing 10 mM ammonium formate with a flow rate of 10 mL/min yielded 53 (RT 4.82 min, 601 mg, 29% yield) as an off-white solid. ESI MS m/z 381.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.49 (s, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.78 (d, J = 3.2 Hz, 1H), 7.61 (s, 2H), 7.40 (s, 1H), 7.29 (s, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 1.77 (s, 3H), 1.69 (s, 3H). 100%ee, [α]26.3D +57.0 (c 0.1, MeOH). The distomer (RT 5.43 min) was not characterized.
3-bromo-N-(3-methoxy-2,6-dimethyl-phenyl)-5-nitro-pyridin-2-amine (127).
To a solution of 125 (20 g, 63.85 mmol) in NMP (120 mL) were added 2,6-dimethylpyridine (11.08 g, 103.4 mmol, 12 mL) and 92 (14 g, 95.2 mmol). The mixture was heated at 130 °C for 18 h. After cooling the mixture to RT, water was added dropwise to yield a precipitate. The suspension was stirred at RT for 20 min, then filtered. The solid was washed with water, dried under vacuum, and the residue was purified by silica gel chromatography eluting with a gradient of 10 to 30% EtOAc in heptane to provide 127 (12 g, 53% yield) as an off-white solid. ESI MS m/z 354.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (s, 1H), 8.80 (d, J = 2.4 Hz, 1H), 8.60 (d, J = 2.5 Hz, 1H), 7.10 (dt, J = 8.3, 0.7 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 3.78 (s, 3H), 2.01 (s, 3H), 1.92 (s, 3H).
2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)-5-nitro-pyrrolo[2,3-b]pyridine-3-carbonitrile (129).
To a solution of malononitrile (4.4 g, 66.6 mmol) in DME (120 mL) was added portion wise NaH (60% dispersion in mineral oil, 2.90 g, 66.9 mmol). After stirring for 5 min, 127 (11.6 g, 32.9 mmol) and Pd(dppf)Cl2•CH2Cl2 (1.34 g, 1.65 mmol) were added. The mixture was heated at 110 °C for 2 h. After cooling to RT, the mixture was diluted with water and extracted twice with EtOAc. The combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 60% EtOAc in hexanes to provide 129 (11 g, 99% yield) as a yellow solid. ESI MS m/z 338.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (d, J = 2.4 Hz, 1H), 8.28 (d, J = 2.4 Hz, 1H), 7.67 (br s, 2H), 7.27 (dt, J = 8.4, 0.8 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 1.79 (d, J = 0.7 Hz, 3H), 1.70 (s, 3H).
tert-butyl N-[3-cyano-1-(3-methoxy-2,6-dimethyl-phenyl)-5-nitro-pyrrolo[2,3-b]pyridin-2-yl]carbamate (131).
To a solution of 129 (1.130 g, 3.35 mmol) in THF (15 mL) were added Et3N (3.37 mmol, 0.47 mL), DMAP (45 mg, 368 μmol) and tert-butoxycarbonyl tert-butyl carbonate (1.47 g, 6.73 mmol). The mixture was stirred at 50 °C for 1h then cooled to RT. Ethylenediamine (500 μL) was added and the mixture was stirred for 2h, then diluted with water and extracted with DCM twice. The combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 20 to 60% EtOAc in to provide 131 (1.27 g, 87% yield). ESI MS m/z 438.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.08 (d, J = 2.4 Hz, 1H), 8.87 (d, J = 2.4 Hz, 1H), 7.23 (dt, J = 8.5, 0.8 Hz, 1H), 7.10 (d, J = 8.5 Hz, 1H), 3.81 (s, 3H), 1.71 (d, J = 0.7 Hz, 3H), 1.61 (s, 3H), 1.38 (s, 9H).
tert-butyl N-[5-amino-3-cyano-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridin-2-yl]carbamate (133).
To a solution of 131 (2.94 g, 6.72 mmol) in DCM (30 mL) and MeOH (30 mL) was added palladium on carbon (10% w/w, 400 mg, 0.376 mmol). The mixture was stirred for 3h under 1 atm of H2 (balloon). The suspension was filtered over a pad of Celite and the filtrate was concentrated under reduced pressure to provide 133 (2.7 g, 99% yield) as an off-white solid. ESI MS m/z 408.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 1H), 7.65 (d, J = 2.5 Hz, 1H), 7.31 – 7.08 (m, 2H), 7.02 (d, J = 8.5 Hz, 1H), 5.16 (s, 2H), 3.78 (s, 3H), 1.71 (d, J = 0.7 Hz, 3H), 1.59 (s, 3H), 1.33 (s, 9H).
tert-butyl N-[5-bromo-3-cyano-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridin-2-yl]carbamate (135).
To a solution of 133 (15.1 g, 37.1 mmol) in a mixture of DMF (60 mL) and ACN (80 mL) was added tert-butyl nitrite (5.72 g, 55.5 mmol, 6.6 mL) followed by Copper(II) bromide (10 g, 44.8 mmol). The mixture was stirred at 60 °C for 20 min, then diluted with water and extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried over Na2SO4, filtered. The filtrate was evaporated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 5% EtOAc in DCM to provide 135 (9.67 g, 55% yield) as an off-white solid. ESI MS m/z 471.2 [M + H]+. 1H NMR (400 MHz, Chloroform-d) δ 8.27 (d, J = 2.2 Hz, 1H), 8.16 (d, J = 2.2 Hz, 1H), 7.22 (d, J = 7.2 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.18 (s, 1H), 3.87 (s, 3H), 1.82 (s, 3H), 1.75 (s, 3H), 1.51 (s, 9H).
2-amino-5-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (137).
To a solution of 135 (1.05 g, 2.23 mmol) in EtOH (15 mL) at 80 °C was added aqueous HCl (6 M, 6 mL). The mixture was stirred for 20 min then concentrated to dryness. The residue was dissolved in MeOH, made alkaline with Et3N and the volatiles were evaporated under reduced pressure. The residue was purified by reverse phase flash chromatography on a C18 cartridge eluting with CH3CN/water/0.1% formic acid to provide 2-amino-5-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carbonitrile (515 mg, 60% yield) as an off-white solid. ESI MS m/z 371.2 [M + H]+. The off-white solid was solubilized in a mixture of EtOH (6 mL) and water (2 mL). LiOH monohydrate (500 mg, 11.9 mmol) and H2O2 (27% w/w aq. solution, 0.65 mL, 21 mmol) were added and the mixture was stirred at 60 °C for 20 min, cooled to RT, diluted with water and the precipitate was recovered by filtration. The solid was washed with water, dried under vacuum to provide 137 (600 mg, 69% yield) as an off-white solid. ESI MS m/z 389.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 2.0 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 7.20(dt, J = 8.4, 0.7 Hz, 1H), 7.13 (s, 2H), 7.05 (d, J = 8.5 Hz, 1H), 6.83 (s, 2H), 3.73 (s,3H), 1.75 (d, J = 0.7 Hz, 3H), 1.65 (s, 3H).
2-amino-5-bromo-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (139).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 8 mL, 8 mmol) was added to a solution of 137 (1.0 g, 2.57 mmol) in DCM (8 mL) and the mixture was stirred for 30 min at RT. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM to provide 139 (605 mg, 62% yield) as an off-white solid. ESI MS m/z 375.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 7.82 (dd, J = 1.8, 0.8 Hz, 1H), 7.54 (dd, J = 1.8, 0.8 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.91 – 6.76 (m, 3H), 6.66 (s, 2H), 2.27 (s, 3H), 1.69 (s, 3H), 1.61 (s, 3H).
2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5-methyl-pyrrolo[2,3-b]pyridine-3-carboxamide (140).
To a solution of 139 (120 mg, 0.31 mmol) in THF (3 mL) was added Pd(t-Bu3P)2 (15 mg, 0.029 mmol). The mixture was flushed with nitrogen and dimethylzinc (2 M, 0.75 mL) was added. The mixture was stirred at 70 °C under nitrogen for 1 h. After cooling to RT, it was diluted with EtOAc (50 mL), washed with water and brine, and the organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with a gradient of 0 to 10% MeOH in DCM to provide 140 (58 mg, 61% yield) as an off-white solid. ESI MS m/z 311.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 7.82 (dd, J = 1.8, 0.8 Hz, 1H), 7.54 (dd, J = 1.8, 0.8 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.91 – 6.76 (m, 3H), 6.66 (s, 2H), 2.27 (s, 3H), 1.69 (s, 3H), 1.61 (s, 3H).
(S)-2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5-methyl-pyrrolo[2,3-b]pyridine-3-carboxamide (54).
SFC separation of 140 (29 mg) using Waters Prep 100 SFC-MS instrument equipped with a Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm column eluting with 45% 1:1 ACN:EtOH with a flow rate of 70 mL/min yielded 53 (RT 3.74 min, 8.5 mg, 30% yield) as an off-white solid. ESI MS m/z 311.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.49 (s, 1H), 7.87 (dd, J = 1.9, 0.8 Hz, 1H), 7.58 (dd, J = 1.9, 0.8 Hz, 1H), 7.04 (d + br s, J = 8.4 Hz, 1H), 6.90 (d, J = 8.3 Hz, 3H), 6.70 (br s, 2H), 2.31 (d, J = 0.8 Hz, 3H), 1.73 (s, 3H), 1.65 (s, 3H). 100%ee, [α]26.3D +32.0 (c 0.1, MeOH). The distomer (RT 4.05 min) was not characterized.
2-amino-5-cyclopropyl-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (141).
To a solution of 2-amino-5-bromo-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (817 mg, 2.10 mmol) in THF (20 mL) were added cyclopropylzinc bromide (0.5 M in THF, 16 mL) and Pd(t-Bu3P)2 (110 mg, 0.22 mmol). The mixture was flushed with nitrogen, then heated to 70 °C for 1 h under nitrogen atmosphere. The mixture was cooled to RT and sat. NH4Cl was added. It was diluted with water, extracted with EtOAc twice. The organic extracts were combined and washed with brine, dried with Na2SO4, and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 30–90% EtOAc in heptane to provide 141 (300 mg, 40% yield) as an off-white solid. ESI MS m/z 337.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.43 (s, 1H), 7.58 (s, 1H), 7.50 (s, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.86 (m, 3H), 6.69 (s, 2H), 1.88 m, 1H), 1.69 (s, 3H), 1.61 (s, 3H), 0.86 (m, 2H), 0.81 – 0.68 (m, 2H).
(S)-2-amino-5-cyclopropyl-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (56).
SFC separation of 141 (300 mg) using Waters Prep 100 SFC-MS instrument equipped with a Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm column eluting with 50% 1:1 ACN:EtOH with a flow rate of 70 mL/min yielded 56 (RT 5.60 min, 90 mg, 30% yield) as an off-white solid. ESI MS m/z 337.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.12 (d, J = 2.2 Hz, 1H), 8.08 – 7.92 (m, 2H), 7.46 (s, 1H), 7.00 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.3 Hz, 1H), 2.03 (m, 1H), 1.70 (s, 3H), 1.59 (s, 3H), 0.96 (m, 2H), 0.68 (m, 2H). 100%ee, [α]25.1D +38.0 (c 0.1, MeOH). The distomer (RT 7.81 min) was not characterized.
3-bromo-N-(3-methoxy-2,6-dimethyl-phenyl)-6-methyl-5-nitro-pyridin-2-amine (128).
92 (9.20 g, 60.8 mmol, 126 (10.11 g, 40.2 mmol), NMP (40 mL) and 2,6-dimethylpyridine (8.58 g, 80.1 mmol, 9.3 mL) were heated to 130 °C for 5 days under nitrogen atmosphere. The mixture was cooled to RT and the resulting paste transferred to a conical flask and 0.5N HCl (500 mL) was added dropwise while stirring, resulting in a sticky brown solid. The supernatant was filtered on a Buchner funnel and the recovered gum was washed with water, dissolved in DCM, and combined with the sticky brown solid which had also been dissolved in DCM (200 mL total). The DCM solution was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% DCM in heptane to provide 128 (10.5 g, 71% yield) as a light yellow solid. ESI MS m/z 368.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.46 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.3 Hz, 1H), 3.74 (s, 3H), 2.38 (s, 3H), 1.98 (s, 3H), 1.89 (s, 3H).
2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)-6-methyl-5-nitro-pyrrolo[2,3-b]pyridine-3-carbonitrile (130).
To a RBF containing sodium hydride (3.13 g, 72.2 mmol, 60% w/w in mineral oil) in DME (150 mL) was added a solution of malononitrile (4.75 g, 71.9 mmol) in DME (50 mL) slowly at RT. After stirring for 30 min, 128 (10.5 g, 28.7 mmol) and Pd(dppf)Cl2•CH2Cl2 (2.31 g, 2.83 mmol) were added. The resulting mixture was degassed by bubbling nitrogen through the solution, equipped with a condenser, and heated to reflux for 1 h. After cooling to RT, the mixture was poured into saturated aqueous NH4Cl and extracted with DCM (3x). The combined organic extracts were washed with water, brine, dried over Na2SO4, filtered, and adsorbed on silica gel. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane. Appropriate fractions were combined, concentrated and the resulting solid was triturated with DCM, filtered, and dried under vacuum, affording 130 (7.97 g, 79% yield) as a bright yellow solid. ESI MS m/z 352.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (s, 1H), 7.46 (s, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 3.80 (s, 3H), 1.75 (s, 3H), 1.67 (s, 3H).
tert-butyl N-[5-amino-3-cyano-1-(3-methoxy-2,6-dimethyl-phenyl)-6-methyl-pyrrolo[2,3-b]pyridin-2-yl]carbamate (134).
To a solution of compound 130 (9.0 g, 25.6 mmol) in THF (120 mL) was added triethylamine (7.99 g, 78.9 mmol, 11 mL), DMAP (312 mg, 2.55 mmol) and tert-butoxycarbonyl tert-butyl carbonate (17.0 g, 77.9 mmol). The mixture was stirred at 50 °C for 40 min. The heating was stopped, ethylenediamine (6.20 g, 103 mmol, 6.90 mL) was added, the mixture was stirred at RT for 45 min, then diluted with water and DCM. The layers were separated, and the aqueous layer was extracted with DCM twice. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 60% EtOAc in heptane to provide impure 132 (13.94 g) as an off-white solid, which was contaminated with tert-butyl N-[2-(tert-butoxycarbonylamino)ethyl]carbamate (50 mol% by 1H NMR). ESI MS m/z 452.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.78 (s, 1H), 7.26 (d, J = 8.5 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 2.66 (s, 3H), 1.77 (s, 3H), 1.66 (s, 3H), 1.40 (s, 9H). To a RBF containing crude 132 (13.94 g, 25.6 mmol) in DCM (280 mL) and MeOH (280 mL) was added palladium on carbon (2.08 g, 1.95 mmol, 10%w/w) as a slurry in some of the solvent mixture. The mixture was flushed with H2 and stirred under H2 atmosphere (balloon) for 18 h. The mixture was flushed with nitrogen, filtered on a celite pad and the pad was rinsed with DCM. The filtrate was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with a gradient of 20 to 100% EtOAc in heptane to afford 134 (9.34 g, 87% yield) as an off-white solid. ESI MS m/z 422.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 7.15 (m, 2H), 7.00 (d, J = 8.4 Hz, 1H), 3.78 (s, 3H), 2.18 (s, 3H), 1.71 (s, 3H), 1.59 (s, 3H), 1.31 (s, 9H).
tert-butyl N-[5-chloro-3-cyano-1-(3-methoxy-2,6-dimethyl-phenyl)-6-methyl-pyrrolo[2,3-b]pyridin-2-yl]carbamate (136).
To a solution of 134 (390 mg, 0.93 mmol) in ACN (3 mL) and DMF (2 mL) was added tert-butyl nitrite (193.64 mg, 1.88 mmol, 0.22 mL), followed by Copper(II) chloride (149 mg, 1.11 mmol). The mixture was transferred to a preheated 60 °C heat block and equipped with a condenser. After 45 min at this temperature, the volatiles were evaporated under reduced pressure and the residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane to provide 136 (156 mg, 38% yield) as a yellow foamy solid. ESI MS m/z 441.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.20 (s, 1H), 7.19 (d, J = 8.5 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 3.80 (s, 3H), 1.71 (s, 3H), 1.59 (s, 3H), 1.34 (s, 9H).
2-amino-5-chloro-1-(3-methoxy-2,6-dimethyl-phenyl)-6-methyl-pyrrolo[2,3-b]pyridine-3-carboxamide (138).
A solution of 136 (135 mg, 0.31 mmol) in NMP (1.3 mL) was heated at 160 °C for 75 min. After cooling to RT, the mixture was diluted with water (3 mL). EtOAc (15 mL) then a saturated aqueous NH4Cl (0.5 mL) were added. The layers were separated, and the aqueous layer was extracted with EtOAc twice. Combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane to provide 2-amino-5-chloro-1-(3-methoxy-2,6-dimethyl-phenyl)-6-methyl-pyrrolo[2,3-b]pyridine-3-carbonitrile (80 mg, 77% yield) as a light beige solid. ESI MS m/z 341.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.58 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.09 (s, 2H), 7.04 (d, J = 8.5 Hz, 1H), 3.79 (s, 3H), 2.30 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). Sulfuric acid (1 mL) was added to the light beige solid. After 30 min, the solution was diluted with cold water (5 mL) and concentrated ammonium hydroxide was added until the pH was slightly basic. The solid recovered by filtration was purified by preparative HPLC (Phenomenex Gemini) eluting with a gradient of CH3CN (30 to 80%) in water both containing 0.1% formic acid. Appropriate fractions were combined and lyophilized to afford 138 (62 mg, 49% yield). ESI MS m/z 359.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (s, 1H), 7.20 (d, J = 8.5 Hz, 1H), 7.04 (d, J = 8.5 Hz, 1H), 6.98 (s, 2H), 6.78 (s, 2H), 3.80 (s, 3H), 2.30 (s, 3H), 1.75 (s, 3H), 1.66 (s, 3H).
2-amino-5-chloro-1-(3-hydroxy-2,6-dimethylphenyl)-6-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide (142).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 9.2 mL, 9.2 mmol) was added to a solution of 138 (1.1 g, 3.07 mmol) in DCM (9.2 mL) and the mixture was stirred for 60 min at RT. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% MeOH in DCM to provide 142 (1.0 g, 90% yield) as an off-white solid ESI MS m/z 345.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 8.07 (s, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.94 (s, 2H), 6.87 (d, J = 8.2 Hz, 1H), 6.76 (s, 2H), 2.31 (s, 3H), 1.70 (s, 3H), 1.62 (s, 3H).
(S)-2-amino-5-chloro-1-(3-hydroxy-2,6-dimethylphenyl)-6-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide (57).
SFC separation of 142 (610 mg) using Waters Prep 100 SFC-MS instrument equipped with a Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm column eluting with 45% 1:1 ACN:EtOH with a flow rate of 70 mL/min yielded 57 (RT 4.22 min, 177 mg, 29% yield) as an off-white solid. ESI MS m/z 345.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 8.07 (s, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.94 (s, 2H), 6.87 (d, J = 8.2 Hz, 1H), 6.76 (s, 2H), 2.31 (s, 3H), 1.70 (s, 3H), 1.62 (s, 3H). 99%ee, [α]25.0D +46.0 (c 0.1, MeOH). The distomer (RT 4.94 min) was not characterized.
3-bromo-5-chloro-N-(3-methoxy-2,6-dimethyl-phenyl)pyridin-2-amine (144).
To a solution of 92 (3.61 g, 23.9 mmol) and 143 (5.02 g, 23.9 mmol) in THF (50 mL) was added LiHMDS solution in THF (1 M, 48 mL) dropwise over 18 min (16 °C exotherm was observed). After 30 min, the mixture was diluted with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was separated, washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with a gradient of 0 to 20% EtOAc in heptane to provide 144 (6.58 g, 81% yield) as a peach solid. ESI MS m/z 343.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 2.3 Hz, 1H), 7.97 (s, 1H), 7.91 (d, J = 2.3 Hz, 1H), 7.04 (dt, J = 8.3, 0.8 Hz, 1H), 6.82 (d, J = 8.4 Hz, 1H), 3.77 (s, 3H), 2.01 (d, J = 0.6 Hz, 3H), 1.91 (s, 3H).
2-amino-5-chloro-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carbonitrile (145).
To a suspension of NaH (60% dispersion in mineral oil, 1.08 g, 24.9 mmol) in DME (60 mL) was added malononitrile (1.62 g, 24.6 mmol) in DME (15 mL) dropwise. After stirring for 30 min, 144 (4.00 g, 11.7 mmol) in DME (15 mL) and Pd(dppf)Cl2•CH2Cl2 (1.08 g, 1.32 mmol) were added. The mixture was flushed with nitrogen, then stirred at 100 °C for 5 h. After cooling to RT, icy water (250 mL) was added dropwise. The resulting precipitate was collected by filtration and washed with water. The solid was air-dried and purified by silica gel chromatography eluting with a gradient of 0 to 100% EtOAc in heptane to afford 145 (3.27 g, 85% yield) as an ivory crystalline solid. ESI MS m/z 327.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.80 (d, J = 2.2 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.32 (br s, 2H), 7.24 (dt, J = 8.5, 0.8 Hz, 1H), 7.09 (d, J = 8.5 Hz, 1H), 3.83 (s, 3H), 1.78 (d, J = 0.7 Hz, 3H), 1.69 (s, 3H).
2-amino-5-chloro-1-(3-methoxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (146).
To a suspension of 145 (7.50 g, 23.0 mmol) in water (60 mL) and EtOH (180 mL) was added LiOH monohydrate (7.22 g, 172 mmol) and H2O2 (27% w/w aq. solution, 9.8 mL). The mixture was stirred at 60 °C for 30 min, then cooled to RT. Water was added dropwise (500 mL) and the solid was collected by filtration, washed with water and air-dried. The filtrate was diluted with more water (500 mL) and a second crop of solid was obtained by filtration. Finally, the filtrate was extracted with EtOAc (3x). The combined organic extracts were dried over Na2SO4, filtered, concentrated, then dried under reduced pressure affording a third crop of solid. The three crops were combined and purified by silica gel chromatography eluting with a gradient of 50 to 100% EtOAc in heptane to provide 146 (3.90 g, 49% yield) as a light-yellow solid. Alternatively, this nitrile hydrolysis could be performed under acidic conditions following the procedure used to prepare 76: a solution of 145 (7.50 g, 23.0 mmol) in sulfuric acid was stirred at RT for 2 h providing 146 (7.9 g, quantitative yield). ESI MS m/z 345.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.15 (d, J = 2.1 Hz, 1H), 7.74 (d, J = 2.1 Hz, 1H), 7.27 – 7.22 (m, 1H), 7.18 (br s, 2H), 7.09 (d, J = 8.5 Hz, 1H), 6.87 (br s, 2H), 3.84 (s, 3H), 1.78 (d, J = 0.7 Hz, 3H), 1.69 (s, 3H).
2-amino-5-chloro-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (147).
Following the procedure used to prepare 3, BBr3 (1M in DCM, 34 mL, 34 mmol) was added to a solution of 146 (3.90 g, 11.3 mmol) in DCM (34 mL) and the mixture was stirred for 2h at RT. The residue was purified by silica gel chromatography using a gradient of 0 to 20% MeOH in DCM to provide (3.54g, 95% yield) as a light beige solid. ESI MS m/z 331.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 8.14 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 2.1 Hz, 1H), 7.14 (br s, 2H), 7.06 (d, J = 8.3 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.86 (br s, 2H), 1.74 (s, 3H), 1.65 (s, 3H).
(S)-2-amino-5-chloro-1-(3-hydroxy-2,6-dimethyl-phenyl)pyrrolo[2,3-b]pyridine-3-carboxamide (55).
SFC separation of 147 (3.54 g) using Waters Prep 100 SFC-MS instrument equipped with a Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm column eluting with 45% 1:1 ACN:EtOH with a flow rate of 70 mL/min yielded 55 (RT 5.37 min, 1.26 g, 36% yield) as an off-white solid. ESI MS m/z 331.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 8.14 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 2.1 Hz, 1H), 7.14 (br s, 2H), 7.06 (dt, J = 8.2, 0.7 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.86 (br s, 2H), 1.74 (d, J = 0.7 Hz, 3H), 1.65 (s, 3H). 100%ee, [α]26.5D +40.0 (c 0.1, MeOH). The distomer (RT 7.79 min) was not characterized.
3-bromo-5,6-dimethylpyridin-2-ol (149).
Sulfuric acid (140 mL) was added slowly to water (1.15 L) and the solution was cooled at 25 °C. 148 (114.80 g, 571 mmol) was added at this temperature and the solution was cooled at 0–5 °C with an ice/water bath to get a suspension. Under vigorous stirring, a solution of sodium nitrite (49.25 g, 714 mmol) in water (175 mL) was added dropwise over 90 minutes. The ice-water bath was removed, and the suspension was warmed up slowly to 11 °C and stirred for 1 h. A solution of sodium hydroxide (175 g, 4.37 mol) in water (400 mL) was added dropwise at 20 °C. The pH of the solution was adjusted to 7 with K2HPO4 (~58 g, 0.33 mol) in water (70 mL). The suspension was filtered at 10 °C. The solid was triturated in water (250 mL) and recovered by filtration. The solid was washed profusely with ice-cold water and dried by vacuum suction. The solid was oven-dried under reduced pressure at 60 °C for 18 h to yield 149 as a light yellow crystalline solid (105.69 g, 92% yield). ESI MS m/z 202.0 204.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.96 (br s, 1H), 7.73 (s, 1H), 2.11 (s, 3H), 1.96 (s, 3H).
2,3-dibromo-5,6-dimethylpyridine (150).
To a solution of 149 (105.3 g, 521 mmol) in DMF (316 mL) and toluene (527 mL) at 90 °C under nitrogen was added phosphorus oxybromide (1.3:1, 56.5% wt/wt in xylenes) (278 mL, 782 mmol) dropwise over 90 min. After the addition was complete, the mixture was stirred at 90 °C for 18 h, cooled to RT, and was slowly added to water (2 L). The flask was washed with 500 mL of water. The combined aqueous phases were extracted with MTBE (3 x 1 L). The organic phases were combined and washed with 0.5N NaOH (1 L), water (3 x 1 L) and brine (1 L) then dried with Na2SO4 and concentrated under reduced pressure. The solid was partially dissolved in MTBE (400 mL) and heptane (300 mL) was added. The volatiles were partially evaporated under reduced pressure to ~175 mL resulting in a suspension that was filtered. The solid was rinsed with heptane and dried under vacuum to afford 149 as a beige solid (114.3 g, 83% yield). The filtrate was concentrated under reduced pressure and filtered as previously to get a second crop (8.7 g, 6.3% yield). ESI MS m/z 264.0, 266.0, 268.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.95 (s, 1H), 2.36 (s, 3H), 2.21 (s, 3H).
3-bromo-N-(3-methoxy-2,6-dimethylphenyl)-5,6-dimethylpyridin-2-amine (151).
A 2 L 4-neck round-bottomed flask was charged with 92 (32.96 g, 218 mmol), DME (750 mL), 150 (55 g, 208 mmol), XantPhos (10.81 g, 18.7 mmol) and cesium carbonate (169.1 g, 519 mmol). The mixture was sonicated for 20 minutes while sparging the suspension with nitrogen. Pd2(dba)3 (8.6 g, 9.3 mmol) was added and the suspension was heated to reflux. After 13 h the mixture was cooled to RT and filtered over a pad of silica gel. The pad was washed with EtOAc (1.2L). The filtrate was partially evaporated under reduced pressure to about ~200 mL and heptane (300 mL) was added. The volatiles were partially evaporated under reduced pressure to ~100 mL resulting in a suspension. The solid was collected by filtration and washed with heptane to get 151 as a light-yellow solid (56.23 g, 81% yield). ESI MS m/z 335.2, 337.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.57 (s, 1H), 7.26 (s, 1H), 7.02 (d, J = 8.6 Hz, 1H), 6.77 (d, J = 8.3 Hz, 1H), 3.76 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.94 (s, 3H).
2-amino-1-(3-methoxy-2,6-dimethylphenyl)-5,6-dimethyl-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (152).
To a degassed solution of malononitrile (33.26 g, 503.5 mmol) in DME (1 L) was added sodium tert-butoxide (46.32 g, 482 mmol) in 4 equal portions. The mixture was stirred 30 min at RT to obtain a solution. 151 (80 g, 238.6 mmol) and Pd(dppf)Cl2•CH2Cl2 (14.91 g, 18.26 mmol) were added in one portion and the suspension was heated to a strong reflux. After 17 h, the mixture was cooled, transferred into a 5 L flask and EtOAc (1.5 L) was added. A solution of N-acetyl-L-cysteine (12.1 g, 74 mmol, 4x Pd mol content) and Na2CO3 (15.7 g, 148 mmol) in water (500 mL) were added. The biphasic solution was stirred for 10 minutes at 60 °C and then cooled slowly to 40 °C over 75 minutes. Inside the 5 L flask, the two layers were separated at 40 °C and the organic phase was washed with water (2x 250 mL), brine (200 mL), and then filtered over a pad of silica gel (185 g). The pad was rinsed with DCM/EtOAc (1:1). The filtrate was evaporated under reduced pressure, and the solvents were switched for EtOAc during rotavap evaporation to get a suspension. The suspension was filtered at RT and the solid was suspended and triturated in 50 mL of ice-cooled EtOAc. The suspension was filtered, and the solid was rinsed with 50 mL of ice-cooled EtOAc. The solid was oven-dried under reduced pressure at 60 °C for 18 h to afford 152 as a light yellow solid (65.38 g, 86% yield). ESI MS m/z 321.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.38 (s, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 8.6 Hz, 1H), 6.76 (br s, 2H), 3.84 (s, 3H), 2.26 (s, 3H), 2.23 (s, 3H), 1.78 (s, 3H), 1.69 (s, 3H).
2-amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide (153).
To methanesulfonic acid (600 mL) was added slowly a solution of sulfuric acid (93 mL) and water (7.0 mL) over 5 minutes at RT. 152 (80 g, 249.7 mmol) was added portion wise over 15 minutes to keep the reaction temperature below 40 °C. The resulting solution was stirred at RT for 90 minutes. DL-methionine (149.0 g, 999 mmol) was added portion wise over 20 minutes below 40 °C. The solution was stirred at 40 °C for 37 h. The mixture was cooled to RT and slowly added over 1.5h to a solution of K2HPO4 (100 g) and NaOH (540 g) in water (5 L). EtOAc (1 L) was added, and the biphasic mixture was stirred for 5 min to get a precipitate. The suspension was filtered. The filtrate was extracted with EtOAc (3 x 1L). The organic phases were combined, dried with Na2SO4, filtered, and concentrated under reduced pressure to ~ 100 mL to get a suspension. The suspension was filtered and rinsed with EtOAc (50 mL). The solids were combined and triturated in water (800 mL) two times. The residue was suspended in EtOAc (500 mL), stirred for 10 min and filtered. The product was oven-dried under reduced pressure to get 67.8 g of a solid that was suspended in DMSO (350 mL, 5 vol) and the mixture was heated to 65 °C to get a solution. The solution was cooled slowly at 28 °C with a water bath. Water (1.05 L) was added dropwise over 2 hours to get a suspension. After 5 minutes of stirring at RT, the suspension was filtered. The solid was triturated in 100 mL of water and filtered. The filter cake was washed with 2 x 100 mL of water. The product was oven dried at 60 °C under reduced pressure to get 153 as a light yellow solid (61.50 g, 75% yield). ESI MS m/z 325.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 7.82 (s, 1H), 7.05 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.71 (br. s, 2H), 6.64 (br. s., 2H), 2.27 (s, 3H), 2.24 (s, 3H), 1.75 (s, 3H), 1.66 (s, 3H).
(S)-2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (RP-6306).
SFC separation of 153 (1.60 g) using Waters Prep 100 SFC-MS instrument equipped with a Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm column eluting with 55% 1:1 ACN:EtOH containing 10 mM ammonium formate with a flow rate of 70 mL/min yielded RP-6306 (RT 3.94 min, 381 mg, 24% yield) as an off-white solid. ESI MS m/z 325.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.72 (s, 2H), 6.65 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). 13C NMR (101 MHz, dmso) δ 168.90, 154.62, 152.27, 145.59, 143.95, 133.10, 128.26, 127.51, 125.66, 124.47, 124.11, 116.59, 115.97, 83.55, 22.22, 19.34, 17.37, 11.35. [a]28D +35.0 (c 5.00, EtOH). Melting point: 273.8 to 279.0 °C. m/z (ESI, +ve ion): 325.1 (M+H)+. HRMS calculated for: C18H21N4O2 325.1665; found 325.1659. 100%ee. The distomer (RT 4.35 min) was not characterized.
Supplementary Material
Acknowledgements
Catalina Suarez for assistance with hepatocyte assay. Adrian Fretland for thoughtful review of this manuscript. We are grateful for the support, training, and custom tools provided by the Chemical Computing Group, Inc., Montreal, Quebec, Canada. This work was supported in part by a CIHR Foundation grant to F.S. and was based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Abbreviations used
- Ac
acetyl
- ATP
adenosine triphosphate
- ACN
acetonitrile
- Ar
aryl
- aq.
aqueous
- Bn
benzyl
- Boc
tert Butyloxycarbonyl
- n-BuLi
n-butyl lithium
- CL
clearance
- DIPEA
diisoproplyethyl amine
- DMAP
dimethylaminopyridine
- DME
dimethoxyethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- ESI
electrospray ionization
- Et3N
trimethylamine
- EtOAc
ethyl acetate
- EtOH
ethanol
- h
hour
- HATU
(1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HPLC
high performance liquid chromatography
- hERG
human Ether-à-go-go-Related Gene
- HMDS
bis(trimethylsilyl)amide
- LCMS
HPLC with mass spectral detection
- M
molar
- mmol
millimole
- MeOH
methanol
- min
minute
- MOM
methoxymethyl
- Ms
methanesulfonyl
- MS
mass spectrometry
- N
normal
- NBS
N-bromosuccinimide
- NMP
N-methyl pyrrolidinone
- NMR
nuclear magnetic resonance spectroscopy
- PdCl2(dppf)
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)
- PdCl2(dppf).CH2Cl2
[1,1′ Bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane
- Pd2(dba)3
tris(dibenzylideneacetone)dipalladium
- Pd-PEPPSI™-SIPr
(1,3-Bis(2,6-diisopropylphenyl)imidazolidene) (3-chloropyridyl) palladium(II) dichloride
- PMB
para-methoxybenzyl
- PPB
plasma protein binding
- rt
room temperature
- sat.
saturated
- SEM
[2-(trimethylsilyl)ethoxy]methyl
- SFC
supercritical fluid chromatography
- SNAr
nucleophilic aromatic substitution
- Tf
trifluoromethanesulfonyl
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TPSA
topological polar susface area
- Ts
p-toluenesulfonyl
- UPLC
ultra-performance liquid chromatography
- Xantphos
4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
Footnotes
Conflict of Interest Disclosure
J. S.; R. P.; E. D.; B. L.; F. V.; M.-E. L.; J. F.; A. L. P.; S. Y. Y.; A. R.; C. G. M.; N. M. D.; R. S.; C. G.; A. B.-F.; R. K.; V. B.; M. G. and W. C. B. are employees of Repare Therapeutics. F. S. is a founder and consultant of Repare Therapeutics. G. M.; V. P.; P. M.; J.-F. T.; M. D.; D. M.; S. O.; P. B.; P. T.; I. K. and Y. M. declare no competing financial interest.
Supporting Information
Images of 28 (crystal structure PDB ID 8D6C) and 41 (crystal structure PDB ID 8D6F) bound to PKMYT1 and detailed 2D plots of the interactions. X-ray data collection, refinement statistics and PDB validation reports for compounds 28, 39, 41, and RP-6306. 1HNMR, 13CNMR and HRMS spectra of RP-6306. UPLC-MS traces of compounds 1–57 and RP-6306. A table of molecular formula stings for each final compound with their PKMYT1 enzymatic and cell-based IC50. Authors will release the atomic coordinates of all four disclosed PDB (8D6C, 8D6D, 8D6E and 8D6F) upon article publication.
REFERENCES
- 1.Sonntag R; Giebeler N; Nevzorova YA; Bangen J-M; Fahrenkamp D; Lambertz D; Haas U; Hu W; Gassler N; Cubero FJ; Müller-Newen G; Abdallah AT; Weiskirchen R; Ticconi F; Costa IG; Barbacid M; Trautwein C; Liedtke. C. Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc. Natl. Acad. Sci 2018, 115, 9282–9287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teixeira LK; Reed SI Cyclin E deregulation and genomic instability. Adv. Exp. Med. Biol 2017, 1042, 527–547. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt M; Rohe A; Platzer C; Najjar A; Erdmann F; Sippl W Regulation of G2/M transition by inhibition of WEE1 and PKMYT1 kinases. Molecules, 2017, 22, 2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu H; George E; Kinose Y; Kim H; Shah JB; Peake JD; Ferman B; Medvedev S; Murtha T; Barger CJ; Devins KM; D’Andrea K; Wubbenhorst B; Schwartz LE; Hwang WT; Mills GB; Nathanson KL; Karpf AR; Drapkin R; Brown EJ; Simpkins F CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models. Cell Rep. Med 2021, 2, 100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow JP; Poon RY The CDK1 inhibitory kinase MYT1 in DNA damage checkpoint recovery. Oncogene 2013, 32, 4778–4788. [DOI] [PubMed] [Google Scholar]
- 6.Gallo D; Young JTF; Fourtounis J; Martino G; Álvarez-Quilón A; Bernier C; Duffy N; Papp R; Roulston A; Stocco R; Szychowski J; Veloso A; Alam H; Baruah P; Bonneau-Fortin A; Bowlan J; Chaudhary N; Desjardins J; Dietrich E; Fournier S; Fugère-Desjardins C; Goullet de Rugy T; Leclaire M-L; Liu B; Melo H; Nicolas O; Pellerin C; Singhania A; Szilard R; Tkáč J; Yin SY; Zinda M; Marshall CG; Durocher D CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature, 2022, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uehling DE; Joseph B; Chung KC; Zhang AX; Ler S; Prakesch MA; Poda G; Grouleff J; Aman A; Kiyota T; Leung-Hagesteijn C; Konda JD; Marcellus R; Griffin C; Subramaniam R; Abibi A; Strathdee CA; Isaac MB; Al-Awar R; Tiedemann RE Design, synthesis, and characterization of 4‑aminoquinazolines as potent inhibitors of the G protein-coupled receptor kinase 6 (GRK6) for the treatment of multiple myeloma. J. Med. Chem 2021, 64, 11129−11147. [DOI] [PubMed] [Google Scholar]
- 8.Grinshtein N; Datti A; Fujitani M; Uehling D; Prakesch M; Isaac M; Irwin MS; Wrana JL; Al-Awar R; Kaplan DR, Small molecule kinase inhibitor screen identifies polo-like kinase 1 as a target for neuroblastoma tumor-initiating cells. Cancer Res 2011, 71, 1385–1395. [DOI] [PubMed] [Google Scholar]
- 9.Trzcinska-Daneluti AM; Nguyen L; Jiang C; Fladd C; Uehling D; Prakesch M; Al-awar R; Rotin D, Use of kinase inhibitors to correct DeltaF508-CFTR function. Mol. Cell. Proteomics 2012, 11, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zegzouti H; Zdanovskaia M; Hsiao K; Goueli SA; ADP-Glo: A Bioluminescent and homogeneous ADP monitoring assay for kinases. Assay Drug Dev. Technol 2009, 7, 560–572. [DOI] [PubMed] [Google Scholar]
- 11.Das J; Chen P; Norris D; Padmanabha R; Lin J; Moquin RV; Shen Z; Cook LS; Doweyko AM; Pitt S; Pang S; Shen DR; Fang Q; de Fex HF; McIntyre KW; Shuster DJ; Gillooly K; Behnia K; Schieven GL; Wityak J; Barrish JC 2-Aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2- hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J. Med. Chem 2006, 49, 6819–6832. [DOI] [PubMed] [Google Scholar]
- 12.Boschelli DH; Ye F; Wang YD; Dutia M; Johnson SL; Wu B; Miller K; Powell DW; Yaczko D; Young M; Tischler M; Arndt K; Discafani C; Etienne C; Gibbons J; Grod J; Lucas J; Weber Boschelli, F. Optimization of 4-Phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J. Med Chem 2001, 44, 3965–3977. [DOI] [PubMed] [Google Scholar]
- 13.Moasser MM; Srethapakdi M; Sachar KS; Kraker AJ; Rosen N; Inhibition of Src kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res 1999, 59, 6145–6152. [PubMed] [Google Scholar]
- 14.Unzue A; Dong J; Lafleur K; Zhao H; Frugier E; Caflisch A; Nevado C Pyrrolo[3,2‑b]quinoxaline derivatives as types I1/2 and II Eph tyrosine kinase inhibitors: structure-based design, synthesis, and in vivo validation. J. Med. Chem 2014, 57, 6834−6844. [DOI] [PubMed] [Google Scholar]
- 15.Zhao H; Huang D Hydrogen bonding penalty upon ligand binding. PloS one, 2011, 6, e19923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bamborough B; Angell RM; Bhamra I; Brown D; Bull J; Christopher JA; Cooper AWJ; Fazal LH; Giordano I; Hind L; Patel VK; Ranshaw LE; Sims MJ; Skone PA; Smith KJ; Vickerstaff E; Washington M N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of Lck: Indazoles as phenol isosteres with improved pharmacokinetics. Bioorg. Med. Chem. Lett 2007, 17, 4363–4368. [DOI] [PubMed] [Google Scholar]
- 17.Dong D; Ako R; Hu M; Wu B Understanding substrate selectivity of human UDP-glucuronosyltransferases through QSAR modeling and analysis of homologous enzymes. Xenobiotica 2012, 42, 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beaudet L; Rodriguez-Suarez R; Venne M-H; Caron M; Bedard J; Brechler V; Parent S; Bielefeld M; AlphaLISA immunoassays: the no-wash alternative to ELISAs for research and drug discovery. Nat. Meth 2008, 5, an8–an9. [Google Scholar]
- 19.Vasta JD; Corona CR; Wilkinson J; Zimprich CA; Hartnett JR; Ingold MR; Zimmerman K; Machleidt T; Kirkland TA; Huwiler KG; Ohana RF; Slater M; Otto P; Cong M; Wells CI; Berger BT; Hanke T; Glas C; Ding K; Drewry DH; Robers MB Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell chemical biology, 2018, 25, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaPlante SR; Fader LD; Fandrick KR; Fandrick DR; Hucke O; Kemper R; Miller SPF; Edwards PJ Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem 2011, 54, 7005–7022. [DOI] [PubMed] [Google Scholar]
- 21.Patricelli MP; Szardenings AK; Liyanage M; Nomanbhoy TK; Wu M; Weissig H; Aban A; Chun D; Tanner S; Kozarich JW Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry, 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- 22.MOE: Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022. [Google Scholar]
- 23.Liu Y; Zhou P; Da H; Jia H; Bai F; Hu G; Zhang B; Fang J An Azo Coupling strategy for protein 3-nitrotyrosine derivatization. Chem. Eur. J 2019, 25, 11228–11232. [DOI] [PubMed] [Google Scholar]
- 24.Brai A; Riva V; Saladini F; Zamperini C; Trivisani CI; Garbelli A; Pennisi C; Giannini A; Boccuto A; Bugli F; Martini M; Sanguinetti M; Zazzi M; Dreassi E; Botta M; Maga G DDX3X inhibitors, an effective way to overcome HIV-1 resistance targeting host proteins. Eur. J. Med. Chem 2020, 200, 112319. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz-Castillo P; Buchwald SL Palladium-catalyzed C-N coupling conditions Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ledneczki I; Tapolcsányi P; Gábor E; Visegrády A; Vass M; Éles J; Holm P; Horváth A; Pocsai A; Mahó S; Greiner I; Krámos B; Béni Z; Kóti J; Káncz AE; Thán M; Kolok S; Laszy J; Balázs O; Bugovits G Némethy, Z. Discovery of novel positive allosteric modulators of the α7 nicotinic acetylcholine receptor: scaffold hopping approach. Eur. J Med Chem 2021, 214, 113189. [DOI] [PubMed] [Google Scholar]
- 27.Jiang M; Xiang H; Zhu F; Xu X; Deng L; Yang C Efficient Pd-catalyzed domino synthesis of 1-phenyl-1H-indol-2-amine and 5-amino-indolo[1,2-a]quinazoline derivatives. Org. Biomol. Chem 2015, 13, 10122–10126. [DOI] [PubMed] [Google Scholar]
- 28.Brooks PR; Wirtz MC; Vetelino MG; Rescek DM; Woodworth GF; Morgan BP; Coe JW; Boron Trichloride/Tetra-n-Butylammonium Iodide: A mild, selective combination reagent for the cleavage of primary alkyl aryl ethers. J. Org. Chem 1999, 64, 9719–9721. [Google Scholar]
- 29.Ghaffar T; Parkins AW A new homogeneous platinum containing catalyst for the hydrolysis of nitriles. Tetrahedron Lett 1995, 36, 8657–8660. [Google Scholar]
- 30.Yamanaka H; Ogawa S; Konno S; Studies on pyrimidine derivatives. XXI. Nucleophilic substitution of 4-chloropyrimidines and related compounds with carbanions. Chem. Pharm. Bull 1981, 29, 98–104. [Google Scholar]
- 31.Schnyder A; Indoleseb AF; Maetzkec T; Wengerd J; Blasera H-U A convenient protocol for the synthesis of hindered aryl malononitriles. Synlett, 2006, 18, 3167–3169. [Google Scholar]
- 32.Demmer CS; Møller C; Brown PMGE; Han L; Pickering DS; Nielsen B; Bowie D; Frydenvang K; Kastrup JS; Bunch L Binding mode of an α-amino acid-linked quinoxaline-2,3-dione analogue at glutamate receptor subtype GluK1. ACS Chemical Neuroscience, 2015, 6, 845–854. [DOI] [PubMed] [Google Scholar]
- 33.Majetich G; Yu J Synthesis of (±)-14-epi-hydroxydolasta-1(15),7,9-triene and (±)-7-epi -acetoxy-14-epi-hydroxydolasta-1(15),8-diene. Can. J. Chem 2012, 90, 75–84. [Google Scholar]
- 34.Ellis GP; Romney-Alexander TM; Cyanation of aromatic halides. Chem. Rev 1987, 87, 779–794 [Google Scholar]
- 35.Miyaura N; Suzuki A Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev 1995, 95, 2457–2483. [Google Scholar]
- 36.Doyle MP; Siegfried B; Dellaria JF Alkyl nitrite-metal halide deamination reactions. 2. Substitutive deamination of arylamines by alkyl nitrites and copper(II) halides. A direct and remarkably efficient conversion of arylamines to aryl halides. J. Org. Chem 1977, 42, 2426–2431. [Google Scholar]
- 37.Bateman KP; Castonguay G; Xu L; Rowland S; Nicoll-Griffith DA; Kelly N; Chan C-C Reduction of animal usage by serial bleeding of mice for pharmacokinetic studies: application of robotic sample preparation and fast liquid chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl 2001, 754, 245–251. [DOI] [PubMed] [Google Scholar]
- 38.Kabsch W XDS. Acta Crystallogr. D. Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD; Afonine PV; Bunkóczi G; Chen VB; Echols N; Headd JJ; Hung LW; Jain S; Kapral GJ; Kunstleve GRW The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morin A; Eisenbraun B; Key J; Sanschagrin PC; Timony MA; Ottaviano M; Sliz P Collaboration gets the most out of software. Elife 2013, 2013, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obafemi CA; Pfleiderer W Synthesis and some reactions of 3-Chloro-2-(cyanomethylene)-1,2-dihydroquinoxalines. Molecules 2004, 9, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szychowski J; Liu B; Dietrich E; Vallée F; Perryman A; Truchon J-F; Papp R; Beaulieu P Compounds, pharmaceuticals compositions, and methods of preparing compounds and of their use 2021. WO2021/195781. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.