

Abstract
Girls with obesity are at increased risk of early puberty. Obesity is associated with insulin resistance and hyperinsulinemia. We hypothesized that insulin plays a physiological role in pubertal transition, and super-imposed hyperinsulinemia due to childhood obesity promotes early initiation of puberty in girls. To isolate the effect of hyperinsulinemia from adiposity, we compared pre-pubertal and pubertal states in hyperinsulinemic, lean muscle (M)-insulin-like growth factor 1 receptor (IGF-1R)-lysine (K)-arginine (R) (MKR) mice to normoinsulinemic WT, with puberty onset defined by vaginal opening (VO). Our results show MKR had greater insulin resistance and higher insulin levels (P < 0.05) than WT despite lower body weight (P < 0.0001) and similar IGF-1 levels (P = NS). Serum luteinizing hormone (LH) levels were higher in hyperinsulinemic MKR (P = 0.005), and insulin stimulation induced an increase in LH levels in WT. VO was earlier in hyperinsulinemic MKR vs WT (P < 0.0001). When compared on the day of VO, kisspeptin expression was higher in hyperinsulinemic MKR vs WT (P < 0.05), and gonadotropin-releasing hormone and insulin receptor isoform expression was similar (P = NS). Despite accelerated VO, MKR had delayed, disordered ovarian follicle and mammary gland development. In conclusion, we found that hyperinsulinemia alone without adiposity triggers earlier puberty. In our study, hyperinsulinemia also promoted dyssynchrony between pubertal initiation and progression, urging future studies in girls with obesity to assess alterations in transition to adulthood.
Keywords: puberty, insulin, insulin receptor, obesity
Introduction
Obesity is associated with early puberty and ovulatory dysfunction in girls (Kaplowitz et al. 2001, Freedman et al. 2002, INSERM Collective Expertise Centre 2007). Obesity affects more than 18% of children ages 6–11 years in the United States (Hales et al. 2017). The consequences of early puberty include increased risk of depression, earlier age of sexual intercourse and first birth, less time spent in education, higher fasting glucose levels, higher blood pressure, higher BMI, and cancers of reproductive organs (Pandeya et al. 2018, Chan et al. 2019).
Puberty is initiated in response to a sophisticated interplay of several regulating factors which activates the hypothalamic–pituitary–ovary (HPO) axis. Up to 43% of the pubertal regulation is thought to be determined by environmental factors (Kaprio et al. 1995, Morris et al. 2011), including internal metabolic cues reflective of sufficient energy stores needed for reproduction and pregnancy. Early clinical observations cite a ‘critical body weight’ that girls must obtain in order to reach reproductive capabilities (Frisch & Revelle 1971), and further studies indicate a strong relationship between adiposity and the pubertal transition in girls (Kaplowitz et al. 2001). Importantly, weight, adiposity, and nutritional status are part of an integrated, dynamic process and individual components. Several hormones and growth factors, including leptin and insulin, increase with adiposity and may serve as metabolic signals of adequate nutrition to the HPO axis (Ahima et al. 1997, Cheung et al. 1997, Elias & Purohit 2013, Cali & Caprio 2008).
Importantly, insulin levels are known to rise during the normal, peri-pubertal transition as a result of an insulin-resistant state mediated by growth hormone and insulin-like growth factor 1 (IGF-1) (Caprio et al. 1989, Goran & Gower 2001, Takano et al. 2001). Insulin crosses the blood–brain barrier (Banks et al. 2012), and insulin receptors (IR) are present in each of the HPO organs (Samoto et al. 1993, Unger & Lange 1997, Obici et al. 2002, DiVall et al. 2015), indicating a role in the regulation of puberty and reproduction. Specifically, within the hypothalamus, IR is present in both the gonadotropin-releasing hormone (GnRH)-secreting neurons and kisspeptin neurons. The onset of puberty is marked by increased GnRH pulsatility, which in turn initiates signaling across the HPO axis (DiVall & Radovick 2009, Hill & Elias 2018). GnRH-secreting neurons are modulated by kisspeptin in response to sex steroid hormones and nutritional status (Hill & Elias 2018, Kumar et al. 2015). In vivo models which delete IR in these neurons show that insulin’s influence on GnRH activity in initiating puberty appears to be through kisspeptin neurons (Qiu et al. 2013), as opposed to direct stimulation of GnRH neurons (DiVall et al. 2010). In the pituitary, insulin may work in concert with GnRH to augment gonadotropin production (Adashi et al. 1981). Moreover, research suggests insulin contributes to early ovarian follicle development, particularly in the gonadotropin-independent phase of follicle growth (Kezele et al. 2002). Together, insulin’s action on each of the organs of the HPO axis supports a role for insulin in the normal physiology of puberty initiation.
We hypothesize that elevated insulin contributes to the initiation of puberty by triggering the HPO axis. We hypothesize that pathophysiological or high levels of insulin, as seen in obesity, stimulate early puberty.
To isolate the effect of insulin and eliminate confounders of other adiposity-driven hormones, adipokines, and inflammation, we investigated the pubertal transition in a mouse model which is hyperinsulinemic yet maintains a normal weight. The muscle (M)-insulin-like growth factor 1 receptor (IGF-1R)-lysine (K)-arginine (R) (MKR) has a transgene mutation causing a dominant-negative IGF1-R mutation specific to skeletal muscle, leading to heterodimerization of mutated IGF-1R to IR in the skeletal muscle (Fernández et al. 2001). As a result, MKR mice have a four-fold elevation of insulin due to insulin resistance in the skeletal muscle. Importantly, leptin, IL-6, TNF-α, and resistin levels of MKR are similar to WT (Fernández et al. 2001, Novosyadlyy et al. 2010, Héron-Milhavet et al. 2004). These mice are able to become pregnant, however, are sub-fertile (unpublished data). To our knowledge, the pubertal transition in MKR mice has not been described, and our work is the first study to isolate the effects of excess insulin stimulation on puberty timing.
Methods
Animals
All animals were housed in the Yale Animal Resources Center and treated according to protocols approved by the Yale University Institutional Animal Care and Use Committee. Mice were maintained on a 12-h light:12-h darkness cycle with free access to food and water, unless otherwise specified for fasting conditions. Calories from chow were 58% carbohydrate, 18% fat, and 24% protein (Tekland Diets, Madison, WI, USA). MKR heterozygous mice (Jackson Laboratories, Farmington, CT, USA) were bred and offspring were genotyped by PCR identification of the IGF-1R mutant and phenotyped by insulin tolerance testing (Fernández et al. 2001). Homozygous MKR and homozygous WT were then bred separately through three generations to ensure consistent metabolic phenotype before involvement in this study.
Pubertal and estrous cycle assessment
First, to characterize pubertal development and compare gene expression across the HPO axis over time, MKR and WT mice were age-matched and sacrificed at the following post-natal ages (in weeks): 3, 3.5, 4, 5, 6, 8. Each age was represented by a group of 3–5 MKR mice (n = 23 total) and 3–5 WT mice (n = 21 total).
In the second phase of this study, mice were compared at identical physiologic developmental milestones. In our study, we used similar descriptors of puberty that have been previously described (Gaytan et al. 2017). The onset of puberty was defined by vaginal opening (VO), a well-established external marker of puberty in mice occurring secondary to an estrogen-mediated apoptosis of the vaginal membrane (Rodriguez et al. 1997, Caligioni 2009). Following weaning at 3 weeks, mice were checked daily for the presence of VO. MKR (n = 34) and WT (n = 32) mice were sacrificed at either developmental milestone of pre-puberty or VO.
In the third phase of this study, female mice were assessed over time to determine the age of VO (MKR, n = 19; WT, n = 21) and first estrus (MKR, n = 12; WT, n = 12). Duration of adult estrous cycles through 12 weeks of age (MKR, n = 12; WT, n = 12) was determined via daily inspection of vaginal cytology for 10–15 days (Byers et al. 2012).
Metabolic phenotyping and hormone testing
Insulin tolerance testing was completed in the morning after 2-h fasting. First, mice were injected intraperitoneal with 1 IU per kg body weight of regular insulin (Novolin R, Novo Nordisk). Next, plasma glucose measurements were made using a glucometer (Bayer, Breeze 2) from tail vein bleeding at 0, 15, 30, 60, 90, and 120 min.
Serum samples were collected from 3- to 5-week-old female mice in random fed or fasted state by unanesthetized tail snips or retro-orbital bleeds under isoflurane anesthesia. ELISA was used to quantify insulin (Mercodia, Winston-Salem, NC, USA) and IGF-1 (R&D). Estradiol (Calbiotech, El Cajon, CA, USA) levels for n = 3 at each age from 3 to 8 weeks for MKR and WT were measured from serum collected from post-mortem cardiac puncture. The lower detection limit of the estradiol assay is 3pg/mL. ELISAs were read with a Bio-Rad iMark Microplate Reader.
Insulin’s effect on serum luteinizing hormone (LH) levels was studied by injecting 3-week-old WT and MKR mice with regular insulin dosed at 1 IU per kg body weight. Blood draws were collected via retro-orbital bleed under isoflurane anesthesia at baseline and by cardiac puncture 30 min after insulin stimulation. LH levels were quantified using a RIA by the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core.
Tissue collection
Immediately prior to sacrifice, mice were assessed for puberty and weighed. At sacrifice, total hypothalamus, pituitary, and ovaries were dissected and placed in RNAlater (Qiagen) and stored at −80°C. Mammary gland four was dissected and placed on a slide for Carmine Alum staining (Sigma Aldrich). Immediately post-mortem, blood was collected via cardiac puncture. Serum was separated from clotting factors during a 1-h incubation at room temperature followed by centrifugation and aspiration of supernatant, which was then stored at −80°C.
Ovarian histology and corpus lutea assessment
The effect of the hyperinsulinemic phenotype on ovarian development was studied by quantitative and qualitative analysis of ovaries from 3-, 3.5-, 4-, 5-, 6-, an-d 8-week-old mice, (n = 5 MKR and n = 5 WT at each time point, 60 mice total). Ovaries were dissected, fixed in 10% formalin or 70% ethanol, dehydrated, and embedded in paraffin. Ten-micrometer serial sections were cut throughout the ovary and stained with hematoxylin and eosin. Every section was examined for the presence or absence of primary follicles, secondary follicles and corpora lutea (CL). Samples which were uninterpretable due to poor quality of histology were unable to be fully analyzed and were therefore excluded. The presence of a CL indicated ovulation had occurred, and in our study, was reflective of coordinated signaling across the HPO axis. In quantitative analysis, one representative slide per ovary was selected for follicle counting.
Mammary gland assessment
The fourth mammary gland, as the most well-differentiated mammary gland suited for whole mount analysis, was collected from 3 to 3.5, 4-, 5-, 6-, and 8-week-old mice, (n = 5 MKR and n = 5 WT at each time point, 60 mice total). Glands were stained with Carmine Alum and underwent blinded histological assessment for growth including ductal extension, defined as the distance between the nipple and the terminal end buds (TEBs) and the distance between the TEBs and lymph node (Hens & Wysolmerski 2005, Cowin & Wysolmerski 2010). The length and pattern of branching are also reported. The length of 20 secondary branches was measured per mouse inside a uniform area between the lymph node and the nipple for 5- and 6-week-old mice (n = 3 MKR and n = 3 WT at each time-point, n = 12 mice total).
RNA isolation and quantitative RT-PCR analysis
Hypothalamus, pituitary, and ovary samples were homogenized with TRIzol (Life Technologies). RNA was isolated with chloroform, precipitated with isopropanol, washed with 75% ethanol, and dissolved in RNase-free water. RNA was treated with DNase I (Qiagen) and purified with RNeasy clean-up kit (Qiagen). RNA concentrations were measured with Nanodrop 2000 (Thermoscientific).
RNA was reverse transcribed to cDNA using iScript kit (Bio-Rad). Mouse primers of IR-A, IR-B, and IGF-1R used in this study were previously validated for quantitative comparison on the same axis (Rowzee et al. 2009). NIH PrimerBlast software was used to design primers for GnRH (F: CTGATGGCCGGCATTCTACT; R: CCTCCTTGCCCATCTCTTGG), GnRH-receptor (F: CAGCTTTCATGATGGTGGTG; R: TAGCGAATGCGACTGTCATC), and LH (F: CAGTCTGCATCACCTTCACCA; R: GGTAGGTGCACACTGGCTG). Primers sequences for Kiss1 are described elsewhere (Torsoni et al. 2016). For all qRT-PCR, samples were run in duplicate using assay-specific primer concentrations, SYBR Green containing deoxynucleoside triphosphates (dNTPs), fluorescein, and DNA polymerase (Bio-Rad), and amplified in a Bio-Rad CFX96 detection system (Bio-Rad) under the following cycling conditions: 95°C for 3 min, 40 cycles of 95°C for 15 s followed by 60°C for 30 s and 72°C for 25 s, 95°C for 1 min, 55°C for 1 min, and an increase to 95°C at 0.5°C increments.
Statistical analysis
Statistical differences between weight, glucose, and serum hormone levels were analyzed via one-way ANOVA after logarithmic conversion. Ovarian morphology measures were analyzed by unpaired Student’s t-test. Ductal extension was evaluated as a whole, using two-way ANOVA. Ductal extension and secondary branching at individual time points were evaluated using unpaired t-tests. The Ct values from qRT-PCR were normalized to β-actin to yield the ΔCt. Relative gene expression was calculated using 2−ΔCt and graphically represented as relative gene expression. Serum LH levels were compared via a two-way ANOVA after log-transformation with Tukey’s correction. Insulin levels were evaluated via two-way ANOVA after log-transformation with Sidak’s correction. Statistical significance for tissue experiments was defined as P ≤ 0.05 and determined using GraphPad Prism (GraphPad Software, Inc).
Results
MKR have early insulin resistance and pubertal hyperinsulinemia despite lean phenotype
The MKR phenotype is lean, with hyperinsulinemia as a consequence of insulin resistance due to a mutation in skeletal muscle IGF-1 receptor (Fernández et al. 2001). We confirmed the phenotype of MKR used in our study. MKR in our study exhibited a 26 and 27% lower body weight than WT, at pre-puberty and at VO (P < 0.001, P < 0.0001, respectively) (Fig. 1A). As expected, both MKR and WT gained weight between pre-puberty and VO (P < 0.01, P < 0.0001, respectively).
Figure 1.
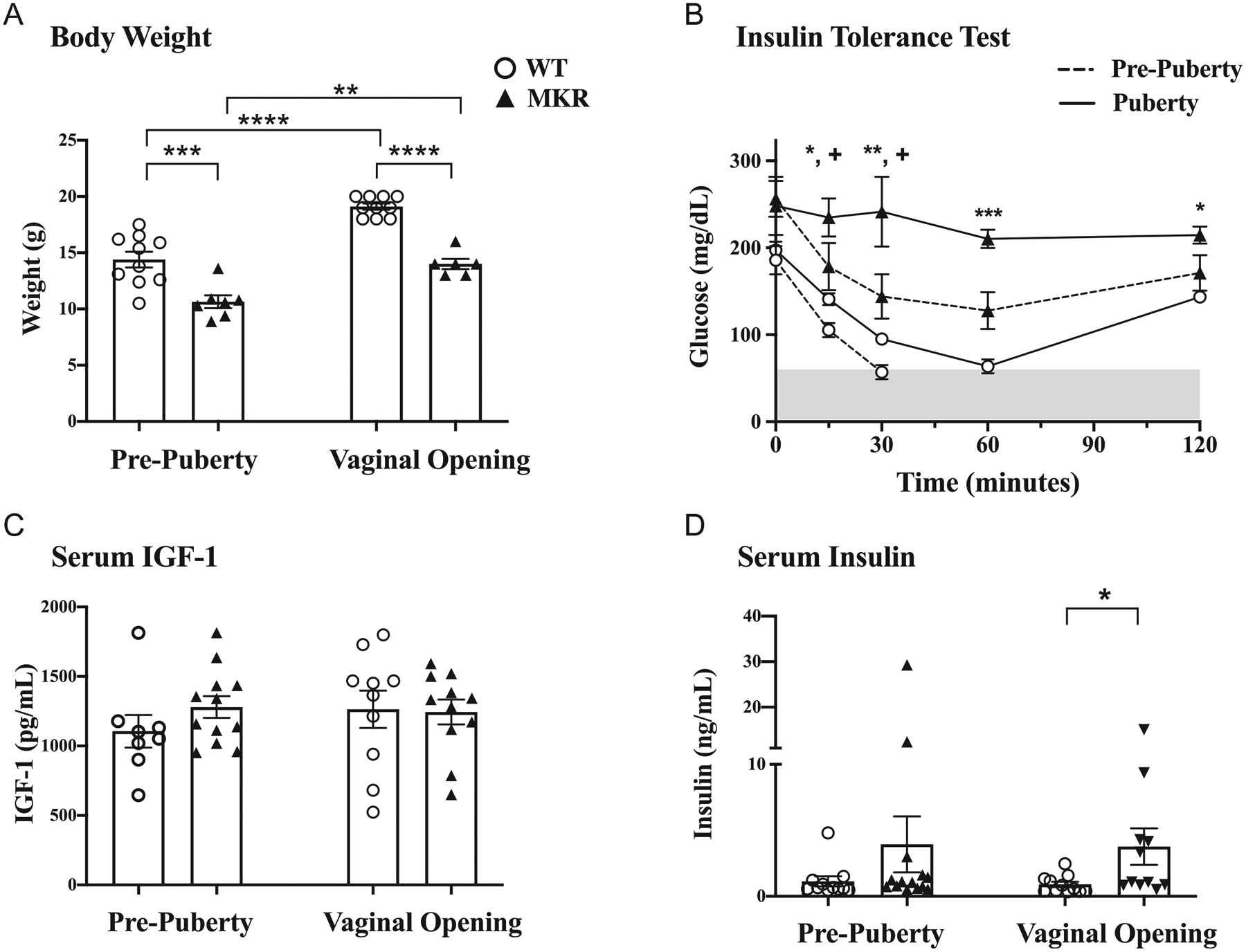
Metabolic phenotyping of MKR and WT mice before and at onset of puberty. (A) Mean body weight ± s.e.m. (g), n = 6–10 mice in each group at each time point. (B) Insulin tolerance test: blood glucose levels following administration of insulin (1 μ/kg body weight), n = 3 mice in each group at each end point. Shaded area represents hypoglycemia, at glucose levels <60 mg/dL. MKR vs WT at pre-puberty: +P < 0.05; WT vs MKR at VO: *P < 0.05, **P < 0.005, ***P < 0.005. (C) IGF-1 levels (pg/mL), n = 8–12 each group at each end point. (D) Insulin levels (ng/mL), n = 11–14 in each group at each end point. ▲MKR, O WT. *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001.
To document the degree of insulin resistance at puberty, we performed insulin tolerance tests in pre-pubertal and peri-pubertal WT and MKR mice (Fig. 1B). Based on initial results from metabolic testing on adult mice, we performed insulin tolerance testing on n = 3 mice in each time point. Pre-pubertal, 3-week-old WT were insulin sensitive, and two of three mice became hypoglycemic (glucose <60 mg/dL) within 30 min of insulin bolus administration (Fig. 1B). At puberty, however, two-thirds of pubertal WT remained euglycemic, with a mean glucose nadir of 64 ± 8 mg/dL, occurring 60 min after insulin bolus. Glucose levels rose thereafter, demonstrating increased insulin resistance at puberty. In contrast to WT, pre-pubertal MKR were insulin resistant, with all mice remaining euglycemic after insulin administration (Fig. 1B), consistent with the MKR phenotype (Fernández et al. 2001). MKR had a similar degree of insulin resistance prior to and at VO (Fig. 1B). Fasting glucose levels were similar between the phases of pre-puberty and VO for both MKR (256 ± 21 mg/dL vs 248 ± 33 mg/dL, P = NS) and WT (186 ± 16 mg/dL vs 197 ± 10 mg/dL, P = NS) and were not significantly different between MKR and WT at either developmental milestone (P = NS, respectively). In pre-pubertal mice, glucose levels were higher in MKR than WT mice 30 min after insulin injection (144 ± 26 mg/dL vs 57 ± 8 mg/dL, P < 0.01). At VO, MKR had higher glucose levels than WT mice 15 min after insulin (235 ± 22 mg/dL vs 141 ±7mg/dL, P < 0.05) and 30 min (242 ± 41 mg/dL vs 95 ± 5 mg/dL, P = 0.005) (Fig. 1B).
Mean serum IGF-1 levels of MKR and WT were similar at both pre-puberty (1280 ± 78 pg/mL vs 1105 ± 117 pg/mL, P = NS) and at VO (1244 ± 90 pg/mL vs 1264 ± 135 pg/mL, P = NS) (Fig. 1C). Insulin levels at VO were 4.2-fold higher in MKR vs WT (3.7 ± 1.4 ng/mL vs 0.9 ±0.2 ng/mL, P < 0.05). In pre-pubertal mice, insulin levels were not significantly different from WT (3.9 ± 2.1 ng/mL vs 1.1 ± 0.4 ng/mL, P = NS) (Fig. 1D).
Hyperinsulinemic MKR initiate puberty earlier
We documented the initiation and natural progression of puberty in MKR and WT by recording the age at VO and first estrus (FE) and confirming ovulation via ovarian histology, similar to established puberty scoring described elsewhere (Gaytan et al. 2017). The mean age of VO in WT (n = 22) was 4.4 ± 0.1 weeks. VO occurred earlier in MKR (n = 19), at a mean age of 3.8 ± 0.1 weeks (P < 0.0001) (Fig. 2A).
Figure 2.
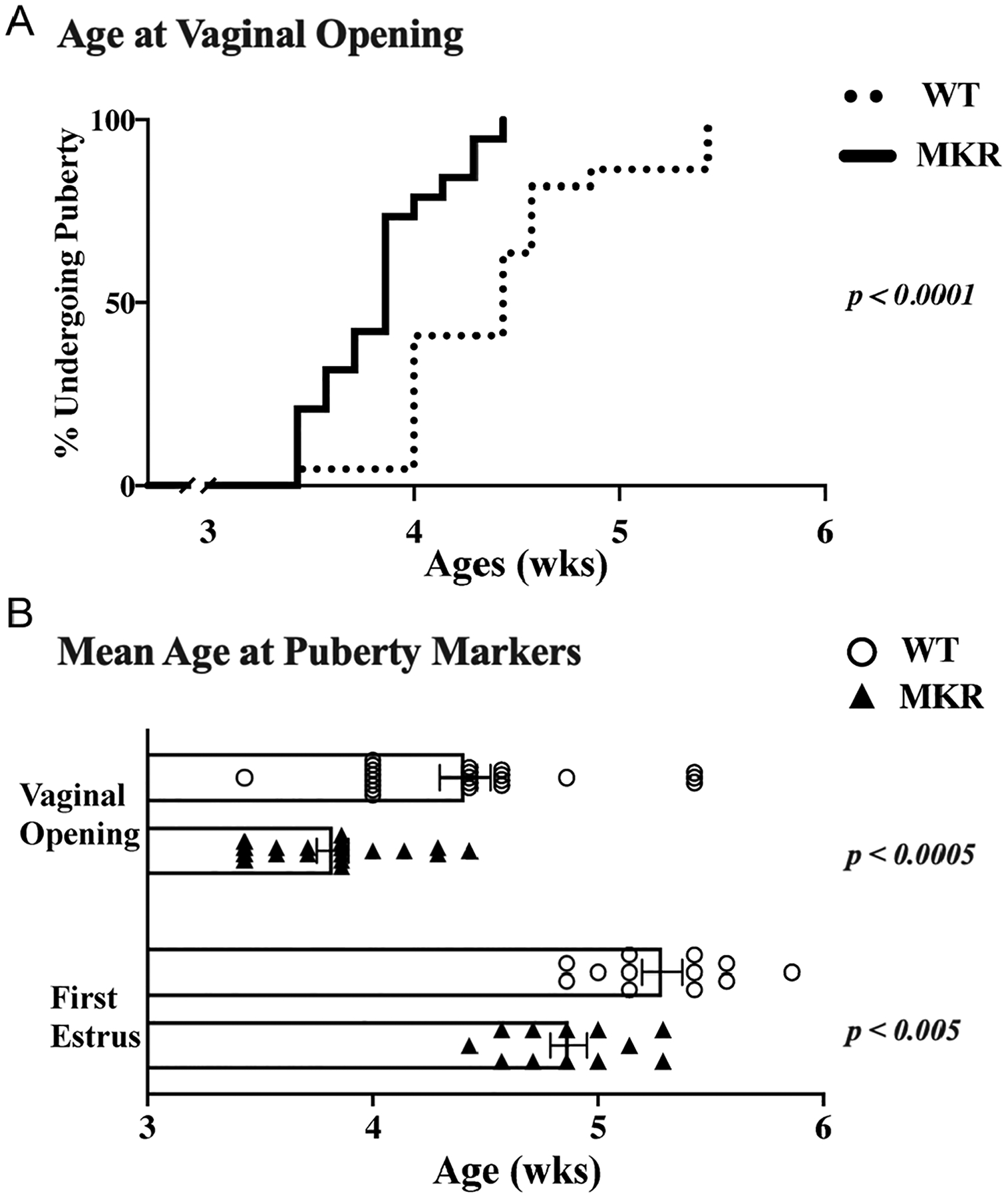
Physiological signs of pubertal initiation. The spontaneous onset of puberty in MKR (n = 19) and WT (n = 22) mice are reported by physical signs and histology of the reproductive organs. Vaginal opening (VO) and first estrus (FE) indicate estrogenization is present. (A) Age of VO, shown as curve of the percent of mice undergoing puberty (MKR n = 19; WT n = 22). (B) Mean age ± s.e.m. (weeks) for vaginal opening (MKR n = 19; WT n = 22) and first estrus (MKR n = 12; WT n = 12). ▲, MKR; O, WT.
After determining MKR had an earlier age at VO and earlier initiation of puberty, we next sought to determine whether they have an altered pubertal progression. We found that on average WT had their FE 7.8 ± 0.5 days after vaginal opening. MKR had a similar time of 7.3 ± 0.4 days between FE and VO (P = NS). However, since MKR developed VO at an earlier age than WT, their FE also occurred earlier at of 4.9 ± 0.1 weeks, whereas WT had FE at 5.3 ± 0.1 weeks (P < 0.003) (Fig. 2B).
MKR have disordered progression of ovarian follicle development
In normal physiology, primary follicles either develop into antral follicles or undergo atresia. The histological comparison of MKR and WT ovaries demonstrates the normal increase in overall size with peri-pubertal age (all images 100× magnification, Fig. 3A). The ovary of 3-week-old MKR was observed to be smaller relative to age-matched WT. Both MKR and WT had primary follicles present during the post-natal ages of 3–8 weeks which were studied (Fig. 3B). The mean number of primary follicles per ovary was similar between MKR and WT through 3.5 weeks (5.8 ± 1.1 vs 7.4 ± 0.4, P = NS). However, by 4 weeks, MKR had 1.9-fold more primary follicles than WT (6.6 ± 0.6 vs 3.5 ± 0.5, respectively, P < 0.05) (Fig. 3B). By 6 weeks of age, WT had a 3.8-fold increase in primary follicles relative to 4 weeks WT cohort. The number of primary follicles decreased between 4- and 6-week MKR. At 6 weeks, MKR had 64% fewer primary follicles relative to WT (4.8 ± 1.6 vs 13.5 ± 2.3, P < 0.05), suggesting that MKR ovaries either had a delay in progression to secondary follicles or more of the primary follicles underwent atresia. Antral follicles were persistently noted in WT ovaries from 3 weeks onward, however, were not observed in MKR ovaries until 4 weeks of age.
Figure 3.
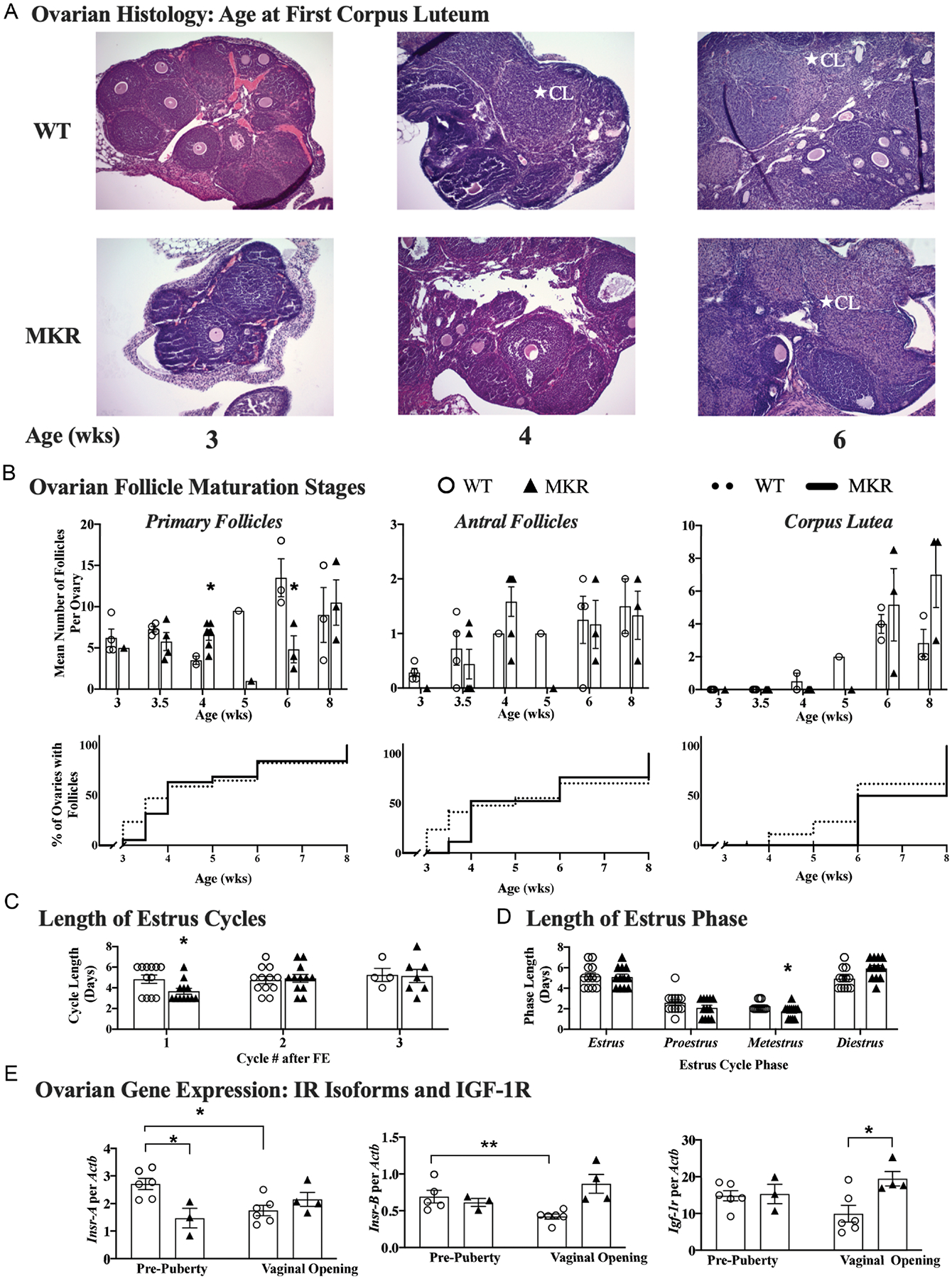
Progression of pubertal development, as demonstrated by ovarian changes reflective of coordinated HPO signaling. Corpus lutea (CL) indicate maturation of the HPO axis. (A) Representation of ovarian morphology of mice at the following post-natal ages: 3 weeks (MKR n = 1; WT n = 4), 4 weeks (MKR n = 6; WT n = 2), and 6 weeks (MKR n = 3; WT n = 3). CL denoted by *. (B) Ovarian follicle counting in MKR vs MKR mice, represented as mean number of follicles per ovary, determined by averaging the number of primary, antral and CL from one representative H&E slides per ovary of MKR vs WT mice at each time point. Data also represented as a survival curve, depicting the percent of ovaries with each type of follicle. (C) Length (days) of serial estrus cycles following first estrus (FE) in MKR vs WT. (D) Length (days) of each phase of estrus cycle over 2 weeks of daily vaginal cytology analysis in MKR vs WT. (E) Ovarian expression of insulin receptor isoforms Insr-A, Insr-B and IGF-1R at pre-puberty (MKR n = 3, WT; n = 5–6) and at VO (MKR n = 3, WT n = 4). ▲, MKR; O, WT. MKR vs WT: *P < 0.05; **P < 0.01. A full color version of this figure is available at https://doi.org/10.1530/JOE-21-0447.
Since ovulation is a marker of coordinated signaling across the HPO axis and requires consistent stimulation by gonadotropins, we assessed ovarian morphology for the presence of CL. WT had the first appearance of CL at 4 weeks old. In contrast, the first CL occurred at 6 weeks of age in MKR.
Estradiol levels for 33 of the 36 WT and MKR at ages 3–8 weeks were below the sensitivity of the ELISA assay, and thus quantitive comparison of estradiol could not be reliably performed (data not shown). Estrus cyclicity was a physiological marker of LH pulsatility and estradiol secretion. After the occurrence of FE, the initial cycle length was found to be shorter in MKR than in WT mice (3.7 ± 0.3 vs 4.8 ± 0.4, P < 0.05, but equalized to a similar cycle duration by 8 weeks of age (5.1 ± 0.6 vs 5.3 ± 0.6, P = NS) (Fig. 3C). MKR had a shorter metestrus phase than WT (1.5 ± 0.3 vs 2.3 ± 0.1 days, P = 0.02) (Fig. 3D).
Insulin receptor expression in ovary is reduced in setting of peripheral insulin resistance
In WT, mean ovarian IR-A expression decreased by 36% between pre-puberty and VO (2.7 ± 0.2 vs 1.7 ± 0.2, P = 0.01) (Fig. 3E). Similarly, mean ovarian IR-B expression was 39% lower at VO than at pre-puberty (0.42 ± 0.03 vs 0.69 ± 0.09, P < 0.01) (Fig. 3E). In MKR, ovarian expression of IR-A and IR-B remained unchanged between pre-puberty (IR-A 1.5 ± 0.4; IR-B 0.61 ± 0.05) and at VO (IR-A 2.2 ±0.3; IR-B 0.87 ± 0.1) (Fig. 3E). Additionally, mean IGF-1R expression remained unchanged between pre-puberty and VO in both WT (14.8 ± 1.4 vs 9.9 ±2.3, P = NS) and MKR (15.3 ± 2.6 vs 19.4 ± 2.0, P = NS) (Fig. 3E).
Mammary gland development is initially accelerated in MKR, but then falters and displaysbranching defect
Since we found significant differences in the pubertal progression of MKR mice, we sought to document the staged progression of the mammary glands, as another organ evolving rapidly during puberty in response to estradiol and other growth factors. Mammary gland development is a step-wise progression, with clear morphological signs of the TEBs traveling from the nipple toward the lymph node, gathering mass and secondary branching as their growth progresses (Cowin & Wysolmerski 2010). Early, by 3–3.5 weeks of age, MKR showed ductal extension which was accelerated relative to WT, as represented by the length of TEBs from the nipple (3.4 ± 0.5 mm vs 2.0 ± 0.4 mm, P = 0.05) (Fig. 4B). However, the next phase of ductal extension after week 4 was significantly stunted in MKR (P = 0.001) (Fig. 4C). By full maturity at week 8, MKR had shorter mammary ductal extension than WT (16.1 ± 1.4 mm vs 19.7 ± 0.3 mm, P = 0.03) (Fig. 4B). In MKR mammary glands, the progression of the TEBs toward then past the lymph node also demonstrated the initial lead at 3–3.5 weeks, before faltering at 4 weeks of age relative to WT (1.66 ± 0.2 mm vs 0.64 ± 0.2 mm, P = 0.01) (Fig. 4C). At this critical juncture, the growth trajectory of MKR and WT mammary glands cross over, with WT leading in progression. From 5–6 weeks of age, the length of the secondary branches increased in WT mice (0.29 ± 0.02 mm vs 0.36 ± 0.02 mm, P = 0.03) (Fig. 4E), however, remained similar in MKR (0.27 ± 0.02 mm vs 0.30 ± 0.02 mm, P = NS) (Fig. 4E). By 6 weeks, the length of secondary branching was shorter in MKR vs WT (0.30 ± 0.02 mm vs 0.36 ± 0.02 mm, P = 0.05)(Fig. 4E), signifying a faltered growth trajectory in MKR. Additionally, MKR mammary glands phenotypically appeared abnormal, and by 6 and 8 weeks of development, MKR mammary glands had well as bulb-like structures at the branching points, suggesting an architectural abnormality that needs further elucidation.
Figure 4.
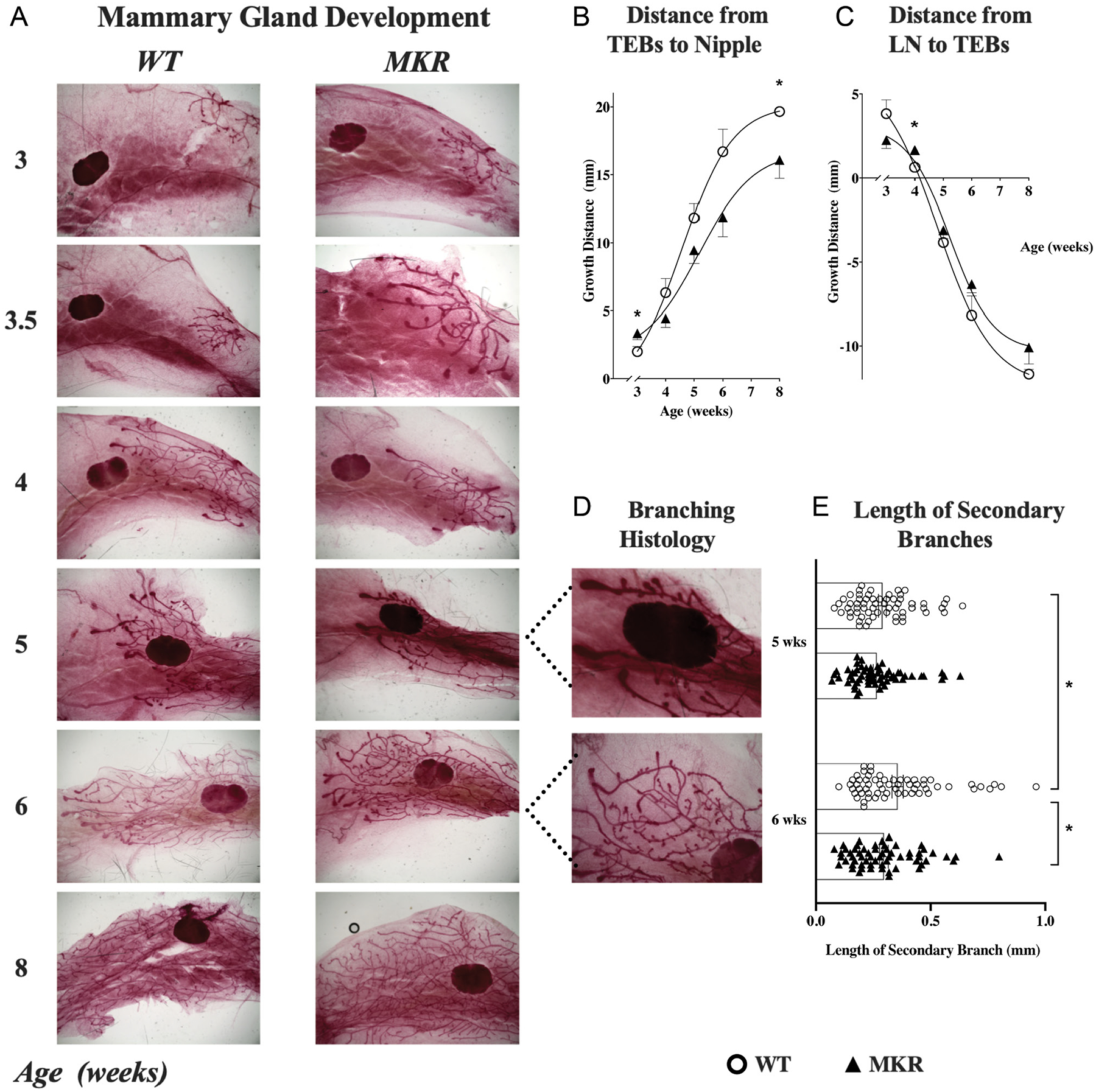
Mammary gland development in MKR mice and WT mice. (A) Representative histology (1×) of the fourth mammary gland at 3, 3.5, 4, 5, 6, and 8 weeks of age from MKR and WT mice, showing the mammary ducts growing from their origination at the nipple toward then past the lymph node. The trajectory of gland development in relation to the nipple and lymph node was quantified in all MKR (▲, n = 30) and WT (O, n = 30) mice. (B) Ductal extension (mean ± s.e.m.) was measured as the distance between the nipple and the terminal end buds (TEBs). MKR mice show an initial accelerated growth spurt before 3.5 weeks of age, which then slows by 4 weeks to a final significant growth delay by 8 weeks. (C) Distance (mean ± s.e.m.) between the TEBs and the lymph node (LN). At 4 weeks of age, the growth trajectory of MKR and WT mammary glands cross over, as MKR growth falters relative to WT. (D) High powered view (4×) of branching pattern in MKR and WT mammary glands at 5 and 6 weeks of age, showing shortened length of branches and a structural abnormality of bulbs at the secondary branching points. (E) Length (mm) of secondary branches at 5 and 6 weeks of age in MKR (▲, n = 6) vs WT (O, n = 6), with 20 branches recorded per mouse.*P ≤ 0.05. A full color version of this figure is available at https://doi.org/10.1530/JOE-21-0447.
Peri-pubertal kisspeptin peak occurs earlier and is higher in MKR hypothalamus
Kisspeptin, a neuropeptide which modulates GnRH pulsatility, has been described in other rodent models as elevated peri-pubertally, with elevated Kiss1 mRNA expression occurring 3–4 days prior to vaginal opening (Takase et al. 2009, Skorupskaite et al. 2014).
When compared on the day of VO, the mean hypothalamic expression of Kiss1 in MKR was 2.8-fold higher than WT (0.42 ± 0.09 vs 0.15 ± 0.003, P = 0.02) (Fig. 5A). In the next phase of our study, kisspeptin expression was compared for age-matched 3-, 3.5-, 4-, and 5-week-old MKR and WT mice. We found that hypothalamic expression of peri-pubertal Kiss1 expression peaked earlier in MKR than in WT (3.4 weeks vs 4.0 weeks of age), consistent with our previously documented earlier VO in MKR (Fig. 5B). In the MKR, we found that Kiss1 peaked 2.8 days prior to the mean age of VO. In WT, Kiss1 also peaked 2.8 days prior to mean VO. Interestingly, the kisspeptin expression remained elevated after VO in MKR and did not return to baseline, as seen in WT.
Figure 5.
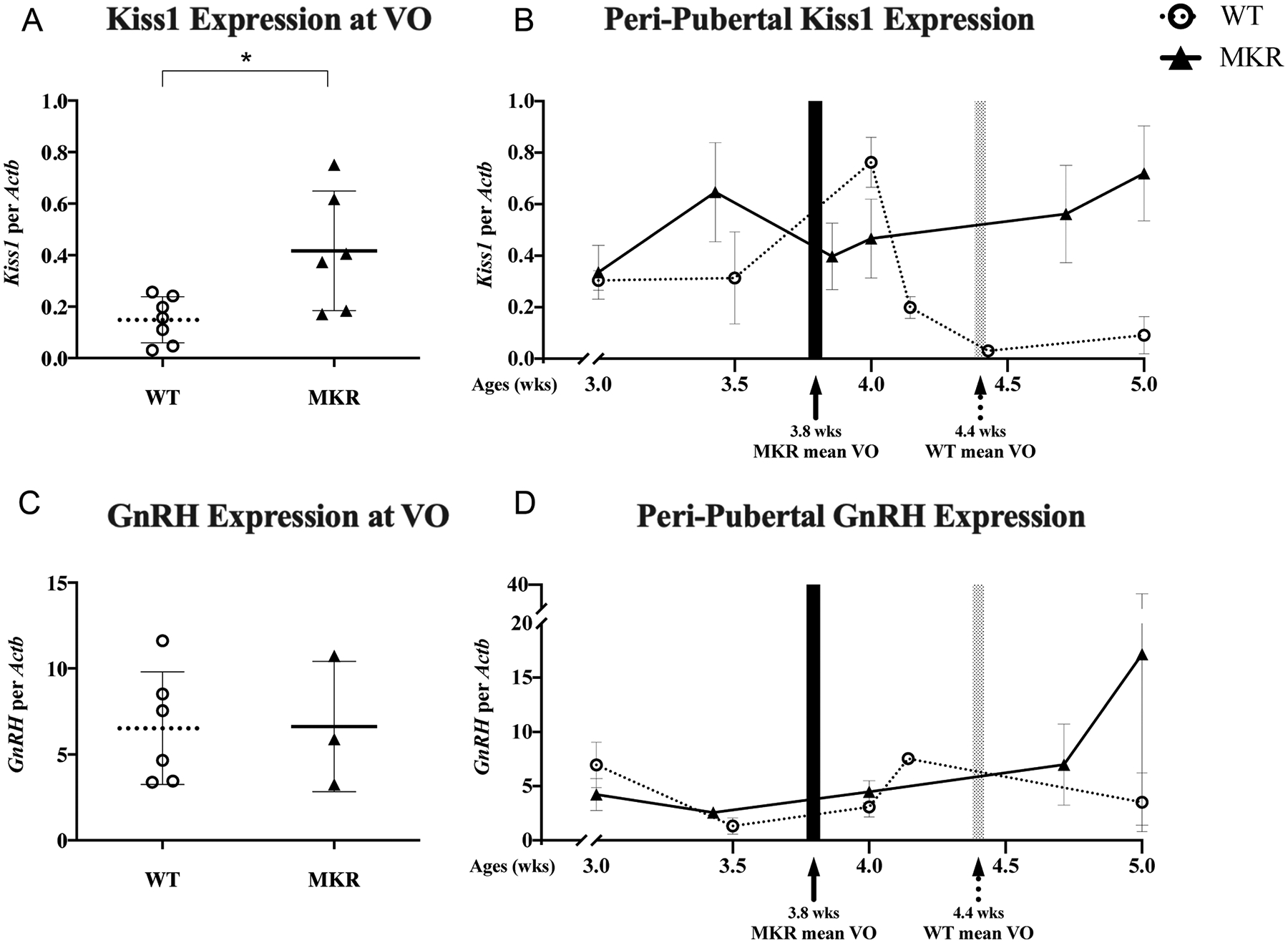
Hypothalamic gene expression. (A) Hypothalamic Kiss1 mRNA expression at the physiological outcome of vaginal opening (VO) in MKR (▲, n = 6) and WT (O, n = 7) mice. (B) The longitudinal, peri-pubertal Kiss1 expression in MKR (▲, n = 23) and WT (O, n = 18) mice by chronological age. (C) Hypothalamic GnRH mRNA expression at the physiological outcome of VO in MKR (▲, n = 3) and WT (O, n = 6) mice. (D) The longitudinal, peri-pubertal GnRH expression in MKR (▲, n = 13) and WT (O, n = 17) mice by chronological age. Mean age of VO, as determined by the physiological cohort of MKR and WT mice, shown in shaded areas. MKR vs WT: *P < 0.05.
We next sought to assess if GnRH expression was modulated in the setting of hyperinsulinemia. On the day of VO, GnRH expression was unchanged between MKR and WT (6.6 ± 2.2 vs 6.5 ± 1.3, P = NS) (Fig. 5C). When comparing age-matched 3-, 3.5-, 4-, and 5-week-old MKR and WT mice, GnRH expression did not increase in the peri-pubertal time period (Fig. 5D). In WT mice at 5 weeks, the GnRH expression values were in a large range, which we suspect is reflective of GnRH pulsatility.
To study HPO sensitivity to insulin, we quantified the expression of IR isoforms IR-A and IR-B across the HPO axis in normal physiology compared to a hyperinsulinemic state. We measured both IR-A and IR-B since each isoform is thought to have different functional effects (Belfiore et al. 2009). In the MKR hypothalamus, the expression of IR-A and IR-B was unchanged in pre-pubertal and pubertal mice (P = NS, respectively) (Fig. 6A). In the WT hypothalamus, expression of IR-A and IR-B was unchanged between pre-pubertal and pubertal mice (P = NS, respectively) (Fig. 6A). In contrast, hypothalamic IGF-1R increased between pre-pubertal and pubertal MKR (7.1 ± 0.7 vs 13.0 ± 0.6, P < 0.001), with a similar trend between developmental stages in WT (8.1 ± 0.5 vs 10.2 ± 1.1, P = NS) (Fig. 6A).
Figure 6.
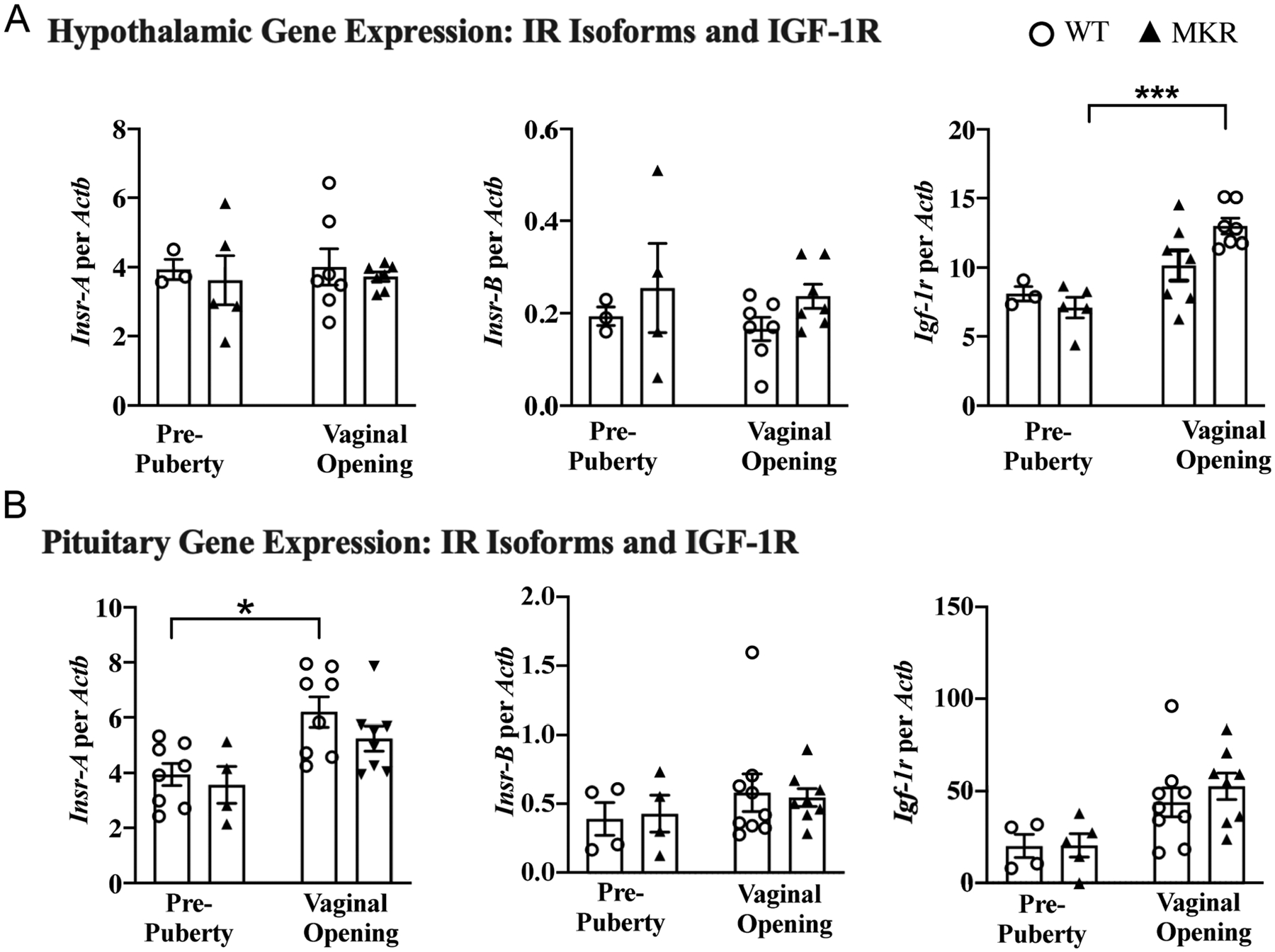
Insulin receptor and insulin-like growth factor-1 gene expression in the hypothalamus and pituitary. (A) Hypothalamic expression of insulin receptor isoforms Insr-A, Insr-B, and IGF-1R at pre-puberty (MKR n = 4–5, WT n = 3) and at VO (MKR n = 7, WT n = 7). (B) Pituitary expression of insulin receptor isoforms Insr-A, Insr-B, and IGF-1R at pre-puberty (MKR n = 4–5, WT; n = 4) and at VO (MKR n = 8, WT n = 8–9. ▲MKR, O WT. *P < 0.05; ***P < 0.001.
Pituitary IR-A increases with normal puberty
In contrast to the hypothalamus, pituitary IR-A increased by 1.6-fold between pre-puberty and puberty in WT (3.9 ± 0.4 vs 6.2 ± 0.5, P = 0.01) and similarly trended upward in MKR (3.6 ± 0.7 vs 5.3 ± 0.5, P = NS) (Fig. 6B). This suggests that pituitary IR-A plays a physiological role in puberty. Interestingly, IR-A levels were not different between MKR and WT at either the pre-puberty or puberty timepoints (P = NS, respectively). Pituitary IR-B was stable between pre-puberty and puberty in both MKR (0.43 ± 0.13 vs 0.55 ± 0.07, P = NS) and WT (0.39 ± 0.11 vs 0.58 ± 0.14, P = NS). No differences were found between MKR and WT at either stage (P = NS, respectively) (Fig. 6B).
In our examination of the pituitary, we found that mean GnRH-receptor expression was unchanged between pre-puberty and puberty, in either the WT or MKR pituitaries. Also, pre-pubertal GnRH-receptor was similar between WT and MKR pituitaries (186 ± 38 vs 143 ± 46, P = NS). At puberty, GnRH-receptor in WT and MKR was also similar (193 ± 51 vs 144 ± 24, respectively; P = NS) (Fig. 7). LH-β mRNA expression MKR and WT were measured at pre-pubertal and pubertal physiological time points. Pituitary mRNA expression of LH-β was similar at VO between MKR and WT (513 ± 87 vs 435 ± 70, P = NS) (Fig. 7).
Figure 7.
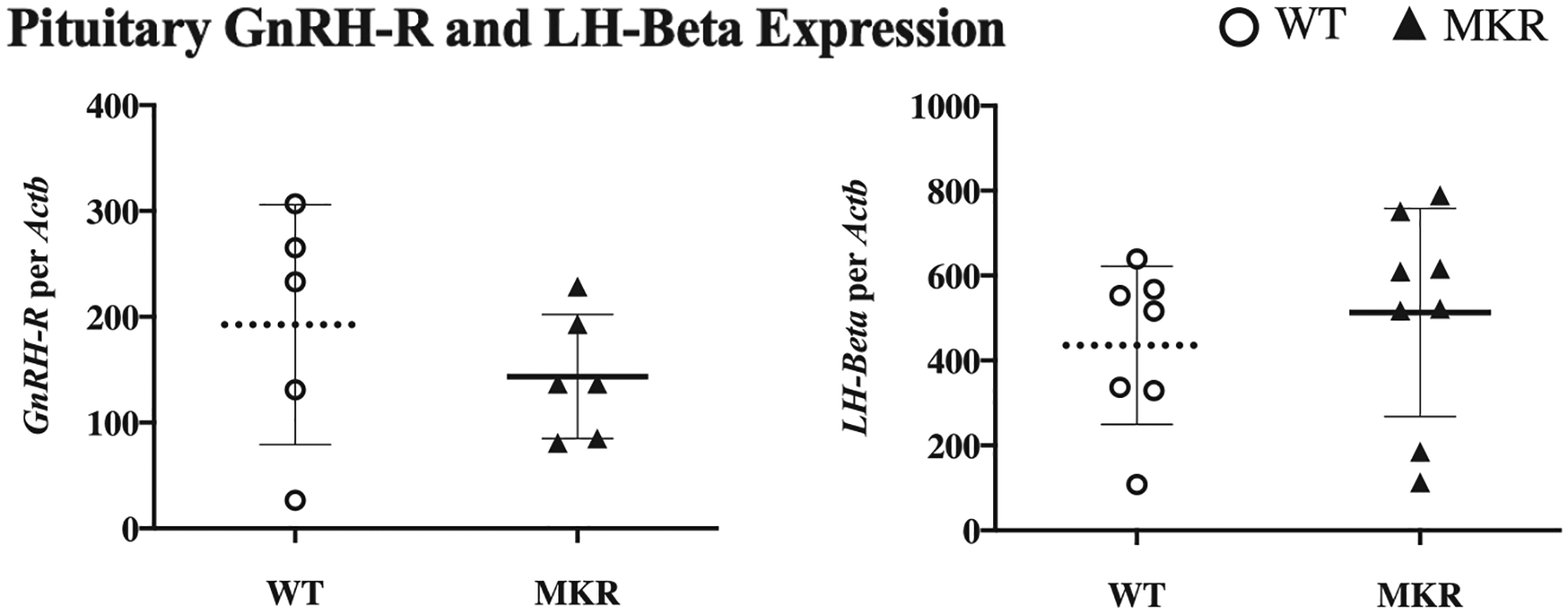
Pituitary expression of gonadotropin-releasing hormone receptor (GnRH-R) and LH-Beta. Pituitary expression of GnRH-R and LH-beta at vaginal opening in MKR (▲, n = 6–8) and WT (O, n = 5–7).
Hyperinsulinemic MKR have higher LH at pre-puberty and insulin induction of LH was replicated in WT
Pre-puberty serum LH levels were 2.2-fold higher in MKR vs WT (0.76 ± 0.14 ng/mL vs 0.35 ± 0.05 ng/mL, P = 0.005) (Fig. 8). Following insulin injection, LH levels increased 2.1-fold in WT (0.75 ± 0.08 ng/mL post-injection vs 0.35 ± 0.05 ng/mL baseline, P < 0.05) (Fig. 8). In MKR, however, LH levels remained unchanged despite insulin injection (0.76 ± 0.14 ng/mL vs 0.86 ± 0.09 ng/mL, P = NS) (Fig. 8). LH levels in WT following insulin injection were similar to the level of baseline, pre-pubertal MKR (0.75 ± 0.08 ng/mL vs 0.76± 0.14 ng/mL, P = NS) (Fig. 8).
Figure 8.
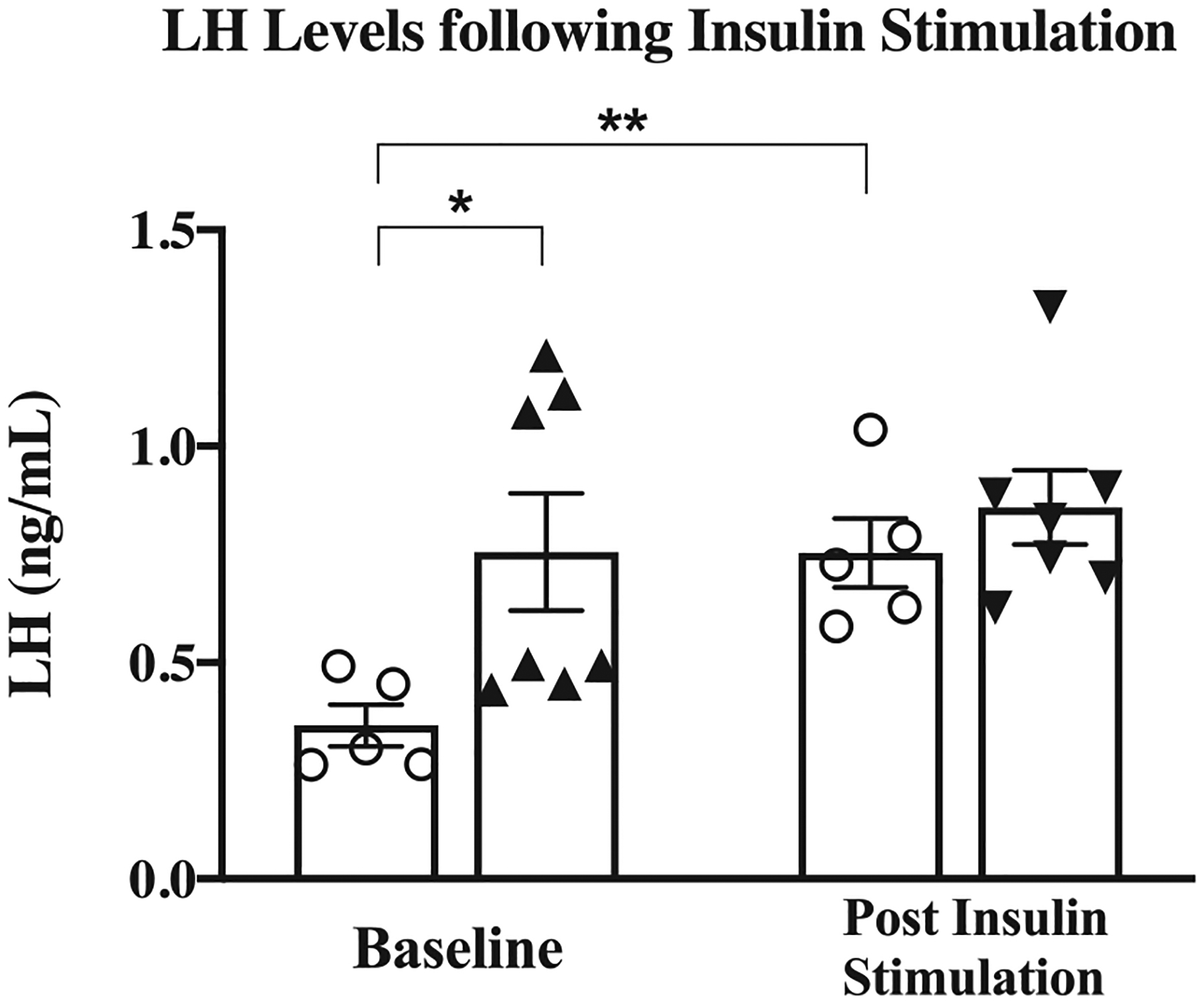
Luteinizing hormone (LH) serum levels following insulin administration. Serum levels of LH pre- and post insulin injection (MKR n = 7, WT; n = 5). ▲MKR, O WT. *, P < 0.05. **, P = 0.005.
Discussion
Our data suggest that early, sustained elevations of insulin in the absence of obesity will initiate puberty. Notably, in the setting of pathophysiologically high insulin levels, this trigger is occurring prematurely when the HPO axis and target organs may not otherwise be prepared or primed to launch a coordinated sequence of development. In our study, hyperinsulinemic MKR mice showed earlier initiation of puberty via estrogen-mediated physiological effects, followed by an abnormal halting sequence of ovarian follicle maturation and abnormal branching patterns of mammary glands. This observed dyssynchrony in the initiation and progression of puberty indicates distinct regulation of each step, in which insulin may accelerate the trigger, but impair the ongoing coordination of HPO activity and the development of target organs.
Our discovery that hyperinsulinemic mice had both an earlier peak and higher expression of kisspeptin suggests that insulin signaling may induce kisspeptin synthesis. Kisspeptin is a positive regulator of GnRH secretion, and our demonstration of similar GnRH expression despite hyperinsulinemia supports the idea of modulated GnRH pulsatility via an insulin-mediated increase in kisspeptin. A multitude of peripheral metabolic cues is integrated through kisspeptin neurons and GnRH neurons with a complexity that makes it difficult to delineate the isolated, direct effect of one hormone. There are two populations of kisspeptin neurons in the rodent hypothalamus which are thought to have opposing responses to estradiol. In the arcuate nucleus, estradiol is suspected to have a positive feedback effect, whereas, in the AVPV, estradiol is thought to signal via negative feedback (Oakley et al. 2009). In our study, we used total hypothalamus tissue and therefore further studies would be needed to specify which population of kisspeptin neurons was affected in MKR and if insulin has differing actions on each cell population. Moreover, our work does not assess the potential influence of insulin on non-neuronal cells, such as glia, which have been shown to regulate GnRH activity (Smedlund & Hill 2020).
In another mouse model, DiVall and colleagues found that females with IR-deletion within the hypothalamic GnRH neurons had normal pubertal timing (DiVall et al. 2010). In our study, we found that GnRH expression was unchanged in hyperinsulinemia, suggesting either the surge was missed in our timepoints due to the fleeting pulsatility, or another mediator is modulating GnRH secretion. Our theory of an insulin-specific effect on kisspeptin is supported by Qiu and colleagues who found delayed VO and FE when IR was deleted from Kiss1 neurons (Qiu et al. 2013). Moreover, LH was also reduced during early puberty in IR-deleted Kiss1 mice, although levels normalized later in adulthood. In this hypothalamic-specific model of IR-deletion in Kiss1 neurons, neither pubertal progression nor fertility was affected, indicating that insulin’s primary role in Kiss1 neurons is to initiate puberty. Our study builds on these aforementioned findings. Herein, we demonstrate that hyperinsulinemic MKR had elevated baseline levels of LH and kisspeptin compared to WT, as well as earlier VO, suggesting insulin-triggered signaling initiated puberty via central regulators. The next step is to study kisspeptin regulation following direct insulin stimulation or by further investigating neuroendocrine mapping of kisspeptin neurons.
The MKR model exposed the same metabolic environment to the hypothalamus, pituitary, and ovary, and further studies are needed to determine at which level insulin had its greatest effect on initiating early puberty. Our model of peripubertal hyperinsulinemia had a larger disturbance of pubertal initiation than the IR-deleted Kiss1 mice (Qiu et al. 2013), and thus insulin’s effect on the pituitary and ovary may also play a role in the early initiation of puberty. At the onset of puberty in normal physiology, we found an increase in pituitary expression of IR-A, suggesting a role for insulin signaling at this timepoint. While we did not see any corresponding change in LH mRNA expression in the pituitary, we noted a significant modulation in LH serum levels due to insulin. We showed that hyperinsulinemic MKR had high serum LH levels. In our WT mouse, we were able to recapitulate the effect of pathophysiologic insulin levels on LH, and serum LH levels in WT mice equaled MKR following insulin injection, further supporting a role for insulin-mediated regulation of gonadotropin release in puberty. Our work aligns with prior studies which have shown that LH levels increased in a diet-induced obesity model, and this effect on LH was abrogated in a pituitary-specific IR deletion model (Brothers et al. 2010), demonstrating insulin is the inciting hormone for excess LH in the setting of obesity. Unfortunately, puberty initiation and progress were not reported in the studies using the pituitary-specific IR deletion mouse model (Brothers et al. 2010), nor in a whole-brain IR deletion model (Nandi et al. 2010). More data are needed to understand any direct role insulin may play in the pituitary toward puberty initiation. Nevertheless, insulin’s effect on the entire HPO axis is supported in further studies which showed that a high-fat diet not only reversed the delayed timing of VO initially observed in the IR-deleted Kiss1 model but prompted an early VO similar to the high-fat-fed mice with IR intact Kiss1 neurons (Qiu et al. 2015).
In the ovary, we observed a decrease in IR isoforms at puberty in the WT mice and in the hyperinsulinemic state of MKR, indicating the ovary reduced local expression of IR in response to physiologic or pathophysiological insulin levels. These results exemplify a tissue-specific regulation of IR in response to peripheral insulin resistance, as occurs during pubertal development or in the setting of obesity. Our findings on the relative abundance of IR-A over IR-B in the mouse ovary are consistent with that found in the human ovary (Phy et al. 2004). In their study, Phy and colleagues found that IR expression did not alter insulin levels, although most women in the cohorts were overweight or obese (Phy et al. 2004). We previously found that IR levels in the endometrium are maintained in the setting of obesity (Flannery et al. 2016) but are decreased when under high insulin exposure alone in vivo and in vitro (unpublished data).
Another mechanism to explain insulin’s contribution to early puberty may be secondary to direct stimulation of the ovary. While IR-deletion within developing mouse oocytes did not alter oocyte growth, differentiation, and maturation (Pitetti et al. 2009), insulin is found to increase estradiol secretion in vitro (Peluso et al. 1991), and in our study, it is possible that high levels of insulin stimulated the ovary to increase estradiol synthesis, both stimulating early mammary gland formation and prompting premature positive feedback to the kisspeptin neurons (Neal-Perry et al. 2014, Hill & Elias 2018). Further, insulin-induced increase in kisspeptin expression may override or disrupt the feedback loop within the HPO axis in a potential mechanism to explain the dyssynchrony and faltering course of pubertal maturation in MKR.
The progression of puberty faltered in the hyperinsulinemic MKR mice, with shorter estrus cycles and a delayed time to the first corpus luteum, indicating ovarian dysfunction. In the same time frame, the mammary gland showed a delay in estradiol-mediated ductal extension and a defect in secondary branching possibly attributed to low progesterone levels (Bocchinfuso et al. 2000). Low progesterone occurs in a setting where ovulation is impaired or irregular. These target organ effects may have occurred due to insufficient steroid hormone levels from abnormal gonadotroph pulsatility or lack of coordination with other regulating factors. Also, since the insulin receptor is present in mammary epithelium (Rowzee et al. 2009), it is possible that insulin acted directly to alter mammary growth trajectory or indirectly to disrupt the activity of estradiol, progesterone, or other regulating factors. Novosyadlyy and colleagues also found that MKR mice had accelerated mammary gland development at 3 weeks of age, and, on their second observation at 15 weeks of age, glands had an increased number of lateral buds and enhanced side branching, relative to WT (Novosyadlyy et al. 2010). Importantly, MKR glands had greater IR and AKT phosphorylation, indicating that insulin may act directly to stimulate growth (Novosyadlyy et al. 2010).
Our model of skeletal muscle insulin resistance and consequential hyperinsulinemia enabled us to examine the effect of insulin on the HPO axis. However, obesity-induced insulin resistance may differ in the organs it affects. Also, even in our model, other factors may be associated with pubertal timing as a result of insulin resistance, and there may be other factors due to insulin resistance or the mutation that were unmeasured and undetected and may be affecting puberty timing (Reinehr et al. 2015).
In summary, puberty can be initiated by elevated insulin met by the physiological insulin resistance of puberty, or earlier in the case of pathophysiological insulin resistance of obesity. Our findings suggest insulin may be acting centrally to trigger puberty, as evidenced by increased hypothalamic kisspeptin expression in hyperinsulinemic mice and the increase in pituitary IR-A in normal puberty. However, our work does not exclude the potential of peripheral action of insulin on the ovaries or even directly on target organs such as the uterus and mammary glands. The integration of metabolic cues or signals within the HPO axis is complex, and likely has redundancy within the multitude of cues received as a result of obesity. However, understanding the specific role of insulin may lead to prevention strategies for premature puberty since multiple therapies for lowering insulin are available.
Acknowledgements
The authors thank several colleagues for conversations, guidance, and valuable insights in physiology and research techniques in their areas of expertise: Hugh Taylor, MD and Shannon Whirledge, PhD on reproductive physiology; John Wysolmerski, MD, PhD on mammary gland development; Emre Seli, MD and Joshua Johnson, PhD on ovarian physiology.
Funding
This project was supported by the Yale Department of Obstetrics, Gynecology, & Reproductive Sciences, the Eunice Kennedy Shriver National Institutes of Child Health and Human Development R01 HD097368 to CAF, and the Yale Diabetes Research Center Pilot Award from the National Institute of Diabetes and Digestive and Kidney Diseases P30DK45735 to CAF.
Footnotes
Declaration of interest
FLS, AAJ, AT, PX, JRH, SLW, and CAF have no conflicts of interest to disclose.
References
- Adashi EY, Hsueh AJW & Yen SSC 1981. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology 108 1441–1449. ( 10.1210/endo-108-4-1441) [DOI] [PubMed] [Google Scholar]
- Ahima RS, Dushay J, Flier SN, Prabakaran D & Flier JS 1997. Leptin accelerates the onset of puberty in normal female mice. Journal of Clinical Investigation 99 391–395. ( 10.1172/jci119172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Owen JB & Erickson MA 2012. Insulin in the brain: there and back again. Pharmacology and Therapeutics 136 82–93. ( 10.1016/j.pharmthera.2012.07.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfiore A, Frasca F, Pandini G, Sciacca L & Vigneri R 2009. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine Reviews 30 586–623. ( 10.1210/er.2008-0047) [DOI] [PubMed] [Google Scholar]
- Bocchinfuso WP, Lindzey JK, Hewitt SC, Clark JA, Myers PH, Cooper R & Korach KS 2000. Induction of mammary gland development in estrogen receptor-α knockout mice. Endocrinology 141 2982–2994. ( 10.1210/endo.141.8.7609) [DOI] [PubMed] [Google Scholar]
- Brothers KJ, Wu S, Divall SA, Messmer MR, Kahn CR, Miller RS, Radovick S, Wondisford FE & Wolfe A 2010. Rescue of obesity-induced infertilityin female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metabolism 12 295–305. ( 10.1016/j.cmet.2010.06.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL & Taft RA 2012. Mouse estrous cycle identification tool and images. PLoS ONE 7 e35538. ( 10.1371/journal.pone.0035538) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali AMG & Caprio S 2008. Obesity in children and adolescents. Journal of Clinical Endocrinology and Metabolism 93 (Supplement 1) S31–S36. ( 10.1210/jc.2008-1363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligioni CS 2009. Assessing reproductive status/stages in mice. Current Protocols in Neuroscience 48 Appendix 4I. ( 10.1002/0471142301.nsa04is48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprio S, Plewe G, Diamond MP, Simonson DC, Boulware SD, Sherwin RS & Tamborlane WV 1989. Increased insulin secretion in puberty:a compensatory response to reductions in insulin sensitivity. Journal of Pediatrics 114 963–967. ( 10.1016/s0022-3476(89)80438-x) [DOI] [PubMed] [Google Scholar]
- Chan YM, Feld A & jonsdottir-Lewis E 2019. Effects of the timing of sex-steroid exposure in adolescence on adult health outcomes. Journal of Clinical Endocrinology and Metabolism 104 4578–4586. ( 10.1210/jc.2019-00569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CC, Thornton JE, Kuijper JL, Weigle DS, Clifton DK & Steiner RA 1997. Leptin is a metabolic gate for the onset of puberty in the female rat. Endocrinology 138 855–858. ( 10.1210/endo.138.2.5054) [DOI] [PubMed] [Google Scholar]
- Cowin P & Wysolmerski J 2010. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harbor Perspectives in Biology 2 a003251. ( 10.1101/cshperspect.a003251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiVall SA & Radovick S 2009. Endocrinology of female puberty. Current Opinion in Endocrinology, Diabetes, and Obesity 16 1–4. ( 10.1097/med.0b013e3283207937) [DOI] [PubMed] [Google Scholar]
- DiVall SA, Williams TR, Carver SE, Koch L, Brüning JC, Kahn CR, Wondisford F, Radovick S & Wolfe A 2010. Divergent roles of growth factors in the GnRH regulation of puberty in mice. Journal of Clinical Investigation 120 2900–2909. ( 10.1172/JCI41069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiVall SA, Herrera D, Sklar B, Wu S, Wondisford F, Radovick S & Wolfe A 2015. Insulin receptor signaling in the GnRH neuron plays a role in the abnormal GnRH pulsatility of obese female mice. PLoS ONE 10 e0119995. ( 10.1371/journal.pone.0119995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF & Purohit D 2013. Leptin signaling and circuits in puberty and fertility. Cellular and Molecular Life Sciences 70 841–862. ( 10.1007/s00018-012-1095-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández AM, Kim JK, Yakar S, Dupont J, hernandez-Sanchez C, Castle AL, Filmore J, Shulman GI & le roith D 2001. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes and Development 15 1926–1934. ( 10.1101/gad.908001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery CA, Saleh FL, Choe GH, Selen DJ, Kodaman PH, Kliman HJ, Wood TL & Taylor HS 2016. Differential expression of Ir-A, IR-Band IGF-1R in endometrial physiology and distinct signature in adenocarcinoma. Journal of Clinical Endocrinology and Metabolism 101 2883–2891. ( 10.1210/jc.2016-1795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman DS, Khan LK, Serdula MK, Dietz WH, Srinivasan SR & Berenson GS 2002. Relation of age at menarche to race, time period, and anthropometric dimensions: the Bogalusa Heart Study. Pediatrics 110 e43. ( 10.1542/peds.110.4.e43) [DOI] [PubMed] [Google Scholar]
- Frisch RE & Revelle R 1971. Height and weight at menarche and a hypothesis of menarche. Archives of Disease in Childhood 46 695–701. ( 10.1136/adc.46.249.695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaytan F, Morales C, Leon S, Heras V, Barroso A, Avendaño MS, Vazquez MJ, Castellano JM, Roa J & tena-Sempere M 2017. Development and validation of a method for precise dating of female puberty in laboratory rodents: the puberty ovarian maturation score (Pub-Score). Scientific Reports 7 46381. ( 10.1038/srep46381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goran MI & Gower BA 2001. Longitudinal study on pubertal insulin resistance. Diabetes 50 2444–2450. ( 10.2337/diabetes.50.11.2444) [DOI] [PubMed] [Google Scholar]
- Hales CM, Carroll MD, Fryar CD & Ogden CL 2017. Prevalence of obesity among adults and youth. United States 2015–2016: NCHS Data Brief. (PMID: https://pubmed.ncbi.nlm.nih.gov/29155689/) [PubMed] [Google Scholar]
- Hens JR & Wysolmerski JJ 2005. Key stages of mammary gland development: molecular mechanisms involved in the formation of the embryonic mammary gland. Breast Cancer Research 7 220–224. ( 10.1186/bcr1306) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Héron-Milhavet L, Haluzik M, Yakar S, Gavrilova O, Pack S, Jou WC, Ibrahimi A, Kim H, Hunt D, Yau D, et al. 2004. Muscle-specific overexpression of CD36 reverses the insulin resistance and diabetes of MKR mice. Endocrinology 145 4667–4676. ( 10.1210/en.2003-1543) [DOI] [PubMed] [Google Scholar]
- Hill JW & Elias CF 2018. Neuroanatomical framework of the metabolic control of reproduction. Physiological Reviews 98 2349–2380. ( 10.1152/physrev.00033.2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- INSERM Collective Expertise center 2007. Growth and puberty secular trends, environmental and genetic factors. INSERM Collective Expert Reports [Internet]. Institut national de la santé et de la recherche médicale. (PMID: https://pubmed.ncbi.nlm.nih.gov/21348172/) [PubMed] [Google Scholar]
- Kaplowitz PB, Slora EJ, Wasserman RC, Pedlow SE & herman-Giddens ME 2001. Earlier onset of puberty in girls: relation to increased body mass index and race. Pediatrics 108 347–353. ( 10.1542/peds.108.2.347) [DOI] [PubMed] [Google Scholar]
- Kaprio J, Rimpela A, Winter T, Viken RJ, Rimpela M & Rose RJ 1995. Common genetic influences on BMI and age at menarche. Human Biology 67 739–753. (PMID: https://pubmed.ncbi.nlm.nih.gov/8543288/) [PubMed] [Google Scholar]
- Kezele PR, Nilsson EE & Skinner MK 2002. Insulin but not insulin-like growth factor-1 promotes the primordial to primary follicle transition. Molecular and Cellular Endocrinology 192 37–43. ( 10.1016/s0303-7207(02)00114-4) [DOI] [PubMed] [Google Scholar]
- Kumar D, Candlish M, Periasamy V, Avcu N, Mayer C & Boehm U 2015. Specialized subpopulations of kisspeptin neurons communicate With GnRH neurons in female mice. Endocrinology 156 32–38. ( 10.1210/en.2014-1671) [DOI] [PubMed] [Google Scholar]
- Morris DH, Jones ME, Schoemaker MJ, Ashworth A & Swerdlow AJ 2011. Familial concordance for age at menarche: analyses from the Breakthrough Generations Study. Paediatric and Perinatal Epidemiology 25 306–311. ( 10.1111/j.1365-3016.2010.01183.x) [DOI] [PubMed] [Google Scholar]
- Nandi A, Wang X, Accili D & Wolgemuth DJ 2010. The effect of insulin signaling on female reproductive function independent of adiposity and hyperglycemia. Endocrinology 151 1863–1871. ( 10.1210/en.2009-0788) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal-Perry G, Yao D, Shu J, Sun Y & Etgen AM 2014. Insulin-like growth factor-I regulates LH release by modulation of kisspeptin and NMDA-mediated neurotransmission in young and middle-aged female rats. Endocrinology 155 1827–1837. ( 10.1210/en.2013-1682) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, Carboni JM, Gottardis MM, Pennisi PA, Molinolo AA, et al. 2010. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Research 70 741–751. ( 10.1158/0008-5472.CAN-09-2141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley AE, Clifton DK & Steiner RA 2009. Kisspeptin signaling in the brain. Endocrine Reviews 30 713–743. ( 10.1210/er.2009-0005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG & Rossetti L 2002. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nature Neuroscience 5 566–572. ( 10.1038/nn0602-861) [DOI] [PubMed] [Google Scholar]
- Pandeya N, Huxley RR, Chung HF, Dobson AJ, Kuh D, Hardy R, Cade JE, Greenwood DC, Giles GG, Bruinsma F, et al. 2018. Female reproductive history and risk of type 2 diabetes: a prospective analysis of 126,721 women. Diabetes, Obesity and Metabolism 20 2103–2112. ( 10.1111/dom.13336) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso JJ, Delidow BC, Lynch J & White BA 1991. Follicle-stimulating hormone and insulin regulation of 17β-estradiol secretion and granulosa cell proliferation within immature rat ovaries maintained in perifusion culture. Endocrinology 128 191–196. ( 10.1210/endo-128-1-191) [DOI] [PubMed] [Google Scholar]
- Phy JL, Conover CA, Abbott DH, Zschunke MA, Walker DL, Session DR, Tummon IS, Thornhill AR, Lesnick TG & Dumesic DA 2004. Insulin and messenger ribonucleic acid expression of insulin receptor isoforms in ovarian follicles from nonhirsute ovulatory women and polycystic ovary syndrome patients. Journal of Clinical Endocrinology and Metabolism 89 3561–3566. ( 10.1210/jc.2003-031888) [DOI] [PubMed] [Google Scholar]
- Pitetti JL, Torre D, Conne B, Papaioannou MD, Cederroth CR, Xuan S, Kahn R, Parada LF, Vassalli JD, Efstratiadis A, et al. 2009. Insulin receptor and IGF1R are not required for oocyte growth, differentiation, and maturation in mice. Sexual Development: Genetics, Molecular Biology, Evolution, Endocrinology, Embryology, and Pathology of Sex Determination and Differentiation 3 264–272. ( 10.1159/000252813) [DOI] [PubMed] [Google Scholar]
- Qiu X, Dowling AR, Marino JS, Faulkner LD, Bryant B, Brüning JC, Elias CF & Hill JW 2013. Delayed puberty but normal fertility in mice with selective deletion of insulin receptors from kiss1 cells. Endocrinology 154 1337–1348. ( 10.1210/en.2012-2056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Dao H, Wang M, Heston A, Garcia KM, Sangal A, Dowling AR, Faulkner LD, Molitor SC, Elias CF, et al. 2015. Insulin and leptin signaling interact in the mouse Kiss1 neuron during the peripubertal period. PLoS ONE 10 e0121974. ( 10.1371/journal.pone.0121974) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinehr T, Elfers C, Lass N & Roth CL 2015. Irisin and its relation to insulin resistance and puberty in obese children: a longitudinal analysis. Journal of Clinical Endocrinology and Metabolism 100 2123–2130. ( 10.1210/jc.2015-1208) [DOI] [PubMed] [Google Scholar]
- Rodriguez I, Araki K, Khatib K, Martinou JC & Vassalli P 1997. Mouse vaginal opening is an apoptosis-dependent process which can be prevented by the overexpression of Bcl2. Developmental Biology 184 115–121. ( 10.1006/dbio.1997.8522) [DOI] [PubMed] [Google Scholar]
- Rowzee AM, Ludwig DL & Wood TL 2009. Insulin-like growth factor type 1 receptor and insulin receptor isoform expression and signaling in mammary epithelial cells. Endocrinology 150 3611–3619. ( 10.1210/en.2008-1473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoto T, Maruo T, ladines-Llave CA, Matsuo H, Deguchi J, Barnea ER & Mochizuki M 1993. Insulin receptor expression in follicular and stromal compartments of the human ovary over the course of follicular growth, regression and atresia. Endocrine Journal 40 715–726. ( 10.1507/endocrj.40.715) [DOI] [PubMed] [Google Scholar]
- Skorupskaite K, George JT & Anderson RA 2014. The kisspeptin-GnRH pathway in human reproductive health and disease. Human Reproduction Update 20 485–500. ( 10.1093/humupd/dmu009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedlund KB & Hill JW 2020. The role of non-neuronal cells in hypogonadotropic hypogonadism. Molecular and Cellular Endocrinology 518 110996. ( 10.1016/j.mce.2020.110996) [DOI] [PubMed] [Google Scholar]
- Takano A, Haruta T, Iwata M, Usui I, Uno T, Kawahara J, Ueno E, Sasaoka T & Kobayashi M 2001. Growth hormone induces cellular insulin resistance by uncoupling phosphatidylinositol 3-kinase and its downstream signals in 3T3-L1 adipocytes. Diabetes 50 1891–1900. ( 10.2337/diabetes.50.8.1891) [DOI] [PubMed] [Google Scholar]
- Takase K, Uenoyama Y, Inoue N, Matsui H, Yamada S, Shimizu M, Homma T, Tomikawa J, Kanda S, Matsumoto H, et al. 2009. Possiblerole of oestrogen in pubertal increase of Kiss1/kisspeptin expression in discrete hypothalamic areas of female rats. Journal of Neuroendocrinology 21 527–537. ( 10.1111/j.1365-2826.2009.01868.x) [DOI] [PubMed] [Google Scholar]
- Torsoni MA, Borges BC, Cote JL, Allen SJ, Mahany E, garcia-Galiano D & Elias CF 2016. AMPKα2 in Kiss1 neurons is required for reproductive adaptations to acute metabolic challenges in adult female mice. Endocrinology 157 4803–4816. ( 10.1210/en.2016-1367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger JW & Lange W 1997. Insulin receptors in the pituitary gland: morphological evidence for influence on opioid peptide-synthesizing cells. Cell and Tissue Research 288 471–483. ( 10.1007/s004410050833) [DOI] [PubMed] [Google Scholar]