

Abstract
Organic cation transporter-3 (OCT3) is widely distributed in the brain with high expression in portions of the stress axis. These high capacity, polyspecific transporters function in monoamine clearance and are sensitive to the stress hormone corticosterone. In rats, withdrawal from chronic amphetamine increases OCT3 expression in specific limbic brain regions involved anxiety and stress responses, including the ventral hippocampus, central nucleus of amygdala (CeA) and dorsomedial hypothalamus. (DMH). Previous studies show that glucocorticoid receptor (GR) agonists increase OCT1 mRNA and OCT2 mRNA expression in non-neural tissues. Thus, we hypothesized that corticosterone increases OCT3 expression in the brain by activating GRs. Male Sprague-Dawley rats were pre-treated daily with the GR antagonist mifepristone (20 mg/kg; sc.) or vehicle followed 45 min later by injections of corticosterone or vehicle for 2 weeks. Corticosterone treatment significantly increased OCT3 expression in the ventral hippocampus and increased anxiety-like behavior. However, these effects were not blocked by mifepristone. Interestingly, treatment with mifepristone alone reduced plasma corticosterone levels and increased serotonin transporter and GR expression in the ventral hippocampus but did not significantly affect OCT3 expression or behavior. No treatment effects on OCT3, serotonin transporter or GR expression were observed in the DMH, CeA or dorsal hippocampus. Our findings suggest that corticosterone increases OCT3 expression in the ventral hippocampus by a mechanism independent of GRs, and that mifepristone and corticosterone can act in an independent manner to affect HPA axis-related physiological and behavioral parameters.
Keywords: Organic cation transporter, corticosterone, mifepristone, stress
1. Introduction
Organic cation transporter 3 (OCT3s), a member of the solute carrier family-22, function as high capacity, low affinity transporters for a variety of cations. The presence of these transporters was characterized in brain tissue in 1998 (Grundermann et al., 1998, Wu et al., 1998). Subsequent studies found that OCT3 is widely distributed in the CNS with high expression levels in a number of brain regions that comprise components of the stress axis (Vialou et al., 2004, Amphoux et al., 2006, Gasser et al., 2009). In the brain, OCTs have been proposed to modify neurotransmission by altering neurotransmitter clearance from extracellular fluid, including the monoamine neurotransmitters, histamine, and acetylcholine (Busch et al., 1996, Grundemann et al., 1998, Koepsell et al., 2007), particularly under conditions during which neurotransmission is enhanced and low capacity, high affinity transporters are saturated (for review, see Daws, 2008, Daws et al., 2013). This function might be especially important in modulating rapid changes in neurotransmission as a response to stress since corticosterone blocks OCT3-mediated transport (Grundermann et al., 1998, Wu et al., 1998, Gasser et al., 2006, Feng et al., 2009, Banganz et al., 2010).
Much of the work implicating OCT3-mediated transport as a modulator of stress has focused on changes in monoaminergic neurotransmission and/or behavior. For example, treatment with corticosterone or decynium-22, both of which can block OCT3 in the rat dorsomedial hypothalamus, which is involved in the integration of the stress response (reviewed in DiMicco et al., 2002) increases extracellular serotonin (Feng et al., 2005; Feng et al., 2009) and alters postsynaptic clearance rates of organic cations, such as serotonin (Gasser et al., 2006). In mice with deficits in serotonin transporter (SERT), OCT3 expression is upregulated in the hippocampus (Schmitt et al., 2002, Baganz et al., 2008). These mice exhibit decreased 5-HT clearance and reduced immobility time when treated with decynium-22, which the authors interpreted as antidepressant-like behavior (Baganz et al., 2008) and the response appears to be corticosterone-dependent (Bagnanz et al., 2010). In the amygdala, OCT3s are expressed in the intercalated cell groups (Hill and Gasser, 2013) and corticosterone-mediated inhibition of OCT3s in this region may play a role in regulating extracellular dopamine. In addition, OCT3 deficient mice exhibit decreased immobility time in forced swim (Kitaichi et al., 2005) and increased open arm/open center time in elevated plus maze (EPM) and open field tests (Wultsch et al., 2009; but see Vialou et al., 2008). Combined, these studies suggest that the behavioral stress response can be modulated by corticosterone blockade of OCT3-mediated monoamine clearance.
In experimental animals, withdrawal from amphetamine is characterized by increases in behavioral measures of anxiety and stress sensitivity (Voung et al., 2010; Barr et al., 2010; Li et al., 2014, Tu et al., 2014). Previously, we reported that acute (24 h) or chronic (2 weeks) withdrawal from chronic amphetamine increases OCT3 expression in the ventral hippocampus of rats (Barr et al., 2013; Solanki et al., 2016), possibly contributing to the reduction in corticosterone or stress-induced increases in extracellular serotonin in rats chronically pre-treated with amphetamine by increasing serotonin clearance (Barr and Forster, 2011; Barr et al., 2013; Li et al., 2014). The decrease in ventral hippocampal serotonin, which normally functions to decrease anxiety (Joca et al., 2003, Joels et al., 2008), is likely to contribute to the presence of heightened anxiety-like behaviors during amphetamine withdrawal (Tu et al., 2014). In addition, we found that OCT3 and serotonin transporter (SERT) expression in the central nucleus of amygdala (CeA) increased following both acute and chronic amphetamine withdrawal and that OCT3 expression increased in the dorsomedial hypothalamus (DMH) following acute (24 h) drug withdrawal (Solanki et al., 2016). Thus, changes in monoamine transport mechanisms may contribute to dysregulation of limbic regions during amphetamine withdrawal that heighten stress susceptibility (Scholl et al., 2010; Li et al., 2014a).
A potential mechanism by which OCT3 and/or SERT expression in limbic brain regions may be altered by stress or drugs of abuse is through glucocorticoid activation of glucocorticoid receptors (GRs). There is evidence that GR activation can alter OCT expression in peripheral tissues. For example, the GR agonist, dexamethasone, upregulates organic cation transporter 1 (OCT1) in human primary hepatocytes (Rulcova et al., 2013). Similarly, glucocorticoid agonists hydrocortisone and dexamethasone upregulate organic cation transporter 2 (OCT2) mRNA and increased OCT2 mediated 14C tetraethylammonium uptake in Madin-Derby canine kidney cells (Shu et al., 2001). However, mechanisms by which stress or amphetamines can alter OCT3 expression in the brain are unknown. Several studies show that stress and psychostimulants, including amphetamine, increase corticosterone (Knych and Eisenberg., 1979; Swerdlow et al., 1993; Parrott et al., 2014; Gomez-Roman et al., 2015; Sial et al., 2015; Qulu et al., 2015). Combined, previous studies suggest that increased corticosterone following amphetamine administration may increase OCT3 expression in limbic brain regions by acting on GRs. In this study, we hypothesized that corticosterone induces increases in OCT3 expression in the brain through the activation of GRs.
2. Material and Methods
2.1. Animals and drug treatment:
Forty-eight male Sprague-Dawley rats were obtained from the Animal Resource Center of the University of the South Dakota. Rats were housed in pairs at 22°C room temperature and 60% relative humidity in a 12 h light: 12 h dark reversed light/dark cycle with lights off at 1000. Food and water were available ad libitum. Procedures were approved by the Institutional Animal Care and Use Committee of the University of South Dakota, and studies were performed during the dark phase of the light cycle.
2.1.1. Mifepristone-Corticosterone treatment:
At 8 weeks of age, rats were divided in four groups and were treated with the respective drugs for 14 consecutive days. All injections were administered during the dark phase, between 1100–1500. Animals were pre-treated with either GR antagonist mifepristone (20 mg/kg; sc.) or propylene glycol vehicle. This concentration of mifepristone blocks GR-mediated effects on amphetamine self-administration (Stairs et al., 2011). Forty-five minutes following pre-treatment, rats were treated with either corticosterone (40 mg/kg, ip.) or vehicle (2-Hydroxypropyl)-β-cyclodextrin (HBC). This concentration of corticosterone administered chronically significantly reduces the time spent in open arms of EPM, which is indicative of elevated anxiety states (Lim et al., 2012).
2.2. Elevated plus maze testing and tissue collection:
Rats were tested for anxiety-like behavior 24 h after the last injection. Anxiety testing was performed using an EPM apparatus (Noldus Information Technology, Wageningen, The Netherlands). The EPM is comprised of four (12 cm wide × 100 cm long) arms, two of which are open and two are enclosed with 40 cm high walls, connected to one another by a central area. To test anxiety-like behavior, a rat was placed in the center space and allowed to freely access all the four arms of EPM for five minutes. Movement of animals was recorded by an automated software (Ethovision XT v5.1, Noldus Technologies) to record the time spent in open and closed arms, and within the center. All behavioral testing was done in the dark phase, between 1100 and 1400.
Four hours following anxiety testing (1500–1800), animals were decapitated; trunk blood and brains were collected and stored on ice at 4°C and on dry ice at −80°C respectively until further use. Blood was centrifuged at 5,000 rpm, supernatant layer of plasma was collected and stored at −80°C.
2.3. Measurement of plasma corticosterone levels:
Plasma corticosterone levels were measured using corticosterone Enzyme Linked Immunosorbent Assay (ELISA) kit (R&D systems, Minneapolis, MN, USA) following manufacturer’s instructions (Forster et al., 2008). Briefly, 10 μL of plasma was mixed with 0.5 μL of steroid displacement reagent, the mixture was then diluted with 990 μL of assay buffer and vortexed. This procedure yielded 100-fold dilution of the original plasma samples. Samples, standards (range 32–20,000 pg/mL), and corticosterone controls were assayed in duplicate. At the end of the assay, corticosterone concentration was determined using colorimetric absorbance scanner (Bio-Tek instruments, Winooski, VA, USA) at the scanning wavelength of 405 nm with wavelength correction set at 595 nm. The sensitivity of this assay was 22 pg/mL and non-specific binding was 5.03%.
2.4. Measurement of OCT3, SERT and GR Expression:
Given that SERT expression often changes concurrently with OCT3 expression in the brain (Solanki et al., 2016), we assessed SERT along with OCT3 expression in the brain regions of interest. These brain regions included the ventral hippocampus, the CeA and the DMH, based on previous findings showing amphetamine treatment increased OCT3 expression and function in these regions (Barr et al., 2013; Solanki et al., 2016). The dorsal hippocampus was included as control region as OCT3 expression is not altered in this region by stimulant treatment (Solanki et al., 2016). Furthermore, we determined whether the glucocorticoid treatment altered the expression of GRs in these same brain regions.
2.4.1. Cryostat brain sectioning and tissue microdissection:
Brains were sectioned in a cryostat (−10°C) at the thickness of 300 μM in the coronal plane. Sections were thaw mounted on glass-slides and stored at −80°C until microdissection. The ventral and dorsal hippocampus, DMH and CeA were identified using the Paxinos & Watson brain atlas (1997). Brain regions were bilaterally microdissected on a freezing stage (Physiotemp, North Central Instruments) and tissue collected in 40 μL of HEPES buffer (1.19%, pH 7.5) containing 0.1 μM of protease inhibitor. Microdissected tissue was sonicated to disrupt the cells (Fisher scientific, PA, ISA) and protein concentration was determined using the Bradford method (Bradford et al., 1976) (BioRad Laboratories, Hercules, CA, USA) and read on microplate reader (Bio-Tek Instruments, Winooski, VT, USA) (Solanki et al., 2016).
2.4.2. Western blot:
Procedures were followed as described in Solanki et al., 2016. Briefly, homogenized samples were mixed with 1.5 M loading buffer containing β-mercaptoethanol followed by 3 min of boiling. Once the samples cooled, they were loaded (50 μg for dorsal and ventral hippocampus; 40 μg for CeA; 35 μg for DMH) on 10% sodium dodecyl sulfate polyacrylamide gel for electrophoresis (BioRad Laboratories) at 90 V for 1.5–2 hrs. Following electrophoresis, proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (0.2 μm, BioRad Laboratories) by a semi dry blotting apparatus (BioRad Laboratories) for 90 min. Then, the PVDF membrane was blocked in 5% skim milk and 1% bovine serum albumin overnight at 4°C. The PVDF membrane was incubated with rabbit polyclonal OCT3 primary antibody (1:1000 dilution; OCT31-A. Alpha Diagnostic International, TX, USA; Solanki et al., 2016; Barr et al., 2013; Gasser et al., 2009) for 24 h at 4°C and washed in tris buffered saline with tween-20 (TBST) 3 times (10 min each). The membranes were then incubated with IRDye800-conjugated goat anti-rabbit IgG secondary antibody (1:5000 dilution; 611-132-122, Rockland, PA) and washed three times in TBST. Membranes were scanned and subsequently washed in TBST and reprobed with Actin (1:2000 dilution; Anti-Actin, clone 4; MAB1501R, Millipore, CA, USA), SERT (1:200 dilution; goat polyclonal against SERT; sc-1458. Santa-Cruz Biotechnology Inc, CA, USA; Solanki et al., 2016; Barr et al., 2013) and GR (1:200 dilution; rabbit polyclonal IgG; sc-1004, Santa-Cruz Biotechnology, USA; Barr & Forster, 2011). The secondary antibodies used to tag above proteins were Cy2 AffiniPure goat anti-mouse secondary IgG antibody (1:5000 dilution; 115-225-003, Jackson Immunoresearch laboratories Inc., PA, USA), AMCA-AffiniPure rabbit anti-goat IgG secondary antibody (1:5000 dilution; 305-155-003, Jackson Immunoresearch, PA, USA) and IRDye800-conjugated goat anti-rabbit IgG secondary antibody (1:5000 dilution; 611-132-122, Rockland, PA). Membranes were scanned using an infrared LiCor imaging system with the 800-channel filter (Odyssey CLx, LiCor Biosciences, NE, USA) to visualize protein brands. Optical densities from each individual sample were corrected against their actin density, and expressed as a percentage of the loading control.
2.5. Statistics:
Separate two-way ANOVA were used to determine the effects of treatment on EPM measures, plasma corticosterone, and OCT3, GR and SERT expression. Significant interactions were followed by Student-Newman-Keul’s (SNK) post hoc test for multiple comparisons. Linear regression analysis were used to assess a relationship between distance moved and time spent in open arms of the EPM. All analyses were performed using SigmaStat v.3.5, with significance determined with P < 0.05.
3. Results
3.1. Effect of corticosterone and mifepristone on anxiety-like behavior:
A significant main effect of corticosterone treatment was observed on anxiety-like behavior within the EPM (Fig. 1A). Specifically, rats treated with corticosterone showed significantly decreased time spent in open arms of the maze (F (1, 30) = 4.771, P = 0.037). There was no main effect of mifepristone pre-treatment (F(1, 30) = 1.432, P = 0.241), nor a significant interaction between mifepristone pre-treatment and corticosterone treatment (F (1, 30) = 0.139, P = 0.712). There was a significant main effect of corticosterone treatment on total distance moved in the maze (F(1,29) = 9.771, P = 0.004; Fig. 1B). Specifically, there was reduced mobility in corticosterone treatment groups relative to the controls. Mifepristone pre-treatment had no effect on total distance moved in the maze (F(1,29) = 0.831, P = 0.370). There was also no significant interaction between mifepristone pre-treatment and corticosterone treatment (F(1,29) = 0.085, P = 0.773). Thus, the decrease in time spent in open arms in corticosterone treated group may partly be due to corticosterone-induced decreased mobility in these groups. Somewhat supportive of this, a weak but non-significant relationship was observed between these two behavioral variables in corticosterone treated groups (r2= 0.165; F(1,16)= 4.36, P= 0.053; Fig. 1C).
Figure. 1:
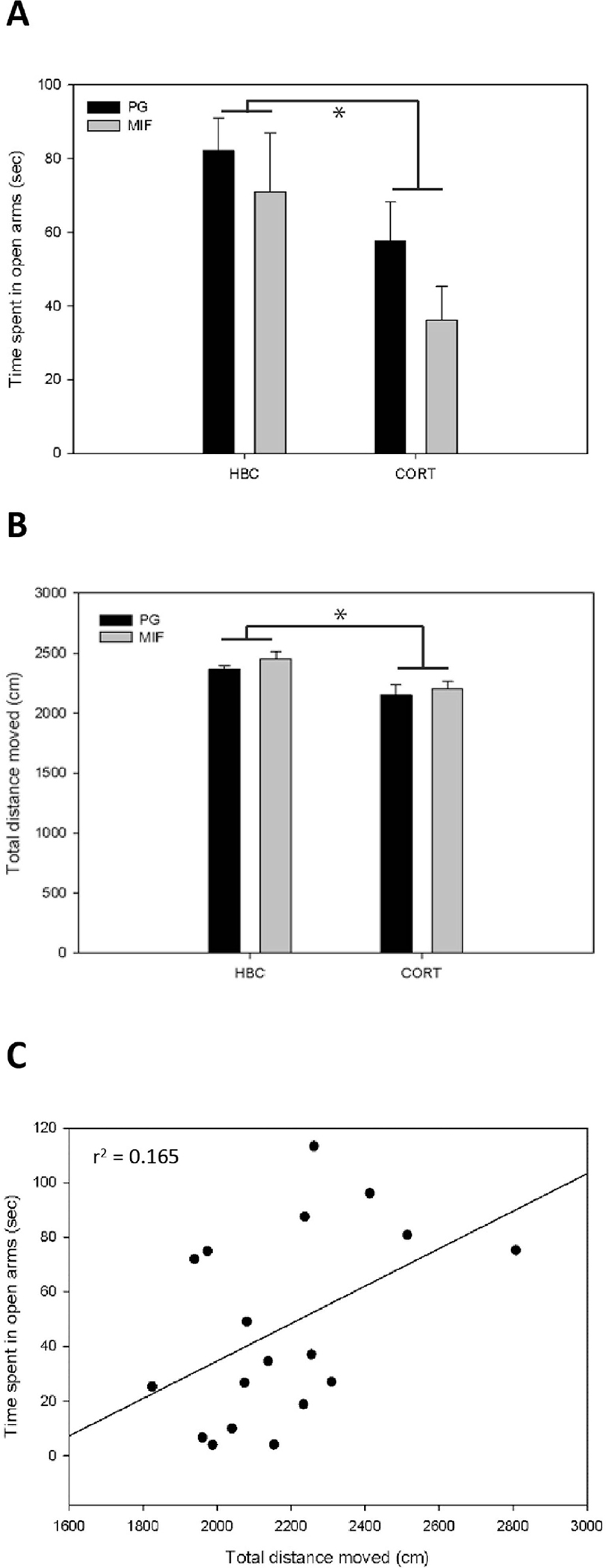
Effect of mifepristone pre-treatment and corticosterone treatment on behavior in the EPM. (A) Corticosterone, but not mifepristone treatment significantly reduced the time spent in the open arms of the maze compared to the HBC-PG vehicle treatment. (B) Corticosterone, but not mifepristone treatment significantly reduced the total distance moved in the maze compared to the HBC-PG vehicle treated group during the 5 min EPM anxiety test. Means expressed as percentage of control, data in the graphs represent Mean ± SEM; *indicates significant difference between groups, P< 0.05. (C) There was a weak non-significant relationship between total distance moved and time spent in open arms within the two corticosterone treated groups, PG/CORT and MIF/CORT. (P> 0.05).
3.2. Effect of corticosterone and mifepristone on circulating plasma corticosterone levels:
There was a significant main effect of mifepristone pre-treatment on circulating plasma corticosterone levels (F(1,43)= 4.637, P = 0.037; Fig. 2). Specifically, mifepristone treatment decreased plasma corticosterone compared to vehicle-treated rats. Corticosterone treatment had no effect on plasma corticosterone levels (F(1,43)= 0.000, P = 0.997) and the interaction between pre-treatment with mifepristone and treatment with corticosterone on plasma corticosterone levels was not significant (F(1,43)= 2.371, P = 0.131).
Figure. 2:
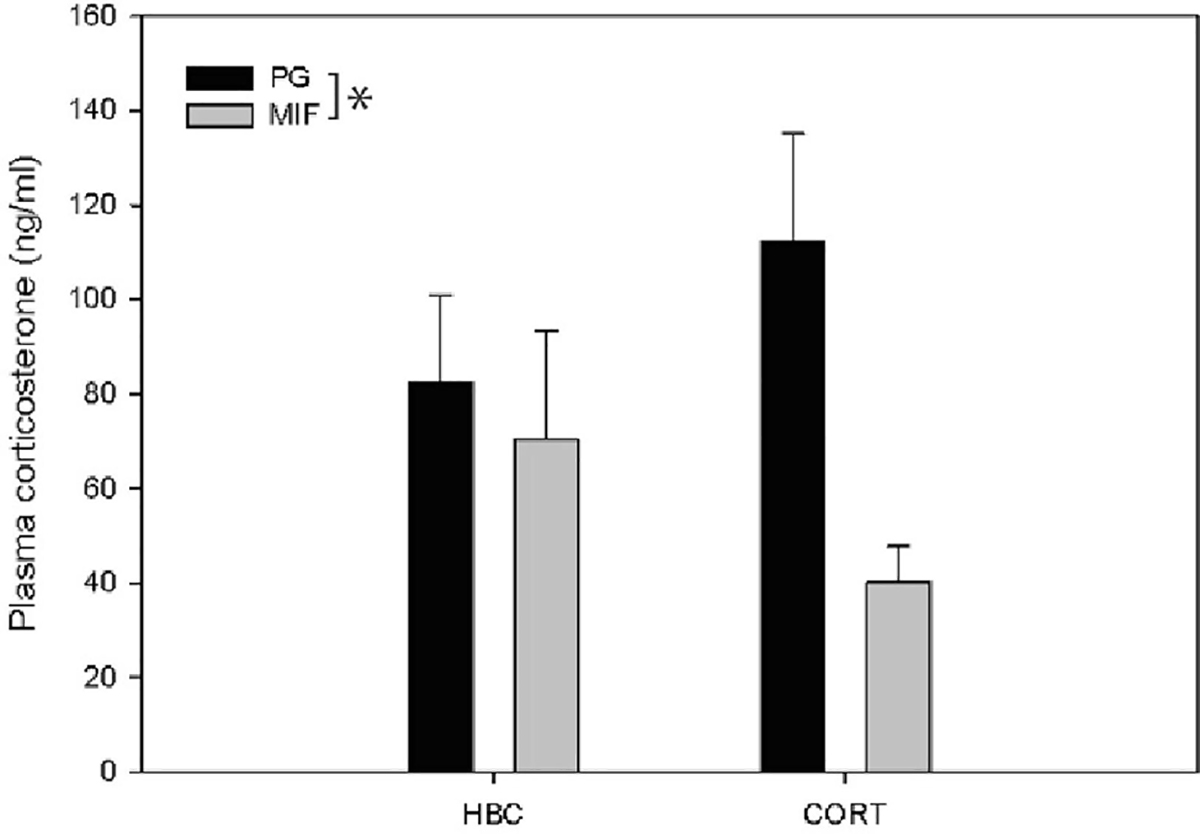
Effect of mifepristone pre-treatment and corticosterone treatment on plasma corticosterone levels as measured by ELISA: Mifepristone but not corticosterone treatment decreased plasma corticosterone levels 28 hours post-treatment (plasma collected 1500–1800; data expressed as Mean ± SEM). *indicates significant difference between groups, P< 0.05.
3.3. Effect of corticosterone and mifepristone treatment on OCT3, SERT and GR protein expression:
There was a significant main effect of corticosterone-treatment on OCT3 expression in the ventral hippocampus (F(1,42) = 4.570, P = 0.038; Fig. 3A), with corticosterone increasing OCT3 immunoreactivity in this region. Mifepristone pre-treatment did not significantly affect OCT3 expression in the ventral hippocampus (F(1,42) = 0.134, P = 0.716; Fig. 3A), and there was no significant interaction between mifepristone and corticosterone on OCT3 expression in the ventral hippocampus (F(1,42) = 0.538, P= 0.467). All OCT3 bands (45kDa) expressed as percent of Actin (40kDa) as the loading control.
Figure 3:
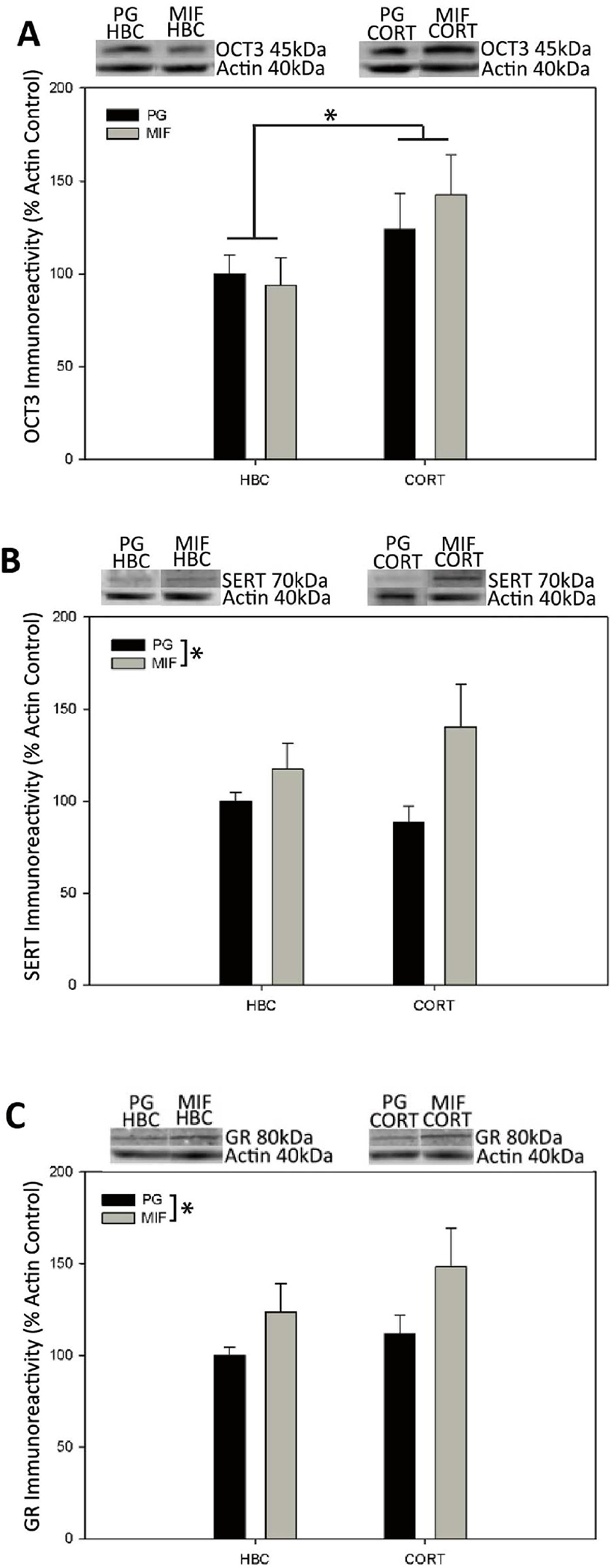
Effect of mifepristone pre-treatment and corticosterone treatment on protein expression in ventral hippocampus as measured by western immunoblot (A) Corticosterone but not mifepristone treatment increased OCT3 expression. Mifepristone but not corticosterone treatment increased SERT (B) and GR expression (C). Means expressed as percentage of control, data in the graphs represent Mean ± SEM; *indicates significant difference between groups, P< 0.05.
OCT3 expression is upregulated by corticosterone in the ventral hippocampus
Mifepristone treatment increases SERT and GR expression in the ventral hippocampus
Regulation of OCT3 expression in the ventral hippocampus is altered by corticosterone
In contrast, there were significant main effects of mifepristone pre-treatment on SERT and GR expression in ventral hippocampus. Mifepristone pre-treatment significantly increased SERT (F(1,41)= 5.309, P= 0.026; Fig. 3B) and GR (F(1,42)= 0.035, P =0.035; Fig. 3C) expression compared to propylene glycol vehicle treatment. Corticosterone treatment did not significantly affect SERT (F(1,41)= 0.135, P =0.715; Fig. 3B) or GR (F(1,42)= 1.731, P =0.195; Fig. 3C) expression in the ventral hippocampus. Furthermore, there was no significant interaction between mifepristone and corticosterone on SERT (F(1,41)= 1.325, P =0.256; Fig. 3B) and GR (F(1,42)= 0.222, P = 0.640; Fig. 3C) expression in the ventral hippocampus. All SERT bands (70kDa) and GR bands (80kDa) expressed as percent of Actin (40kDa) as the loading control.
Corticosterone or mifepristone did not significantly affect OCT3, SERT or GR expression in any of the other brain regions studied (P > 0.05 for all comparisons; Tables 1–3).
Table 1:
OCT3 expression was unchanged in the CeA, DMH and dorsal hippocampus following mifepristone and corticosterone treatment. Data expressed as % of control, P> 0.05.
| Brain region | PG + HBC Mean ± SEM | PG + CORT Mean ± SEM | MIF + HBC Mean ± SEM | MIF + CORT Mean ± SEM |
|---|---|---|---|---|
| CeA | 100.0 ± 7.49 | 108.01 ± 16.71 | 104.64 ± 7.02 | 92.39 ± 8.22 |
| DMH | 100.00 ± 3.63 | 102.16 ± 8.33 | 88.48 + 4.48 | 104.85 ± 11.72 |
| dHipp | 100.00 ± 10.72 | 130.28 ± 22.86 | 99.07 ± 12.00 | 108.25 ± 22.20 |
Table 3:
GR expression was unchanged in the CeA, DMH and dorsal hippocampus following mifepristone and corticosterone treatment. Data expressed as % of control, P> 0.05.
| Brain region | PG + HBC Mean ± SEM | PG + CORT Mean ± SEM | MIF + HBC Mean ± SEM | MIF + CORT Mean ± SEM |
|---|---|---|---|---|
| CeA | 100.00 ± 10.04 | 105.27 ± 13.88 | 85.89 ± 12.46 | 84.02 ± 6.84 |
| DMH | 100.00 ± 6.60 | 127.09 ± 19.93 | 89.95 ± 12.25 | 104.92 ± 18.74 |
| dHipp | 100.00 ± 5.03 | 100.51 ± 15.09 | 109.75 ± 12.35 | 80.11 ± 9.28 |
4. Discussion
Fourteen days of corticosterone treatment increased anxiety-like behavior, and decreased overall activity in the EPM compared to HBC vehicle-treated controls, and pre-treatment with the GR antagonist mifepristone did not block these behavioral effects of corticosterone. Moreover, mifepristone, by itself, did not have any effect on anxiety-like behavior in the EPM, similar to chronic intracerebroventricular administration of mifepristone (Oitzl et al., 1998). Our finding that chronic corticosterone treatment elevates anxiety states adds to other studies suggesting that chronic corticosterone administration is associated with elevated anxiety- and depression-like conditions in rats (Murray et al., 2008; Diniz et al., 2011; Lim et al., 2012). However, we show that corticosterone treatment also significantly reduced total distance moved in open arms. Linear regression analysis showed a weak non-significant relationship (accounting for 16.5% of the variance) between total distance moved and time spent in the open arms. Thus, increases in anxiety measures in corticosterone-treated groups may be in part, due to a decrease in overall movement, which should be examined in more detail in future work.
Acute concentration of circulating plasma corticosterone levels remained unchanged when tested following chronic corticosterone treatment, similar to a previous report in which plasma corticosterone levels were measured 24 h following corticosterone injection (Quadrilatero and Hoffman-Goetz, 2005). Hence, it is likely that the elevated anxiety state observed following chronic corticosterone treatment is not related to this peripheral marker of hypothalamus-pituitary-adrenal (HPA) axis action. However, mifepristone treatment decreased plasma corticosterone levels compared to the both the vehicle-treated control and the corticosterone-treated groups, suggesting enhancement of negative feed-back of the HPA axis. Theoretically, GR activation is thought to negatively regulate HPA axis leading to reduction in plasma corticosterone levels (De Kloet et al., 1998; Smith et al., 2006). Hence, GR antagonism would be predicted to increase plasma corticosterone levels, as shown by some studies (Ghosal et al., 2014; Flores et al., 2006; Lenze et al., 2014) which is opposite to our finding. This difference may be explained by mifepristone acting as a partial agonist at GRs, and the observation that partial agonistic activity may overcome antagonistic effect (Zhang et al., 2007; Gruol et al., 1993; Havel et al., 1996). Thus, it is possible that chronic mifepristone treatment at the dose and period used here results in a net agonistic instead of antagonistic action.
The effects of chronic mifepristone on plasma corticosterone may also be related to mifepristone-induced changes in neural GR expression. Zhang et al. (2007) demonstrated that mifepristone exerted partial agonistic activity on GRs and increased GR expression. Similarly, we observed increased GR expression in the ventral hippocampus following mifepristone treatment, which is likely to increase GR-mediated negative feed-back of the HPA axis to reduce plasma corticosterone levels (Smith et al., 2006; De Kloet et al., 1998).
We also observed differential effects of mifepristone and corticosterone on OCT3 protein expression. Chronic corticosterone treatment increased OCT3 expression in the ventral hippocampus, an effect that was not blocked by GR antagonism by mifepristone. In contrast, neither CORT nor mifepristone alone significantly affected OCT3 expression in the DMH, CeA or dorsal hippocampus. Mifepristone treatment alone increased both SERT and GR expression in the ventral hippocampus and this effect was not observed in any of the other brain regions studied. Expression of SERT and GR were not significantly affected by chronic corticosterone treatment in any brain region studied. Overall, these results indicate that the chronic administration of glucocorticoid ligands has regionally specific effects on the expression of OCT3, SERT and GRs and suggests that the increase in OCT3 expression in the ventral hippocampus is independent of GR activation by corticosterone.
The current findings in the ventral hippocampus are in line with increased OCT3 expression in the ventral hippocampus of rats that exhibit heightened anxiety in response to acute withdrawal from amphetamine (Barr et al., 2013). In contrast, SERT knockout mice exposed to chronic stress showed decreased expression of OCT3 in the hippocampus (Baganz et al., 2010). This finding was explained by suggesting that repeated blockade of OCT3 by stress-induced corticosterone may act similarly to repeated antagonism of other transporters, such as SERT, in down regulating transporter expression (Baganz et al., 2010). It is possible that these disparate results may be related to the SERT knockout mice, which already had a compensatory increased expression of hippocampal OCT3 (Baganz et al., 2010).
Serotonin clearance represents a likely mechanism for OCT3 modulation of stress effects in the hippocampus. The hippocampus is believed to function in adaptive responses to stress (reviewed in McEwen, 2002, Radley et al., 2015). In SERT deficient mice, corticosterone and decynium-22 blockade of OCTs in the CA3 of the dorsal hippocampus decrease serotonin clearance and behaviorally, these animals exhibit antidepressant-like effects (Baganz et al., 2008, 2010). In studies examining the underlying mechanisms associated with amphetamine withdrawal, we found that OCT3 expression was increased in amphetamine-treated rats (Barr et al., 2013, Solanki et al., 2016). In these rats, corticosterone- and decynium-22-induced increases in serotonin the ventral hippocampus were attenuated relative to controls, possibly because higher concentrations of the drugs may be required to counteract serotonin clearance augmented through higher expression of OCT3 relative to controls (Barr and Forster, 2011, Barr et al., 2013). Consistent with this possibility, rats undergoing amphetamine withdrawal exhibit pronounced deficits in stress-induced increases in hippocampal serotonin (Li et al., 2014). The changes in hippocampal serotonin appear to be directly related to heightened anxiety since local hippocampal serotonergic lesions increase anxiety and the increased anxiety-like behavior exhibited during amphetamine withdrawal is reversed by intra-hippocampal infusions blocking serotonin reuptake. Combined, these results suggest that decreases in serotonin in the ventral hippocampus contribute to the increase in anxiety-like behavior exhibited during amphetamine withdrawal (Vuong et al., 2010).
Alterations in protein expression of OCT3, GR and SERT in response to corticosterone or mifepristone treatment was only observed in the ventral hippocampus and not in the CeA, DMH or dorsal hippocampus. The ventral hippocampus contains the highest concentration of GRs in the brain (Sapolsky et al., 1984; Smith et al., 2006) which may make it more vulnerable to chronic corticosterone and mifepristone treatment as compared to other brain regions. In the DMH, OCT3s are highly expressed in the ependymal layer lining the ventricles and are believed to contribute to the corticosterone inhibition of serotonin clearance corticosterone-mediated inhibition of serotonin clearance under conditions of increased serotonergic activity (Feng et al., 2009). In the DMH, OCT3 expression increased at 24 h post-amphetamine withdrawal but not at 2 weeks. In the CeA, OCT3s are present in modest levels (Gasser et al., 2006) including expression in the intercalated cell groups (Hill et al., 2013). In response to both acute and chronic amphetamine withdrawal, expression of both OCT3 and SERT increase in the CeA (Solanki et al., 2016). Our results suggest that these changes in OCT3 and/or SERT following amphetamine withdrawal appear to be independent of GR activation.
Our findings support a role for corticosterone in upregulating OCT3 protein expression in the ventral hippocampus, similar to the glucocorticoid-induced upregulation of OCT1 and OCT2 found in non-neural tissues (Rulcova et al., 2013; Shu et al., 2001). However, it was surprising that the corticosterone-induced increase in OCT3 expression in the ventral hippocampus was not blocked by the GR antagonist mifepristone, suggesting a GR-independent mechanism, or alternatively, a shift from antagonistic effects to agonistic effects (Zhang, 2007) coinciding with the increased GR expression in the vHipp. Our findings also suggest that chronic corticosterone treatment does not regulate SERT expression in any of the regions studied. However, administration of chronic mifepristone upregulated SERT protein expression in the ventral hippocampus. Mifepristone has been shown to block SERT (Li et al., 2014) which may have contributed to the increase in SERT expression. Increases in SERT expression have also been found following chronic social defeat stress, which were prevented by combined treatment of mifepristone and spironolactone (Zhang et al., 2012) suggesting a role for mineralocorticoid receptors rather than GR alone. Combined, the results suggest that chronic mifepristone treatment has the potential to alter serotonergic transmission in the ventral hippocampus.
Conclusions:
Our results suggest that OCT3 expression in the ventral hippocampus is upregulated by chronic corticosterone treatment, an effect that is independent of GR activation. This effect is specific to the ventral hippocampus, and may contribute the increased anxiety states induced by either chronic corticosterone or repeated exposure to stressors. The molecular mechanism by which corticosterone increased OCT3 expression in the ventral hippocampus; and whether blockage of ventral hippocampal OCT3s alleviates anxiety-like behavior is yet to be determined. Furthermore, chronic mifepristone treatment increases SERT and GR expression, again specific to the ventral hippocampus, and the latter finding may underlie reduced plasma corticosterone observed after chronic mifepristone treatment. Mechanisms of mifepristone-mediated reductions in plasma corticosterone and increases in GRs and SERT should be further elucidated as mifepristone is clinically used to treat psychotic major depression in patients with elevated HPA axis activity. Overall, our results contribute to the understanding how central regulation of OCT3 expression is altered by stress exposure, as well as provide new insights into the potential effects of chronic mifepristone treatment.
Table 2:
SERT expression was unchanged in the CeA, DMH and dorsal hippocampus following mifepristone and corticosterone treatment. Data expressed as % of control, P> 0.05.
| Brain region | PG + HBC Mean ± SEM | PG + CORT Mean ± SEM | MIF + HBC Mean ± SEM | MIF + CORT Mean ± SEM |
|---|---|---|---|---|
| CeA | 100.00 ± 7.9 | 104.94 ± 13.81 | 99.92 ± 15.70 | 76.48 ± 6.25 |
| DMH | 100.00 ± 14.22 | 93.23 ± 18.09 | 69.53 ± 11.72 | 100.53 ± 31.89 |
| dHipp | 100.00 ± 5.10 | 96.99 ± 11.37 | 98.79 ± 13.66 | 82.47 ± 6.82 |
Acknowledgements
This work was supported by NIH RO1 DA019921. We thank Mackenzie Mears for help with brain sectioning, Nathan Vinzant for help with the behavioral experiments and Drs. Evelyn Schlenker and Khosrow Rezvani for helpful technical and editing suggestions.
Abbreviations
- CeA
Central nucleus of amygdala
- DMH
Dorsomedial hypothalamu
- EPM
Elevated plus maze
- ELISA
Enzyme-linked immunosorbent assay
- GR
Glucocorticoid receptor
- HPA
Hypothalamus-pituitary-adrenal
- OCT1
Organic cation transporter 1
- OCT2
Organic cation transporter 2
- OCT3
Organic cation transporter 3
- PVDF
Polyvinylidene difluoride
- SERT
Serotonin transporter
- SNK
Student-Newman-Keul’s
- TBST
Tris buttered saline with tween-20
Footnotes
Declarations of Interest
All authors report no declarations of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amphoux A, Vialou V, Drescher E, Bruss M, Mannoury La Cour C, Rochat C, Millan MJ, Giros B, Bonisch H, Gautron S, 2006. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacol. 50, 941–952. [DOI] [PubMed] [Google Scholar]
- Ayala-Lopez N, Jackson WF, Burnett R, Wilson JN, Thompson JM, Watts SW, 2015. Organic cation transporter 3 contributes to norepinephrine uptake into perivascular adipose tissue. Am J Physiol Heart Circ Physiol. 309, H1904–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baganz N, Horton R, Martin K, Holmes A, Daws LC, 2010. Repeated swim impairs serotonin clearance via a corticosterone sensitive mechanism: Organic cation transporter 3, the smoking gun. J Neurosci. 30, 15185–15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JL and Forster GL, 2011. Serotonergic neurotransmission in the ventral hippocampus is enhanced by corticosterone and altered by chronic amphetamine treatment. Neurosci. 182, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JL, Scholl JL, Solanki RR, Watt MJ, Lowry CA, Renner KJ, Forster GL, 2013. Influence of chronic amphetamine treatment and acute withdrawal on serotonin synthesis and clearance mechanisms in the rat ventral hippocampus. Eur J Neurosci. 37, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu EE, Schold C, Schatzberg AF, 2002. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatr. 52, 386–392. [DOI] [PubMed] [Google Scholar]
- Bradford MM, 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chen EC, Liang X, Yee SW, Geier EG, Stocker SL, Chen L, Giacomini KM, 2015. Targeted disruption of organic cation transporter 3 attenuates the pharmacological response to metformin. Mol Pharmacol. 88, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M, 1998. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 19, 269–301. [DOI] [PubMed] [Google Scholar]
- De La Mora MP, Gallegos-Cari A, Arizmendi-Garcia Y, Marcellino D, Fuxe K, 2010. Role of dopamine receptor mechanisms in the amygdaloid modulation of fear and anxiety: structural and functional analysis. Prog Neurobiol. 90, 198–216. [DOI] [PubMed] [Google Scholar]
- Diniz L, Dos Reis BB, Castro De GM, Medalha CC, Viana MB, 2011. Effects of chronic corticosterone and imipramine administration on panic and anxiety related responses. Braz J Med Biol Res. 44, 1048–1053. [DOI] [PubMed] [Google Scholar]
- Duan H and Wang J, 2010. Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther. 335, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N, Telefont M, Kelly KJ, Orchinik M, Forster GL, Renner KJ, Lowry CA, 2009. Local perfusion of corticosterone in the rat medial hypothalamus potentiates D-fenfluramine induced elevations of extra-cellular 5-HT concentrations. Horm Behav. 56, 149–157. [DOI] [PubMed] [Google Scholar]
- Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF, 2006. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacol. 31, 628–636. [DOI] [PubMed] [Google Scholar]
- Forster GL, Pringle RB, Mouw NJ, Vuong SM, Watt MJ, Burke AR, Lowry CA, Summers CH, Renner KJ, 2008. Corticotrophin-releasing factor in in the dorsal raphe nucleus increases medial prefrontal cortical serotonin via type 2 receptors and median raphe nucleus activity. Eur J Neurosci. 28, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser PJ, Lowry CA, Orchinik M, 2006. Corticosterone sensitive monoamine transport in the dorsomedial hypothalamus: Potential role for organic cation transporter 3 in stress induced modulation of monoaminergic neurotransmission. J Neurosci. 26, 8758–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser PJ, Orchinik M, Ilangovan R, Lowry CA, 2009. Distribution of organic cation transporter 3, a corticosterone sensitive monoamine transporter, in the rat brain. J Comp Neurol. 512, 529–555. [DOI] [PubMed] [Google Scholar]
- Ghosal S, Bundzikova-Osacka J, Dolgas CM, Myers B, Herman JP, 2014. Glucocorticoid receptors in the nucleus of the solitary tract (NTS) decrease endocrine and behavioral stress responses. Psychoneuroendocrinol. 45, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glube N, Langguth P, 2008. Caki-1 cells as a model system for the interaction of renally secreted drugs with OCT3. Nephron physiol. 108, 18–28. [DOI] [PubMed] [Google Scholar]
- Gomez-Roman A, Ortega-Sanchez JA, Rotllant D, Gagliano H, Belda X, Delgado-Morales R, Marin-Blasco I, Nadal R, Armario A, 2015. The neuroendocrine response to stress under the effect of drugs: Negative synergy between amphetamine and stressors. Psychoneuroendocrinol. 63, 94–101. [DOI] [PubMed] [Google Scholar]
- Graf EN, Wheeler RA, Baker DA, Ebben AL, Hill JE, McReynolds JR, Robble MA, Vranjknovic O, Wheeler DS, Mantsch JR, Gasser PJ, 2013. Corticosterone acts in the nucleus accumbens to enhance dopamine signaling and potential reinstatement of cocaine seeking. J Neurosci. 33, 11800–11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruol DJ, and Altschmied J, 1993. Synergistic induction of apoptosis with glucocorticoids and 3′,5′-cyclic adenosine monophosphate reveals agonist activity by RU 486. Mol. Endocrinol. 7, 104–113. [DOI] [PubMed] [Google Scholar]
- Grundwald NJ, Brunton PJ, 2015. Prenatal stress programs neuroendocrine stress responses and affective behaviors in second generation rats in a sex-dependent manner. Psychoneuroendocrinol. 62, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel PJ, Busch BL, Curry DL, Johnson PR, Dallman MF, Stern JS, 1996. Predominantly glucocorticoid agonist actions of RU486 in young specific pathogen free Zucker rats. Am J Physiol. 271, R710–R717. [DOI] [PubMed] [Google Scholar]
- Hill JE, Gasser PJ, 2013. Organic cation transporter 3 is densely expressed in the intercalated cell groups of the amygdala: anatomical evidence for a stress hormone sensitive dopamine clearance system. J Chem Neuroanat. 52, 36–43. [DOI] [PubMed] [Google Scholar]
- Horton RE, Apple DM, Owens WA, Baganz NL, Cano S, Mitchell NC, Vitela M, Gould GG, Koek W, Daws LC 2013. Decynium-22 enhances SSRI-induced antidepressant-like effects in mice: uncovering novel targets to beat depression. J Neurosci. 33, 10534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingoglia F, Visigalli R, Rotoli BM, Barilli A, Riccardi B, Puccini P, Dall’Asta V, 2015. Functional characterization of the organic cation transporters (OCTs) in human airway pulmonary epithelial cells. Biochim Biophys Acta. 1848, 1563–1572. [DOI] [PubMed] [Google Scholar]
- Kitaichi K, Morishita Y, Doi Y, Ueyama J, Matsushima M, Zhao YL, Takagi K, Hasegawa T, 2003. Increased plasma concentration and brain penetration of methamphetamine in behaviorally sensitized rats. Eur J Pharmacol. 464, 39–48. [DOI] [PubMed] [Google Scholar]
- Kitachi K, Fekuda M, Nakayama H, Aoyama N, Ito Y, Fujimono Y, Takagi K, Takagi K, Haseqawa T 2005. Behavioral changes following antisense oligonucleotide-induced reduction of organic cation transporter-3 in mice. Neurosci Lett. 382, 195–200. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C, 2007. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 24, 1227–1251. [DOI] [PubMed] [Google Scholar]
- Kwon OB, Lee JH, Kim HJ, Lee S, Lee S, Jeong MJ, Kim SJ, Jo HJ, Ko B, Chang S, Park SK, Choi YB, Bailey CH, Kandel ER, Kim JH, 2015. Dopamine regulation of amygdala inhibitory circuits for expression of learned fear. Neuron. 88, 378–389. [DOI] [PubMed] [Google Scholar]
- Knych ET, Eisenberg RM, 1979. Effect of amphetamine on plasma corticosterone in the conscious rat. Neuroendocrinol. 29, 110–118. [DOI] [PubMed] [Google Scholar]
- Larkin JD, Jenni NL, Floresco SB, 2015. Modulation of risk/reward decision making by dopaminergic transmission within the basolateral amygdala. Psychopharmacol. 233, 121–136. [DOI] [PubMed] [Google Scholar]
- Lenze EJ, Hershey T, Newcomer JW, Karp JF, Blumberger D, Anger J, Dore P, Dixon D, 2014. Antiglucocorticoid therapy for older adults with anxiety and co-occurring cognitive dysfunction: results from a pilot study with mifepristone. Int J Geriatr Psychiatry. 29, 962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Shan L, Li X, Wei L, Li D, 2014. Mifepristone modulates serotonin transporter function. Neural Regen Res. 9, 646–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Scholl JL, Tu W, Hassell JE, Watt MJ, Forster GL, Renner KJ, 2014. Serotonergic responses to stress are enhanced in the central amygdala and inhibited in the ventral hippocampus during amphetamine withdrawal. Eur J Neurosci. 40, 3684–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, He M, Zhou L, Miao X, Wu F, Huang S, Dai X, Wang T, Wu T, 2015b. A solute carrier family 22 member 3 variant rs3088442 G-A associated with coronary heart disease inhibits lipopolysaccharide-induced inflammatory response. J Biol Chem. 290, 5328–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Jang S, Lee Y, Moon S, Kim J, Oh K, 2012. Enhancement of anxiety and modulation of TH and pERK expressions in amygdala by repeated injections of corticosterone. Biomol Ther (Seoul). 20, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips KS, Volk C, Schmitt BM, Pfeil U, Arndt P, Miska D, Ermert L, Kummer W, Koepsell H, 2005. Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am J Respir Cell Mol Biol. 33, 79–88. [DOI] [PubMed] [Google Scholar]
- Marcinkiewcz CA, Devine DP, 2015. Modulation of OCT3 expression by stress, and anti-depressant like activity of decynium-22 in an animal model of depression. Pharmacol Biochem Behav. 131, 33–41. [DOI] [PubMed] [Google Scholar]
- Massmann V, Edemir B, Schlatter E, Al-Monajjed R, Harrach S, Klassen P, Holle SK, Sindic A, Dobrivojevic M, Pavenstadt H, Ciarimboli G, 2013. The organic cation transporter 3 (OCT3) as molecular target of psychotropic drugs: transport characteristics and acute regulation of cloned murine OCT3. European J Physiol. 466, 517–527. [DOI] [PubMed] [Google Scholar]
- McEwen BS 2002. Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging. 23, 921–39. [DOI] [PubMed] [Google Scholar]
- Murray F, Smith DW, Hutson PH, 2008. Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviors in mice. Eur J Pharmacol. 583, 115–127. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Fluttert M, Sutanto W, de Kloet ER 1998. Continuous blockade of brain glucocorticoid receptors facilitates spatial learning and memory in rats. Eur J Neurosci. 10, 3759–3766. [DOI] [PubMed] [Google Scholar]
- Ordjan NE, Pivina SG, Mironova VI, Rakitskaia VV, Akulova VK, 2014. The hypothalamic-pituitary-adrenal axis activity in prenatal stressed female rats in the model of post-traumatic stress disorder. Ross Fiziol Zh Im I M Sechenova. 100, 1409–1420. [PubMed] [Google Scholar]
- Parrott AC, Montgomery C, Wetherell MA, Downey LA, Stough C, Scholey AB, 2014. MDMD, cortisol and heightened stress in recreational ecstasy users. Behav Pharmacol. 25, 458–472. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C., 1997. The Rat Brain in Stereotaxic Coordinates. San Diego, California: Academic Press. [Google Scholar]
- Perusini JN, Meyer EM, Long VA, Rau V, Nocera N, Avershal J, Maksymetz J, Spigelman I, Fanselow MS, 2015. Induction and expression of fear sensitization caused by acute traumatic stress. Neuropsychopharmacol. 41, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrilatero J and Hoffman-Goetz L, 2005. In vivo corticosterone administration at levels occurring with intense exercise does not induce intestinal lymphocyte apoptosis in mice. J Neuroimmunol. 162, 137–148. [DOI] [PubMed] [Google Scholar]
- Qulu L, Daniels WM, Mabandla MV, 2015. Exposure to prenatal stress has deleterious effects on hippocampal function in a febrile seizure rat model. Brain Res. 1624, 506–514. [DOI] [PubMed] [Google Scholar]
- Radley J, Morilak D, Viau V, Campeau S 2015. Chronic stress and brain plasticity: Mechanisms underlying adaptive and maladaptive changes and implications for stress-related CNS disorders. Neurosci Biobehav Rev. 58, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratka A, Sutanto W, Bloemers M, De Kloet ER, 1989. On the role of brain mineralocorticoid (Type I) and glucocorticoid (Type II) receptors in neuroendocrine regulation. Neuroendocrinol. 50, 117–123. [DOI] [PubMed] [Google Scholar]
- Reynolds AR, Saunders MA, Brewton HW, Winchester SR, Elgumati IS, Prendergast MA, 2015. Acute oral administration of the novel, competitive and selective glucocorticoid receptor antagonist ORG34517 reduces the severity of ethanol withdrawal and related hypothalamic pituitary adrenal axis activation. Drug Alcohol Depend. 154, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikov R, Diwan M, Nobrega JN, Hamani C, 2015. Towards a better pre-clinical model of PTSD: characterizing animals with weak extinction maladaptive stress responses and low plasma corticosterone. J Psychiatr Res. 61, 158–165. [DOI] [PubMed] [Google Scholar]
- Rulcova A, Krausova L, Smutny T, Vrzal R, Dvorak Z, Jover R, Pavek P, 2013. Glucocorticoid receptor regulates organic cation transporter 1 (OCT1, SLC22A1) expression via HNF4α upregulation in primary human hepatocytes. Pharmacol Rep. 65, 1322–1335. [DOI] [PubMed] [Google Scholar]
- Sanchez-Covarrubias L, Slosky LM, Thompson BJ, Davis TP, Ronaldson PT, 2014. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Cur Pharm Des. 20, 1422–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS, 1984. Glucocorticoid sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc Natl Acad Sci. 81, 6174–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sata R, Ohtani H, Tsujimoto M, Murakami H, Koyabu N, Nakamura T, Uchiumi T, Kuwano M, Nagata H, Tsukimori K, Nakano H, Sawada Y, 2005. Functional analysis of organic cation transporter 3 expressed in human placenta. J Pharmacol Exp Ther. 315, 888–895. [DOI] [PubMed] [Google Scholar]
- Schatzberg AF, 2015. Anna-Monika award lecture, DGPPN Kongress, 2013: the role of hypothalamic-pituitary-adrenal (HPA) axis in the pathogenesis of psychotic major depression. World J Biol Psych. 16, 2–11. [DOI] [PubMed] [Google Scholar]
- Schmit A, Mössner R, Gossmann A, Fischer IG, Gorboulev V, Murphy DL, Koepsell H, Lesch KP 2003. Organic cation transporter capable of transporting serotonin in up-regulated in serotonin transporter-deficient mice. J Neurosci Res. 71, 701–9. [DOI] [PubMed] [Google Scholar]
- Scholl JL, Vuong SM, Forster GL, 2010. Chronic amphetamine treatment enhances corticotropin releasing factor induced serotonin release in the amygdala. Eur J Pharmacol. 644, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Bello CL, Mangravite LM, Feng B, & Giacomini KM, 2001. Functional characteristics and steroid hormone-mediated regulation of an organic cation transporter in Madin-Darby canine kidney cells. J Pharmacol Exp Ther. 299, 392–398. [PubMed] [Google Scholar]
- Sial OK, Warren BL, Alcantara LF, Parise EM, Bolanos-Guzman CA, 2015. Vicarious social defeat stress: Bridging the gap between physical and emotional stress. J Neurosci Meth. 258, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM and Vale WW, 2006. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses. Dialogues Clin Neurosci. 8, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki RR, Scholl JL, Watt MJ, Renner KJ, Forster GL, 2016. Amphetamine withdrawal alters the expression of organic cation transporter 3 and serotonin transporter in several limbic brain regions. J Exp Neurosci. 10, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stairs DJ, Prendergast MA, Bardo MT, 2011. Environmental-induced differences in corticosterone and glucocorticoid receptor blockade of amphetamine self-administration in rats. Psychopharmacol (Berl). 218, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Koob GF, Cador M, Lorang M, Hauger RL, 1993. Pituitary adrenal axis responses to acute amphetamine in the rat. Pharmacol Biochem Behav. 45, 629–637. [DOI] [PubMed] [Google Scholar]
- Tu W, Cook A, Scholl JL, Mears M, Watt MJ, Renner KJ, Forster GL, 2014. Serotonin modulates the ventral hippocampus anxiety like behavior during amphetamine withdrawal. Neurosci. 281C, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Balasse L, Callebert J, Launay JM, Giros B, Gautron S, 2008. Altered aminergic neurotransmission in the brain of organic cation transporter 3 deficient mice. J Neurochem. 106, 1471–1482. [DOI] [PubMed] [Google Scholar]
- Vuong SM, Oliver HA, Scholl JL, Oliver KM and Forster GL, 2010. Increased anxiety like behavior of rats during amphetamine withdrawal is reversed by CRF2 receptor antagonism. Behav Brain Res. 208, 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Kekuda R, Huang W, Fei YJ, Leibach FH, Chen J, Conway SJ, Ganapathy V, 1998. Identity of the organic cation transporter 3 as the extraneuronal monoamine transporter (uptake 2) and evidence for the expression of the transporter in the brain. J Biol Chem. 273, 32776–32786. [DOI] [PubMed] [Google Scholar]
- Wulsin AC, Herman JP, Solomon MB, 2010. Mifepristone decreases depression like behavior and modulates neuroendocrine and central hypothalamic pituitary adrenocortical axis responsiveness to stress. Psychoneuroendocrinol. 35, 1100–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wultsch T, Grimberg G, Schmitt A, Painsipp E, Wetzstein H, Breitenkamp AF, Grundemann D, Schomig E, Lesch KP, Gerlach M, Reif A, 2009. Decreased anxiety in mice lacking the organic cation transporter 3. J Neural Transm. 116, 689–697. [DOI] [PubMed] [Google Scholar]
- Yee SW, Lin L, Merski M, Keiser MJ, Gupta A, Zhang Y, Chien HC, Shoichet BK, Giacomini KM, 2015. Prediction and validation of enzyme and transporter off-targets for metformin. J Pharmacokinet Pharmacodyn. 42, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Jonklaas J, Danielsen M, 2007. The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptors levels but not on EC50 values. Steroids. 72, 600–608. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fan Y, Li Y, Zhu H, Wang L, Zhu M, 2012. Chronic social defeat up-regulates expression of the serotonin transporter in rat dorsal raphe nucleus and projection regions in a glucocorticoid-dependent manner. J Neurochem. 123, 1054–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]