

Abstract
One of the most significant risk variants for Parkinson’s disease (PD), rs356182, is located at the PD-associated locus near the alpha-synuclein (α-syn) encoding gene, SNCA. SNCA-proximal variants, including rs356182, are thought to function in PD risk through enhancers via allele-specific regulatory effects on SNCA expression. However, this interpretation discounts the complex activity of genetic enhancers and possible non-conical functions of α-syn. Here we investigated a novel risk mechanism for rs356182. We use CRISPR-Cas9 in LUHMES cells, a model for dopaminergic midbrain neurons, to generate precise hemizygous lesions at rs356182. The PD-protective (A/−), PD-risk (G/−) and wild-type (A/G) clones were neuronally differentiated and then compared transcriptionally and morphologically. Among the affected genes was SNCA, whose expression was promoted by the PD-protective allele (A) and repressed in its absence. In addition to SNCA, hundreds of genes were differentially expressed and associated with neurogenesis and axonogenesis—an effect not typically ascribed to α-syn. We also found that the transcription factor FOXO3 specifically binds to the rs356182 A-allele in differentiated LUHMES cells. Finally, we compared the results from the rs356182-edited cells to our previously published knockouts of SNCA and found only minimal overlap between the sets of significant differentially expressed genes. Together, the data implicate a risk mechanism for rs356182 in which the risk-allele (G) is associated with abnormal neuron development, independent of SNCA expression. We speculate that these pathological effects manifest as a diminished population of dopaminergic neurons during development leading to the predisposition for PD later in life.
Introduction
With each consecutive genome-wide association study (GWAS), Parkinson’s disease (PD) is associated with an ever-increasing number of decreasing effect-size single-nucleotide polymorphisms (SNPs) (1,2). The value of the results obtained from these associations has been widely debated (3,4). The reason for this debate is that the mechanisms underpinning the associations are not always clear and most (>90%) of the risk SNPs do not reside in protein coding regions, but instead in presumptive regulatory elements, which affect the expression of often unknown genes (5). Additionally, SNPs at a given locus are often in linkage disequilibrium with one-another making it difficult to identify the ‘functional’ variant. Because of this, descriptions of risk mechanisms require follow-up experiments to determine their functions. For most PD risk loci, the identification of the causal SNP(s) has not been definitive, with few notable exceptions (6–9). A rare example of a risk variant which was investigated for gene-association and risk mechanism involves a SNP within an intron of FTO, associated with increased risk for obesity and type-2 diabetes (10). Despite the assumption that risk was conferred through regulation of FTO, they found that the actual gene target was IRX3, several mega-bases away on linear DNA, establishing precedent for non-presumptive risk variant/gene interactions. A major gap in our ability to understand and utilize PD-GWAS results is the lack of in-depth mechanistic insights. One of the top GWAS-identified PD risk-SNPs, rs356182, is an example of a prominent SNP lacking a confirmed mechanism.
rs356182 has a meta-risk P-value of 1.85 × 10−82, making it one of the most significant associations among non-coding PD risk-SNPs (11). It is an A > G variant, with the risk allele, guanine (G), robustly represented in the population [frequency = 37.22% in the most recent meta-analysis (11)]) and one of the highest odds ratios (1.34) of any PD-associated SNP (11,12). This means the proportion of G alleles in the PD population compared to healthy controls is unusually high even compared to other statistically significant risk signals. Enticingly, this SNP is within a genetic enhancer present in several regions of the brain and located close (~19 kb from the 3′ end of the SNCA transcription on linear DNA) to the gene encoding alpha-synuclein (α-syn) (Fig. 1A), which is one of the first proteins linked to familial PD as well as neuropathologically present as aggregates in Lewy bodies (5,13,14). Additionally, the SNCA locus has at least three independent GWAS risk signals, as determined by conditional analysis (13). These data have led to the assumption that rs356182 confers risk for PD by directly regulating the expression of exclusively SNCA. However, this narrow view of the rs356182 mechanism neglects the potential for additional enhancer–promoter interactions, as well as secondary and tertiary effects of transcription factor (TF) dysregulation.
Figure 1.

An enhancer encompassing rs356182 is active in differentiated LUHMES cells. (A) Histone H3K27ac track for undifferentiated (top) and differentiated (bottom) LUHMES cells. (B) Bright-field images showing the morphology of wild-type undifferentiated (left) and differentiated (right) LUHMES cells. (C) Immunofluorescent (TUJ1 = red and TH = green) and stained images (nuclei DAPI = blue) of differentiated LUHMES cells (Day 6 of differentiation).
Unlike promoters, enhancers are highly dynamic genetic elements, in the sense of functioning both cryptically via multiple TFs and with different gene targets in different cell types and stages. Enhancer variants have continuous effects (graded responses on gene expression) as opposed to the binary effects of gene triplet code variation. Furthermore, enhancers do not necessarily or exclusively interact with the closest gene promoter on linear DNA (SNCA in this case). Up to two-thirds of enhancers skip the nearest gene entirely and interact with distal promoters (15,16). Enhancers may interact with multiple promoters, and a single promoter often interacts with numerous enhancers in the three-dimensional space of chromatin (17,18). Enhancers regulate the expression of target genes by recruiting TFs to specific regulatory binding motifs. In this way, SNPs modulate downstream gene expression by changing the binding affinity of TFs to a particular allele and alter the strength of the enhancer–promoter interactions.
Lund human mesencephalic (LUHMES; a.k.a. MESC2.10) cells offer a unique and useful platform for studying immature human midbrain neurons and the epigenetic architecture present during neuronal development, due to their isolation from 8-week-old fetal mesencephalic tissue (19). This gestational time point coincides with early neurogenesis (20,21). LUHMES cells are immortalized in a stem-like state with the v-MYC oncogene, which, when deactivated allows the cells to enter the differentiation pathway and reach their terminal state within 6 days. In their differentiated state, LUHMES cells have a neuronal phenotype, display long neuronal projections (Fig. 1B) and partially express the rate-limiting enzyme in dopamine production, tyrosine hydroxylase (TH) (Fig. 1C). Beneficial to this research, LUHMES cells are heterozygous for rs356182 with one chromosomal homolog possessing the PD risk-conferring allele (G) and the other homolog possessing the protective allele (A). Interestingly, when differentiated, these cells also contain an enhancer surrounding the rs356182 SNP [7]. This makes LUHMES cells an ideal model for investigating PD pathology or predisposition originating very early in development, including neuronal differentiation-based mechanisms.
The study reported here, seeks to elucidate the mechanism surrounding rs356182 by examining allele-specific gene regulation and changes to differentiated morphology associated with this SNP. To that end, we used CRISPR-Cas9 to generate mono-allelic lesions of rs356182 in LUHMES cells (Fig. 2A) and examined the changes in gene expression and morphology therein. We observed a novel rs356182 risk mechanism not previously attributed to SNCA, pertaining to neuronal differentiation processes.
Figure 2.

Excised rs356182 by CRISPR-Cas9. (A) Schematic depicting the position of rs356182 relative to SNCA on chromosome 4 and the guide-RNA targeting that locus for a double stranded DNA break. (B) The resulting deletions (deleted bases depicted as dashes) from targeting rs356182 with CRISPR-Cas9; position of rs356182 highlighted in red.
Results
rs356182 controls FOXO3 binding, enhancer activity, SNCA expression and genome-wide gene expression
In situ analyses of the TF binding-motifs at the rs356182 region using HaploReg (Supplementary Material, Table S1) and MotifbreakR (Supplementary Material, Table S2) revealed a preferential binding affinity of the protective A-allele over the risk G-allele for the strongest interacting TFs at this locus (Supplementary Material, Tables S1 and S2) (22–24). Notably, the Fox family of TF proteins was identified as strong candidate binders to the A-containing motif, with FOXO3 having the most significant association for the A-allele of all the TFs listed by MotifBreakR (Fig. 3A, Supplementary Material, Table S2). The data suggest that the rs356182 risk-allele (G) disrupts a particularly vital position within the putative binding motif, resulting in diminished binding affinity for FOX TFs, with the potential to reduce enhancer activity. To validate binding affinity of the candidate FOXO proteins, we used ChIP-qPCR with allele-specific amplification (Supplementary Material, Fig. S1). We found that FOXO3 demonstrated allele-specific activity at this locus (Fig. 3B). Additionally, we found that the H3K27ac-bound DNA was significantly enriched for the A-allele over the G-allele, indicating preferential enhancer activity in the presence of the protective A-allele.
Figure 3.

rs356182 controls FOXO3 binding, enhancer activity and downstream gene expression. (A) Results from MotifBreakR depicting the sequence and importance (letter height) of each base within the motif sequence for FOXO3. (B) Box-and-whisker plots showing the relative allele ratio resulting from ChIP for FOXO3 (P = 2.17 × 10−6) and H3K27ac (P = 1.33 × 0−7). (C) Box-and-whisker plot of z-scores for SNCA gene expression; adjusted P-value displayed above each group for comparison. (D) Volcano plot showing the differentially expressed genes between the A/− and G/− clones with the genes proximal to rs356182 annotated; Log2 fold-change cutoff, 1; P-value cutoff, 0.05. (E) Circos plot depicting the significantly modulated genes between the A/− and G/− clones across the entire genome.
After we validated the mechanism by which allele-specific regulation is controlled by rs356182, we next identified potential gene targets regulated by this enhancer. To that end, we created hemizygous clones at this site and therefore possessing a lesion (i.e. short deletion) on one chromosome while maintaining the original major (A) or minor (G) alleles on the other. To achieve this, we used CRISPR-Cas9 to target the PAM site nearest to rs356182 (Fig. 2A and Supplementary Material, Table S3). In individual cloned cells, lesions were randomly formed at either the A-allele or G-allele and were approximately 20 ± 8 bp following DNA repair (Fig. 2B). After screening, three clones for each of three conditions were generated: the PD-protective condition that maintains only the protective allele (A/−), the PD-risk condition that maintains only the risk allele (G/−) or the wild-type control condition in which clones show no edits and maintain both rs356182 alleles (A/G). Statistical analysis comparing the length of lesion replicates in the protective and risk conditions indicated no significant difference in the average deletion size (mean lesion 20 bp, G/− vs A/−, Student’s t-test: P = 0.1176 and Wilcox test: P = 0.20). Interestingly, we were unable to generate any clones which had bi-allelic deletions of rs356182. This may be anecdotal or indicate that bi-allelic deletion in rs356182 is lethal to LUHMES cells. Clonally derived hemizygous (single allele) strains and wild-type (both alleles) controls were differentiated, and allele-specific gene expression was examined using RNA sequencing (RNA-seq). As the deletion method provided slightly different-sized lesions, expression results were analyzed for relation to lesion size but showed no correlation. Looking at the gross gene expression profiles of the samples showed that the experimental groups were segregated along principal components (PC) 1 and 2 with the wild-type controls clustering closely with the A/− clones along PC1 (Supplementary Material, Fig. S2). Based on the TF binding motif analysis (Fig. 3A), we would predict the A-allele to be the functional allele, so it is unsurprising that the lost G-allele is ineffective at separating A/− and WT clones. The separation of experimental groups (A/− and G/−) is maintained when we examined Euclidean clustering method of samples for the top 500 most variable genes across the lesion samples and controls (Supplementary Material, Fig. S3) and the top 50 most variable genes across all samples including the SNCA-KO experiment (Supplementary Material, Fig. S4). Again, we observe that the A/− clones cluster more closely with the wild-type controls. Contrary to what one might expect, SNCA expression was increased in the A/− clones and decreased in the G/− (Fig. 3C), note that the G-allele confers PD risk in GWAS analysis. Additionally, we found many and widespread differentially expressed genes, contrary to the expectation that SNCA would be the primary gene target. While SNCA was significantly modulated in both hemizygous conditions compared to WT (Fig. 3C), neither SNCA nor any other genes near rs356182 were the most significant or highest fold-changed genes affected (Fig. 3D). Our results here corroborate previous results which showed a higher SNCA expression in the A/A genotype and lowest expression in the G/G genotype (25). We observed allele-specific differential gene expression spanning the entire genome (Fig. 3E). Ultimately, both conditions showed several hundred genes significantly altered compared to the wild-type controls, including both up- and down-regulated genes with absolute fold-change greater than two (Supplementary Material, Table S4).
CRISPR-mediated lesions at rs356182 modulate gene ontological enrichment of neurodevelopment, synapse function and cell cycle
Based on the specific activation of the rs356182-containing enhancer during the differentiation of midbrain neurons (Fig. 1A) (5), we hypothesized that the elements at this locus are involved in the differentiation process per se. Neuronal differentiation is a complex biological process and determines cell proliferation, morphology, metabolism and communication (among many other processes). We have previously shown that the differentiation of LUHMES cells has a profound impact on gene expression, with thousands of genes significantly modulated between states of differentiation (5). As expected, gene ontological (GO) analysis of those impacted genes showed enrichment in GO terms associated with neuronal differentiation, neurogenesis (genes up-regulated in the differentiated cells compared to undifferentiated cells) and cell cycle (genes down-regulated in the differentiated cells compared to undifferentiated cells) (5). Relative to the extreme comparison of neuronal vs proliferating LUHMES cells, the lesion strains we generated here had more modest effects on differentiated gene expression (Fig. 4 and Supplementary Material, Table S4). Unexpectedly though, our lesion conditions resulted in differential expression for gene sets enriched for several similar neuronal developmental ontological branches (Fig. 4 and Supplementary Material, Table S5). Specifically, genes up-regulated in the A/− clones (protective condition for PD) compared to the A/G clones participate in the positive regulation of neurogenesis, axonogenesis and neuronal differentiation. Meanwhile, genes down-regulated in the G/− clones (risk condition for PD) compared to A/G clones are enriched in terms associated with axonogenesis, neuron projection guidance and synaptic function, while up-regulated genes are enriched in terms associated with proliferation. We validated that the results were from genotype differences between groups and not driven by individual clones acting as outliers by examining the samples separately for the genes enriched in GO terms (Supplementary Material, Fig. S5 using positive regulation of neuron differentiation as an example). Since the A-allele is the predicted functional (PD-protective) allele, we conclude that the A-allele promotes differentiation, synaptic function and neuronal morphogenesis, while cells lacking the A-allele (G/−) maintain a more proliferative gene expression profile.
Figure 4.

CRISPR-mediated lesions at rs356182 modulate the capacity for differentiation. Volcano plot of RNA-seq data depicting the differentially expressed genes between the lesion clones and wild-type clones. The grey dots represent genes that fail to meet minimum thresholds for adjust P-value (<0.05) and/or log2 fold-change (>1). The left side of the volcano plot show genes which were down-regulated and the associated GO terms, and the right side of the volcano plot shows genes which were up-regulated and the associated GO terms. (A) A/− vs A/G and (B) G/− vs A/G.
The rs356182 risk-allele mediates a pathological morphology change
The GO analysis suggests that this locus is modulating the expression of genes involved in the identity shift from proliferating stem-like cells into differentiating neurons. To determine whether those gene expression changes resulted in obvious differences to morphology or proliferation, we differentiated and did immunofluorescent imaging on wild-type and rs356182-edited clones. The cells were stained with DAPI, as a nuclear marker, and treated with anti-TUJ1 (neuron-specific class III beta-tubulin), which marks neuronal soma and neurites, and anti-TH as an indicator of dopamine production (Fig. 5A). The G/− clones had significantly more cells per well on Day 6 of differentiation than their wild-type counterparts (seeded at equal densities on Day 2 of LUHMES cell differentiation) (Fig. 5B). We believe this is due to a differentiation-specific modulation of proliferative activity because edited clones did not show different growth rates in their undifferentiated state (Supplementary Material, Fig. S6). Additionally, the cell density within each well was more variable in the risk clone samples than in the protective clones or wild-type clones (Fig. 5C). These wells more commonly had clusters of densely packed cells and then sections of sparsely populated cells (Fig. 5A, middle). The TUJ1/DAPI stain ratio is also smaller in the risk (G/−) condition indicating reduced neurite growth per cell, in line with results from GO analysis (Fig. 5D). While deeper analysis of the neurite formation would have been preferred, LUHMES cells are very sensitive to seeding density and do not tolerate differentiation in lower densities, making it difficult to achieve non-overlapped, measurable neurites. Finally, the protective clones had a significantly higher ratio of TH/DAPI well coverage (Fig. 5E) and ratio of TH-expressing cells (Supplementary Material, Fig. S7) on Day 6 of differentiation than either the wild-type controls or the risk clones. The percentage of TH-expressing LUHMES cells seen here is comparable to previously published measures of LUHMES TH expression (26). Interestingly, the RNA-seq analysis revealed that TH was not a significant differentially expressed gene between lesion groups (Supplementary Material, Fig. S8).
Figure 5.

The rs356182 risk-allele mediates a pathological morphology change. (A) Representative immunofluorescent images from wild-type A/G clones (left), risk G/− clones (middle) and protective A/− clones (right); white bar = 50 μm, blue = DAPI, red = TUJ1 and green = TH. (B–E) Quantification of immunofluorescent images by pixel coverage represented in box-and-whisker plots; Wilcoxon comparisons of non-parametric groups, ns: P > 0.05, *: P < =0.05, **: P < =0.01. ***: P < =0.001, ****: P < =0.0001. (B) DAPI coverage per image as a measure of cell density. (C) Standard deviation of inter-well DAPI coverage. (D) Ratio of TUJ1/DAPI as a relative measure of cell body to nucleus. (E) Ratio of TH/DAPI as a normalized measure of TH-expressing cells within the cell population.
The gene expression changes associated with rs356182 are largely independent of SNCA
We have recently published the effects of knocking-out the SNCA gene using the same LUHMES model and similar CRISPR-Cas9 editing techniques (27). Our results provided insight into the normal functions of SNCA/α-SYN in developing midbrain neurons, showing particularly strong regulation of the cell cycle in this model. Knowing that these projects would overlap, we included lesion samples in the differentiation and RNA-seq published previously (28). This allowed us to leverage that data and serve as a control across multiple differentiations. The overlapped samples were included in this analysis and confirmed that the differences we see between groups are due to rs356182-associated changes and not inherent variability between differentiations, as evident by the close clustering of analogous samples on the heatmap (Supplementary Material, Figs S3, S4 and S8). To determine the extent to which SNCA modulation is responsible for the rs356182-associated phenotype observed here, we compared our rs356182-lesion RNA-seq results to the SNCA-KO RNA-seq results (Fig. 6A). Since SNCA was reduced in both the SNCA-KO clones and the G/− clones, we examined the relationship between those differential gene expression profiles and found no overlap in the up-regulated genes in the G/− clones with the SNCA-KO clones. There were 402 genes with decreased expression following KO of SNCA but only 44 genes overlapped with the 591 genes in the G/− down-regulated gene group, representing approximately 7.4% of the total genes in that group (Fig. 6B). If we remove those 44 overlapped genes from the G/− down-regulated gene list and re-examine the GO results, we find that the GO terms are largely conserved (Supplementary Material, Table S8). To further determine if SNCA could account for the changes observed in the G/− clones we examined the fold-change relationship for all genes between the G/− clones or the SNCA-KO clones. We found that there was no correlation between the fold-change of genes between models (Supplementary Material, Fig. S9). The results show that knockout of SNCA was insufficient to reproduce the phenotype from the G/− clones, indicating that cumulative rs356182-driven gene modulation is independent of its regulation of SNCA.
Figure 6.

Gene expression changes associated with rs356182 are largely independent of SNCA. (A) An intersection plot depicting the size of the subset of genes in each group (right-horizntal bar graph) and the degree to which they overlap with each other group (vertical bars). Gene groups separated by condition (G/−, A/− and SNCA-KO) and direction of expression changes (up or down). Intersections were highlighted in red where the genes were decresased in both SNCA-KO and G/−, representing the intersection highlighted in panel B. (B) Euler diagram showing the relative proportion of genes down-regulated in the two conditions where SNCA was inhibited; SNCA-KO and G/−.
Discussion
In this study, we present evidence regarding the mechanism(s) surrounding rs356182 that contradicts the assumption that SNCA is the only rs356182-interacting gene at this locus. The generally accepted assumption is that rs356182 directly regulates the expression of only SNCA, and in this way confers risk for PD. However, we show here by the mono-allelic deletion of rs356182 that hundreds of genes, spanning the entire genome, are affected by rs356182. It remains unclear which of these downstream gene changes are primary, secondary or tertiary events. Furthermore, the affected genes appear to be enriched in GO terms related to the differentiation/proliferation processes (explored in detail below). Comparison of the RNA-seq results from this study to SNCA-knockout data we previously published suggests that rs356182 does not exclusively regulate SNCA and is independently relevant to midbrain neuron physiology via the regulation of many genes.
We observed a neuronal phenotype which was seemingly more differentiated in the PD-protective A/− clones than the risk-associated G/− clones. This was evident by the higher proportion of TH-stained cells in the A/− clones (Fig. 5E and Supplementary Material, Fig. S7) and the decreased beta-tubulin expression per cell in the risk clones (Fig. 5D). The G/− clones produced more cells per well and thus seem more proliferative than A/− and wild-type cells (Fig. 5B). The phenotype changes in the A/− and G/− clones were accompanied by robust gene expression changes. The A/− clones showed increased expression of genes enriched in the GO terms which promote neurogenesis, differentiation, axonogenesis and synapse physiology (Fig. 4A). The G/− clones promoted stem cell proliferation and cell cycle while repressing synapse function and axonogenesis. Finally, the rs356182-encompassing enhancer only became active during differentiation in LUHMES cells (Fig. 1A) and is preferentially active in the presence of the A-allele (Fig. 3B). All these indicate that rs356182 is regulating the activity of the enhancer which in turn regulates the identity shift of LUHMES cells from stem-like proliferative cells into terminally differentiated midbrain neurons. Based on the above observations, coupled with the neurodevelopmental stage of the cell line, we speculate that rs356182 confers risk for PD during neurogenesis by regulating enhancer activation to promote the robust differentiation of midbrain neurons. One caveat to this proposed mechanism is the fact that neurons rely heavily on signaling feedback for complete differentiation. We found that low seeding density resulted in aberrant cell detachment, while others have shown that overcrowding of neuronal stem cell inhibits differentiation (29). As a differentiation-specific gene, SNCA expression would also be indirectly affected by seeding density. Any manipulation which affects the cell growth density (e.g. promotion of proliferative functions in the G/− clones) would indirectly reflect on the differentiation processes. Whichever primary signaling path is directly affected, the result is a more proliferative phenotype in the absence of the functional A-allele.
The most anticipated interaction to examine in this study was the relationship between rs356182 and SNCA expression. Ultimately, we determined that the effects attributed to rs356182 genotype are largely independent of SNCA expression, but we confirmed that SNCA is modulated by this SNP/enhancer (Fig. 6). Due to the known links between SNCA expression and PD, one might expect the risk allele to be associated with an increase in SNCA mRNA; however, we observed the opposite effect in our model (Fig. 3C). This finding is corroborated by brain tissue-specific eQTL data on the Genotype-Tissue Expression project (GTEX) website (https://www.gtexportal.org/home/snp/rs356182), and is mirrored by findings by Cooper et al. (25), which likewise demonstrated that the rs356182 AA-genotype was associated with highest expression of SNCA. There are two things to consider here. First, we concluded that SNCA modulation is not the exclusive risk mechanism of this locus, and while it is interesting that the A-allele would promote SNCA expression, it is perhaps less influential to disease pathogenesis than the cumulative, wide-spread, effects on gene expression. It has even been suggested that α-syn is merely a bystander in the etiology of PD (30), corroborated by the knowledge that alterations of SNCA gene expression is rare in most cases of familial PD (31). Second, there is growing evidence that pathological over-expression of α-syn and the consequential aggregation contribute to inhibition of normal α-syn activity and therefore result in a loss-of-function mechanism (32,33). Although it is associated with PD, α-syn is a normally expressed and functional protein in several tissues including midbrain neurons. In this context, it makes sense that the protective allele would promote normal expression of SNCA and maintain regular function, while the risk allele is associated with lower SNCA expression. Finally, we must consider the origin of LUHMES cells as they were derived from 8-week-old fetal tissue, making it a model for developing midbrain neurons and not mature dopaminergic neurons (19,34). During this gestational time point early neurogenesis has begun and the network of genetic regulatory elements are specific to this developmental stage (20,21). Given the results described presently, it is likely that rs356182 confers risk, at least in part, during embryonic neurodevelopment and in a manner only minimally attributed to SNCA, contributing to one’s propensity to develop PD later in life.
Pihlstrøm et al. (13) concluded that there are at least three independent GWAS risk signals at the SNCA locus. While it might seem that the presence of these independent signals (rs287004 and rs763443) reinforces the idea that SNCA is the primary target for these risk mechanisms, what is true of rs356182 is also true of these SNPS. Specifically, that their proximity to SNCA has shielded them from investigation for their role in regulating other nearby genes. In fact, a deeper look at the tissue-specific eQTL data from GTEX reveals that neither SNP has a significant correlation to SNCA expression in any brain region except for rs763443 in the cerebellar hemisphere (P = 0.02). However, MMRN1 expression is significantly correlated to the rs2870004 genotype in several brain regions, reinforcing our conclusion that interactions peripheral to SNCA may be the primary targets for risk signals stemming from this locus.
The results from the motif analysis of rs356182 (Supplementary Material, Table S1 and Table S2) imply that the A-allele is the primary functional allele and that the FOX family of proteins are the primary TFs binding to the A-containing motif. Fox proteins regulate the expression of genes involved in branching morphogenesis, brain development, axon guidance and maintaining stem-cell pluripotency, among a host of other biological processes (35,36). The FOXA1 motif is known to be particularly vulnerable to variants that disrupt the three consecutive adenines, which is true of rs356182 (Fig. 3A), and was previously shown to result in pathological consequences (37,38). The FOXO family has important roles in cell cycle, apoptosis, oxidative stress and differentiation (39). FOXO3 is pivotal in maintaining hematopoietic stem cell populations (40) and neural stem cell populations (41). Additionally, the FOX family of proteins are known to engage in pioneer factor activity (42). Pioneer factors serve as the first protein to bind to chromatin, allowing it to unwind and exposing enhancers for activation. Since FOX proteins have pioneer TF abilities, they are likely partially or wholly responsible for activation of the encompassing enhancer, resulting in the regulation of many genes by both primary- and secondary-/tertiary-interactions. As the G-allele was predicted and demonstrated to have less efficient binding of the FOXO3 protein, the enhancer would predictably be less active in its presence. This is indeed what was observed, as the allele ratio heavily favored the A-allele in the ChIP-qPCR for H3K27ac (Fig. 3B).
Based on the mechanism proposed above, deletion of the G-allele should have few, or no, consequences on gene expression, but this is not what we observed. In fact, deletion of the G-allele in the PD-protective clones promoted neuronal differentiation and neurogenesis (Fig. 4). The fact that losing one allele increases the expression of certain genes and processes indicates that, contrary to what we predicted from binding-motif analysis, the G-allele is not simply a less functional allele but is counteractively working against the A-allele. Further evidence that this is not a simple dose–response is that ~ 21% of the nearly 25 000 genes in our dataset have opposite expression changes between experimental conditions (i.e. one edited condition increases while the other edited condition decreases in expression compared to the wild-type), an example of which is SNCA itself (Fig. 3C). Additionally, morphological analysis of body-nucleus ratio (Fig. 5D) and the ratio of TH-expressing cells per condition (Fig. 5E and Supplementary Material, Fig. S4) showed that the A/− clones produce more TH-positive cells in culture and more TUJ1 per cell. In other words, the A- and G-alleles impinge on the differentiation–proliferation mechanism by actively working against each other; the A-allele driving the cell towards differentiation and the G-allele holding it back in a more stem-like state. The dual functionality of the rs356182 alleles could explain why this locus has such a strong association with PD.
The PD field has known about the significant association of rs356182 to PD since the earliest GWASs due to its strong association with PD risk. Until now, the mechanism surrounding this risk was attributed to allele-specific expression of SNCA, variation of which would presumably predispose individuals to PD in their later years. However, we have identified a novel mechanism in which rs356182 impinges on the differentiation/proliferation mechanism during development, independently from what could be attributed to SNCA, by regulating enhancer activity through FOXO3 recruitment. These results contribute to the growing body of evidence that PD is, at least in part, a developmental disorder (43,44). We speculate that rs356182 regulates neuronal differentiation and a cascade of related processes, leading to a diminished population of healthy DA midbrain neurons in individuals with the risk allele, making the individual more sensitive to subsequent insults to the DA cell population, and in this way confers risk for PD later in life (45).
Materials and Methods
LUHMES cell model
LUHMES cells, obtained from ATCC (CRL-2927), were cultured essentially as described by Scholz et al. (46). Also, as previously described (5), the cells were incubated in a humidified 37°C, 5% CO2 incubator in flasks pre-coated with 50 mg/mL poly-L-ornithine (Sigma, Cat. # P3655) and 1 mg/mL fibronectin (Sigma, Cat. # F114) in water. The coated flasks were incubated at 37°C overnight, rinsed with water and allowed to dry before seeding cells. Cells were cultured in complete growth medium containing Advanced DMEM:F12 (Thermo Fisher, Cat. # 12634-010) with 2 mm l-glutamine (Thermo Fisher, Cat. # 25030081), 1X N-2 supplement (Thermo Fisher, Cat. # 17502-048) and 0.04 mg/mL bFGF (Stemgent, Cat. # 03-0002). Cells were allowed to reach 80% confluency before passaging with 0.025% trypsin/EDTA. Before differentiation, cells were seeded at 3.5 × 106 per T75 flask containing complete growth medium and incubated at 37°C for 24 h (Day 1). For induction of differentiation, culture medium was changed to freshly prepared DMEM:F12 with 2 mm l-glutamine, 1X N-2, 1 mm cAMP (Carbosynth, Cat. # ND07996), 1 mg/mL tetracycline (Sigma, Cat. # T7660) and 2 ng/mL glial cell line-derived neurotrophic factor (Sigma, Cat. # G1777) (Day 0). LUHMES cells then grow in the differentiation media for 2 days before being passaged, again into differentiation media (Day 2). Finally, cells were harvested on Day 6 after the initial introduction of differentiation media (Day 6).
CRISPR-Cas9 editing
We chose to create hemizygous clones for interrogation of rs356182. As previously stated, LUHMES cells are heterozygous for rs356182 (genotype A/G) meaning that we may investigate each allelic condition separately from each other with minimally invasive editing techniques. Although we attempted to generate bi-allelic rs356182 lesions, we were never able to achieve this condition, indicating either a lethal phenotype or simply an interesting anecdote. This, coupled with the low editing efficiency of homology-dependent repair, made hemizygous clones the next best option.
For each target sequence, double-stranded DNA sequences complementary to the target sequences were generated by PCR and then cloned into the pSpCas9(BB)-2A-GFP (PX458) vector (AddGene, Cat. # 48138) (Supplementary Material, Table S3). Ligated plasmids were delivered into Stbl3 chemically competent Escherichia coli cells and selected from ampicillin-treated agar plates. Transformed E. coli colonies were isolated and expanded in LB media. Expanded plasmids were purified using the QIAprep Spin MiniPrep Kit (Qiagen, Cat. # 27106). LUHMES cells were electroporated and transfected with 2 μg of plasmid using the Amaxa™ Basic Primary Neurons Nucleofector™ Kit and protocol (Lonza, Cat. # VPI-1003). Transfected cells were selected by flow-sorting for DAPI negative, GFP positive singlets into pre-coated 96-well plates (Supplementary Material, Fig. S10). Sorted cells were clonally expanded and screened via Sanger DNA sequencing from Genewiz. Individual allele sequencing was achieved by TOPO-TA cloning (Thermo Fisher, Cat. # K4575J10), followed again by Sanger sequencing. Clones with a confirmed edit disrupting rs356182 were used in the edited conditions, while sorted clones which did not have a confirmed edit at rs356182 were used as the A/G lesion controls (Fig. 2B). In total, nine individually edited, sorted and clonally expanded cell lines were selected for further analysis; three from each of the protective A/− genotype, the risk G/− genotype and unedited wild-type A/G clones.
As a control for the CRISPR-Cas9 technique, we performed CRISPR-Cas9 editing of two additional regions of the genome deemed to be transcriptionally silent in LUHMES cells on chromosome 4 and chromosome 17 (i.e. absent of known genes, promoters or enhancers). In total eight LUHMES clones were expanded (five with confirmed edits of 500–600 bp, and three exposed to CRISPR-Cas9 and cloned but without a confirmed edit). These CRISPR-edited control clones were RNA-sequenced and found to have no significantly modified genes reaching a Benjamini–Hochberg adjusted significance threshold of P < 0.05. While this adds confidence in our results from the other CRISPR-edits we performed, we searched for additional insight by examining a less stringent set of genes (unadjusted P < 0.05). The 17 genes which passed the less-stringent test were analyzed for GO enrichment and determined to be enriched in terms non-specific to neurons or neuronal functions and primarily involved in angiogenesis (Supplementary Material, Table S6).
RNA sequencing
Three individual clones of each of the G/− lesion model and A/− lesion model and parent (wild-type) A/G clones were selected for RNA-seq analysis. Clones were differentiated following standard LUHMES cell differentiation protocol. Clonal RNA was isolated and purified using the QIAGEN QIAshredder (Cat. # 79654) and RNeasy isolation kit (Cat. # 74104). Total RNA was submitted to the Van Andel Research Institute’s Genomics Core for QC, library preparation and paired-end sequencing using the Illumina NovaSeq 6000 in split-lane SP Flowcells. Sequencing results were saved as zipped FASTQ files.
Differential gene expression
Three different datasets were interpreted in this study: the rs356182 lesion samples generated here, the previously published SNCA-knockout samples (27) and the previously published LUHMES wild-type samples as controls (5). These datasets were generated from three separate experiments but their FASTQ files were re-processed for this study. First, Illumina sequencing adapters were trimmed from the raw count files using the TrimGalore software package (version-0.6.0). Next, trimmed reads were aligned to the GRCh37-hg19 reference genome using the STAR software (Spliced Transcripts Aligned to a Reference; version-2.7.8a). After the alignment, sub-threshold count reads were filtered out and raw counts were normalized to count per million and trimmed mean of M values using EdgeR (version-3.32.1). Samples were submitted with a minimum biological triplicate; therefore, genes expressed in fewer than three samples were excluded. A z-score was determined for each gene per sample and fold-change and adjusted P-values were calculated using the limma-voom method to compare genotype groups (version-limma-3.46.0. Detailed scripts on GitHub: https://github.com/jordanprahl/Prahl_et_al_20210629/tree/main. The wild-type LUHMES control cells were used to establish the normal range of variance for each gene within LUHMES cells but were not directly compared to the rs356182-lesion samples or the SNCA-KO samples. It would be inappropriate to directly compare the raw data from the two different experimental groups since these were done at separate times and in separate RNA-seq runs. Rather than directly comparing read counts from the two different experiments, each experimental group was instead analyzed in comparison to their own controls and then later the lists of differentially expressed genes from each experiment were compared.
GO analysis
GO for biological processes was performed using ClusterProfiler (version-3.18.1). The background list of genes in the universe was described as the full list of genes expressed in our LUHMES models. In each case, an adjusted P-value < 0.05 was the maximum threshold for modulated genes. Gene sets were separated by condition and increased/decreased gene expression. An absolute fold-change greater than 2 was used as the minimum threshold for modulated genes. To reduce redundancy of ontological terms and the bias introduced by over-represented genes, terms that have greater than 70% of shared gene annotations were collapsed into one group.
Morphology
For morphological analysis, clones were differentiated following the standard LUHMES cell differentiation protocol. On Day 2 of differentiation, clones were passaged to the standard density of 1.5 × 105 cells/cm2 into pre-coated 24-well plates. Each clone occupies one well of each plate, and four separate differentiations were performed in one plate each. On Day 6 of differentiation, clones are fixed by incubating in 2% formaldehyde solution for 5 min, followed by incubation in 4% formaldehyde solution for 15 min, then stored in DPBS at 4°C for up to 2 weeks. Immunofluorescence was conducted as described by the Invitrogen Human Dopaminergic Neuron Immunocytochemistry Kit (Cat. # A29515). In brief, cells were incubated in perm/block buffer (1% BSA, 0.3% Triton X-100 and DPBS) for 30 min at room temperature (RT). Primary antibody was added directly to the perm/block buffer in the wells and incubated overnight at 4°C. Wells were washed 3× in DPBS for 2 min at RT, and then secondary Ab was added to the cells for 1 h at RT (Supplementary Material, Table S9). Wells were again washed 3× in DPBS. On the third wash, 1–2 drops/ml of DAPI was added to the wash buffer. Cells are then stored in DPBS for < 2 weeks.
After immunohistochemical and DAPI staining, samples were imaged on the Zeiss Celldiscover7 (CD7) at 10× magnification (20 Z-stacks) by the VARI Optical Imaging Core. Composite images were prepared using the ZEN 3.2 (blue edition) software by using automated threshold settings for the DAPI and TUJ1 channels and manually setting the TH channel and Z-stack. The TH channel must be manually set due to the skewed intensity distribution disrupting the automated threshold settings. Split fluorescent channels were converted to binary images in ImageJ and analyzed for pixel coverage and cell count. To account for variability within wells, full images were split into 5 × 5 grids, and five blocks were selected with a random number generator for analysis, each block representing a data point in Fig. 5B, D and E. The inter-well variability of cell density represented in Fig. 5C is a calculation of standard deviation of the selected image blocks. All clones of each genotype were grouped in the analysis. Measurement of cell density was calculated as the percent of the well covered by DAPI stain (binary images: black pixels/total pixels). Neurite development was calculated as the ratio of cell body stain (TUJ1) and nuclei stain (DAPI) (binary images: black pixels/total pixels, red channel/blue channel).
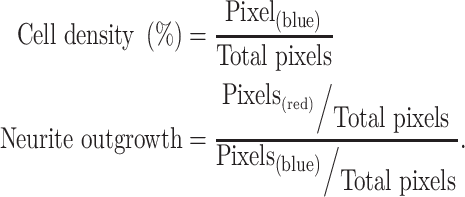 |
ChIP-qPCR
Candidate TFs were selected based on the lists generated by HaploReg and MotifBreakR, the scores associated with each allele and the availability of antibodies specific to each TF. Ultimately, three TFs were selected for further analysis from the FOXO family of proteins: FOXO1, FOXO3 and FOXO4. Positive binding was only observed with FOXO3 (Fig. 3B).
The ChIP was performed essentially as described in the Farnham protocol, and as previously published (5,47,48). Briefly, 1.0 × 107 cultured LUHMES cells were fixed in 1% formaldehyde. Cells were then quenched in glycine, lysed and sonicated until chromatin fragments were approximately 500 bp. For immunoprecipitation, 20–100 μg of chromatin were aliquoted per assay and incubated overnight with each antibody (Supplementary Material, Table S10). The antibody–chromatin complexes were conjugated to magnetic beads and washed in a series of salt buffers before purifying the DNA using the QIAquick PCR Purification kit (Cat. # 28104).
To determine the allele-specific activity we used the TaqMan SNP Genotyping assay for rs356182 (Cat. # 4351379), in the same way described by Soldner et al. in 2016 (6). Briefly, the assay contains a dual fluorophore system with each tag (VIC and FAM) specific to each allele. The qPCR is set up to measure the amplification of each fluorophore separately and then we measure the delta between fluorophore amplification curves using the input control as the standard (Supplementary Material, Fig. S1).
Author contributions
CRediT Taxonomy: J.P., S.E.P., G.A.C. and T.T. did the conceptualization; J.P. and T.T. contributed to the methodology; J.P. and S.E.P. developed the software; J.P. and S.E.P. performed the formal analysis; J.P. did the investigation; J.P., E.J.C.V. and T.T. collected the resources; J.P. made the writing—original draft; J.P., T.T., S.E.P. and G.A.C. made the writing—review and editing; J.P. and S.E.P. did visualization; G.A.C. and T.T. made supervision and G.A.C contributed to the funding acquisition.
Abbreviations
- α-syn
-
alpha-synuclein
CRISPR
clustered regularly interspaced short palindromic repeats
DA
dopaminergic
GTEX
Genotype-Tissue Expression project
GWAS
genome wide association studies
KO
knock-out
LUHMES
Lund human mesencephalic cells
PD
Parkinson’s disease
SNCA
alpha-synuclein gene symbol
SNP
single nucleotide polymorphism
TF
transcription factor
TH
tyrosine hydroxylase
Supplementary Material
Acknowledgements
This research was supported in part by the Van Andel Research Institute (Grand Rapids, MI) Bioinformatics and Biostatistics Core, Flow Cytometry Core, Genomics Core and Optical Imaging Core.
Conflict of Interest statement. The authors have no competing interests to declare.
Contributor Information
Jordan D Prahl, Department of Neurodegenerative Research, Van Andel Institute, 333 Bostwick Ave NE, Grand Rapids MI 49503, USA.
Steven E Pierce, Department of Neurodegenerative Research, Van Andel Institute, 333 Bostwick Ave NE, Grand Rapids MI 49503, USA.
Edwin J C van der Schans, Department of Neurodegenerative Research, Van Andel Institute, 333 Bostwick Ave NE, Grand Rapids MI 49503, USA.
Gerhard A Coetzee, Department of Neurodegenerative Research, Van Andel Institute, 333 Bostwick Ave NE, Grand Rapids MI 49503, USA.
Trevor Tyson, Department of Neurodegenerative Research, Van Andel Institute, 333 Bostwick Ave NE, Grand Rapids MI 49503, USA.
Funding
Funding for this project provided by the Van Andel Institute and Van Andel Institute Graduate School.
References
- 1. Genomes Project, C, Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Consortium, E.P (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey, K.R. and Cheng, C. (2010) Conference scene: the great debate: genome-wide association studies in pharmacogenetics research, good or bad? Pharmacogenetics, 11, 305–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ioannidis, J.P. (2007) Non-replication and inconsistency in the genome-wide association setting. Hum. Hered., 64, 203–213. [DOI] [PubMed] [Google Scholar]
- 5. Pierce, S.E., Tyson, T., Booms, A., Prahl, J. and Coetzee, G.A. (2018) Parkinson's disease genetic risk in a midbrain neuronal cell line. Neurobiol. Dis., 114, 53–64. [DOI] [PubMed] [Google Scholar]
- 6. Soldner, F., Stelzer, Y., Shivalila, C.S., Abraham, B.J., Latourelle, J.C., Barrasa, M.I., Goldmann, J., Myers, R.H., Young, R.A. and Jaenisch, R. (2016) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature, 533, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Heyningen, V. and Yeyati, P.L. (2004) Mechanisms of non-Mendelian inheritance in genetic disease. Hum. Mol. Genet., 13 Spec No 2, R225–R233. [DOI] [PubMed] [Google Scholar]
- 8. Schaffer, A.A. (2013) Digenic inheritance in medical genetics. J. Med. Genet., 50, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Slatkin, M. (2008) Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet., 9, 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smemo, S., Tena, J.J., Kim, K.H., Gamazon, E.R., Sakabe, N.J., Gomez-Marin, C., Aneas, I., Credidio, F.L., Sobreira, D.R., Wasserman, N.F. et al. (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature, 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nalls, M.A., Blauwendraat, C., Vallerga, C.L., Heilbron, K., Bandres-Ciga, S., Chang, D., Tan, M., Kia, D.A., Noyce, A.J., Xue, A. et al. (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol., 18, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nalls, M.A., Pankratz, N., Lill, C.M., Do, C.B., Hernandez, D.G., Saad, M., DeStefano, A.L., Kara, E., Bras, J., Sharma, M. et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet., 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pihlstrom, L., Blauwendraat, C., Cappelletti, C., Berge-Seidl, V., Langmyhr, M., Henriksen, S.P., van de Berg, W.D.J., Gibbs, J.R., Cookson, M.R., International Parkinson Disease Genomics, C et al. (2018) A comprehensive analysis of SNCA-related genetic risk in sporadic parkinson disease. Ann. Neurol., 84, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nott, A., Holtman, I.R., Coufal, N.G., Schlachetzki, J.C.M., Yu, M., Hu, R., Han, C.Z., Pena, M., Xiao, J., Wu, Y. et al. (2019) Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science, 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gusev, A., Ko, A., Shi, H., Bhatia, G., Chung, W., Penninx, B.W., Jansen, R., de Geus, E.J., Boomsma, D.I., Wright, F.A. et al. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet., 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu, Z., Zhang, F., Hu, H., Bakshi, A., Robinson, M.R., Powell, J.E., Montgomery, G.W., Goddard, M.E., Wray, N.R., Visscher, P.M. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
- 17. Chepelev, I., Wei, G., Wangsa, D., Tang, Q. and Zhao, K. (2012) Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res., 22, 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ernst, J. (2012) Mapping enhancer and promoter interactions. Cell Res., 22, 789–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lotharius, J., Barg, S., Wiekop, P., Lundberg, C., Raymon, H.K. and Brundin, P. (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem., 277, 38884–38894. [DOI] [PubMed] [Google Scholar]
- 20. Sarkar, A., Balogun, K., Guzman Lenis, M.S., Acosta, S., Mount, H.T. and Serghides, L. (2020) In utero exposure to protease inhibitor-based antiretroviral regimens delays growth and developmental milestones in mice. PLoS One, 15, e0242513. 10.1371/journal.pone.0242513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Silbereis, J.C., Pochareddy, S., Zhu, Y., Li, M. and Sestan, N. (2016) The Cellular and molecular landscapes of the developing human central nervous system. Neuron, 89, 248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kheradpour, P. and Kellis, M. (2014) Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res., 42, 2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ward, L.D. and Kellis, M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coetzee, S.G., Coetzee, G.A. and Hazelett, D.J. (2015) motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics, 31, 3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cooper, C.A., Jain, N., Gallagher, M.D., Weintraub, D., Xie, S.X., Berlyand, Y., Espay, A.J., Quinn, J., Edwards, K.L., Montine, T. et al. (2017) Common variant rs356182 near SNCA defines a Parkinson's disease endophenotype. Ann. Clin. Transl. Neurol., 4, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harischandra, D.S., Rokad, D., Ghaisas, S., Verma, S., Robertson, A., Jin, H., Anantharam, V., Kanthasamy, A. and Kanthasamy, A.G. (2020) Enhanced differentiation of human dopaminergic neuronal cell model for preclinical translational research in Parkinson's disease. Biochim. Biophys. Acta Mol. basis Dis., 1866, 165533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prahl, J., Pierce, S.E., Coetzee, G.A. and Tyson, T. (2022) Alpha-synuclein negatively controls cell proliferation in dopaminergic neurons. Mol. Cell. Neurosci., 119, 103702. [DOI] [PubMed] [Google Scholar]
- 28. Prahl, J., Pierce, S.E., van der Schans, E.J., Coetzee, G.A. and Tyson, T. (2021) Global effects of a PD risk-SNP at the alpha-synuclein locus. bioRxiv, 2021.2007.2006.451330.
- 29. Saxena, S., Choudhury, S. and Mohan, K.N. (2020) Reproducible differentiation and characterization of neurons from mouse embryonic stem cells. MethodsX, 7, 101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riederer, P., Berg, D., Casadei, N., Cheng, F., Classen, J., Dresel, C., Jost, W., Kruger, R., Muller, T., Reichmann, H. et al. (2019) alpha-Synuclein in Parkinson's disease: causal or bystander? J. Neural Transm. (Vienna), 126, 815–840. [DOI] [PubMed] [Google Scholar]
- 31. Gispert, S., Trenkwalder, C., Mota-Vieira, L., Kostic, V. and Auburger, G. (2005) Failure to find alpha-synuclein gene dosage changes in 190 patients with familial Parkinson disease. Arch. Neurol., 62, 96–98. [DOI] [PubMed] [Google Scholar]
- 32. Benskey, M.J., Perez, R.G. and Manfredsson, F.P. (2016) The contribution of alpha synuclein to neuronal survival and function—implications for Parkinson's disease. J. Neurochem., 137, 331–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kanaan, N.M. and Manfredsson, F.P. (2012) Loss of functional alpha-synuclein: a toxic event in Parkinson's disease? J. Parkinsons Dis., 2, 249–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paul, G., Christophersen, N.S., Raymon, H., Kiaer, C., Smith, R. and Brundin, P. (2007) Tyrosine hydroxylase expression is unstable in a human immortalized mesencephalic cell line—studies in vitro and after intracerebral grafting in vivo. Mol. Cell. Neurosci., 34, 390–399. [DOI] [PubMed] [Google Scholar]
- 35. Tuteja, G. and Kaestner, K.H. (2007) SnapShot: forkhead transcription factors I. Cell, 130, 1160. [DOI] [PubMed] [Google Scholar]
- 36. Zhang, X., Yalcin, S., Lee, D.-F., Yeh, T.-Y.J., Lee, S.-M., Su, J., Mungamuri, S.K., Rimmelé, P., Kennedy, M. and Sellers, R. (2011) FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol., 13, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jia, L., Landan, G., Pomerantz, M., Jaschek, R., Herman, P., Reich, D., Yan, C., Khalid, O., Kantoff, P., Oh, W. et al. (2009) Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Gen., 5, e1000597. 10.1371/journal.pgen.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hazelett, D.J., Coetzee, S.G. and Coetzee, G.A. (2013) A rare variant, which destroys a FoxA1 site at 8q24, is associated with prostate cancer risk. Cell Cycle, 12, 379–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salih, D.A. and Brunet, A. (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol., 20, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miyamoto, K., Araki, K.Y., Naka, K., Arai, F., Takubo, K., Yamazaki, S., Matsuoka, S., Miyamoto, T., Ito, K., Ohmura, M. et al. (2007) Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell, 1, 101–112. [DOI] [PubMed] [Google Scholar]
- 41. Renault, V.M., Rafalski, V.A., Morgan, A.A., Salih, D.A., Brett, J.O., Webb, A.E., Villeda, S.A., Thekkat, P.U., Guillerey, C., Denko, N.C. et al. (2009) FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell, 5, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zaret, K.S. and Carroll, J.S. (2011) Pioneer transcription factors: establishing competence for gene expression. Genes Dev., 25, 2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barlow, B., Coryslechta, D., Richfield, E. and Thiruchelvam, M. (2007) The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod. Toxicol., 23, 457–470. [DOI] [PubMed] [Google Scholar]
- 44. Schwamborn, J.C. (2018) Is Parkinson's disease a neurodevelopmental disorder and will brain organoids help us to understand it? Stem Cells Dev., 27, 968–975. [DOI] [PubMed] [Google Scholar]
- 45. von Linstow, C.U., DeLano-Taylor, M., Kordower, J.H. and Brundin, P. (2020) Does developmental variability in the number of midbrain dopamine neurons affect individual risk for sporadic Parkinson's disease? J. Parkinsons Dis., 10, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scholz, D., Poltl, D., Genewsky, A., Weng, M., Waldmann, T., Schildknecht, S. and Leist, M. (2011) Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem., 119, 957–971. [DOI] [PubMed] [Google Scholar]
- 47. Boyd, K.E. and Farnham, P.J. (1997) Myc versus USF: discrimination at the cad gene is determined by core promoter elements. Mol. Cell. Biol., 17, 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rhie, S.K., Hazelett, D.J., Coetzee, S.G., Yan, C., Noushmehr, H. and Coetzee, G.A. (2014) Nucleosome positioning and histone modifications define relationships between regulatory elements and nearby gene expression in breast epithelial cells. BMC Genomics, 15, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.