

Abstract

Hypervalent chloranes are a class of rare and poorly explored reagents. Their unique electronic properties confer reactivity that is complementary to that of the common iodanes and emerging bromanes. Highly chemo- and regioselective, metal-free, and mild C–C and C–O couplings are reported here. Experimental and computational mechanistic studies elucidate the unprecedented reactivities and selectivities of these systems and the intermediacy of aryne intermediates. The synthetic potential of these transformations is further demonstrated via the post-functionalization of C–C and C–O coupling products obtained from reactions of chloranes with phenols under different conditions.
Introduction
Hypervalent compounds are nowadays fundamental tools in organic chemistry,1,2 both in academia and industry, serving as oxidants, electrophiles, and radical initiators.3−7 Widely investigated, the λ3-iodanes represent the most common hypervalent reagents. On the contrary, their isoelectronic bromine8−11 and chlorine12−14 congeners have remained unexplored for decades as a consequence of a lack of a general and efficient synthesis.15 We recently reported a safe and practical method for the preparation of cyclic diaryl λ3-bromanes.16 The enhanced nucleofugality and the strong electron-withdrawing property of λ3-bromanes arising from the higher ionization potential of Br confer to these compounds a unique reactivity (Figure 1a). Therefore, cyclic diaryl bromanes serve as original aryne precursors (Figure 1b)16,17 and halogen-bond organo-catalysts.18,19 In clear contrast, cyclic diaryl iodines undergo transition metal-catalyzed ortho-functionalizations,20 clearly illustrating the differences between the cyclic diaryl λ3-bromanes and the corresponding iodanes.
Figure 1.

(a) Electronic properties of iodine, bromine, and chlorine; (b) reactivity of cyclic diaryl λ3-iodonium and bromonium salts; (c) common C–O and C–C couplings of phenols; (d) selected examples of biologically active terphenyls; (e) this work.
Considering the higher ionization potential and electronegativity of Cl, translating into a decreased positive charge in hypervalent λ3-chloranes, an amplified reactivity of diaryl λ3-chloranes as aryne precursors could be anticipated.12,21 Such a superior reactivity of Cl(III)-compounds, in comparison with Br(III)-species, is expected to translate into unprecedented reactivity under metal-free conditions. The challenging synthesis of such compounds remains, however, a major impediment in uncovering this reactivity. A general synthetic route furnishing such compounds followed by a demonstration of their chemical behavior would thus open new vistas in the chemistry of emerging hypervalent compounds.
Due to the prevalence of the phenol motif in medicinal chemistry, agrochemistry, and natural products,22−24 the development of site-selective and transition metal-free methodologies for their late-stage functionalization is highly desirable.25 In particular, cross-dehydrogenative coupling (CDC)-type reactions, rapidly expanding molecular complexity on the phenol scaffolds, have attracted considerable attention over the last years (Figure 1c).26,27 Various approaches toward O-arylation via cross-couplings of phenols28 have been disclosed, including C–O coupling between phenols and arynes,29−31 and direct arylation with diaryl iodonium salts.5 In clear contrast, ortho-C-functionalization of phenols via cross-couplings is less developed and mainly limited to electrochemical transformations,32 oxidative couplings, and metal-catalyzed reactions.27 On the contrary, the direct C-arylation of phenols with arynes under transition metal-free and mild conditions remains unprecedented.33,34 Consequently, considering the expected unusual reactivity of λ3-chloranes, combined with the interest in designing chemoselective C–O vs C–C cross-couplings of phenols and the broad diversity of terphenyl compounds in natural and biologically active products (Figure 1d),35 we have embarked on exploring the reactivity of chloranes with phenols.
We report herein: (1) the synthesis and characterization of a large panel of cyclic diaryl λ3-chloranes; (2) their amplified reactivity as aryne precursors as illustrated in metal-free chemoselective C–O couplings; (3) previously unachievable site-selective, metal-free C–C couplings (Figure 1e). Noteworthy, the selectivity issues associated with this transformation are multifaceted and go far beyond the C–O vs C–C chemoselectivity. Aiming at the development of synthetically useful protocols, control of ortho-/meta-functionalization should be reached together with a high site-selectivity, relating to the selective decoration of only one aromatic ring in case of unsymmetrical hypervalent chlorines. Moreover, mechanistic investigations and density functional theory (DFT) calculations rationalized the superior reactivity of chloranes, the solvent-dependent chemoselectivity of C–O vs C–C couplings, and the preferential meta-functionalization.
Results and Discussion
Synthesis of Cyclic Diaryl λ3-Chloranes
We began our investigation by developing a simple and efficient approach for the preparation of cyclic diaryl λ3-chloranes 2. Encouraged by our previously described synthetic route to cyclic diaryl λ3-bromanes, we hypothesized that similar mild reaction conditions, involving tBuONO as an organic oxidant and a Brønsted acid, could enable the preparation of 2 starting from a simple 1,1′-chloro(amino)biphenyl 1(36) (Scheme 1). Remarkably, an aqueous solution of HBF4 assented to isolate 2a-BF4 in 79% yield through simple precipitation with diethyl ether. In analogy, the HPF6, MsOH, and TfOH acids enabled the preparation of 2a in moderate to good yields (52–63%). Subsequently, the generality of this method was evidenced while engaging various 1,1′-chloro(amino)biphenyls 1a-z. A large variety of cyclic diaryl λ3-chloranes 2a-z was thus furnished in excellent to moderate yields. Electron-donating groups, such as methyl, tert-butyl, and methoxy, were well tolerated at the 1-, 2-, 3-, and 4-positions, respectively, delivering the desired products 2b-k in good to excellent yields (46–91%). Substrates bearing various electron-withdrawing substituents, including chlorine, methyl and benzyl esters, as well as methyl ketone, were also compatible, further illustrating an interesting functional group tolerance. Notably, even a radical sensitive −CN moiety could be installed (1l), furnishing 2l in synthetically useful 47% yield. Selected unsymmetric substrates (1q–1z) were successfully submitted to the standard reaction conditions, furnishing highly decorated cyclic diaryl λ3-chloranes. Particularly interesting are the “push–pull” λ3-chloranes, 2q, 2t, and 2x, bearing electron-donating (Me- and MeO-) and electron-withdrawing groups (MeOOC- and Cl-), which were obtained in excellent yields. The robustness and usefulness of the procedure were furthermore demonstrated by performing the synthesis of 2d, 2w, 2x, and 2z at a gram-scale level (1.2 or 2.2 mmol scale).
Scheme 1. Scope of Cyclic Diaryl λ3-Chloranes.

Single crystal X-ray analysis of 2a-BF4 and 2w confirmed the tricyclic structure of the hypervalent cyclic diaryl λ3-chloranes. Considering the scarcity of the hypervalent chlorine-type compounds and targeting their comparison with the corresponding bromanes and iodanes congeners, the crystallographic structures of these hypervalent compounds have been examined (Figure 2). The crystallographic data for the compounds 2a-BF4, 3a-OMs, and 4a-OMs revealed a peculiar structural trend. The bond length between the carbon and the hypervalent halogen atom increases in the order Cl(III) < Br(III) < I(III). Accordingly, a significant enlargement of the C–X–C angle is observed, translating into an almost perfect T-shaped structure for the chlorine compound (4a-OMs, 81.9°; 3a, 86.9°; 2a-BF4, 92.0°), establishing the λ3-hypervalent compounds’ three-center-four-electron bond (3c–4e) feature.
Figure 2.
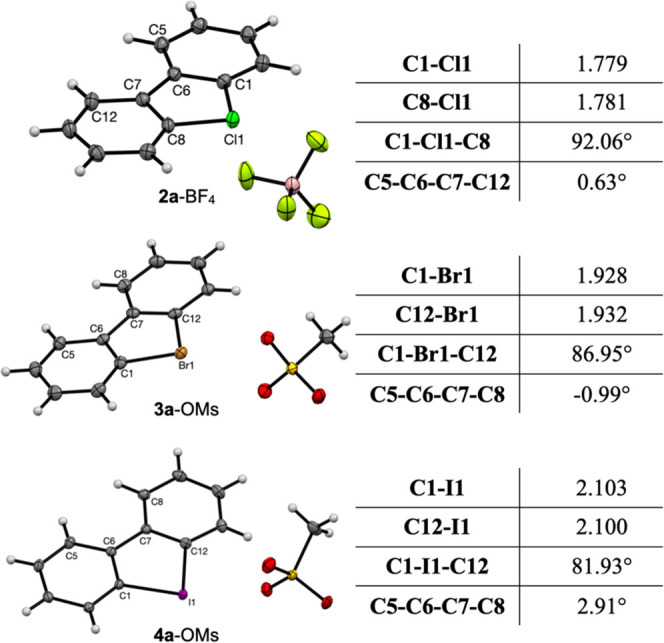
X-ray structure analysis of 2a-BF4 (CCDC 2176956), 3a-OMs (CCDC 2063898) and 4a-OMs (CCDC 2063903).
Reactivity
With the simple and scalable protocol delivering a large family of cyclic diaryl λ3-chloranes 2 in hand, we embarked on exploring their reactivity in sustainable, transition metal-free couplings. Aiming at the development of straightforward and site-selective diversification strategies of phenols, we began our study by investigating the reaction of hypervalent λ3-iodane (4a-OTf), λ3-bromane (3a-OTf), and λ3-chlorane (2a-OTf) with 4-methylphenol 5a in the presence of a weak base, such as cesium or potassium carbonate (Table 1).
Table 1. Reactivity of Cyclic Diaryl Hypervalent λ3-Reagents.

| yieldsb |
||||
|---|---|---|---|---|
| entry | X(III)-OTf | conditionsa | C–C | C–O |
| 1 | 4a-OTf | Cs2CO3; CHCI3 | no conversion | |
| 2 | 3a-OTf | 15% | 70% | |
| 3 | 2a-OTf | 35% | 35% | |
| 4 | 4a-OTf | K2CO3; CHCI3 | no conversion | |
| 5 | 3a-OTf | 26% | 9% | |
| 6 | 2a-OTf | 49% | traces | |
| 7 | 4a-OTf | K2CO3; H2O | no conversion | |
| 8 | 3a-OTf | no conversion | ||
| 9 | 2a-OTf | 13% | 60% | |
Conditions: 0.1 mmol of X(III)-OTf reagent (4a-OTf, 3a-Tf, 2a-OTf), 0.15 mmol of 5a, and 0.3 mmol of Cs2CO3 or K2CO3 in 1 mL of CHCl3 or H2O at room temperature for 16h.
Isolated yields.
While no reaction occurred using the hypervalent λ3-iodine compounds (Table 1, entries 1, 4 and 7), the λ3-bromane and the λ3-chlorane substrates reacted smoothly. Encouragingly, in addition to the expected meta-selective C–O coupling, metal-free dehydrogenative meta-selective C–C bond formation was also observed, delivering product 6a in a significant amount while using 2a-OTf. Remarkably, the nature of the base and the solvent appeared to have a crucial impact on the C–O vs C–C chemoselectivity. Indeed, the hypervalent bromide 3a-OTf in the presence of cesium carbonate provided the C–O coupling product 9a in 70% yield (Table 1, entry 2). In clear contrast, under the same reaction conditions, 2a-OTf showed a complementary reactivity, favoring the formation of the challenging C–C coupling product (6), together with the C–O product (7) (Table 1, entry 3). The simple replacement of the Cs2CO3 base by the potassium salt almost completely inhibited the formation of the ether-derived product, furnishing selectively the direct arylation product, 6a, in 49% yield (Table 1, entry 6). The use of water turned out to be an additional handle to control the reaction outcome. As in the aqueous medium, the reaction between 2a-OTf and 5a provided 7a in 60% isolated yield (Table 1, entry 9), while no coupling occurred when using the hypervalent iodine 4a-OTf and bromane 3a-OTf (Table 1, entries 7 and 8), further highlighting the complemental and potent reactivity of these reagents. Additional control experiments were carried out to elucidate the nature and the role of the base and the solvent (see the SI, Table S-3) identifying potassium carbonate as crucial to avoid the concomitant production of both C–C and C–O coupling products, whereas the choice of the solvent gives a unique handle to reach high chemoselectivity. Noteworthy, in the case of both transformations, the generation of the meta-substituted products is largely favored, and only a trace amount of the ortho-coupling product is observed (see the SI, Table S-2).
Scope of C–C and C–O Couplings
With the optimal reaction conditions in hand, we first investigated the generality of the metal-free dehydrogenative ortho-arylation of phenols (Scheme 2). In all cases, very high meta-selectivity has been observed with regard to the biaryl unit of the hypervalent chlorine precursor. Alkyl substituents (Me, tBu, and Et) at the para-positions were well tolerated, providing 6a, 6b, and 6c in moderate to good yields. Functionalization of the natural product chavicol (5d) afforded 6d in 64% yield, illustrating compatibility with a synthetically useful allyl moiety. Functional groups, such as iodo (5e) and methyl ester (5f) derivatives, were tolerated under these mild reaction conditions, albeit 6e was obtained only in 30% yield. Considering the special role of bioisosteres in medicinal chemistry,37 the 4-adamantyl phenol 5g was also investigated, providing the desired 6g in 72% yield, whereas 5h leads to 6h in moderate amount. Protected 1,4-dihydroxyquinones 5i and 5j conveyed, respectively, 6i and 6j in synthetically useful yields. Couplings with sensitive moieties, such as propargyl 5k, allyl 5l, and cyclopropyl 5m derivatives, occurred smoothly, yielding highly valuable products. Disubstituted phenol 5n furnished a 1:1 mixture of regioisomers of 6n. Interestingly, the introduction of an ortho-substituent on the phenol (5o) does not affect the reaction yield. Furthermore, a remarkable ortho-regioselectivity (with respect to the OH group of the phenol) has been achieved while investigating unsymmetric and sterically hindered substrates, such as 5p, 5q, 5r, and 5s, delivering the desired products as single regioisomers in high yields (up to 76% yield). Finally, the arylation of phenol 5t and 2-naphthol 5u supplied regioisomerically pure 6t and 6u.
Scheme 2. Scope of Metal-Free C–C Bond Formation with Cyclic Diaryl λ3-Chloranes.

Intrigued by the discovery of a solvent-dependent chemoselectivity, we then focused our attention on the development of a water-mediated metal-free C–O coupling (Scheme 3a). The versatility and the robustness of this approach were validated regarding the compatibility of this reaction with a large diversity of ortho-, meta-, and para-functionalized substrates 5. Synthetically useful functional groups, including nitro 5v, iodo 5z, methyl ketone 5x as well as cyanide 5C, were well tolerated, furnishing the corresponding products in good yields. Phenols bearing electron-withdrawing groups, such as 5w and 5y, outcompeted the electron-rich coupling partners, due to the increased acidity. Remarkably, the C–O arylation of the challenging sterically hindered phenol 5H was still possible, albeit 7H was isolated in moderate yield. Moreover, L-tyrosine derivative 5I was submitted to the standard reaction conditions and a good yield of the arylated product 7I was obtained, further illustrating the high versatility of the method.
Scheme 3. Scope of Metal-Free C–O Coupling; (a) with Cyclic Diaryl λ3-Chloranes; (b) with Cyclic Diaryl λ3-Bromanes.

Cyclic diaryl hypervalent λ3-bromane 3a-OTf was also briefly tested for the metal-free carbon–oxygen bond formation (Scheme 3b). Due to the lower reactivity of Br(III)-species under the standard procedure, an alternative protocol involving CHCl3 as solvent and cesium carbonate as base was identified as suitable for various electron-rich and electron-poor phenols affording products 9 in good to excellent yields.
The complexity of the coupling between diaryl λ3-chloranes and phenols does not only rely on the chemoselectivity between C–O and C–C coupling, but an additional degree of selectivity arises from (1) a possible functionalization of the biaryl unit of the λ3-chloranes either in ortho- or meta-positions and (2) selective functionalization of one ring over the other in case of unsymmetrical hypervalent substrates. While using the simple 2a-OTf substrate, meta-selective coupling is highly favored (m/o selectivity from 99:1 to 88:12). To further investigate the meta-/ortho-regioselectivity trends, the reactivity of unsymmetric cyclic diaryl λ3-chlorines was tested (Scheme 4). Under the C–O coupling protocol, the 2-methyl substituted chloranes 2d and 2w were efficiently reacted with the phenol 5w, delivering selectively meta-substituted products 7dw and 7ww, respectively, in 74 and 71% isolated yields. The presence of an electron-withdrawing group at the 3-position, such as methyl ester group (2z), provided 7zw in good yield, but a regioselectivity-switch occurred, translating into formation of the ortho-regioisomer as the major product (m/o 31:69). On the other hand, the impact of the ester substituent, when introduced at the 2-position, is less pronounced, thus generating the meta-product 7yw as the major regioisomer (m/o 68:32). In addition, the unsymmetric λ3-chloranes prone to undergo the functionalization at both, different biaryl aromatic units, tend to be functionalized at the less electron-rich aryl motif.38,39 Indeed, OMe-substituted chlorane 2k delivered 7kw as a mixture of two products in a/b regioselectivity ratio of 73:27. Impressively, a perfect regioseletivity was reached by designing a “push–pull”-type substrate. In case of 2t bearing one electron-rich and one electron-withdrawing substituent, a complete regioselective control was achieved while reacting with 5w. Finally, we extended the regioselectivity studies taking into consideration the effect of different phenols on the transformation with 2w and 2b into the C–C bond formation. Sterically hindered phenols 5u and 5b provided 6wu and 6wb with a similar regioselectivity ratio of 87:13 m/o, while a methyl group (5a) furnished 6wa in 59% yield, albeit with a marginally reduced regioselectivity.
Scheme 4. Unsymmetric Cyclic Diaryl λ3-Chloranes in C–O and C–C Bond Formations.

To demonstrate even further the sustainability of this methodology, the C–O coupling was performed under flow protocol. Reaction of 2a-OTf and the phenol 5a delivered the desired product 7a in 72% isolated yield after a simple liquid–liquid extraction and filtration through silica gel. Remarkably, a drastic reduction of the reaction time (13.3 min of residence time vs 16 h) and an improved chemoselectivity were reached under flow chemistry conditions (see the SI, Scheme S-12).
Mechanistic Studies
Captivated by the singular reactivity of this class of cyclic diaryl λ3-chloranes, mechanistic studies were undertaken, combining both experimental and DFT approaches.
Experimental Studies: Generation of Aryne Intermediate
First, competition tests between λ3-chloranes and λ3-bromanes, in both C–C and C–O coupling, were performed (see the SI, Scheme S-14 and Scheme S-15). Remarkably, C–C coupling is twice as efficient for λ3-chlorane 2a than for λ3-bromane 3a. Even more impressive is the almost 5 times increased reactivity observed for 2a in the C–O coupling reaction. Following this trend, in case of the parallel reactions conducted with 2a-OTf and 3a-OTf, the bromine substate provided the desired products 8b and 9w in only low yields, with 2-bromo-biphenyl being a major decomposition product. On the contrary, 2a-OTf furnished the desired product in good yields, proving the higher and complemental reactivity of the latter.
Subsequently, deuteration studies were conducted (Scheme 5a). C–O coupling between 2a-OTf and 5w, conducted in deuterated solvents, furnished 7w-[D1] in good yield with high deuterium incorporation (up to 95%) at the ortho-position. This result perfectly validates the expected formation of the aryne intermediate and the meta-functionalization. Upon a nucleophilic attack, a carbanion intermediate is formed which can be easily trapped in the presence of [D+]-sources.40
Scheme 5. Mechanistic Studies.

Afterwards, aiming at the determination of the kinetic isotope effect characterizing these metal-free C–C and C–O couplings, deuterated substrate 2a-[D4]-OTf was reacted with 5w and 5b, respectively (Scheme 5b). Both reactions delivered 6a-[D] and 7w-[D] as a mixture of -[D4] and -[D3] adducts with an identical [D4]/[D3] ratio of 77:23, suggesting that the aryne generation features a KIE of 3.3 and might be expected to be the rate-determining step of these transformations.
Generation of the aryne intermediate was also proven by trapping such a highly reactive species (Scheme 6). Taking advantage of highly versatile cycloaddition reactions for the construction of new carbon–carbon and carbon–nitrogen bonds, 2a-OTf was reacted with 2,5-dimethylfuran and benzyl azide, thus delivering 10 in excellent yield and 11 as a separable mixture of products in 58% yield.
Scheme 6. Cyclic Diaryl λ3-Chloranes in Cycloaddition Reactions.

To better understand the aryne formation process, the initial rates of both C–C and C–O coupling were measured (see the SI, Scheme S-20, and Scheme S-21). Similar rates have been observed within 30 min of the reactions, leading to 50% of conversions. These results support a limited influence of the reaction media on the formation of the aryne intermediate step from 2a-OTf, which can further rapidly react with the phenol.
DFT Calculations
To gain further insights into the formation of arynes from hypervalent compounds 2a–4a and their subsequent reaction with phenols 5, density functional theory (DFT) calculations at the ωB97X-D/def2-QZVPP+ SMD(CHCl3)//ωB97X-D/def2-TZVP level of theory including corrections for solvation by chloroform were performed (see the SI for full computational details).41−43
Aryne Generation
The formation of highly reactive aryne intermediates from 2a–4a is proposed to take place via deprotonation with carbonate and concurrent C–X bond cleavage. Coordination of carbonate to intermediate A was found to be thermodynamically favorable (Figure 3), and the larger stabilization of B-I compared to B-Br and of B-Cl can be rationalized by the greater electrostatic stabilization due to the significantly larger positive charge of iodine in A-OTf (+1.20 for I compared to +0.87 for Br, and +0.67 for Cl; see the SI, Figure S-3).
Figure 3.

Calculated energy diagram (in kcal mol–1) for the formation of arynes C from hypervalent halonium 2a (red color), 3a (blue) and 4a (black).
Subsequent deprotonation and C–X bond cleavage occur in a concerted process via seven-membered cyclic transition state TS1, with an activation free energy of 16.2 kcal mol–1 for I, 9.4 kcal mol–1 for Br and only 4.3 kcal mol–1 for Cl with respect to the corresponding intermediate B. The substantially smaller activation free energy as well as the considerably higher stabilization of C agrees well with the experimentally observed preferred aryne formation with hypervalent λ3-bromane and λ3-chlorane compounds compared to well-established λ3-iodane reagents. Furthermore, the experimentally determined increased reactivity of hypervalent λ3-chloranes is consistent with the surprisingly low energy barrier required for aryne formation. The geometries and electronic properties of 3-(o-halophenyl)benzynes C were found to be largely independent of the halide substituent (Figure 4).
Figure 4.

Calculated geometries of aryne intermediates C. Distances are given in Å; values in red correspond to NBO charges.
meta- vsortho-Selectivity
While the slightly larger distortion toward linearity at C2, i.e., the aryne C atom in ortho-position with respect to the ArX substituent, indicates a marginally preferred nucleophilic attack at C2 due to the more pronounced p-orbital character, partial positive charge, and lesser transition state distortion energy,38 we hypothesize that the significant steric bulk of the ArX substituent overrides the predicted selectivity, thus leading to the observed attack at meta-position.44
Chemoselectivity
The C–O vs C–C, condition-controlled chemoselectivity was subsequently investigated. The formation of a solvent-separated potassium and phenolate ion pair is likely to occur in water, whereas in less-polar chloroform, the potassium phenolate ion pair is formed, presumably rendering an attack of the phenolate oxygen onto the benzyne less facile. In contrast, cesium phenolate is expected to form loose ion pairs in chloroform, due to the more complete solvation in part caused by the significantly lower charge density compared to potassium. To shed light on the meta-/ortho-selectivity of the transformation as well as the solvent-dependent switch from C–O to C–C bond formation, we performed a series of calculations at different C–O and C–C bond distances, respectively. These computations were performed because there is no electronic energy barrier to the reaction, although there will be a free-energy barrier due to the decrease of entropy for this bimolecular reaction. The barrier will be low (less than the 8–12 kcalmol−1 −TΔS of a bimolecular reaction). For the reaction of intermediate C-Cl with phenolate, a clear preference for C–O over C–C bond formation can be observed for all relevant bond distances (Figure 5a).45
Figure 5.

Calculated energy profile (in kcal mol–1) for C–O bond (black and red squares) and C–C bond (blue and green triangles) formation with (a) phenolate and (b) potassium phenolate.
In addition, C–O as well as C–C bond formation was found to preferentially occur at the meta-position, which is in good agreement with the experimental observation. In contrast, replacing phenolate with potassium phenolate led to a distinct change in reactivity, with C–C bond formation at the meta-position now preferred over C–O bond formation for bond distances d > 1.8 Å (Figure 5b).
Based on this mechanistic evidence, a plausible reaction mechanism for the formation of C–C and C–O coupling is proposed (Scheme 7). Initially, an anion exchange leads to the energetically more stable 2a-carbonate intermediate I, which through a low-barrier process evolves into a seven-membered ring TS. Concerted base-mediated deprotonation and carbon–halogen cleavage generate the aryne intermediate II. The meta-selective nucleophilic attack onto the benzyne intermediate II results in the formation of the new C–C and C–O bonds, respectively, yielding intermediates IIIa and IIIb. The rearomatization of the intermediate IIIa and the subsequent protonation of the carbanion generate products 6a and 7a.
Scheme 7. Proposed Mechanism.

Post-Functionalization
The transition metal-free C–C and C–O couplings described here provide sustainable and rapid access to highly modular building blocks, straightforwardly diversifiable into various interesting compounds (Scheme 8). To demonstrate several possible post-modifications, large-scale couplings were first performed. The 15 mmol scale synthesis of the cyclic diaryl λ3-chlorane was performed, yielding 3 grams of 2a-OTf in 60% yield within 90 minutes, ensuring a useful and simple approach to these hypervalent reagents. Subsequently, a vast diversification study was carried out to further display the utility of our products. The 2 mmol scale reactions of 2a-OTf with 5a and 5d under the C–C coupling conditions provided 6a and 6d in synthetically useful amounts. Interestingly, exploiting the allyl fragment of 6d, a Grubbs-catalyzed metathesis furnished the highly functionalized dichloro-diphenol compound 12 in 61% yield. A straightforward conversion of the hydroxy group to the corresponding triflate 13 enabled the synthesis of the m-quinquephenyl 14via a double Suzuki reaction in 58% yield, while a Cl-selective palladium-catalyzed Suzuki coupling furnished the product 15 in 59% yield. The peculiar presence of a hydroxyl residue was also exploited for the synthesis of a new phosphine 16, which was prepared in 55% yield. Subsequently, the relevance of C–O coupling product was evaluated. The scale-up reactions up to 3 mmol scales produced the desired products 7y, 7z, and 7J in good to excellent yields (55, 78, and 68%, respectively). Taking advantage of the selective reactivity of the iodine with respect to the chlorine atom, a Suzuki reaction and a lithium base-mediated exchange were performed starting from 7z, supplying, respectively, 17 and 18 in 40 and 50% yields. On the other hand, a palladium-catalyzed C–H activation delivered the dibenzofuran 19 from 7J in 64% yield. Notably, the resulting products contained the residual chlorine substituent as a handle for further post-modifications. Accordingly, a palladium-catalyzed C–N Buchwald–Hartwig reaction and a Suzuki reaction were performed starting from 7y and granting, respectively, access to 20 and 21 in 64 and 83% yields. Finally, a palladium-catalyzed dechlorination process yielded benzofuran 22.
Scheme 8. Post-Functionalization.

(a) Ph2PCl, Et3N, Tol, reflux, 16 h. (b) Tf2O, 2,6-lutidine, DCM, 0 °C to r.t., 2 h. (c) Pd(OAc)2 cat., PCy3 cat., 3,5-OMeC6H3B(OH)2, KF, THF, r.t., 72 h. (d) Pd(OAc)2 cat., PCy3 cat., C6H5B(OH)2, KF, THF, r.t., 72 h. (e) Grubbs I cat., DCM, reflux, 16 h. (f) nBuLi, DMF, THF, −78 °C to r.t., 2 h. (g) Pd(PPh3)2Cl2 cat., NaHCO3, MeOC6H4B(OH)2, DME/H2O, 120 °C, 2 h. (h) Pd2(dba)3 cat., SPhos cat., Cs2CO3, dioxane, 150 °C, 16 h. (i) Pd2(dba)3 cat., SPhos cat., Me2N-NH2, tBuONa, dioxane, 120 °C, 5 h. (j) Pd(PPh3)2Cl2 cat., NaHCO3, MeOC6H4B(OH)2, DME/H2O, 120 °C, 2 h. (k) Pd(OAc)2 cat., P(Cy)3 HBF4 cat., K2CO3, DMA, 130 °C, 16 h.
Conclusions
We have here reported a new, simple, and robust synthetic method for the preparation of novel cyclic diaryl λ3-chloranes 2. The higher reactivity of these compounds proved to be complementary to the well-established chemistry of cyclic diaryl λ3-iodane and more recent λ3-bromane compounds. The increased reactivity of cyclic diaryl λ3-chloranes as aryne precursors enhances the unparalleled metal-free ortho-arylation of phenols under mild base-mediated reaction conditions. Furthermore, experimental and DFT mechanistic investigations elucidated the origins of the reactivities as well as the chemo- and regioselectivity observed in this work. A variety of synthetic applications show the considerable potential of these new reagents in synthetic organic chemistry.
Acknowledgments
We thank the Centre National de la Recherche Scientifique (CNRS), the “Ministère de l’Education Nationale et de la Recherche” (France) for financial support. J.W.D., M.L., and T.S.T. are very grateful to the European Commission for the ERC-Starting Grant “AlCHIMIE” no. 949804. Generous support by the Alexander von Humboldt-foundation (Feodor Lynen fellowship, T.R.) and the National Science Foundation (CHE-1764328 to K.N.H.) is gratefully acknowledged. Calculations were performed on the Hoffman2 cluster at the University of California, Los Angeles. We are grateful to Dr. Lydia Karmazin, Dr. Corinne Bailly, and Dr. Nathalie Gruber for the X-ray diffraction analysis (Service de Radiocrystallographie, CNRS-Université de Strasbourg, Strasbourg, France). The manuscript was written through the contributions of all authors. We thank also Dr. Morgan Donnard (LIMA) and Dr. Armen Panossian (LIMA) for the discussions regarding flow chemistry and arynes.All authors have given approval for the final version of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10090.
General procedure, product characterizations, computational methods, energies and geometries of calculated structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yoshimura A.; Zhdankin V. V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]
- Zhdankin V. V.; Stang P. J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Jr L. F. Jr.; Olofsson B. Hypervalent Iodine Reagents in the Total Synthesis of Natural Products. Nat. Prod. Rep. 2011, 28, 1722. 10.1039/c1np00028d. [DOI] [PubMed] [Google Scholar]
- Morimoto K.; Sakamoto K.; Ohnishi Y.; Miyamoto T.; Ito M.; Dohi T.; Kita Y. Metal-Free Oxidative Para Cross-Coupling of Phenols. Chem. - Eur. J. 2013, 19, 8726–8731. 10.1002/chem.201301028. [DOI] [PubMed] [Google Scholar]
- Stridfeldt E.; Lindstedt E.; Reitti M.; Blid J.; Norrby P.-O.; Olofsson B. Competing Pathways in O-Arylations with Diaryliodonium Salts: Mechanistic Insights. Chem. - Eur. J. 2017, 23, 13249–13258. 10.1002/chem.201703057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E.; Olofsson B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]
- Wang X.; Studer A. Iodine(III) Reagents in Radical Chemistry. Acc. Chem. Res. 2017, 50, 1712–1724. 10.1021/acs.accounts.7b00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterson B.; Patra T.; Wirth T. Hypervalent Bromine(III) Compounds: Synthesis, Applications, Prospects. Synthesis 2022, 54, 1261–1271. 10.1055/a-1675-8404. [DOI] [Google Scholar]
- Ochiai M. Hypervalent Aryl-, Alkynyl-, and Alkenyl-λ3-Bromanes. Synlett 2009, 2009, 159–173. 10.1055/s-0028-1087355. [DOI] [Google Scholar]
- Farooq U.; Shah A.-H. A.; Wirth T. Hypervalent Bromine Compounds: Smaller, More Reactive Analogues of Hypervalent Iodine Compounds. Angew. Chem., Int. Ed. 2009, 48, 1018–1020. 10.1002/anie.200805027. [DOI] [PubMed] [Google Scholar]
- Miyamoto K.; Saito M.; Tsuji S.; Takagi T.; Shiro M.; Uchiyama M.; Ochiai M. Benchtop-Stable Hypervalent Bromine(III) Compounds: Versatile Strategy and Platform for Air- and Moisture-Stable λ3-Bromanes. J. Am. Chem. Soc. 2021, 143, 9327–9331. 10.1021/jacs.1c04536. [DOI] [PubMed] [Google Scholar]
- Miyamoto K.; Uchiyama M. Hypervalent Organo-λ3-Chloranes. Chem. Lett. 2021, 50, 832–838. 10.1246/cl.200849. [DOI] [Google Scholar]
- Nakajima M.; Miyamoto K.; Hirano K.; Uchiyama M. Diaryl-λ3-Chloranes: Versatile Synthesis and Unique Reactivity as Aryl Cation Equivalent. J. Am. Chem. Soc. 2019, 141, 6499–6503. 10.1021/jacs.9b02436. [DOI] [PubMed] [Google Scholar]
- Watanabe Y.; Takagi T.; Miyamoto K.; Kanazawa J.; Uchiyama M. Shelf-Stable (E)- and (Z)-Vinyl-λ3-Chlorane: A Stereospecific Hyper-Vinylating Agent. Org. Lett. 2020, 22, 3469–3473. 10.1021/acs.orglett.0c00924. [DOI] [PubMed] [Google Scholar]
- Sandin R. B.; Hay A. S. Stable Bromonium and Chloronium Salts. J. Am. Chem. Soc. 1952, 74, 274–275. 10.1021/ja01121a524. [DOI] [Google Scholar]
- Lanzi M.; Dherbassy Q.; Wencel-Delord J. Cyclic Diaryl λ3-Bromanes as Original Aryne Precursors. Angew. Chem., Int. Ed. 2021, 60, 14852–14857. 10.1002/anie.202103625. [DOI] [PubMed] [Google Scholar]
- Lanzi M.; Ali Abdine R. A.; De Abreu M.; Wencel-Delord J. Cyclic Diaryl λ3-Bromanes: A Rapid Access to Molecular Complexity via Cycloaddition Reactions. Org. Lett. 2021, 23, 9047–9052. 10.1021/acs.orglett.1c03278. [DOI] [PubMed] [Google Scholar]
- Yoshida Y.; Ishikawa S.; Mino T.; Sakamoto M. Bromonium Salts: Diaryl-λ3-Bromanes as Halogen-Bonding Organocatalysts. Chem. Commun. 2021, 57, 2519–2522. 10.1039/D0CC07733J. [DOI] [PubMed] [Google Scholar]
- Yoshida Y.; Mino T.; Sakamoto M. Chiral Hypervalent Bromine(III) (Bromonium Salt): Hydrogen- and Halogen-Bonding Bifunctional Asymmetric Catalysis by Diaryl-λ3-Bromanes. ACS Catal. 2021, 11, 13028–13033. 10.1021/acscatal.1c04070. [DOI] [Google Scholar]
- a Zhang X.; Zhao K.; Li N.; Yu J.; Gong L.; Gu Z. Atroposelective Ring Opening of Cyclic Diaryliodonium Salts with Bulky Anilines Controlled by a Chiral Cobalt(III) Anion. Angew. Chem., Int. Ed. 2020, 59, 19899–19904. 10.1002/anie.202008431. [DOI] [PubMed] [Google Scholar]; b Li Q.; Zhang M.; Zhan S.; Gu Z. Copper-Catalyzed Enantioselective Ring-Opening of Cyclic Diaryliodoniums and O-Alkylhydroxylamines. Org. Lett. 2019, 21, 6374–6377. 10.1021/acs.orglett.9b02267. [DOI] [PubMed] [Google Scholar]
- Karandikar S. S.; Bhattacharjee A.; Metze B. E.; Javaly N.; Valente E. J.; McCormick T. M.; Stuart D. R. Orbital Analysis of Bonding in Diarylhalonium Salts and Relevance to Periodic Trends in Structure and Reactivity. Chem. Sci. 2022, 13, 6532–6540. 10.1039/D2SC02332F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quideau S.; Deffieux D.; Douat-Casassus C.; Pouységu L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem., Int. Ed. 2011, 50, 586–621. 10.1002/anie.201000044. [DOI] [PubMed] [Google Scholar]
- Scott K. A.; Cox P. B.; Njardarson J. T. Phenols in Pharmaceuticals: Analysis of a Recurring Motif. J. Med. Chem. 2022, 65, 7044–7072. 10.1021/acs.jmedchem.2c00223. [DOI] [PubMed] [Google Scholar]
- The Chemistry of Phenols; Rappoport Z., Ed.; The chemistry of functional groups; Wiley: Hoboken, NJ, 2003. [Google Scholar]
- Huang Z.; Lumb J.-P. Phenol-Directed C–H Functionalization. ACS Catal. 2019, 9, 521–555. 10.1021/acscatal.8b04098. [DOI] [Google Scholar]
- Eisenhofer A.; Hioe J.; Gschwind R. M.; König B. Photocatalytic Phenol-Arene C-C and C-O Cross-Dehydrogenative Coupling: Photocatalytic Phenol-Arene C-C and C-O Cross-Dehydrogenative Coupling. Eur. J. Org. Chem. 2017, 2017, 2194–2204. 10.1002/ejoc.201700211. [DOI] [Google Scholar]
- Tian T.; Li Z.; Li C.-J. Cross-Dehydrogenative Coupling: A Sustainable Reaction for C–C Bond Formations. Green Chem. 2021, 23, 6789–6862. 10.1039/D1GC01871J. [DOI] [Google Scholar]
- Krylov I. B.; Vil’ V. A.; Terent’ev A. O. Cross-Dehydrogenative Coupling for the Intermolecular C–O Bond Formation. Beilstein J. Org. Chem. 2015, 11, 92–146. 10.3762/bjoc.11.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Larock R. C. Facile O-Arylation of Phenols and Carboxylic Acids. Org. Lett. 2004, 6, 99–102. 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]
- Gebara K. S.; Casagrande G. A.; Raminelli C. An Efficient Fluoride-Mediated O-Arylation of Sterically Hindered Halophenols with Silylaryl Triflates under Mild Reaction Conditions. Tetrahedron Lett. 2011, 52, 2849–2852. 10.1016/j.tetlet.2011.03.124. [DOI] [Google Scholar]
- Castillo J.-C.; Quiroga J.; Rodriguez J.; Coquerel Y. Time-Efficient Synthesis of Pyrido [2,3-d]Pyrimidinones via α-Oxoket enes. Eur. J. Org. Chem. 2016, 2016, 1994–1999. 10.1002/ejoc.201600171. [DOI] [Google Scholar]
- Röckl J. L.; Pollok D.; Franke R.; Waldvogel S. R. A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls. Acc. Chem. Res. 2020, 53, 45–61. 10.1021/acs.accounts.9b00511. [DOI] [PubMed] [Google Scholar]
- Truong T.; Daugulis O. Divergent Reaction Pathways for Phenol Arylation by Arynes: Synthesis of Helicenes and 2-Arylphenols. Chem. Sci. 2013, 4, 531–535. 10.1039/C2SC21288A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A.; Fuchs J. M.; Middleton K. R.; Maskaev A. V.; Rohde G. T.; Saito A.; Postnikov P. S.; Yusubov M. S.; Nemykin V. N.; Zhdankin V. V. Pseudocyclic Arylbenziodoxaboroles: Efficient Benzyne Precursors Triggered by Water at Room Temperature. Chem. - Eur. J. 2017, 23, 16738–16742. 10.1002/chem.201704393. [DOI] [PubMed] [Google Scholar]
- a Kitamura S.; Hvorecny K. L.; Niu J.; Hammock B. D.; Madden D. R.; Morisseau C. Rational Design of Potent and Selective Inhibitors of an Epoxide Hydrolase Virulence Factor from Pseudomonas Aeruginosa. J. Med. Chem. 2016, 59, 4790–4799. 10.1021/acs.jmedchem.6b00173. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xu Y.; Wang Y.; Wu D.; He W.; Wang L.; Zhu W. P-Terphenyls From Aspergillus Sp. GZWMJZ-055: Identification, Derivation, Antioxidant and α-Glycosidase Inhibitory Activities. Front. Microbiol. 2021, 12, 654963 10.3389/fmicb.2021.654963. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Guo R.-T.; Cao R.; Liang P.-H.; Ko T.-P.; Chang T.-H.; Hudock M. P.; Jeng W.-Y.; Chen C. K.-M.; Zhang Y.; Song Y.; Kuo C.-J.; Yin F.; Oldfield E.; Wang A. H.-J. Bisphosphonates Target Multiple Sites in Both Cis - and Trans -Prenyltransferases. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10022–10027. 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kovács A.; Vasas A.; Hohmann J. Natural Phenanthrenes and Their Biological Activity. Phytochemistry 2008, 69, 1084–1110. 10.1016/j.phytochem.2007.12.005. [DOI] [PubMed] [Google Scholar]; e Hangeland J. J.; Doweyko A. M.; Dejneka T.; Friends T. J.; Devasthale P.; Mellström K.; Sandberg J.; Grynfarb M.; Sack J. S.; Einspahr H.; Färnegårdh M.; Husman B.; Ljunggren J.; Koehler K.; Sheppard C.; Malm J.; Ryono D. E. Thyroid Receptor Ligands. Part 2: Thyromimetics with Improved Selectivity for the Thyroid Hormone Receptor Beta. Bioorg. Med. Chem. Lett. 2004, 14, 3549–3553. 10.1016/j.bmcl.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Synthesis of 1a was conducted under transition metal-free protocol (see the SI, page 14).
- Tse E. G.; Houston S. D.; Williams C. M.; Savage G. P.; Rendina L. M.; Hallyburton I.; Anderson M.; Sharma R.; Walker G. S.; Obach R. S.; Todd M. H. Nonclassical Phenyl Bioisosteres as Effective Replacements in a Series of Novel Open-Source Antimalarials. J. Med. Chem. 2020, 63, 11585–11601. 10.1021/acs.jmedchem.0c00746. [DOI] [PubMed] [Google Scholar]
- Medina J. M.; Mackey J. L.; Garg N. K.; Houk K. N. The Role of Aryne Distortions, Steric Effects, and Charges in Regioselectivities of Aryne Reactions. J. Am. Chem. Soc. 2014, 136, 15798–15805. 10.1021/ja5099935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner S. M.; Mackey J. L.; Houk K. N.; Garg N. K. Steric Effects Compete with Aryne Distortion To Control Regioselectivities of Nucleophilic Additions to 3-Silylarynes. J. Am. Chem. Soc. 2012, 134, 13966–13969. 10.1021/ja306723r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Li L.; Li Y. O-Silylaryl Triflates: A Journey of Kobayashi Aryne Precursors. Chem. Rev. 2021, 121, 3892–4044. 10.1021/acs.chemrev.0c01011. [DOI] [PubMed] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Cheong P. H.-Y.; Paton R. S.; Bronner S. M.; Im G.-Y. J.; Garg N. K.; Houk K. N. Indolyne and Aryne Distortions and Nucleophilic Regioselectivites. J. Am. Chem. Soc. 2010, 132, 1267–1269. 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previous studies already showed that additions of nucleophiles to arynes often do not display a maximum (i.e. transition state) on the electronic energy surface, especially with anionic nucleophiles, thereby rendering the geometry optimization of a transition state structure for the comparison of activation free energies unfeasible (Refs. 38 and 44).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.