

Abstract
The emergence of clinical metagenomics as an unbiased, hypothesis-free approach to diagnostic testing is set to fundamentally alter the way infectious diseases are detected. Long envisioned as the solution to the limitations of culture-based conventional microbiology, next generation sequencing methods will soon mature, and our attention will inevitably turn to how they can be applied to areas of medicine which need it most urgently. In ophthalmology, the demand for this technology is particularly pressing for the care of infectious corneal ulcers, where current diagnostic tests may fail to identify a causative organism in over half of cases. However, the optimism found in the budding discourse surrounding clinical metagenomics belies the reality that clinicians and scientists will soon be inundated by oppressive volumes of sequencing data, much of which will be foreign and unfamiliar. Therefore, our success in translating clinical metagenomics is likely to hinge on how we make sense of these data, and understanding its implications for the interpretation and implementation of sequencing into routine clinical care. In this consortium-led review, we provide an outline of these data-related issues and how they may be used to inform technical workflows, with the hope that we may edge closer to realizing the potential of clinical metagenomics for this important unmet need.
INTRODUCTION
“The power of population is indefinitely greater than the power in the earth to produce subsistence for man.”
– Thomas Robert Malthus, An Essay on the Principle of Population (1798)1
In the coming years, an evolving group of molecular technologies will challenge current diagnostic paradigms in clinical microbiology. Of these, clinical metagenomics conducted with next generation sequencing (NGS) has commanded the greatest attention, as it enables massive and deep parallel sequencing of nucleic acids found in complex specimens. The theoretical advantages of this unbiased, hypothesis-free approach to pathogen identification lie in its unrivalled potential to facilitate study of antimicrobial susceptibilities, molecular epidemiology, strain typing and monitoring of disease outbreaks, all achievable within the same assay. For ophthalmologists treating infectious ocular diseases, the need to develop more sensitive and rapid diagnostic tests has been felt most acutely for infectious corneal ulcers, the etiologies of which routinely escape detection with conventional culture and stain-based testing.2 However, in anticipating efforts that will be made to integrate NGS workflows into the care of these infections, clinicians, microbiologists and bioinformaticians must be prepared for the deluge of highly complex and computationally intensive sequencing data generated with each study, once described as the Malthusian dilemma of sequencing.3 In this consortium-led, case-study based review, we highlight the inherent data-related obstacles that might stall the urgently needed introduction of NGS for infectious corneal ulcers, with a principle focus on the issues surrounding its interpretation and implementation in clinical care.
1. INFECTIOUS CORNEAL ULCERS: A MATTER OF PUBLIC HEALTH
Infectious corneal ulcers, caused by a variety of viruses, bacteria, fungi and parasites, are a major cause of visual impairment. Worldwide, infectious corneal ulcers were recently estimated to account for up to 3.2% of all cases of blindness.4 Historical estimates of incidence, however, have under-represented their true magnitude because no all-encompassing data exists combining viral and non-viral etiologies. A piecemeal survey of now outdated data reveals at least 1.5 million cases of viral herpetic keratitis5 and a further 2 million cases of non-viral keratitis6 per year worldwide. Capturing the overall burden of disease caused by infectious corneal ulcers has also proven difficult. This is because such data may simply not exist, and/or because existing hospital-based coding systems such as the International Classification of Diseases (ICD) are prone to inconsistent application, human error, and may lack the required clinical specificity for detailed epidemiological study.7 Nonetheless, in the United States, claims from large registry data suggest that corneal infections require up to 1 million healthcare visits, 250,000 hours of dedicated physician care, and at least 175 million dollars of direct healthcare-related expenditure per year.8 It would be reasonable to speculate that the total economic burden associated with the resulting chronic visual disability and lost productivity is many orders of magnitude higher than these direct costs alone.
2. AT A DIAGNOSTIC IMPASSE: THE LIMITS OF CONVENTIONAL MICROBIOLOGY FOR THE CORNEA
Case study
A 37-year-old, otherwise healthy woman presents with a 3-day history of severe left eye pain, photophobia and decreased visual acuity. The patient is a soft contact-lens wearer, endorses appropriate lens-related hygiene, and has no other ocular history. On clinical examination, she is able to perceive only hand motions in her left eye, and by slit lamp biomicroscopy she has a large, central corneal ulcer with moderate corneal thinning and an associated 1mm hypopyon. A clinic-based B-scan does not demonstrate vitreous debris. The history and examination are consistent with a contact-lens related corneal ulcer, and its severity warrants the collection of corneal scrapings for culture. During the procedure, the patient mentions that 3 days prior to her presentation, her left eye was “struck by a tree branch”, while hiking. Understanding that the results of culture are likely to take some days, the clinician requests Gram and calcofluor white stain of a corneal scraping for stat microscopy, but these are unfortunately non-revealing and contain no visible bacterial or fungal elements. Urgent confocal microscopy demonstrates a hyper-reflective sea of inflammatory cells, but nothing further. The clinician is left to ponder the possibility of a fungal etiology, concerned that topical fortified antibiotics may not suffice for the care of this patient.
Current standards of diagnostic care for infectious corneal ulcers
One glaring weakness in our attempts to reduce the burden of infectious corneal ulcers is the perennial lack of innovation in current diagnostic methodologies. Standard-of-care in pathogen detection has not changed significantly in over a half-century. As demonstrated by our case study, these infections represent a true diagnostic conundrum because their microbial etiologies cannot be reliably determined on clinical examination alone.9 This is particularly true for bacterial, fungal, and parasitic ulcers, which often present with non-suggestive signs, and which frequently warrant the collection of corneal cultures. In ophthalmology, sampling ocular tissues and/or fluids is typically constrained by the small volume of inoculum taken from patients. However, the issue of insufficient starting material is particularly problematic for the cornea, and attempts to increase microbial yield (e.g. with biopsy) are not often pursued because iatrogenic injuries to the highly organized architecture of the cornea can be of long-lasting visual consequence. Therefore, to determine the etiology of infectious corneal ulcers, the gold standard of care remains the collection of multiple corneal samples from the site of infection, typically performed with a sterilized surgical blade, spatula, or specialized swabs, which are directly plated onto slides for microscopy and a range of culture media (Figure 1A).10 Expensive, labor intensive, and at times difficult to interpret correctly, this current standard suffers from low sensitivity (~50%2), and as a result cannot be used to guide antimicrobial therapy in over half of patients. The incubation time required to yield actionable results, at times exceeding 30 days, may constitute an unacceptable delay in diagnosis.
FIGURE 1: Overview of current diagnostic paradigms when compared to clinical metagenomics.

Figure 1A demonstrates the current diagnostic workflow for infectious corneal ulcers, a low-resolution, labor-intensive piecemeal approach involving the collection of patient sample to be immediately inoculated or plated on a range of media and slides. The major rate limiting step is pathogen incubation, which is poorly sensitive and may exceed 30 days, as is the case in fungal speciation. Its main advantages include relatively low cost and amenability to high volume testing, as well as the familiar binary or ternary nature of diagnostic reporting (e.g. growth vs. no growth; resistant, intermediately resistant, and susceptible). Figure 1B is a heavily simplified demonstration of a typical clinical metagenomics workflow, which provides a global overview of the nucleic acids found in a single specimen. Computationally processed genomic data may provide relevant data regarding pathogen identification, antimicrobial susceptibilities, typing and molecular epidemiology. Current translational research in ophthalmology is underway regarding the characterization of inherent sequencing and bioinformatics-related biases induced by low-biomass, paucimicrobial samples, and how to overcome these challenges to produce reliable, reproducible, and clinically tractable results.
In addition, one important question is whether culture-negative corneal ulcers harbor viable organisms. In endophthalmitis, for example, most culture-negative samples do not have detectable bacterial DNA using molecular techniques.11 However, in one large study in a South Indian population where both fungal and bacterial pathogens are common, despite a 50% culture-positive rate, over 95% of samples revealed a potential pathogen using directed 16S or 18S PCR.12 Therefore, the low culture-positive rate likely reflects the poor sensitivity of culture for detection of pathogenic organisms from the ocular surface rather than a high incidence of truly pathogen-negative ulcers. As such, it would seem that there is substantial room for improvement in diagnostic technology. In our case study, the primary concern of the clinician is that their patient’s vision may be reduced beyond salvage if empirical antibiotics do not adequately treat the infection. Culture results are not expected to return for some days, and there is certainly no guarantee that the results will be actionable in a way that will inform patient care.
The limits of conventional microbiology
The difficulties in identifying pathogens involved in infectious corneal ulcers broadly illustrate the limitations of conventional diagnostic microbiology, which continue to rely on phenotypic methods for detection and subsequent analysis. These workflows are based on our ability to recover a viable organism(s) in culture, combined with other tests based on microbial morphology (e.g. staining) and biochemical properties (e.g. antigen testing and serology).13 Recent advances have expanded this traditional repertoire, such as the introduction of polymerase chain reaction (PCR) assays and oligonucleotide microarrays. Enhanced automation of laboratory practices has also been a key development, seen with the widespread adoption of matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry for rapid pathogen speciation.13,14 Despite these improvements, the limitations of conventional microbiology remain principally related to the need to culture the organism. For slow-growing, indolent and/or fastidious organisms, cultivation under strict conditions is a time-consuming and costly process.15 Clinically significant pathogens may escape detection altogether if they do not proliferate on the selected culture media (the so-called “great plate anomaly”16), or if growth is suppressed by preceding antimicrobial use. Assuming a pathogen has been grown in culture, its identification marks only the beginning of the diagnostic cascade. In the current era where antibiotic resistance is now common, determination of antimicrobial susceptibilities is almost always required to inform patient treatment, and further typing with multilocus sequencing (MLST), pulsed-field gel electrophoresis (PFGE) and/or microarrays may be necessary for pathogens of public health significance.17 The limitations of current diagnostic paradigms in clinical microbiology for the cornea, therefore, have prompted interest in whether our current armory of tests can be consolidated into a single assay to produce rapid and clinically actionable results, with the added benefit of improving our understanding of infectious ocular diseases epidemiology.
3. THE PROMISE OF NEXT GENERATION SEQUENCING IN CLINICAL MICROBIOLOGY
Case study continued
The patient, diagnosed with an infectious corneal ulcer of uncertain etiology, is commenced on topical fortified antibiotics. She agrees to the addition of topical antifungal therapy, given the severity of the ulcer and history of preceding eye trauma with organic matter. Finally, before discharge from the emergency department, she is enrolled in a research study interrogating a range of molecular technologies for the rapid identification of pathogens involved in corneal ulceration. Among these modalities includes metagenomic next generation sequencing. Following informed consent, one additional swab is collected from the corneal ulcer, placed into a nucleic acid buffer and frozen at −80°C. As part of the research study, IRB-approved provisions have been made to enable disclosure of the results of this study, to the discretion of the laboratory director and treating clinician, in the exceptional circumstance where the results may profoundly alter the nature of the patient’s treatment.
A primer on next generation sequencing
The term “next generation sequencing” (NGS) refers to a broad and evolving range of high-throughput sequencing technologies which enable massive and parallel sequencing of nucleic acids contained within specimens of interest.18,19 Sanger sequencing, the precursor to NGS, was developed in the 1970s based on the DNA polymerase-mediated insertion of dideoxynucleotide chain terminators, now with the addition of fluorescent nucleotides to singular denatured strands.20 Automation of this technique with capillary electrophoresis allowed for completion of the Human Genome Project in 2003.21 However, comparatively high cost, labor intensity, protracted turnaround time, and reaching the limits of sequencing read length (<1000 base pairs) limited the applications of this technology. This changed with the development of microfluidic array-based pyrosequencing by 454 Life Sciences (later acquired by Roche) which ushered in the era of massively parallel short read DNA sequence determination. Unlike the Sanger method, massively parallel sequencing allowed the order of millions of nucleotides in DNA fragments to be determined in a simultaneous and independent fashion,22 ushering in the era of NGS. Sanger sequencing is still used in select circumstances, including as a verification tool to ascertain the accuracy of NGS reads, and in covering regions of the genome which may not be readily amenable to NGS.23 However, the improved automation and vastly reduced cost of NGS technologies have made them the methods of choice for most applications.24
Approaches to next generation sequencing
There are now multiple commercially available sequencing platforms, each with differences in sequencing chemistry, sequencing reagents, read length, error rate, and compatibility with various computational pipelines.25 The basic unit of data generated by metagenomics studies are known as “reads”, which correspond to sequence determinations for DNA/RNA fragments. Broadly, there are two major approaches to diagnostic NGS, each with differing indications, advantages and limitations: targeted amplicon sequencing and whole genome “shotgun” metagenomics. Targeted amplicon sequencing, as its name implies, involves primer-mediated amplification of specific genomic targets (e.g. 16S rRNA for bacteria,26,27 and 18S rRNA and other targets for eukaryotes28,29) suspected of being present in the sample of interest.30 Selective amplification and sequencing can also be useful for probing genomic regions of special interest, such as loci that confer antimicrobial resistance.31 Recently, this targeted approach was successfully leveraged to examine viral genomes in studies of the molecular epidemiology of Zika32,33 and Ebola34 outbreaks in the Americas and West Africa, respectively. While generally less expensive and often providing more depth in complex microbial communities, targeted sequencing using a single primer set is unable to interrogate pathogens across multiple microbial kingdoms. By sequencing only highly conserved genes, such as 16S rRNA, it provides low taxonomic resolution, often restricted to identification of microorganisms at the genus level. Furthermore, targeted assays for low abundance organisms often generate false-positive results.35
In contrast, metagenomic NGS (mNGS) involves the indiscriminate amplification of all nucleic acids contained within biological specimens, and as such is a culture-independent, agnostic approach to clinical diagnostics.36,37 Although a consequence of this untargeted approach is that the vast majority of reads are derived from the host, mNGS with the appropriate depth and coverage may offer quantifiable phylogenetic identification of both known and unknown microorganisms present within a specimen. In the last decade, mNGS has been used as a last-resort diagnostic modality to detect pathogens in patients with a range of severe systemic illnesses,38-43 for whom conventional microbiology has failed, often on multiple occasions, to identify an infectious agent. These studies have also formed the basis of molecular epidemiology studies investigating biogeographical and spatial distributions of pathogens in the context of their metagenome, and have enabled high resolution evolutionary and outbreak tracing.36 More recently, RNA-sequencing methods are now allowing scientists to study transcriptional dynamics and alterations in host and pathogen gene expression which occur in the setting of infection.44-47 Hypothesis-driven efforts are now under way to identify host and/or pathogen-specific biomarkers which may be helpful in the diagnosis and monitoring of infectious processes.48,49 In particular, the analysis of pathogen RNA expression, an indicator of a metabolically active organisms, may allow us to distinguish viable and non-viable or dead microbes found within specimens.
Workflows in next generation sequencing
Sequencing workflows all share the same aim: to produce accurate reads which are later used to reconstruct microbial genomes (Figure 1B). Conceptually, these workflows consist of “wet-lab” and “dry-lab” processes.50-54 Briefly, the wet-lab involves nucleic acid extraction from the specimen of interest, followed by library preparation and sequencing. Following nucleic acid extraction (into DNA, cDNA, RNA, depending on the purpose of the study), the sample must be prepared for sequencing by a process known as library preparation. This involves fragmentation of extracted nucleic acids followed by ligation to sequencer-specific adaptors, and PCR-mediated clonal expansion.50 The sequencing process itself is relatively straightforward and has been the subject of excellent reviews elsewhere.24,55 Most sequencing platforms produce short reads (50 – 1000 base pairs), but a third generation of sequencers, led by Oxford Nanopore and Pacific Biosciences, are now able to produce longer reads. These are typically less accurate but more analytically conducive to de novo microbial genome assembly.56 The bottleneck in most mNGS workflows lies in the computational analysis of sequencing data. A simplistic explanation of this process for clinical metagenomics involves assigning base calls, generation of reads, removal of host nucleic acid sequences and configuring overlapping reads into “contigs” of ever-increasing length.18 Assembling contigs may occur with the assistance of a reference database, or performed de novo if no reference database exists. Particularly for rare pathogens, accurate assembly is important for pathogen identification, classification of taxonomy, and detection of antimicrobial resistance genes. However, the database of sequenced pathogens is growing exponentially, meaning that less and less new sequence information will be required to make accurate calls of both pathogen and resistance phenotype, which for the most common pathogens will be possible by simply matching short k-mer sequences.57 Because of the risk of amplifying and detecting contaminating sequences, strict lab-specific quality control measures are required at every step.
4. VALIDATION AND INTERPRETATION OF NEXT GENERATION SEQUENCING ASSAYS
Case study continued
Fortunately, the patient improves with topical antimicrobial therapy. The final culture results are negative, except for very light growth (1 “C” streak) of coagulase negative staphylococci in the blood agar, considered by the clinician to be a contaminant. With no actionable culture results to determine microbial etiology, the patient is continued on all medications with a plan to wean her as she improves clinically. During a microbial sequencing board meeting with physicians, microbiologists, bioinformaticians and sequencing technicians, the patient’s results are discussed. Following sequencing on the Illumina HiSeq 2500, dry lab analysis with a commonly used computational pipeline, Centrifuge,58 analyzes a total of 34,465,879 reads, of which 30,345,567 (88.0%) are of human origin, 3,101,929 (9.0%) microbial, 72,045 (0.2%) derived from known contaminants also present in the control, and the remaining 946,338 (2.7%) classified as “ambiguous”. Of the microbial reads, 2,894,345 (93.3%) are determined to be of bacterial in origin; 21,990 (0.7%) viral; 42,714 (1.4%) fungal; 782 (0.03%) protozoan and 99 (0.003%) amoebazoan. An additional 141,999 (4.6%) are considered “indeterminate” as they could not be aligned to a reference database. Among the bacterial reads, Pseudomonas aeruginosa is captured by 56.4%, followed by Staphylococcus aureus in 24.3%, and coagulase negative staphylococci by the remaining 19.3%. Curiously, most of the fungal reads are mapped to Verticillium spp., a vanishingly rare cause of human disease, and the majority of viral reads are attributed to the presence of torque teno virus (TTV).11
Interpreting diagnostic metagenomics
One of the inherent challenges facing clinicians is the tension that arises between current diagnostic paradigms and those arising from clinical metagenomics. Traditionally, clinicians have been taught that investigations of any sort should be conducted only if the pre-test probability of a positive (or negative) result surpasses an accepted professional and/or eminence-declared threshold, and if the results of the investigation may alter the patient’s care. Moreover, clinicians, microbiologists and indeed patients, have grown accustomed to the relatively simple output generated by traditional culture-based assays, which is most frequently documented in binary terms (growth vs. no growth; detected vs. not detected; susceptible vs. resistant).59 In developing clinical metagenomics however, we are asked to relinquish our expectations of what an investigation may reveal, and are left vulnerable to unexpected or difficult-to-interpret results. This may complicate, rather than aid, patient care. The embarrassment of riches represented by the sheer volume of data produced, which requires expertise to interpret correctly. In the absence of workflow validation, one might argue that the data cannot be reliably interpreted at all. Amid the potentially overwhelming torrent of various heat maps of read counts and coverage identity plots that may be included in an automated sequencing summary, it is tempting to implicate P. aeruginosa as the cause of this patient’s corneal ulcer. However, there are sources of uncertainty which do not inspire complete confidence in this result. The curse of data-repletion is shown by the breadth of reads aligned to multiple microbial kingdoms and multiple bacteria, and the hint of novel microbes not previously known to cause corneal infection. The task therefore becomes one of disentangling true and false results, a process which requires very careful consideration of the following issues.
The need for validation
The enthusiasm which initially greeted the successful use of mNGS as a last-resort attempt to diagnose rare and potentially lethal systemic infections has now been tempered by growing recognition that rigorous validation standards must be met in order for such tests to enter routine clinical care.60 As a general concept, validation refers to the extent to which a test performs as intended.61-63 In the United States, Laboratory Developed Tests (LDTs) must comply with two complementary sets of regulations set out by the Clinical Laboratory Improvement Amendments (CLIA) program, overseen by the Centers for Medicare and Medicaid Services (CMS), and the Food and Drug Administration (FDA).64 CLIA certification involves a laboratory-specific assessment of the test’s analytical validity, by evaluating a number of performance quality metrics including the limit of detection, precision, interfering substances and accuracy which must be determined in order for these assays to be conducted and interpreted correctly.65,66 On the other hand, the FDA is responsible for performing pre-market assessments of both analytical and clinical validity of in vitro diagnostic tests. A clinically valid test is one which has met pre-established sensitivity and specificity benchmarks in detecting the presence and absence of a given disease or condition. Although no metagenomics assays have yet been FDA-approved for the general market – perhaps reflecting the need for ongoing, iterative validation efforts – several laboratories have now been CLIA-certified to sequence specimens for clinical care.62,67-69 In one recent multicenter review, a University of California San Francisco (UCSF) metagenomics pipeline detected 31/57 (54.3%) of all infectious cases of meningoencephalitis.70 While this figure did not reach the estimated sensitivity of 73% in their laboratory validation study,69 the investigators were still able to demonstrate the important adjunct role of their metagenomics pipeline to detect pathogens which had otherwise escaped detection with conventional culture and PCR.
Limits of detection
In clinical metagenomics, sequencing capability is determined by the so-called “limits of detection” (LOD), which can be thought of as the requisite mass of extracted DNA/RNA required for downstream detection.71 For instance, to define the 95% LOD of a particular assay, one would need to determine the lowest concentration of starting nucleic acid required to detect pathogen-specific reads in 95% of true positive samples.69 The LOD must be determined for the range of pathogens thought to be implicated in a disease process. For the cornea, this will involve establishing the amount of viral, bacterial, fungal and parasitic-derived nucleic acids needed for a particular assay to detect their signal, and enriching this template volume if their expected abundance does not convincingly exceed the established LOD.24 One common method of determining the LOD is by spiking serial dilutions of microbial DNA/RNA into patient samples prior to sequencing and read analysis.72-74 Establishing the LOD for sequencing assays is a time-consuming and laborious process, but without this crucial step, it is not possible to categorically conclude the absence of a pathogen merely because no signal was detected.72 One inherent difficulty in this process relates to the size of microbial genomes when compared to that of humans. For example, one torque teno virion genome is 3.7kb, which is on the order of one millionth of the size of one human haploid genome. In an unenriched environment, 1 million human reads would be expected for every TTV assuming a multiplicity of infection of 1:1. Furthermore, if the LOD for a particular pathogen is greater than expected for a particular assay, then presumably this might lead to an undesirable proportion of false-negative results. In such cases, there would be a need to optimize the sequencing pipeline to lower the LOD to increase the sensitivity of the test to detect reads derived from the pathogen in question.
Diagnostic thresholds: what constitutes a positive test?
As with any diagnostic test, accurate thresholds are required to define true positive findings. However, no universal reference standards exist for mNGS. Clearly, a number or percentage of organism-specific reads is required to identify positive results with confidence,72 but establishing validated diagnostic thresholds may only be possible by performing a multitude of controlled sequencing runs with standardized sample collection and sequencing protocols. Although noticeably absent from many mNGS studies, the meticulous creation of positive and negative controls is a vital part of this process. The favored method to create positive controls is the use of mock microbial communities containing known concentrations of genomic material from multiple taxa, for which well-curated reference databases have been completed.75 Negative controls may include sequencing runs on blank samples, and also samples derived from uninfected sites (e.g. the fellow healthy eye) to inform the baseline microbial consortium of the patient. By comparing the presumed reference standard to the values generated by the assays performed on patient samples, an empirical estimate of uncertainty may be made as to the likelihood of a positive (or negative) test. Spiked-in internal controls may also assist in calibrating sensitivity and specificity thresholds,76 particularly in situations where the microbial burden from a patient sample is expected to be low.65
Establishing reference standards may be particularly difficult for infectious corneal ulcers for several reasons. In our case study, the relative proportions of microbial reads should be interpreted with caution. While it is true that the ocular surface permits direct sampling in a way that avoids the interpretational difficulties associated with specimens which pass through different anatomical zones prior to collection (e.g. stool and sputum), the paucimicrobial nature of the ocular surface (Figures 2 and 3) presents a unique bioinformatic challenge. Paucimicrobial samples are known to introduce artefactual amplification of microbial populations,35 and the effect of this amplification may be compounded by nucleic acid enrichment77 and/or host depletion78,79 during the library preparation phase. In addition, the relative proportions of microbial reads may be the result of bias towards certain organisms within the reference database, or if dry lab analysis cannot distinguish genetically similar taxa.19 If reference genomes are not available, incomplete, and/or inaccurate, it may simply not be possible to determine diagnostic thresholds for certain organisms, the reads of which may be binned as “ambiguous”. In an idealized setting, these thresholds would rely on scalable metagenomics pipelines80 capable of automated updates from genome repositories (e.g. NCBI GenBank), combined with the ability to cross-check other heuristics for result confirmation and/or interrogation of anomalous findings. In the end, reference standards must be sufficiently robust to meet clinically actionable criteria. They must reliably discriminate between reads derived from pathogens and non-pathogens, including commensal organisms,81,82 reagent and/or environmental background,83 and spiked-in controls. They must also be flexible enough to enable confident diagnosis of polymicrobial infections,84 which are likely more common in the cornea than is appreciated. Finally, they must give some indication as to the potential significance of reads attributed to novel pathogens, particularly those not known to be associated with the disease of interest. Sequencing data may be rendered effectively uninterpretable without these stringencies.
FIGURE 2: Rarefaction curves of publicly available metagenomic samples from healthy subjects, retrieved from the NCBI Sequence Read Archive (SRA), and analyzed using the MG-RAST (metagenomics Rapid Annotation using Subsystem Technology)123 bioinformatics pipeline:
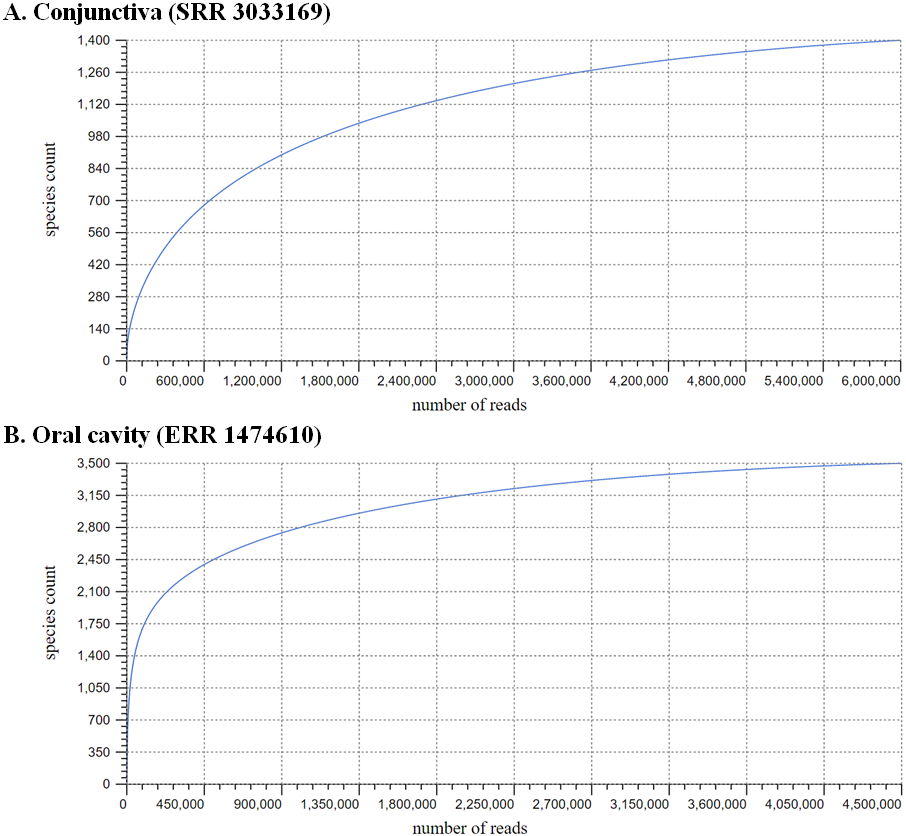
(A) Conjunctival microbiome (SRR3033169; post-quality control sequence count: 4,680,480) and (B) oral salivary microbiome (ERR1474610; post-quality control sequence count: 3,902,367). The respective curves demonstrate the vast discrepancy in species count per read number between the two samples, reflecting the paucimicrobial nature of the conjunctiva compared to the gastrointestinal tract. The MG-RAST-derived alpha-diversity scores of 7 and 84, respectively, are measures of species richness within the samples.
FIGURE 3: Krona124 chart visualizations of bacterial metagenomes retrieved from the:
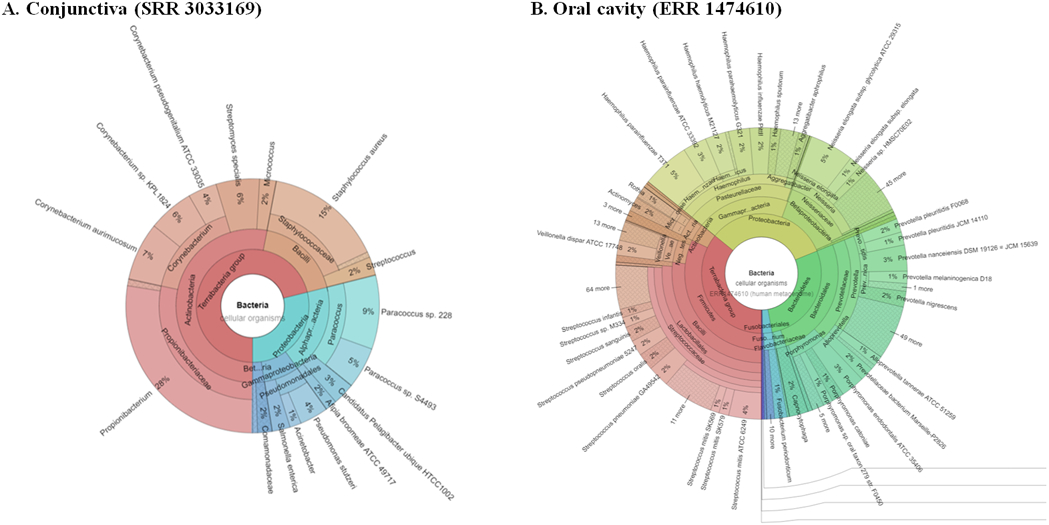
(A) conjunctiva (SRR3033169; left) and (B) oral saliva (ERR1474610; right), offering a bird’s eye view of taxonomic distributions in their respective bacterial communities. The biogeographical distribution of these species is likely the result of multiple factors, including fundamental differences in their environmental and ecological niches, as well as the directness of sampling offered by the ocular surface when compared to the gastrointestinal tract.
Interfering substances: contamination from the reagent-associated microbiome
In recent years, it has become increasingly clear that the reagents and collection devices used in extraction, amplification, and/or library preparation are a significant source of contaminating nucleic acids, usually derived from ubiquitous microbes found in water and soil.83,85,86 For specimens with low microbial load, the possibility of reads sourced from these contaminants may simply overwhelm those derived from the pathogen of interest, distorting the proportion of microbial populations represented in the sample.87 The hallmark features of sequencing contamination include: the presence of common contaminating organisms (for a list, see Salter et al., 201488); batch effects;89 poor intra- and inter-run reproducibility, and results that may defy common understanding of microbial ecology.83 The potential for contamination needs to be thoroughly assessed before any conclusions are reached regarding the taxonomic distributions observed. Past attempts to minimize reagent contamination have utilized various techniques, including radiation90 treatment and restriction enzymes,91 but these methods have been met with mixed success.88
Current recommendations to handle reads generated from sources of contamination involve careful laboratory documentation and creation of batched positive and negative controls at numerous points along the sequencing workflow.88,92 These controls are vital in understanding the nature of background taxa found within each batch of storage media, collection devices, extraction and library preparation kits. Even seemingly innocuous disposables involved in the sequencing process, such as microtiter wells, have been found to be a source of extraneous nucleic acids.93 In addition, several computational pipelines have now made it possible to attribute contaminant-derived reads during the analytical process.94 Results which challenge biological plausibility, but which may still be of interest, require confirmatory testing before reads are called. Such confirmation has been typically performed with best attempts at culture, PCR,95 use of validated fluorescence in-situ hybridization assays,96 or with Sanger sequencing.20,97 More recently, software assisted manual review of sequencing data has become a possibility.98 In our case study, it is conceivable that the presence of nucleic acids attributed to Verticillium spp. could have originated in the reagents or consumables used during sequencing. Without establishing the appropriate diagnostic thresholds, and understanding the nature of background noise, however, the clinician is left to question its true relevance for their patient.
Determining antimicrobial susceptibilities
Sophisticated computational pipelines such as Taxonomer99, SURPI100 and SMART80 have demonstrated the capacity to identify antimicrobial resistance genes from sequencing data, while alternative approaches have involved RNA-based transcriptomic profiling of these genes.101 However, the extent to which the in silico presence of these genes and/or associated transcripts confers phenotypic in vitro resistance is still controversial. Studies of genotypic antimicrobial susceptibilities have identified a spectrum of resistance genes for important organisms such as Staphylococcus aureus102 and Mycobacterium tuberculosis,103,104 but sequencing assays do not always reliably detect these loci with a great deal of confidence.105 Combined with the potential for discrepant phenotypic antimicrobial resistance studies,106,107 these uncertainties may again confound clinical care. One might wonder whether clinicians and microbiologists will be able to accurately interpret the relevance of detected antimicrobial resistance genes, knowing that their full spectrum and importance have yet to be elucidated. To complicate matters, the significance of these markers may once again depend on pre-specified thresholds, and determining the number of associated reads which may be predictive of phenotypic resistance. Reference databases will require routine updates to accurately reflect the dynamic nature of antimicrobial resistance acquisition among clinically important pathogens. Until these issues are resolved, we will continue to rely on other methods of antimicrobial susceptibility testing (e.g. automated MALDI-TOF108,109), which are themselves limited by virtue of being culture-dependent.
5. IMPLEMENTATION OF CLINICAL METAGENOMICS FOR INFECTIOUS CORNEAL ULCERS
Implementation of clinical research in real-time patient care: a question of ethics?
Beyond its technical and interpretational challenges, the sheer volume of data generated by clinical metagenomics may also affect its implementation within clinical practice. The question of whether the results of sequencing-based research should be made available for real-time patient care has now become a topic of intense ethicolegal debate, particularly with regard to incidental findings and the protection of patient privacy. As sequencing technologies become increasingly robust and more powerful, the chances that results may contain clinically pertinent information also increase. In cases where these data may fundamentally alter patient management, a growing trend amongst some academic circles has been to seek explicit permission from their respective ethics committees to make these results available to the clinician. At the same time, the surplus of data afforded by sequencing raises the possibility, or perhaps even certainty, of revealing incidental findings that may not be directly related to the reason why sequencing was performed in the first instance.110 Consider the ethical quandaries which may arise if sequencing uncovers known disease-causing mutations which may be of life-altering consequence to the patient and their family, or if bloodborne infections such as HIV are revealed either by direct detection of the virus and/or its transcriptional footprints. The gravity of these findings are such that clinicians can ill-afford any doubt as to their accuracy, lest erroneous results are released to the patient. While the likelihood of these chance discoveries may be reduced by discarding host reads, and/or depleting host nucleic acids during library preparation, these safeguards should not be viewed as failsafe or even desirable. As discussed earlier, analyzing these reads provides important information regarding the relative abundance of pathogen to host within clinical specimens, and key host-related factors involved in infectious processes.
In addition, one must also consider the perils associated with the public release of clinical metagenomics data, which may contain personally identifiable Health Insurance Portability and Accountability Act of 1996 (HIPAA) information. The risk of inadvertently releasing such information is not solely restricted to consideration of host reads. Pathogen genome sequences, too, may be attached to dates of acquisition, dates of processing, location and other parameters (e.g. single nucleotide polymorphisms) which can be quickly traced back to patients.111,112 Freely available metadata and genome libraries represent a greater threat to cyber-security and patient privacy than generally appreciated, and it is not outside the realm of possibility that sequencing data, in the wrong but capable hands, may compromise the integrity of whole healthcare systems in the future. Regulations surrounding the use and propagation of sequencing data may differ between legal jurisdictions, but the need to share data in the interests of scientific enquiry must always be weighed against the risks this may pose to patient privacy. These issues highlight the importance of appropriate regulatory oversight: the covert risks of “big data” demand comprehensive guidelines related to the acquisition of informed patient consent, responsible handling and storage of data, and provisions on corrective actions that must be undertaken if these standards are breached.
Regulation of clinical metagenomics
Recognizing the unprecedented complexity of technical and ethicolegal issues surrounding sequencing-based diagnostic tests, the US Food and Drug Administration (FDA) recently published draft guidelines on the metrics against which test performance and safety are to be evaluated.113 As stated earlier, the FDA regards sequencing-based tests as in vitro diagnostics (IVD) subject to an array of pre-market and post-market regulatory controls.18 With patient safety being the primary focus of these regulations, diagnostic tests must pass strict FDA analytical and clinical validation standards, and must demonstrate the ability to produce immediate and actionable results with an acceptable false-positive and false-negative error rate. However, some critics have questioned the authority and legitimacy of the FDA in regulating these tests, particularly as researchers are already bound by the tight regulations set by the CMS and CLIA.114 It has been argued that if clinical laboratories are already required to meet CLIA guidelines concerning analytical validation, reproducibility and quality assurance, one might question whether FDA requirements are redundant rather than complementary. One particular fear that has been voiced is that the costs of meeting dual regulations may in the end stifle sequencing innovation,115 particularly as industry giants seek to monopolize the field.
These criticisms ultimately stem from misunderstandings as to the respective mandates of CMS/CLIA and the FDA, and their role in regulating emerging diagnostic technologies. This is because the FDA is exclusively positioned to assess both analytical and clinical validity, and establish inter-laboratory performance and data quality standards in a way laboratory-specific CLIA-certifications do not. For instance, the FDA-led public release of an evolving database of curated microbial genomes, FDA-ARGOS,116 now provides regulatory-grade reference material for infectious diseases diagnostics in a way that relieves the testing burden on resource-limited researchers. Furthermore, it is likely that the FDA will become heavily involved in post-market surveillance of mNGS, which will be vital in establishing requirements regarding proficiency testing, and in uncovering flaws in diagnostic system design and safety. Given the relative infancy of clinical metagenomics however, and the lack of definitive regulatory pathways, one wonders whether the field may benefit from the publication of consensus guidelines by key governing bodies (FDA, CMS), relevant stakeholders such as the Centers of Disease Control and Prevention (CDC), and relevant medical and/or scientific associations such as the College of American Pathologists.117 This collaboration may have the benefit of streamlining benchmark performance thresholds for the seemingly infinite number of sequencing and analytical workflows under current development.118 It may also give rise to much-needed institutionalized norms regarding the ethical and fair use of sequencing data in clinical and research settings, which can be used to inform decision making by ethics review committees.
Costs of sequencing
Although it is true that the per-base and per-run costs of sequencing continue to decrease, most metagenomics assays still remain prohibitively expensive for most applications. As most research endeavors operate under financial constraint, batching samples and sending them to offsite genomic cores has long been the most common approach to sequencing. However, the demand for short turn-around times in diagnostic testing for infectious diseases means that clinical microbiology labs will soon wrestle with the idea of establishing high-cost in-house sequencing facilities. In addition to the costs associated with wet-lab procedures, significant investment must also be made to either purchase and continually update the computational infrastructure and software capable of processing enormous amounts of sequencing data,119 or utilize scalable cloud-based bioinformatics platforms120,121 which are typically more affordable. Furthermore, because current diagnostic paradigms in clinical microbiology are driven by low cost and high volume testing, proposals to introduce molecular tests into clinical care will almost certainly be subject to analysis using the techniques of utilization management.105,122 Sequencing might not ever be as inexpensive as routine microbiological tests, but cost-effectiveness studies might evaluate whether sequencing overhead can be feasibly offset by its ability to achieve rapid and accurate diagnoses, which may in turn prevent costly patient complications. For infectious corneal ulcers, cost utility might be determined by a practical demonstration that quicker, more rapid diagnosis of infectious etiologies results in fewer vision-threatening events, the most severe of which may result in complete vision loss and/or need for lifelong follow-up care. Even then, it is likely that some degree of bureaucratic wrangling will occur between academic institutions, hospitals and medical insurance companies regarding reimbursement policies, which remain heavily tilted towards hypothesis-driven testing.
THE FUTURE OF CLINICAL METAGENOMICS FOR INFECTIOUS CORNEAL ULCERS
As the promise of clinical metagenomics becomes a reality, it is almost certain that attempts will soon be made to introduce sequencing for the diagnosis of a whole spectrum of infections. Invariably, our attention will turn to the application of this technology to characterize infections for which current tests are inadequate. For infectious corneal ulcers, sequencing workflows must address the challenges posed by low volume, paucimicrobial patient samples that are prone to contamination, and developing an understanding of how these factors might affect the diagnostic validation process and interpretation of results. Beyond these technical questions, clinicians and microbiologists may be overwhelmed by the repletion of data afforded by sequencing, an issue which may be generalizable to other infectious ocular diseases and infections overall. This will be of particular importance for the next decade, as we establish the experience base needed to validate this new technology. We must ensure that we are equipped to handle these data in a manner which is beneficial to patient care, and also comply with emerging ethical and legal standards related to the disclosure of incidental findings and safe data sharing. However, over the horizon a day can be envisaged when the rapidly growing sequencing databases are so current and complete, that only a small amount of new sequence data will be required to identify with high accuracy within the database a matching pathogen and its phenotype. The challenge now is to collectively build that database, along with the validating clinical experience, that will inform the calculus that will be used to quickly and simply make accurate pathogen calls in the future. By negotiating the interpretational and implementation-related challenges that stand in the way, perhaps best achieved with a consortium-based approach, we will be better placed to fulfil our collective mandate to save sight in patients affected by this common and often devastating infection.
REFERENCES
- 1.Malthus TR. An Essay on the Principle of Population. 1872. [Google Scholar]
- 2.Ung L, Bispo PJM, Shanbhag SS, Gilmore MS, Chodosh J. The persistent dilemma of microbial keratitis: Global burden, diagnosis, and antimicrobial resistance. Surv Ophthalmol. 2019;64(3):255–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loman N, Pallen M. XDR-TB genome sequencing: a glimpse of the microbiology of the future. 2008. [DOI] [PubMed] [Google Scholar]
- 4.Flaxman SR, Bourne RR, Resnikoff S, et al. Global causes of blindness and distance vision impairment 1990–2020: a systematic review and meta-analysis. The Lancet Global Health. 2017;5(12):e1221–e1234. [DOI] [PubMed] [Google Scholar]
- 5.Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Survey of ophthalmology. 2012;57(5):448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness: a global perspective. Bulletin of the world health organization. 2001;79:214–221. [PMC free article] [PubMed] [Google Scholar]
- 7.Pimentel MA, Browne EN, Janardhana PM, et al. Assessment of the accuracy of using ICD-9 codes to identify uveitis, herpes zoster ophthalmicus, scleritis, and episcleritis. JAMA ophthalmology. 2016;134(9):1001–1006. [DOI] [PubMed] [Google Scholar]
- 8.Collier SA, Gronostaj MP, MacGurn AK, et al. Estimated burden of keratitis—United States, 2010. MMWR Morb Mortal Wkly Rep. 2014;63(45):1027–1030. [PMC free article] [PubMed] [Google Scholar]
- 9.Dahlgren MA, Lingappan A, Wilhelmus KR. The clinical diagnosis of microbial keratitis. American journal of ophthalmology. 2007;143(6):940–944. e941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones DB, Liesegang TJ, Robinson NM, Washington JA. Laboratory diagnosis of ocular infections. Washington DC: American Society for Microbiology; 1981. [Google Scholar]
- 11.Lee AY, Akileswaran L, Tibbetts MD, Garg SJ, Van Gelder RN. Identification of torque teno virus in culture-negative endophthalmitis by representational deep DNA sequencing. Ophthalmology. 2015;122(3):524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim E, Chidambaram JD, Srinivasan M, et al. Prospective comparison of microbial culture and polymerase chain reaction in the diagnosis of corneal ulcer. American journal of ophthalmology. 2008;146(5):714–723. e711. [DOI] [PubMed] [Google Scholar]
- 13.Fournier P-E, Drancourt M, Colson P, Rolain J-M, La Scola B, Raoult D. Modern clinical microbiology: new challenges and solutions. Nature Reviews Microbiology. 2013;11(8):574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertelli C, Greub G. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clinical Microbiology and Infection. 2013;19(9):803–813. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura S, Maeda N, Miron IM, et al. Metagenomic diagnosis of bacterial infections. Emerging infectious diseases. 2008;14(11):1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annual review of microbiology. 1985;39:321–346. [DOI] [PubMed] [Google Scholar]
- 17.Deurenberg RH, Bathoorn E, Chlebowicz MA, et al. Application of next generation sequencing in clinical microbiology and infection prevention. Journal of biotechnology. 2017;243:16–24. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg B, Sichtig H, Geyer C, Ledeboer N, Weinstock GM. Making the leap from research laboratory to clinic: challenges and opportunities for next-generation sequencing in infectious disease diagnostics. MBio. 2015;6(6):e01888–01815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annual Review of Pathology: Mechanisms of Disease. 2019;14:319–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the national academy of sciences. 1977;74(12):5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins FS, Morgan M, Patrinos A. The Human Genome Project: lessons from large-scale biology. Science. 2003;300(5617):286–290. [DOI] [PubMed] [Google Scholar]
- 22.Heather JM, Chain B. The sequence of sequencers: The history of sequencing DNA. Genomics. 2016;107(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagemann IS. Overview of Technical Aspects and Chemistries of Next-Generation Sequencing. Clinical Genomics: Elsevier; 2015:3–19. [Google Scholar]
- 24.Forbes JD, Knox NC, Peterson C-L, Reimer AR. Highlighting clinical metagenomics for enhanced diagnostic decision-making: a step towards wider implementation. Computational and structural biotechnology journal. 2018;16:108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mardis ER. Next-generation sequencing platforms. Annual review of analytical chemistry. 2013;6:287–303. [DOI] [PubMed] [Google Scholar]
- 26.Fadrosh DW, Ma B, Gajer P, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of clinical microbiology. 2007;45(9):2761–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Filippis F, Laiola M, Blaiotta G, Ercolini D. Different amplicon targets for sequencing-based studies of fungal diversity. Appl. Environ. Microbiol 2017;83(17):e00905–00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banos S, Lentendu G, Kopf A, Wubet T, Glöckner FO, Reich M. A comprehensive fungi-specific 18S rRNA gene sequence primer toolkit suited for diverse research issues and sequencing platforms. BMC microbiology. 2018;18(1):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pollock J, Glendinning L, Wisedchanwet T, Watson M. The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol 2018;84(7):e02627–02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefterova MI, Suarez CJ, Banaei N, Pinsky BA. Next-generation sequencing for infectious disease diagnosis and management: a report of the Association for Molecular Pathology. The Journal of Molecular Diagnostics. 2015;17(6):623–634. [DOI] [PubMed] [Google Scholar]
- 32.Faria NR, Quick J, Claro I, et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. 2017;546(7658):406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thézé J, Li T, du Plessis L, et al. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell host & microbe. 2018;23(6):855–864. e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doan T, Akileswaran L, Andersen D, et al. Paucibacterial microbiome and resident DNA virome of the healthy conjunctiva. Investigative ophthalmology & visual science. 2016;57(13):5116–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pallen MJ, Loman NJ, Penn CW. High-throughput sequencing and clinical microbiology: progress, opportunities and challenges. Current opinion in microbiology. 2010;13(5):625–631. [DOI] [PubMed] [Google Scholar]
- 38.Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. New England Journal of Medicine. 2014;370(25):2408–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown JR, Bharucha T, Breuer J. Encephalitis diagnosis using metagenomics: application of next generation sequencing for undiagnosed cases. Journal of infection. 2018;76(3):225–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukui Y, Aoki K, Okuma S, Sato T, Ishii Y, Tateda K. Metagenomic analysis for detecting pathogens in culture-negative infective endocarditis. Journal of Infection and Chemotherapy. 2015;21(12):882–884. [DOI] [PubMed] [Google Scholar]
- 41.Imai A, Gotoh K, Asano Y, et al. Comprehensive metagenomic approach for detecting causative microorganisms in culture-negative infective endocarditis. International journal of cardiology. 2014;172(2):e288–e289. [DOI] [PubMed] [Google Scholar]
- 42.Lelouvier B, Servant F, Delobel P, Courtney M, Elbaz M, Amar J. Identification by highly sensitive 16S metagenomic sequencing of an unusual case of polymicrobial bacteremia. Journal of Infection. 2017;75(3):278–280. [DOI] [PubMed] [Google Scholar]
- 43.Gyarmati P, Kjellander C, Aust C, Kalin M, Öhrmalm L, Giske CG. Bacterial landscape of bloodstream infections in neutropenic patients via high throughput sequencing. PloS one. 2015;10(8):e0135756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Westermann AJ, Förstner KU, Amman F, et al. Dual RNA-seq unveils noncoding RNA functions in host–pathogen interactions. Nature. 2016;529(7587):496. [DOI] [PubMed] [Google Scholar]
- 45.Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nature Reviews Microbiology. 2012;10(9):618. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews genetics. 2009;10(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaas AK, Chen M, Varkey J, et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell host & microbe. 2009;6(3):207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gliddon HD, Herberg JA, Levin M, Kaforou M. Genome-wide host RNA signatures of infectious diseases: discovery and clinical translation. Immunology. 2018;153(2):171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dix A, Vlaic S, Guthke R, Linde J. Use of systems biology to decipher host–pathogen interaction networks and predict biomarkers. Clinical Microbiology and Infection. 2016;22(7):600–606. [DOI] [PubMed] [Google Scholar]
- 50.Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clinical microbiology and infection. 2018;24(4):335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buermans H, Den Dunnen J. Next generation sequencing technology: advances and applications. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2014;1842(10):1932–1941. [DOI] [PubMed] [Google Scholar]
- 52.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 2016;17(6):333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loman NJ, Constantinidou C, Chan JZ, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nature Reviews Microbiology. 2012;10(9):599. [DOI] [PubMed] [Google Scholar]
- 54.Pollard MO, Gurdasani D, Mentzer AJ, Porter T, Sandhu MS. Long reads: their purpose and place. Human molecular genetics. 2018;27(R2):R234–R241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vincent AT, Derome N, Boyle B, Culley AI, Charette SJ. Next-generation sequencing (NGS) in the microbiological world: How to make the most of your money. Journal of microbiological methods. 2017;138:60–71. [DOI] [PubMed] [Google Scholar]
- 56.McCoy RC, Taylor RW, Blauwkamp TA, et al. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PloS one. 2014;9(9):e106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Břinda K, Callendrello A, Cowley L, et al. Lineage calling can identify antibiotic resistant clones within minutes. bioRxiv. 2018:403204. [Google Scholar]
- 58.Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome research. 2016;26(12):1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Köser CU, Ellington MJ, Cartwright EJ, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS pathogens. 2012;8(8):e1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruppé E, Schrenzel J. Messages from the second international conference on clinical metagenomics (ICCMg2). Microbes and infection. 2018;20(4):222–227. [DOI] [PubMed] [Google Scholar]
- 61.Gargis AS, Kalman L, Berry MW, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nature biotechnology. 2012;30(11):1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nature microbiology. 2019;4(4):663. [DOI] [PubMed] [Google Scholar]
- 63.Burd EM. Validation of laboratory-developed molecular assays for infectious diseases. Clin Microbiol Rev. 2010;23(3):550–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Centers for Medicare & Medicaid Services. CLIA Overview. 2013; https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/LDT-and-CLIA_FAQs.pdf. Accessed 9/16/2019, 2019.
- 65.Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Archives of Pathology and Laboratory Medicine. 2017;141(6):776–786. [DOI] [PubMed] [Google Scholar]
- 66.Culbreath K, Melanson S, Gale J, et al. Validation and Retrospective Clinical Evaluation of a Quantitative 16S rRNA Gene Metagenomic Sequencing Assay for Bacterial Pathogen Detection in Body Fluids. The Journal of Molecular Diagnostics. 2019. [DOI] [PubMed] [Google Scholar]
- 67.Hong DK, Blauwkamp TA, Kertesz M, Bercovici S, Truong C, Banaei N. Liquid biopsy for infectious diseases: sequencing of cell-free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagnostic microbiology and infectious disease. 2018;92(3):210–213. [DOI] [PubMed] [Google Scholar]
- 68.Schlaberg R, Queen K, Simmon K, et al. Viral pathogen detection by metagenomics and Pan-viral group polymerase chain reaction in children with pneumonia lacking identifiable etiology. The Journal of infectious diseases. 2017;215(9):1407–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller S, Naccache SN, Samayoa E, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome research. 2019;29(5):831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilson MR, Sample HA, Zorn KC, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. New England Journal of Medicine. 2019;380(24):2327–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pochon X, Bott NJ, Smith KF, Wood SA. Evaluating detection limits of next-generation sequencing for the surveillance and monitoring of international marine pests. PloS one. 2013;8(9):e73935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frey KG, Herrera-Galeano JE, Redden CL, et al. Comparison of three next-generation sequencing platforms for metagenomic sequencing and identification of pathogens in blood. BMC genomics. 2014;15(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moore RA, Warren RL, Freeman JD, et al. The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS One. 2011;6(5):e19838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheval J, Sauvage V, Frangeul L, et al. Evaluation of high-throughput sequencing for identifying known and unknown viruses in biological samples. J Clin Microbiol. Sep 2011;49(9):3268–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hardwick SA, Deveson IW, Mercer TR. Reference standards for next-generation sequencing. Nature Reviews Genetics. 2017;18(8):473. [DOI] [PubMed] [Google Scholar]
- 76.Hasan MR, Rawat A, Tang P, et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. Journal of clinical microbiology. 2016;54(4):919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hall RJ, Wang J, Todd AK, et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. Journal of virological methods. 2014;195:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oyola SO, Gu Y, Manske M, et al. Efficient depletion of host DNA contamination in malaria clinical sequencing. Journal of clinical microbiology. 2013;51(3):745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee AY, Lee CS, Van Gelder RN. Scalable metagenomics alignment research tool (SMART): a scalable, rapid, and complete search heuristic for the classification of metagenomic sequences from complex sequence populations. BMC bioinformatics. 2016;17(1):292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ozkan J, Coroneo M, Willcox M, Wemheuer B, Thomas T. Identification and visualization of a distinct microbiome in ocular surface conjunctival tissue. Investigative ophthalmology & visual science. 2018;59(10):4268–4276. [DOI] [PubMed] [Google Scholar]
- 82.Shivaji S, Jayasudha R, Prashanthi GS, Chakravarthy SK, Sharma S. The Human Ocular Surface Fungal Microbiome. Investigative ophthalmology & visual science. 2019;60(1):451–459. [DOI] [PubMed] [Google Scholar]
- 83.de Goffau MC, Lager S, Salter SJ, et al. Recognizing the reagent microbiome. Nat Microbiol. 2018;3(8):851–853. [DOI] [PubMed] [Google Scholar]
- 84.Salipante SJ, Sengupta DJ, Rosenthal C, et al. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PloS one. 2013;8(5):e65226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends in microbiology. Feb 2019;27(2):105–117. [DOI] [PubMed] [Google Scholar]
- 86.Kim D, Hofstaedter CE, Zhao C, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome. 2017;5(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dekker JP. Metagenomics for clinical infectious disease diagnostics steps closer to reality. Journal of clinical microbiology. 2018;56(9):e00850–00818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC biology. 2014;12(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller RR, Uyaguari-Diaz M, McCabe MN, et al. Metagenomic investigation of plasma in individuals with ME/CFS highlights the importance of technical controls to elucidate contamination and batch effects. PloS one. 2016;11(11):e0165691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deragon JM, Sinnett D, Mitchell G, Potier M, Labuda D. Use of gamma irradiation to eliminate DNA contamination for PCR. Nucleic acids research. Oct 25 1990;18(20):6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mohammadi T, Reesink HW, Vandenbroucke-Grauls CM, Savelkoul PH. Optimization of real-time PCR assay for rapid and sensitive detection of eubacterial 16S ribosomal DNA in platelet concentrates. J Clin Microbiol. Oct 2003;41(10):4796–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bal A, Pichon M, Picard C, et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC infectious diseases. 2018;18(1):537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walker AW. A Lot on Your Plate? Well-to-Well Contamination as an Additional Confounder in Microbiome Sequence Analyses. mSystems. 2019;4(4):e00362–00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martí JM. Recentrifuge: Robust comparative analysis and contamination removal for metagenomics. PLoS computational biology. 2019;15(4):e1006967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Greninger AL, Naccache SN, Federman S, et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. Sep 29 2015;7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Drautz-Moses DI, Little PFR, Williams RBH, Cohen Y. NPJ biofilms and microbiomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu Z, Weng X, Xu C, et al. Metagenomic next-generation sequencing as a diagnostic tool for toxoplasmic encephalitis. Annals of clinical microbiology and antimicrobials. 2018;17(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muzzey D, Kash S, Johnson JI, et al. Software-assisted manual review of clinical next-generation sequencing data: An alternative to routine sanger sequencing confirmation with equivalent results in> 15,000 germline DNA screens. The Journal of Molecular Diagnostics. 2019;21(2):296–306. [DOI] [PubMed] [Google Scholar]
- 99.Flygare S, Simmon K, Miller C, et al. Taxonomer: an interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome biology. 2016;17(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Naccache SN, Federman S, Veeraraghavan N, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome research. 2014;24(7):1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khaledi A, Schniederjans M, Pohl S, et al. Transcriptome profiling of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrobial agents and chemotherapy. 2016;60(8):4722–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gordon N, Price J, Cole K, et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. Journal of clinical microbiology. 2014;52(4):1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Köser CU, Bryant JM, Becq J, et al. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. New England journal of medicine. 2013;369(3):290–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Daum LT, Rodriguez JD, Worthy SA, et al. Next-generation ion torrent sequencing of drug resistance mutations in Mycobacterium tuberculosis strains. Journal of clinical microbiology. 2012;50(12):3831–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Greninger AL. The challenge of diagnostic metagenomics. Expert review of molecular diagnostics. Jul 2018;18(7):605–615. [DOI] [PubMed] [Google Scholar]
- 106.Ocheretina O, Escuyer VE, Mabou M-M, et al. Correlation between genotypic and phenotypic testing for resistance to rifampin in Mycobacterium tuberculosis clinical isolates in Haiti: investigation of cases with discrepant susceptibility results. PloS one. 2014;9(3):e90569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ahmad S, Mokaddas E, Al-Mutairi N, Eldeen HS, Mohammadi S. Discordance across phenotypic and molecular methods for drug susceptibility testing of drug-resistant Mycobacterium tuberculosis isolates in a low TB incidence country. PLoS One. 2016;11(4):e0153563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perez KK, Olsen RJ, Musick WL, et al. Integrating rapid pathogen identification and antimicrobial stewardship significantly decreases hospital costs. Archives of Pathology and Laboratory Medicine. 2012;137(9):1247–1254. [DOI] [PubMed] [Google Scholar]
- 109.Huang AM, Newton D, Kunapuli A, et al. Impact of rapid organism identification via matrix-assisted laser desorption/ionization time-of-flight combined with antimicrobial stewardship team intervention in adult patients with bacteremia and candidemia. Clinical infectious diseases. 2013;57(9):1237–1245. [DOI] [PubMed] [Google Scholar]
- 110.Houldcroft CJ, Beale MA, Breuer J. Clinical and biological insights from viral genome sequencing. Nature Reviews Microbiology. 2017;15(3):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Greninger AL. Societal Implications of the Internet of Pathogens. Journal of clinical microbiology. 2019;57(6):e01914–01918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shean RC, Greninger AL. Private collection: high correlation of sample collection and patient admission date in clinical microbiological testing complicates sharing of phylodynamic metadata. Virus evolution. 2018;4(1):vey005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.U.S. Department of Health and Human Services Food and Drug Administration. Infectious Disease Next Generation Sequencing Based Diagnostic Devices: Microbial Identification and Detection of Antimicrobial Resistance and Virulence Markers - Draft Guidance for Industry and Food and Drug Administration Staff. 2016. FDA-2016-D-0971.
- 114.Luh F, Yen Y. FDA guidance for next generation sequencing-based testing: balancing regulation and innovation in precision medicine. npj Genomic Medicine. 2018/October/03 2018;3(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Evans BJ, Burke W, Jarvik GP. The FDA and genomic tests—getting regulation right. New England Journal of Medicine. 2015;372(23):2258–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sichtig H, Minogue T, Yan Y, et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nature communications. 2019;10(1):3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roy S, Coldren C, Karunamurthy A, et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. The Journal of Molecular Diagnostics. 2018;20(1):4–27. [DOI] [PubMed] [Google Scholar]
- 118.Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clinical Infectious Diseases. 2017;66(5):778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kwong JC, McCallum N, Sintchenko V, Howden BP. Whole genome sequencing in clinical and public health microbiology. Pathology. 2015;47(3):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wall DP, Kudtarkar P, Fusaro VA, Pivovarov R, Patil P, Tonellato PJ. Cloud computing for comparative genomics. BMC Bioinformatics. May 18 2010;11:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schatz MC, Langmead B, Salzberg SL. Cloud computing and the DNA data race. Nature biotechnology. 2010;28(7):691–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ramanan P, Bryson AL, Binnicker MJ, Pritt BS, Patel R. Syndromic panel-based testing in clinical microbiology. Clin Microbiol Rev. 2018;31(1):e00024–00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Meyer F, Paarmann D, D'Souza M, et al. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC bioinformatics. 2008;9(1):386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC bioinformatics. 2011;12(1):385. [DOI] [PMC free article] [PubMed] [Google Scholar]