

Abstract
Pathogenic variants in ATL1 are a known cause of autosomal-dominantly inherited hereditary spastic paraplegia (HSP-ATL1, SPG3A) with a predominantly ‘pure’ HSP phenotype. Although a relatively large number of patients have been reported, no genotype–phenotype correlations have been established for specific ATL1 variants. Confronted with five children carrying de novo ATL1 variants showing early, complex and severe symptoms, we systematically investigated the molecular and phenotypic spectrum of HSP-ATL1. Through a cross-sectional analysis of 537 published and novel cases, we delineate a distinct phenotype observed in patients with de novo variants. Guided by this systematic phenotyping approach and structural modelling of disease-associated variants in atlastin-1, we demonstrate that this distinct phenotypic signature is also prevalent in a subgroup of patients with inherited ATL1 variants and is largely explained by variant localization within a three-dimensional mutational cluster. Establishing genotype–phenotype correlations, we find that symptoms that extend well beyond the typical pure HSP phenotype (i.e. neurodevelopmental abnormalities, upper limb spasticity, bulbar symptoms, peripheral neuropathy and brain imaging abnormalities) are prevalent in patients with variants located within this mutational cluster.
Graphical Abstract
Graphical Abstract.

Introduction
The hereditary spastic paraplegias (HSPs) are a heterogeneous group of neurogenetic disorders characterized by progressive spasticity due to length-dependent degeneration of corticospinal motor neurons (1–3). With well over 80 reported loci (SPG1–86), HSP is one of the genetically most diverse neurologic disorders. While ‘pure’ forms mainly present with isolated lower limb spasticity and pyramidal signs, patients with ‘complex’ forms may exhibit additional symptoms (e.g. neurodevelopmental symptoms, peripheral neuropathy, amyotrophy and cerebellar dysfunction), frequently accompanied by abnormal findings on brain magnetic resonance (MR) imaging (2,3).
ATL1-associated HSP (HSP-ATL1 or SPG3A) is estimated to account for 5–10% of all autosomal dominantly inherited HSPs, second only to SPAST-associated HSP (SPG4) (4,5). ATL1 encodes the membrane-bound GTPase atlastin-1, which upon GTP-binding mediates tethering and fusion of smooth ER tubules through homodimerization, and is crucial for maintaining the normal, polygonal ER network structure (6–11).
While previous studies have found the great majority of patients with HSP-ATL1 to present with a pure HSP phenotype, in recent years a number of pathogenic ATL1 variants have been reported to cause severe, complex HSP with abnormal neurodevelopment (4,12–15). Interestingly, many of these variants occurred de novo. De novo mutations are increasingly recognized as an important cause of neurodevelopmental disease and have been implicated to explain some of the phenotypic pleiotropy seen in children with SPG4 (16,17). Confronted with five patients carrying de novo ATL1 variants and presenting with unusually early, severe and rapidly progressive symptoms, we hypothesized that de novo variants in ATL1 might provide insight into the severe end of the phenotypic spectrum seen in ATL1-associated HSP.
Thus far, de novo ATL1 variants have not been comprehensively analyzed, nor have clear genotype–phenotype correlations been established. To address this, we performed a systematic cross-sectional analysis of 537 patients including 532 published cases, complemented by systematic in silico analyses.
Results
Clinical description
We report five patients with complex HSP and de novo ATL1 variants confirmed by trio-genomic testing (Fig. 1A). Detailed genetic and clinical descriptions are provided in Supplementary Material, Tables S1 and S2. All probands were born at term with no significant ante- or peri-natal complications. Probands 2 and 5 presented first symptoms during the neonatal period in the form of ankle clonus and stiffness of the legs, respectively. Probands 1, 3 and 4 presented with delayed motor development, noticed between five and nine months of age. Delayed motor milestones were universal, with only patient 1 achieving unsupported ambulation. In addition to spasticity and pyramidal signs present in all patients, patients 3, 4 and 5 developed progressive spasticity and hyperreflexia of the upper limbs. These three patients also exhibited significant dysarthria. MR imaging revealed thinning of the corpus callosum and reduced white matter volume in patients 4 and 5, with normal brain MR imaging in patients 1, 2 and 3. Spinal cord MR imaging was normal in all patients (Fig. 1B). These early and severe symptoms observed in all five probands with de novo variants prompted us to reanalyze the genetic spectrum of HSP-ATL1 with a focus on differences between de novo and inherited variants.
Figure 1.

Phenotypes observed in probands and schematic of the atlastin-1 primary protein structure with novel variants and variant distribution. (A) Summary of the phenotypes observed in the five probands with de novo variants. Outer circle: proband number (Supplementary Material, Tables S1 and S2). Second to 11th circles: clinical symptoms, phenotype. White cells represent normal findings/absence of phenotype. (B) MR images of patients 4 and 5. Top row: Axial T2-weighted FLAIR images showing periventricular white matter changes in patient 4 at the age of 3 years and 11 months. Bottom right: Sagittal T1-weighted image showing thinning of the anterior aspects of the corpus callosum in patient 5 at the age of 12 months. (C) Disease-associated variants identified in the literature are annotated along the protein with dot color representing coding impact and dot size representing the number of cases identified. De novo variants are depicted towards the top and non-de novo variants downwards. Variants found in the probands appear in bold red letters. (D) Frequencies of coding impacts in de novo and non-de novo ATL1 variants depicted as percentage of all distinct variants shown in (C). (E) Frequencies of mutations in the functional domains of atlastin-1 depicted as percentage of all distinct variants shown in (C).
Genetic spectrum of HSP-ATL1 (SPG3A)
A systematic literature review identified 69 published studies covering 537 HSP-ATL1 patients harboring 91 distinct ATL1 variants (Supplementary Material, Table S4). Inheritance was determinable for 536 patients based on genetic findings or HSP symptoms present in the patients’ parents. Only 26 patients were reported to carry de novo mutations confirmed by segregation analyses. Including data from the aforementioned five probands, we identified a total of 81 different pathogenic variants occurring as inherited variants (autosomal-dominant/autosomal-recessive) and 24 different variants occurring de novo (Fig. 1C, Supplementary Material, Table S6). Missense variants accounted for the great majority of reported mutations (de novo = 91.7% versus non-de novo = 87.7%), and most variants were located in either the GTPase or the middle domain in both de novo and inherited cases (Fig. 1D and E). Interestingly, while not reaching statistical significance, pathogenic variants tended to be more abundant in the middle domain (50.0% versus 33.3%) and linker region (8.3% versus 2.5%) in patients with de novo variants (Fig. 1E). As previously reported, the majority of pathogenic ATL1 variants, both de novo and inherited, localized within clusters along the primary structure: two of them within the GTPase domain (residues 151–169 and 239–259), one cluster spanning the linker region and N-terminal middle domain (residues 336–355), and one within the middle domain (residues 398–416; Fig. 1C) (17). In line with previous studies, we found p.Arg239Cys, p.Arg416Cys, p.Arg495Trp to be the most common recurrent variants among patients with inherited pathogenic variants. The p.Arg239Cys missense variant was also the most common recurrent variant among patients with de novo pathogenic variants (N = 5), followed by p.Leu157Trp (N = 3). Exploratory in silico analyses using established pathogenicity prediction scores for all possible ATL1-missense variants (CADD PHRED, REVEL, BayesDel and MVP) showed limited power in predicting protein regions with intolerance to amino acid exchange, since only two out of four variant clusters were located in regions predicted to be particularly intolerant to genomic variation (Supplementary Material, Fig. S2).
Patients with de novo ATL1 variants present with early and severe symptoms
Next, we systematically investigated the phenotypic spectrum of patients with pathogenic ATL1 variants. Including the five patients reported in this study, the cohort consisted of 537 patients. Demographic and clinical data are summarized in Supplementary Material, Table S5. Comparison of patients with de novo versus inherited variants revealed differences in the prevalence of clinical manifestations, with 11 out of 25 symptoms significantly enriched in patients with de novo variants (Fig. 2A). These 11 symptoms fall into three major symptom categories: neurodevelopmental abnormality (NdA), bulbar symptoms (BuS) and upper limb symptoms (UlS; Supplementary Material, Fig. S2A). ‘Unsupported walking never achieved’ (64.3% versus 0.7% [9/14 versus 1/148]; estimated OR = 228.8; P = 3.0e−9), upper limb spasticity (40.0% versus 1.3% [8/20 versus 3/249]; estimated OR = 52.0; P = 3.9e−7) and motor delay (63.2% versus 13.7% [12/19 versus 28/204]; estimated OR = 10.6; P = 3.1e−5) were the most important phenotypic differentiators between patients with de novo and non-de novo variants (Fig. 2B and Supplementary Material, Fig. S3A). Noteworthy, bulbar symptoms (dysarthria and dysphagia) were present exclusively in patients with de novo variants. In addition to the severe symptoms beyond the classic, pure HSP phenotype of HSP-ATL1, the onset of symptoms was significantly earlier in patients carrying de novo compared to non-de novo variants (median 12 months [IQR 6.75] versus 36 months [IQR 93]; P = 1.9e-9; Fig. 2C).
Figure 2.

Comparison of the phenotypic spectrum in patients with de novo and non-de novo ATL1 variants. A total of 536 patients (nde novo = 31, nnon-de novo = 505; including the five probands identified in this study) were examined. (A) Frequencies of phenotypic features depicted as percentage of all patients (only patients who were explicitly examined for the feature were included in this retrospective analysis). Rounded percentages are depicted next to each bar. Statistical testing was performed using Fisher’s exact test followed by multiple hypothesis testing correction using the Benjamini–Hochberg procedure. (B) Estimated odds ratios for 11 phenotypic features significantly more frequent in patients with de novo variants compared to patients with inherited variants. Estimated odds ratios depicted to the left of each bar and 95%-confidence intervals depicted to the right end of each line. (C) Patients with de novo variants have a significantly earlier onset of symptoms (nde novo = 26, median = 12 months [IQR = 6.75]) compared to patients with non-de novo variants (nnon-de novo = 257, median = 36 months, [IQR = 93]); Mann–Whitney-U, P = 1.9e−9.
ATL1 variants associated with distinct and severe phenotypes are enriched in a mutational cluster
To identify any potential confounders explaining the phenotypic differences observed between patients with de novo and inherited variants, we performed a set of unbiased comparative analyses. Examining the strongest phenotypic differentiators (‘unsupported walking never achieved’ and ‘upper limb spasticity’) in relation to functional atlastin-1 protein domains, we found that variants associated with these two symptoms were almost exclusively located within the middle domain and linker region (Supplementary Material, Fig. S3B). We therefore examined the frequency distributions of patients positive for at least one feature comprised by the major differentiating symptom categories (NdA, BuS and UlS) with respect to variant localization (Fig. 3A). This revealed an uneven distribution of variants associated with the three symptom complexes between domains (NdA, P = 1.5e-5; BuS, P = 8.2e-3; UlS, P = 1.5e-5), with significant enrichment in the middle domain (nmiddle domain = 189, nother domains = 348; NdA, P = 1.7e-5, est. OR = 4.6; BuS, P = 3.2e-3, est. OR = 15.4; UlS, P = 1.4e-4, est. OR = 3.5; Fig. 3A). Interestingly, within the aforementioned variant hotspots in the middle domain and adjacent linker region variants were associated with NdA and UlS, independently of their pattern of inheritance (Fig. 3B).
Figure 3.

Variants associated with complex phenotypes are unevenly distributed among functional ATL1 domains. (A) Chord diagram depicting association of variant localization with the three symptom categories enriched in patients with de novo variants (Supplementary Material, Fig. S3). Each subsector reflects the absolute number of patients examined for symptoms of the respective symptom category, in relation to respective variant locations. Clockwise order of subsectors for each domain sector: developmental abnormality, bulbar symptoms, upper limb symptoms. Chord widths reflect the absolute number of patients positive for at least one symptom of the respective symptom category. (B) Section of the atlastin-1 primary structure depicting the linker region and N-terminal middle domain. De novo variants are depicted towards the top and non-de novo variants downwards. Colored dots indicate symptoms associated with respective variants. Abbreviations: HVR, hypervariable region; M Dom/3HB, middle domain/3 helix bundle; TM, transmembrane domain; CTH, C-terminal helix.
Next, we examined the spatial distribution of variants using three dimensional structures of prefusion GDP/Pi- bound (Fig. 4A), postfusion GDP-bound (Supplementary Material, Fig. S4A) and predicted full-length atlastin-1 (Supplementary Material, Fig. S4B). Analysis of these structures revealed that the two middle domain clusters along the primary structure localize with close proximity in the prefusion atlastin-1 state, forming a three-dimensional cluster (Fig. 4A). We approximated this cluster by creating the smallest possible sphere enclosing the centroids of residues in this region affected by de novo variants associated with the distinct symptoms (Fig. 4B, I and II). Hereby, non-de novo variants located within the cluster were identified (Fig. 4B, III and C). To test the hypothesis that the distinct phenotypes associated with de novo variants might be explained by variant localization within the cluster, we compared symptom frequencies between patients with inherited variants localized inside and outside the modelled cluster (nnon-de novo|inside cluster = 159, nnon-de novo|outside cluster = 346; Supplementary Material, Fig. S6A). This revealed 11 phenotypic features significantly more frequent among patients with variants inside the cluster (Fig. 5A). While the enriched symptoms in part recapitulated differences observed between de novo and inherited variants, the subgroup analysis yielded additional clinical features (i.e. ‘brain imaging abnormality’, ‘lower limb amyotrophy’, ‘peripheral neuropathy’ and ‘intellectual disability’) previously not deemed distinctive. These were not detected in the first analysis (de novo versus inherited), likely due to being negatively confounded by the mode of inheritance (Fig. 2A and Supplementary Material, Fig. S6A). Despite the small subgroup size (nde novo|inside cluster = 13, nde novo|outside cluster = 18), when omitting Benjamini–Hochberg procedure, nine phenotypic features were significantly enriched in patients harboring de novo variants located in the cluster, closely matching the results of the analogous subgroup analysis in patients with inherited variants (Supplementary Material, Fig. S5A and B). Therefore, we reanalyzed the cohort, stratifying for variant localization by comparing patients with variants located inside versus outside the cluster independent of inheritance (ninside cluster = 172, noutside cluster = 364). This yielded significant enrichment of 17 symptoms in patients with variants located within the cluster. The distinct, cluster-specific phenotypical features can be assigned to five major categories: neurodevelopmental abnormality, bulbar symptoms, upper limb symptoms, lower limb symptoms and sensory symptoms. Interestingly, brain imaging abnormalities also emerged as features significantly enriched in patients with cluster variants, with 60% (15/25) of patients who underwent MR imaging showing brain imaging abnormalities; the most frequent findings were thinning of the corpus callosum in 86.7% (13/15) followed by white matter abnormalities in 20% (3/15).
Figure 4.
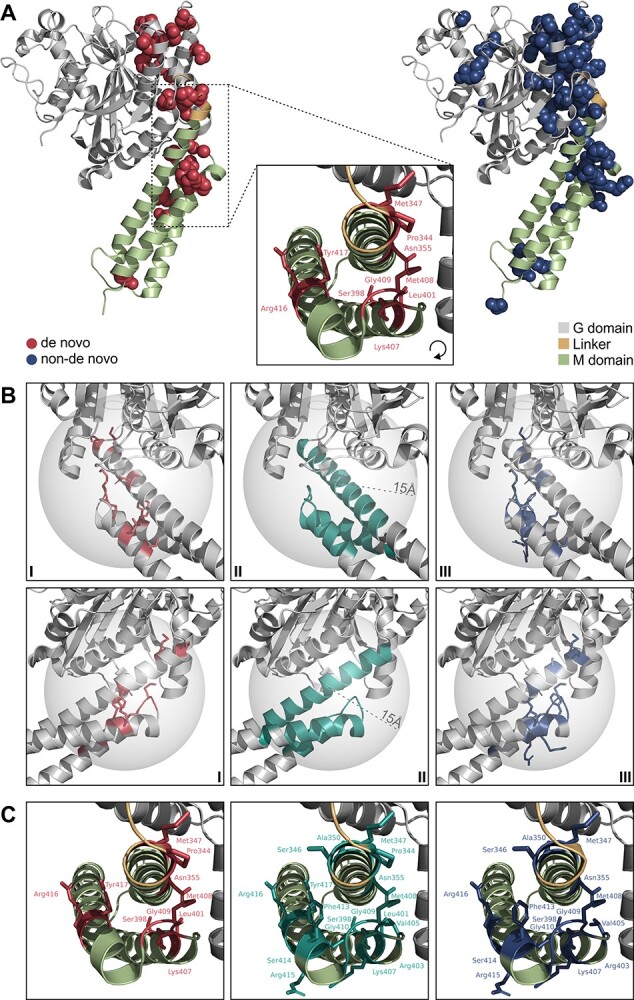
Variant distribution in three dimensional atlastin-1 structures. De novo variants associated with distinct, severe phenotypes, three-dimensionally cluster in the linker region and N-terminal M domain. (A) Wildtype prefusion GDP/Pi- and Mg2+-bound atlastin-1 (PDB 3Q5E, residues 1–447) with sites of de novo variants depicted as red spheres (left) and non-de novo variants as blue spheres (right). Black box indicating zoom-in view of the linker/middle (M) domain region, where variants associated with severe phenotypes cluster. (B) Localization of the cluster is approximated by creating the smallest possible sphere (radius 15 Å) containing all de novo variants associated with severe symptoms in this region (I, II). Hereby, non-de novo variants located within the cluster are identified (III). Protein region depicted from two different viewing angles (first and second row of panels). (C) Pathogenic variants located within the cluster. De novo variants depicted in red (left), non-de novo variants in blue (right) and all pathogenic variants within the cluster in teal (middle).
Figure 5.
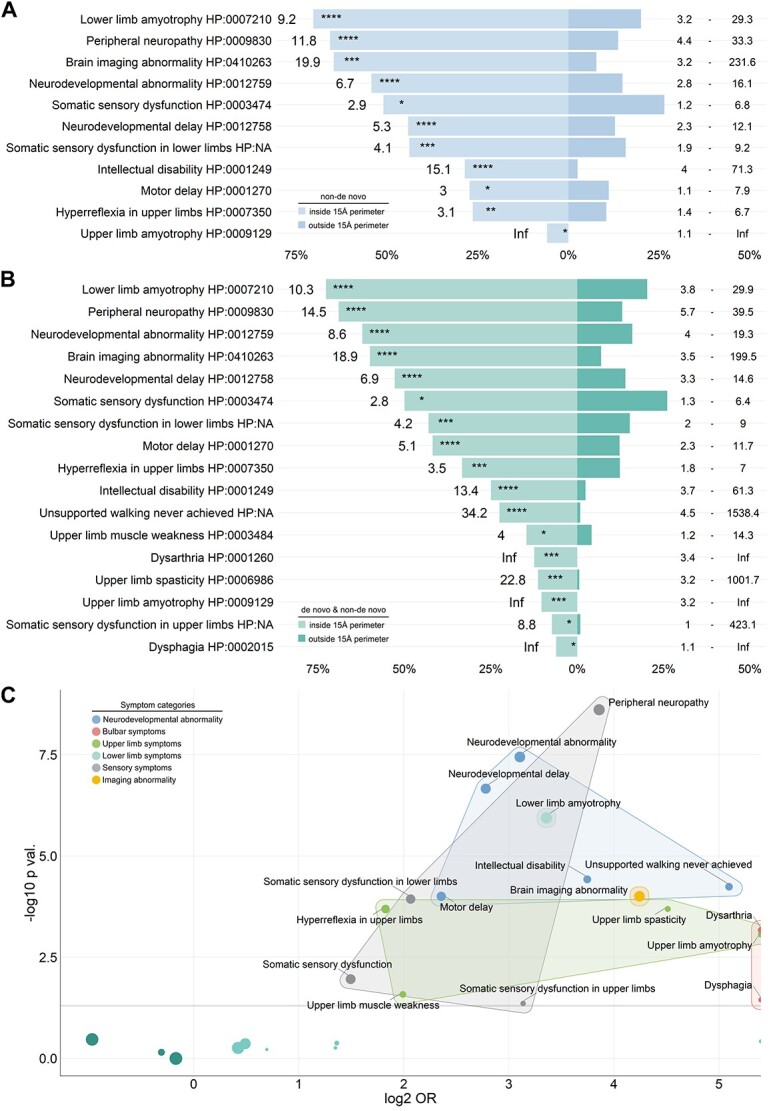
ATL1 variants located within the modelled cluster are associated with distinct and severe phenotypes. (A) Frequencies and odds ratios for phenotypic features significantly more frequent in patients with non-de novo variants located inside compared to variants located outside the perimeter of the modelled cluster. Estimated odds ratios depicted to the left and 95%-confidence intervals depicted to the right of each bar. Bar lengths reflect the percentages of patients exhibiting the respective clinical feature (only patients who were explicitly examined for the feature were included; nnon-de novo|inside cluster = 159, nnon-de novo|outside cluster = 346). (B) Frequencies and odds ratios for phenotypic features significantly enriched in patients with variants located inside compared to variants located outside the perimeter of the modelled cluster. Estimated odds ratios depicted to the left and 95%-confidence intervals depicted to the right of each bar. Bar lengths reflect the percentages of patients exhibiting the respective clinical feature (only patients who were explicitly examined for the feature were included; ninside cluster = 172, noutside cluster = 364). (C) Clinical features significantly enriched in patients with variants located within the modelled cluster can be assigned to five major categories. Estimated odds ratios plotted against P values for phenotypic features in patients with variants located inside compared to variants located outside the perimeter of the modelled cluster (significantly enriched features are labelled).
Discussion
In this study, we describe five patients with de novo ATL1 variants who developed early-onset severe motor and non-motor symptoms during the neonatal period or infancy. Given the early onset and complex symptoms diverging from the pure HSP phenotype predominantly reported for HSP-ATL1, as well as the lack of comprehensive studies on de novo variants or general genotype–phenotype correlations in the literature, we systematically analyzed the genotypic and phenotypic spectrum of HSP-ATL1. We performed a systematic cross-sectional analysis of 537 patients, including a total of 31 patients with de novo variants. This revealed enrichment of symptoms of complex HSP (neurodevelopmental abnormality, bulbar symptoms, upper limb symptoms) in patients with de novo variants. Further subgroup and in silico analyses, however, unveiled that the distinct phenotypes are largely explained by variant location within a mutational cluster and are similarly prevalent in patients with inherited variants. The distinct phenotypic signature of variants located within the cluster encompasses neurodevelopmental abnormalities, bulbar symptoms, upper limb and certain lower limb symptoms, as well as peripheral neuropathy and abnormal findings on brain MRI.
Our findings are corroborated by results from previous studies focusing on mechanisms underlying atlastin-1-mediated membrane fusion. Briefly, two membrane-bound ATL1 monomers form a dimer upon GTP-binding, thereby tethering two membrane sections. Triggered by GTP hydrolysis, the interaction between the middle and G domains within each monomer is released, allowing for the monomers to cross dimerize and mediate fusion of the tethered membrane sections (6–8). The mutational cluster we identified based on distinct clinical features, colocalizes: (1) with the top of the middle domain three-helix bundle, which in the prefusion state of atlastin-1 binds to the adjacent G domain, and (2) with the linker region, which upon GTP hydrolysis has been shown to undergo substantial conformational changes, enabling crossover dimerization. Several residues that locate within the cluster and are affected by variants associated with distinct phenotypes, have either been demonstrated to mediate the interaction of the middle and G domain in the prefusion state (Met347, Met408) or to serve as a pivot point during conformational transition (Pro344) (6–8). In the case of p.Arg416_Tyr417insAsn (N417ins; located within the cluster), a relative gain-of-function for the prefusion atlastin-1 state was proposed, based on increased propensity of G domain release due to altered middle and G domain interaction (18). Similar interference with the coordinate catalytic process seems conceivable for other variants within the cluster as well, especially those affecting middle and G domain interaction.
Despite sharing the same distinct clinical features, patients carrying de novo compared to inherited variants within the cluster appeared to be more severely affected, for example with higher frequencies of upper limb and bulbar symptoms. The lack of detailed quantitative data on symptom severity and functional impairment reported in the literature, however, prevented us from further investigating and quantifying this observation. The question arises whether, in addition to variant localization, the mode of inheritance might constitute a genetic modifier influencing expressivity, i.e. due to concomitant de novo variants in noncoding regions in the case of de novo clusters (19,20). Conversely, it might be possible that—owing to the cross-sectional nature of the collected data—very severe phenotypes appear to be enriched among patients with de novo variants, due to symptom severity making transmission of such variants less likely. While potentially resulting in a form of mutational survivorship bias affecting inherited variants, as has been reported for other genes (21), this also emphasizes the important role that careful evaluation of de novo variants and associated phenotypes can play in identifying relevant genotype–phenotype correlation. Although the mode of inheritance has been previously implicated to partially account for the heterogeneity of clinical presentations observed in HSP-SPAST (SPG4) (17), no mechanistic explanation has been reported thus far. In contrast, genotype–phenotype correlations based on variant location in relation to functional protein domains or coding impact have been described and functionally characterized for several HSP subtypes, including autosomal-dominant HSP-KIF5A (SPG10) and autosomal-recessive HSP-PNPLA6 (SPG39) (22–24).
Our findings establish a genotype–phenotype correlation for ATL1-associated HSP, provide the basis for future longitudinal studies, and highlight the importance of specific protein domains to understand phenotypic pleiotropy in HSP and more broadly in monogenic diseases linked to pathogenic missense variants.
More comprehensive and quantitative data are needed to allow for further exploration of unsupervised clustering approaches to the genotypic and phenotypic landscape of HSP-ATL1 in order to increase the precision of mutational cluster mapping based on phenotypic signatures. In addition, prospective studies are needed to confirm our findings in a larger cohort of de novo cases and functional studies are needed to elucidate the effects of single variants on atlastin-1 catalytic function to disentangle potentially unique disease mechanisms underlying distinct phenotypes.
Materials and Methods
Study participants, genetic testing and clinical characterization
This study was approved at Boston Children’s Hospital (IRB-P00033016) and written informed consent was obtained. Trio-exome-sequencing was performed at GeneDx (Gaithersburg, MD) and trio-genome-sequencing at Fulgent Genetics (Temple City, CA). Probands were examined by board-certified pediatric neurologists at the Boston Children’s Hospital Movement Disorders Program.
Systematic literature review and annotation of Human Phenotype Ontology terms
A systematic PubMed search on HSP-ATL1 was performed (Supplementary Material, Table S3). References of selected publications were screened for additional articles. Two independent reviewers assessed and selected 69 studies (Supplementary Material, Table S4) published between 2001 and 2022, which evaluated molecular findings and genotype–phenotype correlation in patients with ATL1-associated HSP. Pathogenicity of reported variants was assigned based on ACMG criteria. Following extraction from original reports, phenotypic information was transferred to standardized Human Phenotype Ontology (HPO) terms (v1.7.15, release 2022/02/14; Supplementary Material, Fig. S1) as previously described (25,26). A total of 21 HPO terms were assigned and the HPO ancestor term ‘Neurodevelopmental abnormality HP:0012759’ was retrieved for further quantification. In addition, three phenotypic terms further specifying the HPO terms ‘Somatic sensory dysfunction HP:0003474’ (Somatic sensory dysfunction in upper/lower limbs) and ‘Motor delay HP:0001270’ (Unsupported walking never achieved) for which no HPO terms have been defined as of version 1.7.5 were included. Only phenotypic features for whom a patient was explicitly reported to have been evaluated for, or where such information could unequivocally be derived from the clinical description (e.g. a patient reported to ambulate without assistance was considered to having achieved unsupported walking) were annotated present/absent and included in the retrospective analysis. Annotation of the HPO term ‘Peripheral neuropathy HP:0009830’ was based on the presence/absence of abnormal electrophysiological findings only in patients for whom electrophysiological evaluation was reported. For the annotation of ‘Brain imaging abnormality HP:0410263’ only MR imaging studies were considered, in order to reduce the risk of false negative annotation due to subtle structural changes being missed on less sensitive imaging modalities.
In silico analyses
Information on the human ATL1 protein sequence was obtained from the Universal Protein Resource (ID: Q8WXF7) and AlphaFold Protein Structure (v2.0, DeepMind Technologies, London, UK) databases (27,28), as well as published data accessed through RCSB Protein Data Bank (ID: 3Q5E, 4IDO) (7,8). Protein structures were visualized using Pymol (v2.5.0, Schroedinger, Inc., New York, NY). CADD PHRED, REVEL, BayesDel addAF and MVP scores of all possible base substitutions of the ATL1 transcript were computed using dbNSFP (v4.3, Liu Lab, University of South Florida) (29,30) and mapped to the corresponding protein sequence. All variants were harmonized to NM_015915.5/GRCh38/hg38. Modelling of the ATL1 primary protein structure and variants was done in R (v4.2.0) and RStudio (v2022.02.2; RStudio, Inc., Boston, MA) as previously described (31).
Statistical analysis
Differences between two groups were evaluated using Mann–Whitney-U test for nonparametric distributions and Fisher’s exact test for evaluating frequency distributions. Following Fisher’s exact test, P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg procedure and adjusted odds ratios and 95%-confidence intervals were estimated using conditional maximum likelihood estimation (32). Statistical analyses were performed in R (v4.2.0) and RStudio (v2022.02.2; RStudio, Inc., Boston, MA). P < 0.05 was considered significant. P values are denoted as follows: P < 0.05 (*), P < 0.01 (**), P < 0.001 (***) and P < 0.0001 (****).
Supplementary Material
Acknowledgement
The authors thank the patients and their families who participated in this study.
Conflict of Interest statement. S.S. has received consulting fees from GLG, Guidepoint (which connected to a client, Fortress Biotech), Novartis, ExpertConnect and Orchard Therapeutics. D.E.F. received speaker honoraria from the Movement Disorder Society, publishing royalties from Cambridge University Press and research funding through a joint research agreement with Astellas Pharmaceuticals Inc. The other authors report no conflict of interest.
Contributor Information
Julian E Alecu, Department of Neurology, F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA; Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, 91054, Germany.
Afshin Saffari, Department of Neurology, F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Catherine Jordan, Department of Neurology, F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Siddharth Srivastava, Department of Neurology, F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Craig Blackstone, Movement Disorders Unit, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Darius Ebrahimi-Fakhari, Department of Neurology, F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA; Movement Disorders Program, Department of Neurology, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA; Intellectual and Developmental Disabilities Research Center, Boston Children’s Hospital, Boston, MA 02115, USA; The Manton Center for Orphan Disease Research, Boston Children’s Hospital, Boston, MA 02115, USA.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (German Research Foundation, 270949263/GRK2162 to J.E.A.), scholarships by the German Academic Exchange Service (DAAD), the German National Academic Foundation and the Max Weber-Program of the State of Bavaria (all to J.E.A); the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, SA 4171/1-1 to A.S.); the National Institute of Health/National Institute of Neurological Disorders and Stroke (K23NS119666 to S.S.); the CureAP4 Foundation, the CureSPG50 Foundation, the Spastic Paraplegia Foundation USA, the Manton Center for Orphan Disease Research, the National Institutes of Health/National Institute of Neurological Disorders and Stroke (1K08NS123552-01) and the BCH Office of Faculty Development (all to D.E.F.). The BCH Intellectual and the Developmental Disabilities Research Center is supported by the National Institutes of Health (BCH IDDRC, NIH P50 HD105351).
Data and Code Availability
The data and code that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Lange, L.M., Gonzalez-Latapi, P., Rajalingam, R., Tijssen, M.A.J., Ebrahimi-Fakhari, D., Gabbert, C., Ganos, C., Ghosh, R., Kumar, K.R., Lang, A.E. et al. (2022) Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society Task Force—an update. Mov. Disord. First published on April 28, 2022, 37, 905–935. [DOI] [PubMed] [Google Scholar]
- 2. Shribman, S., Reid, E., Crosby, A.H., Houlden, H. and Warner, T.T. (2019) Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol., 18, 1136–1146. [DOI] [PubMed] [Google Scholar]
- 3. Elsayed, L.E.O., Eltazi, I.Z., Ahmed, A.E. and Stevanin, G. (2021) Insights into clinical, genetic, and pathological aspects of hereditary spastic paraplegias: a comprehensive overview. Front. Mol. Biosci. First published on November 26, 2021, 8. 10.3389/fmolb.2021.690899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Erfanian Omidvar, M., Torkamandi, S., Rezaei, S., Alipoor, B., Omrani, M.D., Darvish, H. and Ghaedi, H. (2021) Genotype-phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J. Neurol. First published on November 19, 2019, 268, 2065–2082. [DOI] [PubMed] [Google Scholar]
- 5. Namekawa, M., Ribai, P., Nelson, I., Forlani, S., Fellmann, F., Goizet, C., Depienne, C., Stevanin, G., Ruberg, M., Dürr, A. et al. (2006) SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology, 66, 112–114. [DOI] [PubMed] [Google Scholar]
- 6. Bian, X., Klemm, R.W., Liu, T.Y., Zhang, M., Sun, S., Sui, X., Liu, X., Rapoport, T.A. and Hu, J. (2011) Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. U. S. A. First published on February 22, 2011, 108, 3976–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Byrnes, L.J. and Sondermann, H. (2011) Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc. Natl. Acad. Sci. U. S. A. First published on January 10, 2011, 108, 2216–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Byrnes, L.J., Singh, A., Szeto, K., Benvin, N.M., O'Donnell, J.P., Zipfel, W.R. and Sondermann, H. (2013) Structural basis for conformational switching and GTP loading of the large G protein atlastin. EMBO J. First published on January 18, 2013, 32, 369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rismanchi, N., Soderblom, C., Stadler, J., Zhu, P.-P. and Blackstone, C. (2008) Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. First published on February 12, 2008, 17, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu, X., Guo, X., Niu, L., Li, X., Sun, F., Hu, J., Wang, X. and Shen, K. (2019) Atlastin-1 regulates morphology and function of endoplasmic reticulum in dendrites. Nat. Commun. First published on February 04, 2019, 10. 10.1038/s41467-019-08478-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu, P.-P., Hung, H.-F., Batchenkova, N., Nixon-Abell, J., Henderson, J., Zheng, P., Renvoisé, B., Pang, S., Xu, C.S., Saalfeld, S. et al. (2022) Transverse endoplasmic reticulum expansion in hereditary spastic paraplegia corticospinal axons. Hum. Mol. Genet., First published on March 26, 2022. 10.1093/hmg/ddac072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fusco, C., Frattini, D., Farnetti, E., Nicoli, D., Casali, B., Fiorentino, F., Nuccitelli, A. and Della Giustina, E. (2010) Hereditary spastic paraplegia and axonal motor neuropathy caused by a novel SPG3A de novo mutation. Brain Dev. First published on September 06, 2009, 32, 592–594. [DOI] [PubMed] [Google Scholar]
- 13. Haberlová, J., Claeys, K.G., Zámecník, J., de Jonghe, P. and Seeman, P. (2008) Extending the clinical spectrum of SPG3A mutations to a very severe and very early complicated phenotype. J. Neurol. First published on April 30, 2008, 255, 927–928. [DOI] [PubMed] [Google Scholar]
- 14. Yonekawa, T., Oya, Y., Higuchi, Y., Hashiguchi, A., Takashima, H., Sugai, K. and Sasaki, M. (2014) Extremely severe complicated spastic paraplegia 3A with neonatal onset. Pediatr. Neurol. First published on July 24, 2014, 51, 726–729. [DOI] [PubMed] [Google Scholar]
- 15. Méreaux, J.-L., Banneau, G., Papin, M., Coarelli, G., Valter, R., Raymond, L., Kol, B., Ariste, O., Parodi, L., Tissier, L. et al. (2022) Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain First published on January 04, 2022, 145, 1029–1037. [DOI] [PubMed] [Google Scholar]
- 16.Deciphering Developmental Disorders Study. (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature First published on January 04, 2022, 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schieving, J.H., de Bot, S.T., van de Pol, L.A., Wolf, N.I., Brilstra, E.H., Frints, S.G., van Gaalen, J., Misra-Isrie, M., Pennings, M., Verschuuren-Bemelmans, C.C. et al. (2019) De novo SPAST mutations may cause a complex SPG4 phenotype. Brain, 142, e31. [DOI] [PubMed] [Google Scholar]
- 18. Kelly, C.M., Zeiger, P.J., Narayanan, V., Ramsey, K. and Sondermann, H. (2021) A novel insertion mutation in atlastin 1 is associated with spastic quadriplegia, increased membrane tethering, and aberrant conformational switching. J. Biol. Chem. First published on November 19, 2021, 298. 10.1016/j.jbc.2021.101438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goldmann, J.M., Veltman, J.A. and Gilissen, C. (2019) De novo mutations reflect development and aging of the human germline. Trends Genet. First published on October 11, 2019, 35, 828–839. [DOI] [PubMed] [Google Scholar]
- 20. Turner, T.N. and Eichler, E.E. (2019) The Role of De novo noncoding regulatory mutations in neurodevelopmental disorders. Trends Neurosci. First published on December 15, 2018, 42, 115–127. 10.1016/j.tins.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bermúdez-Guzmán, L., Jimenez-Huezo, G., Arguedas, A. and Leal, A. (2020) Mutational survivorship bias: The case of PNKP. PLoS One First published on December 17, 2020, 15, e0237682. 10.1371/journal.pone.0237682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Boer, E.M.J., van Rheenen, W., Goedee, H.S., Kamsteeg, E.-J., Brilstra, E.H., Veldink, J.H., van den Berg, L.H. and van Es, M.A. (2021) Genotype-phenotype correlations of KIF5A stalk domain variants. Amyotroph. Lateral Scler. Frontotemporal Degen., First published on April 08, 2021. 10.1080/21678421.2021.1907412. [DOI] [PubMed] [Google Scholar]
- 23. Synofzik, M., Gonzalez, M.A., Lourenco, C.M., Coutelier, M., Haack, T.B., Rebelo, A., Hannequin, D., Strom, T.M., Prokisch, H., Kernstock, C. et al. (2014) PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain First published on December 19, 2013, 137, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kretzschmar, D. (2022) PNPLA6/NTE, an evolutionary conserved phospholipase linked to a group of complex human diseases. Metabolites First published on March 24, 2022, 12, 2022. 2022, 10.3390/metabo12040284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Köhler, S., Gargano, M., Matentzoglu, N., Carmody, L.C., Lewis-Smith, D., Vasilevsky, N.A., Danis, D., Balagura, G., Baynam, G., Brower, A.M. et al. (2021) The Human Phenotype Ontology in 2021. Nucleic Acids Res., 49, D1207–D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saffari, A., Schröter, J., Garbade, S.F., Alecu, J.E., Ebrahimi-Fakhari, D., Hoffmann, G.F., Kölker, S., Ries, M. and Syrbe, S. (2021) Quantitative retrospective natural history modeling of WDR45-related developmental and epileptic encephalopathy—a systematic cross-sectional analysis of 160 published cases. Autophagy First published on November 24, 2021, 18, 1715–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature First published on July 15, 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., Yuan, D., Stroe, O., Wood, G., Laydon, A. et al. (2022) AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res., 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu, X., Jian, X. and Boerwinkle, E. (2011) dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat., 32, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu, X., Li, C., Mou, C., Dong, Y. and Tu, Y. (2020) dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. First published on December 02, 2020, 12, 103. https://doi.org/10.1186/s13073-020- 00803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alecu, J.E., Saffari, A., Jumo, H., Ziegler, M., Strelko, O., Brownstein, C.A., Gonzalez-Heydrich, J., Rodan, L.H., Gorman, M.P., Sahin, M. and Ebrahimi-Fakhari, D. (2022) Novel CAPN1 missense variants in complex hereditary spastic paraplegia with early-onset psychosis. Ann. Clin. Transl. Neurol. First published on March 16, 2022, 9, 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benjamini, Y. and Hochberg, Y. (1995) Controlling the False Discovery Rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B, 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and code that support the findings of this study are available from the corresponding author upon reasonable request.