

Abstract
RNA labeling in situ has enormous potential to visualize transcripts and quantify their levels in single cells, but it remains challenging to produce high levels of signal while also enabling multiplexed detection of multiple RNA species simultaneously. Here, we describe clampFISH 2.0, a method that uses an inverted padlock design to efficiently detect many RNA species and exponentially amplify their signals at once, while also reducing the time and cost compared with the prior clampFISH method. We leverage the increased throughput afforded by multiplexed signal amplification and sequential detection to detect 10 different RNA species in more than 1 million cells. We also show that clampFISH 2.0 works in tissue sections. We expect that the advantages offered by clampFISH 2.0 will enable many applications in spatial transcriptomics.
Methods to label RNA in its cellular context have enabled researchers to visualize different aspects of gene expression. Initially, these approaches were dependent on an abundant and densely packed target to generate sufficient signal1, although developments in probe synthesis and fluorescence microscopy enabled labeling and detection efficiencies sufficient to identify individual RNA molecules in fixed cells and tissue2,3, a set of methods collectively termed single-molecule RNA fluorescence in situ hybridization (RNA FISH). These single-molecule RNA FISH methods localize multiple fluorescent dye molecules to a target RNA, typically using complementary DNA probes that, in early designs, were directly labeled with fluorescent dyes2,3. This labeling approach, however, produces only weak fluorescent signals, which hinders its use in high-background tissue sections and also necessitates long imaging times. To amplify the signal, there are now multiple single-molecule RNA FISH methods that build molecular scaffolds on the target RNA to provide a larger addressable sequence for fluorescent labeling. Each of these amplified methods, however, requires compromises in accuracy, multiplexing capacity or cost. We recently described a method called clampFISH that is highly accurate and offers very high signal amplification, but is time-consuming, costly and was not validated for multiplexing beyond three targets. Here, we present clampFISH 2.0, which solves these issues while retaining the strengths of the original method. Amplification methods that build molecular scaffolds on their RNA do so using one of two approaches: the enzyme method or the hybridization method, each with their own drawbacks. Using multiple enzymes, rolling circle amplification generates a repeating DNA scaffold that can then be labeled fluorescently. However, although there have been some reported improvements4-7, enzymatic inefficiencies can leave many messenger RNAs undetected8. Alternatively, a number of methods forgo the use of enzymes and instead use hybridization-based approaches, such as hybridization chain reaction (HCR)9-11, bDNA/RNAscope12, FISH-STICs13, SABER-FISH14 and clampFISH15. Although each of these hybridization-based methods offers various advantages, such as high signal gain or ease of repurposing for new targets, none can offer these features while offering rapid multiplexing beyond the 3–5 targets possible with spectrally distinguishable dyes.
Higher degrees of multiplexing beyond 3–5 targets require the removal of the previous imaging cycle’s signal and iteratively re-probing a new set of targets8. The means by which this iterative procedure can be accomplished is constrained by the molecular scaffold design for each method. With HCR, given that the scaffold is built with dye-conjugated probes, higher degrees of multiplexing use DNAse digestion16 of the previous cycle’s probes and repeat the entire protocol, thus substantially increasing the protocol time. Furthermore, the design of orthogonal HCR hairpin sets for multiplexing is complicated by the limited sequence diversity available for their short toehold sequences, such that to date, a maximum of 4–5 HCR amplifier probes have been used at once10,11. Unlike HCR, HiPlex RNAscope and non-branching SABER decouple the time-consuming amplification steps, which are done for all targets in parallel, and the short readout steps, in which fluorescently labeled readout probes bind to a subset (for example, 3–5) of amplified scaffolds. After imaging in multiple fluorescent channels, the previous cycle’s signal is removed, and only the readout and imaging steps are repeated until all of the targets’ scaffolds have been probed. This approach is faster for higher degrees of multiplexing, but it must also manage to remove the fluorescent signal without removing the remaining scaffolds. Thus, although the dye-coupled readout probes could otherwise be dissociated from their scaffolds with a stringent wash, this would also dissociate the scaffold components themselves, which are constructed via DNA hybrids. Hence, in lieu of a simple high-stringency wash, many published protocols call for photobleaching17 or fluorophore cleavage17,18 to eliminate the previous cycle’s signal. Although non-branching SABER scaffolds (with up to ~10-fold amplification) can withstand the stringent wash to remove fluorescent readout probes, this approach has not been validated for the higher-gain branching SABER scaffolds (with up to ~450-fold amplification)14, probably because the shorter binding sequences of the branches could lead to their dissociation during the stringent washes. In contrast, clampFISH scaffolds could, in principle, withstand multiple stringent washes owing to their covalently locked configuration15, although the other weaknesses (high probe cost, long protocol time) have limited the potential of this multiplexing approach. Thus, there remains a need for an amplified RNA FISH method that permits accurate, high-gain, and flexible multiplexing. We reasoned that the original clampFISH15, herein referred to as clampFISH 1.0, offered an excellent starting point for further development owing to a number of favorable attributes: its enzyme-free and step-wise amplification, its de-coupled amplification and readout, and its click chemistry-enabled boost in performance beyond the constraints imposed by oligonucleotide binding kinetics.
ClampFISH 1.0 probes are designed to form a looping scaffold off a target nucleic acid molecule (for example, RNA). Each probe begins as a linear DNA oligonucleotide, which, upon hybridization to its target, forms a ‘C’ shape, the 5′ and 3′ ends of which are kept in close proximity via complementarity to a second sequence. Because the two ends are modified with alkyne (5′) and azide (3′) moieties, they can then be covalently linked using a click chemistry reaction (copper(I)-catalyzed azide–alkyne cycloaddition), which replaces the low-yield enzymatic ligation in situ19. The click reaction circularizes the probe and, because of the helical structure of nucleic acid hybrids, stably loops it around its complementary strand15.
The clampFISH 1.0 protocol begins by hybridizing multiple primary probes to the target, followed by multiple alternating amplification steps with secondary probes and tertiary probes. Successive rounds of amplification, in which two amplification probes can bind to each probe from the previous round, leads to a looping scaffold that grows exponentially in size (the 2:1 amplifier binding ratio means that if the amplifier probes bind with 100% efficiency, then the scaffolds would double in addressable sequence each round). After every two steps, a click reaction is used to circularize the probes, thus stably linking the probes to one another. This amplified scaffold can then be labeled by hybridizing a fluorescent dye-coupled DNA readout probe to the many secondary or tertiary probes in the scaffold, thus localizing many fluorophores to the target to produce a single bright spot. Although clampFISH 1.0 offered a means for accurate, high-gain amplification, it had a number of shortcomings. First, owing to multiple time-consuming amplification steps, clampFISH often took ~2.5–3 days to perform. Second, the method was plagued by many bright, mostly extracellular, non-specific spots that complicated downstream image analysis. Third, the high probe costs prohibited its widespread use, and these costs scaled poorly with additional gene targets, thus placing a practical limit on the method’s multiplexing potential. Last, it was unknown whether higher degrees of multiplexing, beyond spectral (three-gene) multiplexing, would be problematic, for example due to amplifier cross-reactivity or issues while removing readout probes. Here, we introduce clampFISH 2.0, which addresses these shortcomings through multiple improvements to the original method. With an updated probe design and synthesis protocol, we greatly reduced the overall method cost, increased its scalability, and eliminated the extracellular non-specific spots. We optimized the method for faster amplification, reducing the pre-readout protocol time from ~2.5 days to ~18 hours, of which only 8 hours require hands-on engagement, and we validated a 35 minute readout hybridization and stripping protocol for rapid 10-gene multiplexing. We show that clampFISH 2.0 is a fast, accurate, cheap and flexible technology for multiplexed RNA detection in situ.
Results
Probe re-design and protocol optimization
ClampFISH 1.0’s primary probes were assembled with two gene-specific oligonucleotides that each required chemical modifications, substantially adding to the method’s cost. We therefore asked whether we could invert the orientation of the primary probes, such that the gene-specific RNA-binding oligonucleotide components could remain unmodified, and therefore cheaper, while incorporating the click chemistry modifications into a reusable, gene-independent oligonucleotide. In this scheme, we do not change the orientation of the secondary and tertiary probes, the 5′ and 3′ ends of which are still brought together in close proximity upon hybridization. To aid the circularization of the primary probe during the click reaction, we add a ‘circularizer oligo’ (concurrently with the secondary probe), the sequence complementarity of which is designed to bring the 5′ and 3′ ends of the primary probe in close proximity (Fig. 1a). This new primary probe design had the additional benefit of permitting larger-scale probe synthesis, given that all of a gene’s primary probes could be ligated to an amplifier-specific oligonucleotide in a single pooled reaction, rather than requiring separate three-part ligations for each of 10+ primary probes for a given gene (Fig. 1b and Supplementary Fig. 1). As a potential downside, the benefits of the new design could, in principle, come at the expense of specificity: the lack of a proximity ligation mediated by the target RNA molecule could allow for more non-specific probe self-ligation because the proximity ligation is typically thought to increase specificity. Along with a re-design of the primary probes, we also shortened the length of the secondary and tertiary probes (collectively referred to as ‘amplifier probes’), such that they can be made from a single commercially produced oligonucleotide, thus simplifying their formerly three-part synthesis. When comparing the costs of the two methods at a low and high experimental scale (from ~100 experiments to ~10,000 experiments), the probe design and synthesis changes reduced the primary probe costs, respectively, by ~9-fold to ~27-fold (assuming 10 clampFISH 1.0 probes and 30 clampFISH 2.0 probes per primary probe set) and reduced the amplifier probe costs by ~2-fold to ~14-fold. With all major costs considered, including readout probes and enzymes used for probe synthesis (the concentrations of which were optimized for synthesis speed rather than cost), clampFISH 2.0 is ~4–5-fold cheaper than the original clampFISH 1.0. At current commercial costs, multiplexing of 10 gene targets with clampFISH 2.0 (using 10 primary probe sets and 10 amplifier probe sets) can be performed for ~400 experiments for a total of ~US$11,000 (versus ~US$42,000 for clampFISH 1.0). Furthermore, once the amplifier and readout probe sets have been purchased, purchasing primary probes for a different 10-gene panel would cost only ~US$3,300 (versus ~US$30,000 for clampFISH 1.0) (see Supplementary Tables 9-11 for details).
Fig. 1 ∣. ClampFISH 2.0 enables fast, cost-effective, exponential amplification of multiplexed RNA FISH signal in situ.
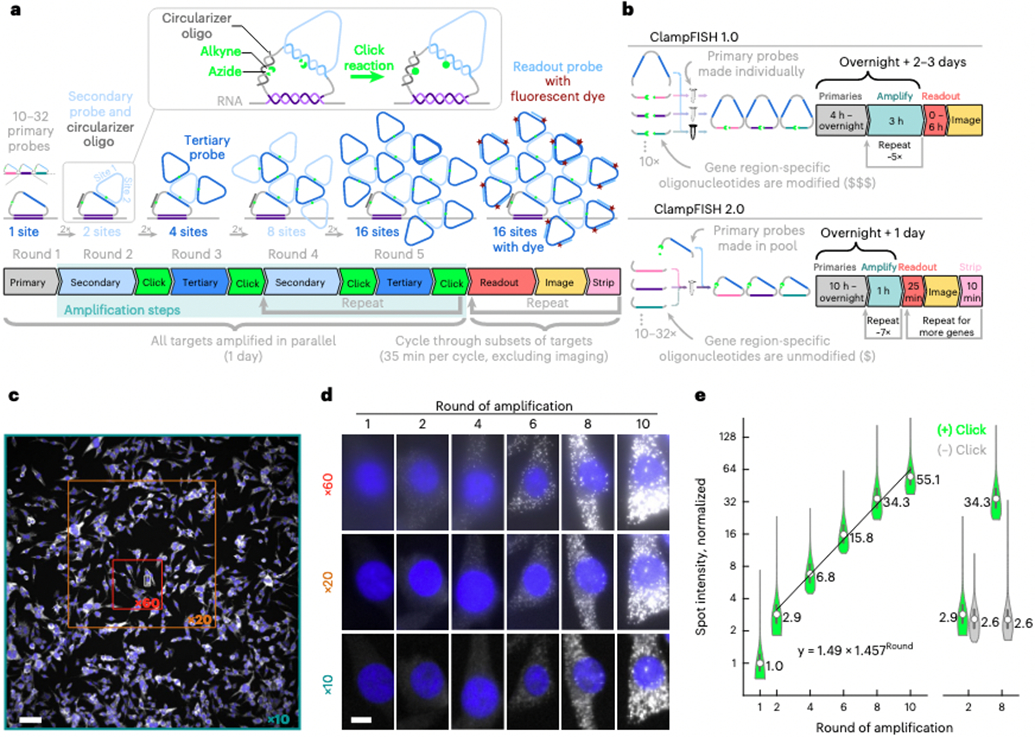
a, Schematic diagram of clampFISH 2.0. b, Comparison of clampFISH 2.0 and clampFISH 1.0: in clampFISH 2.0 the primary probes feature an inverted design, in which oligonucleotides modified for use with click chemistry can be re-used for all probes in any primary probe set. c, UBC clampFISH 2.0 at round 10 in WM989 A6-G3 cells, imaged with a ×10 objective, with the sizes of the smaller ×20 and ×60 fields of view overlaid. Scale bar, 100 μm. d, UBC clampFISH 2.0 in WM989 A-G3 cells shown at progressively higher rounds of amplification at ×60, ×20 and ×10 magnification. The experiment was performed twice with similar results. Scale bar, 10 μm. e, Left: UBC clampFISH 2.0 spot intensity (normalized to the median intensity from round 1) over progressively higher rounds of amplification, with the normalized median intensity from rounds 2, 4, 6, 8 and 10 fitted to an exponential curve (n = 70,800, 64,440, 47,520, 49,800, 53,760 and 53.280 spots from each of rounds 1, 2, 4, 6, 8 and 10, respectively, from the same experiment). Right: spot intensity at rounds 2 and 8 when the copper catalyst is included (green) or not included (gray) in the click reaction (n = 59,520 and 57,120 spots in rounds 2 and 8, respectively, when the copper catalyst was not included, from the same experiment). The circles represent the median, and the bounds of the inner box plots represent the 25th and 75th percentiles. The experiment was performed twice. See Extended Data Fig. 4 and Supplementary Fig. 3 for data associated with all targets and amplifier sets from both replicates.
In addition to its high cost, the clampFISH 1.0 protocol was time-consuming, in large part because each round of amplification required ~3 hours. For example, the amplification protocol would require 2 days with 4–5 rounds of amplification, or 3 days for 6–8 rounds of amplification. To reduce the time of amplification, we sought to reduce the hybridization time by reducing secondary structure, as previously reported18,20,21. With these new probe designs and additional optimization of the wash steps, click reaction, and buffer compositions, we reduced the time for a round of amplification from 3 hours to only 1 hour, which includes a 30 minute amplifier hybridization. This three-fold speed improvement in amplification enables the full protocol, up to readout probe hybridization and imaging, to be performed with an overnight primary incubation (10 h) and ~8 hours the next day (Fig. 1b).
We wondered whether this updated scheme would still produce a specific, amplified RNA FISH signal. We made primary probes for each of two separate mRNA targets (green fluorescent protein (GFP) mRNA, 10 probes; and human epidermal growth factor receptor (EGFR) mRNA, 30 probes) and tested their performance on a mixture of two cell lines known to express different RNAs: a WM989 A6-G3 H2B-GFP line, expressing the GFP sequence as mRNA, and a WM989 A6-G3 RC4 line grown in drug-containing media that we have shown to express high levels of EGFR mRNA22-24. We observed bright, amplified spots for the mRNAs specifically in the cells that were expected to express them (GFP spots in WM989 A6-G3 H2B-GFP cells, and EGFR spots in much greater numbers in WM989 A6-G3 RC4 cells; Extended Data Figs. 1 and 2), and good co-localization with conventional single-molecule RNA FISH probes targeting non-overlapping regions of the same RNAs (Extended Data Figs. 1, 2 and Supplementary Fig. 2) as was similarly done for clampFISH 1.0 (ref. 15), confirming the method’s specificity despite the lack of an RNA-splinted proximity ligation in the new primary probes. Furthermore, by introducing a number of centrifugation steps to the probe synthesis protocol whereby we discard any material at the bottom of the tube, we largely eliminated the bright, non-specific spots seen with clampFISH 1.0 (Extended Data Fig. 3). Although the primary probe and amplifier probe synthesis protocols both include three such centrifugation steps (Supplementary Fig. 1), it is unclear whether fewer centrifugations could achieve the same result.
We next sought to determine whether clampFISH 2.0 could exponentially amplify the signal to a level that is detectable with lower-powered (×20/0.75 numerical aperture (NA) and ×10/0.45 NA) air objective lenses. We ran the clampFISH 2.0 protocol to varying stopping points: 1 round (primaries), 2 rounds (primaries and secondaries), 4 rounds (primaries, secondaries, tertiaries and secondaries again), 6 rounds, 8 rounds and 10 rounds, and hybridized readout probes to these scaffolds. Using low-powered magnification with large fields of view, we can reliably detect spots after amplification, thus demonstrating the capacity of clampFISH 2.0 for high-throughput RNA detection (Fig. 1c,d). Furthermore, we observed an exponential rate of amplification, measured to be 1.406–1.678-fold per round (Fig. 1e and Extended Data Fig. 4), implying that the amplifier binding efficiency is 70–84% of the theoretical doubling of intensity per round. This exponential rate of growth did not appreciably slow down, even at the maximum number of rounds tested (round 10), where we detected up to ~100-fold signal amplification (Extended Data Fig. 4), suggesting that an even brighter signal could be achievable with additional amplification. When we removed the copper catalyst from the click reaction we saw no amplification from round 2 to round 8 (Fig. 1e and Supplementary Fig. 3), suggesting that the scaffold’s growth requires looping of circularized amplifier probes to one another. Despite the greatly increased signal from amplification, the size of the spots did not appreciably grow (Extended Data Fig. 5), such that at round 10 the median spot sizes (full width at half maximum) were only ~264 nm and ~316 nm for Atto 488- and Atto 647N-labeled readout probes, respectively, which is the same size as conventional single-molecule RNA FISH spots (Extended Data Fig. 6). We additionally tested a one-pot amplification approach (adding secondaries, tertiaries and the click reaction simultaneously), although this did not produce amplified spots (Supplementary Fig. 4). To achieve a higher degree of multiplexing, we needed to have a number of orthogonal sets of amplifier probes that had high gain and low off-target activity. We thus screened 15 amplifier probe sets, each used with primary probes targeting GFP mRNA or EGFR mRNA. Of these, we chose 10 sets of amplifier probes (1, 3, 5, 6, 7, 9, 10, 12, 14 and 15) with high gain and low off-target activity (amplifier set 11 was excluded based on its high number of off-target spots). We observed that the gain of an amplifier probe set for one RNA target strongly correlated with its gain on the other RNA target, suggesting that amplifiers can be used in a modular fashion with any set of primary probes without substantial primary probe-specific effects on performance (Supplementary Figs. 5 and 6). We also confirmed that amplifier probes do not cross-react with one another by showing that the spot intensities were equivalent when amplifier sets were used individually versus when they were used in a pooled mixture (Supplementary Fig. 7). We did not explicitly test whether amplifier probes interact with primary probes for which they were not designed.
Given the method’s capacity for fast, flexible multiplexed RNA detection, we next characterized its quantitative accuracy when used at low magnification, a capability useful for high-throughput imaging of large sample areas. We performed clampFISH 2.0 to round 8 (one round of primary probes and seven rounds of amplifier probes) targeting three human mRNAs (EGFR, AXL and DDX58) with a range of expression levels. After the clampFISH 2.0 protocol, we hybridized conventional, unamplified single-molecule RNA FISH probes as a gold standard3, which were designed to bind to non-overlapping sites on the same mRNA. We were able to observe many of the same spots with clampFISH 2.0 at ×20 magnification that we saw using conventional single-molecule RNA FISH at ×60 high magnification, confirming the method’s high sensitivity and specificity (Fig. 2a). In addition to the ninefold larger field of view and greater depth of field offered by ×20 magnification compared with ×60 magnification, we detected clampFISH 2.0 spots at ×20 using shorter exposure times (100 ms for EGFR, 250 ms for AXL and 500 ms for DDX58) in comparison with the 2 s exposure time used with conventional single-molecule RNA FISH. On comparison of the spot counts for multiple targets between clampFISH 2.0 at ×20 magnification and conventional single-molecule RNA FISH at ×60 magnification, we observed a clampFISH 2.0 detection efficiency between 49% and 73% (see Methods) and a high correlation in spot counts (Fig. 2b), demonstrating that clampFISH 2.0 can be used as a higher-throughput replacement for conventional single-molecule RNA FISH. Even for a target (DDX58) expressed at low levels in a subset of cells, we were able to accurately identify cells with three or more RNAs (41–53% sensitivity, 97–99% specificity), thus supporting the ability of clampFISH 2.0 to reliably quantify even low-expression genes at ×20 magnification. We observed no particular decrease in clampFISH 2.0 detection efficiency with increasing conventional single-molecule RNA FISH counts (that is, AXL, EGFR), suggesting that under-counting of spots at ×20 due to optical crowding is minimal in this range of expression levels. Comparing spot counts from clampFISH 2.0 at ×10 magnification with conventional single-molecule RNA FISH at ×60 magnification, we saw a reduction in the correlation strength (for example, when targeting AXL we observed an R2 of 0.740–0.773 at ×10 magnification, versus an R2 of 0.891–0.899 for ×20 magnification; Fig. 2b and Extended Data Fig. 7), suggesting that more accurate quantification using ×10 magnification may require additional rounds of amplification beyond round 8. Although the majority of clampFISH 2.0 spots lie in the cytoplasm, as expected, we also detected spots in the nucleus (Fig. 3b) and, unlike with clampFISH 1.0 (ref. 15), at transcription sites (Supplementary Figs. 8 and 9), a feature of clampFISH 2.0 that enables high-throughput analyses involving RNA localization.
Fig. 2 ∣. ClampFISH 2.0 accurately quantifies RNA spot counts at low-powered magnification.
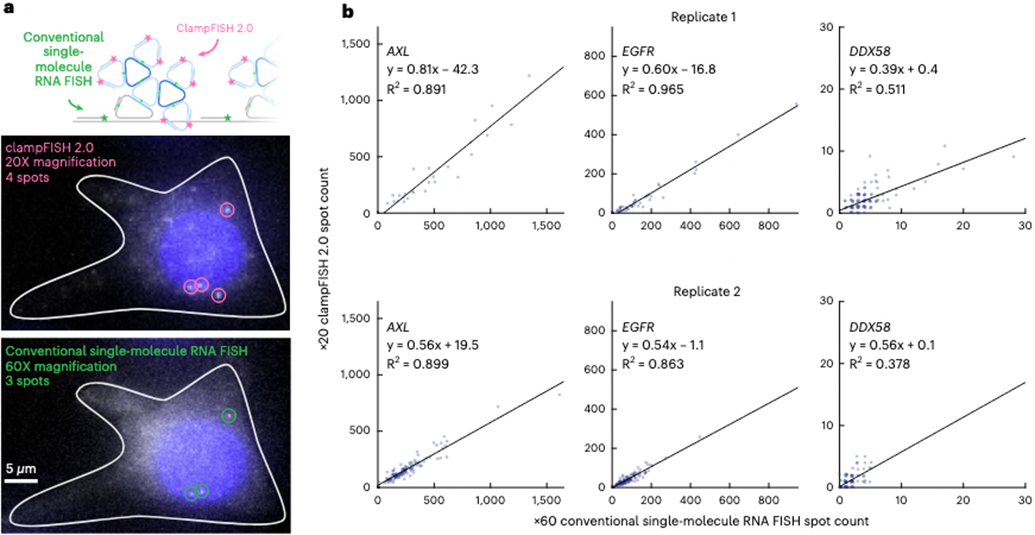
a, Top: schematic diagram of labeling of the same RNA with clampFISH 2.0 and conventional single-molecule RNA FISH, probing non-overlapping regions of the RNA. Middle: image of DDX58 clampFISH 2.0 spots with readout probes labeled in Alexa Fluor 594 and imaged at ×20 magnification. Bottom: image of conventional single-molecule RNA FISH (labeled with Cy3) targeting non-overlapping regions of DDX58 at ×60 magnification in the same cell. Scale bar, 5 μm. b, Comparison of the spot counts between clampFISH 2.0 at ×20 magnification and conventional single-molecule RNA FISH at ×60 magnification. We performed clampFISH 2.0 for 10 genes, amplified the 10 scaffolds in parallel to round 8, then added a single pair of readout probes to label a scaffold corresponding to AXL (left; in drug-resistant WM989 A6-G3 RC4 cells). EGFR (middle; in drug-resistant WM989 A6-G3 RC4 cells), or DDX58 (right; in drug-naive WM989 A6-G3 cells). In two biological replicates we counted spots for clampFISH 2.0 at ×20 magnification and conventional single-molecule RNA FISH at ×60 magnification, which targeted non-overlapping regions of the same RNAs, as shown in a.
Fig. 3 ∣.
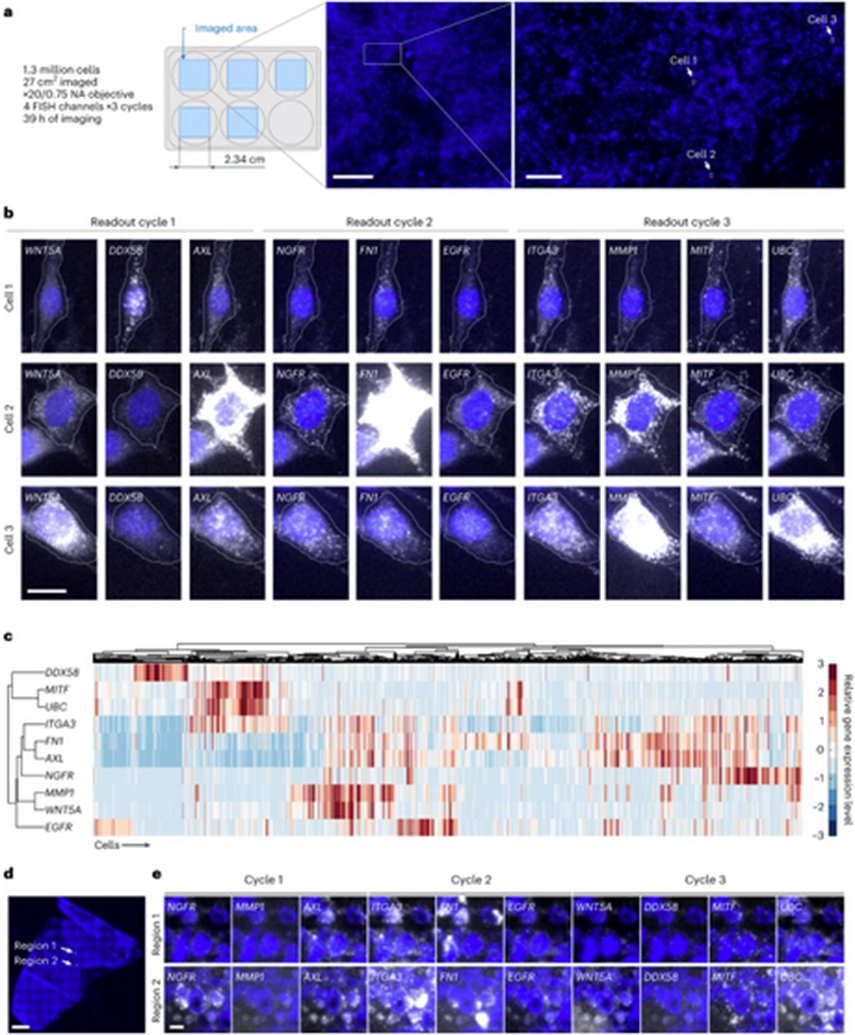
a, In the high-throughput profiling experiment, clampFISH 2.0 was performed for 10 genes in 13 million drug-naive WM989 A6-G3 cells. b, Images at ×20 magnification of three example cells from a that had high levels of expression of one or more of eight cancer marker genes (WNT5A, DDX58, AXL, NGFR, FN1, EGFR, ITCA3 and MMP1), showing spots for these genes as well as MITFand UBC, for a total of 10 genes probed throughout three readout cycles. c, Hierarchical clustering for 42,802 cells (5.9% of the 722,298 cells passing quality control checks) that had high levels of expression of one or more of the eight cancer marker genes. The experiment in a–c was repeated twice, with replicate 2 performed in a single well, with similar results (Supplementary Fig. 16). d, A ×20 magnification scan of DAPI in a fresh frozen tumor model using human WM989-A6-G3-Cas9-5a3 cells injected into a mouse that was fed chow containing a BRAFV600E inhibitor. e, ×20 magnification images of clampFISH 2.0 spots in the same tissue section as in d, probing for the same 10 genes as in b. The experiment in d,e was performed twice (see the Supplementary Methods for details). Scale bars: a, 5 mm (zoom out, 500 μm); b, 20 μm; d, 1 mm; e, 20 μm.
As an additional measure of the quantitative performance of clampFISH 2.0, we compared the average clampFISH 2.0 spot count for 10 human gene targets with their relative abundance (transcripts per million) as detected by bulk RNA sequencing, and found a moderate correlations in two melanoma cell lines (R2 between 0.256 and 0.607; Supplementary Fig. 10). We observed that FN1 and MMP1, both of which have a lower mean clampFISH 2.0 spot count than would be expected from the trend of the remaining genes, are expressed at particularly high levels in a subset of cells (Fig. 3b), suggesting that optical crowding at ×20 magnification may contribute to their under-counting by clampFISH 2.0. Correlations between the ×20 magnification spot count and mean fluorescence intensity suggested that for cells with particularly high RNA copy numbers, the cell’s mean fluorescence intensity might be useful to correct for under-counting due to optical crowding, even though mean fluorescence intensity is not an accurate proxy for spot counts with typical RNA copy numbers (Supplementary Figs. 11 and 12).
Multiplexing with iterative readout in 1.3 million cells
A crucial advantage of clampFISH 2.0 is its potential for rapid multiplexing through iterative hybridization of readout probes. Iterative hybridization refers to schemes for multiplexing beyond the spectral capabilities of conventional fluorescence microscopes25. The basic idea is to detect RNA FISH signal from a small number (typically 3–4) of RNA targets using spectrally distinct fluorophores for each target. To measure RNA FISH signal from more targets in the same cells, the signal from the current set of targets is removed and then another round of hybridization to the next set of targets is performed, enabling detection of another set of RNA species. ClampFISH 2.0 is, in principle, ideally suited for such iterative schemes because all of the scaffolds can be generated at once before any readout steps, and the short readout probes could be stripped and re-probed very rapidly.
An important first step for iterative hybridization is the ability to remove the fluorescence signal from the sample after imaging. Thus, we first tested whether the readout probes could be reliably stripped from their scaffolds with a simple high-stringency wash. We probed the mRNA from 10 genes (each with its own primary probe set) with one of 10 amplifier-specific sequences (pairing gene 1 with amplifier set 1, gene 2 with amplifier set 2 and so on), and generated scaffolds by amplifying to round 8. With these scaffolds generated in three separate wells, we then hybridized four spectrally distinct sets of readout probes (coupled to Atto488, Cy3, Alexa Fluor 594 or Atto 647N), each binding to a specific amplifier set, such that each readout probe set contained a secondary-targeting probe and a tertiary-targeting probe (which must be distinct due to design constraints; see Methods). Thus, we visualize four genes simultaneously per well (10 genes in total, with the scaffolds for one housekeeping gene, UBC, probed in all three wells). After imaging these spots we then stripped off the readout probes with 30% formamide in 2X SSC, re-imaged the samples, and noted that nearly all of the spots were removed (Extended Data Fig. 8 and Supplementary Fig. 13). Given that the clampFISH 2.0 scaffolds are constructed of interlocking loops, we expected the scaffolds to remain stably attached to the mRNA targets despite the dissociation of the readout probes. Indeed, when we re-probed the same scaffolds after multiple cycles of readout hybridization and stripping, we observed the same spots as in the initial readout cycle (Extended Data Fig. 9) and found high correlations in spot counts when compared with the initial readout cycle (for example, R2 of 0.833 (replicate 1) and 0.846 (replicate 2) for WNT5A; Supplementary Figs. 14 and 15), thus demonstrating the stability of the scaffolds and their ability to be repeatedly probed. And even after leaving a sample refrigerated for 4 months and again re-probing the same scaffolds, we still observed the same spots (Extended Data Fig. 9) and a similarly high correlation in spot counts to the initial readout cycle (R2 of 0.758 (replicate 1) for WNT5A; Supplementary Fig. 14), thus demonstrating flexibility in the timing of readout and imaging.
Having demonstrated the ability to strip off readout probes, we then attempted to detect the mRNA from 10 different genes simultaneously in individual cells. We targeted the transcripts of the genes WNT5A, DDX58, AXL, NGFR, FN1, EGFR, ITGA3, MMP1, MITF and UBC at the same time in the WM989 A6-G3 melanoma cell line22 (and WM989 A6-G3 RC4 cells; see Methods for details). We imaged cells spread over five wells of a six-well culture dish with three cycles of imaging. Each imaging cycle consisted of detection in four readout probe channels, with UBC mRNA probed in every cycle as a control for consistency. The amplified signal allowed for a typical exposure time of 250 ms with a ×20/0.75 NA objective lens, enabling us to detect 10 genes in 1.3 million cells in 39 hours of imaging (Fig. 3a,b), demonstrating the ability to perform multiplex gene expression analysis via iterative hybridization across a large number of individual cells.
As a demonstration of the type of analysis that such high-throughput multiplexed RNA quantification enables, we analyzed the co-expression of these genes in the rare subpopulations that express them. These genes have been shown to be expressed highly only in rare cells (1:50–1:500), and it is these rare cells with high expression that are the ones that survive targeted drug therapies22,23,26. Many of these genes co-express in single cells22, but the precise co-expression relationships have been difficult to decipher due to the rarity of the expression. We reasoned that the much higher number of cells that we could image with multiplex clampFISH 2.0 (~1.3 million versus ~8,700 for conventional single-molecule RNA FISH22) would enable us to measure these relationships. Using automated cell segmentation27 and a spot-detection pipeline, we identified 42,802 cells with one or more marker genes positively associated with drug resistance out of a total pool of 722,298 cells. This sample size was large enough that we could observe distinct clusters of co-expression (Fig. 3c; for a technical replicate see Supplementary Fig. 16), including subsets of cells associated with distinct resistance phenotypes23, demonstrating the types of analysis that are now possible with the high throughput of clampFISH 2.0.
ClampFISH 2.0 detects RNA in tissue sections
An important application of image-based gene expression detection methods is in multicellular organisms and tissues. To demonstrate that clampFISH 2.0 could work in this context as well, we used the same 10-gene panel described above in fresh frozen tumor sections with a thickness of 6 μm. These sections came from the injection of WM989-A6-G3-Cas9-5a3 cells into mice, which subsequently grew into tumors and were then treated with the BRAFV600E inhibitor PLX4720 (samples first used in ref. 28; see that paper for details). We observed a clampFISH 2.0 signal in many of the cells, including a consistent UBC signal in virtually all of the cells that we assessed as being of presumptive human origin but minimal signal in those of presumptive mouse origin (species distinguished by two distinct nuclear morphologies; Extended Data Fig. 10), confirming that clampFISH 2.0 was able to detect RNA in tissue sections (Fig. 3d,e). We also performed clampFISH 2.0 in a formalin-fixed paraffin-embedded (FFPE) tissue section, in which we noted a dimmer UBC clampFISH 2.0 signal (Supplementary Fig. 17). There were regions of the FFPE tissue section that were completely devoid of signal, perhaps due to sample degradation or other unknown factors. We found that ITGA3 spot intensities in a fresh frozen tissue section were of a similar intensity to those in a cell line, while the spots in a FFPE tissue section were ~20% dimmer (Supplementary Fig. 18).
Discussion
We have here described the development of an improved version of clampFISH, termed clampFISH 2.0. Two key updates to the original method are the inverted probe design, which makes probe synthesis far more cost-effective and time-efficient, and the increased speed of the protocol. In particular, the efficiencies for probe synthesis are critical for multiplexing applications in which multiple RNA species are targeted at the same time. While we expect that most use cases will be better served by clampFISH 2.0, clampFISH 1.0 might, in principle, be preferable for probing splice junctions, in which the proximity ligation of the primary probe could be optimized to discriminate between exon pairs.
One important aspect of amplified signal is that lower-powered optics can be used, in particular at lower magnification. By using a ×20 (or ×10) objective, a 20–25-fold (40–75-fold) increase can be obtained in throughput (number of cells imaged per unit time) as compared with conventional single-molecule RNA FISH imaged using a ×60 objective. These order-of-magnitude increases in throughput can enable many new applications, especially in the detection of rare cell types. It is possible that other imaging improvements may be facilitated by the dramatically increased signal afforded by signal amplification.
While here we have demonstrated a straightforward iterative hybridization scheme for multiplex RNA detection, it is possible that clampFISH 2.0 could be used for more complex combinatorial multiplex schemes as well16-18,25,29-31. Many of those schemes rely on the detection of the same RNA in a specified subset of iterative detection rounds. ClampFISH 2.0 could be particularly well-suited for such schemes because combinations of readout probes could be used in each round to detect specific RNA species. That readout probes can be re-hybridized to the same scaffolds offers added flexibility in sequential encoding schemes. For example, whereas the sequential barcode is normally encoded by the library of RNA-binding probes, which cannot be modified after their construction, each gene might instead have a single associated amplifier set, where the choice of each imaging cycle’s subset of readout probes would define the barcode.
Another potential benefit of clampFISH 2.0 for such sequential barcoding schemes is the small optical size of the spots, ~264 nm and ~316 nm full width at half maximum for Atto 488- and Atto 647N-labeled readout probes, respectively. Both HCR and rolling circle amplification produce spots that are larger (up to ~1 μm)18,29,32 than diffraction-limited spots, which contributes to optical crowding: when visualizing a large number of spots, they can overlap, making it difficult to discriminate neighboring spots. This makes it particularly difficult to co-localize spots through multiple rounds of hybridization and imaging. Other benefits of a diffraction-limited spot size are that it is suitable for accurate super-resolution structural analysis by, for example, STORM33, DNA-PAINT34-38 or STED39, and also that many image analysis tools assume diffraction-limited spots.
ClampFISH 2.0’s combination of high amplification, rapid and flexible multiplexing, small spot sizes and low cost enables very high-throughput and quantitative RNA detection. In potential further extensions of the method, clampFISH 2.0 could serve as a platform for higher-throughput sequential labeling schemes and super-resolution imaging.
Methods
Probe design and construction
ClampFISH 2.0 primary probe design and construction.
We constructed clampFISH 2.0 primary probes as follows. First, we designed a set of 30mer RNA-targeting probe sequences for each target gene with custom MATLAB software3 and added a flanking 10mer 5′ sequence (AAGTGACTGT) and a 10mer 3′ sequence (ACATCATAGT) to each of those respective ends, producing a 50mer sequence (Supplementary Table 1). The 50mer sequences were run through a custom MATLAB script using BLAST40 for alignment to the human transcriptome and NUPACK41-44 to predict binding energies of the off-target transcriptomic hits. We kept only the hits with binding energy less than −14 kcal mol−1, and then assigned to each of these hits the maximum fragments per kilobase of transcript per million (FPKM) from a set of 13 human RNA sequencing (RNA-seq) datasets from the ENCODE portal45,46 (https://www.encodeproject.org/; see Supplementary Table 6 for file identifiers). For each gene we selected 24–32 primary probes per gene target, with a preference for probes targeting the coding region, and for which the sum of FPKM values from its predicted off-target hits was minimized. For probes targeting GFP mRNA, we used 10 probes for which the 30mer primary probe sequences were taken from ref. 15. The 50mer sequences were ordered from Integrated DNA Technologies (IDT) and pooled together for a given gene. For each gene-specific pool, an azido-deoxyadenosine-triphosphate (azido-dATP) (N6-(6-Azido)hexyl-3′-dATP, Jena Bioscience, NU-1707L) was added to the 3′ ends of the probes with Terminal Transferase (New England Biolabs, M0315L), which adds a single azido-dATP molecule. Then, the 5′ ends were phosphorylated with T4 Polynucleotide Kinase (New England Biolabs, M0201L). Each gene-specific pool of 51mer oligonucleotides was mixed with a 20mer ligation adapter (ACAGTCACTTCAACACTCAG) and a 58mer oligonucleotide, which were both ordered from IDT. The 58mer oligonucleotide was ordered with a 5′ alkyne modification (5′ hexynyl) and was designed with the following sequences, in 5′ to 3′ order: a universal 18mer sequence (AGACATTCTCGTCAAGAT), an amplifier-specific 30mer sequence (serving as a landing pad upon which a secondary probe can bind), and a universal 10mer sequence (CTGAGTGTTG). Then, T7 DNA Ligase (New England Biolabs, M0318L) was added to ligate a complete 109mer (50 + 1 + 58) primary probe. We then added ammonium acetate to a 2.5 M concentration, centrifuged twice at 17,000 ×g (each time pipetting all but the bottom 20 μl of solution to a new tube), ethanol precipitated the probes, resuspended the probes in nuclease-free water, centrifuged the tube at 17,000 ×g and pipetted all but the bottom 5 μl into a new tube. See Supplementary Tables 1, 2, 4 and 7 for sequences and information related to the primary probes.
ClampFISH 2.0 amplifier probes design and construction.
ClampFISH 2.0 amplifier probes (secondary probes and tertiary probes) were constructed as follows. To design amplifier probe sets 1 and 2, two 30mer ‘landing pad’ sequences (one for the secondary, one for the tertiary) were manually generated with approximately 50% GC content and AT at the center, and the 30mer was then concatenated to itself to form a 60mer backbone sequence. We added 15mer arms on each end of the 60mer secondary backbones, such that arms were reverse complements to their paired tertiary backbone, and similarly added 15mer arms to each tertiary backbone to be reverse complements to their paired secondary backbone, thus completing each amplifier probe’s full 90mer probe sequence. Hence, the sequences of the 90mer secondary probe and 90mer tertiary probe pair are entirely determined by two 30mer landing pad sequences. For the remaining amplifier series we generated 500,000 random 30mers, then replaced the middle two bases with AT. We retained sequences for which the percent GC content of the left 15 nucleotides and the right 15 nucleotides were both between 45% and 55%, and then concatenated the remaining two 30mers to create a 60mer backbone sequence. Backbone sequences with stretches of three or more C, three or more G, or five or more G or C bases were discarded. For amplifier series 3–7 we then selected backbones for which the free energy of each backbone’s folded structure was greater than −2 kcal mol−1 as predicted using the DINAMelt web server47, selected those without hits against the human transcriptome using BLAST (https://blast.ncbi.nlm.nih.gov), added two 15mer arms to each backbone as before to generate a 90mer amplifier probe, and then selected the five 90mer amplifier probe pairs for which the free energy of folding was the least negative as predicted using DINAMelt. For amplifier series 8–15 we followed the same steps to generate 60mer backbones (using a different random number generator seed), and then used NUPACK to predict the minimum free energy of its folded structure, accepting those with a value greater than −1.5 kcal mol−1. We designated half of the 60mer sequences as secondary backbones and the other half as tertiary backbones and paired each secondary backbone with a tertiary backbone. We again used NUPACK to retain only those with a minimum free energy greater than −2.0 kcal mol−1. We checked for off-target binding against the human transcriptome using BLAST, using both a spliced transcriptome database and a custom-generated transcriptome database with unspliced transcripts, used NUPACK to retain only those with strong off-target binding to RNAs, then took the sum of the RNA transcripts’ maximum FPKM from the ENCODE RNA-seq datasets to generate an off-target FPKM for each secondary and tertiary probe. We chose secondary and tertiary probe pairs for which each probes’ FPKM sum is ≤500 when using the spliced transcript database and ≤2,500 when using the unspliced transcript database. We then dropped any amplifier sets with probes hitting genomic repeats using repeat masker (https://www.repeatmasker.org/). We used NUPACK to simulate binding against other probes of the same probe type (each secondary against other secondaries, each tertiary against other tertiaries), and discarded four amplifier sets for which the predicted binding energy to another probe was < −23 kcal mol−1. All amplifier probe sequences are listed in Supplementary Table 2.
We ordered amplifier probes from IDT as 89mers with a 5′ hexynyl modification for 15 amplifier sets in total (15 secondaries and 15 tertiaries). In separate reactions for each amplifier probe, we added an azido-dATP (N6-(6-Azido)hexyl-3’-dATP, Jena Bioscience, NU-1707L) to the 3′ ends of the probes with Terminal Transferase, thus completing the 90mer amplifier sequence. We then added ammonium acetate to 2.5 M and magnesium chloride to 10 mM, then centrifuged twice at 17,000 ×g (and each time pipetting all but the bottom 10 μl of solution to a new tube). We then ethanol precipitated the probes, resuspended the probes in 200 μl nuclease-free water, centrifuged the tube at 17,000 ×g and pipetted all but the bottom 20 μl into a new tube. Probe design and synthesis protocols are shown in Supplementary Fig. 1.
ClampFISH 2.0 readout probe design and construction.
For the amplifier screen experiment we used a 20 nucleotide readout probe that was designed to bind to the center of the 30mer landing pad sequences of each secondary probe. The readout probes were ordered from IDT with a 3′ Amino modifier (/3AmMO/), coupled to Atto 647N NHS-ester (ATTO-TEC, AD 647N-31), ethanol precipitated, purified by high-performance liquid chromatography (HPLC)3 and resuspended in TE pH 8.0 buffer (Invitrogen, AM9849).
For all other experiments we designed two readout probes for each amplifier set: one to bind to the secondary probe and one to bind to the tertiary probe, and each was designed to bind roughly to the center of the probe’s 30mer landing pad sequences. These two readout probes must be distinct because the secondary landing pad and tertiary landing pad must, by design, be distinct from one another (otherwise, for example, the two arms of a secondary probe could also bind to its own landing pad). Readout probe sequences were chosen such that the Gibbs free energy of binding to their target amplifier backbone (DNA:DNA binding) was −22 kcal mol−1 or −24 kcal mol−1, as calculated using the MATLAB oligoprop function (based on the parameters from ref. 48), and then ordered from IDT with a 3′ Amino modifier. The two readout probes targeting a given amplifier set were pooled and then coupled to one of four NHS-ester dyes (Atto 488, ATTO-TEC, AD 488-31; Cy3, Sigma-Aldrich, GEPA23001; Alexa Fluor 594, ThermoFisher, A20004; or Atto 647N, ATTO-TEC, AD 647N-31), ethanol precipitated, purified by HPLC and resuspended in TE pH 8.0 buffer, except for readout probes coupled to Atto 488, which were not pooled until after the HPLC steps. All readout probe sequences are listed in Supplementary Table 3.
Conventional single-molecule RNA FISH probes.
We designed conventional single-molecule RNA FISH probes for GFP, AXL, EGFR and DDX58 as previously described3, but selected a subset of probes not overlapping with the clampFISH 2.0 primary probes for these genes. We coupled probes to the NHS-ester dyes Cy3 (for the AXL, EGFR and DDX58 probe sets) and Alexa Fluor 555 (Invitrogen, A-20009; for the GFP probe set). All conventional single-molecule RNA FISH probe sequences are listed in Supplementary Table 5.
All oligonucleotide sequences are listed in Supplementary Tables 1-5 with recommended amplifier sets listed in Supplementary Table 13. The scripts used to generate probe sequences are available at https://www.dropbox.com/sh/q51kmcphoyi9yi3/AAB4g1a6ODDHaphsvbmBJAy-a?dl=0
Cell culture and tissue processing
A single-cell bottlenecked human melanoma cell line (WM989 A6-G3 cells) and cell lines derived from it (WM989 A6-G3 H2B-GFP cells and WM989 A6-G3 RC4 cells) were cultured and placed on no. 1 or no. 1.5 cover glass. See Supplementary Methods for details of the culturing and plating of the cell lines.
We fixed cell lines at room temperature by rinsing cells once in 1X PBS (Invitrogen, AM9624), incubating for 10 min in 3.7% formaldehyde (Sigma-Aldrich, F1635-500ML) in 1X PBS, then rinsing twice in 1X PBS. Cells were permeabilized in 70% ethanol and stored at 4 °C for at least 8 h. Nuclease-free water (Invitrogen, 4387936) was used in all buffers for fixation onwards, including permeabilization, probe synthesis and all RNA FISH steps.
In brief, for the fresh frozen tissue experiment, sample preparation steps included injection of human WM989-A6-G3-Cas9-5a3 cells into a mouse, collection of the tumor, freezing in optimal cutting temperature compound, cryo-sectioning onto a microscope slide, fixation with 3.7% formaldehyde in PBS, and permeabilization. In brief, for the FFPE tissue experiment, sample preparation steps included implantation and growth of patient-derived tissue (WM4505-1 or WM4298-2) in a mouse, collection of the tumor, fixation with 10% neutral-buffered formalin, permeabilization, paraffin embedding, sectioning to a thickness of 5 μm, placement on a microscope slide, sealing with paraffin, storage at room temperature, deparaffinization, post-fixation, re-hydration, and antigen retrieval. Both fresh frozen and FFPE tissue samples were then cleared with 8% sodium dodecyl sulfate. See Supplementary Methods for details of the tissue experiment sample preparation.
The mouse experiments were approved by the Wistar Institutional Animal Care and Use Committee and were performed in accordance with institutional and national guidelines and regulations.
ClampFISH 2.0 protocol
ClampFISH 2.0 primary probe steps.
We performed clampFISH 2.0 in eight-well chambers as follows. First, we aspirated the 70% ethanol (or 2X SSC for tissue sections), rinsed with 10% wash buffer (10% formamide, 2X SSC), then washed with 40% wash buffer (40% formamide, 2X SSC) for 5–10 min. We mixed primary probes with 40% hybridization buffer (40% formamide, 10% dextran sulfate, 2X SSC) such that each probe’s final concentration was 0.1 ng μl−1 (~2.8 nM), added this mixture to the well, covered and spread it out with a coverslip, and then incubated it overnight (for ≥10 h) in a humidified container at 37 °C. See Supplementary Methods for details of the clampFISH 2.0 primary probe steps.
The following day we pre-warmed all wash buffers to 37 °C: 10% wash buffer, 30% wash buffer (30% formamide, 2X SSC), and 40% wash buffer. We first added warm 10% wash buffer, removed the coverslips and aspirated the solution, and washed again with warm 10% wash buffer. We then washed twice for 20 min each with warm 40% wash buffer on a hotplate set to 37 °C (the temperature setting used throughout the protocol). After removing the chamber from the hotplate, we added 10% wash buffer before beginning the amplification steps.
ClampFISH 2.0 amplification steps.
For amplification we first mixed together all the secondary probes with 10% hybridization buffer with Triton-X (10% formamide, 10% dextran sulfate, 2X SSC and 0.1% Triton-X (Sigma-Aldrich, T8787-100ML)) to a final ~20 nM concentration per probe (range, ~13 nM to 25 nM) with a 27mer circularizer oligonucleotide (TCTTGACGAGAATGTCTTACTATGATG) at a 40 nM final concentration. We also mixed together all tertiary probes with 10% hybridization buffer with Triton-X at the same concentration, but without the circularizer oligonucleotide. In preparation for multiple click reaction steps, we prepared the tubes each with an appropriate volume of pre-warmed 2X SSC with Triton-X and dimethylsulfoxide (DMSO; 2X SSC, 0.25% Triton-X, 10% DMSO) for the amplification step, and warmed them to 37 °C. We also aliquoted sodium ascorbate (Acros, AC352680050) into 1.5 ml tubes, ready to be dissolved fresh with each click step. We prepared a CuSO4 (Fisher Scientific, S25289) and BTTAA ( Jena Bioscience, CLK-067-100) mixture in a 1:2 CuSO4:BTTAA molar ratio, enough to use for all the click reactions throughout the rounds of amplification. We added the secondary probe and circularizer oligo-containing 10% hybridization buffer with Triton-X to the well, covered it with a coverslip, and incubated it for 30 min in a 37 °C incubator. After taking the chamber out of the incubator, we added warm 10% wash buffer, removed the coverslips, washed twice for 1 min each with warm 10% wash buffer, and then washed again with warm 10% wash buffer for 10 min on the hotplate. We then took the chamber off the hotplate and added 2X SSC before the click reaction. The click reaction mixture was then prepared by first mixing the CuSO4 and BTTAA mixture with the pre-warmed 2X SSC with Triton-X and DMSO buffer. Working quickly, we added nuclease-free water to an ascorbic acid aliquot and vortexed it until it had dissolved. We aspirated the 2X SSC solution from the well plate and quickly added aqueous sodium ascorbate to the CuSO4 + BTTAA + 2X SSC with Triton-X and DMSO mixture (final concentrations: 150 μM CuSO4, 300 μM BTTAA, 5 mM sodium ascorbate, ~2X SSC, ~0.25% Triton-X, ~10% DMSO) and briefly mixed it by swirling the tube by hand. We immediately added the click reaction solution to the wells and incubated it on the hotplate for 10 min. Next, we aspirated the click reaction mixture and washed the sample with warm 30% wash buffer for 5 min on the hotplate. The above steps (amplifier probe hybridization, 10% wash buffer steps, click reaction, and 30% wash buffer step) constitute a single round of amplification and take ~1 h when accounting for pipetting time.
Before beginning the next round of amplification we then replaced the 30% wash buffer with warm 10% wash buffer. (If, alternatively, a breakpoint was needed in between rounds of amplification, we instead replaced the 30% wash buffer with 2X SSC and stored the sample at room temperature for up to 2 h or at 4 °C for up to 24 hours.) We then proceeded to the next round of amplification, this time using tertiary probes instead of secondary probes. We term the completion of the primary step as having performed clampFISH 2.0 to ‘round 1’, the first secondary step as ‘round 2’, the first tertiary step as ‘round 3’, the next secondary step as ‘round 4’ and so on. We ran all amplifications to round 8, involving one primary probe round and seven amplification rounds, except where noted differently. At the end of the last amplification round we stored the sample at 4 °C in 2X SSC until the readout probe steps (typically the samples were stored overnight for readout and imaging the subsequent day). See Supplementary Methods for details of the clampFISH 2.0 amplification steps.
Readout cycle steps.
The following day, either directly following amplification or the subsequent conventional RNA FISH, we performed a readout probe cycle as follows. First, samples were brought to room temperature and rinsed once with room temperature 2X SSC. For each amplifier set (each of which corresponds to a particular gene target) to be probed, two readout probes were hybridized (with one binding to the secondary and one binding to the tertiary), both coupled to the same fluorescent dye. A set of readout probes for each of four spectrally distinguishable dyes could be included in a given readout cycle. Each readout probe was hybridized at a 10 nM final concentration in 5% ethylene carbonate hybridization buffer (5% ethylene carbonate, 10% dextran sulfate, 2X SSC, 0.1% Triton-X) for 20 min at room temperature. We then aspirated the solution, washed twice for 1 min each with 2X SSC with Triton-X (2X SSC, 0.1% Triton-X), 1 min with 2X SSC buffer, 5 min with 2X SSC with 50 ng ml−1 DAPI, then replaced the solution with 2X SSC before imaging.
After imaging a given readout cycle we stripped off the readout probes by incubating twice for 5 min each at 37 °C with 30% wash buffer pre-warmed to 37 °C, then added 2X SSC before starting another readout cycle. If we imaged the post-strip sample we incubated it for 5 min with 2X SSC with 50 ng ml−1 DAPI, and replaced the solution with 2X SSC before imaging.
For the conventional single-molecule RNA FISH comparison experiment, after stripping the readout probes we proceeded with conventional single-molecule RNA FISH, as described above, but instead we used probes for AXL, EGFR or DDX58 without any additional readout probes.
Imaging and image analysis
Imaging.
For imaging we used a Nikon Ti-E inverted microscope controlled by NIS-Elements v5.11.01 on a personal computer running Windows 10 and equipped with an ORCA-Flash4.0 V3 sCMOS camera (Hamamatsu, C13440-20CU), a SOLA SE U-nIR light engine (Lumencor), and a Nikon Perfect Focus System. We used ×100 (1.45 NA) Plan-Apo λ (Nikon, MRD01905), ×60 (1.4 NA) Plan-Apo λ (Nikon, MRD01605), ×20 (0.75 NA) Plan-Apo λ (Nikon, MRD00205) and ×10 (0.45 NA) Plan-Apo λ (Nikon, MRD00105) objectives and filter sets for DAPI, Atto 488, Cy3, Alexa Fluor 594 and Atto 647N (see Supplementary Table 8 for filter sets used; filter sets can also be viewed at https://www.fpbase.org/microscope/455WNQygW6268avMhrTNx8/). All ×60 images were taken using 2 × 2 camera binning, while ×100, ×20 and ×10 images used 1 × 1 binning. Based on the camera’s pixel size of 6.5 μm, the image pixel size for each magnification level is 65 nm (×100, 1 × 1 binning), 216.7 nm (×60, 2 × 2 binning), 325 nm (×20, 1 × 1 binning) and 650 nm (×10, 1 × 1 binning).
Image analysis.
We converted ND2 files to Tiff files using NIS-Elements and bio-formats 6.5.1 for MATLAB (bfmatlab, from www.openmicros-copy.org). Segmentation of cellular outlines was performed manually using rajlabimagetools or using Cellpose27 (downloaded on 16 March 2021), running python 3.7.10 (64-bit for MacOS from www.anaconda.com). When overlapping images were taken in a grid format, the scans were stitched and registered using the pixyDuck repository. Spots were automatically detected using rajlabimagetools for z-stack data and with dentist2 for scan data. See Supplementary Methods for details on the image analysis.
All code to generate contrasted images are located in the paper/scripts/showImagesScripts directory at https://www.dropbox.com/sh/q51kmcphoyi9yi3/AAB4g1a6ODDHaphsvbmBJAy-a?dl=0. We provide the minimum and maximum values of the display look up tables in the code itself or, when many panels are used, in tables located in the example images directory (paper/exampleImages). We used a look up table with γ = 0.7 for displaying the images used in Fig. 2a (maximum intensity projections) and γ = 1 for all other images.
All box-and-whisker plots show the median (center line), upper and lower quartiles (box), and the most extreme values (whiskers) that are not considered outliers, defined as values with a distance of more than 1.5-fold the interquartile range above the upper quartile or below the lower quartile. All violin plots contain an inner box plot showing the median (circle) and upper and lower quartiles (box). All R2 values are unadjusted coefficients of determination. All trendlines shown are least-squares linear regressions.
RNA sequencing
We performed bulk RNA sequencing as described in ref. 24. See Supplementary Methods for details on the RNA sequencing.
Extended Data
Extended Data Fig. 1∣. clampFISH 2.0 amplifies GFP RNA FISH signal with high specificity.
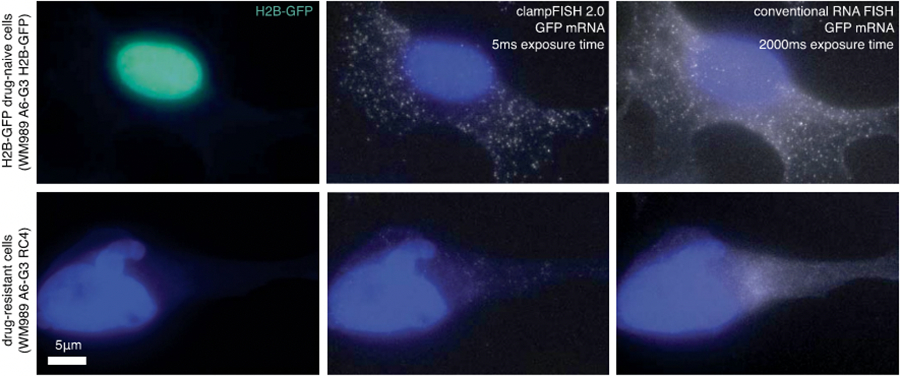
Images of GFP clampFISH 2.0 spots in drug-naive H2B GFP VVM989 A6-G3 cells (top) and vemurafenib-resistant WM989 A6-G3 RC4 cells (bottom) with a 20 nucleotide secondary-targeting readout probe (labeled with Atto 647N) and conventional single-molecule RNA FISH probes (labeled with Alexa 555) targeting different regions of the same RNA. Shown are maximum intensity projections of 9 z-planes at 60X magnification. Shown are data using amplifier set 1, where 15 amplifier sets were tested in total. The experiment was performed once. See Supplementary Fig. 2 for co-localization quantification of all amplifier sets. For details, see Supplementary Methods description of amplifier screen experiment. As expected, we observed bright GFP clampFISH 2.0 spot counts in cells with nuclear-localized GFP signal, but not in cells without the H2B-GFP construct.
Extended Data Fig. 2∣. clampFISH 2.0 amplifies EGFR RNA FISH signal with high accuracy.
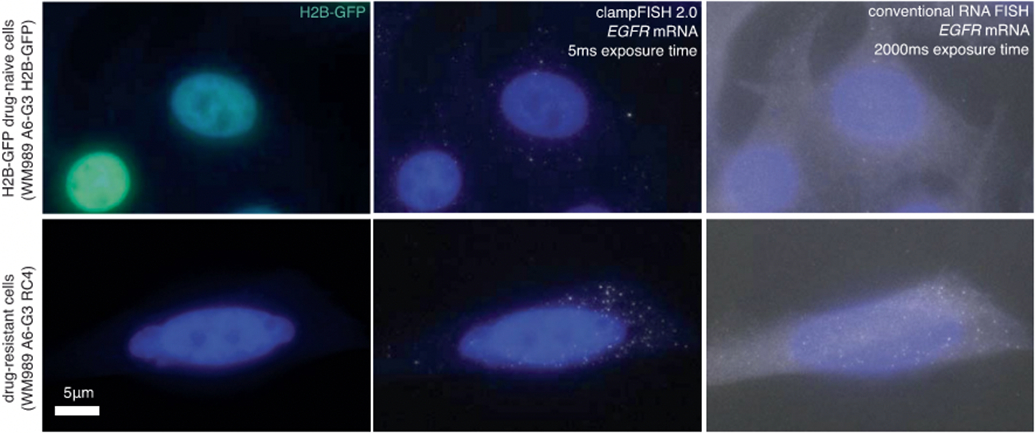
Images of EGFR clampFISH 2.0 spots in drug-naive H2B-GFP WM989 A6-G3 cells (top) and vemurafenib-resistant WM989 A6-G3 RC4 cells (bottom) with a 20 nucleotide secondary-targeting readout probe (labeled with Atto 647N) and conventional single-molecule RNA FISH probes (labeled with Cy3) targeting different regions of the same RNA. Shown are maximum intensity projections of 9 z-planes at 60X magnification. Shown are data using amplifier set 1, where 15 amplifier sets were tested in total. The experiment was performed once. For details, see Supplementary Methods description of amplifier screen experiment. As expected from bulk RNA sequencing data, we observed many more EGFR clampFISH 2.0 spots in the vemurafenib-resistant cells than in the drug-naive cells.
Extended Data Fig. 3 ∣. clampFISH 2.0 eliminates the bright, non-specific fluorescent spots that were observed in clampFISH 1.0.
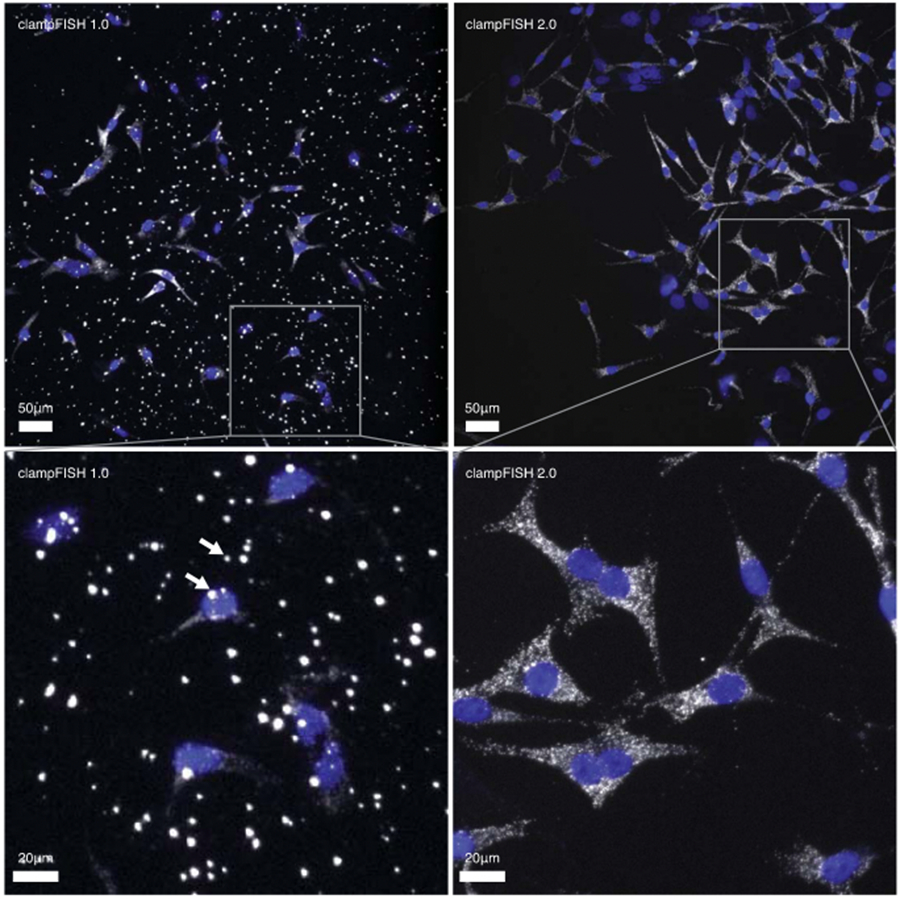
Top left: clampFISH 1.0 targeting GFP in WM983b GFP melanoma cells, amplified to round 6 with amplifier probes containing an internal Cy5 dye and imaged at 20X with a 3 second exposure time using a cooled CCD camera with a 13μm pixel size (image from Rouhanifard et al. 2018; see that paper for further details). The two arrows point to two of the non-specific spots. Top right: clampFISH 2.0 targeting GFP in a mixed population of cells (a majority of WM989 A6-G3 H2B GFP cells and fewer WM989 A6 G3 RC4 cells), amplified to round 8 with readout probes labeled with Atto 647N and imaged at 20X with a 1 second exposure time using a sCMOS camera with a 6.5μm pixel size. Image shown is from the present work’s ‘pooled amplifier experiment’, which was performed once. For all experiments performed in this work, we observed similar results to those depicted here. Bottom: zoomed in views of the top images. We found that we could eliminate the bright non-specific spots by introducing a number of centrifugation steps to both the primary probe and amplifier probe synthesis protocols. To perform this step, we centrifuge the solution in 1.5 mL tubes at 17.000 g for 20 minutes and transfer the top portion of the solution to a new tube and discarded the bottom portion. We perform this step twice after the enzymatic steps are complete, and once after ethanol precipitation (see Supplementary Fig. 1). Additionally, we found that by adding the centrifugation step to completed clampFISH 1.0 probe solutions, we could similarly reduce the non-specific spots seen in that method.
Extended Data Fig. 4∣. clampFISH 2.0 amplifies signal exponentially.
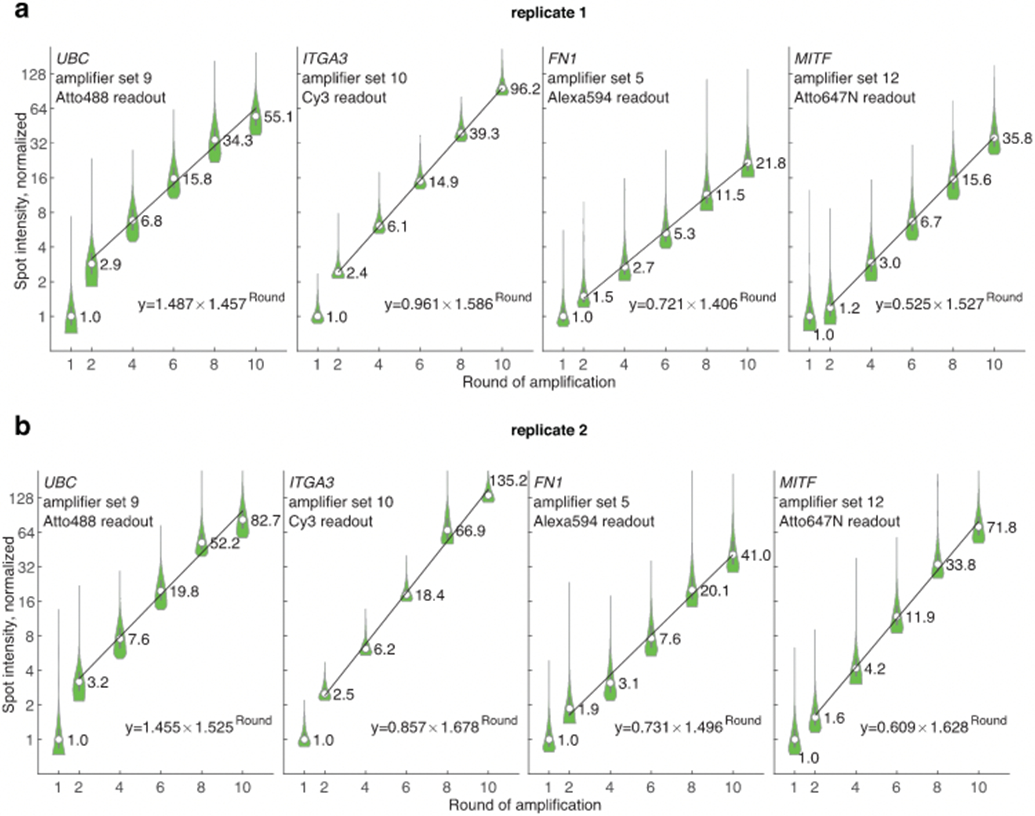
(a) In an amplification characterization experiment, we performed clampFISH 2.0 with amplification to varying rounds (round 1, 2, 4, 6, 8, and 10) and then hybridized four readout probes to measure the spot intensities, with the median intensity from rounds 2, 4, 6, 8 and 10 fit to an exponential curve (labeled values are median intensities). We found that every round the spot intensities grew by a factor of 1.457, 1.586, 1.406, and 1.527 for each probe set respectively. With a hypothetical 2:1 binding ratio of each amplifier probe to the previous probe, these factors suggest a per-probe binding efficiency of 73%, 79%, 70%, and 76%, respectively. (b) Replicate 2 of the same experiment as in (a), where the spot intensities grew by a factor of 1.525, 1.678, 1.496, and 1.628, suggesting per-probe binding efficiencies of 76%, 84%, 75%, and 81%, respectively. For spot counts associated with each condition in panels a-b, see Supplementary Table 12. Circles are median values and bounds of boxes are 25th and 75th percentiles.
Extended Data Fig. 5∣. clampFISH 2.0 spot sizes remain similar throughout the rounds of amplification.
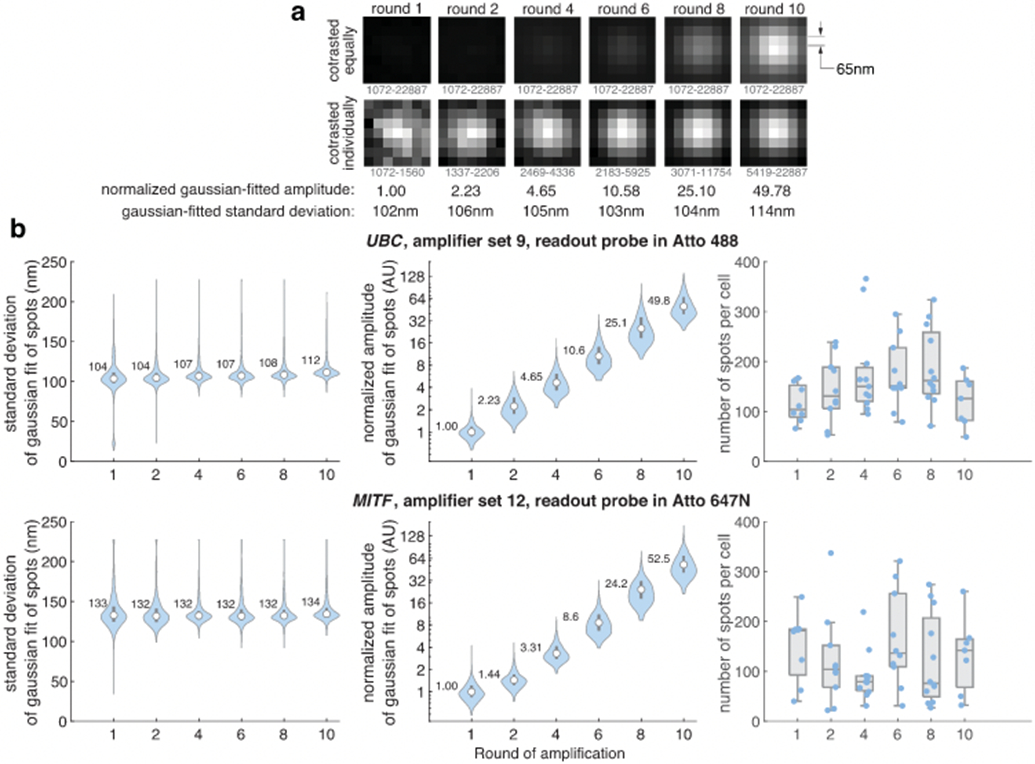
(a) Cropped images of spots from UBC clampFISH 2.0 with readout probes in Atto 488 at varying levels of amplification (from left to right: round 1, 2, 4, 6, 8, and 10) imaged with a 100X/1.45NA objective (65 nm pixel sizes). A spot with a representative (median) fitted amplitude was chosen for display. The minimum intensity (black) and maximum intensity (white) used for contrasting are shown below the images. Contrasting is applied equally to all images (top row) or set to each image’s minimum and maximum values (bottom row). (b) ClampFISH 2.0 was performed to varying rounds of amplification using primary probes targeting UBC mRNA, amplifier set 9, and readout probes labeled in Atto 488 (top panels) or using primary probes targeting MITF. amplifier set 12, and readout probes labeled in Atto 647N (bottom panels). Samples were imaged with a 100X/1.45NA objective (65 nm pixel sizes) and each called spot was fit at its maximal-intensity z-plane to a 2D Gaussian distribution. Shown are the standard deviation of each spot’s Gaussian fit (left panels), amplitude of each spot’s Gaussian fit normalized to the round 1 median amplitude (middle panels), and each segmented cell’s spot count (right panels). For the left and middle panels, circles and numbers shown are median values and bounds of boxes are 25th and 75th percentiles. For UBC data, n = 923, 1437, 1968, 1737, 2251, 846 spots and for MITF data n = 1206, 1219, 994, 1634, 1450. and 930 for rounds 1, 2, 4, 6, 8, and 10, respectively. For the right panel, circles are median values, bounds of boxes are 25th and 75th percentiles, and whiskers extend to non-outlier minima and maxima, where data falling more than 1.5 times the interquartile range beyond the box bounds are considered outliers. Theoretical standard deviations of Gaussian approximations of diffraction-limited spots (0.21λ/NA; with paraxial optics assumptions) with wavelengths at the midpoints of the emission filters (535 nm for Cy3; 667 nm for Atto 647N) are 77.5 nm (Cy3) and 96.6 nm (Atto 647N).
Extended Data Fig. 6∣. clampFISH 2.0 spot sizes are similar to conventional single-molecule RNA FISH spot sizes.
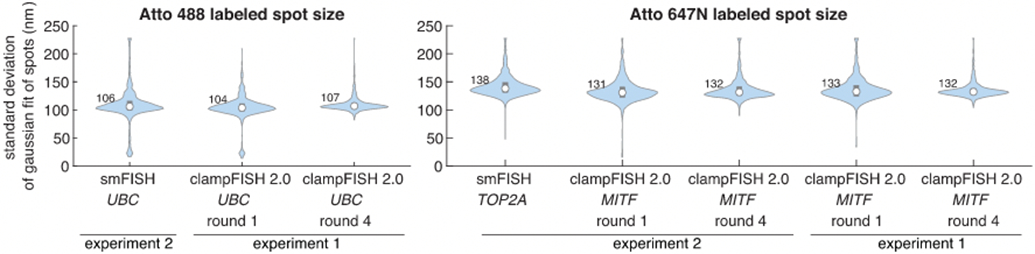
Conventional single-molecule RNA FISH (smFISH) spot sizes are compared to clampFISH 2.0 spots imaged on the same day (experiment 2; see method section for description of positive control for ‘one-pot’ experiment) and to clampFISH 2.0 spots in a previous experiment (experiment 1, also depicted in Extended Data Fig. 5; see method section description of ’amplification characterization’ experiment). We imaged the samples with a 100X/1.45NA objective at 1×1 camera binning (65 nm pixel size) and fit the pixel values in the neighborhood of each spot to a 2D Gaussian distribution. Left: standard deviation of Gaussian-fitted spots for UBC smFISH labeled in Atto 488 and UBC clampFISH 2.0 amplified to round 1 or round 4 with readout probes labeled in Atto 488. Right: standard deviation of Gaussian-fitted spots for TOP2A smFISH labeled in Atto 647N and MITF clampFISH 2.0 amplified to round 1 or round 4 with readout probes labeled in Atto 647N. Values shown are the median standard deviations. For the left and right panels, circles and numbers shown are median values and bounds of boxes are 25th and 75th percentiles. For Atto 488 data (left panel), n = 1053, 923, and 1968 (from left to right) and for Atto 647N data (right panel) n = 1875, 1230, 2254, 1206, and 994 (from left to right).
Extended Data Fig. 7∣. clampFISH 2.0 quantifies RNA spot counts at 10X magnification.
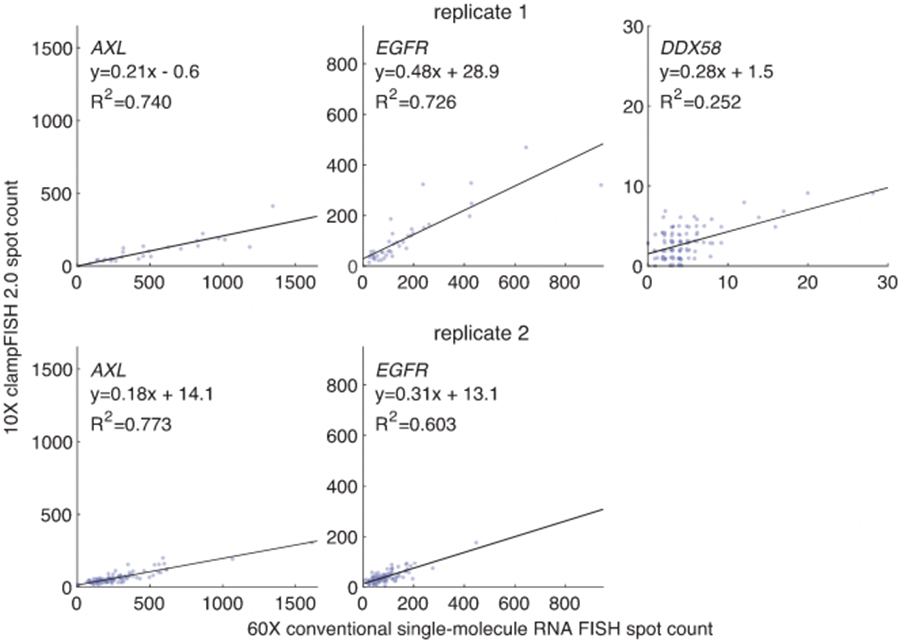
Depicting the same data as in Fig. 2b, but with clampFISH 2.0 spots imaged at 10X magnification. We performed clampFISH 2.0 for 10 genes, amplified the 10 scaffolds in parallel to round 8, then added a single pair of readout probes to label a scaffold corresponding to AXL (left; in drug-resistant WM989 A6-G3 RC4 cells). EGFR (middle; in drug-resistant WM989 A6-G3 RC4 cells), or DDX58(right; in drug-naïve VVM989 A6-G3 cells). In two biological replicates (top: replicate 1; bottom: replicate 2), we counted spots for clampFISH 2.0 at 10X magnification (y-axis) and conventional single-molecule RNA FISH at 60X magnification (x-axis), which targeted non-overlapping regions of the same RNAs. In replicate 2, imaging at 10X of DDX58 spots before conventional single-molecule RNA FISH was not performed.
Extended Data Fig. 8∣. Signal from the previous readout cycle is removed after a high-formamide strip.
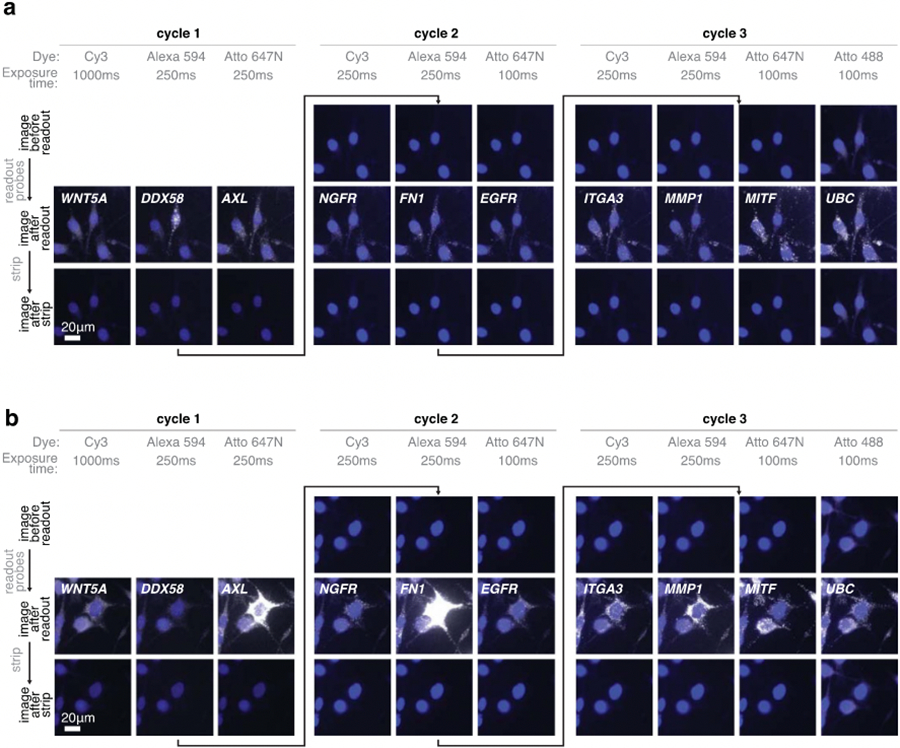
(a) Example images of clampFISH 2.0 spots at 20X magnification before the readout probe hybridization (top row), after adding readout probes (middle row), and after stripping off readout probes (bottom row). The first three columns are from readout cycle 1, the next three are from readout cycle 2, and the last 4 columns are from readout cycle 3. Each column’s images are from the same channel (with the corresponding readout probe dye indicated), exposure time (as indicated in milliseconds), and are contrasted identically. (b) Example images as in (a) at a different position on the plate. The experiment was performed twice with similar results.
Extended Data Fig. 9∣. clampFISH 2.0 scaffolds remain stably bound after multiple rounds of readout stripping and storage at 4 °C for 4 months.
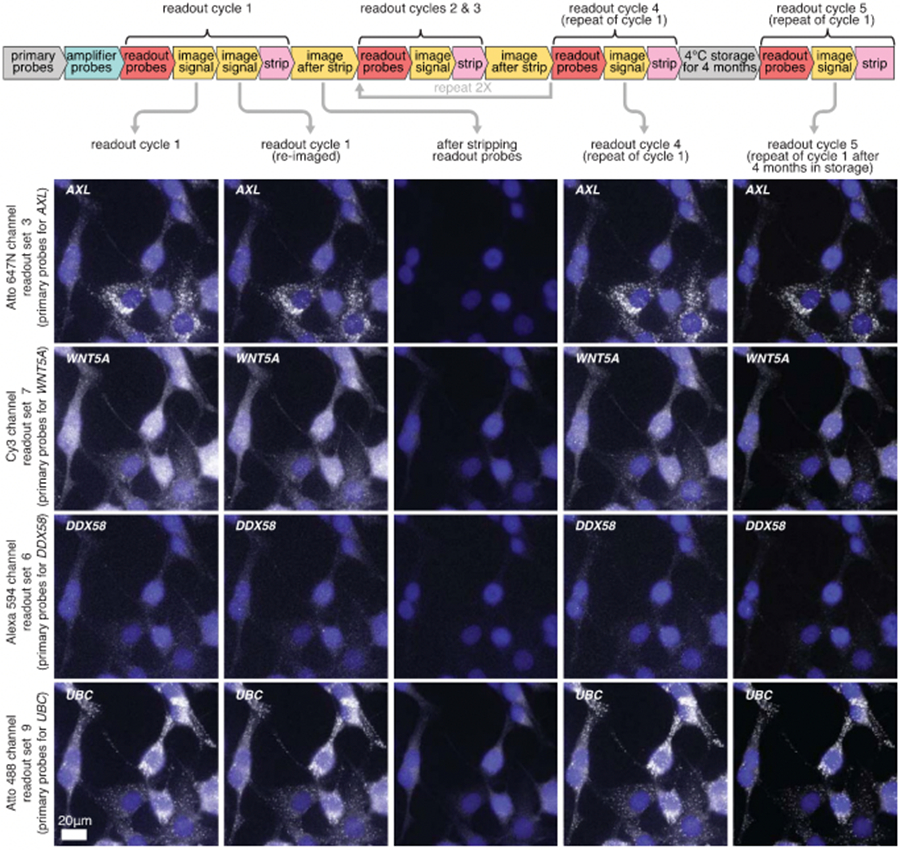
Images of clampFISH 2.0 spots from a 20X objective over readout cycles where we repeatedly use 4 sets of readout probes which label (from top to bottom) AXL, WNTSA, DDX58, and UBC clampFISH 2.0 scaffolds. Column 1: readout cycle 1. Column 2: readout cycle 1, re-imaged after removing the sample from the microscope stage and stored overnight at 4 °C. Column 3: after stripping off readout probes from readout cycle 1. Column 4: readout cycle 4, where we repeat readout cycle 1 after readout cycles 2 and 3 (where different sets of genes were labeled). Column 5: readout cycle 5, performed after storing the sample at 4 °C in 2X SSC for 4 months. DAPI overlay is contrasted separately for each column. Each row of readout cycle S (column 5) is contrasted with 180% the intensity range of the first four columns. The cycle S signal presumably appeared brighter due to changes in the microscope’s optics during that time frame (for example, greater sample illumination or increased transmission to the sensor). The experiment was performed twice with similar results, except for column 5 data (after 4 months storage) which was performed once. See Supplementary Figs. 14 and 15 for quantification of each experimental replicate.
Extended Data Fig. 10 ∣. clampFISH 2.0 detects RNAs In presumptive human cells in tissue.
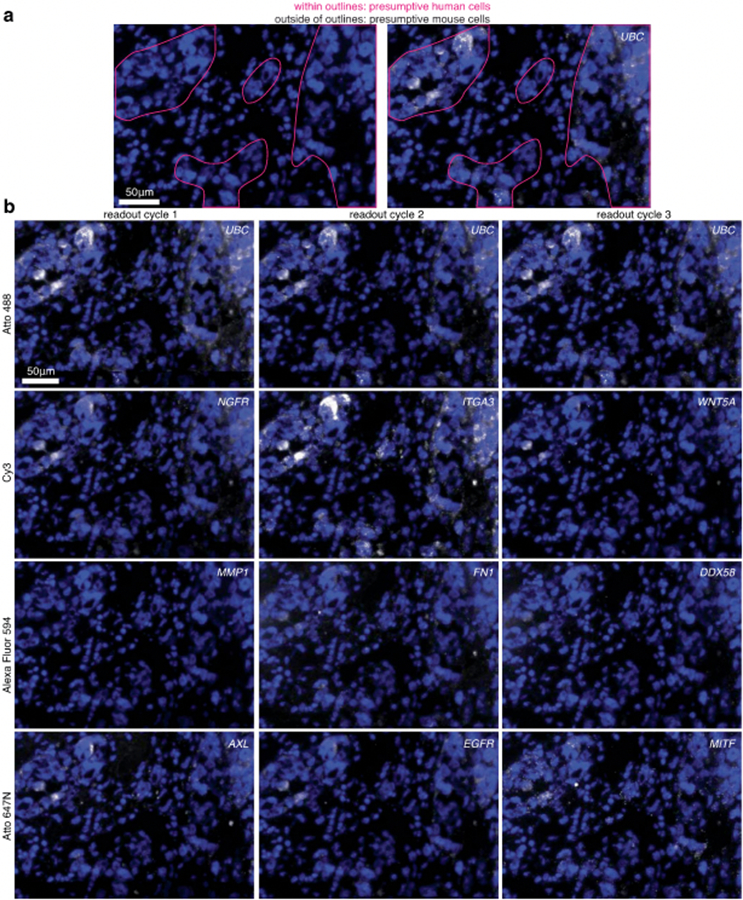
clampFISH 2.0 was performed in a 6μm fresh frozen tissue section of a dissected tumor, derived from human WM989-A6-G3-Cas9-5a3 cells injected into a mouse and fed chow containing the BRAFV600E inhibitor PLX4720. Shown are stitched maximum intensity projections of 20X Image stacks with 5 z-planes at 1.2μm z-step Increments. (a) Pink outlines around regions containing mostly presumptive human cells, demarcated based on nuclear morphology, showing DAPI staining alone (left) and DAPI with UBC clampFISH 2.0 signal overlaid (right), where images are from readout cycle 2. (b) clampFISH 2.0 scaffolds for 10 genes were probed across readout cycles 1 (left), 2 (middle), and 3 (right), where the UBC scaffold was probed each round as a positive control. The dyes on each readout probe set were (top to bottom): Atto488, Cy3, Alexa Fluor 594, and Atto647N. The experiment was performed twice with similar results.
Supplementary Material
Acknowledgements
The authors thank A. Coté and I.A. Mellis for helpful input on image analysis; the ENCODE Consortium and the laboratory of J.M. Cherry for RNA-seq datasets used for probe design; the Penn Center for Musculoskeletal Disorders Histology Core (P30 AR069619) for their guidance on tissue cryo-sectioning; and the Wistar Institute’s Histotechnology facility (supported by Cancer Center Support Grant P30 CA010815) for processing the FFPE tissue samples. The authors acknowledge support from the National Institutes of Health (NIH) grants F30 CA236129, T32 GM007170 and T32 HG000046 (to B.L.E.); a Career Award at the Scientific Interface from BWF and the Schmidt Science Fellowship in partnership with the Rhodes Trust (to Y.G.); training grants NIH F30 HG010822, NIH T32 DK007780 and NIH T32 GM007170 (to C.L.J.); NIH K00-CA-212437-02 (to A.K.); the Chan Zuckerberg Initiative (to S.H.R.); R01CA174746 and R01CA207935 (to M.E.F. and A.T.W); a Team Science Award from the Melanoma Research Alliance and P01 CA114046 (to A.T.W.); NIH grants RO1 CA238237, U54 CA224070, PO1 CA114046, P50CA174523 and the Dr Miriam and Sheldon G. Adelson Medical Research Foundation (to M.H.); 5-U2C-CA-233285-04, NIH 4DN U01 HL129998 and NIH 4DN U01DK127405 (to I.D. and A.R.); and the NIH Center for Photogenomics RM1 HG007743, NIH Director’s Transformative Research Award R01 GM137425, and the Penn Epigenetics Institute (to A.R.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592-022-01653-6.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
I.D., B.L.E. and A.R. have filed a patent application related to this work. A.R. receives patent royalties from LGC/Biosearch Technologies related to Stellaris mRNA FISH products. A.R. is on the scientific advisory board of Spatial Genomics. A.T.W. is on the Board of Directors at ReGAIN therapeutics. All other authors have no competing interests.
Extended data are available for this paper at https://doi.org/10.1038/s41592-022-01653-6.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41592-022-01653-6.
Data availability
All imaging and RNA sequencing data are publicly available on Dropbox https://www.dropbox.com/sh/q51kmcphoyi9yi3/AAB4g1a6ODDHaphsvbmBJAy-a?dl=0, with a description of how data were associated with each figure in the README.docx and ExperimentList.xlsx files. RNA sequencing data are also deposited on Gene Expression Omnibus (GEO accession GSE211491). Source data are provided with this paper.
Code availability
All MATLAB data analysis code, including repositories for rajlabimagetools, dentist2, pixyDuck, bfmatlab, Cellpose, and scripts for raw data processing, image data extraction and plotting, is publicly available on Dropbox (https://www.dropbox.com/sh/q51kmcphoyi9yi3/AAB4g1a6ODDHaphsvbmBJAy-a?dl=0), with instructions in the README.docx file and also on GitHub (https://github.com/iandarr/clampFISH2allcode). Code was run with MATLAB R2021a (64-bit, maci64), available from www.mathworks.com.
References
- 1.Singer RH & Ward DC Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc. Natl Acad. Sci. USA 79, 7331–7335 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Femino AM, Fay FS, Fogarty K & Singer RH Visualization of single RNA transcripts in situ. Science 280, 585–590 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Sun Y-C, Church GM, Lee JH & Zador AM Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 46, e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S et al. Barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses. Nucleic Acids Res. 49, e58 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng R, Zhang K, Sun Y, Ren X & Li J Highly specific imaging of mRNA in single cells by target RNA-initiated rolling circle amplification. Chem. Sci 8, 3668–3675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider N & Meier M Efficient in situ detection of mRNAs using the Chlorella virus DNA ligase for padlock probe ligation. RNA 23, 250–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lein E, Borm LE & Linnarsson S The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Dirks RM & Pierce NA Triggered amplification by hybridization chain reaction. Proc. Natl Acad. Sci. USA 101, 15275–15278 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi HMT, Beck VA & Pierce NA Next-generation in situ hybridization chain reaction: higher gain, lower cost, greater durability. ACS Nano 8, 4284–4294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi HMT et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn 14, 22–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinnamon JR & Czaplinski K RNA detection in situ with FISH-STICs. RNA 20, 260–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kishi JY et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat. Methods 16, 533–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouhanifard SH et al. ClampFISH detects individual nucleic acid molecules using click chemistry-based amplification. Nat. Biotechnol 10.1038/nbt.4286 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah S, Lubeck E, Zhou W & Cai L In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffitt JR et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl Acad. Sci. USA 113, 11046–11051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia C, Babcock HP, Moffitt JR & Zhuang X Multiplexed detection of RNA using MERFISH and branched DNA amplification. Sci. Rep 9, 7721 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagunavicius A et al. Novel application of Phi29 DNA polymerase: RNA detection and analysis in vitro and in situ by target RNA-primed RCA. RNA 15, 765–771 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, Wolf LK & Georgiadis RM Secondary structure effects on DNA hybridization kinetics: a solution versus surface comparison. Nucleic Acids Res. 34, 3370–3377 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Revyakin A, Grimm JB, Lavis LD & Tjian R Single-molecule tracking of the transcription cycle by sub-second RNA detection. Elife 3, e01775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaffer SM et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emert BL et al. Variability within rare cell states enables multiple paths toward drug resistance. Nat. Biotechnol 39, 865–876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goyal Y et al. Pre-determined diversity in resistant fates emerges from homogenous cells after anti-cancer drug treatment. Preprint at 10.1101/2021.12.08.471833 (2021). [DOI] [Google Scholar]
- 25.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuh L et al. Gene networks with transcriptional bursting recapitulate rare transient coordinated high expression states in cancer. Cell Syst. 10, 363–378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Torre EA et al. Genetic screening for single-cell variability modulators driving therapy resistance. Nat. Genet 53, 76–85 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah S et al. Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development 143, 2862–2867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moffitt JR et al. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl Acad. Sci. USA 113, 14456–14461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc 10, 442–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rust MJ, Bates M & Zhuang X Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auer A, Strauss MT, Schlichthaerle T & Jungmann R Fast, background-free DNA-PAINT imaging using FRET-based probes. Nano Lett. 17, 6428–6434 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Lee J, Park S & Hohng S Accelerated FRET-PAINT microscopy. Mol. Brain 11, 70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giannone G et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J 99, 1303–1310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharonov A & Hochstrasser RM Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl Acad. Sci. USA 103, 18911–18916 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoen I, Ries J, Klotzsch E, Ewers H & Vogel V Binding-activated localization microscopy of DNA structures. Nano Lett. 11, 4008–4011 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Hell SW Toward fluorescence nanoscopy. Nat. Biotechnol 21, 1347–1355 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Camacho C et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dirks RM & Pierce NA An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J. Comput. Chem 25, 1295–1304 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Dirks RM, Bois JS, Schaeffer JM, Winfree E & Pierce NA Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev. 49, 65–88 (2007). [Google Scholar]
- 43.Dirks RM & Pierce NA A partition function algorithm for nucleic acid secondary structure including pseudoknots. J. Comput. Chem 24, 1664–1677 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Fornace ME, Porubsky NJ & Pierce NA A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synth. Biol 9, 2665–2678 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Davis CA et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markham NR & Zuker M DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 33, W577–W581 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto N, Nakano S, Yoneyama M & Honda K Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 24, 4501–4505 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All imaging and RNA sequencing data are publicly available on Dropbox https://www.dropbox.com/sh/q51kmcphoyi9yi3/AAB4g1a6ODDHaphsvbmBJAy-a?dl=0, with a description of how data were associated with each figure in the README.docx and ExperimentList.xlsx files. RNA sequencing data are also deposited on Gene Expression Omnibus (GEO accession GSE211491). Source data are provided with this paper.
All MATLAB data analysis code, including repositories for rajlabimagetools, dentist2, pixyDuck, bfmatlab, Cellpose, and scripts for raw data processing, image data extraction and plotting, is publicly available on Dropbox (https://www.dropbox.com/sh/q51kmcphoyi9yi3/AAB4g1a6ODDHaphsvbmBJAy-a?dl=0), with instructions in the README.docx file and also on GitHub (https://github.com/iandarr/clampFISH2allcode). Code was run with MATLAB R2021a (64-bit, maci64), available from www.mathworks.com.