

Abstract
Lipid peroxidation and iron accumulation are closely associated with neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, or neurodegeneration with brain iron accumulation disorders. Mitochondrial dysfunction, lipofuscin accumulation, autophagy disruption, and ferroptosis have been implicated as the critical pathomechanisms of lipid peroxidation and iron accumulation in these disorders. Currently, the connection between lipid peroxidation and iron accumulation and the initial cause or consequence in neurodegeneration processes is unclear. In this review, we have compiled the known mechanisms by which lipid peroxidation triggers iron accumulation and lipofuscin formation, and the effect of iron overload on lipid peroxidation and cellular function. The vicious cycle established between both pathological alterations may lead to the development of neurodegeneration. Therefore, the investigation of these mechanisms is essential for exploring therapeutic strategies to restrict neurodegeneration. In addition, we discuss the interplay between lipid peroxidation and iron accumulation in neurodegeneration, particularly in PLA2G6-associated neurodegeneration, a rare neurodegenerative disease with autosomal recessive inheritance, which belongs to the group of neurodegeneration with brain iron accumulation disorders.
Keywords: 4-hidroxynonenal, ferroptosis, iron, lipid peroxidation, lipofuscin, neurodegeneration, neurodegeneration with brain iron accumulation, oxidative stress, PLA2G6-associated neurodegeneration
Introduction
Neurodegenerative diseases are caused by complex neuronal cell death mechanisms involving iron accumulation and lipid peroxidation in different areas of the brain (Guiney et al., 2017). Neuronal cell death frequently occurs by ferroptosis, a cell death process dependent on iron-mediated lipid peroxidation. Moreover, abnormal iron homeostasis is associated with iron overload, which destroys proteins and lipids through Fenton reactions (Ke and Qian, 2007; Wu et al., 2019). Thus, iron is essential in the initiation and progression of neurodegenerative diseases (Ward et al., 2014). Excessive iron accumulation and lipid peroxidation in these diseases are frequently accompanied by oxidative stress, mitochondrial dysfunction, increased iron content in lipofuscin, and autophagy dysregulation (Ke and Qian, 2007; Álvarez-Córdoba et al., 2019; Corti et al., 2020).
We have attempted to answer the following questions in this systematic review, aiming to clarify the role of lipid peroxidation in iron accumulation and neurodegenerative processes: How does lipid peroxidation induce iron accumulation? How does lipid peroxidation affect autophagy and the renewal of cell organelles? How does lipid peroxidation affect mitochondria? Are both processes interconnected? How is the vicious cycle between lipid peroxidation and iron accumulation established? How is it linked to cell death by ferroptosis? Is iron accumulated in the form of lipofuscin?
Peroxidation of membrane lipids affects a variety of functions, resulting in increased membrane rigidity and consequently, affects the activities of cellular functions, including transport along cell membranes, mitochondrial bioenergetics, vesicular traffic, and autophagy/mitophagy machineries. In addition to phospholipids damage, increased free radical species can directly react with membrane proteins and induce lipid-protein and protein-protein crosslinking, which cause altered membrane integrity and function of the associated proteins (Farooqui and Horrocks, 1998). Hence, it can be hypothesized that alterations of all the aforementioned functions are interconnected and cause neuronal cell death in neurodegeneration.
Neurodegeneration with brain iron accumulation (NBIA) is a group of rare inherited neurological movement disorders characterized by an abnormal iron accumulation in the brain, and progressive degeneration of the nervous system (Hayflick et al., 2018). These disorders present with genetic and clinical heterogeneity, which are challenging to diagnose. The suspicion of NBIA is generally based on brain magnetic resonance imaging that reveals increased iron accumulation in the basal ganglia and the presence of clinical features, including movement disorders, neuropsychiatric symptoms, seizures, visual disturbances, and cognitive deficits (Gregory et al., 2009). Furthermore, the diagnosis requires sequential testing to determine the specific disorder. The most common NBIA disorders are Pantothenate kinase-associated neurodegeneration (PKAN), beta-propeller protein-associated neurodegeneration (BPAN), and Phospholipase A2-associated neurodegeneration (PLAN) due to mutations in PANK2, WDR45 and PLA2G6, respectively (Hayflick et al., 2018). Genetics, pathophysiology, and clinical features of NBIA disorders have recently been reported (Spaull et al., 2021; Svetel et al., 2021).
PLAN has been reported as a reliable model for investigating the pathogenesis of neurodegeneration (Beck et al., 2011, 2016; Kinghorn et al., 2015; Kinghorn and Castillo-Quan, 2016). A Pla2g6 knock-out mouse model exhibited several features of the human disease and demonstrated cerebellar atrophy with loss of Purkinje cells, pronounced neuroinflammation, and accumulation of spheroids, though not iron accumulation (Malik et al., 2008), and characteristic ultrastructural alterations of mitochondria. In subsequent studies, an age-dependent iron accumulation in the regions of the brain was reported in Pla2g6–/– mice, which revealed enhanced lipid peroxidation and mitochondrial dysfunction (Beck et al., 2011). These alterations preceded neuronal death, suggesting that iron-dependent reactive oxygen species (ROS) formation may be a primary cause of neurodegeneration.
In flies, a P-element insertion in iPLA2-VIA was demonstrated to impair gene expression, and homozygous animals were viable and exhibited locomotor defects. In addition, mitochondrial abnormalities and degeneration of mitochondrial cristae were documented (Kinghorn et al., 2015). These phenotypes recapitulated some of the defects observed in mice and humans.
Similarly, PKAN and PLAN patient-derived fibroblasts and induced neurons generated by direct reprogramming have been proposed as noteworthy biological models for studying the implications of lipid peroxidation and iron accumulation in neurodegeneration (Álvarez-Córdoba et al., 2019; Villalón-García et al., 2022). In PLAN, both fibroblasts and induced neurons exhibited the key pathological hallmarks of the disease, and can be used as models to describe the interaction between lipid peroxidation and iron/lipofuscin accumulation (Villalón-García et al., 2022). Lipid peroxidation disrupts normal mitochondrial function, autophagy, and other vesicle-based processes promoting lipofuscinogenesis and iron accumulation. In turn, lipofuscin and iron accumulation may increase oxidative stress and lipid peroxidation, propagating and aggravating neuronal damage.
Search Strategy
Studies on lipid peroxidation, iron, and neurodegeneration published from 1988 to 2022 were identified from the PubMed database. The search method was based on the following terms: “lipid peroxidation”; “lipid peroxidation” AND “neurodegeneration”; “iron” AND “lipid peroxidation”; “4-hydroxy-2E-nonenal”; “4-HNE” AND “PPAR”; “lipid peroxidation” and “iron”; “lipid peroxidation” AND “Alzheimer’s disease” OR “Parkinson’s disease”; “ferroptosis”; “lipid peroxidation” AND “PLA2G6-Associated Neurodegeneration”; “NBIA”; “BPAN” AND “ferroptosis”; “lipid peroxidation” AND “lipofuscin”. The results were screened by titles and abstracts; finally, 85 articles were included, the majority of which were published in the last 5 years.
Lipid Peroxidation
Lipids are vital in biological processes and are the main structural units of subcellular organelles and cellular membranes, creating a selective barrier that protects the cell’s interior from the outside environment. In addition, they have important cellular functions, including cell signaling, molecular transport, proliferation, energy storage, secretion, and survival. Lipids’ fatty acid composition, specifically the degree of unsaturation, chain length, hydroxylation, and double bond position of fatty acid in phospholipids, determines the biophysical properties of membranes. Thus, mono-unsaturated and polyunsaturated fatty acids provide increased membrane fluidity, while short-chain saturated fatty acids contribute to rigid membranes (Ayala et al., 2014; Hu et al., 2017; Carvalho and Caramujo, 2018). Furthermore, lipids govern the folding, organization, and final structure of all membrane proteins. Lipids directly modulate the function of the membrane and numerous other proteins that reversibly interact with the membrane surface (Bogdanov et al., 2008).
Moreover, membrane lipid composition is crucial for mitochondrial function and energy production because of its influence on the biogenesis, activity, and stability of respiratory chain supercomplexes and it creates a specific environment to promote the organization and function of mitochondrial proteins (Mårtensson et al., 2017).
Owing to the biological relevance of lipids, alterations in cellular lipid composition are involved in the pathomechanisms of different diseases including cancer, diabetes, autoimmune diseases, and neurodegenerative disorders (Codreanu and Liebler, 2015; Hu et al., 2017; Ramana et al., 2019). Hydroxy radical (OH·), superoxide anion (O2–), nitric oxide radical (NO·), and hydrogen peroxide (H2O2) among others, comprise a group of oxidant molecules termed ROS (Yadav and Ramana, 2013; Latunde-Dada, 2017; Su et al., 2019). ROS produced in normal cells through physiological processes, such as the mitochondrial electron-transport chain or the activity of NADPH oxidases and Xanthine oxidase, participate in cell signaling (Latunde-Dada, 2017; Su et al., 2019); even lipid peroxides, under homeostatic conditions, act as mediators of inflammation signaling (Gaschler and Stockwell, 2017). In homeostatic conditions, ROS are neutralized by endogenous antioxidant systems (enzymatic and nonenzymatic) (Latunde-Dada, 2017; Su et al., 2019). Imbalance between free radicals and antioxidants results in disruption of redox homeostasis and excessive oxidative stress and causes oxidative damage in cell structures such as DNA, proteins, and lipids (Yadav and Ramana, 2013; Ayala et al., 2014; Gaschler and Stockwell, 2017; Su et al., 2019). Lipid peroxidation is caused by the attack of oxidants on lipids (Ayala et al., 2014; Schaur et al., 2015; Gaschler and Stockwell, 2017; Su et al., 2019); it compromises membrane integrity, causes loss of cell function, and may result in severe cytotoxicity (Yadav and Ramana, 2013). Mammalian cell membrane lipids (glycerophospholipids and phosphatidylinositol) carry saturated, mono-unsaturated (esterified in sn-1 position), and polyunsaturated fatty acids (esterified in sn-2 position), which are most sensitive to ROS and hence, are primary targets of peroxidation. Thus, primary and secondary lipid peroxidation by-products with high reactivity are formed (Ayala et al., 2014; Davies and Guo, 2014; Codreanu and Liebler, 2015; Gaschler and Stockwell, 2017; Hu et al., 2017; Su et al., 2019).
Lipid peroxidation mechanisms
Lipid peroxidation can be produced by two mechanisms: enzymatic and nonenzymatic processes. In the enzymatic peroxidation, lipid peroxidation is catalyzed by lipoxygenases whose main substrates are arachidonic and linoleic acid, and generate a phospholipid with sn-2 position hydroxylated fatty acids (Ayala et al., 2014; Davies and Guo, 2014; Codreanu and Liebler, 2015; Gaschler and Stockwell, 2017; Su et al., 2019). Other enzymatic peroxidation processes include the oxygenation of free fatty acids by lipoxygenase or cytochrome P450 enzymes, which add an oxygen molecule to arachidonic acid at specific double bonds, or controlled peroxide synthesis by cyclooxygenases, which produce lipid endoperoxides from linoleic acid (Ayala et al., 2014; Davies and Guo, 2014; Gaschler and Stockwell, 2017).
Conversely, nonenzymatic peroxidation is carried out by ROS (Yadav and Ramana, 2013; Davies and Guo, 2014; Su et al., 2019), mainly hydroxyl (HO·) and hydroperoxyl (HO·2) radicals that can strongly initiate polyunsaturated fatty acids chain oxidation (Ayala et al., 2014; Bilska-Wilkosz et al., 2022). The other ROS initiators of lipid peroxidation, include ozone (O3), nitrogen oxide (NO), nitrogen dioxide (NO2), and sulphur dioxide (Bilska-Wilkosz et al., 2022). Nonenzymatic peroxidation can be divided into three phases: initiation, propagation, and termination (Table 1) (Yadav and Ramana, 2013; Gaschler and Stockwell, 2017; Maiorino et al., 2018; Su et al., 2019; Bilska-Wilkosz et al., 2022). The initiation phase consists of the generation of fatty acid radicals that can propagate the peroxidation to other molecules (Gaschler and Stockwell, 2017). First, a free radical subtracts a hydrogen atom situated between two double bonds from a fatty acid part of a phospholipid; this reaction produces a lipid radical with an unpaired electron (L·). In the propagation phase, the lipid radical reacts with O2 or with a transition-metal forming a peroxyl radical (LOO·), which in turn maintains the chain reaction, reacting with a new unsaturated fatty acid and producing a new L· and lipid hydroperoxide (LOOH). These primary products of lipid peroxidation can decompose into secondary products. In the termination phase, the interaction of a radical with a molecule with antioxidant capacity, such as α-tocopherol (vitamin E), that terminates peroxidation by inhibiting the propagation of chain reactions. The reaction between a lipid radical with a lipid peroxide or by the combination of two lipid peroxide molecules is another method of termination; both processes result in relatively stable nonreactive molecules (Yadav and Ramana, 2013; Ayala et al., 2014; Schaur et al., 2015; Su et al., 2019; Bilska-Wilkosz et al., 2022).
Table 1.
Nonenzymatic lipid peroxidation steps
| Step | Reactions |
|---|---|
| Initiation | LH + HO· → L· + H2O |
| Propagation | L· + O2 → LOO· |
| LOO· + LH → LOOH + L· | |
| LOOH → LO·, LOO·, HO· | |
| LOOH + Mn → LO· + HO· + Mn+1 | |
| LOOH + Mn+1 → LOO· + H· + Mn | |
| Termination | L· + L· → L+L |
| L· + LOO· → LOOL | |
| LOO· + LOO· → LOOL + O2 | |
| LOO· + α-tocopherol → LOOH |
L+L: Fatty acid dimer; L·: lipid radical; LH: lipid containing carbon-carbon double bound; LO·: alkoxyl radical; LOO·: lipid peroxy radical; LOOH: lipid hydroperoxide; LOOL: peroxide bridge dimer; M: transition metal; OH·: hydroxyl radical.
Glutathione peroxidase (GPX) enzymes catalyze the reduction of lipid peroxides using glutathione (GSH) as a co-substrate and prevent lipid peroxidation. GPX, particularly GPX4, participates in the 2-electro reduction of peroxides or intermediate radicals to less cytotoxic compounds (Ayala et al., 2014; Gaschler and Stockwell, 2017).
Lipid peroxidation products
Various oxidation products with high biological activity are produced by lipid peroxidation (Figure 1; Ayala et al., 2014; Su et al., 2019). Lipid peroxidation alters membrane permeability and fluidity, and ion gradients (Gaschler and Stockwell, 2017). In turn, lipid peroxidation by-products can interact with proteins or lipids creating protein-lipid adducts responsible for protein aggregation, enzyme inactivation, and cytotoxicity (Ayala et al., 2014; Gaschler and Stockwell, 2017; Bilska-Wilkosz et al., 2022).
Figure 1.
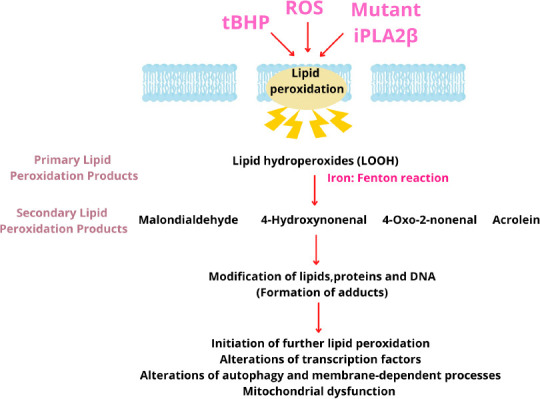
Mechanism of membrane lipid peroxidation of polyunsaturated fatty acids leading to the generation of various reactive aldehydes.
The reactive aldehydes trigger several redox signaling pathways and cellular alterations. iPLA2β: Calcium-independent phospholipase A2 group VI; ROS: reactive oxygen species; tBHP: tert-butylhydroperoxide.
Primary lipid peroxidation products
Hydroperoxides produced during the propagation phase are the primary lipid peroxidation products. LOOH is more stable at moderate conditions of low temperatures and absence of ions, when compared with free radicals. LOOH can be converted to LOO· and LO· through interaction with transition-metal ions or with free radicals causing widespread oxidative damage (Ayala et al., 2014; Davies and Guo, 2014; Bilska-Wilkosz et al., 2022).
Secondary lipid peroxidation products
Secondary products are generated as a result of degradation of primary lipid peroxides through enzymatic and nonenzymatic pathways and by β-fragmentation or β-scission processes (Davies and Guo, 2014; Codreanu and Liebler, 2015; Hu et al., 2017; Bilska-Wilkosz et al., 2022). The major degradation products are 4-hydroxynonenal (4-HNE) and malondialdehyde (Gaschler and Stockwell, 2017); the others include reactive electrophiles such as aldehydes and alkenals, including α, β-unsaturated aldehydes (acrolein), oxoalkenals, epoxyalkenals, and γ-ketoaldehydes.
Malondialdehyde, a highly reactive and toxic dialdehyde, forms adducts with primary amines on proteins or DNA, and has been described as the most mutagenic lipid peroxidation product and a reliable marker of oxidative stress (Ayala et al., 2014; Gaschler and Stockwell, 2017).
4-HNE, the major by-product of lipid peroxidation, is an α, β-unsaturated electrophilic compound. Under homeostatic conditions it is a protective signaling molecule; however, at high concentrations, it acts as a cytotoxic molecule inhibiting gene expression (Yadav and Ramana, 2013; Ayala et al., 2014; Zhong and Yin, 2015). The other molecules from the hydroxyalkenals family are 4-hydroxy-2E-hexenal, generated by nonenzymatic n-3 PUFA peroxidation, and 4-hydroxy-2E,6Z-dodecadienal, produced from the action of 12-lipoxygenase (Hu et al., 2017).
Reactive electrophiles act as secondary messengers of lipid peroxidation by reacting with thiol groups and cysteine, histidine, and lysine of nucleophile amino acids forming covalent adducts; these modifications result in advanced lipoxidation end-products (Codreanu and Liebler, 2015; Schaur et al., 2015; Gaschler and Stockwell, 2017; Hu et al., 2017; Bilska-Wilkosz et al., 2022). Protein adducts, generated by Michael and Schiff base adducts (Codreanu and Liebler, 2015; Schaur et al., 2015; Gallo et al., 2020; Bilska-Wilkosz et al., 2022), are involved in cellular responses related to the resistance or susceptibility to diseases such as diabetes, atherosclerosis, and cancer and neurodegenerative disorders (Ayala et al., 2014; Codreanu and Liebler, 2015; Zhong and Yin, 2015). Accumulation of hydroxyalkenal adducts is associated with liver inflammation, cardiovascular and autoimmune diseases, renal failure, aging, and neurodegenerative disorders (Hu et al., 2017). In particular, protein modification by 4-HNE in neurodegeneration has been reported. Hallmarks of neurodegeneration, including energy metabolism disruption, reduced antioxidant defenses, and mitochondrial dysfunction may be caused by 4-HNE (Ayala et al., 2014), which may be accumulated in membrane rich organelles such as mitochondria and endoplasmic reticulum (Codreanu and Liebler, 2015). It has been observed that 4-HNE induces disruption of the mitochondrial respiratory chain in different tissues, proteasomal dysfunction, and endoplasmic reticulum stress (Gallo et al., 2020) and modifies protein and lipid membrane conformation, interfering with membrane fluidity (Schaur et al., 2015).
Different detoxification mechanisms to transform 4-HNE to a less reactive compound include enzymatic process carried out by glutathione S-transferase enzymes, aldehyde dehydrogenase, and aldose reductase (Schaur et al., 2015; Zhong and Yin, 2015; Gallo et al., 2020).
Lipid peroxidation by-products may act as signaling molecules depending on their concentrations. They regulate several physiological functions or may act as toxic secondary messengers inducing cell/tissue injury by regulating several transcription factors (Yadav and Ramana, 2013; Su et al., 2019). 4-HNE, at moderate levels, regulates several transcription factors with protective functions, such as Nrf2 that binds with the antioxidant-response element (Ayala et al., 2014; Dodson et al., 2019) or upregulates AP-1 transcription factor that increases GSH content (Ayala et al., 2014). It has been reported that the Nrf2-antioxidant-response element pathway is crucial in different pathological processes, including neurodegenerative diseases (Ayala et al., 2014; Dodson et al., 2019). Moreover, 4-HNE can up- or downregulate nuclear factor-kappa B, a transcription factor that regulates genes involved in stress responses, inflammation, and apoptosis, depending on the cell type and concentration (Yadav and Ramana, 2013; Ayala et al., 2014; Su et al., 2019; Gallo et al., 2020). The other superfamilies of nuclear receptors modulated by 4-HNE are perosyxomel proliferator activated receptors (PPARs) (PPARα, β/δ, and γ), which regulate antioxidant defense, lipid metabolism, and mitochondrial biogenesis (Ayala et al., 2014; Sasson, 2017; Gallo et al., 2020). The action of 4-HNE on protein kinases such as MAPKs, protein kinases C, and Akt is being studied (Yadav and Ramana, 2013; Ayala et al., 2014; Su et al., 2019).
Lipid peroxidation in mitochondria
The impairment of mitochondrial function, including dysregulation of lipid metabolism and increased ROS production, may be a mechanism of cellular bioenergetic alterations. The majority of mitochondrial lipids are synthesized in the endoplasmic reticulum and transported to the mitochondria; however, some lipids (cardiolipin, phosphatidylethanolamine) are synthesized within the inner membrane of the mitochondria and are critical for maintaining the unique architecture of the mitochondrial membranes, the cristae (Schlame and Greenberg, 2017).
An increase in ROS production may induce mitochondrial dysfunction, partly via lipid peroxidation and oxidation of respiratory chain proteins (Ademowo et al., 2017). Furthermore, mitochondrial dysfunction and excessive ROS production are common features of neurodegeneration (Paradies et al., 2014).
Role of Iron in Lipid Peroxidation
Iron is an essential element for human life that is required for the synthesis of oxygen transport proteins, in particular hemoglobin and myoglobin, and for the formation of heme enzymes and other iron-containing enzymes involved in electron transfer and oxidation-reduction reactions (Rockfield et al., 2018). However, free iron is a highly reactive element; therefore, it is stored in a less reactive form in different cell compartments, such as ferritin in the cytoplasm or inside the mitochondrial matrix (Bertrand, 2017; Rockfield et al., 2018). Despite this, intracellular free iron or labile iron pool, is capable of producing ROS by the Fenton reaction, i.e., the interaction between Fe++ with H2O2 and the subsequent formation of Fe+++ and hydroxide anion. Hence, iron content must be strictly regulated (Latunde-Dada, 2017; Feng and Stockwell, 2018; Maiorino et al., 2018; Rockfield et al., 2018).
In addition, nonenzymatic lipid peroxidation can be mediated by transition-metals such as Fe++, Fe+++, and less frequently copper, nickel, cobalt, and vanadium. Free iron may initiate and propagate peroxidation through two mechanisms: by direct interactions with lipid hydroperoxide (LOOH) or by a reaction with hydrogen peroxide (H2O2). Fe++ can react with LOOH producing Fe+++ and LO· via the Fenton reaction. In addition, the interaction of Fe+++ with LOOH generates LOO· via Haber-Weiss reaction (Table 1) (Yadav and Ramana, 2013; Ayala et al., 2014; Gaschler and Stockwell, 2017; Maiorino et al., 2018; Rockfield et al., 2018; Su et al., 2019; Bilska-Wilkosz et al., 2022). Moreover, iron is implicated in enzymatic peroxidation because lipoxygenases are iron dependent enzymes (Rockfield et al., 2018; Zhao et al., 2021). In addition, free iron participates in the decomposition of secondary lipid peroxidation by-products by generating electrophilic compounds (Zhao et al., 2021).
Role of Lipid Peroxidation in Neurodegeneration
The etiopathogenesis of neurodegeneration is diverse, and risk factors include aging, genetics, nutrition, pollution, and alcohol consumption (Peña-Bautista et al., 2019). Some pathogenic signs of neurodegeneration are inflammation, mitochondrial dysfunction, oxidative stress, iron accumulation, and lipid peroxidation (Urrutia et al., 2014). Lipid peroxidation is a well-studied molecular pathway involved in the pathogenesis of several neurodegenerative diseases, such as Parkinson’s, Alzheimer’s, and Huntington’s diseases and amyotrophic lateral sclerosis, and mitochondrial dysfunction (Peña-Bautista et al., 2019; Angelova et al., 2021; Cháfer-Pericás, 2021), and ferroptosis may be a pathomechanism (Hambright et al., 2017; Peña-Bautista et al., 2019; Cháfer-Pericás, 2021). However, whether lipid peroxidation products are the cause or consequence of neurodegeneration remains unclear (Ayala et al., 2014). It has been reported that 4-HNE-induced neuronal loss and α-synuclein (α-Syn) aggregation is commonly associated with neurodegenerative disorders (Peña-Bautista et al., 2019). Furthermore, it has been reported that oligomeric α-Syn induces lipid peroxidation (Angelova et al., 2015). Recently, Angelova et al. (2020) have reported that oligomeric α-Syn produces an iron-dependent increase in ROS and lipid peroxidation, which in turn induce α-Syn aggregation in membranes, disrupt calcium flux, and lead to ferroptosis. These findings suggest a crucial role of lipid peroxidation and ferroptosis in neurodegenerative diseases (Angelova et al., 2020). Moreover, a positive feedback loop of iron accumulation, mitochondrial dysfunction, and ROS damage that causes α-Syn aggregation, changes in mitochondrial dynamics, proteasomal dysfunction, and activation of cell death pathways has been reported (Urrutia et al., 2014). Studies have reported that lipid peroxidation by-products, 4-HNE and malondialdehyde among others, produce selective protein-modifications by Michael adduction in Alzheimer’s disease (Sultana et al., 2013). This indicates that lipid peroxidation is an early event in Alzheimer’s disease progression and that the levels of lipid peroxidation by-products and protein adducts may be used as biomarkers of disease progression and severity. Moreover, relationship between lipid peroxidation in Alzheimer’s disease and transition-metals through its interaction with amyloid-β, the major component of senile plaques, has been established. Amyloid-β can chelate transition-metals forming aggregates that have pro-oxidant properties (Arlt et al., 2002). In addition, neuronal death in Alzheimer’s disease has been associated with ferroptosis (Angelova et al., 2021).
Role of Lipid Peroxidation in Iron Accumulation
Although the role of iron in lipid peroxidation has been well-elucidated (Gaschler and Stockwell, 2017; Maiorino et al., 2018; Rockfield et al., 2018; Su et al., 2019; Zhao et al., 2021; Bilska-Wilkosz et al., 2022), the effect of lipid peroxidation in iron overload has not adequately been addressed. Hence, biological models of increased lipid peroxidation due to genetic or toxic causes can be used to verify whether lipid oxidation intrinsically induces intracellular iron accumulation. Recently, this relationship has been examined in cellular models of PLAN (Villalón-García et al., 2022), a rare genetic neurodegenerative disease within the group of NBIA disorders, caused by mutations in the PLA2G6 gene that encodes the group VI calcium-independent Phospholipase A2 (iPLA2β), an enzyme responsible for scission of polyunsaturated fatty acids at sn-2-position of membrane glycerophospholipids (Hayflick et al., 2018; Hinarejos et al., 2020). Owing to PLA2G6 mutations, lipid peroxidation (Kinghorn et al., 2015; Arber et al., 2016; Villalón-García et al., 2022) and iron accumulation have been described as significant events in the pathomechanism of PLAN (Beck et al., 2016; Villalón-García et al., 2022). It is hypothesized that excessive lipid peroxidation in cell membranes damages lipids and proteins that alter both, normal function of organelles such as mitochondria and membrane-dependent processes such as vesicular traffic and autophagy/mitophagy. These alterations may trigger iron accumulation in the form of iron-rich lipofuscin (Brunk and Terman, 2002; Milward et al., 2012; Frolova et al., 2015; Davids et al., 2016; Dodson et al., 2017; Moreno-García et al., 2018; Ilie et al., 2020; Villalón-García et al., 2022).
iPLA2β deficiency causing progressive lipid peroxidation in mitochondrial membranes rapidly propagated by the presence of high iron content in mitochondria is a plausible pathogenic mechanism in PLAN. Thus, a significant increase of lipid peroxidation in mitochondrial membranes was detected in PLA2G6 mutant fibroblasts. Moreover, the evaluation of mitochondrial membrane potential (ΔΨm) and mitochondrial network in these fibroblasts revealed a significant reduction in ΔΨm and mitochondrial network disruption with reduced mitochondrial elongation, and an increased number of rounded depolarized mitochondria. Additionally, mitochondrial depolarization was observed in PLAN induced neurons (Villalón-García et al., 2022). These findings in PLAN cellular models corroborated previous investigations in PLAN fibroblasts (homozygous p-R747W) and iPLA2-VIA–/– fly brains (Kinghorn et al., 2015). Electron microscopy of PLAN mutant cells revealed condensation of mitochondrial membranes with disruption of the internal mitochondrial cristae (Villalón-García et al., 2022), supporting these findings. In addition, PLAN cellular models manifested altered vesicle-based processes such as autophagy/mitophagy (Arber et al., 2016) and lysosomal dysfunction (Lin et al., 2018) causing defective mitochondrial degradation in autolysosomes and increasing lipofuscinogenesis. Thus, these interconnected mechanisms may participate in lipofuscin formation.
Studies describing that iron accumulation in lipofuscin may originate from damaged mitochondria have been previously reported (Frolova et al., 2015; Álvarez-Córdoba et al., 2019; Kakimoto et al., 2019). In addition, lipofuscin recruits iron resulting in a redox-active surface that catalyzes the Fenton reaction and increases the formation of free radicals (Höhn and Grune, 2013), and consequently, enhance lipid peroxidation. The ability of lipofuscin to inhibit the degradation of oxidized proteins by competitively binding and sequestering the proteasome, a major characteristic, has previously been demonstrated (Sitte et al., 2001).
Villalón-García et al. (2022) assessed iron and lipofuscin accumulation by the induction of lipid peroxidation with tert-Butyl peroxide, a radical initiator of lipid peroxidation, with the aim of replicating PLAN pathophysiology. They observed that the induction of lipid peroxidation in healthy control cells promoted iron/lipofuscin accumulation and the supplementation of the control cells with iron-induced lipid peroxidation (Villalón-García et al., 2022). Based on these findings from PLAN and control cellular models, they concluded that lipid peroxidation essentially induces iron/lipofuscin accumulation and iron/lipofuscin accumulation induces lipid peroxidation in a vicious cycle.
Vitamin E, a blocker of lipid peroxidation propagation and an efficient ferroptosis inhibitor, has been reported to reduce lipid peroxidation and iron/lipofuscin accumulation and correct mitochondrial dysfunction and the main pathological alterations in PLAN cellular models (Villalón-García et al., 2022).
Lipid Peroxidation, Iron Accumulation and Lipofuscin
Lipofuscin is a pigmented by-product of failed intracellular catabolism found inside lysosomes and cytosol of aging postmitotic cells (Snyder and Crane., 2021). Lipofuscin formation has been explained through different hypotheses, such as their lysosomal and mitochondrial origin. According to the lysosomal hypothesis, lipid peroxidation damages the lysosomal membranes, which causes membrane dysfunction and increased iron permeability. Intralysosomal iron catalyzes the Fenton reaction within the lysosome and produces hydroxyl radicals, which cause oxidation of lipid and proteins that accumulate in undegradable aggregates (Kakimoto et al., 2019; Ilie et al., 2020; Tonolli et al., 2020; Călin et al., 2021). Lipid peroxidation by-products react with lipids and proteins forming adducts with Schiff bases; these have emission in the 450–470 nm region when excited by ultraviolet light, which could explain the fluorescent properties of lipofuscin (Călin et al., 2021). In addition, the fusion of damaged secondary lysosomes with other damaged lysosomes can lead to lipofuscin accumulation (Kakimoto et al., 2019; Ilie et al., 2020; Tonolli et al., 2020; Villalón-García et al., 2022). Furthermore, the lysosomal and mitochondrial origins of lipofuscin are related because damaged mitochondrial turnover is affected by low mitophagy and the release of iron by mitochondria in autolysosomes, which trigger more Fenton reactions and lipid peroxidation (Moreno-García et al., 2018; Ilie et al., 2020). Mitophagy, a selective form of mitochondrial degradation, eliminates ROS-damaged mitochondria, while impaired mitophagy leads to the accumulation of damaged proteins and lipids in mitochondria (Mijaljica et al., 2010).
ROS can regulate autophagy or “self-eating”, a protective role in eliminating ROS-damaged organelles and cell components, such as mitochondria, proteins, and lipids, respectively. However, excessive ROS-induced autophagy can lead to autophagic cell death. In addition, lipid peroxidation can induce autophagic dysfunction, triggering autophagic cell death in certain conditions through the inhibition of AMP-activated protein kinase, and consequently, the activation of the mammalian target of rapamycin pathway (Su et al., 2019). Lipid peroxidation by-products form adducts with proteins involved in autophagosome and autolysosome formation thereby decreasing autophagic flux (Dodson et al., 2017), and hinder the timely renewal of organelles such as mitochondria. Moreover, lipid peroxidation may induce lysosomal dysfunction and lipofuscinogenesis, contributing to decreased autophagy (Su et al., 2019). The defective degradation of damaged organelles can facilitate iron accumulation and lipofuscin formation (Moreno-García et al., 2018). A deficient autophagy/mitophagy with incomplete lysosomal degradation has been associated with lipofuscin accumulation, while the enhancement of autophagy/mitophagy alleviates lipofuscin accumulation (Li et al., 2021). The Mitochondrial-Lysosomal Axis Theory, hypothesized by Brunk and Terman in 2002, accurately describes the cross-talk between both origins of lipofuscinogenesis (Brunk and Terman, 2002; Snyder and Crane., 2021).
Although both mitochondrial and lysosomal lipofuscinogenesis are related processes, lipofuscin formation can be produced by impaired mitochondrial fission without alteration of autophagosome-lysosome system (Frolova et al., 2015; Kakimoto et al., 2019). The mitochondrial origin of lipofuscin is based on the fact that a mitochondrion contains a large reserve of cellular iron (Paul et al., 2017), which contributes to mitochondrial lipid peroxidation along with NADPH and ADP and alters mitochondrial integrity and function. In turn, lipid peroxidation by-products disrupt the mitochondrial architecture and contribute to lipofuscin formation (Bindoli, 1988). The different formation pathways of lipofuscin formation in mitochondria, as a result of lipid peroxidation without lysosomal participation, have been described in the literature (Frolova et al., 2015).
Ferroptosis Induced by Lipid Peroxidation and Iron Accumulation
Programmed cell death, in the context of homeostasis maintenance, depends on specific molecular machinery and can be modulated by genetic or pharmacological interventions (Hirschhorn and Stockwell, 2019). Apoptosis, autophagy-dependent cell death, and necrosis were the earlier known types of cell death (Kroemer et al., 2009; Hirschhorn and Stockwell, 2019); however, other types of regulated cell death such as necroptosis, pyroptosis, parthanatos, and ferroptosis, have been described over the years (Latunde-Dada, 2017).
Ferroptosis, coined by Dixon et al. (2012), is described as morphologically, genetically, and biochemically different from other forms of regulated cell death (Dixon et al., 2012; Bertrand, 2017; Latunde-Dada, 2017; Chen et al., 2020; Li et al., 2020). It is a regulated cell death caused by lipid peroxidation and iron accumulation (Hirschhorn and Stockwell, 2019; Chen et al., 2020; Li et al., 2020). Ferroptosis is associated with 3 main biochemical characteristics: (i) generation and accumulation of ROS and lipid peroxidation damage, (ii) reduction of GSH and GPX4 activity, and (iii) accumulation of iron with reactive ability through the Fenton reaction (Bertrand, 2017; Latunde-Dada, 2017; Maiorino et al., 2018; Hirschhorn and Stockwell, 2019; Ge et al., 2022). It has been hypothesized that each of these key events needs to simultaneously occur, since the arrest of any one event prevents ferroptosis. Moreover, each event is amplified by positive feedback loops (Bertrand, 2017). Plasma membrane and subcellular locations (mitochondria, ER, and lysosomes) have been hypothesized as important sites of lipid peroxidation during ferroptosis (Feng and Stockwell, 2018). Morphologically, ferroptosis is characterized by a reduced mitochondrial volume, disappearance of mitochondrial cristae, and increased density of bilayer membrane. However, the cell membrane and nucleus maintain their integrity. Moreover, ferroptosis is regulated by different genes that involve lipid peroxidation metabolism and iron homeostasis (Li et al., 2020).
Accumulation of lipid peroxidation and ferroptosis have been associated with various pathological conditions, including cancer, neurodegeneration, kidney injury, steatohepatitis, and ischemic diseases (Feng and Stockwell, 2018; Hirschhorn and Stockwell, 2019; Nishizawa et al., 2021).
Ferroptosis mechanisms
Ferroptosis is iron-dependent; when the antioxidant capacity of cells is exceeded and ROS and lipid peroxidation are accumulated, a non-apoptotic cell death phenotype is triggered (Bertrand, 2017; Latunde-Dada, 2017). Under oxidative stress conditions, release of redox-active iron from iron compounds could be induced by high superoxide levels (Latunde-Dada, 2017). Ferritinophagy (autophagic releasing of iron from ferritin) has been implicated in ferroptosis, through the regulation of iron homeostasis (Latunde-Dada, 2017; Park and Chung, 2019). Lipid peroxidation by-products have been described as inducers of autophagy and an excess of this process promotes ferroptosis, suggesting a complex feedback between autophagy, ferroptosis, and organelle damage (Tang et al., 2021). In addition, the role of lipoxygenases and Cytochrome P450 oxidoreductase in initiation of lipid peroxidation and ferroptosis has been suggested (Maiorino et al., 2018; Zou et al., 2020; Jiang et al., 2021).
The inhibition of cysteine import into the cell by direct inactivation of System Xc– reduces GSH levels and GPX activity, triggering the accumulation of ROS and lipid peroxidation involved in oxidative damage and ferroptosis, has been reported (Li et al., 2020). GPX4 enzyme specifically neutralizes lipid peroxidation using GSH as a co-substrate, and in turn, cysteine is involved in the GSH synthesis; hence, inhibition of cysteine uptake indirectly affects GPX4 activity, increasing lipid peroxidation accumulation (Latunde-Dada, 2017; Li et al., 2020). As mentioned earlier, excessive oxidative stress accumulation may increase free iron (Latunde-Dada, 2017); however, iron accumulation through Fenton reactions could cause GSH depletion during ferroptosis (Bertrand, 2017), while Fenton reactions cause lipid peroxidation (Bertrand, 2017).
Simultaneous occurrence of all these events causes ferroptosis because the inhibition of any one of these events prevents ferroptosis (Bertrand, 2017). The reduction of iron accumulation by iron-chelators, such as deferoxamine (Bertrand, 2017; Latunde-Dada, 2017; Chen et al., 2020), or the inhibition of an iron metabolism transcription factor such as iron-responsive element-binding protein 2, inhibit ferroptosis (Li et al., 2020). Furthermore, the action of vitamin E, suppressing the propagation of lipid peroxidation, induces a protective effect against ferroptosis (Latunde-Dada, 2017). The presence of iron accumulation needs the simultaneous depletion of GSH because in cells with high free iron levels, the inhibition of System Xc– is needed to decrease GSH levels to undergo ferroptosis (Bertrand, 2017). In summary, the presence of redox-active iron is simultaneously needed with lipid peroxidation accumulation, low levels of GSH, and decreased GXP4 activity to trigger ferroptosis (Bertrand, 2017). In addition to their simultaneous presence, these events can be mutually exacerbated. Lipid peroxidation increases permeability of lysosomal membranes resulting in the leakage of free iron into the cytosol and causing Fenton reactions that cause oxidative stress and more lipid peroxidation (Bertrand, 2017). Lipid peroxidation by-products such as 4-HNE contribute to depletion of GSH, since 4-HNE is detoxified by the interaction with GSH through its Cys residue (Sultana et al., 2013). Concurrently, Fenton radicals have been associated with the depletion of GSH reserves (Bertrand, 2017).
Ferroptosis in neurodegenerative diseases
Recent studies have indicated the vital role of ferroptosis in the pathophysiological process of multiple organ systems, including the nervous system (Zhou et al., 2022). Ferroptosis is crucially involved in neurological diseases, including neurodegeneration, neurotrauma, and stroke (Reichert et al., 2020). Cellular features of ferroptosis have been observed in dopaminergic neurons in Parkinson’s disease (Do Van et al., 2016), Alzheimer’s disease (Yan and Zhang, 2019), and in neuronal cell death in Huntington’s disease (Zhou et al., 2022).
It has recently been reported that massive iron accumulation in PKAN-derived neurons and astrocytes associated with higher levels of lipid peroxidation, and a lower amount of GPX4 in PKAN-derived astrocytes, revealed signs of ferroptosis (Santambrogio et al., 2022).
Mutations in the WDR45 (WD Repeat Domain 45) gene leads to the impairment of autophagy, which is associated with BPAN. The intracellular iron content was increased in cells overexpressing mutant WDR45; this further promoted cell ferroptosis, demonstrated by an increased lipid peroxidation and ROS, and decreased GPX4 and cell viability (Xiong et al., 2021).
Programmed cell death processes such as apoptosis and autophagic cell death play a role in neurodegenerative diseases; the interplay between them and ferroptosis in neuronal cell death is an emerging field of research (Dang et al., 2022).
Conclusions
In summary, lipid peroxidation, iron accumulation, and ferroptosis are pathomechanisms in neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases and NBIA disorders. In this systematic review, we have compiled available literature on the role of lipid peroxidation in iron accumulation, including the vicious cycle between them in neurodegeneration (Figure 2). Lipid peroxidation in iron-rich organelles such as mitochondria and defective membrane-dependent processes such as autophagy/mitophagy or vesicle traffic may lead to iron accumulation in lipofuscin, which in turn increases lipid peroxidation. This negative series of events that reinforce each other may aggravate and accelerate the progression of neurodegenerative diseases. Membrane antioxidants, including vitamin E, can break this vicious cycle in neurodegenerative diseases by blocking lipid peroxidation propagation; hence, discovering protective treatments will be a significant breakthrough in the management of neurodegeneration. Further investigations are required to reveal the effects of lipid peroxidation on autophagy/ferritinophagy/mitophagy; to provide insights into the molecular mechanisms involved in iron accumulation and the precise mechanisms by which iron accumulation occurs in lipofuscin. Finally, further research on the mechanism of ferroptosis is needed to clarify its exact role in neurodegeneration and provide newer therapeutic approaches.
Figure 2.
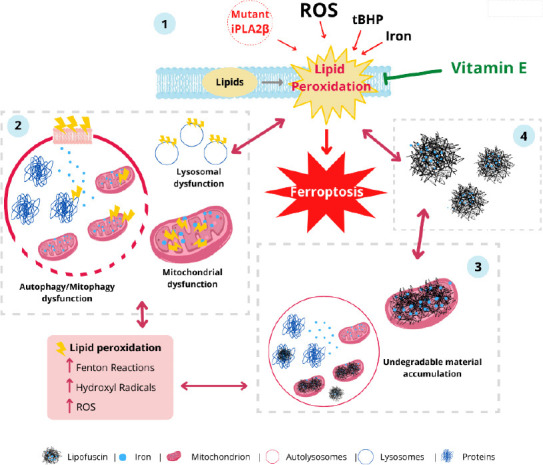
Iron/lipofuscin accumulation and lipid peroxidation: a feedback loop in neurodegenerative diseases.
1. Lipid peroxidation in mitochondrial, lysosomal, endoplasmic reticulum and plasma membranes caused by reactive oxygen species (ROS), tert-butylhydroperoxide (tBHP), calcium-independent phospholipase A2 group VI (iPLA2β) mutations, or iron. 2. Lipid peroxidation is enhanced by ROS and Fenton reaction leading to autophagy, lysosomal and mitochondrial dysfunction. 3. Aggregation of peroxided protein and lipids lead to lipofuscinogenesis. 4. Iron-rich lipofuscin granules increase lipid peroxidation. Eventually, neurons die by ferroptosis.
Acknowledgments:
We acknowledge the support of “Ayudas para la Formación de Profesorado Universitario” (FPU/MINECO), FEDER (Federación Española de Enfermedades Raras), Fundación MERCK Salud and Fundación MEHUER/ Colegio Oficial de Farmacéuticos.
Footnotes
Funding: This work was supported by FIS PI16/00786 (2016) and FIS PI19/00377 (2019) grants, the Ministerio de Sanidad, Spain and the Fondo Europeo de Desarrollo Regional (FEDER Unión Europea), Spanish Ministry of Education, Culture and Sport. This activity has been co-financed by the European Regional Development Fund (ERDF) and by the Regional Ministry of Economic Transformation, Industry, Knowledge and Universities of the Junta de Andalucía, within the framework of the ERDF Andalusia operational program 2014-2020 Thematic objective “01 - Reinforcement of research, technological development and innovation” through the reference research project CTS-5725 and PY18-850 (to JASA).
Conflicts of interest: The authors declare no conflicts of interest.
C-Editors: Zhao M, Zhao LJ, Qiu Y; T-Editor: Jia Y
References
- 1.Ademowo OS, Dias HKI, Burton DGA, Griffiths HR. Lipid (per) oxidation in mitochondria:an emerging target in the ageing process? Biogerontology. 2017;18:859–879. doi: 10.1007/s10522-017-9710-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Álvarez-Córdoba M, Khoury AF, Villanueva-Paz M, Gómez-Navarro C, Villalón-García I, Suárez-Rivero JM, Povea-Cabello S, Mata Mdl, Cotán D, Talaverón-Rey M, Pérez-Pulido AJ, Salas JJ, Pérez-Villegas EM, Díaz-Quintana A, Armengol JA, Sánchez-Alcázar JA. Pantothenate rescues iron accumulation in pantothenate kinase-associated neurodegeneration depending on the type of mutation. Mol Neurobiol. 2019;56:3638–3656. doi: 10.1007/s12035-018-1333-0. [DOI] [PubMed] [Google Scholar]
- 3.Angelova PR, Horrocks MH, Klenerman D, Sonia Gandhi AYA, Shchepinov MS. Lipid peroxidation is essential for α-synuclein-induced cell death. J Neurochem. 2015;133:582–589. doi: 10.1111/jnc.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angelova PR, Choi ML, Berezhnov AV, Horrocks MH, Hughes CD, De S, Rodrigues M, Yapom R, Little D, Dolt KS, Kunath T, Devine MJ, Gissen P, Shchepinov MS, Sylantyev S, Pavlov EV, Klenerman D, Abramov AY, Gandhi S. Alpha synuclein aggregation drives ferroptosis:an interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020;27:2781–2796. doi: 10.1038/s41418-020-0542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angelova PR, Esteras N, Abramov AY. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration:Finding ways for prevention. Med Res Rev. 2021;41:770–784. doi: 10.1002/med.21712. [DOI] [PubMed] [Google Scholar]
- 6.Arber CE, Li A, Houlden H, Wray S. Review:Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation:unifying theories. Neuropathol Appl Neurobiol. 2016;42:220–241. doi: 10.1111/nan.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arlt S, Beisiegel U, Kontush A. Lipid peroxidation in neurodegeneration:new insights into Alzheimer's disease. Curr Opin Lipidol. 2002;13:289–294. doi: 10.1097/00041433-200206000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation:production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck G, Sugiura Y, Shinzawa K, Kato S, Setou M, Tsujimoto Y, Sakoda S, Sumi-Akamaru H. Neuroaxonal dystrophy in calcium-independent phospholipase A2βdeficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci. 2011;31:11411–11420. doi: 10.1523/JNEUROSCI.0345-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck G, Shinzawa K, Hayakawa H, Baba K, Yasuda T, Sumi-Akamaru H, Tsujimoto Y, Mochizuki H. Deficiency of calcium-independent phospholipase A2 beta induces brain iron accumulation through upregulation of divalent metal transporter 1. PLoS One. 2016;10:e0141629. doi: 10.1371/journal.pone.0141629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertrand RL. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med Hypotheses. 2017;101:69–74. doi: 10.1016/j.mehy.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 12.Bilska-Wilkosz A, Iciek M, Gorny M. Chemistry and biochemistry aspects of the 4-hydroxy-2,3-trans-nonenal. Biomolecules. 2022;12:145. doi: 10.3390/biom12010145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bindoli A. Lipid peroxidation in mitochondria. Free Radic Biol Med. 1988;5:247–261. doi: 10.1016/0891-5849(88)90018-4. [DOI] [PubMed] [Google Scholar]
- 14.Bogdanov M, Mileykovskaya E, Dowhan W. Lipids in the assembly of membrane proteins and organization of protein supercomplexes:implications for lipid-linked disorders. Subcell Biochem. 2008;49:197–239. doi: 10.1007/978-1-4020-8831-5_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging:accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 16.Călin EF, Popescu SIP, Cernat CCC, Patoni C, Popescu MN, Muşat O. Lipofuscin:a key compound in ophthalmic practice. Rom J Ophthalmol. 2021;65:109–113. doi: 10.22336/rjo.2021.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cháfer-Pericás C. Lipid peroxidation in neurodegeneration. Antioxidants. 2021;10:484. doi: 10.3390/antiox10030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226. doi: 10.3389/fcell.2020.590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Codreanu SG, Liebler DC. Novel approaches to identify protein adducts produced by lipid peroxidation. Free Radic Res. 2015;49:881–887. doi: 10.3109/10715762.2015.1019348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corti O, Blomgren K, Poletti A, Beart PM. Autophagy in neurodegeneration:New insights underpinning therapy for neurological diseases. J Neurochem. 2020;154:354–371. doi: 10.1111/jnc.15002. [DOI] [PubMed] [Google Scholar]
- 21.Dang X, Huan X, Du X, Chen X, Bi M, Yan C, Jiao Q, Jiang H. Correlation of ferroptosis and other types of cell death in neurodegenerative diseases. Neurosci Bull. 2022;38:938–952. doi: 10.1007/s12264-022-00861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davids M, Kane MS, He M, Wolfe LA, Li X, Raihan MA, Chao KR, Bone WP, Boerkoel CF, Gahl WA, Toro C. Disruption of Golgi morphology and altered protein glycosylation in PLA2G6-associated neurodegeneration. J Med Genet. 2016;53:180–189. doi: 10.1136/jmedgenet-2015-103338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies SS, Guo L. Lipid peroxidation generates biologically active phospholipids including oxidatively N-modified phospholipids. Chem Phys Lipids. 2014;0:1–33. doi: 10.1016/j.chemphyslip.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Carvalho CCCR, Caramujo MJ. The various roles of fatty acids. Molecules. 2018;23:2583. doi: 10.3390/molecules23102583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR. Ferroptosis:an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi: 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Dodson M, Wani WY, Redmann M, Benavides GA, Johnson MS, Ouyang X, Cofield SS, Mitra K, Darley-Usmar V, Zhang J. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy. 2017;13:1828–1840. doi: 10.1080/15548627.2017.1356948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farooqui AA, Horrocks LA. Lipid peroxides in the free radical pathophysiology of brain diseases. Cell Mol Neurobiol. 1998;18:599–608. doi: 10.1023/A:1020625717298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng H, Stockwell BR. Unsolved mysteries:how does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16:e2006203. doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frolova MS, Surin AM, Braslavski AV, Vekshin NL. Degradation of mitochondria to lipofuscin upon heating and illumination. Biofizika. 2015;60:1125–1131. [PubMed] [Google Scholar]
- 32.Gallo G, Sprovieri P, Martino G. 4-hydroxynonenal and oxidative stress in several organelles and its damaging effects on cell functions. J Physiol Pharmacol. 2020;71:15–33. doi: 10.26402/jpp.2020.1.07. [DOI] [PubMed] [Google Scholar]
- 33.Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ge C, Zhang S, Mu H, Zheng S, Tan Z, Huang X, Xu C, Zou J, Zhu Y, Feng D, Aa J. Emerging mechanisms and disease implications of ferroptosis:potential applications of natural products. Front Cell Dev Biol. 2022;9:774957. doi: 10.3389/fcell.2021.774957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S. Ferroptosis and cell death mechanisms in Parkinson's disease. Neurochem Int. 2017;104:34–48. doi: 10.1016/j.neuint.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17. doi: 10.1016/j.redox.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayflick SJ, Kurian MA, Hogarth P. Neurodegeneration with brain iron accumulation. Handb Clin Neurol. 2018;147:293–305. doi: 10.1016/B978-0-444-63233-3.00019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinarejos I, Machuca-Arellano C, Sancho P, Espinós C. Mitochondrial dysfunction, oxidative stress and neuroinflammation in neurodegeneration with brain iron accumulation (NBIA) Antioxidants (Basel) 2020;9:1020. doi: 10.3390/antiox9101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–143. doi: 10.1016/j.freeradbiomed.2018.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Höhn A, Grune T. Lipofuscin:formation, effects and role of macroautophagy. Redox Biol. 2013;1:140–144. doi: 10.1016/j.redox.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu C, Wangb M, Hana X. Shotgun lipidomics in substantiating lipid peroxidation in redox biology:methods and applications. Redox Biol. 2017;12:946–955. doi: 10.1016/j.redox.2017.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ilie OD, Ciobica A, Riga S, Dhunna N, McKenna J, Mavroudis I, Doroftei B, Ciobanu A-M, Riga D. Mini-review on lipofuscin and aging:focusing on the molecular interface, the biological recycling mechanism, oxidative stress, and the gut-brain axis functionality. Medicina (Kaunas) 2020;56:626. doi: 10.3390/medicina56110626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang X, Stockwell BR, Conrad M. Ferroptosis:mechanisms, biology, and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kakimoto Y, Okada C, Kawabe N, Sasaki A, Tsukamoto H, Nagao R, Osawa M. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci Rep. 2019;9:3304. doi: 10.1038/s41598-019-40250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ke Y, Qian ZM. Brain iron metabolism:neurobiology and neurochemistry. Prog Neurobiol. 2007;83:149–173. doi: 10.1016/j.pneurobio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Kinghorn KJ, Castillo-Quan JI, Bartolome F, Angelova PR, Li L, Pope S, Cochemé HM, Khan S, Asghari S, Bhatia KP, Hardy J, Abramov AY, Partridge L. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain. 2015;138:1801–1816. doi: 10.1093/brain/awv132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinghorn KJ, Castillo-Quan JI. Mitochondrial dysfunction and defects in lipid homeostasis as therapeutic targets in neurodegeneration with brain iron accumulation. Rare Dis. 2016;4:e1128616. doi: 10.1080/21675511.2015.1128616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nuñez G, Peter ME, Tschopp J, Yuan J, Piacentini M, et al. Classification of cell death:recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Latunde-Dada GO. Ferroptosis:role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 51.Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G. Ferroptosis:past, present and future. Cell Death Dis. 2020;11:88. doi: 10.1038/s41419-020-2298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li WW, Wang HJ, Tan YZ, Wang YL, Yu SN, Li ZH. Reducing lipofuscin accumulation and cardiomyocytic senescence of aging heart by enhancing autophagy. Exp Cell Res. 2021;403:112585. doi: 10.1016/j.yexcr.2021.112585. [DOI] [PubMed] [Google Scholar]
- 53.Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, Lin WW, Wang L, Bellen HJ. Phospholipase PLA2G6, a Parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to α-synuclein gain. Cell Metab. 2018;28:605–618. doi: 10.1016/j.cmet.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 54.Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death:discoveries, rediscoveries, and open issues. Antioxid Redox Signal. 2018;29:61–74. doi: 10.1089/ars.2017.7115. [DOI] [PubMed] [Google Scholar]
- 55.Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, Kotzbauer PT. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol. 2008;172:406–416. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mårtensson CU, Doan KN, Becker T. Effects of lipids on mitochondrial functions. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:102–113. doi: 10.1016/j.bbalip.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 57.Mijaljica D, Prescott M, Devenish RJ. Mitophagy and mitoptosis in disease processes. In: Bross P, Gregersen N, editors. Protein misfolding and cellular stress in disease and aging:concepts and protocols (Methods and Protocols) Totowa, NJ: Humana Press; 2010. pp. 93–106. [DOI] [PubMed] [Google Scholar]
- 58.Milward E, Acikyol B, Bassett B, Williams E, Graham R, Delima R, Trinder D, Olynyk J, Johnstone D. Brain changes in iron loading disorders. In: Linert W, Kozlowski H, editors. Metal ions in neurological systems. Vienna: Springer-Verlag; 2012. pp. 17–29. [Google Scholar]
- 59.Moreno-García A, Kun A, Calero O, Medina M, Calero M. An overview of the role of lipofuscin in age-related neurodegeneration. Front Neurosci. 2018;12:464. doi: 10.3389/fnins.2018.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishizawa H, Matsumoto M, Chen G, Ishii Y, Tada K, Onodera M, Kato H, Muto A, Tanaka K, Igarashi K. Lipid peroxidation and the subsequent cell death transmitting from ferroptotic cells to neighboring cells. Cell Death Dis. 2021;12:332. doi: 10.1038/s41419-021-03613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:14205–14218. doi: 10.3748/wjg.v20.i39.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822. doi: 10.1038/s41419-019-2064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paul BT, Manz DH, Torti FM, Torti SV. Mitochondria and iron:current questions. Expert Rev Hematol. 2017;10:65–79. doi: 10.1080/17474086.2016.1268047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peña-Bautista C, Vento M, Baquero M, Cháfer-Pericás C. Lipid peroxidation in neurodegeneration. Clin Chim Acta. 2019;467:178–188. doi: 10.1016/j.cca.2019.07.037. [DOI] [PubMed] [Google Scholar]
- 65.Ramana KV, Srivastava S, Singhal SS. Lipid peroxidation products in human health and disease 2019. Oxid Med Cell Longev. 2019;2019:7147235. doi: 10.1155/2019/7147235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reichert CO, Freitas FAd, Sampaio-Silva J, Rokita-Rosa L, Barros PdL, Levy D, Bydlowski SP. Ferroptosis mechanisms involved in neurodegenerative diseases. Int J Mol Sci. 2020;21:8765. doi: 10.3390/ijms21228765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rockfield S, Chhabra R, Robertson M, Rehman N, Bisht R, Nanjundan M. Links between iron and lipids:implications in some major human diseases. Pharmaceuticals (Basel) 2018;11:113. doi: 10.3390/ph11040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Santambrogio P, Ripamonti M, Cozzi A, Raimondi M, Cavestro C, Meo ID, Rubio A, Taverna S, Tiranti V, Levi S. Massive iron accumulation in PKAN-derived neurons and astrocytes:light on the human pathological phenotype. Cell Death Dis. 2022;13:185. doi: 10.1038/s41419-022-04626-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sasson S. 4-Hydroxyalkenal-activated PPARd mediates hormetic interactions in diabetes. Biochimie. 2017;136:85–89. doi: 10.1016/j.biochi.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 70.Schaur RJ, Siems W, Bresgen N, Eckl PM. 4-Hydroxy-nonenal—A bioactive lipid peroxidation product. Biomolecules. 2015;5:2247–2337. doi: 10.3390/biom5042247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlame M, Greenberg ML. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:3–7. doi: 10.1016/j.bbalip.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sitte N, Merker K, Grune T, Zglinicki Tv. Lipofuscin accumulation in proliferating fibroblasts in vitro:an indicator of oxidative stress. Exp Gerontol. 2001;36:475–486. doi: 10.1016/s0531-5565(00)00253-9. [DOI] [PubMed] [Google Scholar]
- 73.Snyder AN, Crane JS. Histology, Lipofuscin. Treasure Island (FL): StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 74.Spaull RVV, Soo AKS, Hogarth P, Hayflick SJ, Kurian MA. Towards precision therapies for inherited disorders of neurodegeneration with brain iron accumulation. Tremor Other Hyperkinet Mov (N Y) 2021;11:51. doi: 10.5334/tohm.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, Jiang F, Peng ZY. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843. doi: 10.1155/2019/5080843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration:a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157–169. doi: 10.1016/j.freeradbiomed.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Svetel M, Dragašević N, Petrović I, Novaković I, Tomić A, Kresojević N, Stanković I, Kostić V. NBIA syndromes:a step forward from the previous knowledge. Neurol India. 2021;69:1380–1388. doi: 10.4103/0028-3886.329603. [DOI] [PubMed] [Google Scholar]
- 78.Tang D, Chen X, Kang R, Kroemer G. Ferroptosis:molecular mechanisms and health implications. Cell Res. 2021;31:107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tonolli PN, Martins WK, Junqueira HC, Silva MN, Severino D, Santacruz-Perez C, Watanabe I, Baptista MS. Lipofuscin in keratinocytes:production, properties, and consequences of the photosensitization with visible light. Free Radic Biol Med. 2020;160:277–292. doi: 10.1016/j.freeradbiomed.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 80.Urrutia PJ, Mena NP, Núñez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 2014;5:38. doi: 10.3389/fphar.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Villalón-García I, Álvarez-Córdoba M, Povea-Cabello S, Talaverón-Rey M, Villanueva-Paz M, Luzón-Hidalgo R, Suárez-Rivero JM, Suárez-Carrillo A, Munuera-Cabeza M, Salas JJ, Falcón-Moya R, Rodríguez-Moreno A, Armengol JA, Sánchez-Alcázar JA. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-associated neurodegeneration. Neurobiol Dis. 2022;165:105649. doi: 10.1016/j.nbd.2022.105649. [DOI] [PubMed] [Google Scholar]
- 82.Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–1160. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu Y, Song J, Wang Y, Wang X, Culmsee C, Zhu C. The potential role of ferroptosis in neonatal brain injury. Front Neurosci. 2019;13:115. doi: 10.3389/fnins.2019.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiong Q, Li X, Li W, Chen G, Xiao H, Li P, Wu C. WDR45 mutation impairs the autophagic degradation of transferrin receptor and promotes ferroptosis. Front Mol Biosci. 2021;8:645831. doi: 10.3389/fmolb.2021.645831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yadav UC, Ramana KV. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid Med Cell Longev. 2013;2013:690545. doi: 10.1155/2013/690545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan N, Zhang J. Iron metabolism, ferroptosis, and the links with Alzheimer's disease. Front Neurosci. 2019;13:1443. doi: 10.3389/fnins.2019.01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao T, Guo X, Sun Y. Iron accumulation and lipid peroxidation in the aging retina:implication of ferroptosis in age-related macular degeneration. Aging Dis. 2021;12:529–551. doi: 10.14336/AD.2020.0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhong H, Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer:focusing on mitochondria. Redox Biol. 2015;4:193–199. doi: 10.1016/j.redox.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou Y, Lin W, Rao T, Zheng J, Zhang T, Zhang M, Lin Z. Ferroptosis and its potential role in the nervous system diseases. J Inflamm Res. 2022;15:1555–1574. doi: 10.2147/JIR.S351799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, Schreiber SL. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–309. doi: 10.1038/s41589-020-0472-6. [DOI] [PMC free article] [PubMed] [Google Scholar]