

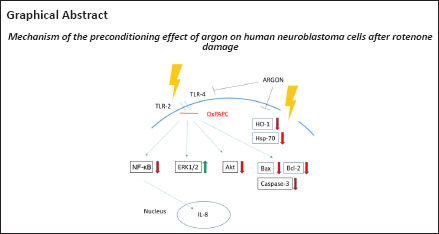
Keywords: apoptosis, inflammation, interleukin-8, neuroprotection, rotenone, SH-SY5Y, Toll-like receptor 2, Toll-like receptor 4
Abstract
The noble gas argon has the potential to protect neuronal cells from cell death. So far, this effect has been studied in treatment after acute damage. Preconditioning using argon has not yet been investigated. In this study, human neuroblastoma SH-SY5Y cells were treated with different concentrations of argon (25%, 50%, and 74%; 21% O2, 5% CO2, balance nitrogen) at different time intervals before inflicting damage with rotenone (20 µM, 4 hours). Apoptosis was determined by flow cytometry after annexin V and propidium iodide staining. Surface expressions of Toll-like receptors 2 and 4 were also examined. Cells were also processed for analysis by western blot and qPCR to determine the expression of apoptotic and inflammatory proteins, such as extracellular-signal regulated kinase (ERK1/2), nuclear transcription factor-κB (NF-κB), protein kinase B (Akt), caspase-3, Bax, Bcl-2, interleukin-8, and heat shock proteins. Immunohistochemical staining was performed for TLR2 and 4 and interleukin-8. Cells were also pretreated with OxPAPC, an antagonist of TLR2 and 4 to elucidate the molecular mechanism. Results showed that argon preconditioning before rotenone application caused a dose-dependent but not a time-dependent reduction in the number of apoptotic cells. Preconditioning with 74% argon for 2 hours was used for further experiments showing the most promising results. Argon decreased the surface expression of TLR2 and 4, whereas OxPAPC treatment partially abolished the protective effect of argon. Argon increased phosphorylation of ERK1/2 but decreased NF-κB and Akt. Preconditioning inhibited mitochondrial apoptosis and the heat shock response. Argon also suppressed the expression of the pro-inflammatory cytokine interleukin-8. Immunohistochemistry confirmed the alteration of TLRs and interleukin-8. OxPAPC reversed the argon effect on ERK1/2, Bax, Bcl-2, caspase-3, and interleukin-8 expression, but not on NF-κB and the heat shock proteins. Taken together, argon preconditioning protects against apoptosis of neuronal cells and mediates its action via Toll-like receptors. Argon may represent a promising therapeutic alternative in various clinical settings, such as the treatment of stroke.
Introduction
Interruption of oxygen and glucose supply to neuronal cells, as can occur during ischemic stroke or cardiovascular arrest, leads to an inevitable deterioration of neuronal function. The consequences for patients are devastating, ranging from permanent neurological impairment with a physical disability to death. The pathophysiological responses to cerebral ischemia include cell membrane depolarization, tissue acidosis, and glutamate release, which in turn results in massive calcium release (Lo et al., 2003; Moro et al., 2005; Yuan, 2009; Auriel and Bornstein, 2010; Wang and Qin, 2010). This subsequently activates several different inflammatory and apoptotic signaling pathways via protein phosphorylation, interference with mitochondrial function, and generation of free radicals (Chan, 2001; Huang et al., 2006).
Attempts to mitigate this destructive process pharmaceutically have failed so far. However, in recent years, the noble gas argon has moved into the focus of scientific research in this area. The inhaled use of this odorless, non-toxic and inexpensive argon could be advantageous because, in contrast to xenon, it is not sedating and has had no adverse side effects described to date. In fact, promising studies now indicate that argon mediates neuroprotective effects (Ulbrich et al., 2016; Ma et al., 2019; Goebel et al., 2021).
The beneficial effect of argon in moderating brain damage after cardiac arrest was demonstrated by Brucken et al. (2013), who showed that rats had a better neurological outcome with less neuronal damage, especially in the neurocortex and hippocampal C3/4 region, after one hour of inhalation with 70% argon (30% oxygen). Similarly, Ristagno et al. (2014) confirmed the beneficial effect of argon inhalation after ischemic cardiac arrest in pigs. They observed that 4-hour ventilation with a mixture of 70% argon/30% O2, initiated immediately after resuscitation, led to a marked improvement in neurological function, together with reduced levels of serum neuron-specific enolase. In another study, this group also reported that a 4-hour treatment with 70% argon (30% O2) ventilation after prolonged ischemic cardiac arrest provided more pronounced neuroprotection than 50% argon (also with 30% O2). Histopathological analyses revealed significantly less neuronal degeneration and fewer reactive microglia with argon treatment than in the untreated control group (Fumagalli et al., 2020).
Argon has also been studied in experimental stroke models. For example, Ryang et al. (2011) exposed rats to a 2-hour occlusion of the middle cerebral artery. One hour after ischemia induction, 50% argon (balance O2) was administered by inhalation via a face mask for 1 hour. Compared with the untreated control group, the argon inhalation group showed a significant reduction in infarct size and improved neurologic outcome, although the 24-hour survival was not improved (Ryang et al., 2011). In a more recent study, Ma et al. (2019) tested the effect of 24-hour argon (70% and 30% O2) inhalation after middle cerebral artery occlusion. In that study, argon also improved neurologic outcomes and reduced infarct volume (Ma et al., 2019). In our previous studies, we found that argon protected neuronal ganglion cells after retinal ischemia-reperfusion injury in a time- and dose-dependent manner (Ulbrich et al., 2014).
The underlying mechanism by which argon exerts this protective effect is not yet fully understood. An in vitro study by Fahlenkamp et al. (2012) showed that argon influences protein kinase 1/2 in neuronal cells and reduces LPS-induced inflammation in microglial cells. We were able to demonstrate that argon inhibits Toll-like receptors 2 and 4 (TLR2 and 4) and reduces nuclear transcription factor-κB (NF-κB) activation in both an in vitro model after damaging human neuroblastoma cells with rotenone and in an in vivo retinal ischemia-reperfusion injury model (Ulbrich et al., 2015a, 2016). A review comprising the up-to-date known parts of the mechanism also named interleukin-8 (IL-8) and other proteins (Ulbrich and Goebel, 2016).
All the investigations carried out to date on the neuroprotective effect have administered argon during or after damage. However, the neuroprotective effect of preconditioning with argon (i.e., argon administration before damage) has not yet been investigated. In the present study, we investigated the effect of argon preconditioning in an in vitro damage model using human neuroblastoma cells.
Methods
Reagents
The TLR2+4 inhibitor OxPAPC (#tlrl-oxpap1) was purchased from InvivoGen (San Diego, CA, USA). Rotenone, dimethyl sulfoxide (DMSO), ionomycin, and phorbol-myristate acetate (PMA) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Rotenone was freshly prepared and dissolved in DMSO before the experiments. The concentration of 20 µM used in the following experiments was obtained in a previously conducted dose-finding study (data not shown). The DMSO concentration in the cell culture media did not exceed 0.5%. Cells were exposed to a gas mixture consisting of argon (Air Liquide, Kornwestheim, Germany), oxygen (O2), carbon dioxide (CO2), and nitrogen (N2). The gas mixture was generated and controlled in a designated incubator (BioSpherix, Parish, NY, USA) at final concentrations of 5% CO2, 21% O2, argon (25%, 50%, or 74% [v/v]), and the balance N2.
Cell culture and treatment
Neuroblastoma cells (cell line SH-SY5Y) were obtained directly from ATCC (Cat# CRL-2266, RRID: CVCL_019, Manassas, VA, USA), cultured in DMEM/F12 medium (GIBCO Life Technologies, Darmstadt, Germany) supplemented with 1% penicillin/streptomycin and 10% fetal calf serum and incubated in a humidified atmosphere containing 5% carbon dioxide at 37°C constant temperature until the cells reached 80% confluence. The cells for experiments were seeded in 6-well culture plates at a density of approximately 1.5 × 105 per well and cultured for a further 48 hours. Prior to rotenone treatment, the cells were exposed to gas mixtures containing argon at 25%, 50%, or 74% for 2 or 4 hours in a designated incubator with a humidified atmosphere. The gas delivery and mixing were done automatically, and the pre-set target values were reached in less than 60 seconds. The temperature was maintained at 37°C during all exposures. Subsequently, the cells were transferred to media containing 1% fetal calf serum to prevent the inactivation of rotenone by protein binding. Then, the rotenone treatment (20 µM) was performed. OxPAPC (30 µg/mL) was added 60 minutes before the argon treatment in the specific experiments. Cells were collected immediately after 4-hour rotenone treatment for FACS analysis and protein quantification.
Flow cytometry of Annexin V/propidium iodide and TLR2 and TLR4
Cells were washed in cold phosphate-buffered saline (PBS), trypsinized, and resuspended in 100 μL binding buffer (0.01 M HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 0.14 M NaCl, 2.5 mM CaCl2, pH 7.4). Staining was performed by adding 5 μL FITC (fluorescein isothiocyanate) annexin V and propidium iodide (PI; Becton Dickinson, Heidelberg, Germany). Staining for TLR2 and TLR4 was done using FITC-conjugated TLR4 antibody (1:40; mouse; Abcam, Cambridge, UK, Cat# ab45126, RRID: AB_778502) and TLR2 antibody (1:20; mouse; Abcam, Cat# ab114070, RRID: AB_10901879). Samples were incubated at room temperature for 15 minutes before adding 400 μL binding buffer. Flow cytometry analysis was performed using an acoustic focusing cytometer (Attune®, Life Technologies, Darmstadt, Germany). Unstained and single-stained cells served as negative controls for background fluorescence and for setting compensation and quadrants.
Western blot analysis
Western blotting was performed as previously described (Ulbrich et al., 2014). Briefly, equal amounts of cell protein extract (30 μg) were boiled in 5× sodium dodecyl sulfate loading dye (50% glycerol, 0.5 M dithiothreitol, 350 mM sodium dodecyl sulfate, 7.5 mM bromphenol blue, 250 mM TRIS, pH 6.8) for 5 minutes and subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After protein transfer to polyvinylidene difluoride membranes (Immobilon-P; Millipore, Schwalbach, Germany), the membranes were blocked with 5% skim milk in Tween20/PBS (TBST) or BSA (bovine serum albumin) and incubated in the recommended dilution of protein-specific antibody (p44/42 MAP kinase (phospho-ERK1/2 [extracellular-signal regulated kinase]) (Thr202/Tyr204), rabbit, 1:2000, Cell Signaling Technology, Danvers, MA, USA, Cat# 9101, RRID: AB_331646; phospho-NF-κB (Ser536) (93H1), rabbit, 1:1000, Cell Signaling Technology, Cat# 3033, RRID: AB_331284; phospho-Akt (protein kinase B) antibody (Ser473), rabbit, 1:1000, Cell Signaling Technology, Cat# 9271, RRID: AB_329825; Bax antibody, rabbit, 1:1000, Cell Signaling Technology, Cat# 2772, RRID: AB_10695870; Bcl-2 antibody, rabbit, 1:1000, Cell Signaling Technology, Cat# 2876, RRID: AB_2064177; and cleaved caspase-3 antibody (Asp175), rabbit, 1:500, Cell Signaling Technology, Cat# 9661, RRID: AB_2341188; NF-E2-related factor 2 (Nrf2) antibody [nuclear factor erythroid 2-related factor 2, phosphor-S40], rabbit, 1:60000, Abcam, Cat# ab76026, RRID: AB_1524049). After incubation with a horseradish peroxidase-conjugated anti-rabbit secondary antibody (anti-rabbit IgG, goat, 1:5000, Cell Signaling Technology, Cat# 7074, RRID: AB_2099233), the proteins were visualized using an enhanced chemiluminescence western blotting detection reagent (Western Lightning plus enhanced chemiluminescence, Cat# NEL103001EA; PerkinElmer, Waltham, MA, USA), following the manufacturer’s instructions. Images were generated with Fusion Fx® imaging system (PEQLAB Biotechnologie GmbH, Erlangen, Germany). For normalization, blots were re-probed with p44/42 MAPK (mitogen-activated protein kinase, ERK1/2) antibody (rabbit, 1:1000, Cell Signaling Technology, Cat# 9102, RRID: AB_330744), NF-κB p65 (D14E12) (rabbit, 1:1000, Cell Signaling Technology, Cat# 8242, RRID: AB_10859369), Akt antibody (rabbit, 1:1000, Cell Signaling Technology, Cat# 9272, RRID: AB_329827), caspase-3 antibody (rabbit, 1:4000, Cell Signaling Technology, Cat# 9662, RRID: AB_331439), β-actin antibody (rabbit, 1:1000, Cell Signaling Technology, Cat# 4967, RRID: 330288), and NFE2L2 (rabbit, 1:1000, Abcam, Cat# ab62352, RRID: AB_944418).
Real-time polymerase chain reaction
Approximately 2 × 106 cells were washed, trypsinized, and processed as described above. Reverse transcription was performed with 50 ng of total RNA using random primers (High Capacity cDNA Reverse Transcription Kit; Thermo Fisher Scientific, Waltham, MA, USA). Real-time polymerase chain reactions (RT-PCR) were done with TaqMan® probe-based detection kit (TaqMan® PCR universal mastermix; Thermo Fisher Scientific). The following primers were used: HSP70 (heat shock protein 70) Hs00359163_s1, HO1 Hs01110250_m1, L-8 Hs00174103_m1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) Hs02786624_g1 (all from Thermo Fisher Scientific). The PCR assays were then performed on an RT-PCR System (StepOne™; Thermo Fisher Scientific) under the following conditions: 95°C for 10 minutes, 40 cycles of 95°C for 10 seconds, and 60°C for 1 minute. Reaction specificity was confirmed by running appropriate negative controls. The cycle threshold (CT) values for each gene of interest were normalized to the corresponding CT values for GAPDH (ΔCT).
Immunohistochemical staining
Cells were grown to approximately 50% confluency, and then stained with a DoubleStain IHC kit (rabbit/mouse; 3,3′-diaminobenzidine & FastRed, Cat# ab210062, Abcam) to visualize TLR2 antibody (mouse, 5 µg/mL, Abcam, Cat# ab16894, RRID: AB_44353) and TLR4 antibody (mouse, 5 µg/mL, Abcam, Cat# ab13556, RRID: 300457). The assay was performed according to the manufacturer’s instructions. Thereafter, the cells were stained with hematoxylin (Merck, Darmstadt, Germany) for background visualization and embedded in a mounting medium. For IL-8 single staining, washing with TBST was followed by blocking with endogenous peroxidase/AP (Dako Dual Endogenous Enzyme Block for Autostainer Cat# S2003, Agilent, Santa Clara, CA, USA). After another washing step and blocking with goat serum (10%) and BSA (1%), the primary antibody was applied (anti-IL-8 antibody, rabbit, 1 µg/mL, Abcam, Cat# ab84995). After 1 hour and another wash, the secondary antibody was added (goat anti-rabbit IgG H&L-HRP, 1:1000, Abcam, Cat# ab6721). After 30 minutes, horseradish peroxidase (HRP) substrate 3,3′-diaminobenzidine (Dako K3467, Agilent) was added. After rinsing with distilled water, the cells were counterstained with Mayer’s-hemalum solution (Merck, Darmstadt, Germany), and mounting medium (mounting medium for IHC, Abcam, Cat# ab64230) was added. Images were obtained using Axio-Vision Software (Zeiss, Oberkochen, Germany). Quantification of immunohistochemistry was done using the ImageJ software (version 1.43u, NIH, Bethesda, MD, USA; Schneider et al., 2012) and the histogram tool. Photographs of 5 individual experiments (10 fields of vision for each experiment) were taken and the histogram was analyzed in a blinded fashion to the observer.
Statistical analysis
A statistics software program (SigmaPlot Version 11.0, Systat Software Inc., San Jose, CA, USA) was used to analyze the data. After checking the normal distribution of the data with the Shapiro-Wilk test, the results were presented as mean ± SD. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test. A value of p < 0.05 was considered statistically significant.
Results
Argon preconditioning reduces rotenone-induced cell damage in a dose-dependent but not time-dependent manner
We added rotenone (20 µM for 4 hours) to human neuroblastoma cells and performed annexin V/PI staining to determine the percentage of apoptotic cells. Exposure to rotenone increased the percentage of apoptotic cells (annexin V/PI positive) (P < 0.001; Figure 1A). Preconditioning of the cells with argon (74%, 50%, or 25%) prior to rotenone at different time intervals (2 or 4 hours) resulted in a reduction in apoptotic cells at all argon concentrations (rotenone vs. argon 50% 2 hours + rotenone, argon 50% 4 hours + rotenone, argon 25% 2 hours + rotenone, argon 25% 4 hours + rotenone; all P < 0.001; Figure 1A–D), with the strongest effect seen with for 74% argon (rotenone vs. argon 74% 2 hours + rotenone, argon 74% 4 hours + rotenone; both P < 0.001; Figure 1B). The time interval of application had no effect on the outcome at all concentrations (Figure 1A–C).
Figure 1.
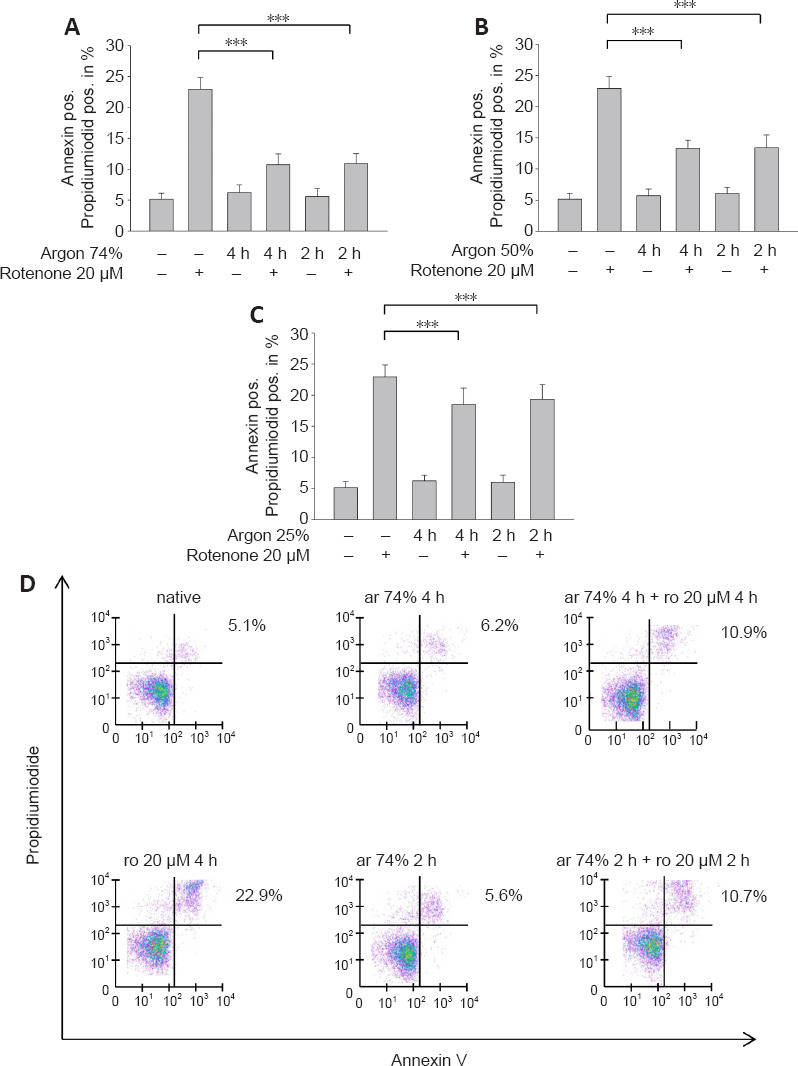
Argon preconditioning attenuates rotenone-induced apoptosis in a dose-dependent manner, irrespective of treatment time.
(A) The percentage of annexin and porpidiumiodide positive (pos.) cells, indicating late apoptosis. Exposure to rotenone (ro) increased the percentage of apoptotic cells. Two and four hours of preconditioning with argon (ar) 74% reduced apoptosis. (B) The effect was similar at a lower argon concentration (argon 50%). (C) Preconditioning with argon 25% also showed a significant but attenuated effect. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; ***P < 0.001). (D) Flow cytometry representation of dot plots for the interventions with argon 74% (2 and 4 hours). The purple points represent single cells. Clustering leads to a change of the color spectrum to green. Rotenone created a cluster predominately in the upper right quadrant, which decreased due to argon preconditioning.
Argon preconditioning inhibits rotenone-induced TLR2 and 4 receptor expression
A previous series of experiments demonstrated the involvement of TLRs in argon responses (Ulbrich et al., 2015a); therefore, we now investigated whether TLR2 and 4 might also be involved in the anti-apoptotic effect of argon preconditioning. The flow cytometry results showed that rotenone treatment increased the TLR2 and 4 receptor expression (TLR2: native vs. rotenone, P < 0.05; TLR4: native vs. rotenone; P < 0.001; Figure 2A and B). Preconditioning with 74% argon showed the strongest anti-apoptotic effect and significantly reduced TLR2 and 4 receptor expression, with a stronger effect on TLR4 (TLR2: rotenone vs. argon 74% 2 hours, P < 0.05; TLR4: rotenone vs. argon 74% 2 hours, P < 0.001; Figure 2C and D). Immunohistological analyses confirmed these results (Figure 3). In the native samples, a weak red TLR4 staining was observed only in the cytoplasmic region (Figure 3A), and this was also evident in the samples in which the cells were treated with argon only (Figure 3C). The application of rotenone resulted in a significant increase in red color intensity in the cytoplasmic region (Figure 3B). After rotenone treatment, brown staining (TLR2) dominated, especially in the peripheral region of the nucleus (Figure 3B). A prior application of argon strongly reduced both the red cytoplasmic red staining and the brown staining at the periphery of the nucleus (Figure 3D). This effect was reversed by OxPAPC (Figure 3E). The quantification showed that argon preconditioning reduced the expression of TLR2 and 4 after treatment with rotenone, while OxPAPC abrogated this effect (P < 0.001; Figure 3F and G).
Figure 2.
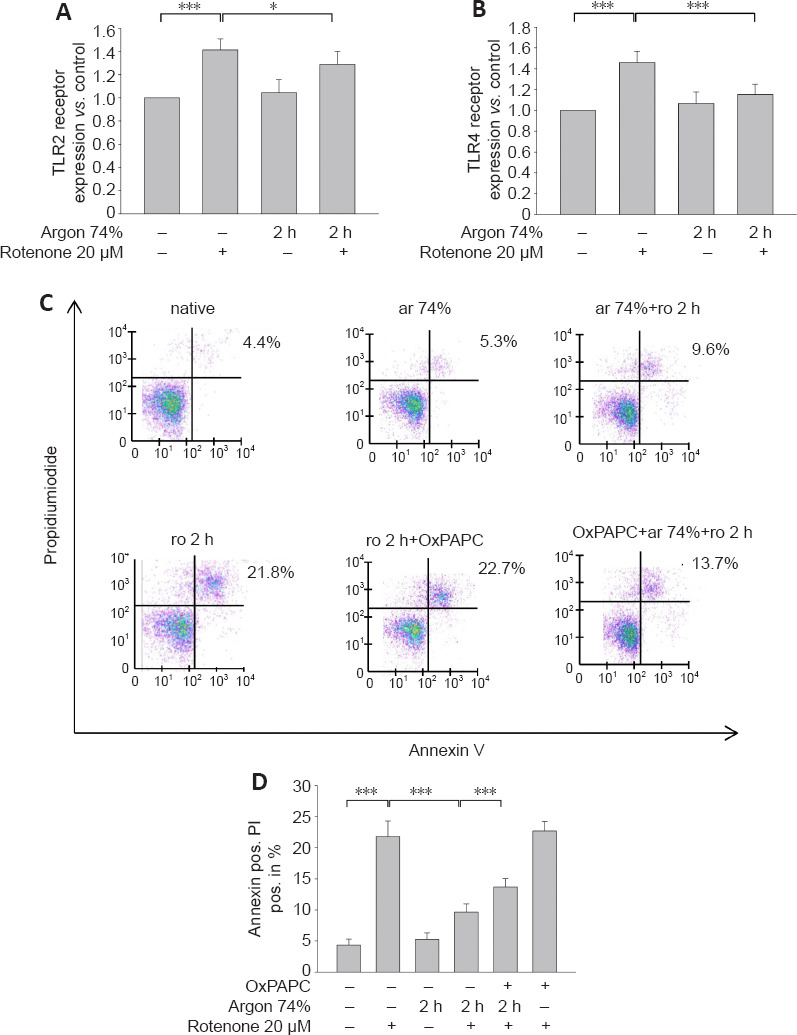
Argon preconditioning reduces TLR2 and TLR4 receptor density and the TLR2 and TLR4 inhibitor OxPAPC attenuates the effect of argon preconditioning.
(A) Fluorescence-activated cell sorting (FACS) analyses of TLR2 receptor expression after rotenone (ro) treatment and argon (ar) preconditioning. Rotenone increased the TLR2 receptor expression, while argon preconditioning reduced the receptor density. (B) Similarly, treatment with rotenone increased TLR4-expression and Argon abolished this effect. (C) Representative dot blots for the interventions with argon 75% (2 and 4 hours) and inhibition with OxPAPC. The purple points represent single cells. Clustering leads to a change of the color spectrum to green. Rotenone created a cluster predominately in the upper right quadrant, which decreased due to argon preconditioning. The treatment with OxPAPC reduced the argon effect. (D) Annexin V and PI-positive cells after rotenone-induced apoptosis, preconditioning with argon, and inhibition with the TLR2 and 4 receptor antagonist OxPAPC. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; *P < 0.05, ***P < 0.001). TLR: Toll-like receptor
Figure 3.
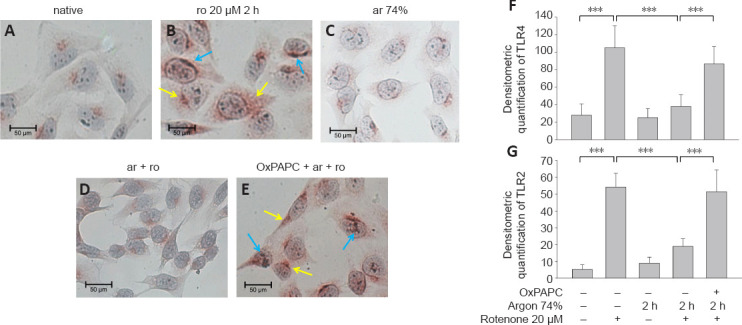
Argon preconditioning before rotenone exposure inhibits TLR2 and TLR4 expression.
(A–E) Representative histological images after different types of treatment. Blue arrows: TLR2 expression (brown), yellow arrows: TLR4 expression (red). Staining was performed with a DoubleStain IHC kit. (A) In the native samples, a weak red TLR4 staining was observed only in the cytoplasmic region. (B) The application of rotenone resulted in a significant increase in red color intensity in the cytoplasmic region and brown staining (TLR2) dominanted, especially in the peripheral region of the nucleus. (C) The staining of the samples treated with argon only was comparable to the native samples. (D) Argon preconditioning before rotenone strongly reduced both the red cytoplasmic red staining and the brown staining at the periphery of the nucleus. (E) This effect was reversed by OxPAPC. (F, G) Quantification of TLR2 (F) and TLR4 (G). Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 5; mean ± SD; ***P < 0.001).
The TLR2 and 4 receptor antagonist OxPAPC partially abolishes the argon preconditioning effect
Annexin V and PI staining showed that the TLR2 and 4 antagonist OxPAPC partially abolished the anti-apoptotic effect of argon (argon 74% 2 hours + rotenone vs. argon 75% 2 hours + rotenone + OxPAPC; P < 0.001; Figure 2C and D). OxPAPC treatment alone did not have any effect on apoptosis by rotenone (Figure 2C and D).
Argon preconditioning induces ERK1/2 and inhibits NF-κB phosphorylation
We also examined the effect of argon preconditioning on phosphorylation of the transcription factors ERK1/2 and NF-κB. Argon increased ERK1/2 phosphorylation (rotenone vs. argon 74% 2 hours + rotenone; P < 0.01; Figure 4A and B), but this effect was partially abolished by OxPAPC treatment (argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC; P < 0.05). By contrast, argon significantly reduced the rotenone-induced increase in NF-κB phosphorylation (rotenone vs. argon 74% 2 hours + rotenone; P <0.01; Figure 4C and D). However, this effect was not significantly reversed by OxPAPC antagonism of TLR2 and 4 (P > 0.05).
Figure 4.
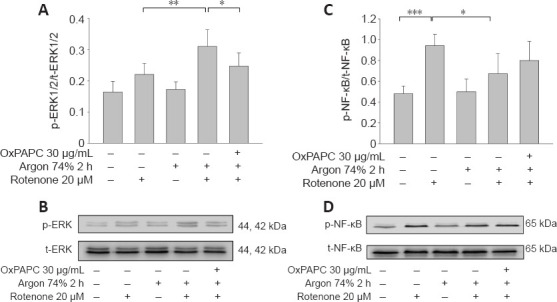
Argon preconditioning activates ERK1/2 and inhibits NF-κB phosphorylation – OxPAP partially reverses the argon effect part.
Evaluation of ERK1/2 and NF-κB phosphorylation by western blotting. (A) Densitometric analysis of western blots of ERK1/2 phosphorylation, normalized against total-ERK1/2. (B) Representative western blot image of six independent experiments that showed similar results demonstrating the influence of rotenone, argon preconditioning, and inhibition with OxPAPC on ERK1/2 phosphorylation. (C) Densitometric analysis of western blots of NF-κB phosphorylation, normalized against total NF-κB. (D) Representative western blot image of six independent experiments that showed similar results demonstrating the influence of rotenone, argon preconditioning, and inhibition with OxPAPC on NF-κB phosphorylation. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; *P < 0.05, **P < 0.01). ERK1/2: Extracellular-signal regulated kinase; NF-κB: nuclear transcription factor-κB.
Argon inhibits Akt phosphorylation but has no effect on Nrf-2
Argon preconditioning suppressed the rotenone-induced increase in Akt phosphorylation (rotenone vs. argon 74% 2 hours + rotenone; P < 0.001; Figure 5A and B) and this effect was totally eliminated by OxPAPC treatment (argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC; P < 0.05). By contrast, Nrf-2 phosphorylation was not affected by rotenone or argon treatments (argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC; P > 0.05; Figure 5C and D).
Figure 5.
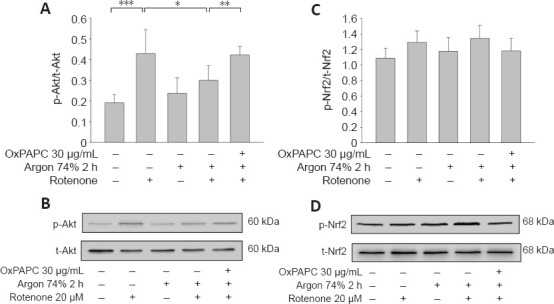
Argon preconditioning reduces Akt phosphorylation but has no effect on Nrf2 expression.
Evaluation of Akt and Nrf-2 phosphorylation by western blotting. (A) Densitometric analysis of western blots of Akt phosphorylation, normalized against total Akt. (B) Representative western blot image of six independent experiments that showed similar results demonstrating the influence of rotenone, argon preconditioning, and inhibition with OxPAPC on Akt phosphorylation. (C) Densitometric analysis of western blots of Nrf-2 phosphorylation, normalized against total-Nrf-2. (D) Representative western blot image of six independent experiments that showed similar results demonstrating no effect after the interventions directed against Nrf-2 phosphorylation. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001). Akt: Protein kinase B; Nrf2: nuclear factor erythroid 2-related factor 2.
Argon reduces the rotenone-induced increase in Bax and caspase-3 and increases Bcl-2
Western blot analysis showed that rotenone treatment induced an increase in Bax and a decrease in Bcl-2 (Bax: native vs. rotenone; P < 0.001; Figure 6A and B). Argon treatment attenuated the increase in Bax, but this argon effect was reversed by OxPAPC treatment (rotenone vs. argon 74% 2 hours + rotenone, P < 0.001 and argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC, P < 0.001). By contrast, the argon treatment increased Bcl-2, but this effect was reduced by OxPAPC (rotenone vs. argon 74% 2 hours + rotenone; P < 0.01 and argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC, P < 0.05). The Bax/Bcl-2 ratio was reduced from 3 to 1.4 after argon preconditioning compared to cells injured with rotenone. As with the pro-apoptotic Bax, the observed changes in caspase 3 further supported the anti-apoptotic effect of argon (Figure 7; native vs. rotenone, P < 0.001, rotenone vs. argon 74% 2 hours + rotenone, P < 0.01 and argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC, P < 0.05).
Figure 6.
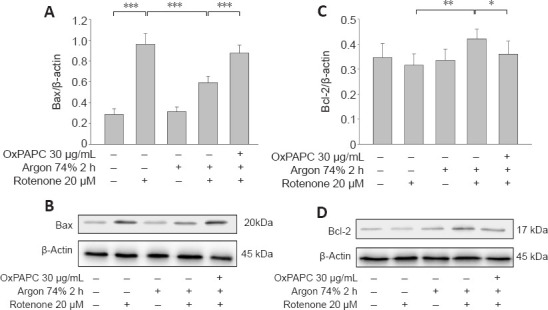
Argon preconditioning reduces rotenone-induced protein expression of Bax and increases Bcl-2, but the effect is abolished by OxPAPC.
(A, C) Densitometric analysis of western blots for Bax (A) and Bcl-2 (C) protein expression. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001). (B, D) Representative western blot images of six independent experiments that showed similar results demonstrating the influence of rotenone, argon preconditioning, and inhibition with OxPAPC on Bax (B) and Bcl-2 (D).
Figure 7.
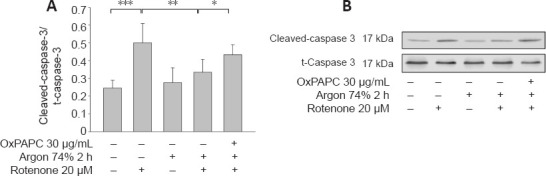
Effect of Argon preconditioning on caspase-3 cleavage.
(A) Evaluation of caspase-3 cleavage by western blotting. Densitometric analysis of western blots of caspase-3 cleavage, normalized against uncleaved caspase-3. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001). (B) Representative western blot image of six independent experiments that showed similar results demonstrating the influence of rotenone, argon preconditioning, and inhibition with OxPAPC on caspase-3 cleavage.
Argon preconditioning inhibits the heat shock response independent of TLR-signaling
Rotenone induced the expression of HSP-70 and heme oxygenase 1 (HO-1) mRNA (Figure 8A and B; HSP-70: native vs. rotenone and HO-1: native vs. rotenone; both P < 0.001), and this expression was attenuated by argon (HSP-70: rotenone vs. argon 74% 2 hours + rotenone; HO-1: rotenone vs. argon 74% 2 hours + rotenone; both P < 0.05). However, OxPAPC had no effect and could not counteract the argon response (P > 0.05).
Figure 8.
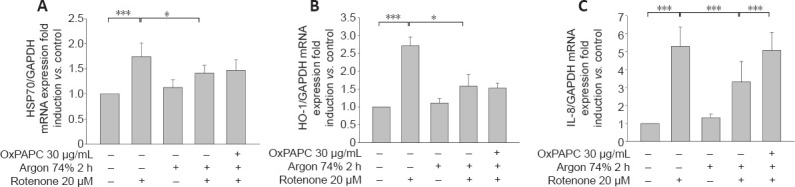
OxPAPC treatment does not influence the effect of argon preconditioning on HO-1 and HSP-70.
(A, B) Analysis of HSP70 (A) and HO-1 mRNA (B) expression using qPCR. Fold induction of HSP70 and HO-1 mRNA expression compared to GAPDH in relation to untreated cells after treatment with rotenone, preconditioning with argon, and inhibition with OxPAPC. Argon attenuates the rotenone-induced release of the cytokine IL-8 and OxPAPC counteracts this effect. (C) Analysis of IL-8 mRNA expression using qPCR. Fold induction of IL-8 mRNA expression compared to GAPDH in relation to untreated cells after treatment with rotenone, preconditioning with argon, and inhibition with OxPAPC. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 6; mean ± SD; *P < 0.05, ***P < 0.001). GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HSP70: Heat shock protein 70; IL-8: interleukin-8; qPCR: real-time polymerase chain reaction.
Argon preconditioning suppresses IL-8 expression
Argon preconditioning had an impressive effect on the IL-8 expression induced by rotenone (native vs. rotenone; P < 0.001; Figure 9F), as argon treatment resulted in a suppression of IL-8 expression (rotenone vs. argon 74% 2 hours + rotenone; P < 0.001). This argon effect could be reversed by the application of OxPAPC (argon 74% 2 hours + rotenone vs. argon 74% 2 hours + rotenone + OxPAPC; both P < 0.001). Immunohistological quantification confirmed these findings (Figure 9): Untreated cells and cells only treated with argon alone showed only slight brown staining in the cytoplasm (Figure 9A and C). By contrast, rotenone-induced a visible increase in color intensity, which was also evident in the quantification (Figure 9B and F). Preconditioning with argon significantly attenuated this effect, while OxPAPC treatment significantly increased the color (IL-8 untreated vs. rotenone; rotenone vs. argon 75% 2 hours + rotenone; argon 75% 2 hours + rotenone vs. OxPAPC+ argon 75% 2 hours; all P < 0.001; Figure 9D–F).
Figure 9.
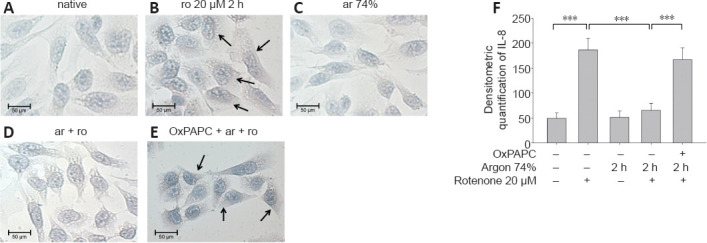
Argon treatment before rotenone exposure inhibits IL-8 immunoreactivity in human SH-SY5Y cells.
(A–E) Representative histological images after different types of treatment. Black arrow: IL-8-immunoreactive cells (brown). (A) Untreated cells and cells treated with argon alone (C) showed only slight brown staining in the cytoplasm. (B) By contrast, rotenone-induced a clearly visible increase in color intensity. (D) Preconditioning with argon significantly attenuated the effect, while OxPAPC treatment (E) markedly increased the color. (F) Analysis of histogram quantification for IL-8. Between-group comparisons were performed with a one-way analysis of variance using the post hoc Holm Sidak test (n = 5; mean ± SD; ***P < 0.01). ar: Argon; IL-8: interleukin-8; ro: rotenone;
Discussion
The main findings of this study are that argon preconditioning: 1) reduces apoptosis of human neuroblastoma cells in a dose-dependent manner irrespective of treatment time; 2) mediates its protective effect at least in part via TLRs 2 and 4, and its impact can be partially abrogated by the TLR2+4 antagonist OxPAPC; 3) induces ERK1/2, inhibits Akt and NF-κB phosphorylation, and has no effect on Nrf-2 phosphorylation; 4) reduces the levels of both apoptosis-inducing proteins and heat shock proteins; and 5) suppresses the expression of the cytokine IL-8.
Among the gaseous molecules that provide cell- and organ protection (CO, H2S, and NO) (Scheid et al., 2021), argon emerges as a potential therapeutical agent especially regarding neuroprotection. To our knowledge, the effects of argon application prior to neuronal cell damage have not yet been studied. One study analyzing argon preconditioning on airway epithelial cells showed a lung-protective effect. Here, A549 cells were treated with a cytotoxic concentration of H2O2. Preconditioning with argon 50% for 30, 45, and 180 minutes and argon 30% for 180 minutes resulted in significant protection of A549 cells against H2O2-induced apoptosis (Hafner et al., 2016). A protective effect of argon preconditioning has also been demonstrated in cardiomyocytes. Mayer et al. (2016) treated primary isolated cardiomyocytes with 50% argon for 1 hour before hypoxia. In these cells, preconditioning in an early time window (0–3 hours) before hypoxia had no effect, but a time interval of 24 hours before hypoxia significantly increased the percentage of viable cells. In an in vivo study, rats were treated with 3 short cycles of argon (50%, 5 minutes) before isolated hearts were subjected to cold ischemia and evaluated after a period of reperfusion. Preconditioning the rats with argon resulted in significant recovery of cardiac output (Kiss et al., 2018).
It has been shown that noble gases such as xenon and argon mediate neuroprotective effects (Jawad et al., 2009). In the present study, argon preconditioning showed protective effects at concentrations of 25%, 50%, and 74%, with the best effect seen with argon at 74%. By contrast, the duration of argon treatment (2 hours vs. 4 hours) prior to rotenone administration had no influence on this protective argon effect.
Inflammatory processes are well known to play a major role in cerebral ischemia-reperfusion injury, such as stroke. TLRs are part of the innate immune response and generate pro-apoptotic and pro-inflammatory mediators that enhance brain injury after ischemia (Wang et al., 2013). TLRs 2 and 4 and their downstream signaling cascade play a pivotal role in triggering neuronal cell death in the first hours after a stroke (Tang et al., 2007). Our group has previously shown that argon postconditioning induces protective effects in vitro and in vivo by downregulating TLR2 and 4 expression (Ulbrich et al., 2015a). The present findings show that argon preconditioning (74%, 2 hours) likewise reduces the expression of these same TLRs to trigger an anti-apoptotic effect which can be partially abrogated by OxPAPC. Although the exact function of OxPAPC is controversial and seems contradictory (Bochkov et al., 2010), OxPAPC itself did not mediate either a pro- or anti-apoptotic effect in the present study. Instead, OxPAPC blocked the protective argon signaling cascade mediated by TLRs 2 and 4 inside the cells.
The TLRs trigger an inflammatory cascade via downstream mitogen-activated protein kinases and transcription factors. The major pathways involved in TLR activation are mediated by NF-κB and MAPK: TLR activation promotes the recruitment of various adaptive proteins to activate NF-κB, which then induces the expression of proinflammatory genes and inflammatory cytokines such as IL-8 (Akira, 2006; Leitner et al., 2019). The transcription factor NF-κB also regulates the expression of many genes involved in cell death by promoting the expression of pro-apoptotic genes that appear to be involved in various pathological conditions of the central nervous system (Yang et al., 2007). MAP kinases, such as ERK1/2, are also activated in the further course of the TLR-mediated pathway (Kawai and Akira, 2007). Stimulation of TLR4 activates the p65 NF-κB pathway to trigger the upregulation of pro-inflammatory cytokines, while also activating the Akt pathway (Okun et al., 2009; Wen et al., 2019). After an ischemia/reperfusion injury, TLRs can respond to various endogenous cellular and extracellular components, including HSP70 and HO-1, whose structure has been altered by released cellular enzymes.(Arumugam et al., 2009; Sharp et al., 2013) TLR4 signaling also affects the expression of the apoptosis-related proteins Bax, Bcl-2, and effector caspase-3 to induce apoptosis via activation of transcription of inflammatory factors downstream of TLR4 (Wang et al., 2016).
Zhao et al. (2016c) performed in vivo studies in rat pups treated with 70% argon and hypothermia after unilateral ligation of the carotid artery and found a reduced infarct size as well as suppression of NF-κB in the cortex and hippocampus. We demonstrated that argon preconditioning led to a reduction in NF-κB phosphorylation. However, this reduction was not affected by OxPAPC treatment, suggesting that other mechanisms are also involved in the protective effect of argon preconditioning, such as mitogen-activated protein kinases.
The preconditioning results available to date show that argon can protect airway epithelial cells from apoptotic cell death via activation of the MAP kinases JNK, p38, and ERK1/2, but not via activation of the Akt pathway (Hafner et al., 2016). Argon treatment protected cardiomyocytes from hypoxic injury mediated by an increase in the expression of heat shock protein HSP27, superoxide dismutase 2 (SOD2), vascular endothelial growth factor, and inducible nitric oxide synthase. In that study, no activation was detected for either ERK1/2 or Akt (Mayer et al., 2016).
We demonstrated that argon preconditioning activates ERK1/2 and inhibits Akt phosphorylation but has no effect on Nrf-2. The mitogen-activated protein kinase ERK1/2 may be involved in both cell survival and cell death events. Studies on the protective effects of argon have already demonstrated the activation of ERK1/2 (Sawe et al., 2008). Somewhat surprising, however, is the reduction in Akt phosphorylation by argon, since Akt is normally essential for cell survival and inhibits transcription factors that inhibit the expression of cell death genes (Zhao et al., 2016a). However, some studies have shown that activation of ERK1/2 suppresses the PI3/Akt signaling pathway, and this could explain the reduced phosphorylation (Zhou et al., 2015). Activation of the transcription factor Nrf2 upregulates the expression of numerous antioxidant enzymes under oxidative stress, thereby mediating a protective effect (Farina et al., 2021). It has been demonstrated that argon protects against hypoxic-ischemic brain injury through the activation of Nrf-2 (Zhao et al., 2016b). We were unable to confirm this in our in vitro study, as we did not observe any significant increase in Nrf-2 phosphorylation. We speculate that argon preconditioning might decrease the production of reactive oxygen species (ROS), thereby suppressing Nrf-2 activation.
Reactive oxygen radicals are generated by the mitochondrial electron transport chain, which contains at least four mitochondrial ROS-generating sites (Liu and Schubert, 2009). Rotenone blocks the mitochondrial respiratory chain, thereby mimicking the damage that occurs during cerebral ischemia. Mitochondrial dysfunction triggers an oxidative stress response that leads to massive ROS production, with consequent activation of mitochondrial apoptotic signaling pathways and aggravation of ischemic cell death (Bakthavachalam and Shanmugam, 2017).
Among the pathways involved in mitochondrial apoptosis are effector caspase 3 and the Bcl-2 family proteins (Kluck et al., 1997; Li et al., 1997). In the present study, we demonstrated that argon preconditioning reduced the expression of the caspase-3 cleavage product and the pro-apoptotic Bax. The fact that the effect was partially abolished by OxPAPC suggests that preconditioning already provides most of the protection against the development of mitochondrial apoptosis. In accordance with this study, Spaggiari et al. (2013) demonstrated that in vitro treatment of human sarcoma cells with argon protects against mitochondrial apoptosis after damage with staurosporine. In a model of neonatal asphyxia in rats, argon had a neuroprotective effect, showing increased expression of Bcl-2 and a reduction of Bax in brain tissue (Zhuang et al., 2012).
The expression of heat shock proteins is a natural response to cellular stress and occurs in neurons in the context of stroke, where it plays an important role in cell survival (Sharp et al., 1999; O’Sullivan et al., 2007). One stress response protein, HO-1, can be induced by a variety of factors, such as ROS, NF-κB, and other transcription factors (Sharp et al., 2013). While HSP-70 and HO-1 are thought to have protective functions, we demonstrated that argon preconditioning reduced the expression of heat shock proteins. Interestingly, the effect could not be abolished with OxPAPC. Argon showed a protective effect in an in vivo rat retinal ischemia-reperfusion injury model by inhibiting HSP-70, HSP-90, and HO-1, and this effect could be abolished by the ERK1/2 inhibitor PD98059 (Ulbrich et al., 2015b).
In a study that investigated the wound healing process in diabetic wounds in mice, argon was shown to mediate a pro-survival effect and lead to increased expression of the cytoprotective mediator Bcl-2. However, in contrast to this study, argon increased the expression of HO-1 (Ning et al., 2019).
The pro-inflammatory chemokine interleukin-8 is produced and released during cell stress, including in hypoxic conditions. It also acts as a chemotactic agent for polymorphonuclear leukocytes in ischemic stroke, causing inflammation and edema, and has been suggested as a prognostic marker (Hoffmann et al., 2002; Kozak et al., 2017; Zhang et al., 2021). Argon significantly reduces IL-8 during preconditioning in retinal ischemia-reperfusion injury (Ulbrich et al., 2016), and IL-8 presumably contributes significantly to the protective effect. Since OxPAPC abrogates the argon-mediated reduction of MAPK ERK1/2, mitochondrial apoptosis, and IL-8, a mechanism of action via this axis is conceivable.
This study has some limitations to consider. One is that we used the immortalized cell line SH-SY5Y instead of primary neuronal cells. While investigations with SH-SY5Y cells are well established and these cells are widely used in Parkinson´s disease research, cells in culture certainly do not share the same characteristics as cells in tissue. We also chose a well-known in vitro model in which human neuroblastoma cells were injured with rotenone. Rotenone blocks the respiratory chain in mitochondria, thereby simulating the effect of an insufficient oxygen supply to brain tissue, as occurs in stroke or cerebral hemorrhage. Nevertheless, this in vitro model has only a limited ability to reproduce the pathology of acute cerebral ischemia.
Conclusion
In this study, we investigated the preconditioning effect of argon on human neuroblastoma cells after rotenone injury. Argon protects against apoptosis and modulates the cell response to stress via TLRs 2 and 4. Argon treatment also affects the expression of apoptotic and inflammatory signaling proteins, the heat shock response, and the expression of IL-8. OxPAPC attenuates the effect on MAPK ERK1/2, mitochondrial apoptosis, and IL-8, suggesting a possible mechanism of argon via the TLRs. For this reason, argon could be interesting for the treatment of a variety of diseases, such as stroke.
Acknowledgments:
The authors thank Heide Marniga (technical assistant) for the support.
Footnotes
Funding: This work was financially supported by the Department of Anesthesiology and Critical Care, Medical Center – University of Freiburg, Germany. The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding program Open Access Publishing.
Conflicts of interest: There are no competing interests.
C-Editors: Zhao M, Liu WJ; S-Editor: Li CH; L-Editors: Li CH, Song LP; T-Editor: Jia Y
References
- 1.Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. doi: 10.1007/3-540-32636-7_1. [DOI] [PubMed] [Google Scholar]
- 2.Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 3.Auriel E, Bornstein NM. Neuroprotection in acute ischemic stroke--current status. J Cell Mol Med. 2010;14:2200–2202. doi: 10.1111/j.1582-4934.2010.01135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakthavachalam P, Shanmugam PST. Mitochondrial dysfunction - Silent killer in cerebral ischemia. J Neurol Sci. 2017;375:417–423. doi: 10.1016/j.jns.2017.02.043. [DOI] [PubMed] [Google Scholar]
- 5.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2010;12:1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brucken A, Cizen A, Fera C, Meinhardt A, Weis J, Nolte K, Rossaint R, Pufe T, Marx G, Fries M. Argon reduces neurohistopathological damage and preserves functional recovery after cardiac arrest in rats. Br J Anaesth 110 Suppl. 2013;1:i106–112. doi: 10.1093/bja/aes509. [DOI] [PubMed] [Google Scholar]
- 7.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Fahlenkamp AV, Rossaint R, Haase H, Al Kassam H, Ryang YM, Beyer C, Coburn M. The noble gas argon modifies extracellular signal-regulated kinase 1/2 signaling in neurons and glial cells. Eur J Pharmacol. 2012;674:104–111. doi: 10.1016/j.ejphar.2011.10.045. [DOI] [PubMed] [Google Scholar]
- 9.Farina M, Vieira LE, Buttari B, Profumo E, Saso L. The Nrf2 pathway in ischemic stroke:a review. Molecules. 2021;26:5001. doi: 10.3390/molecules26165001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fumagalli F, Olivari D, Boccardo A, De Giorgio D, Affatato R, Ceriani S, Bariselli S, Sala G, Cucino A, Zani D, Novelli D, Babini G, Magliocca A, Russo I, Staszewsky L, Salio M, Lucchetti J, Maisano AM, Fiordaliso F, Furlan R, et al. Ventilation with argon improves survival with good neurological recovery after prolonged untreated cardiac arrest in pigs. J Am Heart Assoc. 2020;9:e016494. doi: 10.1161/JAHA.120.016494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goebel U, Scheid S, Spassov S, Schallner N, Wollborn J, Buerkle H, Ulbrich F. Argon reduces microglial activation and inflammatory cytokine expression in retinal ischemia/reperfusion injury. Neural Regen Res. 2021;16:192–198. doi: 10.4103/1673-5374.290098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hafner C, Qi H, Soto-Gonzalez L, Doerr K, Ullrich R, Tretter EV, Markstaller K, Klein KU. Argon preconditioning protects airway epithelial cells against hydrogen peroxide-induced oxidative stress. Eur Surg Res. 2016;57:252–262. doi: 10.1159/000448682. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- 14.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 15.Jawad N, Rizvi M, Gu J, Adeyi O, Tao G, Maze M, Ma D. Neuroprotection (and lack of neuroprotection) afforded by a series of noble gases in an in vitro model of neuronal injury. Neurosci Lett. 2009;460:232–236. doi: 10.1016/j.neulet.2009.05.069. [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Kiss A, Shu H, Hamza O, Santer D, Tretter EV, Yao S, Markstaller K, Hallström S, Podesser BK, Klein KU. Argon preconditioning enhances postischaemic cardiac functional recovery following cardioplegic arrest and global cold ischaemia. Eur J Cardiothorac Surg. 2018;54:539–546. doi: 10.1093/ejcts/ezy104. [DOI] [PubMed] [Google Scholar]
- 18.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria:a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 19.Kozak HH, Uğuz F, Kılınçİ, Uca AU, Serhat Tokgöz O, Akpınar Z, Özer N. Delirium in patients with acute ischemic stroke admitted to the non-intensive stroke unit:Incidence and association between clinical features and inflammatory markers. Neurol Neurochir Pol. 2017;51:38–44. doi: 10.1016/j.pjnns.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Leitner GR, Wenzel TJ, Marshall N, Gates EJ, Klegeris A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin Ther Targets. 2019;23:865–882. doi: 10.1080/14728222.2019.1676416. [DOI] [PubMed] [Google Scholar]
- 21.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Schubert DR. The specificity of neuroprotection by antioxidants. J Biomed Sci. 2009;16:98. doi: 10.1186/1423-0127-16-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 24.Ma S, Chu D, Li L, Creed JA, Ryang YM, Sheng H, Yang W, Warner DS, Turner DA, Hoffmann U. Argon inhalation for 24 hours after onset of permanent focal cerebral ischemia in rats provides neuroprotection and improves neurologic outcome. Crit Care Med. 2019;47:e693–699. doi: 10.1097/CCM.0000000000003809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer B, Soppert J, Kraemer S, Schemmel S, Beckers C, Bleilevens C, Rossaint R, Coburn M, Goetzenich A, Stoppe C. Argon induces protective effects in cardiomyocytes during the second window of preconditioning. Int J Mol Sci. 2016;17:1159. doi: 10.3390/ijms17071159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moro MA, Almeida A, Bolanos JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005;39:1291–1304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 27.Ning J, Zhao H, Chen B, Mi EZ, Yang Z, Qing W, Lam KWJ, Yi B, Chen Q, Gu J, Ichim T, Bogin V, Lu K, Ma D. Argon mitigates impaired wound healing process and enhances wound healing in vitro and in vivo. Theranostics. 2019;9:477–490. doi: 10.7150/thno.29361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Sullivan JC, Yao XL, Alam H, McCabe JT. Diazoxide, as a postconditioning and delayed preconditioning trigger, increases HSP25 and HSP70 in the central nervous system following combined cerebral stroke and hemorrhagic shock. J Neurotrauma. 2007;24:532–546. doi: 10.1089/neu.2006.0128. [DOI] [PubMed] [Google Scholar]
- 29.Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ristagno G, Fumagalli F, Russo I, Tantillo S, Zani DD, Locatelli V, De Maglie M, Novelli D, Staszewsky L, Vago T, Belloli A, Di Giancamillo M, Fries M, Masson S, Scanziani E, Latini R. Postresuscitation treatment with argon improves early neurological recovery in a porcine model of cardiac arrest. Shock. 2014;41:72–78. doi: 10.1097/SHK.0000000000000049. [DOI] [PubMed] [Google Scholar]
- 31.Ryang YM, Fahlenkamp AV, Rossaint R, Wesp D, Loetscher PD, Beyer C, Coburn M. Neuroprotective effects of argon in an in vivo model of transient middle cerebral artery occlusion in rats. Crit Care Med. 2011;39:1448–1453. doi: 10.1097/CCM.0b013e31821209be. [DOI] [PubMed] [Google Scholar]
- 32.Sawe N, Steinberg G, Zhao H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res. 2008;86:1659–1669. doi: 10.1002/jnr.21604. [DOI] [PubMed] [Google Scholar]
- 33.Scheid S, Goeller M, Baar W, Wollborn J, Buerkle H, Schlunck G, Lagrèze W, Goebel U, Ulbrich F. Hydrogen sulfide reduces ischemia and reperfusion injury in neuronal cells in a dose- and time-dependent manner. Int J Mol Sci. 2021;22:10099. doi: 10.3390/ijms221810099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ:25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- 36.Sharp FR, Zhan X, Liu DZ. Heat shock proteins in the brain:role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl Stroke Res. 2013;4:685–692. doi: 10.1007/s12975-013-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spaggiari S, Kepp O, Rello-Varona S, Chaba K, Adjemian S, Pype J, Galluzzi L, Lemaire M, Kroemer G. Antiapoptotic activity of argon and xenon. Cell Cycle. 2013;12:2636–2642. doi: 10.4161/cc.25650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ulbrich F, Schallner N, Coburn M, Loop T, Lagreze WA, Biermann J, Goebel U. Argon inhalation attenuates retinal apoptosis after ischemia/reperfusion injury in a time- and dose-dependent manner in rats. PLoS One. 2014;9:e115984. doi: 10.1371/journal.pone.0115984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ulbrich F, Kaufmann K, Roesslein M, Wellner F, Auwarter V, Kempf J, Loop T, Buerkle H, Goebel U. Argon mediates anti-apoptotic signaling and neuroprotection via inhibition of Toll-like receptor 2 and 4. PLoS One. 2015a;10:e0143887. doi: 10.1371/journal.pone.0143887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ulbrich F, Kaufmann KB, Coburn M, Lagreze WA, Roesslein M, Biermann J, Buerkle H, Loop T, Goebel U. Neuroprotective effects of argon are mediated via an ERK-1/2 dependent regulation of heme-oxygenase-1 in retinal ganglion cells. J Neurochem. 2015b;134:717–727. doi: 10.1111/jnc.13115. [DOI] [PubMed] [Google Scholar]
- 42.Ulbrich F, Goebel U. The molecular pathway of argon-mediated neuroprotection. Int J Mol Sci. 2016;17:1816. doi: 10.3390/ijms17111816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ulbrich F, Lerach T, Biermann J, Kaufmann KB, Lagreze WA, Buerkle H, Loop T, Goebel U. Argon mediates protection by interleukin-8 suppression via a TLR2/TLR4/STAT3/NF-κB pathway in a model of apoptosis in neuroblastoma cells in vitro and following ischemia-reperfusion injury in rat retina in vivo. J Neurochem. 2016;138:859–873. doi: 10.1111/jnc.13662. [DOI] [PubMed] [Google Scholar]
- 44.Wang X, Sun Y, Yang H, Lu Y, Li L. Oxidized low-density lipoprotein induces apoptosis in cultured neonatal rat cardiomyocytes by modulating the TLR4/NF-κB pathway. Sci Rep. 2016;6:27866. doi: 10.1038/srep27866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–1402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Ge P, Zhu Y. TLR2 and TLR4 in the brain injury caused by cerebral ischemia and reperfusion. Mediators Inflamm. 2013;2013:124614. doi: 10.1155/2013/124614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen Y, He J, Xue X, Qiu J, Xu Y, Tang Z, Qian H, Qin L, Yang X. β-arrestin2 inhibits apoptosis and liver inflamation induced by ischemia-reperfusion in mice via AKT and TLR4 Pathway. Arch Med Res. 2019;50:413–422. doi: 10.1016/j.arcmed.2019.10.012. [DOI] [PubMed] [Google Scholar]
- 48.Yang L, Tao LY, Chen XP. Roles of NF-kappaB in central nervous system damage and repair. Neurosci Bull. 2007;23:307–313. doi: 10.1007/s12264-007-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–477. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Xu D, Zhang T, Hou W, Yixi L. Correlation between interleukin-6, interleukin-8, and modified early warning score of patients with acute ischemic stroke and their condition and prognosis. Ann Palliat Med. 2021;10:148–155. doi: 10.21037/apm-20-2200. [DOI] [PubMed] [Google Scholar]
- 51.Zhao EY, Efendizade A, Cai L, Ding Y. The role of Akt (protein kinase B) and protein kinase C in ischemia-reperfusion injury. Neurol Res. 2016a;38:301–308. doi: 10.1080/01616412.2015.1133024. [DOI] [PubMed] [Google Scholar]
- 52.Zhao H, Mitchell S, Ciechanowicz S, Savage S, Wang T, Ji X, Ma D. Argon protects against hypoxic-ischemic brain injury in neonatal rats through activation of nuclear factor (erythroid-derived 2)-like 2. Oncotarget. 2016b;7:25640–25651. doi: 10.18632/oncotarget.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao H, Mitchell S, Koumpa S, Cui YT, Lian Q, Hagberg H, Johnson MR, Takata M, Ma D. Heme oxygenase-1 mediates neuroprotection conferred by argon in combination with hypothermia in neonatal hypoxia-ischemia brain injury. Anesthesiology. 2016c;125:180–192. doi: 10.1097/ALN.0000000000001128. [DOI] [PubMed] [Google Scholar]
- 54.Zhou J, Du T, Li B, Rong Y, Verkhratsky A, Peng L. Crosstalk between MAPK/ERK and PI3K/AKT signal pathways during brain ischemia/reperfusion. ASN Neuro. 2015;7:1759091415602463. doi: 10.1177/1759091415602463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhuang L, Yang T, Zhao H, Fidalgo AR, Vizcaychipi MP, Sanders RD, Yu B, Takata M, Johnson MR, Ma D. The protective profile of argon, helium, and xenon in a model of neonatal asphyxia in rats. Crit Care Med. 2012;40:1724–1730. doi: 10.1097/CCM.0b013e3182452164. [DOI] [PubMed] [Google Scholar]