

Abstract
2022 saw a 25% drop in new drug approvals and a less predictable regulator.

Brain light / Alamy Stock Photo
New drug approvals in the US were down in 2022, by about 25% — to the lowest level since 2016. Innovation hasn’t slowed, though: new modalities such as antibody–drug conjugates, bispecific proteins, and cell and gene therapies accounted for about a third of 2022’s approvals, helping push biologics approvals ahead of small molecules for the first time (Fig. 1 and Table 1).
Fig. 1. Historical FDA approvals.

Approvals were down after a string of strong years. *Figures for 2022 as of 14 December.
Table 1.
Selected FDA approvals 2022
| Drug name | Generic name | Indication | Molecule |
|---|---|---|---|
| Adstiladrin | Nadofaragene firadenovec-vncg | Bladder cancer | Adenovirus-5 vector-based gene therapy, containing an interferon-α2b transgene with Syn3, an excipient to improve transfection |
| Carvykti | Ciltacabtagene autoleucel | Multiple myeloma | CAR-T created ex vivo using a lentiviral vector to deliver two BCMA-targeting scFvs, a 4-1BB co-stimulatory domain and a CD3ζ signaling cytoplasmic domain |
| Daxxify | DaxibotulinumtoxinA-lanm | Wrinkles | Purified botulinum toxin type A molecule with TransMTS peptide |
| Elaherea | Mirvetuximab soravtansine-gynx | Ovarian cancer | Antibody–drug conjugate consisting of the cytotoxic maytansinoid DM4 covalently linked to the humanized mAb M9346A, which binds folate receptor 1 |
| Enjaymo | Sutimlimab-jome | Autoimmune hemolytic anemia | Humanized IgG4 mAb with subnanomolar affinity for total complement component 1 |
| Hemgenix | Etranacogene dezaparvovec-drlb | Hemophilia | AAV5 vector with the FIX-Padua mutant |
| Imjudo | Tremelimumab-actl | Hepatocellular cancer, NSCLC | CTLA-4-blocking human IgG2 mAb |
| Kimmtrak | Tebentafusp-tebn | Uveal melanoma | Bispecific fusion protein comprising a high-affinity TCR specific to a peptide from the gp100 antigen, fused to an anti-CD3 single-chain antibody fragment |
| Lunsumio | Mosunetuzumab-axgb | Follicular lymphoma | Full-length bispecific antibody that targets CD20 on B cells and CD3 on T cells. |
| Opdualag | Nivolumab relatlimab-rmbw | Melanoma | Fixed dose combination of two checkpoint inhibitor IgG4ҡ mAbs: relatlimab, which targets LAG-3, and nivolumab, an approved mAb that targets PD-1 |
| Rolvedon (Rolontis) | Eflapegrastim-xnst | Neutropenia, leukopenia | Long-acting peptide G-CSF receptor agonist produced by covalent coupling of a human G-CSF analog (18.6 kDa) and an Fc fragment of human IgG4 (49.8 kDa) via a 3.4-kDa polyethylene glycol linker |
| Skyrizi | Risankizumab-rzaa | Crohn’s disease | Humanized IgG1 mAb that selectively binds p19 subunit of human IL-23 cytokine and inhibits its interaction with IL-23 receptor |
| Skysona | Elivaldogene autotemcel | Adrenoleukodystrophy | Autologous CD34+ hematopoietic stem cells transduced with lentiviral vector, Lenti-D, encoding the human ABCD1 cDNA. |
| Spevigo | Spesolimab-sbzo | Psoriasis | Humanized IgG1 anti-IL-36 receptor mAb |
| Spikevax | COVID-19 vaccine, mRNA | COVID-19 | mRNA vaccine against coronavirus SARS-CoV-2 encoding a pre-fusion stabilized form of the spike protein |
| Tecvaylia | Teclistamab-cqyv | Multiple myeloma | Bispecific BCMA-directed CD3 T cell engager; a humanized immunoglobulin G4-PAA antibody |
| Tzield | Teplizumab-mzwv | Type 2 diabetes | Humanized, non-Fc-receptor-binding mAb against the CD3ε chain expressed on mature T lymphocytes |
| Vabysmo | Faricimab-svoa | Wet age-related macular degeneration | Humanized bispecific IgG1 mAb that binds VEGF-A on one arm and angiopoietin-2 on the other |
| Xenpozyme | Olipudase alfa-rpcp | Acid sphingomyelinase deficiency | Recombinant human acid sphingomyelinase |
| Zynteglo | Betibeglogene autotemcel | β-thalassemia | Autologous CD34+ cells transduced with human β-(A-T87Q)-globin gene under control of β-globin enhancer and locus control region |
| Notable new molecular entities | |||
| Terlivaz | Terlipressin | Hepatorenal syndrome | 1-Triglycyl-8-lysine-vasopressin, a 12-amino-acid peptide |
| Amvuttra | Vutrisiran | hATTR amyloidosis | Double-stranded 2′-fluoro,2′-methoxy-modified phosphorothioate siRNA conjugated to GalNAc |
| Vtama | Tapinarof | Psoriasis | Aryl hydrocarbon receptor agonist |
| Mounjaro | Tirzepatide | Diabetes mellitus type 2 | Peptide receptor agonist of GIP and GLP-1; a 39-amino-acid modified peptide based on the GIP sequence, containing the non-canonical amino acid aminoisobutyric acid in positions 2 and 13, a C-terminal amide, and a lysine at position 20 that is attached to 1,20-eicosanedioic acid via a linker |
aAccelerated Approval. CAR-T, chimeric antigen receptor T cell; scFv, single-chain variable fragment; BCMA, B cell maturation antigen, mAb, monoclonal antibody; IgG, immunoglobulin G; PAA, proline, alanine, alanine; mAb, monoclonal antibody; CTLA-4, cytotoxic T lymphocyte-associated antigen 4; TCR, T cell receptor; G-CSF, granulocyte colony-stimulating factor; Fc, immunoglobulin constant fragment; IL, interleukin; LAG-3, lymphocyte activating gene 3; ABCD1, ATP binding cassette subfamily D member 1; VEGF-A, vascular endothelial growth factor-A; Ang-2, angiopoietin-2; hATTR, hereditary transthyretin-mediated; GalNAc, N-acetylgalactosamine; GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon-like peptide-1.
The US Food and Drug Administration (FDA) toughened its stand on Accelerated Approvals and submissions based on China-only data. But it also waved through some drugs despite negative votes or previous rejections, published new plans to speed up access to treatments for rare neurodegenerative diseases, and continues to expand faster approval options (Box 1). The agency also appears to be loosening the reins on cheaper biosimilars.
Box 1 FDA flip-flops
Two reversals captured the mood of uncertainty in 2022. Amylyx Pharmaceuticals’ neurodegenerative disease drug Relyvrio (sodium phenylbutyrate and taurursodiol) was approved in September 2022 for patients with amyotrophic lateral sclerosis (ALS) after a negative advisory committee vote in March 2022 and a positive one, from the same committee, in September. In between the votes, Amylyx submitted new analyses, and regulators reconsidered on the basis of the unmet need and disease severity. The FDA also, in June 2022, launched a five-year action plan for advancing development of treatments for rare neurodegenerative diseases like ALS, including a public–private partnership co-launched with the National Institutes of Health, along with efforts to improve disease characterization, discover biomarkers and enhance clinical trial access and infrastructure. The compounds in Relyvrio work together to help prevent motor neuron damage and death by blocking stress signals within cells’ mitochondria and endoplasmic reticulum.
The regulatory path for Ardelyx’s tenapanor has also been winding. The drug was approved for irritable bowel syndrome in 2019, as Ibsrela. In July 2021, the company received a complete response letter for its application in a second indication, to control serum phosphorus in adults with chronic kidney disease on dialysis. The company appealed, and by November 2022, tenapanor had received a positive vote from the cardiovascular and renal drugs advisory committee. The drug inhibits the sodium hydrogen exchanger 3 (NHE3) in the gut to reduce phosphate absorption through the paracellular pathway. Ardelyx claims almost 80% of patients on dialysis need this treatment as they can’t achieve target serum phosphatase, and it has carried out two phase 3 trials in 1,200 patients. The company awaits the result of the appeal. If approved in this indication, the drug would be taken as a 30-mg tablet twice daily, rather than the 50-mg twice daily regimen in irritable bowel syndrome.
“Ferociously unpredictable” is how one clinician-oncologist summed up 2022’s FDA — a sentiment shared by many biopharma executives. Clinical holds were abundant at the start of 2022, with 15 full or partial holds reported in the first 8 weeks, according to BioMedTracker. Yet by year’s end, the total (44) was short of 2021’s full year count (49). Complete response letters (rejections) were trending similarly to 2021 (35 versus 36) and slightly below the 40 reported in 2020, according to BioMedTracker.
Widening the lens to the past decade makes 2022’s approval tally look more like reversion to the norm. Between 2012 and 2021, FDA approved on average 44 drugs per year, skewed upwards by the 50+ annual tally between 2018 and 2021 (Fig. 1). Overall, it was a golden decade, with 445 new biologics and small molecule medicines, up 76% from the previous ten years. Expedited approvals — including Fast Track, Priority Review and Accelerated Approval, as well as Breakthrough Therapy or Orphan Drug designations — have also grown to about 70% of approvals in 2021 and 2022 (Table 2).
Table 2.
Drug approvals with expedited review in 2022
| Breakthrough | Fast Track | Orphan | Rare disease | Accelerated Approval | ||
|---|---|---|---|---|---|---|
| Amvuttra (vutrisiran) | Hereditary transthyretin (hATTR) amyloidosis with polyneuropathy | ■ | ■ | ■ | ||
| Camzyos (mavacamten) | Hypertrophic cardiomyopathy | ■ | ■ | |||
| Carvykti (ciltacabtagene autoleucel) | Multiple myeloma | ■ | ■ | ■ | ||
| Cibinqo (abrocitinib) | Atopic dermatitis (eczema) | ■ | ||||
| Elahere (mirvetuximab soravtansine-gynx) | Ovarian cancer | ■ | ■ | ■ | ■ | |
| Enjaymo (sutimlimab-jome) | Autoimmune hemolytic anemia | ■ | ■ | ■ | ||
| Hemgenix (etranacogene dezaparvovec-drlb) | Hemophilia B | ■ | ■ | ■ | ||
| Imjudo (tremelimumab-actl) | Hepatocellular cancer (including secondary metastases) | ■ | ■ | |||
| Kimmtrak (tebentafusp-tebn) | Uveal melanoma | ■ | ■ | ■ | ||
| Krazati (adagrasib) | Multiple myeloma | ■ | ||||
| Lunsumio (mosunetuzumab-axgb) | Follicular lymphoma | ■ | ■ | |||
| Lytgobi (futibatinib) | Biliary tract cancer | ■ | ■ | ■ | ■ | |
| Pluvicto (lutetium (177Lu) vipivotide tetraxetan) | Prostate cancer | ■ | ||||
| Pyrukynd (mitapivat) | Pyruvate kinase deficiency | ■ | ■ | ■ | ||
| Rebyota (fecal microbiota, live-jslm) | C. difficile-associated diarrhea | ■ | ■ | ■ | ||
| Rezlidhia (olutasidenib) | Acute myeloid leukemia | ■ | ■ | |||
| Sezaby (phenobarbital sodium) | Seizure disorders | ■ | ||||
| Skysona (elivaldogene autotemcel) | Adrenoleukodystrophy | ■ | ■ | ■ | ■ | |
| Spevigo (spesolimab-sbzo) | Psoriasis | ■ | ■ | |||
| Sunlenca (lenacapavir) | HIV/AIDS | ■ | ■ | |||
| Takecab/Voquezna (vonoprazan fumarate) | Helicobacter pylori infection | ■ | ||||
| Tecvayli (teclistamab-cqyv) | Multiple myeloma | ■ | ■ | ■ | ■ | |
| Terlivaz (terlipressin) | Hepatorenal syndrome | ■ | ■ | ■ | ||
| Tzield (teplizumab-mzwv) | Type 1 diabetes mellitus | ■ | ■ | |||
| Vivjoa (oteseconazole) | Non-systemic fungal infections | ■ | ||||
| Vonjo (pacritinib) | Myelofibrosis | ■ | ■ | ■ | ■ | |
| Xenpozyme (olipudase alfa) | Niemann–Pick disease | ■ | ■ | ■ | ||
| Ztalmy (ganaxolone) | Seizure disorders | ■ | ■ | |||
| Zynteglo (betibeglogene autotemcel) | β-thalassemia | ■ | ■ | ■ | ■ |
But industry faced a new source of uncertainty in 2022: the Inflation Reduction Act. This law, signed in August, introduces US drug price negotiations and caps on annual price rises, with harsh penalties for flouters. Its provisions are already affecting drug development and regulatory submission decisions, with several unintended consequences (Box 2).
Box 2 Inflation Reduction Act introduces US drug price negotiations
The Inflation Reduction Act, signed into US law in August 2022, sets a path to price negotiations for some of the most costly Medicare drugs. It also curbs annual above-inflation price increases for all Medicare treatments from 2023. The Act is expected to save the government $100 billion over ten years and has provoked strong pushback from industry, which says it will dampen innovation and reduce the number of drugs in development.
Price negotiations will begin in 2026 for small molecules, and two years later for biologics. Yet they apply only to the (initially ten) highest-cost Medicare drugs without generic or biosimilar competition. One controversial aspect of the law is that price negotiations won’t kick in until 13 years after approval for eligible biologics, but only 9 years after approval for small molecules, potentially dis-incentivizing small molecule R&D. New drugs like Novartis’s small interfering RNA cholesterol drug Leqvio (inclisiran), categorized as a small molecule, may come up against pricing curbs sooner than anticipated. Skewing development of medicines further toward expensive large molecules — if that occurs — would likely raise the overall bill for medicines — the opposite of what the law intended.
Industry representatives say the act is already influencing pipeline prioritization decisions. For example, Alnylam says the provision to exempt single-indication orphan drugs from price negotiations led it to delay development of a second orphan indication for its rare-disease treatment Amvuttra while it evaluates the potential impact of the law.
Another unintended consequence of the IRA is likely to be near-term increases in some list prices as companies seek to compensate for lower-than-hoped annual hikes down the line. Biosimilars and generics may also be negatively affected, though the law’s impact on generics remains unclear. Deals between innovator and biosimilar firms may occur, seeking to game the part of the law that allows biologic price negotiations to be delayed for up to two years if biosimilar competition is imminent.
Most policy experts expect legal challenges to the IRA, and many questions remain about its implementation. But it isn’t going away. Neither US political party wants to be seen favoring pharma over patient access to medicines, and other parts of the Act, like a $2,000 annual cap on out-of-pocket costs for Medicare patients, are broadly popular with voters.
Accelerated Approval clampdown
The Accelerated Approvals controversy continued in 2022, amid concerns that the FDA had tilted too far toward early access at the expense of robust evidence. Accelerated Approvals are granted on the basis of surrogate markers and limited clinical data, on condition that sponsors follow up with confirmatory trials. Many don’t. A study published in September 2022 by the US Department of Health and Human Services found that almost 40% of sponsors of the 278 drugs approved between 1992 and 2021 hadn’t completed them.
Biogen’s Alzheimer’s drug Aduhelm (aducanumab) ignited the Accelerated Approval debate in 2021. Covis Pharma’s preterm drug Makena (hydroxyprogesterone caproate injection) fanned the flames in 2022. Makena received Accelerated Approval in 2011 and has since failed a confirmatory study and twice been voted off the market by FDA advisory committees. The second vote to pull the drug was in October 2022. The FDA had yet to make its final decision as Nature Biotechnology went to press.
Until recently, FDA didn’t have sufficient authority — or will — to punish sponsors who failed to generate the required data. Once a drug is on the market and some patients appear to benefit from it, withdrawal can be lengthy and difficult. It also becomes harder to recruit patients into confirmatory trials with a placebo or comparator arm, especially in rare and/or severe conditions with limited alternatives.
Legislation to sharpen FDA’s claws failed to get into the new five-year User Fee Reauthorization Act, signed at the very last minute on 30 September 2022. But modest reforms to Accelerated Approval were squeezed into a broader year-end spending bill. These allow the FDA to pull an approval if confirmatory studies fall short and to require that a confirmatory trial be underway before Accelerated Approval is granted. They also require sponsors to submit six-monthly trial progress reports.
Several events in 2022 showed that the FDA is clamping down anyway. Seven new drugs were granted Accelerated Approval — the 2022 count includes Mirati Therapeutics’ adagrasib (Krazati) for KRAS G12C-mutated NSCLC, approved 12 December — against nine the year before. One of 2021’s approvals, TG Therapeutics’ lymphoma treatment Ukoniq (umbralisib), was withdrawn in June 2022, after less than 18 months, owing to safety concerns. One of 2020’s Accelerated Approval cohort, GlaxoSmithKline’s antibody–drug conjugate Blenrep (belantamab mafodotin-blmf), was withdrawn in November 2022, two weeks after reporting a failed phase 3 confirmatory trial in relapsed or refractory multiple myeloma. (An early trial showed a third of patients responding to the drug, but it did not appear to extend progression-free survival in the confirmatory trial. GSK says it will continue the trial program and try to find a way forward.) In late November, Basel, Switzerland-based Roche pulled Tecentriq (atezolizumab) for urothelial carcinoma, a type of bladder cancer, six years after its first approval.
The FDA’s tightening appears to be deterring Accelerated Approval submissions. ADC Therapeutics said it wouldn’t pursue one for its Hodgkin’s lymphoma drug camidanlumab tesirine because the FDA had required a phase 3 program be planned and underway before the review started. Section 506(c) of the Federal Food, Drug and Cosmetic Act allows the FDA to make such a request, but the wording of the procedural guidance leaves wiggle room, which FDA officials wanted to limit. The new legislation helps with that. Commissioner Robert Califf spoke at The Organisation for Professionals in Regulatory Affairs annual symposium in October about industry’s failure to provide confirmatory evidence and the need for reform “in one way or another.” Richard Pazdur, head of the FDA’s Oncology Center of Excellence, reported in a perspective paper in the New England Journal of Medicine that having a trial underway at approval shortens the time to withdrawal (if needed) by three-and-a-half years on average.
Titles on an FDA list of planned procedural guidances for 2022 included “Civil monetary penalties for failure to meet accelerated post-marketing requirements” (not yet published) and “Considerations for rescinding breakthrough therapy designations” (draft guidance published in June 2022).
An FDA that holds drug sponsors to account for confirmatory trial data and timelines is good for patients and payers. It also makes sense “for the long-term health of the industry,” tweeted Atlas Venture partner Bruce Booth in response to GSK’s news. (The European Medicines Agency’s conditional approval pathway mandates follow-up studies within defined timelines, with the threat of regulatory action if sponsors fail to comply.)
Accelerated Approval reforms aside, expedited pathways are here to stay. The FDA is pursuing several programs to expand and refine speedy reviews, including through new regulator-sponsored meetings, the Oncology Center of Excellence’s Project Confirm (designed to bring greater transparency and discussion to Accelerated Approvals in cancer) and an expansion of the Split Real Time Application Review (STAR) pilot beyond oncology. Other commitments (spelled out in the 2023–2027 User Fee Reauthorization Act goals) include advancing novel efficacy endpoint development for rare disease drugs and continuing the use of real-world evidence to support labeling or postapproval study requirements.
Biologics outpace small molecules
By year-end 2022, the number of biologic approvals narrowly outpaced that of small-molecule new molecular entities (NMEs), a landmark in biologics’ steady rise since the end of the twentieth century. Like small molecules, they’re being combined and finding new indications: AstraZeneca’s CTLA-4 inhibitor Imjudo (tremelimumab-actl) was approved in October 2022 for unresectable liver cancer and, the following month, in combination with Imfinzi (durvalumab) and chemotherapy in patients with metastatic non-small-cell lung cancer (NSCLC) not harboring epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) mutations. Merck’s anti-PD-1 (programmed cell death protein-1) behemoth Keytruda (pembrolizumab) in 2022 added advanced endometrial cancer to its 30+ tally of approved indications, with another lined up for 2023 after an overall survival hit in a phase 3 trial in patients with human epidermal growth factor receptor 2 (HER2)-negative gastroesophageal junction adenocarcinoma. The company is running over a dozen trials of Keytruda in early-stage disease.
Another force driving biologics’ growth is their evolution beyond simple antibodies, peptides or enzymes. Newer modalities including bispecific proteins, antibody–drug conjugates, and cell or gene therapies accounted for about half of biologic approvals through the end of November 2022, up from less than a third in 2021.
Oxford, UK-based ImmunoCore’s Kimmtrak (tebentafusp-tebn), a bispecific T cell engager protein approved for unresectable or metastatic uveal melanoma, was first out of the gate in January 2022. It comprises a soluble T cell receptor fused to an anti-CD3 immune effector that redirects T cells to target glycoprotein 100 (gp100)-expressing melanoma cells. Kimmtrak, the first therapy for metastatic uveal melanoma, benefited from Breakthrough designation and Real Time Oncology Review and was assessed concurrently by other health authorities, including European Medicines Agency, under Project Orbis. (It was approved in Europe in April 2022.)
Kimmtrak paved the way for other bispecifics designed to bind specific cancer and immune system targets to generate a more powerful anticancer response. Johnson & Johnson’s Tecvayli (teclistamab-cqyv), approved in October for advanced multiple myeloma, is a bispecific T cell engager antibody that latches onto T cells, via the surface CD3 receptor, and to B cell maturation antigen (BCMA) expressed on myeloma cells. The big pharma on 9 December submitted another bispecific, talquetamab, which targets CD3 and GPRC5D, a new multiple myeloma target.
In the closing weeks of 2022, the FDA approved Roche’s bispecific antibody Lunsumio (mosunetuzumab) for follicular lymphoma that has failed least two previous therapies. It targets CD20 on B cells and CD3 on T cells, redirecting the latter to engage and destroy the B cells. Lunsumio received conditional approval in Europe in June. Another Roche bispecific, Vabysmo (faricimab-svoa) was approved in 2022 for wet age-related macular degeneration and diabetic macular edema. Vabysmo targets the angiopoietin-2 and vascular endothelial growth factor-A pathways that contribute to vision loss in these conditions.
Bispecifics may provide a more convenient alternative to CAR-T cell therapies, whose complex, expensive manufacture and administration have limited their reach and commercial fortunes. Earlier in 2022, Johnson & Johnson and partner Legend Biotech won approval for Carvykti (ciltacabtagene autoleucel), a BCMA-directed CAR-T cell therapy, in the same indication as Tecvayli: adults having received at least four lines of previous therapy, including a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody.
Carvykti became the sixth CAR-T cell therapy in the United States (Table 3). The CAR-T cell R&D pipeline is huge, including sponsors seeking more convenient allogeneic (off the shelf) varieties and ‘point of care’ manufacturing facilities to expand access, as well as those seeking to address solid tumors. In October, Europe’s Committee for Medicinal Products for Human Use recommended approval of what could be the first ever allogeneic T cell therapy, Atara Biotherapeutics’ tabelecleucel (Tab-cel) for Epstein–Barr virus (EBV)-positive post-transplant lymphoproliferative disease. Tab-cel uses EBV-directed T cells from healthy donors. A US biologic license application has been delayed owing to comparability issues between commercial and pivotal clinical trial product.
Table 3.
FDA-approved CAR-T therapies
| Drug/company | Year approved | Target | Number of approvals |
|---|---|---|---|
| Carvykti (ciltacabtagene autoleucel)/Johnson & Johnson | 2022 | B cell maturation antigen | 1 |
| Abecma (idecabtagene vicleucel)/Bristol Myers Squibb | 2021 | B cell maturation antigen | 1 |
| Breyanzi (lisocabtagene maraleucel)/Bristol Myers Squibb | 2021 | CD19 | 2 |
| Tecartus (brexucabtagene autoleucel)/Gilead Sciences | 2021 | CD19 | 2 |
| Yescarta (axicabtagene ciloleucel)/Gilead Sciences | 2017 | CD19 | 2 |
| Kymriah (tisagenlecleucel)/Novartis | 2017 | CD19 | 3 |
Another new modality arrived 30 November, when the FDA approved the first fecal microbiome product, Rebyota (fecal microbiota, live-jslm), for people with recurrent Clostridioides difficile infections following antibiotic treatment. The product is prepared from stool donated by healthy individuals, screened for pathogens, and administered as a one-time enema to restore a healthy balance of bacteria in the colon. The approval, granted to Saint-Prex, Switzerland-based Ferring Pharmaceuticals, follows several setbacks across the field and could open the gates for other similar treatments. Seres Therapeutics completed a rolling submission of its orally administered microbiome therapeutic, also for C. difficile, in September 2022. Fecal microbiota transplants — doses of untampered stool — have been used to manage this condition, but regulation and quality control are difficult.
Gene therapies fuel pricing debate
A triplet of gene and cell therapies approved in the US during 2022 underscored the continued growth of this class. Yet, with each one costing over $2 million, they also fueled the drug pricing debate.
In August, bluebird bio’s Zynteglo (betibeglogene autotemcel) became the first FDA-approved stem cell-based gene therapy for patients with β-thalassemia requiring regular transfusions. It genetically modifies patients’ bone marrow stem cells to produce functional β-globin, and helped 89% of patients in two multi-center trials to achieve transfusion independence for at least a year.
In Europe, where Zynteglo was approved in 2019, German and other payers refused to pay the approximately $1.7 million price tag (even with a pay-for-performance clause), so bluebird retreated from the continent two years later. In the United States, Zynteglo was priced at $2.8 million, with a deal to refund 80% for patients failing to maintain transfusion independence up to two years later. (The US-based Institute for Clinical and Economic Review (ICER) said $2.1 million, with some restitution depending on outcome, would represent value-for-money.)
Zynteglo became the most expensive drug ever — for about a month. Then a second bluebird gene therapy, Skysona (elivaldogene autotemcel), received Accelerated Approval to slow neurologic dysfunction in boys aged 4–17 with cerebral adrenoleukodystrophy (CALD), a rare neurological disorder. It was priced at $3 million. Skysona involves adding functional copies of the ABCD1 gene, which is mutated in CALD, to patients’ blood stem cells using a lenti-D viral vector, then re-infusing the engineered cells. These help patients break down very long chain fatty acids that build up because of the mutations. A confirmatory study was underway at the time of the approval.
Skysona faced an FDA clinical hold in 2021 due to a patient developing myelodysplastic syndrome (possibly due to the viral vector) and carries a warning for hematologic malignancy. It was approved in Europe in 2021 but withdrawn when bluebird pulled out of the region.
CSL Behring and licensee UniQure, based in Amsterdam, took the title for the most expensive drug in November with their $3.5 million price tag on Hemgenix (etranacogene dezaparvovec-drlb), the first FDA-approved gene therapy for hemophilia B. Patients with this bleeding disorder lack clotting factor IX and require regular blood transfusions. Hemgenix uses an adeno-associated virus vector to deliver a factor IX gene to eligible patients on prophylaxis therapy or at risk of hemorrhage or spontaneous bleeding.
One-time therapies like Hemgenix can lead to significant system savings if they work. The problem is that payers on the hook for up-front costs don’t always benefit from the downstream savings. ICER said Hemgenix would offer value priced up to $2.9 million, given high transfusion costs.
There are now over a dozen FDA-approved cell and gene therapies (Ferring’s gene therapy Adstiladrin (nadofaragene firadenovec-vncg) got over the line for bladder cancer in mid-December), with more coming. BioMarin’s gene therapy Roctavian (valoctocogene roxaparvovec) for severe hemophilia A was resubmitted in October 2022 after a rebuff in 2020; a decision is expected in March 2023.
The European Medicines Agency granted conditional approval to Roctavian in August 2022 — another example, along with Lunsumio and bluebird’s gene therapy duo, of Europe-first approvals for next-generation biologics. Most drugs are submitted and approved initially in the United States for commercial reasons: Europe’s tough reimbursement hurdles mean early approval doesn’t always translate into faster access. US and European regulators have historically agreed on whether to approve most new drugs and continue to cooperate.
Yet regulators everywhere need time to properly investigate and understand the tools, approaches and manufacturing requirements underpinning many new medicines. The FDA in August 2022 put the brakes on some next-generation gene editing programs, including BEAM Therapeutics’ BEAM-201, an anti-CD7 multiplex-edited allogeneic CAR-T cell program for advanced T cell acute lymphoblastic leukemia and lymphoma. BEAM’s technology allows several single bases to be edited at the same time, and the FDA wanted further information on off-target editing before allowing BEAM-201 to progress into humans. On 7 November, Verve Therapeutics’ gene editing medicine for heterozygous familial hypercholesterolemia was also held up before entering the clinic. It involves a single A-to-G base change that disrupts PCSK9 protein production, lowering low-density lipoprotein cholesterol in the blood.
These delays are usually temporary. The hold on BEAM-201 came off on 2 December. A previous hold on first-generation CRISPR players Vertex Pharmaceuticals and Zug, Switzerland-based CRISPR Therapeutics was also lifted within months.
The FDA issued four sets of cell and gene therapy guidance for industry in 2022, including draft guidance on CAR-T cell therapy development and the development of gene therapy products incorporating gene editing.
NMEs: antisense, targeted radioligands
Not all 2022’s cohort of new modalities were biologics. Alnylam Pharmaceuticals’ next-generation small interfering RNA (siRNA) drug Amvuttra (vutrisiran) in June became the fifth FDA-approved siRNA treatment. Amvuttra is indicated for polyneuropathy in hereditary transthyretin-mediated amyloidosis (ATTR), a rare debilitating condition that affects nerves, the heart and other organs. It’s a more convenient version of Alnylam’s Onpattro (patisiran), delivered as a quarterly subcutaneous injection rather than as an intravenous infusion every three weeks, thanks to enhanced stability and N-acetylgalactosamine conjugation.
Both drugs are also in development for the more common ATTR with cardiomyopathy, where the only approved therapy, Pfizer’s transthyretin-stabilizing non-NSAID benzoxazole derivative Vyndaqel (tafamidis meglumine) had $2 billion in sales in 2021. Alnylam plans a supplementary new drug application in 2022 for Onpattro; the plan to expand Amvuttra’s indications may be affected by the Inflation Reduction Act (Box 2). AstraZeneca and Ionis Pharmaceuticals plan to file their once-monthly antisense oligonucleotide eplontersen for the polyneuropathy ATTR subtype during 2022, following interim phase 3 results.
Targeted radiopharmaceutical (or radioligand) therapies is another emerging NME class that gained prominence in 2022. These link tumor-targeting molecules to cancer-destroying radioactive isotopes to create precise radiation therapy. Basel-based Novartis’s Pluvicto (lutetium (177Lu) vipivotide tetraxetan) was approved in March for prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer after other therapies. The vipivotide tetraxetan latches onto the PSMA found on the surface of many prostate cancer cells and lutetium-177 provides the targeted radiation. It was approved with a complementary diagnostic imaging agent that uses gallium (68Ga) gozetotide injection to identify PSMA-positive lesions via position emission tomography (PET) scan.
Pluvicto is the first FDA approval of a targeted radioligand since Novartis’s Lutathera (lutetium 177Lu dotatate) in 2018 for gastroenteropancreatic neuroendocrine tumors (NETs). Unconjugated radiotherapeutics had arrived five years earlier, with Leverkusen, Germany-based Bayer’s prostate cancer treatment Xofigo (radium-223 dichloride). Xofigo doesn’t include a separate ligand-targeting molecule; the radium itself, which is chemically like calcium, is attracted to actively growing bone tissue such as prostate cancer bone metastases.
Lutathera and Pluvicto came from Novartis’s 2018 acquisitions of Advanced Accelerator Applications and Endocyte, respectively. 2022 saw action among other radiopharmaceuticals-focused biotechs: in November, Lantheus paid $260 million up front to Point Biopharma for licenses to a phase 3 177Lu-based PSMA-targeted therapy like Pluvicto and to PNT2003 (lutetium-177 octreotate), which, like Lutathera, targets the somatostatin receptor in gastroenteropancreatic NETs. ITM Isotope Technologies Munich’s phase 3 candidate ITM-11, comprising lutetium-177 and edotreotide, a NET-targeting synthetic form of somatostatin, received FDA Fast Track designation in October.
Biosimilars’ tailwind
Fittingly perhaps, in the same year that new biologic approvals surpassed those of small molecules, FDA loosened the reins on biosimilars. Two such drugs — lower-cost copies of reference biologics — were granted ‘interchangeable’ status in 2022, meaning they can be substituted by pharmacists in place of the originator drug, without physician say-so. There were two interchangeable designations in 2021 too, but the more recent duo — Coherus’s Cimerli (ranibizumab-eqrn), mirroring Genentech’s Lucentis, and Eli Lilly’s Lantus biosimilar Rezvoglar (insulin glargine-aglr) — did not require switching studies to achieve the stamp. (Switching studies are designed to show that patients achieve the same outcome whether they switch between biosimilar and reference drug or remain on the reference drug throughout.)
This more relaxed approach to studies brings the United States closer in line with Europe, where, for this and other reasons, biosimilar penetration and resulting savings are much more significant. Drug pricing and value decisions are not in the FDA’s remit. But the agency is authorized under the 2021 Advancing Education on Biosimilars Act to educate consumers and providers on biologic products, including biosimilars. The agency also meets regularly with the Centers for Medicare and Medicaid Services on implementing the Inflation Reduction Act, according to Sarah Yim, director of the FDA’s Office of Therapeutic Biologics and Biosimilars, speaking at an Association for Accessible Medicines conference in early November (Box 2).
The FDA’s 2023–2027 goals include a regulatory science pilot to improve the efficiency of biosimilar development. “We’re really focusing on how to develop an interchangeable product,” said FDA Center for Drug Evaluation and Research deputy director Jacqueline Corrigan-Curay at the same conference. A bill designed to remove switching study requirements for interchangeability was introduced into the US Senate in November.
Anti-China?
While encouraging biosimilar development, FDA put the brakes on another strategy to help reduce the rising cost burden of biologics. Companies like EQRx hoped to develop anti-PD-1 and anti-PD-L1 (programmed cell death ligand-1) monoclonal antibodies and other me-too biologics in China, where development costs are lower, and sell them relatively cheaply in the United States. But in 2022, the FDA pushed back strongly on China-only submissions — a stance that several drug sponsors feel is less about clinical science and more to do with the frosty geopolitical situation.
An application from Lilly and Jiangsu, China-based Innovent Biologics for PD-1 inhibitor sintilimab was stopped in March 2022 because its China-only NSCLC trial was deemed not generalizable to the US population owing in part to the chemotherapy comparator. Sintilimab is sold in China as Tyvyt for Hodgkin’s disease, and Lilly had promised to offer the drug at a steep discount in the United States. In May, another complete response letter went to Coherus and Shanghai-based Junshi Bioscience’s PD-1 inhibitor toripalimab for nasopharyngeal carcinoma because of a requested quality process change, with COVID-19-linked delays to China site inspections pushing the Prescription Drug User Fee Act date to 23 December. Also in May, the FDA told Hong Kong-based Hutchmed that it would need a multiregional trial to gain US approval for the small-molecule angiogenesis inhibitor and immune modulator surufatinib in pancreatic and extrapancreatic NETs, having not made that demand initially. Two positive phase 3 studies in China and a (prediscussed) US bridging study were deemed insufficient — with the choice of comparator again an issue.
So EQRx in September 2022 abandoned plans to submit China-developed PD-L1 inhibitor sugemalimab in the United States as a first-line NSCLC drug for use along with chemotherapy. A US trial against another approved checkpoint inhibitor, rather than placebo, was economically unfeasible. “We were caught in a geopolitical maelstrom,” said EQRx CEO Melanie Nallicheri at the Jefferies Global Healthcare Conference in London on 17 November.
It’s fair enough for the FDA to demand data representative of the US population and medical practice. Defending its position, the FDA points out that international multiregional trials are standard for registrational studies, per International Council for Harmonisation good clinical practice guidelines. Yet the agency’s stand was interpreted — rightly or wrongly — as backtracking on earlier comments from the FDA welcoming China-developed medicines. In 2019, Beijing-based BeiGene’s Brukinsa for mantle cell lymphoma, a much smaller indication than NSCLC, was approved on the basis of data predominantly — but not exclusively — from China.
European and UK regulators appear comfortable with China-only data, according to EQRx. The group expects to submit sugemalimab to the UK’s Medicines and Healthcare Products Regulatory Agency this year and to the European Medicines Agency in 2023 for stage 4 NSCLC. “The applicability issue — whether it’s the right SOC [standard of care] or right degree of racial diversity” — only arises in the United States, said Nallicheri. The company is still discussing a US submission for sugemalimab in a much smaller indication, extranodal natural killer cell/T cell lymphoma.
Trial diversity and obesity
FDA made broader moves to increase racial and ethnic diversity in clinical trials. In April, the agency issued a new guidance to help sponsors develop plans to enroll more participants from under-represented racial and ethnic populations, with recommendations to submit race and ethnicity diversity plans early in the development cycle and to plan public education and outreach campaigns.
One aspect of trial diversity and representativeness may have far greater repercussions across drug R&D. Obesity now affects over 40% of the US adult population, and even more in some ethnic groups, according to the Centers for Disease Control and Prevention. Yet patients with obesity are routinely excluded from trials. “The implications of obesity cannot be overstated. Yet there’s a deficit of evidence about medicines in obese patients,” said the FDA’s Califf on November 9. He was introducing a workshop, co-hosted by FDA and the University of Maryland, to review the implications of obesity for drug safety, efficacy, dosing and disposition.
The FDA doesn’t currently require patient weight to be evaluated as part of dosing recommendations. But at least one invited expert, David Greenblatt from the Tufts University School of Medicine, made clear his support for “mandated study of obesity” for all new drugs. Actionable pharmacokinetic, pharmacodynamic and dosing data in individuals with obesity already exists in the medical literature, he pointed out. Studies dating back several decades show that it can take longer for them to achieve effective drug concentrations and full wash-out, and that half-lives of some treatments can be over five times longer in those with obesity than those with healthy weight. “We know what to do. We just need to put it in the regulations,” he said.
Mandated inclusion of patients with obesity in clinical trials — if it happens — would add to already-rising per-patient trial costs. Such patients often have comorbidities and underlying inflammation, and there are complex social, ethnic and cultural hurdles to overcome in ensuring they are adequately represented in trials. Workshop participants discussed using cost-efficient technologies such as in silico modeling and virtual patients to generate drug-specific dosing guidelines.
Califf is a long-time proponent of improving evidence generation and trial design more broadly, calling for more accessible, simpler trials, better data systems and data sources, and greater use of real-world data. In a JAMA Internal Medicine Viewpoint published in October 2022, he outlined the benefits of simplified “point-of-care” trials run in clinical settings and lessons learned from the UK RECOVERY trial of COVID-19 medicines. Califf and Pazdur have also spoken out against over-reliance on single-arm trials in oncology, especially in cancers where there are existing treatments and randomized controlled trials are possible.
More drugs for big diseases
Cancer continued to dominate in 2022, accounting for about a third of all new drug approvals (Fig. 2). Yet new products came online for other widespread, underserved conditions, including autoimmune disorders (psoriasis and atopic dermatitis), diabetes and heart disease.
Fig. 2. FDA approvals by disease group.
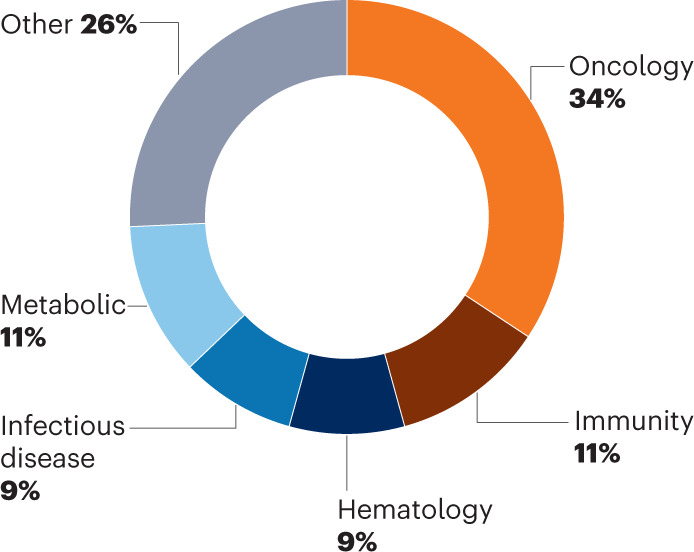
Oncology always leads.
The first new drug to slow the onset of type 1 diabetes, Provention Bio’s Tzield (teplizumab-mzwv), was approved in November. This anti-CD3 monoclonal antibody, given as a daily intravenous infusion for 14 days, binds to T lymphocytes and dampens their attack on insulin-producing pancreatic beta cells. In trials, Tzield delayed the onset of more severe stage 3 diabetes (when typical symptoms including fatigue and ketoacidosis appear) by about two years, relative to placebo.
Progress in the quest for new drugs to combat obesity continued in 2022 with the arrival of Lilly’s dual glucagon-like peptide-1 (GLP-1)/glucose-dependent insulinotropic polypeptide (GIP) agonist Mounjaro (tirzepatide). The drug was approved for type 2 diabetes but is also fast-tracked for obesity, in which Lilly plans to initiate a rolling submission before year end.
Judging by its clinical results, Mounjaro may be one of the most effective obesity drugs yet. It helped nearly two-thirds of patients with obesity but without diabetes in the phase 3 SURMOUNT-1 trial to achieve at least 20% weight loss, appearing even more effective than Bagsvaerd, Denmark-based Novo Nordisk’s GLP-1 agonist Wegovy (semaglutide), whose manufacturer was unable to meet demand for the drug after its June 2021 approval. GLP-1 and GIP are incretin hormones that stimulate insulin release and expand pancreatic beta cell mass; GIP may also help improve lipid handling and enhance the appetite-suppressing effects of GLP-1. Mounjaro’s 2028 sales forecasts are close to $11 billion, according to consensus figures from Evaluate Pharma; the drug is also in development for non-alcoholic steatohepatitis (NASH), heart failure and obstructive sleep apnea.
Bristol Myers Squibb and MyoKardia’s Camzyos (mavacamten) gained approval for certain types of obstructive hypertrophic cardiomyopathy, the most common genetic heart disease (affecting about 0.2% of the population). It’s the first allosteric and reversible inhibitor of the excessive cardiac myosin–actin cross-bridge formation that marks the condition. Camzyos carries a risk evaluation and mitigation strategy for heart failure risk because it reduces left ventricular ejection fraction.
The year also saw positive data readouts in two other major under-served illnesses: Alzheimer’s disease and schizophrenia. Biogen and Tokyo-based Eisai’s Aduhelm follow-up lecanemab, an amyloid-β protofibril-targeting antibody that the FDA accepted in July 2022 for Accelerated Approval and Priority Review, reported positive phase 3 data showing a 27% reduction in the rate of cognitive decline versus placebo over 18 months among patients with early Alzheimer’s. The FDA’s decision is expected 6 January 2023; Eisai also plans to push ahead with a full approval submission, which is more likely to support Medicare and Medicaid reimbursement. (Aduhelm isn’t reimbursed other than for patients in clinical trials.)
Lilly also reported positive phase 3 results at six months for its anti-amyloid-β donanemab at the Clinical Trials on Alzheimer’s Disease conference on November 30. In a head-to-head trial versus Aduhelm, it reduced amyloid plaque by 65.2% — almost four times the 17% Aduhelm achieved over the same period. Full trial results are expected in mid-2023; FDA granted Priority Review and Accelerated Approval in August.
Roche was unable to equal the feat with gantenerumab, which targets amyloid-β oligomers and fibrils. The antibody failed to meet primary endpoints in two 1,000-patient studies, but the company is pursuing phase 2 development of a version of the antibody that crosses the blood–brain barrier more easily.
In schizophrenia, Karuna Therapeutics in August 2022 reported positive top-line phase 3 results for its muscarinic agonist–antagonist combination xanomeline–trospium. The muscarinic agonist xanomeline preferentially stimulates M1 and M4 muscarinic receptors in the brain and nervous system while antagonist trospium, whose positive charge means it can’t cross the blood–brain barrier, helps limit the gastrointestinal side effects resulting from peripheral muscarinic receptor stimulation. Neither molecule is new: xanomeline was discovered in the 1990s, but trials in Alzheimer’s and schizophrenia were discontinued because of adverse events; trospium is used for overactive bladder. Yet together, they led to a statistically significant reduction in the Positive and Negative Syndrome Scale (PANSS) relative to placebo. A submission is planned for mid-2023; if approved, the product — protected by method, formulation and composition patents — would be the first new treatment class for schizophrenia in half a century, according to CEO and chairman Steve Paul.
There were also new treatments for rare conditions: Paris-based Sanofi’s Enjaymo (sutimlimab-jome), a complement C1s-targeting humanized monoclonal antibody, was approved to counter hemolysis in cold agglutinin disease and thereby reduce the need for transfusions. Agios Pharmaceuticals’ oral Pyrukynd (mitapivat) became the first approved disease-modifying agent for hemolytic anemia in pyruvate kinase deficiency. Marinus Pharmaceuticals’ oral Ztalmy (ganaxolone), a neuroactive steroid γ-aminobutyric acid receptor A (GABAA) modulator, was approved for seizures in cyclin-dependent kinase-like-5 deficiency disorder.
2023 is hard to call
FDA appeared jittery in 2022, but a more positive reading puts it in ‘reset’ mode after a tumultuous two years. Califf, who took on his role in February 2022, faced a huge in-tray. Alongside drug reviews, site inspections and cleaning up Accelerated Approvals came an infant formula supply crisis exacerbated by global events and the tail end of COVID-19. (The latest extension to the US public emergency, issued by the Department of Health and Human Services, runs to 11 January 2023.) The FDA issued three initial Emergency Use Authorizations for COVID-19 vaccines or treatments in 2022 and later revised one of those, for Lilly’s neutralizing immunoglobulin G1 monoclonal antibody bebtelovimab, because it lacked efficacy against what had become the most prevalent Omicron subvariants.
With over 130 active biologic license applications and new drug applications underway, according to BioMedTracker, there is plenty of scope for US approvals to pick up again in 2023. Yet a more stringent FDA, as well as the specter of drug price negotiations, make that an uncertain bet.