

Abstract
The distinctive morphology characteristics of microfold cells (M cells) allow the vaccine antigen not only to interact with immune cells directly, but also to effectively stimulate mucosal immune responses via receptors on its apical surface. Human prion protein, a transmembrane receptor for Brucella abortus Hsp60, is highly expressed on the M cell surface. Nonetheless, this protein tends to express in inclusion body in prokaryotic hosts. In this study, the shorter interacting regions of human prion protein were identified via computational methods such as docking and molecular dynamics simulations to minimize its aggregation tendency. The computational calculations revealed three novel human prion protein-interacting regions, namely PrP125, PrP174, and PrP180. In accordance with in silico prediction, the biologically synthesized peptides fusing with GST tag demonstrated their specific binding to Hsp60 protein via pull-down assay. Hence, this finding laid the groundwork for M-cell targeting candidate validation through these newly identified interacting regions.
Keywords: Hsp60 protein, Homology modeling, Human prion protein, M cell, Molecular docking, Molecular dynamics (MDs), Pull-down assay
Introduction
Gastrointestinal infection is one of the top 10 global leading causes of death [1]. The roots of this disease are the infection of bacteria, viruses, and parasites, whereby acute diarrhea is a frequent symptom [2]. This symptom results in death in people with not only weak immune system, such as the elderly over the age of 70, but also immature immune system, such as children (under 5 years). According to a World Health Organization (WHO) report, diarrhea is ranked second in the causes of death in under-5-year-old children [3]. Annually, there are merely 1.7 billion new cases and approximately 525,000 death cases in this age group [3]. In fact, death cases have frequently occurred in developing countries, for instance, Asia, Africa, and Latin America [4]. Therefore, an essential preventive measure is an unmet need.
Oral vaccines stimulate mucosal immune responses and secrete IgA against pathogens at the infection site. However, there are several obstacles to overcome in the development of the oral vaccine. Firstly, antigen vaccines could be dispersed over a larger surface area of the intestine (nearly 32 m2) [5]. Furthermore, the digestive tract is exposed to thousands of foreign antigens on a daily basis through the food route, resulting in immune tolerance. Therefore, nowadays, researchers have focused on targeting microfold cell strategies to develop oral administration.
Microfold cells (M cells) are considered as “gateways” of mucosal immunity, which are found in follicle-associated epithelium—FAE and located overlying Peyer’s Patch (PP). This cell lacks microvilli on the apical surface and has a thinner glycocalyx layer in comparison with epithelium cells [6]. Moreover, M cells form a dome-like structure at the basolateral surface where the immune cells such as B cells, T cells, dendritic cells, and macrophages reside. Thus, vaccine antigens targeting M cells could be effortlessly captured by immune cells and mount the mucosal immune system.
On the M cell surface, there is a plethora of receptors in which human prion protein (huPrP) is highly expressed [7, 8]. In a previous study, 60-kDa heat-shock protein (Hsp60) from Brucella abortus has been experimentally proven as a ligand for murine prion protein expressed on M cells [9]. Hence, this receptor has been focused on the application of ligand evaluation in oral vaccines development. Nonetheless, huPrP is a transmembrane protein with a disulfide bond in this structure, leading to the tendency of inclusion bodies expression in prokaryotic expression system like Escherichia coli.
In this study, we utilized bioinformatic tools to identify the interacting regions of huPrP that utterly retain the interaction ability with their cognate ligand, Hsp60 protein. The predicted regions were then constructed and expressed in E. coli. Afterward, the interaction of Hsp60 protein with the recombinant regions was assessed. The findings of this study would be used to evaluate the binding of recombinant guiding peptides derived from Hsp60 protein for oral vaccine development.
Materials and Methods
Web-Based Homology Modeling, Molecular Docking, and Dynamics Simulations
Molecular Modeling of B. abortus Hsp60 Protein
The crystal structure of B. abortus Hsp60 protein has not been determined by any experimental methods; therefore, it is necessary to construct its 3D structure based on a closely related protein family. The complete B. abortus Hsp60 protein sequence, consisting of 546 amino acids, was retrieved from the UniProt KnowledgeBase (UniProtKB) database with the accession number B2SCZ4. The protein sequence was used as a query sequence for homology modeling using SWISS-MODEL server [10]. This tool used BLAST [11] to identify suitable templates from Protein Data Bank (PDB) respiratory. The structure with sufficient query sequence coverage, sequence identity, and crystal structure resolution was selected. The quality of the constructed model was validated through QMEAN score and local quality estimate plot. PROCHECK [12] and MolProbity programs [13] were used to evaluate the stereochemical quality of the predicted model through Ramachandran Plot.
Molecular Docking of the Predicted B. abortus Hsp60 Protein with Human Prion Protein (huPrP)-Derived Peptides
Human prion protein (huPrP) with full NMR structure (from amino acid 125 to 228, PDB ID: 1QLX) was chopped into four different fragments using PyMOL based on its secondary structures and the results of Edenhofer et al. [14]—125 to 173, 144 to 156, 174 to 223, and 180 to 210. Following the preparation of all huPrP-derived peptides and Hsp60 protein, molecular docking studies were carried out using High Ambiguity Driven DOCKing (HADDOCK) server [15]. In HADDOCK web portal, Hsp60 model was uploaded as the first molecule, and all peptides were uploaded separately as the second molecule. The putative binding site of Hsp60 protein was predicted to be on the apical domain using CASTp server [16]. Binding affinity (ΔG) of docked complexes was predicted using PRODIGY server [17]. The docked conformation from the top cluster with the highest Z score and binding affinity in kcal/mol was selected for the following molecular dynamics (MD) simulations.
Molecular Dynamics Simulations (MDs)
Molecular dynamics simulations were performed on the huPrP-derived peptides and Hsp60 complex obtained from previous studies using GROningen MAchine for Chemical Simulations (GROMACS) 2022.2 package [18] for 100 ns. Physical forces were implemented using the ff14sb protein force field to carry out MD simulations. Structures were solvated in a cubic box filled with TIP3P water molecules, followed by ionization and neutralization of the simulation system with Na+ and Cl− ions. The peptide–protein complexes were minimized in 50,000 steps using the steepest descent method. After minimization, 500 ps NVT (isothermal-isochoric) and 1 ns NPT (isothermal-isobaric) ensembles with the V-rescale temperature coupling and Parrinello-Rahman pressure coupling were used to equilibrate the system at 310 K and 1 atm. Leap-frog integrator was used with a step size of 2 fs. Bond lengths were constrained with the LINCS algorithm. The short-range van der Waals cutoff was 1.0 nm. Finally, three 100 ns molecular dynamics simulations were carried out for all complexes. Following the simulations, the output data were analyzed for root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF). The relative binding free energy for all complexes was computed using MMPBSA approach with gmx_MMPBSA package [19].
Bacterial Strains and Plasmids
E. coli strain DH5α [F- endA1 hsdR17 (rk−/mk−) supE44 thi λ-recA1 gyrA96 ΔlacU169 (φ80 lacZ ΔM15)] was used for cloning, whereas E. coli strain BL21 [F + ompT hsdSB (rB− mB−) gal dcm(DE3)] and SHuffle® Express Competent E. coli (NEB) were used for protein expression. These host strains were grown in Luria–Bertani (LB) medium. B. abortus genome and pET-hprp plasmid were kindly provided by Prof. Kim Suk (Institute of Animal Medicine, College of Veterinary Medicine, Gyeongsang National University, Korea) and Dr. Thanh-Hoa Tran (VNUK Institute for Research and Executive Education, Vietnam), respectively. Genes coding for the interacting regions of huPrP protein and Hsp60 protein were in turn inserted into pGEX-5X1 and pET22b vector.
Recombinant Plasmid Constructions
Constructions of pGST-hprp, pGST-prp125, pGST-prp174, and pGST-prp180
Genes coding for huPrP protein and its peptides (PrP125, PrP174, and PrP180) were obtained from pET-hprp via PCR with specific forward and reverse primer pairs containing restriction sites of BamHI and XhoI (Thermo Scientific), respectively (Table 1). PCR products and pGEX-5X1 plasmid were treated with double enzymes to create compatible sticky ends and then ligated using T4 ligase (Thermo Scientific). After transforming the ligated products into competent E. coli DH5α cells via heat-shock method, bacterial transformations were spread onto a LB agar plate containing 100 μg/ml ampicillin [20]. The target plasmids were confirmed by PCR cloning and sequencing.
Table 1.
List of primers used in this study
| Name of primer pair | Sequence (5′–3′) | Gene | Size of amplicon (bp) |
|---|---|---|---|
|
499F 500R |
ggatccccaagaagcgcccgaagcc ctcgagtcacgatcctctctggtaata |
hprp | 624 |
|
205F 206R |
ggatccccctcggtggttatatgttaggc ctcgagctaattctgattgctatattcatcc |
prp125 | 147 |
|
207F 208R |
ggatccccaattttgttcacgactgcg ctcgagctactgtgattctctttcatac |
prp174 | 150 |
|
207F 209R |
ggatccccaattttgttcacgactgcg ctcgagctacaccactctttccatcattttc |
prp180 | 111 |
|
581F 846R |
tctagaaataattttgtttaactttaag ctcgaggaagtccatgccgcccatgcc |
hsp60 | 1647 |
Underlined letters indicate recognition sites of restriction enzymes
Constructions of pET22b-hsp60
B. abortus genome was used as a template for amplifying hsp60 gene using specific primer pairs via PCR (Table 1). Hsp60 gene was cut by XbaI and XhoI (Thermo Scientific) and then cloned into predigested pET22b plasmid with compatible sticky ends. The ligation was also delivered into competent E. coli DH5α cells via chemical transformation and screened by selective medium, LB agar containing 100 ng/μl ampicillin [20]. The growth colonies were confirmed via PCR colony and sequencing.
Expression of Recombinant Proteins
pGST-prp125, pGST-prp174, pGST-prp180, and pET22b-hsp60 plasmid were transformed into E. coli BL21(DE3) for recombinant protein expression, whereas pGST-hprp plasmid was chemically introduced into SHuffle® Express Competent E. coli (NEB) for expression. Before sub-culturing at a 1:20 ratio (v/v), the strains containing targeted plasmids were cultivated into LB medium with antibiotic at 37 °C. Subsequently, protein expressions were induced at 25 °C for 4 h with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) (Biobasic) for Hsp60, PrP125-GST, PrP174-GST, PrP180-GST proteins, and 16 °C overnight with huPrP-GST protein. Harvested cells were disrupted in PBS 1X, pH 7.4 on ice using sonication to obtain total, soluble, and insoluble fractions. All fractions were diluted in sample buffer 6X (0.375 M Tris, pH 6.8, 12% SDS, 60% Glycerol, 0.6 M DTT, 0.06% Bromophenol Blue) for protein expression analysis via SDS-PAGE and Western blot [20].
SDS-PAGE and Western Blot
Protein samples were analyzed on 15% SDS-PAGE gels and stained with Coomassie Brilliant Blue. Following electrophoresis, gels were transferred onto nitrocellulose membrane and probed either with anti-GST tag conjugated HRP (ProteinTech) for recombinant human PrP and its derived peptides, and anti-His-tag conjugated HRP (ProteinTech) for Hsp60 protein [20].
Purification of Recombinant Human Prion Protein and Its Peptide
Soluble forms of huPrP-GST, PrP125-GST, PrP174-GST, and PrP180-GST protein were collected and filtered by 0.2 μm membrane before loading into GSTrap 4B column (Cytiva) using ÄKTA protein purification system (Cytiva). Then, the contaminants were removed by binding buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4); following that, the target proteins were eluted with elution buffer (50 mM Tris–HCl, 10 mM reduced glutathione, pH 8.0) [21].
Purification of Hsp60 Protein
Similarly, the dissolved phase of Hsp60 protein was also filtered by a 0.2-μm membrane. The Cytiva ÄKTA protein purification system (Cytiva) used immobilized metal ion affinity chromatography (IMAC) via Histrap FF column (Cytiva). Afterward, Hsp60 protein was efficiently eluted with elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4) [20].
After purification, all samples were treated with sample buffer 6X (0.375 M Tris, pH 6.8, 12% SDS, 60% Glycerol, 0.6 M DTT, 0.06% Bromophenol Blue) and then analyzed by SDS-PAGE and silver staining. The concentration of proteins was determined via Bradford assay using BSA protein as a standard protein.
In Vitro Binding of Prion Peptides to Hsp60
Pull-down assay was performed to evaluate the interaction between Hsp60 protein and prion peptides using Pierce™ Glutathione Spin Columns. Firstly, human prion protein and its peptides were loaded into the spin column and mixed for 60 min at room temperature via a rotator platform, then equilibrated with washing buffer (50 mM Tris, 150 mM NaCl containing 0.05% Tween20, pH 8.0.). Secondly, Hsp60 protein was added individually with a ratio of 1:1 (mol:mol) and incubated at 4 °C overnight. After centrifugation (2500 rpm, 5 min) to remove unbound Hsp60 protein, the columns were loaded with elution buffer (50 mM Tris, 150 mM NaCl containing 10 mM reduced glutathione, pH 8.0) [14]. All fractions were collected and analyzed on SDS-PAGE gels.
Results
Web-based Homology Modeling, Molecular Docking, and Dynamics Simulations
Molecular Modeling of B. abortus Hsp60 Protein
Bacterial chaperonin GroEL (PDB ID: 4V43) was selected as a template structure for modeling B. abortus Hsp60 protein with a sequence identity of 68.33% and coverage of 99% compared to other templates. The SWISS-MODEL template library (SMTL version 2021-07-07, PDB release 2021-07-02) was searched with BLAST and HHblits for evolutionary-related structures matching the target sequence. Overall, 930 templates were found. The modeled structure obtained from SWISS-MODEL server (Fig. 1A, B) has a − 0.32 QMEAN score. The Ramachandran plot was drawn for this model using MolProbity webserver. The modeled structure contains 93.97% of its residues in favored regions, 2.24% in outlier regions, and 2.70% in rotamer outlier region (Fig. 1C). The plot of the predicted local similarity to target against the residue number of the predicted 3D structure of the modeled protein was graphically represented (Fig. 1D). Most of the residues had values close to 1, indicating that the predicted model's local quality assessment of the residues was good. Residues with values less than 0.5 were deemed low quality; also, the modeled protein structure fell within the range of other PDB protein structures (Fig. 1E). This demonstrated that the predicted model was highly reliable and could be used for the next docking studies.
Fig. 1.

Predicted B. abortus Hsp60 protein (cyan) structure based on the structure of GroEL (orange) obtained from SWISS-MODEL server and structure quality assessment. A In tetradecamer; B in monomer; C Ramachandran plot of the model; D Local Quality Estimate plot; E normalized QMEAN score plot
Molecular Docking of the Predicted B. abortus Hsp60 Protein with Human Prion Protein (huPrP)-Derived Peptides
All peptide–protein docking structures were selected based on HADDOCK score, Z score, and cluster size (Fig. 2). All four peptides interacted with Hsp60 at the region contained two α-helix forming a socket, Q231 to T243, and E257 to R268. HADDOCK scores, Z score, and binding affinity of four peptides ranged from − 84.8 ± 6.1 to − 62.1 ± 2.0, − 2.3 to − 1.4, and − 9.1 to − 6.3, respectively. Other interaction forces like Van der Waals (Evdw) and electrostatic (Eelec) for all peptides ranged from − 64.1 ± 1.1 to − 31.2 ± 2.6 kcal/mol and − 334.4 ± 38.5 to − 161.7 ± 14.0 kcal/mol, respectively. Among four fragments of huPrP, fragments 125–173, 174–223, and 180–210 showed good binding affinity (ΔG) of − 8.6, − 9.0, and − 9.1 kcal/mol, respectively. The complex structures of these three peptides with Hsp60 protein were subjected to molecular dynamics simulations to validate their stability and analyze their binding free energy. All the results are presented in Table 2.
Fig. 2.

HADDOCK results of all peptide-Hsp60 complexes. Hsp60 protein, huPrP-derived peptides, and interacting residues were colored green, cyan, and magenta, respectively. A huPrP125-173; B huPrP144-156; C huPrP174-223; D huPrP180-210
Table 2.
HADDOCK docking statistics and PRODIGY server predicted binding affinity of huPrP-derived peptides
| Peptides | HADDOCK score | Z score | Evdw | Eelec | Edesol | EAIR | ΔG |
|---|---|---|---|---|---|---|---|
| huPrP | − 82.9 ± 8.3 | − 2.6 | − 47.8 ± 2.4 | − 291.4 ± 17.7 | − 6.8 ± 2.6 | 164.7 ± 28.39 | − 9.9 |
| huPrP125-173 | − 79.1 ± 4.9 | − 1.5 | − 58.8 ± 6.1 | − 193.3 ± 34.1 | − 28.6 ± 6.4 | 469.4 ± 80.21 | − 8.6 |
| huPrP144-156 | − 62.1 ± 2.0 | − 1.4 | − 31.2 ± 2.6 | − 161.7 ± 14.0 | − 11.4 ± 3.0 | 127.9 ± 37.12 | − 6.6 |
| huPrP174-223 | − 84.8 ± 6.1 | − 2.0 | − 59.2 ± 6.7 | − 334.4 ± 38.5 | − 10.0 ± 2.4 | 432.8 ± 61.58 | − 9.0 |
| huPrP180-210 | − 83.5 ± 2.0 | − 2.3 | − 64.1 ± 1.1 | − 210.8 ± 19.4 | − 7.8 ± 1.3 | 439.5 ± 13.48 | − 9.1 |
All energy terms were calculated in kcal/mol
Evdw Van der Waals energy, Eelec electrostatic energy, Edesol desolvation energy, EAIR restraints violation energy, ΔG PRODIGY binding affinity
Molecular Dynamics Simulations (MDs)
Analysis of RMSD values for Cα atoms was explored to understand the structural rigidity and the stability of the peptide–protein complex. Based on the RMSD plot (Fig. 3A), huPrP125-173-Hsp60 complex had high fluctuations throughout three simulations and tended to keep increasing in Run 3; on the contrary, huPrP174-223 and huPrP180-210 were equilibrated well within 100 ns in all three simulations with RMSD values below 0.75 nm, indicating overall stability of these two peptides. We also conducted RMSF analysis to understand which regions of protein complexes display higher levels of flexibility (Fig. 3B). Overall, the flexible regions were mainly found after residue 546, and this region corresponded to the huPrP-derived peptides whereas the Hsp60 region was stable during the simulation time. As expected, huPrP125-173 fragment highly fluctuated throughout three runs. This was explained by this peptide’s secondary structure which mainly consists of loops, and its flexibility became the main cause of differences in the RMSF values. Binding free energy is a reliable measure of protein–peptide interactions. We computed the binding energy (kJ/mol) using the gmx_MMPBSA package. Both the huPrP174-223 and huPrP180-210 fragments remained stable and in close contact with the Hsp60 binding surface throughout the 100 ns MD simulations. Average total binding energy through three simulations, along with the energy components: Van der Waals, electrostatic, polar contributions, and solvation-free energy, are presented in Table 3. Among the three fragments of huPrP, huPrP174-223 and huPrP180-210 fragments gave the most favorable estimated binding free energy. Despite the results from MDs, all three peptides were used for the following in vitro studies of PrP125, PrP174, and PrP180 peptides.
Fig. 3.

Root-mean-square deviation (RMSD) of the Cα atoms as a function of simulation time (A) and root-mean-square fluctuation (RMSF) of the Cα atoms versus residue number (B) of peptides-Hsp60 complexes.1, huPrP125-173; 2, huPrP174-223; 3, huPrP180-210
Table 3.
Average binding free energy predicted using MMPBSA approach for B. abortus Hsp60 complexed with three different peptides derived from huPrP
| huPrP125-173 | huPrP174-223 | huPrP180-210 | |
|---|---|---|---|
| ΔTOTAL | − 59.1 ± 0.52 | − 82.2 ± 0.38 | − 80.1 ± 0.14 |
| ΔVDWAALS | − 98.3 ± 0.30 | − 127.4 ± 0.57 | − 100.7 ± 0.45 |
| ΔEEL + ΔEGB | 50.7 ± 0.61 | 63.8 ± 0.55 | 58.6 ± 0.59 |
| ΔESURF | − 13.78 ± 0.02 | − 14.88 ± 0.05 | − 11.29 ± 0.03 |
Expression of huPrP, PrP125-GST, PrP174-GST, PrP180-GST, and Hsp60 Protein in E. coli Strains
Genes coding for human prion protein, its peptides, and Hsp60 protein were successfully amplified via PCR and the results were visualized using 1.5% agarose gel electrophoresis. Hprp, prp125, prp174, prp180, and hsp60 gene were 624 bp, 147 bp, 150 bp, 111 bp, and 1638 bp, respectively, as shown in Fig. 4.
Fig. 4.

Agarose electrophoresis of gene amplification with specific primer pairs; lane 1: Generuler 1 kb DNA ladder (Thermo Scientific); lane 2: negative control; lane 3: hsp60 gene; lane 4: 100 bp DNA ladder (NEB); lane 5: hprp gene; lane 6: prp125 gene; lane 7: prp174 gene; lane 8: prp180 gene
Constructed plasmids were sequenced and aligned with the corresponding sequences used in the molecular docking and dynamics simulations studies (B2SCZ4 for B. abortus Hsp60 protein and 1QLX for human prion). All cloned genes showed completely homologous to the corresponding queries. Subsequently, the constructed plasmids were then extracted and transformed into expression strains. Simultaneously, E. coli BL21(DE3)/pGEX-5X1 would be induced at the same condition with prion peptides, and GST protein (lane 3–5, Fig. 5B–E) was used as a positive control for protein expression, and a negative control for the following pull-down assay. Furthermore, in the protein expression, E. coli BL21(DE3) strain was also induced and used as a negative control (lane 2, Fig. 5B–E). The results indicated that Hsp60 protein and huPrP fused GST tag were overexpressed at approximately 60 kDa and 49 kDa, respectively (lane 3, Fig. 5A and lane 6, Fig. 5B). Besides that, there was a similarity in the molecular weight of prion peptides, merely 32 kDa for both PrP125-GST, PrP174-GST, and 30 kDa for PrP180-GST (lane 6, Fig. 5C–E). In comparison with insoluble phase, all human prion protein, its peptides fused GST tag and Hsp60 protein were overexpressed in soluble phase (lane 3–5, Fig. 5A and lane 6–8, Fig. 5B–E). Additionally, anti-GST antibody identified GST-fused proteins, and anti-6His antibody was bound to Hsp60 protein. Blotting results confirmed the successful expression of these proteins.
Fig. 5.

Expression of human prion protein, its peptides fused with GST tag and Hsp60 via SDS-PAGE (left) and Western blot (right) A, Hsp60 protein; B, huPrP-GST protein; C, PrP125-GST protein; D, PrP174-GST protein; E, PrP180-GST protein; lane 1: low range weight protein marker, 97–14.4 kDa (Cytiva) (SDS-PAGE) and Pierce™ prestained protein molecular weight marker, 20–120 kDa (Western blot); lane 2: induced E. coli BL21(DE3); lane 3–6 of A total cellular protein, soluble, insoluble fractions of E. coli BL21(DE3)/pET22b-hsp60, respectively; lane 3–5 of B, C, D, E total cellular protein, soluble, insoluble fractions of induced E. coli BL21(DE3)/ pGEX-5X1, respectively; lane 6–8: total cellular protein, soluble, insoluble fractions of target proteins, respectively
Evaluation the Interaction Ability Between Prion Peptides and Hsp60 Protein
According to Fig. 6, there was a band at approximately 60 kDa in eluted fraction of prion proteins (upper band, lane 2–5, Fig. 6) that corresponded to the molecular weight of Hsp60 protein. Besides, human prion protein and its predicted peptides (PrP125, PrP174, and PrP180) also were eluted at 45 kDa (middle band, lane 2, Fig. 6) and merely 30 kDa, (lower band, lane 3–5, Fig. 6), whereas Hsp60 protein was not observed in eluted fraction of GST alone and there was an only band at 30 kDa of GST protein that confirmed no specific binding between GST and Hsp60 protein.
Fig. 6.
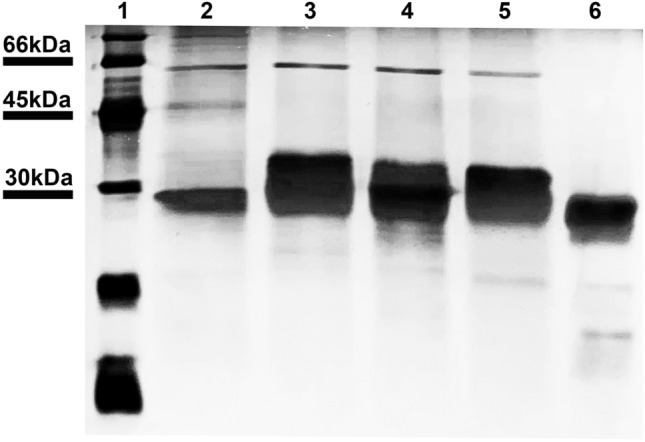
Pull-down assays for Hsp60 protein and prion proteins interaction using Pierce™ Glutathione Spin Columns; lane 1: low range weight protein marker, 97–14.4 kDa (Cytiva); lane 2: HuPrP-GST, lane 3: PrP125-GST; lane 4: PrP174-GST; lane 5: PrP180-GST; lane 6: GST protein. Data present three independent experiments
Discussion
Brucella abortus Hsp60 and human prion proteins are a prospective pair in the application of M cell targeting vaccine development. In this study, we have modeled three peptides derived from the huPrP protein that were PrP125, PrP174, and PrP180 peptides. The latter one was experimentally identified as the main interaction site for huPrP on B. abortus Hsp60 in the work of Edenhofer et al. [14]. PrP180 peptide is located within PrP174; however, two peptides bound differently to the apical domain of Hsp60 protein. PrP174 peptide is primarily bound to Hsp60 protein at the α-helix, whereas PrP180 peptide interacted with Hsp60 protein at the loop connecting the two helixes. This resulted in a more favorable binding energy for the PrP174 peptide, which is longer in sequence compared to PrP180. Molecular dynamics simulations performed on the complex further supported our results. RMSD and RMSF graphs revealed that PrP174 and PrP180 peptides in the complex with Hsp60 protein remained stable throughout simulation times. The MM-GBSA method was applied for binding energy calculation. The negative number indicated favorable binding. According to MD simulation, these two peptides were tightly bounded with Hsp60 protein and could be used as screening ligands.
To validate the in silico results, PrP125, PrP174, and PrP180 peptides were fused to GST tag and biologically synthesized. Additionally, full-length prion protein fused to GST and GST alone were also expressed to use as a positive and negative controls, respectively. In Weiss et al. work [22], cellular prion protein for Syria golden hamster fused to GST was employed in three individual expression systems, including E. coli with several reagents, whereas in our study, SHuffle® Express Competent E. coli strain would be used to express overnight at a low temperature (16 °C) without any chemical agents. This strain would assist in correctly folding proteins and enhance the expression capacity of soluble form. Nevertheless, human prion protein expressed in SHuffle® E. coli strain might be unstable and tended to be degraded to other bands alongside target band protein (lower band, lane 2, Fig. 6).
According to in silico and in vitro interactions, shortened predicted peptides derived from human prion protein (including PrP125, PrP174, and PrP180) were capable of specifically binding to Hsp60 protein. Aside from PrP180 peptide, which was also identified by Edenhofer et al. study [14], we discovered two more novel peptides, namely PrP125, PrP174. When compared to full-length human cellular prion protein, those shortened peptides have both effortless condition and shorter induction time at 25 °C in only 4 h instead of 16 °C overnight. It would decelerate the time required to evaluate and screen Hsp60-derived M cell-targeted ligands. All these predicted peptides could become potential receptors in lieu of full-length human cellular prion protein in the long run for oral vaccine development.
Acknowledgements
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. The authors express gratitude to the Faculty of Information Technology, University of Science, VNU-HCM for accessing the high-performance computer with GPU acceleration.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hoang-Anh Le-Dao and Thuan-Thien Dinh have contributed equally to this work.
References
- 1.Organization, W.H. (2020) The top 10 causes of death; Published online at: [who.int]. Retrieved from https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death
- 2.Hodges K, Gill R. Infectious diarrhea: Cellular and molecular mechanisms. Gut Microbes. 2010;1:4–21. doi: 10.4161/gmic.1.1.11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Organization, W.H. (2017). Diarrhoeal disease; Published online at: [who.int]. Retrieved from https://www.who.int/news-room/fact-sheets/detail/diarrhoeal-disease
- 4.Manetu WM, M’masi S, Recha CW. Diarrhea disease among children under 5 years of age: A global systematic review. Open Journal of Epidemiology. 2021;11:207–221. doi: 10.4236/ojepi.2021.113018. [DOI] [Google Scholar]
- 5.Helander HF, Fandriks L. Surface area of the digestive tract—Revisited. Scandinavian Journal of Gastroenterology. 2014;49:681–689. doi: 10.3109/00365521.2014.898326. [DOI] [PubMed] [Google Scholar]
- 6.Corr SC, Gahan CC, Hill C. M-cells: Origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunology and Medical Microbiology. 2008;52:2–12. doi: 10.1111/j.1574-695X.2007.00359.x. [DOI] [PubMed] [Google Scholar]
- 7.Nakato G, et al. New approach for m-cell-specific molecules screening by comprehensive transcriptome analysis. DNA Research. 2009;16:227–235. doi: 10.1093/dnares/dsp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandit J, Sultana Y, Kalam M. Newer technologies in oral vaccine delivery. Current Drug Therapy. 2015;9:173–187. doi: 10.2174/1574885509666141202232803. [DOI] [Google Scholar]
- 9.Nakato G, et al. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. The Journal of Immunology. 2012;189:1540–1544. doi: 10.4049/jimmunol.1103332. [DOI] [PubMed] [Google Scholar]
- 10.Schwede T, et al. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Research. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camacho C, et al. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:1–9. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laskowski RA, et al. PROCHECK: A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 13.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edenhofer F, et al. Prion protein PrPc interacts with molecular chaperones of the Hsp60 family. Journal of Virology. 1996;70:4724–4728. doi: 10.1128/jvi.70.7.4724-4728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Zundert GCP, et al. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. Journal of Molecular Biology. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Tian W, et al. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Research. 2018;46:W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue LC, et al. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32:3676–3678. doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- 18.Abraham MJ, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 19.Valdés-Tresanco MS, et al. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. Journal of Chemical Theory and Computation. 2021;17:6281–6291. doi: 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- 20.Vo-Nguyen HV, et al. Recombinant human SCARB2 expressed in Escherichia coli and its potential in enterovirus 71 blockage. Iranian Journal of Science and Technology, Transactions A: Science. 2021;45:455–461. doi: 10.1007/s40995-020-01025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen P, et al. Cloning, expression, purification, and assessment of Vibrio parahaemolyticus-agglutinating C-type lectin from Litopenaeus vannamei fused to GST tag. SSR Institute of International Journal of Life Sciences. 2020;6:2544–2551. doi: 10.21276/SSR-IIJLS.2020.6.3.3. [DOI] [Google Scholar]
- 22.Weiss S, et al. Overexpression of active Syrian golden hamster prion protein PrPc as a glutathione S-transferase fusion in heterologous systems. Journal of Virology. 1995;69:4776–4783. doi: 10.1128/jvi.69.8.4776-4783.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]