

Summary
During metastasis, cancer cells invade, intravasate, enter the circulation, extravasate, and colonize target organs. Here, we examined the role of interleukin (IL)-22 in metastasis. Immune cell-derived IL-22 acts on epithelial tissues, promoting regeneration and healing upon tissue damage, but it is also associated with malignancy. Il22-deficient mice and mice treated with an IL-22 antibody were protected from colon-cancer-derived liver and lung metastasis formation, while overexpression of IL-22 promoted metastasis. Mechanistically, IL-22 acted on endothelial cells, promoting endothelial permeability and cancer cell transmigration via induction of endothelial aminopeptidase N. Multi-parameter flow cytometry and single-cell sequencing of immune cells isolated during cancer cell extravasation into the liver revealed iNKT17 cells as source of IL-22. iNKT-cell-deficient mice exhibited reduced metastases, which was reversed by injection of wild type, but not Il22-deficient, invariant natural killer T (iNKT) cells. IL-22-producing iNKT cells promoting metastasis were tissue resident, as demonstrated by parabiosis. Thus, IL-22 may present a therapeutic target for prevention of metastasis.
Keywords: tissue resident cells, IL-22, metastasis formation, cancer cell extravasation, extravasation, endothelial cells, ANPEP, metastasis
Graphical abstract
Highlights
-
•
Il22-deficient mice are protected against metastasis formation
-
•
IL-22 neutralization blocks cancer cell extravasation
-
•
IL-22 acts on endothelial cells, promoting cancer cell extravasation via ANPEP induction
-
•
Tissue resident iNKT17 cells are the key IL-22 source during cancer cell extravasation
Interleukin-22 (IL-22) is produced by immune cells and promotes tissue repair and regeneration; however, in malignancy, IL-22 can promote tumor growth. Giannou et al. find that tissue resident iNKT17 cells produce IL-22 and promote cancer cell extravasation through regulation of aminopeptidase N. Neutralization of IL-22 inhibits metastasis formation, suggesting therapeutic avenues for cancer treatment.
Introduction
Metastasis is a common cause of cancer-related death. What is known as the “metastatic cascade” describes the multi-step process of metastasis formation: invasion, intravasation, circulation, extravasation, and colonization of the target organs.1 The crosstalk among the immune system, cancer cells, and tissue-specific stromal cells is thought to be the critical determinant in metastatic progression,2 although specific insight into the cells and factors mediating this communication is limited. Tissue resident immune cells lodge in non-lymphoid tissues, providing continuous immune surveillance, and enable a prompt and efficient in situ response upon pathogen invasion.3,4 Furthermore, tissue resident immune cells, and specifically CD8+ T cells, innate lymphoid cells (ILCs), and innate-like T cells, perform an anti-tumorigenic function in the primary tumor in mice.5,6 Tissue resident invariant natural killer T (iNKT) cells can also contribute to the anti-tumor response.7 There are distinct subsets of iNKT cells, namely iNKT1, iNKT2, and iNKT17 cells, whose transcriptional profiles and signature cytokines are analog to Th1, Th2, and Th17 cell subsets among CD4+ T cells. However, the role of tissue resident immune cells in the metastatic cascade is poorly understood.
Interleukin (IL)-22, a cytokine belonging to the IL-10 family, can be produced by tissue resident immune cells.8 Furthermore, iNKT cells, and specifically iNKT17 cells, are able to produce IL-22.9,10,11,12 IL-22 is also released by several other immune cell types, including ILCs and CD4+ T lymphocytes.13 Interestingly, cancer cells can directly induce IL-22 production in vitro and in vivo14 and IL-22-producing Th17 cells accumulate in colon cancer tissues.15 Accordingly, the number of infiltrating CD4+ IL17A+ IL-22+ T cells is higher in human colorectal cancer (CRC), while CD4+IL17A−IL-22+ T cells are not increased compared to healthy adjacent tissue.16,17,18 Unlike other cytokines, IL-22 binds to non-immune cells expressing its specific receptor, IL-22RA1.19 Thus, IL-22 may mediate the communication between the immune system and the tissue.
Interestingly, IL-22 has dual functions: short-term IL-22 exposure promotes intestinal integrity and protects intestinal stem cells against genotoxic stress.19,20 However, uncontrolled and prolonged IL-22 activity promotes intestinal tumorigenesis.21,22,23 IL-22 promotes cancer cell stemness24,25 and facilitates tumor growth.26,27 These data are further supported by human studies, which indicate a detrimental role of uncontrolled IL-22 activity in cancer.28,29 Despite these data, the role of IL-22 during metastasis formation is still unclear.
In this study, we examined the role of IL-22 in metastasis formation. We found that tissue-resident iNKT17 cell-derived IL-22 acted directly on endothelial cells, thereby facilitating cancer cell extravasation via induction of endothelial aminopeptidase N (ANPEP) and, subsequently, liver metastasis formation. Our data present a rationale for targeting IL-22 in the clinic.
Results
Il22-deficiency in mice protects against liver metastasis
First, we analyzed the role of IL-22 using a forced metastatic mouse model,30 namely intrasplenic (i.s.) injection of CRC cells (MC38, murine colon adenocarcinoma cells). Il22-deficient mice were protected from metastasis formation compared to littermate controls (Figures 1A–1C). Likewise, IL-22 blockade protected mice from developing liver metastasis (Figures 1D and 1E). We then used a gain-of-function approach: Il22tg8Tg transgenic mice overexpress IL-22 in the liver and also have increased serum IL-22 levels (≈6,000 pg/ml).31 Indeed, we found an increased number of liver metastases in Il22tg8Tg transgenic mice compared to littermate controls upon induction of forced liver metastasis (Figure S1A). The pro-metastatic function of IL-22 was seen also in Balb/c wild-type (WT) mice after hydrodynamic overexpression of IL-22 or control plasmid administration (eGFP) in the liver followed by an i.s. induction of liver metastasis with the CT26GAG-Luc colon cancer cell line (Figures 1F–1H and S1B–S1F).
Figure 1.

Il22-deficient mice are protected against liver metastasis
(A) Schematic overview of the experiment (i.s. injection of MC38 cells).
(B) Representative pictures, number of macroscopic liver metastases, and liver weight of Il22+/+ mice compared to Il22−/− mice. n ≥ 11 mice per group.
(C) H&E staining and metastatic foci. n = 5 mice per group.
(D) Schematic overview of the experiment (i.s. injection of MC38 cells).
(E) Liver weight and number of macroscopic liver metastases in mice treated with an α-IL-22 or IgG control antibody. n ≥ 8 mice per group.
(F) Schematic overview of the experiment (i.s. injection of CT26CAG-Luc cells).
(G) Ex vivo bioluminescent imaging of livers after hydrodynamic overexpression of eGFP or Il22eGFP plasmid. Bioluminescent scale: 2 × 107–2 × 108 photons/sec/cm2/sr. Liver weight and number of macroscopic liver metastases in Balb/c mice after hydrodynamic overexpression of eGFP or Il22eGFP plasmid. n ≥ 6 mice per group.
(H) Bioluminescent imaging of Balb/c mice after hydrodynamic overexpression of eGFP or Il22eGFP plasmid. Bioluminescent scale: 5 × 107–5 × 108 photons/sec/cm2/sr. n ≥ 6 mice per group. Scale bar: 2 mm.
(I) Schematic overview of the experiment (injection of LLC cells into the cecum).
(J) Representative pictures and number of macroscopic liver metastases in Il22+/+ and Il22−/− mice. n ≥ 6 mice per group.
(K) Schematic overview of the experiment (adenoviral infection of colonic epithelium for orthotopic colon cancer induction as a model of spontaneous liver metastasis).
(L) Endoscopic score of primary colon cancer of Apc15lox;KrasG12D mice. n ≥ 6 mice per group.
(M) Il22 mRNA levels in total liver of Apc15lox;KrasG12D mice upon hydrodynamic liver overexpression of eGFP or Il22eGFP plasmid. n = 6 mice per group.
(N) Representative pictures and percentage of Apc15lox;KrasG12D mice that developed macroscopic metastases. n = 6 mice per group. Scale bar: 2 mm. Data presented as mean ± SEM, not significant (ns): p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by Mann-Whitney U test or Fischer’s exact test (K).
We next examined the role of IL-22 in spontaneous metastasis models. To this end, we first injected C57BL/6 Lewis lung adenocarcinoma syngeneic cancer cells (Lewis lung carcinoma [LLC] cell line) into the cecum of Il22-deficient mice and littermate controls. Il22-deficient mice were largely protected from metastasis formation compared to littermate controls (Figures 1I and 1J). As a second model, we used Apc15lox;KrasG12D mice,32,33 a genetic model which spontaneously develops CRC and liver metastasis upon adenovirus, Ad-Cre (see STAR Methods), injection. Upon hydrodynamic overexpression of IL-22 in the liver, 60% of the mice developed liver metastases compared to no detectable metastases in mice with control (Egfp) gene transfer (Figures 1K–1N). There was no difference in the formation of the primary tumor in the colon. As a third spontaneous model, we used an intramucosal injection of CT26GAG-Luc cells in order to mimic human CRC development. This experimental setup resulted in liver metastasis formation only in Balb/c WT mice overexpressing IL-22 in their liver, while Balb/c Il22-deficient mice with a control hydrodynamic gene transfer did not develop any liver metastases (Figure S1G).
Thus, in various experimental models, IL-22 promoted liver metastasis formation.
IL-22 affects the host endothelium, thereby promoting metastasis
The next step was to identify the target cells of IL-22. We found that tumor cells (MC38 and LCC) express IL-22RA1 and can respond to IL-22 in vitro (Figures S2A–S2D). Furthermore, IL-22 increased cancer cell stemness, evidenced by enhanced tumor sphere formation in vitro (Figure S2E). To assess the relevance of this finding in vivo, we injected LLC-GFP (LLC-Green Fluorescent Protein) cancer cells into the cecum of mice or MC38-GFP cells intrasplenically. Il22-deficient mice showed lower circulating tumor cell (CTC) numbers in both models (Figures S2F–S2I). In order to test whether IL-22 acts directly on tumor cells in vivo, we silenced the expression of IL-22RA1 in MC38 cells (Figures S2J and S2K). We found no difference upon injection of Il22ra1-deficient and control MC38 cells when we used WT mice as recipients, suggesting that endogenous IL-22 levels are not sufficient to promote metastasis by acting on tumor cells directly. However, we observed reduced metastatic sites in Il22tg8Tg transgenic mice receiving Il22ra1-deficient MC38 cells compared to the control group (Figures 2A and 2B), thus indicating that IL-22 in high concentrations can act on tumor cells in vivo. On this basis, we aimed to assess whether IL-22 could also act on non-tumor cells in vivo. To this end, we injected WT MC38 cells (which express IL-22RA1) intrasplenically into Il22ra1-deficient19 and littermate control mice. Il22ra1-deficient mice exhibited lower liver weight and macroscopic metastases compared to WT littermate controls (Figures 2C and 2D). Thus, IL-22 can act on non-tumor cells, thereby promoting metastasis formation.
Figure 2.

IL-22 affects the host endothelium, thereby promoting metastasis
(A) Schematic overview of the experiment (i.s. injection of MC38shC and MC38shIl22ra1 cells in Il22tg(wt) and Il22tg8(Tg) mice).
(B) Liver weight and number of macroscopic liver metastases. n ≥ 7 mice per group.
(C) Schematic overview of the experiment (i.s. injection of MC38 cells in WT and Il22ra1−/− mice).
(D) Liver weight and number of macroscopic liver metastases. n ≥ 7 mice per group.
(E) IL-22RA1 expression in CD45+ cells and LSECs.
(F) Immunostaining of LSECs with CD31 (green) and IL-22RA1 (red). Scale bar, 50 μm.
(G) pSTAT3 levels in LSECs isolated from 6 WT mice and treated with recombinant murine (rm) IL-22.
(H) Schematic overview of the experiment (i.s. MC38 cell injection in Il22ra1wt/wt;AlbCre+ and Il22ra1flox/flox;AlbCre+ mice).
(I) Liver weight and number of macroscopic liver metastases. n ≥ 7 mice per group.
(J) Schematic overview of the experiment (i.s. MC38 cell injection in Il22ra1wt/wt;Cdh5Cre+ and Il22ra1flox/flox;Cdh5Cre+ mice).
(K) Liver weight and number of macroscopic liver metastases. n ≥ 14 mice per group. Scale bar, 2 mm (B, D, I, and K). Data presented as mean ± SEM, ns: p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by Mann-Whitney U test (C, D, I, and K) or one-way ANOVA with Bonferroni post hoc tests (B and G).
We next aimed to identify the non-tumor cell that would respond to IL-22. We did not detect expression of the IL-22RA1 receptor in CD45+ (immune) cells. However, we found that the liver sinusoidal endothelial cells (LSECs) highly expressed the IL22RA1 receptor (Figures 2E and 2F). Furthermore, IL-22 induced STAT3 phosphorylation in LSECs in vitro (Figure 2G). We further used a mouse model with conditional deletion of Il22ra1 (Figures S2L, S2M, and S2O). This was crossed with Cdh5Cre mice to obtain an endothelial-specific deletion,34 and we observed Cre recombination in approximately 60% of endothelial cells (Figure S2O). As control, we used mice with a hepatocyte-specific driver, Albumin Cre (AlbCre), because hepatocytes can also respond to IL-22.35 We did not observe a difference in the number of metastatic sites in Il22ra1flox/flox;AlbCre+ mice compared to littermate controls upon i.s. injection of MC38 cells inducing liver metastasis (Figures 2H and 2I). In contrast, Il22ra1flox/flox Cdh5Cre+ mice phenocopied Il22ra1-deficient mice by showing reduced forced liver metastases (Figures 2J and 2K).
Taken together, these findings indicate that IL-22 acts on the host endothelium, thereby promoting metastasis formation.
IL-22 signaling in endothelial cells promotes cancer cell extravasation
Next, we aimed to uncover the mechanism by which IL-22 signaling would promote metastasis formation. We hypothesized that this is due to altered endothelial adhesion of cancer cells, increased permeability of the endothelial layer allowing enhanced extravasation of cancer cells, or a combination of both. To test this, we used an in vitro adhesion assay and found that IL-22 did not increase the adhesion of CRC cells, HT-29 or MC38, to human umbilical vein endothelial cells (HUVECs) or LSECs, respectively (Figures S3A–S3C). As positive control, we used IL-1α and tumor necrosis factor (TNF)-α (Figures S3A–S3C). Additionally, IL-22 did not affect the expression of adhesion molecules such as E-Sel, P-Sel, ICAM-1, and VCAM-1 (Figures S3D–S3F). Next, we tested whether IL-22 would increase endothelial permeability and thus cancer cell extravasation. We found that IL-22 signaling enhanced the extravasation of tumor cells into the liver parenchyma, whereas this was decreased upon IL-22 blockade (Figures 3A–3F and S3G–S3K). Likewise, Il22ra1flox/flox;Cdh5Cre+ mice lacking IL-22 signaling on the endothelium showed reduced extravasation (Figures 3G–3J). Finally, humanized MISTRG mice (Rag2-/-Il2rg-/- mice in which the genes encoding human M-CSF, human IL-3 and GM-CSF, and human TPO are knocked in to their respective mouse loci; they also include a BAC-transgene encoding human SIRPα) that were engrafted with hCD34+ cells showed reduced extravasating human cancer cells (HCT-116, human CRC cells) into the liver parenchyma upon treatment with Fezakinumab, a human IL-22 antibody (Figures S3L and S3M).
Figure 3.

IL-22 signaling in endothelial cells is required for cancer cell extravasation
(A) Schematic overview of the experiment (i.s. injection of MC38 GFP-labelled cells in WT, Il22−/−, and Il22ra1−/− mice [extravasation assay]). See also Figure S3.
(B) Representative FACS (fluorescence-activated cell sorting) plots and number of extravasated cancer cells 24 h post i.s. cancer cell injection. n ≥ 4 mice per group.
(C) Representative images of immunostaining of MC38 Cherry-labeled cells and endomucin (endothelial marker; green) 24 h post i.s. cancer cell injection. Scale bar, 50 μm.
(D) Number of extravasated cancer cells 24 h post i.s. injection. n = 6 mice per group.
(E) Schematic overview of the experiment (injection of MC38 Cherry-labeled cells in WT
mice receiving an anti-IL-22 or IgG control antibody).
(F) Representative FACS plots and number of extravasated cancer cells from (E). n ≥ 5 mice per group.
(G) Schematic overview of the experiment (i.s. injection of MC38 GFP-labeled cells in Il22ra1wt/wt;Cdh5Cre+ and Il22ra1flox/flox;Cdh5Cre+ mice [extravasation assay]).
(H) Representative FACS plots and number of extravasated cancer cells 24 h post i.s. cancer cell injection. n ≥ 8 mice per group.
(I) Schematic overview of the experiment (i.s. injection of MC38 cells in Il22ra1wt/wt;Cdh5Cre+ and Il22ra1flox/flox;Cdh5Cre+ mice [extravasation assay]).
(J) Immune cell infiltration in the liver 24 h post i.s. injection. n ≥ 7 mice per group. Data presented as mean ± SEM, ns p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by one-way ANOVA with Bonferroni post hoc tests (B and D) or Mann-Whitney U test (F, H, and J).
Thus, these findings indicate that IL-22 blockade can be a therapeutic approach to prevent extravasation of tumor cells into the target organ.
IL-22 promotes increased endothelial expression of ANPEP and thereby cancer cell extravasation
In order to identify the molecular mechanisms underlying the effect of IL-22, we first used an in vitro assay. We found that LSEC exposure to rmIL-22 particularly induced the expression of three transcripts, namely Anpep, Epas1, and Fgfr3 in vitro (Figures 4A and S4A–S4E). We decided to focus on the aminopeptidase ANPEP, since ANPEP has been implicated in metastasis formation before36 and the increased expression in LSECs could be verified by qPCR and immunoblotting (Figures 4B, 4C, and S4A–S4E). Additionally, MC38 cell i.s. injection in WT mice induced the expression of Anpep in LSECs 12 h after injection compared to Il22-deficient mice (Figure S4F). Indeed, ANPEP staining of WT LSECs isolated from a murine liver at the steady state revealed that the majority of CD31+ endothelial cells are positive for ANPEP (Figure 4D). Subsequently, we used two different in vitro endothelial cell assays. These experiments showed that IL-22 acts on the endothelium and affects cancer cell transmigration and vascular permeability (Figures 4E and 4F). We then silenced the ANPEP expression on HUVEC cells using lentiviral transduction (Figure 4H). This resulted in reduced cancer cell transmigration compared to respective controls (Figure 4I). Next, we used Ubenimex, which blocks aminopeptidase activity. Ubenimex-treated mice exhibited reduced metastatic sites compared to respective controls (Figures S4I and S4J). However, Ubenimex treatment also resulted in reduced tumor cell proliferation (Figures S4G and S4H). Thus, ubenimex might also act on cancer cells and have ANPEP-independent effects. Therefore, we next intrasplenically injected MC38 cells into ANPEP-deficient mice (Cd13−/− mice) and WT littermate controls. ANPEP-deficient mice showed reduced metastasis formation compared to littermate controls (Figures S4K and S4L). In order to further examine the role of the IL-22-ANPEP axis in the extravasation of tumor cells, we treated Cd13−/− mice with an anti-IL-22 neutralizing antibody and assessed the extravasation of tumor cells. WT mice, but not Cd13−/− mice, receiving anti-IL-22 neutralizing antibody showed a decreased extravasation of tumor cells compared to mice receiving IgG antibody (Figures 4J–4K).
Figure 4.

IL-22 increases endothelial ANPEP expression, thereby promoting endothelial permeability and cancer cell transmigration
(A) Gene expression analysis of LSECs upon rm IL-22 or PBS stimulation.
(B) Anpep expression in LSECs upon stimulation with rmIL-22.
(C) Immunoblotting analysis and quantification of ANPEP in LSECs upon rmIL-22 stimulation.
(D) Confocal microscopy pictures of in vitro cultured LSECs showing the CD31 (green) and ANPEP (red) expression.
(E) In vitro extravasation model of HT29 GFP-labeled cancer cells through HUVEC cells.
(F) TEER of HUVEC cell layers upon rmIL-22 stimulation.
(G) ANPEP RNA levels upon rmIL-22 stimulation.
(H) ANPEP RNA expression after shRNA silencing of ANPEP in different HUVEC clones.
(I) In vitro extravasation model of HT29 GFP-labeled cancer cells through shC or shANPEP HUVEC cells. n = 2 independent experiments.
(J) Schematic overview of the experiment (i.s. injection of MC38 Cherry cells in Cd13+/+ or Cd13−/− mice receiving an α-IL-22 or IgG control antibody).
(K) Representative FACS plots and statistics. n ≥ 10 mice per group. Data presented as mean ± SEM, ns p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by one-way ANOVA with Bonferroni post hoc tests (D and K) or Mann-Whitney U test (B, E, F, and I).
Thus, IL-22 regulates ANPEP expression in endothelial cells in vitro and in vivo. Furthermore, the effect of IL-22 on cancer cell extravasation seems to depend at least in part on ANPEP.
IL-22 promotes lung metastasis by promoting cancer cell extravasation
On the basis of our data obtained in the liver, we next aimed to extend our findings to lung metastasis. We used two lung metastasis models and found in both models that Il22-deficient mice were protected from metastasis formation (Figures 5A and 5B). In contrast, Il22tg8Tg mice showed increased lung metastasis (Figure S5A). In this model of forced lung metastasis, 60% of Il22tg8Tg mice also developed liver metastasis, while this was not the case in any of their littermate controls (Figure S5B).
Figure 5.

IL-22 promotes lung metastasis by promoting cancer cell extravasation
(A) Schematic overview of the experiment (i.v. injection of MC38 cells for forced lung metastasis), representative pictures and number of macroscopic metastases, H&E staining, and metastatic foci percentage of Il22+/+ and Il22−/− mice. n ≥ 10 mice per group.
(B) Schematic overview of the experiment (flank injection of MC38 cells for spontaneous lung metastasis), representative pictures, and number of macroscopic metastases. n ≥ 10 mice per group. Scale bar, 2 mm.
(C) Schematic overview of the experiment (i.v. injection of MC38 for forced lung metastasis in Il22ra1wt/wt;Cdh5Cre+ and Il22ra1flox/flox;Cdh5Cre+ mice).
(D) Number of macroscopic lung metastases. n ≥ 5 mice per group.
(E) Schematic overview of the experiment (i.v. MC38 cell injection following single left orthotopic lung transplantation using WT mice as recipients and Il22ra1−/− mice as donors).
(F) Representative pictures and quantification of lung metastases. n = 7 mice per group.
(G) Schematic overview of the experiment (i.v. injection of MC38 Cherry-labeled cells in WT, Il22−/− and Il22ra1−/− mice [extravasation assay]).
(H) Representative FACS plots and number of extravasated cancer cells 24 h post i.v. injection. n = 4 mice per group.
(I) Schematic overview of the experiment (i.v. injection of MC38 GFP-labeled cells in WT mice receiving an anti-IL-22 or IgG control antibody).
(J) Representative FACS plots and number of extravasated cancer cells. n ≥ 11 mice per group.
(K) Schematic overview of the experiment (i.v. injection of MC38 GFP-labeled cells in Il22ra1wt/wt;Cdh5Cre+ and Il22ra1flox/flox;Cdh5Cre+ mice [extravasation assay]).
(L) Representative FACS plots and number of extravasated cancer cells from Il22ra1wt/wt;Cdh5Cre+ and Il22ra1flox/flox;Cdh5Cre+ mice 24 h post i.v. injection. n = 5 mice per group. Data presented as mean ± SEM, ns p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by Mann-Whitney U test (A–D, J, and L), one-way ANOVA with Bonferroni post hoc tests (H), or two-tailed Wilcoxon matched-pairs signed rank test (F).
In line with our data in liver metastasis, Il22ra1flox/flox;Cdh5Cre+ mice (Figure S2) were also protected in a forced lung metastasis model (Figures 5C and 5D). To test the hypothesis that IL-22 acts on the endothelial cells in the target organ, we transplanted a single left lung from Il22ra1-deficient mice (Il22ra1−/−) orthotopically into C57BL/6N Il22ra1-proficient mice (Il22ra1+/+). Thus, an Il22ra1-proficient lung and an Il22ra1-deficient lung were in the same host. The mice subsequently underwent the forced metastasis model by intravenous (i.v.) administration of MC38 cells. We found that the orthotopically transplanted Il22ra1-deficient left lung from the donor was protected from metastasis compared to the host Il22ra1-proficient right lung (Figures 5E and 5F), indicating that IL-22 acts on the endothelial cells of the target organ.
To test whether IL-22 would increase the permeability of the endothelial layer, thereby allowing enhanced extravasation of cancer cells, we performed an in vivo extravasation assay. We found that Il22- and Il22ra1-deficient mice as well as mice treated with an α-IL-22 antibody showed less cancer cell extravasation compared to littermate WT controls (Figures 5G–5J). Il22ra1flox/flox;Cdh5Cre+ mice lacking IL-22 signaling on the endothelium showed reduced extravasation into the lung (Figures 5K and 5L). Finally, we exposed mouse primary lung endothelial cells to rmIL-22, which resulted in increased Anpep expression (Figure S5C).
Next, we aimed to investigate the identity of IL-22-producing cells that participate in cancer cell extravasation into the lung. Using flow cytometry, we found that the relative contribution of T and γδ T cells of all IL-22-producing cells increased 12 h post cancer cell injection (Figure S5D). On this basis, we performed an extravasation assay in WT and Rag1−/− mice, which lack adaptive immune cells. We observed higher cancer cell extravasation in the lungs of WT compared to Rag1−/− mice, suggesting that adaptive immune cells affect cancer cell extravasation (Figures S5E and S5F). We then used Il22flox/flox;CD4Cre+ mice and found that T cell-derived IL-22 does not impact extravasation into the lung (Figures S5G and S5H). To test the role of iNKTs (invariant natural killer T cells), we used Ja18−/− mice, injected intravenously with MC38 Cherry-labeled cancer cells. No difference was observed in mice lacking iNKT cells compared to littermate controls (Figures S5I and S5J). Finally, we used diphtheria toxin-induced γδ T cell-deficient mice (TcrdGDL/GDL [GDL: GFP, human diphtheria toxin receptor, and luciferase knocked-in]) and found that these mice had reduced cancer cell extravasation into the lungs compared to controls (Figures S5K and S5L).
Taken together, IL-22 also acts on the lung endothelium, thereby inducing ANPEP and, consequently, cancer cell transmigration and metastasis formation.
IL-22-producing iNKT cells facilitate cancer cell extravasation into the liver parenchyma
Our next aim was to identify the source of IL-22 at the extravasation step of liver metastasis formation. To this end, we used an IL-22 reporter mouse model (IL-22sgBFP;IL-17AeGFP;Foxp3mRFP) and analyzed IL-22 expression 12 h post i.s. cancer cell injection. IL-22 was already expressed in steady state and was further upregulated 12 h post cancer cell injection (Figures 6A and 6B). The highest IL-22 frequency was observed in CD8+ T cells, but several other immune cells including CD3+CD4−CD8−immune cells (Figures 6C, 6D, and S6A) and ILCs expressed IL-22 (Figures S6B–S6G). To test whether ILCs are a relevant source of IL-22 in cancer cell extravasation, we performed an extravasation assay in Rag1−/− mice, which lack adaptive immune cells, and WT mice with IgG or a neutralizing IL-22 antibody. Treatment of WT but not Rag1−/− mice with the neutralizing IL-22 antibody resulted in reduced cancer cell extravasation (Figures 6E and 6F). In line with these data, both Rag1−/− and Rag1−/−Il22−/− mice showed a similar liver metastasis burden 21 days post cancer cell injection (Figure S6H). This finding suggests that adaptive immune cells promote cancer cell extravasation via IL-22. On this basis, we used Il22flox/flox;CD4Cre+ mice and found that IL-22 derived from CD4+ or CD8+ T cells does not impact cancer cell extravasation into the liver (Figures 6G and 6H). Based on these data, we hypothesized that a CD3+CD4−CD8−cell, which is not present in Rag1−/− mice, might be the functionally relevant source of IL-22. This could, for example, be iNKT or γδ T cells. To test this hypothesis, we engrafted Rag1−/−Il22−/− mice with the total population of iNKT or γδ T cells isolated from the livers of WT (Il22+/+) or Il22-deficient mice (Figures 6I and S6I–S6L). Indeed, we found that Rag1−/−Il22−/− mice harboring WT (Il22+/+) iNKT cells exhibited increased extravasation compared to mice harboring Il22−/− iNKT cells 24 h upon i.s. injection of labeled tumor cells (Figures 6I and 6J). On the contrary, Rag−/−Il22−/− mice receiving Il22+/+ or Il22−/− γδ T cells did not show a difference in cancer cell extravasation (Figures S6M and S6Q). Thus, in contrast to our observations in lung tissues, absence of γδ T cells had no impact on cancer cell extravasation into the liver parenchyma (Figures S6M–S6Q). Next, we used Ja18-deficient mice, which lack iNKT cells. These mice were largely protected from metastasis formation compared to littermate controls (Figure S6R–S6S). We then engrafted Ja18-deficient mice with iNKT cells. Engraftment with iNKT cells isolated from the livers of WT (Il22+/+) mice resulted in increased extravasation compared to mice harboring Il22−/− iNKT cells (Figures 6K and 6L). Finally, we aimed to examine whether iNKT cells would also express IL-22 in human liver metastasis (Figures S6T–S6V). Using flow cytometric analysis of human perimetastatic liver and liver metastasis from patients with primary CRC (M1, TNM staging), we found that iNKT cells were also able to produce IL-22 in human liver metastasis, particularly in the perimetastatic region (Figure S6V).
Figure 6.

iNKT cells promote cancer cell extravasation via IL-22
(A) Schematic overview of the experiment (i.v. injection of MC38 cells in IL-22 reporter mice).
(B) Il22 mRNA levels in the liver at steady state, 12 and 24 h post i.s. MC38 cell injection.
(C) Representative concatenated FACS plots indicating mean and statistics of CD45+, CD3−, CD3+CD4+, CD3+CD8+, and CD3+CD4−CD8− IL-22-producing cells in the liver at 0 and 12 h post i.s. MC38 cell injection.
(D) Il22 mRNA levels from indicated cell populations sorted from the liver at 0 and 12 h post i.s. MC38 cell injection.
(E) Schematic overview of the experiment (i.s. injection of MC38 GFP-labeled cells in WT and Rag1−/− mice, receiving an α-IL-22 or IgG control antibody).
(F) Representative FACS plots and statistics. n ≥ 4 mice per group.
(G) Schematic overview of the experiment (i.s. injection of MC38 GFP-labeled cells in Il22wt/wt;CD4Cre+ and Il22flox/flox;CD4Cre+ mice). n ≥ 5 mice per group.
(H) Representative FACS plots and number of extravasated cancer cells 24 h post i.s. injection. n ≥ 4 mice per group.
(I) Schematic overview of the experiment (i.s. injection of MC38 GFP-labeled cells in Rag−/−Il22−/− mice engrafted with Il22+/+or Il22−/− iNKT cells).
(J) Representative FACS plots and number of extravasated cancer cells 24 h post i.s. injection. n ≥ 10 mice per group.
(K) Schematic overview of the experiment (i.s. injection of MC38 Cherry-labeled cells in Ja18−/− mice engrafted with Il22+/+or Il22−/− iNKT cells).
(L) Representative FACS plots and number of extravasated cancer cells 24 h post i.s. injection. n ≥ 10 mice per group. Data presented as mean ± SEM, ns p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by one-way ANOVA with Bonferroni post hoc tests (B and F) or Mann-Whitney U test (C, D, H, J, and L).
Thus, iNKT cells in the liver can produce IL-22 in mice and humans. Furthermore, IL-22 derived from iNKT cells plays a key functional role during cancer cell extravasation into the liver parenchyma.
Tissue resident iNKT17 IL-22-producing cells facilitate cancer cell extravasation
A tissue resident phenotype could explain why IL-22 is expressed in the target organ prior to metastasis formation. To test this, WT and IL-22 reporter mice were injected with MC38 cancer cells intrasplenically. 12 h post cancer cell injection, the mice received an α-CD45-AF700 antibody intravenously in order to label circulating immune cells and were sacrificed 2–3 min later. Using flow cytometric analysis, we found that the frequency of CD45-PeCy7 single-positive (infiltrating) cells increased upon cancer cell injection (Figures 7A and 7B). We then aimed to assess the relative contribution of IL-22-producing iNKT cells to the total amount of IL-22-producing CD3+ cells. We found that the relative contribution of iNKT cells accounted for about 13% of all CD3+IL-22-producing cells and increased to about 21% 12 h post cancer cell injection (Figures 7C and S7). There was a higher frequency of infiltrating iNKT cells producing IL-22 compared to circulating ones (Figures 7C and S7). We further assessed the contribution of IL-22-producing iNKT cells within IL-22-producing CD45+ cells. The frequency of IL-22-producing iNKT cells, in particular the frequency of infiltrating IL-22-producing iNKT cells, also increased within this cell fraction, while the frequency of γδ T cells, which was measured as a control, stayed the same (Figures 7D, 7E, and S7). In order to better characterize the iNKT cells 12 h post i.s. injection, we used flow cytometry and single-cell sequencing. Using flow cytometry, we found that among iNKT cells, the production of IL-22 was almost exclusive to iNKT17 cells. Only a very small percentage of iNKT1 or iNKT2 cells produced IL-22. Furthermore, the single cell sequencing data demonstrated that iNKT cells express tissue resident markers, namely Tmem176a and Tmem176b (Figures 7F, 7G, and S7).37 We could not test the iNKT17 phenotype of the iNKT cells using the single-cell sequencing data, since Il17a and Il22 were barely detectable and only a very small percentage of Rorc-positive cells were found (Figure S7). Finally, we performed parabiosis experiments, thereby creating parabiotic pairs between CD45.2 and CD45.½ mice.38 Fourteen days post connection (Figure 7H), we injected MC38 cells or PBS into the portal vein of the CD45.2 members of the pairs (Figures 7I and 7J). Twelve hours post injection, we found that the majority of liver CD45+IL-22+ cells were in fact tissue resident cells (Figure 7K). In addition, we created pairs consisting of either two WT (littermate controls of Il22-deficient mice) mice or WT and Il22-deficient mice (Figure 7L). After 14 days, MC38 GFP-labeled cells were injected into the portal vein of both mice of the pair, which were then sacrificed 24 h later. Il22-deficient mice showed less extravasation compared to WT mice of the same pair (Figures 7M and 7N). WT mice paired with WT (littermate controls of Il22−/− mice) mice showed an equal extravasation number, suggesting that tissue-resident cell-derived IL-22 promotes extravasation of cancer cells.
Figure 7.

IL-22-producing iNKT17 cells are tissue resident and facilitate cancer cell extravasation
(A) Injection of an α-CD45 antibody intravenously after i.s. injection of MC38 cells.
(B) Representative FACS plots and statistics 0 and 12 h post i.s. injection. n ≥ 10 mice per group.
(C) Pie charts showing the proportion of infiltrating and circulating immune subsets among CD3+IL-22+ cells in murine liver 0 and 12 h post i.s. injection.
(D) Concatenated FACS plots of iNKT and γδ T cells gated on CD45+IL-22+ cells, including in vivo and in vitro CD45 staining. n ≥ 8 mice per group.
(E) Statistics of iNKT and γδ T cells gated on CD45+IL-22+ cells, including in vivo and in vitro CD45 staining. n ≥ 8 mice per group.
(F) Representative FACS plots showing the immune characterisation of iNKT cells gated on CD1d-tetramer+IL-22+ cells 12 h upon i.s. cancer cell injection.
(G) Representative FACS plots and statistics showing the immune subtypes (iNKT1, iNKT2, and iNKT17) of iNKT cells gated on CD1d-tetramer+IL-22+ cells 12 h upon i.s. cancer cell injection.
(H) CD45.2 reporter mice were connected together with CD45.1 (n = 3 mice per group) mice and the established blood chimerism was checked 14 days later.
(I) CD45.2 reporter mice were connected together with CD45.½ mice (n = 12 mice per group), followed by intraportal cancer cell injection after established blood chimerism.
(J) Representative FACS plots of CD45.½ and CD45.2 cells from parabiotic pairs.
(K) The proportion of CD45.½ and CD45.2 cells of parabiotic pairs was quantified.
(L) WT mice were connected with WT or Il22−/− mice (n ≥ 8 mice per group), followed by intraportal injection of MC38 GFP-labeled cells.
(M and N) Mice were sacrificed and the number of extravasated cancer cells was quantified. Data presented as mean ± SEM, ns p > 0.05; ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 as assessed by Mann-Whitney U test (B, E, and K) or two-tailed Wilcoxon matched-pairs signed rank test (N).
Taken together, tissue resident iNKT17 cells are the functional relevant source of IL-22 during cancer cell extravasation.
Discussion
Metastasis is responsible for the majority of cancer-related deaths worldwide.39 The secondary tumor development and growth are influenced by tumor-infiltrating immune cells,40 which can either promote or inhibit metastasis development.41,42,43 However, the role of the immune cells that reside at the target site prior to its colonization with cancer cells in metastasis formation remains elusive. Thus, it is unclear whether tissue resident immune cells facilitate or inhibit metastasis. Consequently, it is unknown whether these cells and their cytokines could be targets for novel immunotherapies.
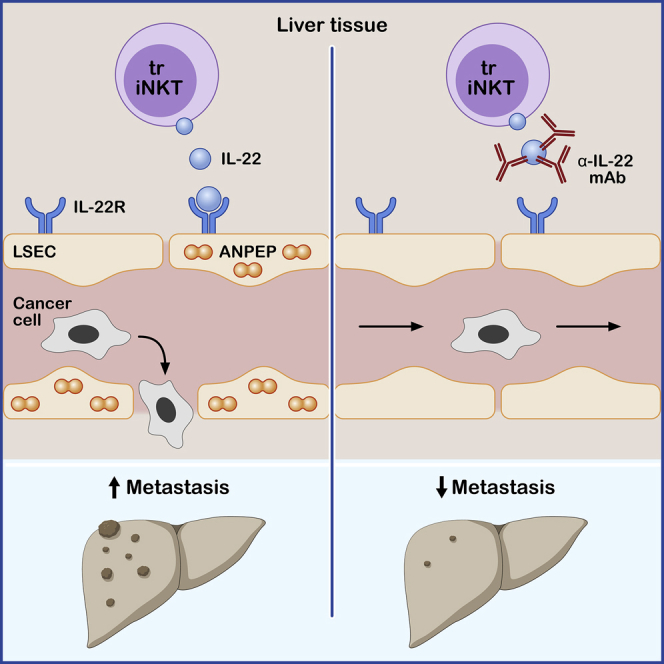
We report here that IL-22 is produced by tissue resident iNKT17 cells and mediates the immune system – cancer cell – tissue-specific stromal cell crosstalk, thereby facilitating cancer cell extravasation and thus liver metastasis formation. IL-22 increased endothelial permeability, thereby facilitating cancer cell extravasation. The consequence was tissue invasion, the critical step in metastasis formation. This effect was mediated by an IL-22-mediated upregulation of ANPEP expression in LSECs (see graphical abstract).
ANPEP is an aminopeptidase produced by a plethora of cells.44 Interestingly, myeloid-derived ANPEP expression facilitates metastasis formation because Anpep-deficient mice show reduced lung metastases compared to Anpep-deficient mice transplanted with bone marrow from WT donors.36 We extend this finding and we suggest that ANPEP aminopeptidase is an IL-22 target gene that increases endothelial permeability when expressed in the murine endothelium.
Several studies have reported that iNKT cells interact with liver sinusoids via the CXCL16-CXCR6 axis.45,46 Interestingly, CXCL16 secretion by liver sinusoids has been linked to anti-tumor and anti-metastatic activity of iNKT cells in the liver.46 Thus, we assessed whether this pathway also correlates with IL-22 production. However, this was not the case. Therefore, it seems that while the anti-tumorigenic functions of iNKT cells are linked to the CXCL16-CXCR6 axis, the pro-metastatic functions of iNKT cells shown here are linked to the IL-22-ANPEP axis. Interestingly, this effect of IL-22 as a promoter of cancer cell extravasation was not only found in the liver, but also in lung. The liver and the lungs both have extended endothelium, but their endothelial cells are markedly different.47 In contrast to the lung, the liver is characterized by unique capillaries that differ from other capillaries in the body because of the presence of open pores. However, the endothelial permeability cannot fully explain the higher metastatic rates and metastatic organotropism exhibited by various forms of cancer. For example, primary CRC exhibits high metastatic rates both in the liver and the lung.48 Regardless of these differences, our findings indicate that an IL-22 blockade could serve as a therapy to prevent both liver and lung metastasis. Indeed, from a therapeutic point of view, the extravasation process where the circulating cancer cells interact with the endothelium of the target organ is likely a rate-limiting step of metastasis formation. Here, cancer cell – endothelial cell interaction is required, and endothelial intercellular junctions are loosened in order to allow cancer cell transmigration into the stroma of the host organ.49 IL-22 blockade could therefore have a major therapeutic effect. Indeed, we could show that Fezakinumab, a human monoclonal IL-22 antibody, which is already tested in Phase II trials for atopic dermatitis,50 was able to block cancer cell extravasation in a humanized mouse model.51
Of note, cancer cells, like the endothelial cells, were responsive to IL-22, indicating that IL-22 can also mediate the communication between the immune system and the cancer cells. Indeed, IL-22RA1 silencing in cancer cells reduced the metastatic sites in an environment with high IL-22 levels. This finding shows an additive effect of IL-22 on cancer cells that express IL-22RA1. Thus, IL-22 promotes cancer cell extravasation. However, this does not exclude the fact that IL-22 has additional pro-metastatic effects. Indeed, other studies reported a direct tumorigenic effect of IL-22 on cancer cells, thereby promoting survival and proliferation.52
Many different cell types are able to produce IL-22 in liver metastasis. However, we found that tissue resident iNKT cells represent one major functionally relevant source of IL-22 in the early extravasation step of cancer cells. In this phase, iNKT17-cell-derived IL-22 acts on endothelial cells and increases their permeability to extravasating cancer cells. Importantly, our data indicate that iNKT cells control metastasis formation in the liver but not in the lungs. Indeed, we found low frequencies of IL-22-producing iNKT cells in the lung. Instead, we identified that γδ T cells represent the relevant cellular source of IL-22 in this setting and control the extravasation of cancer cells into the lungs. Thus, we speculate that, depending on the target organ, different cells can regulate the extravasation step of metastasis formation by producing IL-22. Interestingly, it has been reported before that NKT cells can have tissue residency and an effector-memory phenotype.53,54 Indeed, we found here that IL-22-producing iNKT17 cells are tissue resident and play an essential role during cancer cell extravasation. It is tempting to speculate that this specific effect of iNKT17 cells is due to their tissue residency and presence in the liver even prior to the arrival of the circulating cancer cells.
NKT cells were best known for their anti-tumorigenic function.46 However, emerging data involving innate-like T cells (NKT and MAIT [mucosal-associated invariant T cells]) indicate that these cells might also have a tumor-promoting function.55 For example, it was published that invariant NKT cells are pro-tumorigenic in the colon,56 but this activity disappears when a-GalCer is administered to the mice.57 Furthermore, Molgora et al. showed that Il1r8−/− mice were protected from the development of liver metastasis.58 Of note, these mice had increased numbers of NK cells and also a more mature NK cell phenotype. Furthermore, the adoptive transfer of Il1r8−/− NK cells also protected mice from the development of MC38 liver metastasis. Thus, Il1r8−/−NK cells seem to play a protective role in metastasis. However, this study did not specifically investigate the role of tissue resident iNKT cells or the effect of IL-22 in this process. For our study, we wanted to address two points that remained open: whether IL-22-producing iNKT17 cells are tissue resident and, if so, whether they contribute to metastasis formation. Indeed, we found both to be affirmative. However, the mechanisms regulating IL-22 production in iNKT17 cells require further investigation.
In conclusion, our findings allowed us to identify the mechanisms by which tissue resident immune cells mediate the immune system – cancer cell – tissue-specific stromal cell crosstalk and revealed how tissue-resident iNKT-cell-derived IL-22 promotes extravasation during metastasis formation by acting on endothelial cells and inducing ANPEP expression. We would like to stress that the pro-metastatic effect of IL-22 can in fact be targeted therapeutically. When we neutralized IL-22 using an antibody, it blocked cancer cell extravasation and thus prevented metastasis formation in the target organ both in a murine model and also in a humanized mouse model. Based on these data, we propose targeting IL-22, e.g., by using Fezakinumab, as immunotherapy in patients suffering from established or impending metastasis.
Limitations of the study
We found that IL-22 promotes both lung and liver metastasis. However, the cellular mechanism involving iNKT cells as IL-22 producers in aminopeptidase regulation on endothelial cells has only been shown for the hepatic environment. Therefore, further studies are critical to extend this finding to other organs. Indeed, in the lung, γδ T cells seem to be the functionally relevant source of IL-22 during metastasis. Thus, depending on the target organ, different cells may regulate metastasis formation. Further studies are required to test whether this difference is just due to different number, location, or function of iNKT and γδ T cells.
Other genes, besides ANPEP, known to promote cancer cell invasion and angiogenesis might also contribute to the effects of IL-22. We found that, besides ANPEP, NRP1 was also impacted by IL-22. Furthermore, we found that IL-22 can regulate VEGF (vascular endothelial growth factor) expression in vitro. Thus, we cannot exclude that other factors besides ANPEP may play an additional role in mediating the pro-metastatic effect of IL-22, and further studies will be critical in order to address this.
Blockade of IL-22 protected WT mice from developing forced liver metastasis. Testing this therapeutic regimen in Apc15lox;KrasG12D mice, which develop spontaneous cancer, was not feasible, since only mice with hydrodynamic overexpression of IL-22 in the liver and no control mice developed liver metastasis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD45 | BioLegend | Cat# 304010; RRID: AB_314398 |
| Anti-human CD3 | BD Bioscience | Cat# 564307; RRID: AB_2744390 |
| Anti-human CD4 | BioLegend | Cat# 357410; RRID: AB_2565662 |
| Anti-human IL-22 | eBioscience | Cat# 12-7229-42; RRID: AB_1834463 |
| Human PBS57-loaded CD1d tetramer | NIH tetramer core facility | N/A |
| Anti-mouse CD45 | BioLegend | Cat# 103149; RRID: AB_2564590 |
| Anti-mouse CD45 | BioLegend | Cat# 103114; RRID: AB_312979 |
| Anti-mouse CD45 | BioLegend | Cat# 147716; RRID: AB_2750449 |
| Anti-mouse CD45.1 | BioLegend | Cat# 110714; RRID: AB_313503 |
| Anti-mouse CD45.2 | BioLegend | Cat# 109830; RRID: AB_1186098 |
| Anti-mouse CD3 | BioLegend | Cat# 100216; RRID: AB_493697 |
| Anti-mouse CD3 | BioLegend | Cat# 100206; RRID: AB_312663 |
| Anti-mouse CD3 | eBioscience | Cat# 35-0031-82; RRID: AB_11219266 |
| Anti-mouse CD8 | BioLegend | Cat# 100722; RRID: AB_312761 |
| Anti-mouse CD4 | BD Bioscience | Cat# 563790; RRID: AB_2738426 |
| Anti-mouse TCR γ/δ | BioLegend | Cat# 118116; RRID: AB_1731813 |
| Anti-mouse IL-22RA1 | R&D | Cat# FAB42941P; RRID: AB_1964624 |
| Anti-mouse IgG | R&D | Cat# IC006P; RRID: AB_357256 |
| Anti-mouse pSTAT3 | BD Bioscience | Cat# 560312; RRID: AB_1645327 |
| Anti-mouse Lin-1 | BioLegend | Cat# 133313; RRID: AB_2715571 |
| Mouse PBS57-loaded CD1d tetramer | NIH tetramer core facility | N/A |
| Anti-mouse CD90.1 | BioLegend | Cat# 202526; RRID: AB_1595470 |
| Anti-mouse CD90.1 | BioLegend | Cat# 202537; RRID: AB_2562644 |
| Anti-mouse CD90.1 | BioLegend | Cat# 202518; RRID: AB_1659223 |
| Anti-mouse CD90.1 | BioLegend | Cat# 202529; RRID: AB_10899572 |
| Anti-mouse CD90.2 | BioLegend | Cat# 105320; RRID: AB_493725 |
| Anti-mouse CD90.2 | BioLegend | Cat# 105343; RRID: AB_10643586 |
| Anti-mouse CD90.2 | BioLegend | Cat# 140310; RRID: AB_2632889 |
| Anti-mouse IL-7Ra | BioLegend | Cat# 135020; RRID: AB_1937209 |
| Anti-mouse IL-7Ra | BioLegend | Cat# 135014; RRID: AB_1937265 |
| Anti-mouse CD3 | BioLegend | Cat# 100303; RRID: AB_312668 |
| Anti-mouse CD4 | BioLegend | Cat# 100507; RRID: AB_312710 |
| Anti-mouse CD5 | BioLegend | Cat# 100603; RRID: AB_312732 |
| Anti-mouse CD8 | BioLegend | Cat# 100703; RRID: AB_312742 |
| Anti-mouse CD11b | BioLegend | Cat# 101203; RRID: AB_312786 |
| Anti-mouse CD11c | BD Bioscience | Cat# 553800; RRID: AB_395059 |
| Anti-mouse CD19 | BioLegend | Cat# 115503; RRID: AB_313638 |
| Anti-mouse CD49b | BioLegend | Cat# 103521; RRID: AB_2566365 |
| Anti-mouse GR-1 | BioLegend | Cat# 108403; RRID: AB_313368 |
| Anti-mouse NK1.1 | BioLegend | Cat# 108703; RRID: AB_313390 |
| Anti-mouse TCR gd | BioLegend | Cat# 118103; RRID: AB_313827 |
| Anti-mouse TCRb | BioLegend | Cat# 109203; RRID: AB_313426 |
| Anti-mouse Ter119 | BioLegend | Cat# 116204; RRID: AB_313705 |
| Anti-Fc- γ receptors | Samuel Huber Lab, UKE, Germany | clone 2.4G2 |
| Streptavidin | BD Bioscience | Cat# 564176 |
| Anti-GFP rabbit | Life-technologies | Cat# A11122; RRID: AB_221569 |
| AF488 donkey anti-mouse IgG | Life-technologies | Cat# A21202; RRID: AB_141607 |
| AF488 goat anti-rat IgG | Invitrogen | Cat# A21208; RRID: AB_141709 |
| AF568 goat anti-chicken IgG | Life-technologies | Cat# A11041; RRID: AB_2534098 |
| AF568 goat anti-rat IgG | Life-technologies | Cat# A11077; RRID: AB_2534121 |
| AF568 donkey anti-goat IgG | Life-technologies | Cat# A11057; RRID: AB_2534104 |
| AF568 donkey anti-rabbit IgG | Life-technologies | Cat# A10042; RRID: AB_2534017 |
| AF633 goat anti-rat IgG | Life-technologies | Cat# A21094; RRID: AB_2535749 |
| CD31 | Abcam | Cat# ab24590; RRID: AB_448167 |
| Endomucin | Santa Cruz | Cat# sc-53941 RCID: AB_2100037 |
| rat anti-mouse CD31 | BD Bioscience | Cat# 550274; RRID: AB_393571 |
| CD31 Rat anti-mouse | BD Bioscience | Cat# 553369; RRID: AB_394815 |
| anti-IL-22 ab | Genentech | Cat# 8E11.9; RRID: AB_2651129 |
| IgG control ab | Genentech | Cat# 10E7 |
| GAPDH (6C5) mouse | Santa Cruz Biotechnology | Cat# sc-32233; RRID: AB_627679 |
| anti-rabbit IgG HRPlinked Antibody | Cell Signaling | Cat# 7074; RRID: AB_2099233 |
| Stat3 (124H6) mouse | Cell Signaling | Cat# 9139; RRID: AB_331757 |
| pStat3 (Y705) rabbit | Cell Signaling | Cat# 9131; RRID: AB_331586 |
| ANPEP | Merck Millipore | Cat# MABC950 |
| Polyclonal Swine anti-Rabbit Immunoglobulins/HRP | Dako | Cat# P0214 |
| Polyclonal Rabbit anti-Mouse Immunoglobulins/HRP | Dako | Cat# P0161 |
| Bacterial and virus strains | ||
| Stellar™ Competent Cells | Clontech | Cat# 636763 |
| Adenovirus Cre | Cell Biolabs | ADV-005 |
| Chemicals, peptides, and recombinant proteins | ||
| rmIL-22 (recombinante murine IL-22) | PeproTech | Cat# 210-22 |
| rmIL-6 (recombinante murine IL-6) | BioLegend | Cat# 715202 |
| IL-1α | PeproTech | Cat# 211-11A |
| TNF-α | Peprotech | Cat# 315-01A |
| Agarose | Biozym Scientific GmbH | Cat# 849004 |
| IGEPAL® CA-630 | Sigma-Aldrich | Cat# I3021 |
| Formaldehyde | Sigma-Aldrich | Cat# F8775 |
| Ubenimex | Sigma-Aldrich | Cat# B8385 |
| BSA | Biomol | Cat# 9048-46-8 |
| Lumisensor Chemiluminescent HRP Substrate | Genscript | Cat# L00221V300 |
| DNase | AppliChem | Cat# A3778 |
| Collagenase | Sigma-Aldrich | Cat# C2139 |
| AccuCount Blank Particles 8.0–12.9 μm | Spherotech | Cat# ACBP-100-10 |
| Ionomycin | Sigma-Aldrich | Cat# I9657 |
| Monensin A | BioLegend | Cat# 420701 |
| PMA (phorbol 12-myristate 13-acetate) | Sigma-Aldrich | Cat# P8139 |
| peqGOLD TriFast™ | Peqlab | Cat# 30-2010 |
| peqGOLD TriFast™ FL | Peqlab | Cat#30-2110 |
| DMSO | Invitrogen | Cat# C34557 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Luciferin | Biosynth | Cat# FL08608 |
| Hoechst 33,258 | Sigma-Aldrich | Cat# 94403 |
| Zombie UV™ Fixable Viability | BioLegend | Cat# 423108 |
| Fixable Viability Dye eFluor™ 506 | Invitrogen | Cat# 65-0866-14 |
| EcoRI | New England Biolabs | Cat# R0101S |
| Superscript III reverse transcriptase | Invitrogen | Cat# 18080044 |
| Phusion® Hot Start Flex DNA polymerase | New England Biolabs | Cat#M0535S |
| In-Fusion® HD Cloning Plus | Takara | Cat# 638916 |
| TransIT®-LT1 reagent | Mirus | Cat# MIR2300 |
| Transwell Permeble Supports | Costar | Cat# 3464 |
| Transwell Permeble Supports | Costar | Cat# 3470 |
| Macherey-Nagel™ NucleoSpin™ Gel and PCR Clean-up Kit | Macherey-Nagel™ | Cat# 740609.50 |
| GeneJet plasmid mini prep kit | ThermoFischer | Cat# K0503 |
| Percoll | GE Healthcare | Cat# GE17-0891-01 |
| Polyvinylidene difluoride membranes | Merck Millipore | Cat# IPFL00010 |
| IL-22-ELISA | Peprotech | Cat# 900-K257 |
| ECL substrate | Merck Millipore | Cat# WBULS0100 |
| Liberase | Roche Diagnostics | Cat# 05578566001 |
| O.C.T. | Sakura | Cat# 4583 |
| FBS | Life Technologies | Cat# 16000044 |
| Critical commercial assays | ||
| CytoSelect Tumor-endothelium adhesion assay | Cell Biolabs | Cat# CBA-215 |
| RT2 Profiler™ PCR Array Mouse Angiogenesis | Qiagen | PAMM-024Z |
| Colonoscopy system | Karl Storz | custom-made |
| stereoscope | Olympus Corporation | custom-made |
| Axio Vert.A1- inverted microscope | Zeiss | custom-made |
| SP5-confocal microscope | Leica | custom-made |
| small-animal ventilator | UNO Apparatus | Cat# 55-0000 |
| epithelial Ohm-voltmeter | Merck Millipore | Cat# MERS00002 |
| ibidiTreat μ-slide IV0.4 | ibidi GmbH | Cat# 80606 |
| Experimental models: Cell lines | ||
| Mouse: MC38 | Brunner Lab Neumaier Lab |
University of Konstanz, Germany University of Heidelberg |
| Mouse: MC38-eGFP (peGFP-C1) | This paper | N/A |
| Mouse: MC38-mCherry (LeGO-C2) | This paper | N/A |
| Mouse: MC38shC | This paper | N/A |
| Mouse: MC38shIl22ra1 clone 1 | This paper | N/A |
| Mouse: MC38shIl22ra1 clone 1 | This paper | N/A |
| Mouse: CT26-Luc-GFP-12- | TRON-University Mainz | N/A |
| Mouse: LLC | ATCC | CRL-1642, RRID: CVCL_4358 |
| Human: HT29 | Schumacher Lab – UKE Hamburg, Germany | HTB-38, RRID: CVCL_0320 |
| Human: HT29GFP (pEGFPC1) | This paper | N/A |
| Human: HUVEC | Loges Lab – UKE Hamburg, Germany | #CRL-1730, RRID: CVCL_2959 |
| Human: HUVECshC | This paper | N/A |
| Human: HUVECshANPEP1 | This paper | N/A |
| Human: HUVECshANPEP2 | This paper | N/A |
| Human: HUVECshANPEP3 | This paper | N/A |
| Human: HUVECshANPEP4 | This paper | N/A |
| Human: HUVECshANPEP5 | This paper | N/A |
| Human: HUVECshANPEP6 | This paper | N/A |
| Human: HEK293T | ATCC | #CRL-3216, RRID: CVCL_0063 |
| Human: HEK293T-Il22eGFP (pIl22eGFP) | This paper | N/A |
| Human: HEKT293GFP (peGFPC1) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | Stock:000,664; RRID:IMSR_JAX:000,664 |
| Mouse: BALB/c: Balb/cAnNCrl | Charles River | Stock:028; RRID:IMSR_CRL:028 |
| Mouse: Il22−/−: B6;129SvF1-Il22tm1Flv/J | Flavell Lab – Yale University, USA | RRID:MGI:3761616 |
| Mouse: Rag1−/−: B6.129S7-Rag1tm1Mom/J | Flavell Lab – Yale University, USA | Stock:002,216; RRID:MGI:3582299 |
| Mouse: CD4Cre+: B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | Flavell Lab – Yale University, USA | Stock:022,071; RRID:IMSR_JAX:022,071 |
| Mouse: Il22flox/flox: B6;129S5-Il22tm1.1Lex/Mmucd | MMRRC, Lexicon Genetics | Stock:036745-UCD; RRID: MMRRC_036745-UCD |
| Mouse: Il17ACre+: B6.129(SJL)-Il17atm1.1(icre)Stck/RthsnJ | Flavell Lab – Yale University, USA | Stock:034,347; RRID:IMSR_JAX:034,347 |
| Mouse: Il22ra1flox/flox: B6.Cg-Il22ra1tm1.1Koll/J | Strowig Lab - Helmholtz-Centre for Infection Research, Germany | Stock:031,003; RRID:IMSR_JAX:031,003 |
| Mouse: Foxp3RFP: B6.Cg-Foxp3tm1Flv/J | Flavell Lab – Yale University, USA | Stock:008,374; RRID:IMSR_JAX:008,374 |
| Mouse: Il17aeGFP: B6.Cg-Il17atm1.1Flv/J | Flavell Lab – Yale University, USA | RRID:MGI:5006666 |
| Mouse: Il17aFP635 | Flavell Lab – Yale University, USA | N/A |
| Mouse: Il22sgBFP | Flavell Lab – Yale University, USA | N/A |
| Mouse: Il10eGFP: B6.Cg-Il10tm1Flv/J | Flavell Lab – Yale University, USA | Stock:008,379; RRID:IMSR_JAX:008,379 |
| Mouse: Cdh5Cre+: B6.FVB-Tg(Cdh5-cre)7Mlia/J (backcrossed with C57BL/6 for at least 12 generations) | Herkel Lab – UKE Hamburg, Germany | Stock:006,137; RRID:IMSR_JAX:006,137 |
| Mouse: AlbCre+: B6.FVB(129)-Tg(Alb1-cre)1Dlr/J (backcrossed with C57BL/6 for at least 12 generations) | Herkel Lab – UKE Hamburg, Germany | Stock:016,832; RRID:IMSR_JAX:016,832 |
| Mouse: KrasG12D: B6.129S4-Krastm4Tyj/J | Jackson Laboratories | Stock:008,179; RRID:IMSR_JAX:008,179 |
| Mouse: Apc15lox: B6.129P2-Apctm1Rsmi/RfoJ | Ron Smits - Erasmus University Medical Center | Stock:029,275; RRID:IMSR_JAX:029,275 |
| Mouse: RosaYFP: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | Jackson Laboratories | Stock:006,148; RRID:IMSR_JAX:006,148 |
| Mouse: Il22tg8(Tg): | (B6N Background) | Gao, NIH |
| Mouse: Cd13−/− | Renata Pasqualini Lab- Rutgers Cancer Institute of New Jersey and Division New Jersey, USA | N/A |
| Mouse: Ja18−/−: B6(Cg)-Traj18tm1.1Kro/J | Paolo Dellabona Lab- Experimental Immunology Unit, San Raffaele Scientific Institute, Milano | Stock:030,524 RRID:IMSR_JAX: 030,524 |
| Mouse: Il22ra1−/−: | Till Strowig Lab-Helmholtz Center for Infection Research, Braunschweig, Germany | N/A |
| Mouse: CD45.1: B6.SJL-PtprcaPepcb/BoyJ | Flavell Lab – Yale University, USA | Stock:002,014; RRID:IMSR_JAX:002,014 |
| Mouse: MISTRG: C; 129S4-Rag2tm1.1FlvCsf1tm1(CSF1)FlvCsf2/Il3tm1.1(CSF2,IL3)FlvThpotm1.1(TPO)FlvIl2rgtm1.1Flv Tg(SIRPA)1Flv/J | Flavell Lab – Yale University, USA | Stock:017,712 RRID:IMSR_JAX:017,712 |
| Oligonucleotides | ||
| Oligonucleotides for qPCR, PCR and cloning | This paper | See Table S1 |
| Oligonucleotides for sgRNA cloning | This paper | See Table S2 |
| Recombinant DNA | ||
| Plasmid: pCMV-EGFP C1 | Clontech | This plasmid has been discontinued by Clontech |
| Plasmid: peGFP-C1 | Clontech | This plasmid has been discontinued by Clontech |
| Plasmid: pIl22eGFP | This paper | N/A |
| Plasmid: pGEM®-T Easy vector | Promega | A1360 |
| Plasmid: GAC.Luc-puro | Addgene30 | RRID:Addgene_74409 |
| Plasmid: LeGO-C2 | Addgene59 | RRID:Addgene_27339 |
| Software and algorithms | ||
| Adobe Illustrator CC 2017 | Adobe | https://www.adobe.com/de/products/illustrator.html |
| Flowjo 10 | Tree Star | https://www.flowjo.co |
| GraphPad Prism 8.0 | GraphPad | https://www.graphpad.com |
| ImageJ 1.52. | ImageJ | https://imagej.nih.gov/ij/ |
| Image Lab 5.2 | BioRad | https://www.bio-rad.com/ |
| CapImage software V8.6 | Dr. Heinrich Zeintl | http://www.drzeintl.de |
| SteponeSoftware 2.1 | Thermfischer | https://www.thermofisher.com/de/de/home.html |
| Microsoft Office 2016 | Microsoft | https://products.office.com |
| BD FACSDiva | BD Biosciences | https://www.bdbiosciences.com/en-in |
| Snapgene | GSL Biotech | https://www.snapgene.com/ |
| SnapGene Viewer | GSL Biotech | https://www.snapgene.com/ |
| G∗power | HHU Düsseldorf | http://www.gpower.hhu.de/ |
| Cell Ranger v3.1.0 | 10x Genomics | https://www.10xgenomics.com |
| R | R Core | https://www.r-project.org/ |
Resource availability
Lead contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact, Anastasios Giannou (a.giannou@uke.de).
Materials availability
All data necessary to understand and assess the conclusions of the manuscript are available in the body of the paper and in the supplementary materials. Il22−/− (VG437) mice are available under a material transfer agreement with Regeneron. Il22flox mice are available under a material transfer agreement with Lexicon and Genentech. α-IL-22 treatment antibody was kindly provided by Genentech under a material transfer agreement with the University Medical Center Hamburg-Eppendorf. Apc15lox mice are available under a material transfer agreement with Prof. Dr. Ron Smits from Erasmus University Medical Center. Ja18−/− mice were kindly provided by Dr. Paolo Dellabona at Ospedale San Raffaele - Milano (Italy). MISTRG (humanized mice) were kindly provided by Dr. Richard Flavell as part of an ongoing collaboration. All reagents generated or used in this study are available on request from the lead contact with a completed Materials Transfer Agreement. Information on reagents used in this study is available in the key resources table.
Further information and requests for resources and reagents should be directed to Anastasios Giannou (a.giannou@uke.de) and Samuel Huber (s.huber@uke.de).
Method details
Reagents
IL-22 neutralizing antibodies and IgG2a control were provided by Genentech (San Francisco, CA). rmIL-22 and rmTNF-α were from Peprotech (London, UK), rmIL-1a and rhIL-22 from R&D Systems (Minneapolis, MN), ELISA kits from Peprotech, R&D or Antigenix America and Boyden chambers from Millipore (Billerica, MA).
Cells
Lewis lung carcinoma (LLC), human cell line expressing mutant version of the SV40 large T antigen (HEK293T) (ATCC, Manassas, VA), colon adenocarcinoma (MC38) syngenic for C57BL/6 mice, colon adenocarcinoma (CT26) syngenic for Balb/c mice,60,61 human colon carcinoma (HT29)62 and Human Umbilical Vein Endothelial (HUVEC) cells63 were cultured and tested as described in the Supplemental Experimental Procedures.
Animals
C57BL/6J, C57BL/6N, Balb/c Il22+/+, Il22−/−, Il22ra1−/−, Il22tg8Tg, Apc15lox;LSL-KrasG12D, IL-22sgBFP;IL-17AsgGFP;Foxp3mRFP, IL-22sgBFP;IL-17AKatushka;Foxp3mRFP;IL-10sgGFP, Il22ra1flox/flox;AlbCre+, Il22ra1flox/flox;Cdh5Cre+, Cd13−/−(Anpep knock out), MISTRG (humanized mice), Il22ra1flox/flox;Cdh5Cre+;R26YFP, Il22flox/flox;CD4Cre+, Ja18−/−, Rag−/− and Rag−/−;Il22−/−, TcrdGDL/GDL and Tcrd+/+ mice64,65,66 were bred and housed under specific pathogen-free conditions in the animal facility of the University Medical Center Hamburg Eppendorf. Ja18−/− mice were kindly provided by Dr. Paolo Dellabona at Ospedale San Raffaele - Milano (Italy). MISTRG (humanized mice) were kindly provided by Dr. Richard Flavell as part of an ongoing collaboration. Age- and sex-matched littermates between 8 and 16 weeks of age were used. Animal experiments were carried out in accordance with the Institutional Animal Care and Use Committee of Yale University or the Institutional Review Board “Behörde für Justiz und Verbraucherschutz, Lebensmittelsicherheit und Veterinärwesen” (Hamburg, Germany).
Isolation of hematopoietic cells from murine and human liver metastasis
Hematopoietic cells were isolated from macroscopically healthy human liver, perimetastatic liver and human liver metastasis or murine liver or lung metastasis. Human tissues were obtained freshly after surgical removal of metastases from patients diagnosed with CRC and liver metastasis. Human studies were approved by the local ethical committee (Ethik-Kommission der Ärztekammer Hamburg). To isolate the lymphocytes, the human or murine tissues were cut into small pieces and minced using a scalpel. The healthy liver or metastasis tissue was incubated for 30 min at 37°C on a shaking incubator in HBSS (with Ca2+ and Mg2+) with Collagenase (1 mg/mL) and DNase I (10 U/ml) and supernatant was collected. Leukocytes were further enriched by Percoll gradient centrifugation (GE Healthcare, Chicago, IL).35
Fluorescence-activated cell sorting
Fc-γ receptors were blocked using a mAb (clone 2.4G2). The cells were stained with fluorochrome-conjugated antibodies. BD LSRFortessa and FACSAria (BD Biosciences, San Jose, CA) were used for cell analysis and cell sorting, respectively. Data were analyzed using FlowJo v.6.1 (TreeStar, Ashland, OR).
Mouse models of forced metastasis
Intrasplenic (i.s.) injection (forced liver metastasis). For induction of forced liver metastases, mice received 250 μL PBS containing 3x105 cancer cells i.s. The injection was performed in hemi-spleen which was removed 3 min after cancer cell injection. The mice were sacrificed after 3 weeks. Macroscopic liver metastases were counted using a stereoscope (Olympus Corporation, Germany).30,67
Intraportal (in.p.) injection (forced liver metastasis). Mice were anesthetized using continuous isoflurane, and their abdomens were sterilized. After administration of analgesic agents, median laparotomy (10 mm) was performed, and the incision site was held open using a retractor. After exposure of the peritoneal cavity, the intestines were located and exteriorized onto a sterile field surrounding the incision site to visualize the portal vein. Throughout the procedure, the intestines were kept hydrated with sterile PBS that was pre-warmed at 37°C. For intraportal injection, sterile PBS or MC38 cancer cells (3x105 suspended in 250 μL of sterile PBS) were injected into the portal vein via a 30-gauge needle. Successful injection was confirmed by partial blanching of the liver. After the injection, a sterile coagulation gauge was then held over the injection site for 1 min to ensure that no injected contents would leak into the peritoneal cavity. Afterward, the intestines were placed back into the peritoneal cavity, and the peritoneum and skin were closed with a suture and autoclips, respectively. The mice were sacrificed after 24 h. Livers were harvested and an extravasation assay was performed.
Intravenous (i.v.) injection (forced lung metastasis). For induction of forced lung metastases, mice received 100 μL PBS containing 2.5x105 cancer cells i.v. The mice were sacrificed after 3 weeks. Macroscopic metastases in harvested lungs were counted using a stereoscope.
Mouse models of spontaneous metastasis
Flank model (spontaneous lung and liver metastasis). For induction of solid tumors, mice were anesthetized using isoflurane inhalation and received subcutaneous injection of 100 μL PBS containing 5x105 LLC cells. Three vertical tumor dimensions (δ1, δ2, and δ3) were monitored longitudinally and tumor volume was calculated using the formula π ∗δ1 ∗δ2 ∗δ3/6. Both models were described elsewhere.30 The tumors were resected after 2 weeks. The mice were sacrificed 4 weeks after tumor resection and macroscopic metastases in lungs were counted using a stereoscope.
Cecum model (spontaneous liver metastasis). The cecum of anesthetized mice was exteriorized through an abdominal laparotomy. 1×106 LLC cells were injected into the cecal wall between the mucosa and the muscularis externa layers using a 30-gauge needle. A proper implantation into the cecum was confirmed at day 0 by a localized bubble in the cecal wall. The mice were sacrificed after 4 weeks and macroscopic metastases in livers were counted using a stereoscope.68
Orthotopic colonic sub-mucosal implantation of CRC cells
Mice were anesthetized using isoflurane. Sub-mucosal injections were accomplished using stainless flexible stainless steel; 8 inch long, 30 gauge and 45° bevel hypodermic needles custom made according to our specification (Cadence Inc. U.S.A). The needle was inserted through Luer lock (Söllner, GmbH) screwed on the working channel of the scope to avoid air leakage. Subsequently, the scope was inserted into the mouse colon and following its inflation the needle was brought through the working channel to the scope’s front. The CRC cell implantation procedure was performed by two coordinated persons; one person was navigating the colonoscopy while the other person was operating the injection maneuver. The injection was performed under observation by a very gentle sub-mucosal penetration with the open side of the bevel heading up in a flat angle. 50 μL PBS containing 1.5x105 CRC tumor cells were then injected into the colonic sub-mucosa.
Adenoviral infection of colonic epithelium for orthotopic colon cancer induction
Apc15lox;LSL-KrasG12D mice were fasted overnight and anesthetized using 2% isoflurane. A midline incision was performed and the distal colon was clamped 3 cm from the anus. After washing with PBS, 100 μL trypsin were injected into the colon for 10 min. The lining of the distal colon was then mechanically abraded using a small caliber brush. After washing with PBS, 109 pfu of adenovirus in 100 μL PBS were injected into the colon for 30 min. For all incubations, a second clamp was placed 1 cm from the anus to ensure localization during the entire incubation period. After the infection period, both clamps were removed and the abdominal wall was closed in two layers. These procedures were well tolerated by all animals. Ad5CMVCre and adenoviruses were obtained from the Cellbiolabs (San Diego, USA). Mice developed CRC after 6– 8 weeks and were sacrificed after 8 months. The primary tumors and macroscopic liver metastases were quantified by colonoscopy or using a stereoscope, respectively.
Mouse colonoscopy
Mice were fasted overnight and anesthetized using 2% isoflurane. A custom-made colonoscopy system (Karl Storz) was used as previously described.69 Air was carefully insufflated into the colon to allow full visualization but avoid perforation. Endoscopic images and movies were saved for later offline analysis with ImageJ software to calculate the ratio of the tumor area relative to that of the lumen. Tumor sizes were graded from 1 to 5. Tumors observed during endoscopy were counted to obtain the total number of tumors per animal. The total tumor score per mouse was calculated as the sum of all tumor sizes.
Immunoblotting
To analyze protein activation and expression, LSECs or hepatocyte lysates were separated in a 10% SDS-PAGE assay, transferred to polyvinylidene difluoride membranes (Merck Millipore), probed with specific antibodies, and were visualized by enhanced chemiluminescence. Band density was determined by incubation with the appropriate HRP-conjugated secondary Abs (Dako, Carpintera, CA) and were visualized with the ECL substrate (Merck Millipore).
Hematoxylin and eosin (H&E) staining
Liver or lung specimens were fixed in 4% buffered formalin, embedded in paraffin or OCT (Sakura, Tokyo, Japan), and stored at −80°C. Tissue sections (4 μm) were prepared and stained with H&E. Metastatic lesion areas were quantified using ImageJ (ImageJ, U.S. National Institutes of Health, Bethesda, MD).
Plasmids and transfections
The coding sequences for IL-22 were cloned via reverse transcription and PCR from murine C57BL/6 intestinal total RNA with the corresponding primers (primer set A), Superscript III reverse transcriptase (Invitrogen) and Phusion Hot Start Flex DNA polymerase (New England Biolabs). After cleanup and addition of 3′ A overhangs, the PCR product was cloned in pGEM-T Easy vector (Promega). A second PCR product using the pGEM-T Easy-IL-22 construct template with Phusion Hot Start Flex DNA polymerase and primer set B was performed that was sub-cloned into pCMV-EGFP C1 (Clontech) mammalian expression vector by the InFusion ligation system (Takara). The IL22 expressing construct was verified by standard Sanger sequencing.
The construct expressing the sgRNA against IL-22RA1 was a kind gift of Dr Sebastian Kobold. Chemiluminescent MC38 cells stably expressing the sgRNA against IL-22RA1 protein were obtained as follows: MC38 CRC cells were transfected with TransIT-LT1 reagent (Mirus) instructions and a mixture of 1:5 of GAC.Luc-puro Plasmid (Addgene Number# 74409): Il22ra1sgRNA encoding vector. Briefly, 1.5x105/mL MC38 cells were plated in full DMEM medium for overnight incubation in 5 cm diameter cell culture plates. On the next day, the plasmids and TransIT-LT1 reagent were each diluted in 100 μL OptiMEM (Gibco) and left for 5 min at room temperature (RT). The plasmid and reagent solutions were then combined, mixed briefly by vortex and were left for 15 min at RT. The medium was removed from MC38 cells leaving approximately 800 μL at the plate. The plasmid and reagent solution was carefully overlaid on the cells and the plates were incubated overnight at 37°C in a 5% CO2 humidified incubator. 24 h later, 1–3 mL DMEM medium supplemented with 10% FBS was added into the cell culture medium. Cells were collected 48 h after transfection, washed twice with PBS, trypsinized and splitted into three 10 cm diameter plates containing 5, 10 and 15 μg/ml puromycin. After 5 days, stable visible clones were individually picked into a 96-well plate and assessed for pSTAT3 and chemiluminescence. Correct clones were expanded for liver metastasis induction.
Isolation of primary hepatocytes
For the isolation of primary hepatocytes, the liver was digested with Liberase (Roche Diagnostics) and was then gently disrupted to free residual cells. Single-cell suspension was filtered through a 100-μm cell strainer and the cells were allowed to settle by gravity for 20 min. Subsequently, parenchymal cells were separated by 10 min centrifugation in a 90% Percoll gradient (GE Healthcare). For primary hepatocyte culture, William’s E + GlutaMAX-I medium (Life Technologies, Karlsruhe, Germany) was supplemented with 10% FBS (Life Technologies), 1% penicillin/streptomycin (Life Technologies) and 1% L-glutamine (Life Technologies). The cells were incubated overnight at 37°C, 40% O2. Subsequently, the hepatocytes were washed and incubated with 100 ng/mL rmIL-22 or rmIL-6 for 15 min (eBioscience; R&D Systems; Peprotech, respectively). Finally, the cells were harvested and the RNA was extracted.
Immunofluorescence
Primary LSECs were fixed for 10 min in 4% paraformaldehyde at RT. Sections (5 μm) were washed with PBS and incubated in PBS-Triton 0.3% for 5 min. After washing, sections or cells were incubated for 60 min in blocking buffer. Samples were stained overnight with specific antibodies at 4°C. After washing, secondary antibody staining was performed (1 h, RT) followed by 5 min staining with Hoechst 33,258 (1:5000). As control, the primary Ab was omitted. Bright-field and fluorescent microscopy was carried out using either an Axio Vert.A1 (Zeiss, Jena, Germany) inverted microscope or an SP5 (Leica, Heidelberg, Germany) confocal microscope.
Isolation of LSECs
For the isolation of LSECs, a slit was cut in the left ventricle of the heart. The liver was perfused with PBS and then with∼ 5 mL 0.05% collagenase through the portal vein and vena cava, until it was clear of blood. After perfusion, the liver tissues were collected in complete medium on ice. Afterward, the liver was sliced into small pieces of approximately 0.5 cm. Two livers per genotype were digested in collagenase with 1% collagen and 0.83% DNase at 37°C for 25 min, while shaking. The remaining liver pieces were filtered through a 200-μm cell strainer and further centrifuged to remove the hepatocytes. The supernatant was again centrifuged. The pellet was resolved with maximum 5 mL PBS and added to 2 mL Optiprep. On top, 1 mL PBS was carefully pipetted and then centrifuged. The white interphase was collected in a new tube and washed with PBS. Single-cell suspension was filtered through a 40-μm cell strainer and centrifuged. For isolating the LSECs, the magnetic activated cell sorting was used. IMDM complete medium was used in order to culture the LSECs on collagen-coated plates at 37°C.
IL-22 ELISA
IL-22 serum levels were measured using a mouse IL-22 ELISA kit (Antigenix America) according to the manufacturer’s instructions.
Adoptive transfer
INKT cells were isolated from livers using fluorescent activated cell sorting (see Sorting panel in Figures S6H and S6I). Rag1−/−Il22−/− or Ja18−/− mice were engrafted with 105 iNKT or γδ T cells by intraportal injection. After 5–7 days, an in vivo extravasation assay was performed.
Laminar flow adhesion assay
The dynamic adhesion of human HT29 and mouse MC38 tumor cells to endothelial cells under physiological flow conditions was analyzed in a laminar flow adhesion assay as previously described.70 In short, HUVEC cells (PromoCell, Heidelberg) or LSECs were seeded in ibidiTreat μ-slide IV0.4 (ibidi GmbH, Germany) flow chambers and after confluence to monolayers, endothelial cells were stimulated with 10 ng/mL recombinant human or murine IL-1α (R&D Systems, Minneapolis, MN), 10 ng/mL recombinant human or murine TNFα or 20 to 100 ng/mL human or murine IL-22 for 4 h prior to the flow assay (tumor cell suspension: 1x105 cells/ml; flow rate: 8.5 mL/h; shear stress: 0.25 dyn/cm2). Non-stimulated endothelial cells served as negative controls. Data were acquired and evaluated with CapImage software (version 8.6, Dr. Heinrich Zeintl, Heidelberg). The adhesive events on the endothelium were distinguished into firm adhesion, rolling and tethering and counted in three separate regions of interest for 1 min each.
Extravasation assay
For the in vivo extravasation assay, mice received 250 μL PBS containing 1x106 MC38 GFP-labelled cells i.s. The mice were sacrificed after 24 h and their livers were weighed. To isolate the MC38 GFP-labelled cells, the murine livers were cut in small pieces and minced using a scalpel. The tissue was incubated for 30 min at 37°C on a shaking incubator in HBSS (with Ca2+ and Mg2+) with Collagenase (1 mg/mL) and DNase I (10 U/ml) and supernatant was collected. Then, the supernatants were centrifuged for 4 min at 40 g. We repeated this step twice more. After 3 centrifugations, the hepatocytes were removed and the cells were diluted in PBS 1x. One-fifth of the cells were mixed with beads in 1:10 dilution and were analyzed by flow cytometry. In every extravasation assay, we used a mouse injected with not labeled cells in order to set up the gate of GFP + or Cherry + cells.
Circulating tumor cell isolation and quantification
6 mL Ficoll (Ficoll Paque PLUS, endotoxin tested, Cytiva, Uppsala, Sweden) were placed in a 15 mL Falcon tube and overlayed with the mouse blood (2 mL)/PBS (5 mL) mixture. After centrifugation for 30 min at 400 x g (rotor brake inactivated), supernatant and interphase were carefully removed and transferred to a 50 mL Falcon tube using a serological pipette and mixed with 30 mL PBS. Erythrocytes were lysed by adding 1 mL Flow Cytometry Human Lyse Buffer (R&D systems), mixing and incubating for 1 min. Subsequently, the sample was centrifuged for 10 min at 400 x g (rotor brake activated). Supernatant was discarded and the cell pellet was resuspended in 3 mL PBS. Cytospins with 500.000 cells per slide were prepared by spinning the appropriate volume for 3 min at 150 x g (Swing out rotor 1626, 1100 rpm, Hettich ROTOFIX 32) and air-drying. All steps were performed at RT.
Administration of antibody and aminopeptidase inhibitor treatment
For administration of antibodies, the abdomen of mice was sterilized, and anti-IgG or anti-IL-22 (50 μg in 200 μL PBS; Genentech, California, USA) were suspended in 200 μL of sterile PBS. For administration of Ubenimex, 20 mg/kg was diluted and injected for 7 days.
IV injection of CD45 Ab
To distinguish between circulating CD45+ cells and CD45+ cells infiltrating into the liver parenchyma, we injected fluorochrome labeled antibody specific for PeCy7 conjugated anti-CD45 Ab (30-F11) into the bloodstream of mice. Briefly, mice received 5 μg PeCy7 conjugated anti-CD45 Ab (30-F11) in PBS intravenously, and were euthanized after 3 min. For in vitro CD45 staining, an A-700 conjugated anti-CD45 antibody was used. In all the experiments blood staining controls were used and around 96–99% of CD45+ T cells were labeled.
Parabiosis
In short, females were anesthetized with a mixture of ketamine/xylazine (100 mg/kg and 10 mg/kg, respectively). After shaving the corresponding lateral aspects of each mouse, matching skin incisions were made from behind the ear to the hip and sutured together with nylon and collagen (5-0 Ethicon, Vicryl) absorbable suture. Then, these areas were clipped with 7-mm stainless-steel wound clips (Reflex7).38
Orthotopic single left lung transplantation (LuTx)
Orthotopic LuTxes were performed as described previously with minor modifications.71 No immunosuppression was applied to any of the transplanted mice. Il22ra1−/− mice were used as donors, and WT mice were used as recipients. Briefly, donors were anesthetized with isoflurane anesthesia. The pulmonary artery, bronchus, and pulmonary vein were carefully separated from one another with blunted forceps, prior to cuffing with 24-, 20-, and 22-gauge cuffs, respectively. The left lung graft was stored for less than 20 min before its implantation. The recipient mouse was anesthetized with isoflurane anesthesia, intubated and ventilated using a small-animal ventilator (UNO Apparatus) at a respiratory rate of 120 bpm and a tidal volume of 300 μL. The chest was opened on the left side between the third and fourth ribs and the native left lung was retracted with a clamp. The hilar structures were carefully separated from one another with blunted forceps. After arrest of the blood and air flow toward the left lung, the cuffed graft pulmonary artery, bronchus, and pulmonary vein were inserted into the recipient counterparts and ligated with 10-0 sutures. The native left lung was removed and the incision in the chest was closed with a 6-0 suture, after removing all potential air bubbles from the chest. The mice were extubated. After the operation, the recipient mice were allowed to recover at 30°C overnight and received buprenorphine for 3 days. The mice were sacrificed at indicated time points according to the experimental plan.
Endothelial gene assay
Endothelial gene RNA levels were measured using a RT2 profiler PCR assay (Qiagen) according to the manufacturer’s instructions. The altered genes were verified by qPCR analysis
Measurement of the transendothelial electrical resistance (TEER)
HUVEC cells were seeded onto collagen-coated supported polycarbonate and polyester porous (0.4 μm pores) membranes (Transwell and Transwell Clear; Corning-Costar, Corning, NY) and cultured at 37°C and 5% CO2 in HUVEC complete medium to promote proliferation. Media were changed every 2 days and TEER was measured using an epithelial Ohm-voltmeter (Millicell ERS 2, Merck Millipore, Darmstadt, Germany). After reaching a plateau of TEER, we exposed HUVEC cells to recombinant human (rh) IL-22 (100 ng/ml). 0 and 24 h after exposure to rh IL-22, TEER was measured in every well.
In vitro transmigration assay
HUVEC cells (1x105) were seeded onto collagen-coated supported polycarbonate and polyester porous (0.4 μm pores) membranes (Transwell and Transwell Clear; Corning-Costar, Corning, NY) and cultured at 37°C and 5% CO2 in HUVEC complete medium to promote proliferation and creating monolayer. Media were changed every 2 days and TEER) was measured using an epithelial Ohm-voltmeter (Millicell ERS 2, Merck Millipore, Darmstadt, Germany) to check the monolayer induction. 5x104 HT-29 GFP-labelled cells were then seeded in the transwell inserts with an endothelial monolayer in HUVEC medium with 2% FBS in presence of 0.1% BSA or rh IL-22 (100 ng/ml). 24 h after seeding, cells were washed and fixed with 4% paraformaldehyde. Cells on the apical side of each insert were scraped off and the migration to the basolateral side was visualized using a Zeiss fluorescence microscope.
Lentiviral transfer of shRNAs into HUVEC cells
Lentiviral vectors expressing shRNAs under control of the human U6 promoter (MISSION pLKO.1-puro) directed against human ANPEP (TRCN0000050238, TRCN0000050239, TRCN0000050240, TRCN0000050241, TRCN0000050242, TRCN0000435991, TRCN0000418482, TRCN0000425830) and a non-targeted control shRNA (SHC002) were obtained from Sigma-Aldrich (Munich, Germany). Production of lentiviral particles was described in detail earlier59 and protocols are available online (http://www.LentiGO-Vectors.de). Selection of successfully transduced cells with 1 μg/mL puromycin in the culture medium was started two days after transduction.
Bioluminescence imaging
Cells and mice were imaged after addition of 300 μg/mL D-luciferin to culture media or i.v. delivery of 1 mg D-luciferin on a Xenogen Lumina II. Data were analyzed on Living Image v.4.2 (Perkin-Elmer, Waltham, MA).72
Hydrodynamic injection of plasmid
For hydrodynamic injection of plasmid, mice were anesthetized using continuous isoflurane, and their tail was sterilized. Subsequently, 10 μg of plasmid (eGFP or Il22eGFP) diluted in PBS (volume of 9% of mouse weight) were injected into the tail vein.
Depletion of γδ T cells in TcrdGDL/GDL mice
γδ T cells in TcrdGDL/GDL mice were depleted using a single intraperitoneal diphtheria toxin injection (50 ng/g bodyweight). Experiments using these mice always included WT littermates injected with diphtheria toxin and TcrdGDL/GDL mice injected with PBS as correct controls.
RNA analysis
Total RNA was extracted from tissue and cells of colon, lymph nodes, liver and spleen using TRIzol Reagent (Invitrogen). The High capacity cDNA synthesis Kit (Applied Biosystems) was used for cDNA synthesis. Primers and probes were purchased from Applied Biosystems. Real-time PCR was performed using the Kapa Probe Fast qPCR Master Mix (Kapa Biosystems) on the StepOne Plus system (Applied Biosystems). For both human and mouse, relative expression was normalized to HPRT and calculated using the 2−ΔΔCt method.
scRNA-seq
Droplet-based single-cell RNA sequencing (10x Chromium single cell 5′ solution v1.1, 10x Genomics) experiments were performed according to the manufacturer’s protocol. In brief, after sorting, cells were centrifuged and loaded onto the chip for GEM generation. The droplets were recovered and reverse transcription as well as cell barcoding were performed. The cDNA was recovered using Dynabeads MyOne SILANE (ThermoFisher Scientific). Afterward, cDNA was amplified and the quality was checked with a Bioanalyzer 2100 (Agilient Technologies). For the gene expression library construction, cDNA was fragmented and end-repaired, followed by further amplification with index primers. The libraries were sequenced on a Illumina NovaSeq 6000. After demultiplexing, the Cellranger pipeline (v. 3.0.1, 10x Genomics) was used to process fastq files, aligning the reads to the mm10 mouse genome draft. The resulting barcode-read count matrices were processed in R, using the Seurat package v. 4.0.4.73 Briefly, low quality and suspected doublet cells were removed, resulting in 7065 CD45+IL-22− cells and 277 CD45+IL-22+ cells before injection, as well as 9730 CD45+ IL-22− cells and 1302 CD45+ IL-22 + cells 12 h after injection. All four samples were normalized using the SCTransform function,74 followed by integration as described previously.75 PCA was ran on the integrated expression values, followed by UMAP based on the top 30 PCs and shared nearest neighbor prediction on the top 20 PCs. 23 clusters were found which were grouped into 11 higher level clusters based on cell type markers (see heatmap in Figure S7). To compute the tissue residency score, the AddModuleScore function was used based on the following marker genes: Cd69, Cxcr3, Cxcr6, Itgal, Itga1, Pdcd1, Cd101, Selplg, Itgb2, Ccr10, Zfp683, Prdm1, Runx3.76
Quantification and statistical analysis
Sample size was calculated using G∗power (http://www.gpower.hhu.de/) assuming α = 0.05, β = 0.8, and ρ = 0.3. No data were excluded. Animals were allocated to treatments by alternation and transgenic animals were enrolled case-control-wise. Data acquisition was blinded on samples previously coded by a non-blinded investigator.
Statistical analysis was performed using GraphPad Prism Software (GraphPad Software, San Diego, CA, USA). For paired group comparison, the non-parametric two-sided Mann-Whitney test was used. Multiple comparisons testing was examined by one-way ANOVA with Bonferroni post-hoc tests. Paired group comparison test was carried out using two-tailed Wilcoxon matched-pairs signed rank test. Sample size (n) refers to biological replicates. The mRNA expression of the cytokines was transformed using a base 10 logarithm. The significance level α was set to 0.05.
Acknowledgments
The authors thank Elaine Hussey for reading the manuscript; Cathleen Haueis, Sandra Wende, and Tom Blankenburg for technical assistance; Gerrit Wolters-Eisfeld for Western blot antibodies; and Francesco Siracusa, the in vivo Optical Imaging Core Facility and the FACS Core Sorting Unit at the University Medical Center Hamburg-Eppendorf for their technical assistance. The PBS57-CD1d-tetramer was provided by the NIH tetramer core facility. Prof. Dr. Ron Smits, Erasmus University Medical Center, provided the Apc15lox mice. We thank Dr. Guido Hegasy (www.hegasy.de) for designing the graphical abstract. This work was supported in part by the Deutsche Forschungsgemeinschaft (DFG) (SFB841 to S.H., N.G., A.W.L., K.R. [via B. Fehse]; SFB1328 to S.H., N.G.; grant 50552-1, SFB-TRR 338/1 2021–452881907 to S.K.; LA3373/6-1 to T.L.), the European Research Council (CoG 865466 to S.H., StG 756017 to S.K., ERC StG 715271 to N.G.), Else Kröner Memorial Stipendium (A.D.G.), Erich und Gertrud Roggenbuck-Stiftung (A.D.G.), Hamburger Krebsgesellschaft Stiftung (A.D.G.), the Jung Foundation for Science and Research (A.D.G.), and the Howard Hughes Medical Institute (R.A.F.). N.G. receives research grants from F. Hoffmann-La Roche and DZIF. S.H. has an endowed Heisenberg-Professorship awarded by the Deutsche Forschungsgemeinschaft. S.G.Z. acknowledges the Alexander von Humboldt Stiftung for an Experienced Researcher Fellowship. F.C. is supported by an iCARE-2 fellowship issued by AIRC and the European Union’s Horizon 2020 grant 800924). K.P. received funding from the European Union Horizon 2020, grant No 765492, ERA-NET EU/TRANSCAN 2 JTC 2016 PROLIPSY, Deutsche Krebshilfe (Nr. 70112504), DFG SPP2084 μBone, and ERC Advanced Investigator Grant (Nr. 834974).
Author contributions
A.D.G., J.K., and A.M.S. conceived, designed, and carried out most experiments, analyzed data, and wrote the manuscript; J. Lücke, L.Z., and T.Z. carried out in vivo experiments and flow cytometry assays; D.E.Z. did immune labeling, confocal microscopy, in vitro transmigration assays and endothelial assays; F.C. helped with NKT characterization; K.R. and T.A. constructed expression vectors and performed transfection experiments; A.D.G., J.S.L., and M.C.A.V. carried out parabiosis experiments; H.X. carried out sorting; E.K. engrafted humanized mice; L.G.-P. performed ILC staining; A.D.G., Y.Y., and W.J. carried out single-left orthotopic lung transplantations; E.Z. established the intramucosal injection; K.F.K., L.K., K.S., J.V.F., M.R., and J.M. provided the human liver metastasis specimens; B.S. and J.W. carried out single-cell sequencing; A.C. isolated murine LSECs; P.M. and D.B. provided us with CRISPR for Il22ra1 silencing; L.B. and T.B. performed fluorescence-activated cell sorting; S. Soukou and T.L. performed in vitro adhesion assays; C.K. and S.R. carried out CTC experiments; P.P., M.B., and M.S. performed in vitro experiments; F.J.H. performed in vivo experiments; S. Soukou characterized the Il22ra1flox/flox;Cdh5Cre+;R26YFP mice; A.W. performed genotyping of Il22ra1−/−, Il22ra1flox/flox;AlbCre+ and Il22ra1flox/flox;Cdh5Cre+ mice; C.M. performed experiments of orthotopic colon cancer induction; B.G. provided the Il22tg8+/− mice; M.R., J.K.G., R.W., H.S., B.-O.S., M.F.A., A.D., G.P., I.C.M., D.P., P.S., M.T., T.G., A.H., J. Li, and N.M. provided the human liver metastasis specimens; S.G.Z. established the transendothelial electrical resistance (TEER) assay; T.S. provided Il22ra1−/− and Il22ra1flox/flox mice and provided critical intellectual input; R.P. and W.A. provided Anpep−/− mice and provided critical intellectual input; J.S.G. provided Il22flox/flox;CD4Cre+ mice and critical intellectual input; P.C.A., S.K., A.H.G., M.T., T.G., A.H., J. Li, N.M., O.M., J.R.I., K.P., U.S., A.W.L., and R.A.F. provided critical intellectual input; N.G. designed experiments, provided critical intellectual input, and edited the paper; and S.H. conceived the idea and supervised the study, designed experiments, and wrote the manuscript. All authors reviewed and concur with the submitted manuscript.
Declaration of interests
S.K. declares honoraria from GSK, BMS, Novartis, and TCR2, Inc.; license fees from TCR2, Inc. and Carina Biotech; and research support from TCR2, Inc., Plectonic GmbH, Tabby Therapeutics, and Arcus Biosciences.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.12.014.
Contributor Information
Anastasios D. Giannou, Email: a.giannou@uke.de.
Samuel Huber, Email: shuber@uke.de.
Supplemental information
Data and code availability
All the data supporting the findings of the article are available within the main text or supplemental information. The published article includes datasets generated during this study. Original single cell RNA-seq data has been deposited in GEO: GSE217223.
References
- 1.Bertocchi A., Carloni S., Ravenda P.S., Bertalot G., Spadoni I., Lo Cascio A., Gandini S., Lizier M., Braga D., Asnicar F., et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell. 2021;39:708–724.e11. doi: 10.1016/j.ccell.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Garner H., de Visser K.E. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat. Rev. Immunol. 2020;20:483–497. doi: 10.1038/s41577-019-0271-z. [DOI] [PubMed] [Google Scholar]
- 3.Bromley S.K., Thomas S.Y., Luster A.D. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 2005;6:895–901. doi: 10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Ruiz D., Ng W.Y., Holz L.E., Ma J.Z., Zaid A., Wong Y.C., Lau L.S., Mollard V., Cozijnsen A., Collins N., et al. Liver-resident memory CD8(+) T cells form a front-line defense against malaria liver-stage infection. Immunity. 2016;45:889–902. doi: 10.1016/j.immuni.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Park S.L., Buzzai A., Rautela J., Hor J.L., Hochheiser K., Effern M., McBain N., Wagner T., Edwards J., McConville R., et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature. 2019;565:366–371. doi: 10.1038/s41586-018-0812-9. [DOI] [PubMed] [Google Scholar]
- 6.Savas P., Virassamy B., Ye C., Salim A., Mintoff C.P., Caramia F., Salgado R., Byrne D.J., Teo Z.L., Dushyanthen S., et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018;24:986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 7.Salio M., Silk J.D., Jones E.Y., Cerundolo V. Biology of CD1- and MR1-Restricted T Cells. Annu. Rev. Immunol. 2014;32:323–366. doi: 10.1146/annurev-immunol-032713-120243. [DOI] [PubMed] [Google Scholar]
- 8.Matos T.R., O'Malley J.T., Lowry E.L., Hamm D., Kirsch I.R., Robins H.S., Kupper T.S., Krueger J.G., Clark R.A. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J. Clin. Invest. 2017;127:4031–4041. doi: 10.1172/JCI93396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paget C., Ivanov S., Fontaine J., Renneson J., Blanc F., Pichavant M., Dumoutier L., Ryffel B., Renauld J.C., Gosset P., et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J. Biol. Chem. 2012;287:8816–8829. doi: 10.1074/jbc.M111.304758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godfrey D.I., Stankovic S., Baxter A.G. Raising the NKT cell family. Nat. Immunol. 2010;11:197–206. doi: 10.1038/ni.1841. [DOI] [PubMed] [Google Scholar]
- 11.Goto M., Murakawa M., Kadoshima-Yamaoka K., Tanaka Y., Nagahira K., Fukuda Y., Nishimura T. Murine NKT cells produce Th17 cytokine interleukin-22. Cell. Immunol. 2009;254:81–84. doi: 10.1016/j.cellimm.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Yang W., Yu T., Huang X., Bilotta A.J., Xu L., Lu Y., Sun J., Pan F., Zhou J., Zhang W., et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020;11:4457. doi: 10.1038/s41467-020-18262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutz S., Eidenschenk C., Ouyang W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013;252:116–132. doi: 10.1111/imr.12027. [DOI] [PubMed] [Google Scholar]
- 14.Voigt C., May P., Gottschlich A., Markota A., Wenk D., Gerlach I., Voigt S., Stathopoulos G.T., Arendt K.A.M., Heise C., et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA. 2017;114:12994–12999. doi: 10.1073/pnas.1705165114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doulabi H., Rastin M., Shabahangh H., Maddah G., Abdollahi A., Nosratabadi R., Esmaeili S.-A., Mahmoudi M. Analysis of Th22, Th17 and CD4+cells co-producing IL-17/IL-22 at different stages of human colon cancer. Biomed. Pharmacother. 2018;103:1101–1106. doi: 10.1016/j.biopha.2018.04.147. [DOI] [PubMed] [Google Scholar]
- 16.Perez L.G., Kempski J., McGee H.M., Pelzcar P., Agalioti T., Giannou A., Konczalla L., Brockmann L., Wahib R., Xu H., et al. TGF-β signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat. Commun. 2020;11:2608. doi: 10.1038/s41467-020-16363-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui G. TH9, TH17, and TH22 cell subsets and their main cytokine products in the pathogenesis of colorectal cancer. Front. Oncol. 2019;9:1002. doi: 10.3389/fonc.2019.01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyerich S., Eyerich K., Pennino D., Carbone T., Nasorri F., Pallotta S., Cianfarani F., Odorisio T., Traidl-Hoffmann C., Behrendt H., et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 2009;119:3573–3585. doi: 10.1172/jci40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gronke K., Hernández P.P., Zimmermann J., Klose C.S.N., Kofoed-Branzk M., Guendel F., Witkowski M., Tizian C., Amann L., Schumacher F., et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019;566:249–253. doi: 10.1038/s41586-019-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindemans C.A., Calafiore M., Mertelsmann A.M., O’Connor M.H., Dudakov J.A., Jenq R.R., Velardi E., Young L.F., Smith O.M., Lawrence G., et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528:560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamanaka M., Huber S., Zenewicz L.A., Gagliani N., Rathinam C., O'Connor W., Jr., Wan Y.Y., Nakae S., Iwakura Y., Hao L., Flavell R.A. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J. Exp. Med. 2011;208:1027–1040. doi: 10.1084/jem.20102149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber S., Gagliani N., Zenewicz L.A., Huber F.J., Bosurgi L., Hu B., Hedl M., Zhang W., O'Connor W., Jr., Murphy A.J., et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirchberger S., Royston D.J., Boulard O., Thornton E., Franchini F., Szabady R.L., Harrison O., Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kryczek I., Lin Y., Nagarsheth N., Peng D., Zhao L., Zhao E., Vatan L., Szeliga W., Dou Y., Owens S., et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40:772–784. doi: 10.1016/j.immuni.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khosravi N., Caetano M.S., Cumpian A.M., Unver N., De la Garza Ramos C., Noble O., Daliri S., Hernandez B.J., Gutierrez B.A., Evans S.E., et al. IL22 promotes Kras-mutant lung cancer by induction of a protumor immune response and protection of stemness properties. Cancer Immunol. Res. 2018;6:788–797. doi: 10.1158/2326-6066.cir-17-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang R., Tan Z., Deng L., Chen Y., Xia Y., Gao Y., Wang X., Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–909. doi: 10.1002/hep.24486. [DOI] [PubMed] [Google Scholar]
- 27.Jiang R., Wang H., Deng L., Hou J., Shi R., Yao M., Gao Y., Yao A., Wang X., Yu L., Sun B. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer. 2013;13:59. doi: 10.1186/1471-2407-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCuaig S., Barras D., Mann E.H., Friedrich M., Bullers S.J., Janney A., Garner L.C., Domingo E., Koelzer V.H., Delorenzi M., et al. The interleukin 22 Pathway interacts with mutant KRAS to promote poor prognosis in colon cancer. Clin. Cancer Res. 2020;26:4313–4325. doi: 10.1158/1078-0432.ccr-19-1086. [DOI] [PubMed] [Google Scholar]
- 29.Wen Z., Liao Q., Zhao J., Hu Y., You L., Lu Z., Jia C., Wei Y., Zhao Y. High expression of interleukin-22 and its receptor predicts poor prognosis in pancreatic ductal adenocarcinoma. Ann. Surg Oncol. 2014;21:125–132. doi: 10.1245/s10434-013-3322-x. [DOI] [PubMed] [Google Scholar]
- 30.Giannou A.D., Marazioti A., Kanellakis N.I., Giopanou I., Lilis I., Zazara D.E., Ntaliarda G., Kati D., Armenis V., Giotopoulou G.A., et al. NRAS destines tumor cells to the lungs. EMBO Mol. Med. 2017;9:672–686. doi: 10.15252/emmm.201606978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D., Wang Y., Wang H., Weng H., Kong X., Martin-Murphy B.V., Li Y., Park O., Dooley S., Ju C., Gao B. Acute and chronic effects of IL-22 on acetaminophen-induced liver injury. J. Immunol. 2014;193:2512–2518. doi: 10.4049/jimmunol.1400588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hung K.E., Maricevich M.A., Richard L.G., Chen W.Y., Richardson M.P., Kunin A., Bronson R.T., Mahmood U., Kucherlapati R. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc. Natl. Acad. Sci. USA. 2010;107:1565–1570. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robanus-Maandag E.C., Koelink P.J., Breukel C., Salvatori D.C.F., Jagmohan-Changur S.C., Bosch C.A.J., Verspaget H.W., Devilee P., Fodde R., Smits R. A new conditional Apc -mutant mouse model for colorectal cancer. Carcinogenesis. 2010;31:946–952. doi: 10.1093/carcin/bgq046. [DOI] [PubMed] [Google Scholar]
- 34.Alva J.A., Zovein A.C., Monvoisin A., Murphy T., Salazar A., Harvey N.L., Carmeliet P., Iruela-Arispe M.L. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 35.Kleinschmidt D., Giannou A.D., McGee H.M., Kempski J., Steglich B., Huber F.J., Ernst T.M., Shiri A.M., Wegscheid C., Tasika E., et al. A protective function of IL-22BP in ischemia reperfusion and acetaminophen-induced liver injury. J. Immunol. 2017;199:4078–4090. doi: 10.4049/jimmunol.1700587. [DOI] [PubMed] [Google Scholar]
- 36.Dondossola E., Rangel R., Guzman-Rojas L., Barbu E.M., Hosoya H., St John L.S., Molldrem J.J., Corti A., Sidman R.L., Arap W., Pasqualini R. CD13-positive bone marrow-derived myeloid cells promote angiogenesis, tumor growth, and metastasis. Proc. Natl. Acad. Sci. USA. 2013;110:20717–20722. doi: 10.1073/pnas.1321139110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drujont L., Lemoine A., Moreau A., Bienvenu G., Lancien M., Cens T., Guillot F., Bériou G., Bouchet-Delbos L., Fehling H.J., et al. RORγt+ cells selectively express redundant cation channels linked to the Golgi apparatus. Sci. Rep. 2016;6:23682. doi: 10.1038/srep23682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amezcua Vesely M.C., Pallis P., Bielecki P., Low J.S., Zhao J., Harman C.C.D., Kroehling L., Jackson R., Bailis W., Licona-Limón P., et al. Effector TH17 cells give rise to long-lived TRM cells that are essential for an immediate response against bacterial infection. Cell. 2019;178:1176–1188.e15. doi: 10.1016/j.cell.2019.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fares J., Fares M.Y., Khachfe H.H., Salhab H.A., Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020;5:28. doi: 10.1038/s41392-020-0134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian B.-Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bindea G., Mlecnik B., Tosolini M., Kirilovsky A., Waldner M., Obenauf A.C., Angell H., Fredriksen T., Lafontaine L., Berger A., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Coffelt S.B., Kersten K., Doornebal C.W., Weiden J., Vrijland K., Hau C.-S., Verstegen N.J.M., Ciampricotti M., Hawinkels L.J.A.C., Jonkers J., de Visser K.E. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mlecnik B., Bindea G., Angell H.K., Maby P., Angelova M., Tougeron D., Church S.E., Lafontaine L., Fischer M., Fredriksen T., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 44.Guzman-Rojas L., Rangel R., Salameh A., Edwards J.K., Dondossola E., Kim Y.-G., Saghatelian A., Giordano R.J., Kolonin M.G., Staquicini F.I., et al. Cooperative effects of aminopeptidase N (CD13) expressed by nonmalignant and cancer cells within the tumor microenvironment. Proc. Natl. Acad. Sci. USA. 2012;109:1637–1642. doi: 10.1073/pnas.1120790109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geissmann F., Cameron T.O., Sidobre S., Manlongat N., Kronenberg M., Briskin M.J., Dustin M.L., Littman D.R. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma C., Han M., Heinrich B., Fu Q., Zhang Q., Sandhu M., Agdashian D., Terabe M., Berzofsky J.A., Fako V., et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paik D.T., Tian L., Williams I.M., Rhee S., Zhang H., Liu C., Mishra R., Wu S.M., Red-Horse K., Wu J.C. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. 2020;142:1848–1862. doi: 10.1161/circulationaha.119.041433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J., Li S., Liu Y., Zhang C., Li H., Lai B. Metastatic patterns and survival outcomes in patients with stage IV colon cancer: A population-based analysis. Cancer Med. 2020;9:361–373. doi: 10.1002/cam4.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stoletov K., Kato H., Zardouzian E., Kelber J., Yang J., Shattil S., Klemke R. Visualizing extravasation dynamics of metastatic tumor cells. J. Cell Sci. 2010;123:2332–2341. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guttman-Yassky E., Brunner P.M., Neumann A.U., Khattri S., Pavel A.B., Malik K., Singer G.K., Baum D., Gilleaudeau P., Sullivan-Whalen M., et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018;78:872–881.e6. doi: 10.1016/j.jaad.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S.V., Teichmann L.L., Saito Y., Marches F., Halene S., Palucka A.K., et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu T., Cui L., Liang Z., Liu C., Liu Y., Li J. Elevated serum IL-22 levels correlate with chemoresistant condition of colorectal cancer. Clin. Immunol. 2013;147:38–39. doi: 10.1016/j.clim.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Thomas S.Y., Scanlon S.T., Griewank K.G., Constantinides M.G., Savage A.K., Barr K.A., Meng F., Luster A.D., Bendelac A. PLZF induces an intravascular surveillance program mediated by long-lived LFA-1–ICAM-1 interactions. J. Exp. Med. 2011;208:1179–1188. doi: 10.1084/jem.20102630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salou M., Legoux F., Gilet J., Darbois A., du Halgouet A., Alonso R., Richer W., Goubet A.-G., Daviaud C., Menger L., et al. A common transcriptomic program acquired in the thymus defines tissue residency of MAIT and NKT subsets. J. Exp. Med. 2019;216:133–151. doi: 10.1084/jem.20181483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan J., Allen S., McDonald E., Das I., Mak J.Y.W., Liu L., Fairlie D.P., Meehan B.S., Chen Z., Corbett A.J., et al. MAIT cells promote tumor initiation, growth, and metastases via tumor MR1. Cancer Discov. 2020;10:124–141. doi: 10.1158/2159-8290.CD-19-0569. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y., Sedimbi S., Löfbom L., Singh A.K., Porcelli S.A., Cardell S.L. Unique invariant natural killer T cells promote intestinal polyps by suppressing TH1 immunity and promoting regulatory T cells. Mucosal Immunol. 2018;11:131–143. doi: 10.1038/mi.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., Springfield R., Chen S., Li X., Feng X., Moshirian R., Yang R., Yuan W. α-GalCer and iNKT cell-based cancer immunotherapy: realizing the therapeutic potentials. Front. Immunol. 2019;10:1126. doi: 10.3389/fimmu.2019.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Molgora M., Bonavita E., Ponzetta A., Riva F., Barbagallo M., Jaillon S., Popović B., Bernardini G., Magrini E., Gianni F., et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. 2017;551:110–114. doi: 10.1038/nature24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weber K., Bartsch U., Stocking C., Fehse B. A multicolor panel of novel lentiviral “Gene Ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 2008;16:698–706. doi: 10.1038/mt.2008.6. [DOI] [PubMed] [Google Scholar]
- 60.Grabinger T., Bode K.J., Demgenski J., Seitz C., Delgado M.E., Kostadinova F., Reinhold C., Etemadi N., Wilhelm S., Schweinlin M., et al. Inhibitor of apoptosis protein-1 regulates tumor necrosis factor–mediated destruction of intestinal epithelial cells. Gastroenterology. 2017;152:867–879. doi: 10.1053/j.gastro.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 61.Nittka S., Krueger M.A., Shively J.E., Boll H., Brockmann M.A., Doyon F., Pichler B.J., Neumaier M. Radioimmunoimaging of liver metastases with PET using a 64Cu-labeled CEA antibody in transgenic mice. PLoS One. 2014;9:e106921. doi: 10.1371/journal.pone.0106921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brodbeck T., Nehmann N., Bethge A., Wedemann G., Schumacher U. Perforin-dependent direct cytotoxicity in natural killer cells induces considerable knockdown of spontaneous lung metastases and computer modelling-proven tumor cell dormancy in a HT29 human colon cancer xenograft mouse model. Mol. Cancer. 2014;13:244. doi: 10.1186/1476-4598-13-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bauer R., Udonta F., Wroblewski M., Ben-Batalla I., Santos I.M., Taverna F., Kuhlencord M., Gensch V., Päsler S., Vinckier S., et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018;78:3220–3232. doi: 10.1158/0008-5472.CAN-17-3415. [DOI] [PubMed] [Google Scholar]
- 64.Esplugues E., Huber S., Gagliani N., Hauser A.E., Town T., Wan Y.Y., O'Connor W., Jr., Rongvaux A., Van Rooijen N., Haberman A.M., et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huber S., Gagliani N., Esplugues E., O'Connor W., Jr., Huber F.J., Chaudhry A., Kamanaka M., Kobayashi Y., Booth C.J., Rudensky A.Y., et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3⁻ and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–565. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wan Y.Y., Flavell R.A. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. USA. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soares K.C., Foley K., Olino K., Leubner A., Mayo S.C., Jain A., Jaffee E., Schulick R.D., Yoshimura K., Edil B., Zheng L. A preclinical murine model of hepatic metastases. JoVE. 2014:51677. doi: 10.3791/51677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao H.-W., Hsu J.-M., Xia W., Wang H.-L., Wang Y.-N., Chang W.-C., Arold S.T., Chou C.-K., Tsou P.-H., Yamaguchi H., et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Invest. 2015;125:4529–4543. doi: 10.1172/JCI82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pelczar P., Witkowski M., Perez L.G., Kempski J., Hammel A.G., Brockmann L., Kleinschmidt D., Wende S., Haueis C., Bedke T., et al. A pathogenic role for T cell–derived IL-22BP in inflammatory bowel disease. Science. 2016;354:358–362. doi: 10.1126/science.aah5903. [DOI] [PubMed] [Google Scholar]
- 70.Richter U., Schröder C., Wicklein D., Lange T., Geleff S., Dippel V., Schumacher U., Klutmann S. Adhesion of small cell lung cancer cells to E- and P-Selectin under physiological flow conditions: implications for metastasis formation. Histochem. Cell Biol. 2011;135:499–512. doi: 10.1007/s00418-011-0804-4. [DOI] [PubMed] [Google Scholar]
- 71.Jungraithmayr W.M., Korom S., Hillinger S., Weder W. A mouse model of orthotopic, single-lung transplantation. J. Thorac. Cardiovasc. Surg. 2009;137:486–491. doi: 10.1016/j.jtcvs.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 72.Giannou A.D., Marazioti A., Spella M., Kanellakis N.I., Apostolopoulou H., Psallidas I., Prijovich Z.M., Vreka M., Zazara D.E., Lilis I., et al. Mast cells mediate malignant pleural effusion formation. J. Clin. Invest. 2015;125:2317–2334. doi: 10.1172/JCI79840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szabo P.A., Miron M., Farber D.L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 2019;4:eaas9673. doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data supporting the findings of the article are available within the main text or supplemental information. The published article includes datasets generated during this study. Original single cell RNA-seq data has been deposited in GEO: GSE217223.