

ABSTRACT
Calcium ions (Ca2+) are the basis of a unique and potent array of cellular responses. Calmodulin (CaM) is a small but vital protein that is able to rapidly transmit information about changes in Ca2+ concentrations to its regulatory targets. CaM plays a critical role in cellular Ca2+ signaling, and interacts with a myriad of target proteins. Ca2+-dependent modulation by CaM is a major component of a diverse array of processes, ranging from gene expression in neurons to the shaping of the cardiac action potential in heart cells. Furthermore, the protein sequence of CaM is highly evolutionarily conserved, and identical CaM proteins are encoded by three independent genes (CALM1-3) in humans. Mutations within any of these three genes may lead to severe cardiac deficits including severe long QT syndrome (LQTS) and/or catecholaminergic polymorphic ventricular tachycardia (CPVT). Research into disease-associated CaM variants has identified several proteins modulated by CaM that are likely to underlie the pathogenesis of these calmodulinopathies, including the cardiac L-type Ca2+ channel (LTCC) CaV1.2, and the sarcoplasmic reticulum Ca2+ release channel, ryanodine receptor 2 (RyR2). Here, we review the research that has been done to identify calmodulinopathic CaM mutations and evaluate the mechanisms underlying their role in disease.
KEYWORDS: Calmodulin, calmodulinopathy, cardiac arrhythmia, channelopathy, long QT syndrome
Introduction
In 1957, Hodgkin and Keynes noted that Ca2+ ions microinjected into a squid giant axon were not free to move in the presence of an externally applied electric field[1] and postulated the existence of an intracellular agent capable of binding Ca2+. Later work by Cheung et al. [2–4] as well as Kakiuchi and Yamazaki [5] converged on an endogenous protein factor identified in brain tissue that could enhance the Ca2+ sensitivity of particular enzymes. This protein was soon recognized to be calmodulin (CaM), and over the following decades it was identified as an indispensable component of signal transduction pathways that target a large array of structural proteins, receptors, enzymes, and ion channels [6,7]. By acting on these diverse targets, CaM impacts numerous cellular processes including cell differentiation and division, gene transcription, membrane fusion, and muscle contraction.
CaM is a ubiquitously expressed Ca2+-sensor molecule found in all eukaryotic cells. CaM is a 17-kDa 149 amino acid protein that contains two distinct yet broadly similar regions, the N- and C-lobes, linked by a flexible helix. Each lobe of CaM contains two canonical Ca2+ binding motifs, termed EF hands. Each lobe of CaM exhibits a distinct Ca2+ binding affinity, and the KD has been measured to be approximately 2.4 µM for the C-lobe, and 16 µM for the N-lobe [8]; however, the presence of a target protein may have a significant impact on these values. Interestingly, the N- and C-lobes of CaM can each contribute separable effects on a target protein, a concept that highlights the functional bipartition of CaM lobes. The first glimpse of this phenomenon was observed in Paramecium. Deficits in the Ca2+-binding properties of CaM’s N-lobe leads to sluggish behavior in paramecia, while the same type of deficit in the Ca2+-binding properties in the C-lobe leads to hyperactive behavior [9]. Thus, each lobe of CaM is capable of a unique interaction with, and impact on, a target protein, amplifying the modulatory capability of this Ca2+ sensor. In this way, CaM confers a critical Ca2+ sensitivity to hundreds of substrates [10].
In humans, CaM is encoded by three genes (CALM1, CALM2, and CALM3) that vary in nucleotide sequence, yet nonetheless encode identical protein products. Each CaM gene resides on a distinct chromosome, with CALM1 located at chr14q31, CALM2 at chr2p21 and CALM3 at chr19a13. Many of the mechanisms underlying how these three distinct genes regulate CaM expression remain unknown. While all three genes appear to function across different tissues, the level of transcript expression may not be equal. In human heart, the relative abundance of CALM3 has been shown to be largest, with all three genes producing significant levels of transcript across multiple developmental timepoints [11]; however, transcript levels measured in induced pluripotent stem cell (iPSC)-derived cardiomyocytes showed higher levels of CALM1 [12]. Moreover, the expression of all three transcripts has been shown to increase over the course of development [11,13].
CaM is highly conserved across species and is essential for normal physiological function. It was therefore once thought that non-synonymous mutations in CALM genes were incompatible with life [14]. Starting in 2012, the discovery of disease-associated human CaM mutations overturned this idea [11,15] and initiated a wave of research into the mechanisms of disrupted CaM regulation.
CaM regulation in diverse cellular contexts
Either through activation of third-party enzymes or direct binding, CaM modulates a diverse range of targets that span across multiple organ systems, including the immune system, the nervous system, smooth muscle, and the heart.
CaM regulation in the immune system
In the immune system, Ca2+/CaM is required to activate T lymphocytes, one of the major mechanisms of adaptive immunity [16,17]. Upon binding to the antigen-presenting cell, intracellular Ca2+ in T lymphocytes rises due to the opening of IP3 receptors and store-operated Ca2+ channels, STIM and ORAI. The calcified CaM then binds to and activates calcineurin, which in turn dephosphorylates the transcription factor, nuclear factor of activated T-cells (NFAT). This modification permits NFAT to enter the nucleus and initiate the production of a cytokine responsible for activating downstream defense against the pathogen.
CaM regulation in smooth muscle
In smooth muscle, CaM controls both initiation and cessation of contraction. Ca2+/CaM binds and activates myosin light chain kinase (MLCK), which phosphorylates the light chain of myosin, and thus initiates smooth muscle contraction [18]. In addition, Ca2+/CaM also activates Ca2+/CaM-dependent kinase II (CaMKII) which phosphorylates MLCK, lowering its sensitivity to Ca2+ and thus promoting muscle relaxation. In contrast, within skeletal muscle, CaM modulates the contraction rather than initiating or ceasing it. Ca2+/CaM activates MLCK, which in turn phosphorylates the regulatory light chain of myosin, making it more sensitive to Ca2+ and thus generating more force [19]. Moreover, CaMKII and CaMKIV play a major role in muscle plasticity. These two enzymes encode frequency-dependent muscle usage by transducing the changes in frequency and magnitude of Ca2+ transients into changes in protein expression [20]. It is through dysregulation of this mechanism that CaM mutations in Drosophila can lead to skeletal muscle dysfunction [21].
CaM regulation in the brain
In the nervous system, Ca2+/CaM-dependent enzymes regulate neural development, memory, and learning. The classic example includes long-term potentiation in hippocampal CA1 neurons, which involves phosphorylation of AMPA and NMDA receptors by CaMKII [22]. Calcineurin, a Ca2+/CaM-dependent phosphatase, is also proposed to be involved in long-term depression in hippocampal neurons [23,24] and to control the polymerization of actin in dendrites, which ultimately leads to dendritic growth or retraction [25,26].
CaM regulation in the heart
In the heart, Ca2+/CaM-dependent enzymes shape electrical and Ca2+ activities of cardiomyocytes both during physiological and pathological responses. For instance, responding to the rise in intracellular Ca2+, CaMKII phosphorylates numerous Ca2+ signaling proteins includingRyR2, phospholamban, sarcoplasmic reticulum Ca2+ ATPase (SERCA), and L-type Ca2+ channels [27,28]. During pathologic adaptation such as in heart failure, calcineurin dephosphorylates numerous targets including the transcription factor NFAT, which leads to altered expression levels of proteins involved in cardiac hypertrophy [29,30].
CaM regulation of Ion channels
CaM regulation of voltage-gated calcium channels
A critical form of CaM modulation is the direct binding of apoCaM or Ca2+/CaM to targets, particularly ion channels. By physically associating with ion channels, CaM is capable of imparting multifunctional Ca2+-dependent regulatory processes on channel biophysical properties. For the CaV1 and CaV2 families of voltage-gated Ca2+ channels (VGCCs), at low cytosolic levels of Ca2+, apoCaM is bound to the IQ motif within the carboxy-tail of the channel, positioning CaM as a resident Ca2+ sensor. Upon Ca2+ elevation, Ca2+ binding to this resident CaM initiates either of two distinct forms of feedback regulation. Ca2+-dependent facilitation (CDF) occurs within select cannels, where Ca2+ entry through the channel initiates an increase in current amplitude [31]. Alternatively, Ca2+ binding to CaM often results in Ca2+ dependent inactivation (CDI), where a rise in cytosolic Ca2+ reduces the Ca2+ influx through the channels. As each lobe of CaM is able to impart its own independent form of channel regulation, each channel type may exhibit one or both forms of regulation. CaV2.2 and CaV2.3 each display CDI driven by Ca2+ binding to the N-lobe of CaM [32], while CaV2.1 exhibits CDI in response to Ca2+ binding to the N-lobe of CaM, and CDF in response to C-lobe Ca2+ binding [31,33]. Finally, the L-type channels (CaV1.2, CaV1.3, and CaV1.4) each display CDI in response to Ca2+ binding to either the N- or C-lobe of CaM, with distinct kinetics corresponding to signaling from each lobe [34–41]. The exception to this CaM regulatory scheme is the CaV1.1 channel, where apoCaM binds poorly to the carboxy-tail of the channel [42], resulting in a lack of CDI or CDF in most physiological systems [43]. Nonetheless, fusion of CaM to the channel enhances trafficking and open probability [44], generalizing much of the mechanism of CaM regulation across the L-type channel family. Functionally, CDF has been shown to be an important element in synaptic plasticity [45], CDI can reduce Ca2+-dependent and activity-dependent toxicity, as well as tune the temporal activity of Ca2+ flux to shape action potentials (APs) in excitable cells, especially those in cardiac tissue [14].
Unlike CaV1-2 channels, CaV3 channels lack a carboxy-terminal IQ motif, eliminating the Ca2+/CaM regulatory scheme described above. Despite this, several reports have described a role for CaM in the function of these channels, including binding to the gating brake of the I–II linker of CaV3.1, CaV3.2 and CaV3.3 channels [46], activity-dependent association of CaM with the CaV3.1 channel [47], and Ca2+-dependent regulation of the CaV3.3 channel [48], providing a potential role for calmodulin within this channel family.
CaM regulation of voltage-gated sodium channels
Parallel to its modulation of VGCCs, apoCaM also binds to select voltage-gated sodium channels [49,50]. In particular, apoCaM has been shown to bind to the C-tail of sodium channels in an analogous manner to its binding with voltage-gated Ca2+ channels [51]. In contrast to the dual-lobe regulation of VGCCs, it is the C-lobe that tethers apoCaM to NaV1.5 [52], and disruption of this interaction both reduces the peak open probability of the channel and increases the amount of late sodium current [53]. Moreover, it has been shown that the N-lobe of Ca2+/CaM drives CDI in the skeletal muscle isoform of Na channels (NaV1.4), however the same process does not appear to exist in the cardiac NaV1.5 channels [50].
CaM regulation of potassium channels
Slowly activating delayed rectifier K channels (KCNQ1/KCNE1) that generate IKs currents in cardiac myocytes are also proposed to be modulated by CaM. Like for VGCCs, CaM appears to constitutively associate with KCNQ channels and is required for voltage-dependent opening. ApoCaM may help in channel folding, assembly, and trafficking of this channel to the plasmalemma [54,55]. However, the impact of Ca2+/CaM dependent regulation on IKS remains under debate. Despite this, it has been established that CaM association with the channel is critical to modulate the channel’s activity [56]. Moreover, neuronal M-currents (KCNQ2/KCNQ3) were proposed to be regulated by Ca2+/CaM- either by the promotion of channel assembly and trafficking [57] or by acute current suppression in response to a rise in the intracellular Ca2+. Finally, [58]the Ca2+ activated SK potassium channel represents one of the first channels in which CaM was identified as the Ca2+ sensor [59]. ApoCaM pre-associates with the cytosolic tail of SK1,2 and 3 channels [60], with each domain channel capable of binding, resulting in four CaM proteins per channel. For SK2 channels, it has been shown that the N-lobe of CaM drives the Ca2+ dependence of the channel [61].
CaM regulation of intracellular ion channels
In addition to regulating ion channels on the plasma membrane, CaM also modulates the properties of intracellular ion channels such as ryanodine receptors. For the skeletal form of the ryanodine receptor (RyR1), CaM increases the open probability of the channels at submicromolar Ca2+ concentrations, i.e. in the apoCaM state, while the C-lobe of CaM inhibits channel opening under calcified conditions [62]. Conversely, both apoCaM and Ca2+/CaM decrease the open probability of the cardiac isoform of RyR (RyR2) [62,63], while only the C-lobe of Ca2+/CaM increases the termination threshold of spontaneous Ca2+ release from the sarcoplasmic reticulum [64]. Another type of intracellular Ca2+ release channel is the inositol 1,4,5 triphosphate receptor (IP3R). Both apoCaM and Ca2+/CaM decrease the sensitivity of the channel to IP3, and thus inhibit the IP3-induced Ca2+ release in type I IP3Rs [65,66].
Importantly, CaM in its apo versus calcified form can exert opposite modulatory effects on channels. Mutations in the EF hands of CaM can alter the Ca2+ binding affinity without altering the structure of CaM in the Ca2+-free state, thus keeping the apoCaM modulation intact despite disrupted Ca2+/CaM modulation. Thus, mutations that alter distinct properties of CaM may have unique effects on different targets.
Described here are only a few examples of a vast array of targets of calmodulation. A more comprehensive list can be found elsewhere [67,68].
LQTS-associated CaM mutations
Long-QT syndrome (LQTS) is characterized by an abnormal prolongation of the QT interval of an ECG, resulting from a prolongation of the cardiac action potential. Multiple forms of LQTS have been identified and are classified based on the gene associated with the phenotype. The majority of these genes are either ion channels, or proteins that modulate ion channels. In 2013, Crotti et al. identified a new form of LQTS, where infants exhibiting severe LQTS and recurrent cardiac arrest were found to harbor mutations in CaM (Figure 1) [11]. These mutations span a broad phenotypic spectrum with various degrees of penetrance and severity. In addition to frequent ventricular fibrillations brought on by adrenergic stimulation, the patients also exhibited neurological and/or developmental delay and seizure. Whole-exome sequencing of these probands revealed de novo CaM mutations, D130G and F142L in CALM1, as well as D96V in CALM2. Patients harboring these mutations displayed severe clinical pathology early in life. Moreover, these patients were resistant to conventional treatments for LQTS such as beta blockers, sodium channel blockers, calcium channel blockers, and implantable cardioverter defibrillators.
Figure 1.
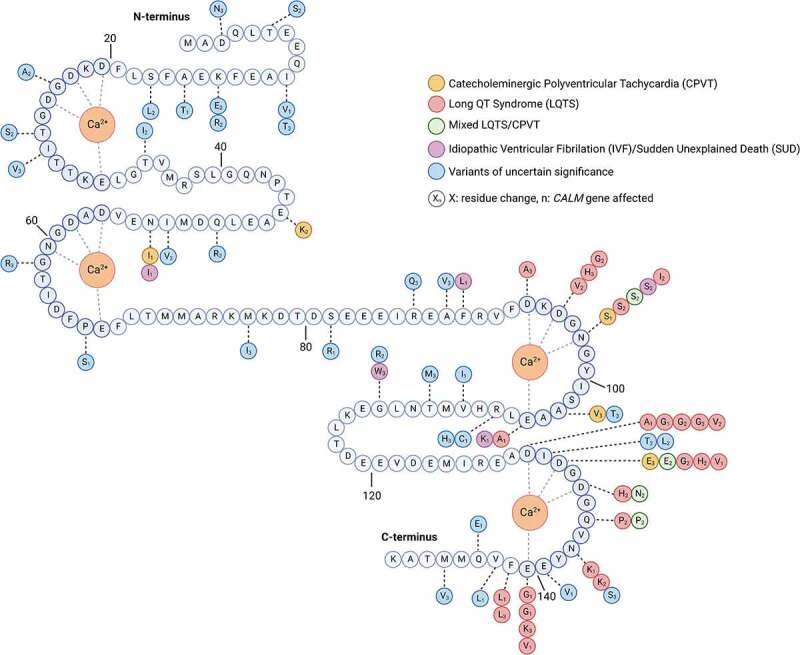
Missense Mutations Identified Within Human Calmodulin. Schematic featuring the amino acid sequence of CaM, with the location of identified pathogenic mutations indicated. Calcium ions (Orange) are coordinated within each of the four EF hands (shaded blue). Mutations in residues identified within patients suffering from CPVT are in Orange, LQTS residues are in red, mixed CPVT and LQTS are in green, IVF and/or SUD are in purple, and variants of uncertain significance that are logged in Gnomad are in blue. Numbers within each mutation circle indicate the specific CALM gene where the mutation was identified. Figure created with BioRender.com.
Since this first description, numerous mutations in CaM have been described in patients with LQTS (Figure 1, Table 1). In 2014, Makita et al. performed whole-exome sequencing on over 200 LQTS patients and discovered de novo CaM mutations, all within the CALM2 gene [69]. Three cases (N98S, N98I, and D134H) had a diagnosis of LQTS alone, while the two other cases (D132E and Q136P) had a mixed diagnosis of LQTS and CPVT. The onset of symptoms occurred in early childhood, and these symptoms were relatively well controlled by treatment with beta blockers and sodium channel blockers. Moreover, none of these patients were reported to exhibit developmental delays or neurological symptoms. Interestingly, the same D130G mutation identified originally in CALM1 [11] was subsequently identified as producing LQTS when present in both CALM3 [70,71] and CALM2 [72]. Thus, all three CALM genes are able to produce LQTS, leading to the designation of LQT type 14, 15 and 16 corresponding to mutations within CALM1, CALM2 and CALM3, respectively. Moreover, this same D130 residue has been identified in CALM2 with different changes at the same amino acid, such that D130V [72], and D130A [73] have both been associated with LQTS. Likewise, the mutation E141G in CALM1 was identified as associated with LQTS by two groups [72,74], and subsequently the same locus with a distinct E141V mutation was identified [71]. As the number of known CaM mutations continues to grow, additional clusters of mutations continue to be identified, sometimes with as many as four distinct residue changes at a single locus (Figure 1). The majority of these calmodulinopathy hotspots occur within or directly adjacent to the EF-hand motifs, demonstrating the critical nature of Ca2+ binding to CaM.
Table 1.
Summary of identified missense CaM mutations.
| Mutation | CALM gene | Clinical Phenotype | Reference | Variant ID (Gnomad/ClinVar) | Polyphen | SIFT | ACMG Classification |
|---|---|---|---|---|---|---|---|
| D3N | 3 | Uncertain | Gnomad[75] | 19–47109087-G-A | Benign | tolerated | |
| T6S | 2 | Uncertain | Gnomad[75] | 2–47397890-G-C | Benign | tolerated | |
| I10T | 3 | Uncertain | Gnomad[75] | 14–90866433-A-G | Benign | deleterious | |
| I10V | 1 | Uncertain | Gnomad[75] | 19–47109109-T-C | Benign | tolerated | |
| K14E | 2 | Uncertain | Gnomad[75] | 2–47389796-T-C | Benign | deleterious | |
| K14R | 2 | Uncertain | Gnomad[75] | 2–47389796-T-C | Benign | tolerated | |
| A16T | 1 | Uncertain | Gnomad[75] | 14–90867614-G-A | Possibly damaging | deleterious | |
| S18L | 2 | Uncertain | Gnomad[75] | 2–47389783-G-A | Benign | tolerated | |
| G24A | 2 | Uncertain | Gnomad[75] | 2–47389765-C-G | Benign | deleterious | |
| T27S | 2 | Uncertain | Gnomad[75] | 2–47389756-G-C | Benign | tolerated | |
| I28V | 3 | Uncertain | Gnomad[75] | 19–47111501-A-G | Benign | deleterious | |
| T35I | 2 | Uncertain | Gnomad[75] | 2–47389732-G-A | Possibly damaging | tolerated | |
| E46K | 2 | CPVT | Crotti et al 2019[73] | Likely Pathogenic | |||
| Q50R | 2 | Uncertain | Gnomad[75] | 2–47389687-T-C | Benign | tolerated | |
| I53V | 1 | Uncertain | Nykamp et al 2017[76] | VCV000475380.4 | |||
| N54I | 1 | CPVT | Nyegaard et al 2012[15] | VCV000039757.5 | |||
| N54I | 1 | CPVT, IVF/SUD | Crotti et al 2019[73] | VCV000039757.5 | Pathogenic | ||
| G62R | 3 | Uncertain | Gnomad[75] | 19–47111744-G-A | Probably damaging | deleterious | |
| P67S | 1 | Uncertain | Gnomad[75] | 14–90870226-C-T | Benign | tolerated | |
| M77I | 3 | Uncertain | Gnomad[75] | 19–47111791-G-A | Benign | tolerated | |
| S82R | 1 | Uncertain | Gnomad[75] | 14–90870273-T-G | Benign | tolerated | |
| R87Q | 3 | Uncertain | Mirsadeghi et al 2021[77] | ||||
| A89V | 3 | Uncertain | Gnomad[75] | 19–47111826-C-T | Benign | deleterious | |
| F90L | 1 | IVF/SUD | Marsman et al 2013[85] | VCV000183232.2 | Pathogenic | ||
| D94A | 3 | LQTS | Nykamp et al 2017[76] | VCV000409871.6 | Likely Pathogenic | ||
| D96G | 2 | LQTS | Crotti et al 2019[73] | Likely Pathogenic | |||
| D96H | 3 | LQTS | Chaix et al 2016[83] | VCV000409870.6 | Pathogenic | ||
| D96V | 2 | LQTS | Crotti et al 2013[11] | VCV000183233.5 | Pathogenic | ||
| N98I | 2 | LQTS | Makita et al 2014[69] | VCV000096721.4 | Likely Pathogenic | ||
| N98S | 1 | CPVT | Nyegaard et al 2012[15] | VCV000039758.7 | Pathogenic | ||
| N98S | 2 | LQTS | Makita et al 2014[69] | VCV000096720.7 | Pathogenic | ||
| N98S | 2 | Mixed LQTS + CPVT, SUD | Jiménez-Jáimez et al, 2016[78] | VCV000096720.7 | Pathogenic | ||
| A103V | 3 | CPVT | Gomez-Hurtado et al, 2016[84] | VCV000579223.4 | Likely Pathogenic | ||
| A103T | 3 | Uncertain | Gnomad[75] | 19–47112124-G-A | Possibly damaging | deleterious | |
| E105A | 1 | LQTS | Takahashi et al 2016[104] | Likely Pathogenic | |||
| E105K | 1 | Atypical | Crotti et al 2019[73] | VCV000388500.2 | Likely Pathogenic | ||
| R107C | 1 | Uncertain | Gnomad[75] | 14–90870756-C-T | Benign | tolerated | |
| R107H | 3 | Uncertain | Gnomad[75] | 19–47112137-G-A | Benign | tolerated | |
| V109I | 1 | Uncertain | Gnomad[75] | 14–90870762-G-A | Benign | tolerated | |
| T111M | 3 | Uncertain | Gnomad[75] | 19–47112149-C-T | Possibly damaging | deleterious | |
| G114 R | 2 | Uncertain | Brohus et al 2020[79] | Likely pathogenic (in-silico analysis) | |||
| G114W | 3 | IVF | Crotti et al 2019[73] | Likely Pathogenic | |||
| D130A | 1 | LQTS | Crotti et al 2019[73] | Likely Pathogenic | |||
| D130G | 1 | LQTS | Crotti et al 2013[11] | VCV000183230.3 | Pathogenic | ||
| D130G | 2 | LQTS | Boczek et al 2016[72] | VCV000812710.7 | Pathogenic | ||
| D130G | 3 | LQTS | Reed et al 2015[70] | VCV000812676.1 | Pathogenic | ||
| D130V | 2 | LQTS | Boczek et al 2016[72] | Likely Pathogenic | |||
| I131T | 2 | Uncertain | Gnomad[75] | 2–47388891-A-G | Benign | tolerated | |
| I131L | 2 | Uncertain | Gnomad[75] | 2–47388892-T-G | Benign | tolerated | |
| D132E | 2 | Mixed LQTS + CPVT | Makita et al 2014[69] | VCV000096722.3 | Pathogenic | ||
| D132E | 3 | CPVT | Crotti et al 2019[73] | VCV000423164.2 | Pathogenic | ||
| D132H | 2 | LQTS | Pipilas et al 2016[80] | Likely Pathogenic | |||
| D132G | 2 | LQTS | Zahavich et al 2018[81] | VCV000955533.6 | Likely Pathogenic | ||
| D132V | 1 | LQTS | Pipilas et al 2016[80] | VCV000948239.5 | Likely Pathogenic | ||
| D134H | 2 | LQTS | Makita et al 2014[69] | VCV000096723.1 | Likely Pathogenic | ||
| D134N | 2 | Mixed LQTS + CPVT | Crotti et al 2019[73] | VCV000448971.4 | Likely Pathogenic | ||
| Q136P | 2 | Mixed LQTS + CPVT | Makita et al 2014[69] | VCV000096724.1 | Likely Pathogenic | ||
| Q136P | 2 | LQTS | Crotti et al 2019[73] | VCV000096724.1 | Likely Pathogenic | ||
| N138K | 2 | LQTS | Crotti et al 2019[73] | VCV000488476.2 | Likely Pathogenic | ||
| N138K | 3 | LQTS | Kato et al 2022[91] | ||||
| N138S | 3 | Uncertain | Gnomad[75] | 19–47112230-A-G | Benign | tolerated | |
| E140V | 1 | Nervous system abnormality | Retterer et al 2016[105] | VCV000418107.2 | |||
| E141G | 1 | LQTS | Boczek et al 2016[72] | VCV000974612.1 | Pathogenic | ||
| E141G | 2 | LQTS | Crotti et al 2019[73] | Pathogenic | |||
| E141K | 3 | LQTS | Wren et al 2019[71] | Likely Pathogenic | |||
| E141V | 1 | LQTS | Wren et al 2019[71] | VCV000974613.1 | Pathogenic | ||
| F142L | 1 | LQTS | Crotti et al 2013[11] | VCV000183231.4 | Pathogenic | ||
| F142L | 3 | LQTS | Chaix et al 2016[83] | Pathogenic | |||
| V143L | 1 | Uncertain | Gnomad[75] | 14–90870762-G-A | Benign | deleterious | |
| Q144E | 1 | Uncertain | Gnomad[75] | 14–90871041-C-G | Benign | deleterious | |
| M146V | 3 | Uncertain | Gnomad[75] | 19–47112396-A-G | Benign | deleterious |
While all three CALM genes have been implicated in the same overall LQTS phenotype, differences in severity and age of onset point to possible differences between the genes. CALM1 mutations have been described as having a higher likelihood of identification among patients younger than 10 years old [72]. This finding aligns well with a later age of symptom onset reported for patients with mutations reported in CALM2 [69] as compared to CALM1 [11]. Thus, it may be possible that relative expression levels of CALM1 and CALM2 at different timepoints during development, or distinct tissue distribution of the CALM genes, may impact the severity and onset of symptoms. Moreover, while the majority of reported missense mutations within CaM are located within CALM1 and CALM2 genes, LQTS-associated mutations have also been observed in CALM3. The first of these mutations described was a D130G mutation, which produced severe LQTS with 2:1 AV block and fetal bradycardia in an infant [82]. In addition, the patient presented somewhat younger than most calmodulinopathy patients, with symptoms identified at birth. Additional CALM3 mutations associated with LQTS, F142L and D96H, were identified in patients presenting with fetal bradycardia [83]. The patients each survived for over a decade and a half at the time the article was published, with both responding suitably to traditional LQTS therapies including beta blockers and pacemakers. Thus, there may be some subtle differences in phenotype dependent on the specific CALM gene affected. However, the de-novo nature of most CaM mutations also brings along the possibility of mosaicism, where the mutation occurs at a point in development such that expression occurs only in a subset of cells or tissues. This has been identified in some parents of calmodulinopathy patients, where mosaicism in the parent prevented overt symptoms but enabled genetic transmission of the mutation [71].
CPVT-associated CaM mutations
The first set of CaM mutations associated with CPVT was discovered in 2012 by Nyegaard et al. [15]. The study reported a case of dominant inherited CPVT-like syndrome in a Swedish family with a history of ventricular arrhythmias, syncope, and sudden death, typically brought on by physical exertion and/or emotional stress during childhood. Despite these CPVT-like symptoms, genetic screening showed no mutations in RYR2 or CASQ2, the typical genetic loci for CPVT-associated mutations. Linkage analysis identified the first human CaM mutation, N54I, in the CALM1 gene dominantly inherited with 100% penetrance in this family. Having identified a mutation in CALM1 as a likely cause of this CPVT phenotype in a single family, the researchers probed whether mutations in CALM1 might underlie CPVT in a group of patients who had previously tested negative for all other known arrhythmia-associated genes. Sequencing of this cohort revealed a de novo N98S mutation in CALM1. Both the N54I and N98S mutations produced a phenotype that was unresponsive to beta blockers, the conventional treatment of CPVT, indicating a potentially new mechanism of CPVT pathogenesis. In the following year, targeted sequencing in over 100 patients with genotype-negative LQTS, CPVT, IVF, and sudden death in the young (SUDY) identified an A103V mutation within CALM3 which was associated with CPVT [84]. Thus, mutations in CaM became a newly recognized cause of CPVT.
Interestingly, while the N98S mutation in CALM1 was associated with CPVT in one patient [15], the same point mutation in CALM2 was associated with LQTS in another patient, as described earlier [69]. Moreover, the phenotypic divergence is complicated by a sometimes mixed phenotype of CPVT and LQTS, as observed for D132E and Q136P mutations in the CALM2 gene. Although the mechanism behind this intriguing phenomenon is yet unknown, it has been hypothesized that the different clinical phenotypes could stem from cell type-specific expression levels of CALM1 vs. CALM2, or different genetic backgrounds [69]. Another consideration may be the age of onset of the symptoms. While LQTS onset is earlier in life (i.e. during infancy) with more severe symptoms, the CPVT phenotype seems to surface during late childhood with slightly milder symptoms.
IVF/SCD-associated CaM mutations
A second heritable CaM mutation was discovered in 2014 in a Moroccan family with a history of idiopathic ventricular fibrillation (IVF) manifesting in childhood and adolescence [85]. Upon exome sequencing of members of this family, Marsman and colleagues discovered a heterozygous F90L mutation in CALM1 in the affected individuals. However, there were also individuals in the family carrying the F90L mutation without presentation of symptoms. Since this first description, several other CaM mutations have been associated with IVF (Figure 1). Given that IVF is one of the most common causes of sudden cardiac death (SCD), it is not surprising that SCD has been associated with calmodulinopathies. Moreover, there is considerable overlap between these phenotypes. Both LQTS and CPVT are associated with distinct forms of ventricular fibrillation and can cause SCD, thus this category of mutations may not be entirely distinct from those described above.
Overall, CaM mutations span a broad genotypic and phenotypic spectrum. For example, mutations have been discovered in all three CALM genes, affecting both the N- and C-lobes of CaM, and can be either hereditary or de novo in nature. Moreover, in addition to various degrees of penetrance, calmodulinopathies also display different levels of expressivity among affected individuals. While some mutations lead to severe symptoms emerging during infancy, others lead to milder symptoms that do not manifest until childhood or adolescence. Furthermore, the same mutation can manifest as CPVT in one individual and LQTS in another. Thus, uncovering the mechanisms underlying the different phenotypes of this array of CaM mutations has begun to offer insights into both disease pathophysiology and the development of therapies.
Mechanistic studies of LQT-associated CaM mutations
As the number of known calmodulinopathic mutations has grown, a general pattern has emerged that has informed studies into the structure–function relationship of the protein. The majority of the documented LQTS-associated CaM mutations, including CaM D96V, D130G, and F142L, reside in or near the Ca2+-binding EF hand motifs of the C-lobe (Figure 1). This observation points to a potential disruption of the Ca2+ binding affinity of the C-lobe of CaM. This prediction has been confirmed experimentally. While the Ca2+ binding affinity of the N-lobe remains intact [86], CaM D96V, D130G, and F142L cause a 13.6-, 53.6-, and 5.4-fold reduction in Ca2+ binding affinity of the C-lobe, respectively, [11]. The greater reduction in Ca2+-binding displayed by CaM D96V and D130G as compared to CaM F142L likely stems from the fact that D96 and D130 act as Ca2+-coordinating residues of the EF hands, while F142 resides one position adjacent to the Ca2+ binding loop. Moreover, the locus of the LQTS mutations on CaM (Figure 1) leads one to postulate that the LQTS phenotype is associated with target proteins that are modulated by the C-lobe of CaM.
While CaM has numerous targets, the known importance of CaM regulation of the LTCC CaV1.2 in controlling the cardiac action potential (AP) positions these channels as likely targets for LQTS associated calmodulinopathy mutations [14,87]. The mechanism for this CaV1.2 mediated LQTS is illustrated in Figure 2. As a resident Ca2+ sensor, CaM is bound to the C-tail of the CaV1.2 channel (Figure 2a). A mutation in the C-lobe of CaM reduces Ca2+ binding (Figure 2a, red). Upon Ca2+ entry through the channel, CaV1.2 normally exhibits robust CDI (Figure 2b, black); however, CDI is significantly blunted in channels harboring a mutant CaM (Figure 2b, red). This loss of inactivation results in added Ca2+ entry during the plateau phase of the cardiac AP, significantly prolonging the duration of the AP (Figure 2c, red) as compared to cardiomyocytes harboring a WT CaM (Figure 2c, black). This AP prolongation at the level of the ventricular myocyte corresponds to a prolongation of the QT interval as seen on a patient ECG (Figure 2d). Moreover, the increased influx of Ca2+ through CaV1.2 under these conditions also leads to increased calcium-induced calcium release (CICR) from the SR, further enhancing the propensity for arrhythmia in response to these mutations.
Figure 2.
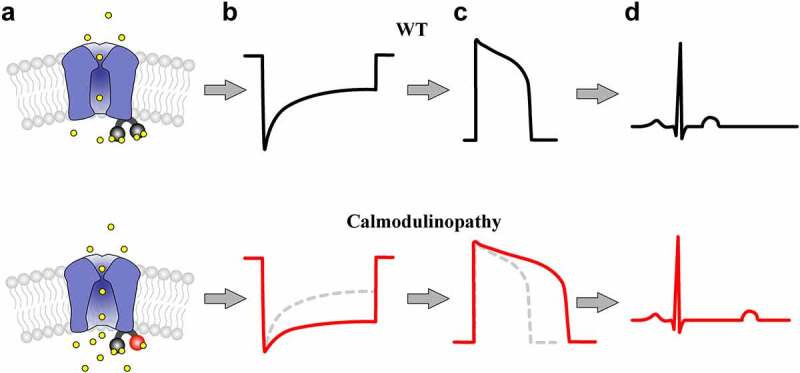
Proposed Impact of a Reduction in CaV1.2 CDI in the Heart. (a), Cartoon illustrating CaV1.2 channels pre-associated with WT (top) vs. mutant (bottom) calmodulin. A mutation in the EF-hand of the C-lobe of CaM (bottom, red) reduces the ability of CaM to bind Ca2+ (yellow) (b), Illustration of CaV1.2 Ca2+ current in response to a step depolarization from channels harboring a WT (top, black) vs. calmodulinopathic (bottom, red) CaM. The CaM mutation causes a reduction in CDI (bottom, red), as compared to WT CaM (top, black; bottom dashed gray). (c), The resulting cardiac AP for WT (top, black) vs. mutant (bottom, red) CaM. Excess calcium due to loss of channel inactivation leads to the characteristically prolonged action potential, as seen in the red trace. WT AP (top, black) is reproduced in the bottom panel as the dashed gray line for comparison. (d), ECG schematic illustrating the effect of CaM for WT (top, black) vs. calmodulinopathic (bottom, red) conditions. The prolonged action potential results in a pathologically extended QT interval (bottom).
This mechanism has been evaluated in a cardiomyocyte expression system. Overexpression of mutant CaM (CaM D96V, CaM D130G, and CaM F142L) in adult Guinea pig ventricular myocytes (aGPVMs) severely blunted CDI of the Ca2+ current from these cells (Figure 3a) [87]. Likewise, similar deficits in CDI were demonstrated in induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) derived from a patient harboring the CaM F142L mutation [12] and the D130G mutation [88] (Figure 3b). Consistent with the proposed impact of reduced inactivation of CaV1.2 (Figure 2), overexpression of mutant CaM within aGPVMs produced severe prolongation of the AP and proarrhythmic events including electrical alternans and after depolarizations, as well as increased Ca2+ transient magnitude and duration, increased diastolic Ca2+, and increased SR load [87]. In addition, aGPVMs expressing LQTS-associated CaM displayed increased beat-to-beat heterogeneity in both AP and Ca2+ transient morphology, a critical element in creating a proarrhythmic condition. In concordance with these findings, Yin et al. observed slower Ca2+ current decay and similar Ca2+ cycling disturbances upon overexpression of LQTS-associated mutant CaM in fetal mouse ventricular myocytes [89]. Thus, evidence suggests that disruption of CDI of CaV1.2 channels by mutant CaM may be sufficient to produce an LQTS phenotype.
Figure 3.
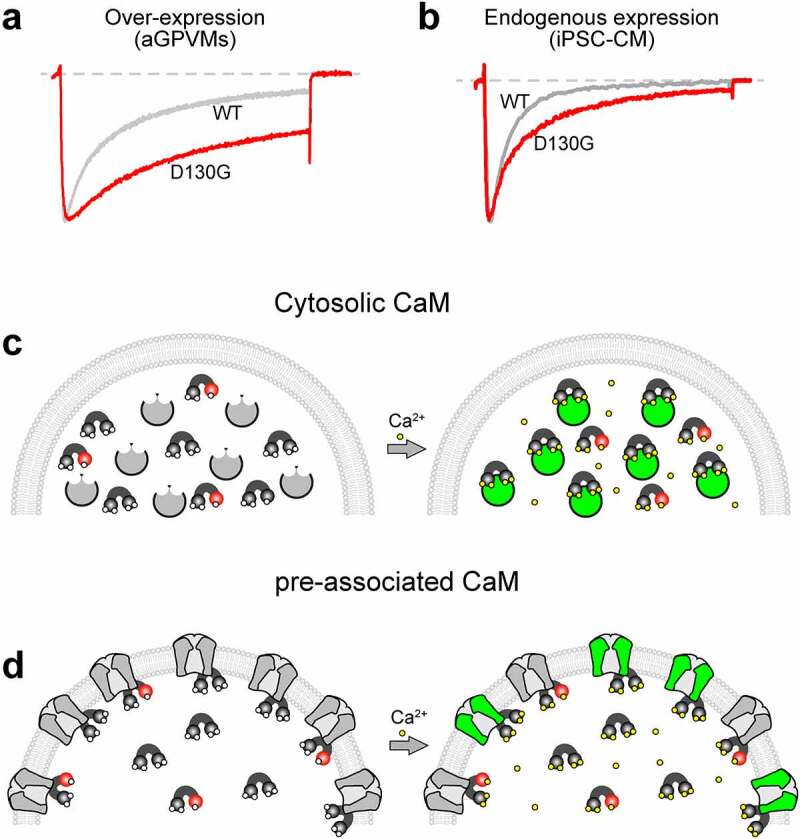
The Implications of Mixed Mutant and WT CaM Expression. (a), Exemplar Ca2+ current in response to a 30 mV depolarizing step recorded from aGPVMs overexpressing CaM WT (gray) vs. CaM D130G (red). The D130G mutations causes a loss of CDI, as seen by the lesser decay of the Ca2+ current (red) as compared to WT (gray). This figure is reproduced from Limpitikul et. al. (2014) [87] with permission. (b), Exemplar Ca2+ current recording in response to a 30 mV depolarizing step from iPSC-CMs derived from a patient harboring a D130G CaM mutation (red) as compared to iPSC-CMs from an unaffected individual (gray) demonstrating a reduction in CDI. Data is reproduced from Limpitikul et. al. (2017) [88] with permission. (c), Cartoon illustrating the impact of mutant CaM under conditions where CaM remains cytosolic until Ca2+ is bound, resulting in Ca2+/CaM binding to the target. Green indicates a modulated target protein. Mutant CaM (red) inhibits Ca2+ (yellow) from binding, thus preventing mutant CaM from interacting with the target protein. (d), Cartoon illustrating the impact of mutant CaM under conditions where CaM is pre-associated to its target in the apo state. Mutant CaM (red) inhibits Ca2+ (yellow) from binding, thus preventing the associated target protein from undergoing Ca2+ dependent modulation (target remains gray).
However, Ca2+ channels are only one of many potential targets of CaM that may be disrupted in calmodulinopathies, and additional targets have been considered. Particular focus has been aimed at ion channels with known CaM interactions, and which have been identified as substrates of genetic forms of LQTS. As such, the cardiac sodium channel, NaV1.5, has been considered as a potential pathogenic element in the LQTS phenotype of calmodulinopathies [89]. Modulation of apoCaM binding via mutations within the C-tail of the channel has been shown to be causative of LQTS [53], making NaV1.5 a potential target in the pathogenesis of calmodulinopathies despite the lack of CDI identified within these channels [50]. However, heterologous expression of a human adult splice variant of NaV1.5 along with an LQTS-associated CaM demonstrated no significant difference in persistent sodium current under both low and high Ca2+ conditions [89]. A slight increase in persistent current was observed upon co-expression of CaM D130G with the fetal variant of NaV1.5 under high Ca2+ conditions in tsA201 cells, but this finding was not confirmed in mouse ventricular myocytes. Overall, this study concluded that NaV1.5 was not the primary target of LQTS-associated CaM mutations. In contrast, work in a heterologous expression system raised the possibility that mild enhancement of NaV1.5 late current was possible alongside overexpression of the CaM mutant E141G [72]. Thus, mutant CaM acting on NaV1.5 may represent a contributory pathway in the pathogenesis of LQTS in the context of calmodulinopathies, but may not be sufficient on its own to explain the full phenotype.
Finally, potassium channels may participate in the mechanisms underlying LQTS in calmodulinopathies. The small-conductance Ca2+-activated K+ (SK) channel has been identified as a possible element of pathogenesis. Co-expression of LQTS-associated mutant CaM in HEK 293 cells demonstrated a reduction in activation of SK2 channels [90], pointing to a possible role of these channels in arrhythmogenesis. Furthermore, IKS may play a role in modulating the pathogenesis of select mutations. Recent work has shown that a somewhat milder LQTS phenotype results from an N138K CaM mutation, and may be caused by a moderate loss of CaV1.2 CDI, combined with an enhancement of current through the KCNQ1 channel [91]. Thus, combinatorial effects of mutations on multiple CaM targets may further impact the severity or presentation of calmodulinopathy phenotypes.
Mechanistic studies of CPVT-associated CaM mutations
As with LQTS-associated CaM mutations, numerous studies have aimed to elucidate the mechanism underlying the CPVT phenotype of calmodulinopathy patients. In the case of CaM N98S, the mutated residue itself participates in Ca2+ binding of the third EF hand motif, and the mutation causes a 1.6–3.3-fold reduction in Ca2+ binding affinity of C-lobe CaM, with no effect on the N-lobe [15,86]. On the other hand, the mutation N54I lies outside the Ca2+-binding portion of the CaM EF hands and does not alter Ca2+ binding affinity of either the N- or C-lobe [15,86]. Nonetheless, both mutations caused an increase in Ca2+ wave frequency with a slightly smaller amplitude in adult mouse ventricular myocytes incubated with either mutant or WT CaM [86]. These results are similar to those obtained by an established CPVT mouse model (Casq2 knockout) [92], demonstrating a mechanistic overlap between previously studied forms of CPVT and calmodulinopathies.
As CPVT is known to involve RyR2 dysregulation [93], it seems reasonable that disruption of CaM binding to RyR2 might represent a potential pathological mechanism. Indeed, it has been shown that the N98S mutation produced a significant reduction in binding affinity of apoCaM to one of the CaM binding domains (CaM binding domain 2: CaMBD2) of RyR2; however, binding was restored following calcification of CaM [15]. At the same time, there was no disruption in the binding between CaM N54I and CaMBD2 either at low or high Ca2+ concentrations. Since both apoCaM and Ca2+/CaM decrease the open probability of RyR263, Nyegaard et al. hypothesized that CaM N98S may contribute to CPVT via a diminished ability to inhibit RyR2 due to decreased apoCaM binding. However, since there was no change in the binding of CaM N54I to RyR2, they proposed that this mutant CaM might act through another mechanism to cause CPVT. Similarly, the binding affinity of mutant CaM to full-length RyR2 was examined in SR vesicles extracted from porcine cardiomyocytes [86]. Interestingly, both CaM N54I and CaM N98S were shown to exhibit enhanced binding to RyR2 as compared to WT CaM, when evaluated in the apo state (30 nM Ca2+). However, binding was equivalent between mutant and WT CaM in the calcified state (30 μM Ca2+). The apparent discrepancy with previous study may stem from the use of a shortened RyR2 peptide containing only the CaMBD2 binding domain [15] as compared to full-length RyR2 [86]. However, these are not the only studies to identify variable effects of mutant CaM on RyR2 binding. The CPVT-associated mutation A103V did not appear to increase affinity for RyR2 [84]. However, a later study observed a significant decrease in binding to RyR2 for both A103V and CaM N54I [94]. Thus, measured changes in binding affinity between CaM and RyR2 have not indicated a consistent mechanism leading to CPVT.
Despite the lack of consensus in terms of binding affinity changes, the impact of mutant CaM on RyR2 function appears to be more consistent. Generally, CaM mutants associated with CPVT have increased Ca2+ release from the SR. Both N54I and N98S CaM mutations induced higher spark frequency and longer spark duration in permeabilized ventricular myocytes preincubated with CaM at low (50 nM) Ca2+ concentrations. This effect was amplified upon addition of cAMP to mimic adrenergic stress. Corresponding to the high Ca2+ efflux from RyR2, the SR content was also lower in cells with mutant compared to WT CaM [86]. Furthermore, CaM N54I enhanced the open probability of RyR2 in both low and high Ca2+ conditions, and CaM N98S increased the open probability of RyR2 in the presence of Ca2+. Moreover, CPVT-associated mutation A103V promoted spontaneous Ca2+ wave and spark activity by enhancing SR Ca2+ release, even when expressed at a ratio of 1:3 mutant to WT CaM, demonstrating an ability to disrupt Ca2+ dynamics even in the presence of WT CaM [84], as would occur in a calmodulinopathy patient. Likewise, Hwang et al. demonstrated an increase in Ca2+ wave frequency in cardiac cells even when mutant CaM was expressed with WT CaM at a ratio of 1:8. Overall, increased spontaneous Ca2+ release from the SR appears to be a common mechanism underlying the pathogenesis of CPVT in calmodulinopathies, consistent with other forms of CPVT that are associated with increased Ca2+ leak through RyR2.
The impact of CaM pre-association
In all reported cases, calmodulinopathy patients harbor a heterozygous mutation in CaM, sometimes with somatic mosaicism. Given that there are three different CALM genes (CALM1, 2, and 3) encoding the identical protein, how can a relatively small amount of mutant CaM produce severe disease phenotypes while in the presence of relatively abundant WT CaM? To resolve this question, one can consider the binding state of apoCaM, which can either exist in a free form floating in the cytoplasm, or can pre-associate with a target protein. The latter option provides an avenue by which a relatively small amount of mutant CaM can lead to the large pathological effects observed on cellular function (Figure 3c,d)
First, consider the case where apoCaM is distributed throughout the cell (Figure 3c, left), with a fraction of the population harboring a mutation that reduces the Ca2+-binding affinity (Figure 3c, red). Upon the rise of intracellular Ca2+, only WT CaM fully binds the ion and is thus able to bind to the Ca2+/CaM effector site on the target (Figure 3c, right). Because there is an abundance of WT CaM, all target proteins are effectively modulated (Figure 3c, green), with mutant CaM unable to compete for the target protein. Thus, when the primary effect of the mutation is a loss of Ca2+ binding to CaM, targets without pre-association are unlikely to represent pathogenic elements. Examples of CaM targets that follow this model include CaMKII, MLCK, and calcineurin.
On the other hand, consider the case where apoCaM is pre-associated with its target (Figure 3d). If apoCaM can bind to its target at least as well as WT CaM, i.e. the mutations do not diminish the binding affinity of apoCaM to its target, then a fraction of targets will be occupied by mutant CaM even at basal Ca2+ levels (Figure 3d, left). Upon the rise of intracellular Ca2+, mutant CaM is unable to fully bind Ca2+, leaving the fraction of target proteins associated with these mutant proteins unmodulated (Figure 3d, right, gray). This fraction of unmodulated targets results in an overall decrease in the magnitude of Ca2+/CaM regulation. Thus, pre-association enables a smaller amount of mutant CaM to have a larger effect on cellular function. Indeed, multiple ion channels exhibit such apoCaM pre-association, including the CaV1.2 channel that has been strongly implicated in the pathogenesis of calmodulinopathic LQTS [87,95]. Moreover, it has been demonstrated that for most calmodulinopathic mutations, apoCaM binding to the channel is preserved, or in some cases enhanced, validating the ability of the mutant CaM to pre-associate with the channel [87,96]. As a result of this pre-association, it has been shown that small amounts of mutant CaM, even in the presence of an overabundance of WT CaM, can reduce the CDI of CaV1.2 channels in heterologous systems [87]. This translates to a marked effect of endogenously expressed mutant CaM on CDI when measured in iPSC-derived cardiomyocytes derived from calmodulinopathy patients (Figure 3b) [12,88,89]. Interestingly, this pre-association also means that the effect of any CaM mutation can be modulated if the mutation also alters the binding affinity between apoCaM and its target. This appears particularly relevant for select mutations such as F142L, which have been shown to bind CaV1.2 with higher affinity as compared to WT CaM [87,96], thus increasing the number of channels harboring a mutant CaM and thereby enhancing the pathogenic impact of the mutation. Conversely, mutation Q136P appears to reduce apoCaM binding to CaV1.2, thus likely reducing the pathogenic burden of the mutation [97]. Thus, in addition to impacting the Ca2+ binding affinity of CaM, pathogenesis may be modulated by other changes to the protein structure resulting in altered interactions with the target of interest.
Potential effects outside the heart
CaM is ubiquitously expressed and modulates multiple targets across various organ systems. Alterations in the levels of CaM protein have been implicated in several disease states, including heart failure [98], schizophrenia [99], and Parkinson’s disease [100–103]. Thus, pathogenic CaM mutations may be predicted to impair cellular function in tissues beyond the heart. In fact, multiple reports include extra-cardiac symptoms among calmodulinopathy patients. In the original description of LQTS-associated calmodulinopathy, multiple patients were reported to suffer from seizures and neuro-developmental delay [11]. Subsequent reports have identified similar effects. The E105A mutation in CALM1 is associated with both LQTS and developmental delay [104], and neurological features were identified in 13 calmodulinopathy patients in an international calmodulinopathy registry [73]. Likewise, CaM variant E140V was detected in a patient with a described abnormality of the nervous system [105]. Although it remains possible that some of these neurological symptoms could be caused by hypoxic brain injuries secondary to cardiac arrest, in at least one case cardiac influence was ruled out [73]. Thus, mutant CaM may directly impact neuronal targets, leading to these neurological symptoms.
To date, few studies have considered the functional impact of mutant CaM on the brain; however, ion channels that pre-associate with apoCaM may be considered potential candidates for neurological pathogenicity in calmodulinopathy patients. In fact, both RyR2 and CaV1.2, which have been shown to be critical to the pathogenesis of calmodulinopathic CPVT and LTS respectively, are also highly expressed in the brain. Thus, similar pathogenic mechanisms involving these two proteins may well underly the neurological phenotypes of calmodulinopathies. Moreover, the complement of ion channels in the brain includes a variety of channels not found in the heart. In particular, multiple voltage-gated Ca2+ channels that are known to pre-associate with apoCaM are found in the brain. Given the greater abundance of C-lobe CaM mutations (Figure 1) channels with strong C-lobe CaM regulation may be of interest. In particular, CaV1.3 channels exhibit both N- and C-lobe CaM mediated CDI. On the other hand, CaV2.1 is known to display N-lobe mediated CDI, and C-lobe mediated CDF [32]. Thus, the potential impact of mutant CaM on these channels may actually be in the opposite direction, causing a disruption of facilitation rather than inactivation. Overall, the potential impact of these and other channels in the context of disease-associated CaM variants remains unknown and may describe an interesting future direction in calmodulinopathy research.
Therapeutic implications
Treatment options for calmodulinopathies have remained limited as most patients are refractory to conventional therapies. For those patients suffering from LQTS, beta-adrenergic blockers, Ca2+ channel blockers (CCBs), and Na+ channel blockers have often failed to alleviate symptoms [73]. The lack of efficacy by CCBs, is worth further consideration. Given that LQTS in calmodulinopathy patients appears to result largely from a loss of inactivation of the CaV1.2 LTCC, it might be reasonable to expect CCBs to provide some symptom relief due to their inhibition of LTCCs. However, the CCB verapamil failed to correct the LQTS phenotype of one patient, instead causing electrical instability, resulting in discontinuation of the drug [11]. Subsequently, a combination of a beta-blocker with verapamil reduced the occurrence of cardiac arrest, but still did not correct the QT prolongation [106]. This lack of efficacy may stem directly from the inactivation deficits produced by the mutant CaM. CCBs are known to be use-dependent blockers, where the drug binds preferentially to channels in the open and inactivated states [107,108]. Thus, as mutant CaM reduces channel inactivation, it may also reduce CCB block. Such a mechanism has been shown for mutations on the CaV1.2 channel itself, where loss of inactivation correlated with decreased efficacy of CCBs [109]. Thus, currently available CCBs may not be adequate for the treatment of calmodulinopathic LQTS.
Similar challenges have been found in the treatment of calmodulinopathic CPVT. Beta-adrenergic blockers, typically a first-line therapy for CPVT, have often been ineffective in the context of calmodulinopathies [15,69]. As a result, additional options have been sought. The sodium channel blocker flecainide was shown to suppress the Ca2+ wave frequency in permeabilized myocytes incubated with CaM N54I and N98S, providing hope for use of this drug in calmodulinopathy patients [86]. In fact, flecainide was shown to be efficacious in multiple CPVT disease models [110], providing evidence that its effect might be mutation-independent. Another potential candidate that has been suggested for the treatment of calmodulinopathic CPVT is the benzothiazepine derivative, K201, also called JTV519 [111,112]. K201 has been shown to stabilize the closed state of RyR2, thereby restoring channel function in a RyR-centric disease model [113]. Additionally, a related compound S107, also called rycal, has been shown to stabilize RyR2-calstabin2 interactions and thereby reduce diastolic SR Ca2+ leak, as well as reduce arrhythmias, infarct size, and ventricular remodeling in a rat ischemia–reperfusion model [114,115]. While it remains to be seen if these therapies can provide long-term benefit in patients, the research indicates that treatment options may exist for the CPVT phenotype of calmodulinopathies.
More broadly, targeted ablation of alleles containing disease-associated CaM mutations has been proposed as a potential avenue for therapy. Multiple frameshift mutations have been identified that can result in the near-complete loss of a CaM allele without apparent clinical deficits [116], leaving open the possibility that a single CALM gene could be targeted without detrimental side effects. CRISPR/Cas9 deletion of a CALM gene harboring a deleterious mutation has been evaluated in iPSC derived cardiomyocytes derived from calmodulinopathy patients [88,117]. In these studies, ablation of a single CALM gene harboring either a D130G [88] or an N98S [117] mutation restored normal CDI and AP morphology in iPSC derived cardiomyocytes. Thus, a genetic approach to the treatment of calmodulinopathies may provide a path forward for the treatment of LQTS in these patients.
Funding Statement
This work is funded by and R21 from The National Institute of Neurological Disorders and Stroke (NINDS) [R21NS127294].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Hodgkin AL, Keynes RD.. Movements of labelled calcium in squid giant axons. J Physiol. 1957;138(2):253–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cheung WY. Cyclic 3’,5’-nucleotide phosphodiesterase. Demonstration of an activator. Biochem Biophys Res Commun. 1970;38(3):533–538. [DOI] [PubMed] [Google Scholar]
- [3].Cheung WY. Cyclic 3‘,5‘-nucleotide phosphodiesterase. Effect of divalent cations. Biochim Biophys Acta. 1971;242(2):395–409. [DOI] [PubMed] [Google Scholar]
- [4].Cheung WY. Calmodulin plays a pivotal role in cellular regulation. Science. 1980;207(4426):19–27. [DOI] [PubMed] [Google Scholar]
- [5].Kakiuchi S, Yamazaki R. Calcium dependent phosphodiesterase activity and its activating factor (PAF) from brain studies on cyclic 3,’5’-nucleotide phosphodiesterase (3). Biochem Biophys Res Commun. 1970;41(5):1104–1110. [DOI] [PubMed] [Google Scholar]
- [6].Bhattacharya S, Bunick CG, Chazin WJ. Target selectivity in EF-hand calcium binding proteins. Biochim Biophys Acta. 2004;1742(1–3):69–79. [DOI] [PubMed] [Google Scholar]
- [7].Berchtold MW, Villalobo A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta. 2014;1843(2):398–435. [DOI] [PubMed] [Google Scholar]
- [8].Sondergaard MT, Sorensen AB, Skov LL, et al. Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 2015;282(4):803–816. [DOI] [PubMed] [Google Scholar]
- [9].Kink JA, Maley ME, Preston RR, et al. Mutations in paramecium calmodulin indicate functional differences between the C-terminal and N-terminal lobes in vivo. Cell. 1990;62:165–174. [DOI] [PubMed] [Google Scholar]
- [10].Westerlund AM, Delemotte L, Nussinov R. Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput Biol. 2018;14(4):e1006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127(9):1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rocchetti M, Sala L, Dreizehnter L, et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc Res. 2017;113(5):531–541. [DOI] [PubMed] [Google Scholar]
- [13].Weinman J, Della Gaspera B, Dautigny A, et al. Developmental regulation of calmodulin gene expression in rat brain and skeletal muscle. Cell Regul. 1991;2(10):819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alseikhan BA, DeMaria CD, Colecraft HM, et al. Engineered calmodulins reveal the unexpected eminence of Ca2+ Ca 2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99(26):17185–17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nyegaard M, Overgaard MT, Sondergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91(4):703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and Orai. Annu Rev Immunol. 2010;28(1):491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Oh-hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol. 2008;20(3):250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Webb RC. Smooth muscle contraction and relaxation. Adv Physiol Educ. 2003;27(4):201–206. [DOI] [PubMed] [Google Scholar]
- [19].Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. A J Physiol. 1993;264(5):C1085–95. [DOI] [PubMed] [Google Scholar]
- [20].Chin ER. Role of Ca2+/calmodulin-dependent Ca 2+ /calmodulin-dependent kinases in skeletal muscle plasticity. J Appl Physiol. 2005;99(2):414–423. [DOI] [PubMed] [Google Scholar]
- [21].Wang B, Sullivan KM, Beckingham K. Drosophila calmodulin mutants with specific defects in the musculature or in the nervous system. Genetics. 2003;165(3):1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14(3):318–327. [DOI] [PubMed] [Google Scholar]
- [23].Mulkey RM, Endo S, Shenolikar S, et al. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369(6480):486–488. [DOI] [PubMed] [Google Scholar]
- [24].Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2(7):461–474. [DOI] [PubMed] [Google Scholar]
- [25].Casanova JR, Nishimura M, Swann JW. The effects of early-life seizures on hippocampal dendrite development and later-life learning and memory. Brain Res Bull. 2014;103:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen LJ, Wang YJ, Chen JR, et al. NMDA receptor triggered molecular cascade underlies compression-induced rapid dendritic spine plasticity in cortical neurons. Exp Neurol. 2015;266C:86–98. [DOI] [PubMed] [Google Scholar]
- [27].Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007;73(4):641–647. [DOI] [PubMed] [Google Scholar]
- [28].Rokita AG, Anderson ME. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. 2012;126(17):2125–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65(1):45–79. [DOI] [PubMed] [Google Scholar]
- [30].Wilkins BJ, Dai YS, Bueno OF, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94(1):110–118. [DOI] [PubMed] [Google Scholar]
- [31].DeMaria CD, Soong TW, Alseikhan BA, et al. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411(6836):484–489. [DOI] [PubMed] [Google Scholar]
- [32].Liang H, DeMaria CD, Erickson MG, et al. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39(6):951–960. [DOI] [PubMed] [Google Scholar]
- [33].Lee A, Zhou H, Scheuer T, et al. Molecular determinants of Ca(2+)/calmodulin-dependent Ca2+/calmodulin-dependent regulation of Ca(v)2.1 Ca v 2.1 channels. Proc Natl Acad Sci U S A. 2003;100(26):16059–16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Qin N, Olcese R, Bransby M, et al. Ca2+-induced Ca 2+ -induced inhibition of the cardiac Ca2+ Ca 2+ channel depends on calmodulin. Proc Natl Acad Sci U S A. 1999;96(5):2435–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zuhlke RD, Reuter H. Ca2+-sensitive Ca 2+ -sensitive inactivation of L-type Ca2+ Ca 2+ channels depends on multiple cytoplasmic amino acid sequences of the alpha1C α 1C subunit. Proc Natl Acad Sci U S A. 1998;95(6):3287–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Peterson BZ, DeMaria CD, Adelman JP, et al. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999;22(3):549–558. [DOI] [PubMed] [Google Scholar]
- [37].Zuhlke RD, Pitt GS, Deisseroth K, et al. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399(6732):159–162. [DOI] [PubMed] [Google Scholar]
- [38].Yang PS, Alseikhan BA, Hiel H, et al. Switching of Ca2+-dependent inactivation of Ca(v)1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26(42):10677–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Singh A, Hamedinger D, Hoda JC, et al. C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat Neurosci. 2006;9(9):1108–1116. [DOI] [PubMed] [Google Scholar]
- [40].Wahl-Schott C, Baumann L, Cuny H, et al. Switching off calcium-dependent inactivation in L-type l -type calcium channels by an autoinhibitory domain. Proc Natl Acad Sci U S A. 2006;103(42):15657–15662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dick IE, Tadross MR, Liang H, et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451(7180):830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ohrtman J, Ritter B, Polster A, et al. Sequence differences in the IQ motifs of CaV1.1 and CaV1.2 strongly impact calmodulin binding and calcium-dependent inactivation. J Biol Chem. 2008;283(43):29301–29311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bannister RA, Beam KG. Ca(V)1.1: the atypical prototypical voltage-gated Ca(2)(+) channel. Biochim Biophys Acta. 2013;1828(7):1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Niu J, Yang W, Yue DT, et al. Duplex signaling by CaM and Stac3 enhances Ca(V)1.1 function and provides insights into congenital myopathy. J Gen Physiol. 2018;150(8):1145–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Adams PJ, Rungta RL, Garcia E, et al. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc Natl Acad Sci U S A. 2010;107(43):18694–18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chemin J, Taiakina V, Monteil A, et al. Calmodulin regulates Ca(v)3 T-type channels at their gating brake. J Biol Chem. 2017;292(49):20010–20031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Asmara H, Micu I, Rizwan AP, et al. A T-type channel-calmodulin complex triggers alphaCaMKII activation. Mol Brain. 2017;10(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lee N, Jeong S, Kim KC, et al. Ca(2+) Ca 2+ regulation of Ca(v)3.3 Ca v 3.3 T-type Ca(2+) Ca 2+ channel is mediated by calmodulin. Mol Pharmacol. 2017;92(3):347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Adams PJ, Ben-Johny M, Dick IE, et al. Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell. 2014;159(3):608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ben-Johny M, Yang PS, Niu J, et al. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell. 2014;157(7):1657–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Feldkamp MD, Yu L, Shea MA. Structural and energetic determinants of apo calmodulin binding to the IQ motif of the Na(V)1.2 voltage-dependent sodium channel. Structure. 2011;19(5):733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yoder JB, Ben-Johny M, Farinelli F, et al. Ca(2+)-dependent regulation of sodium channels NaV1.4 and NaV1.5 is controlled by the post-IQ motif. Nat Commun. 2019;10(1):1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kang PW, Chakouri N, Diaz J, et al. Elementary mechanisms of calmodulin regulation of NaV1.5 producing divergent arrhythmogenic phenotypes. Proc Natl Acad Sci U S A. 2021;118(21):e2025085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Roden DM. A new role for calmodulin in ion channel biology. Circ Res. 2006;98(8):979–981. [DOI] [PubMed] [Google Scholar]
- [55].Shamgar L, Ma L, Schmitt N, et al. Calmodulin is essential for cardiac IKS I KS channel gating and assembly: impaired function in long-QT mutations. Circ Res. 2006;98(8):1055–1063. [DOI] [PubMed] [Google Scholar]
- [56].Kang PW, Westerlund AM, Shi J, et al. Calmodulin acts as a state-dependent switch to control a cardiac potassium channel opening. Sci Adv. 2020;6(50):eabd6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu W, Devaux JJ. Calmodulin orchestrates the heteromeric assembly and the trafficking of KCNQ2/3 (Kv7.2/3) channels in neurons. Mol Cell Neurosci. 2014;58:40–52. [DOI] [PubMed] [Google Scholar]
- [58].Gamper N, Shapiro MS. Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol. 2003;122(1):17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Orlov SN, Kravtsov GM. Participation of calmodulin in the regulation of plasma membrane electric potential by intracellular calcium. Biokhimiia. 1983;48(9):1447–1455. [PubMed] [Google Scholar]
- [60].Xia XM, Fakler B, Rivard A, et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395(6701):503–507. [DOI] [PubMed] [Google Scholar]
- [61].Keen JE, Khawaled R, Farrens DL, et al. Domains responsible for constitutive and Ca(2+)-dependent Ca 2+ -Dependent interactions between calmodulin and small conductance Ca(2+)-activated Ca 2+ -activated potassium channels. J Neurosci. 1999;19(20):8830–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fruen BR, Black DJ, Bloomquist RA, et al. Regulation of the RYR1 and RYR2 Ca2+ Ca 2+ release channel isoforms by Ca2+-insensitive Ca2+-insensitive mutants of calmodulin. Biochemistry. 2003;42(9):2740–2747. [DOI] [PubMed] [Google Scholar]
- [63].Xu L, Meer G. Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys J. 2004;86(2):797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jiang D, Xiao B, Yang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ Ca 2+ release (SOICR). Proc Natl Acad Sci U S A. 2004;101(35):13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Patel S, Morris SA, Adkins CE, et al. Ca2+-independent Ca2+-independent inhibition of inositol trisphosphate receptors by calmodulin: redistribution of calmodulin as a possible means of regulating Ca2+ Ca2+ mobilization. Proc Natl Acad Sci U S A. 1997;94(21):11627–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sienaert I, Nadif Kasri N, Vanlingen S, et al. Localization and function of a calmodulin-apocalmodulin-binding domain in the N-terminal part of the type 1 inositol 1,4,5-trisphosphate receptor. Biochem J. 2002;365(1):269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Klee CB, Crouch TH, Richman PG. Calmodulin. Annu Rev Biochem. 1980;49(1):489–515. [DOI] [PubMed] [Google Scholar]
- [68].Yap KL, Kim J, Truong K, et al. Calmodulin target database. J Struct Funct Genomics. 2000;1(1):8–14. [DOI] [PubMed] [Google Scholar]
- [69].Makita N, Yagihara N, Crotti L, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc genet. 2014;7(4):466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Reed GJ, Boczek NJ, Etheridge SP, et al. CALM3 mutation associated with long QT syndrome. Heart Rhythm. 2015;12(2):419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wren LM, Jimenez-Jaimez J, Al-Ghamdi S, et al. Genetic Mosaicism in Calmodulinopathy. Circ Genom Precis Med. 2019;12(9):375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Boczek NJ, Gomez-Hurtado N, Ye D, et al. Spectrum and prevalence of CALM1-CALM1 -, CALM2-CALM2 -, and CALM3-encoded CALM3 -encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated syndrome–associated calmodulin missense variant, E141G. Circ Cardiovasc genet. 2016;9(2):136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Crotti L, Spazzolini C, Tester DJ, et al. Calmodulin mutations and life-threatening cardiac arrhythmias: insights from the International Calmodulinopathy Registry. Eur Heart J. 2019;40(35):2964–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gomez-Hurtado N, Kryshtal DO, Hwang HS, et al. Calmodulin mutation (CALM1-e141g associated with long qt syndrome disrupts calmodulin calcium binding and impairs l-type ca channel inactivation). Heart Rhythm. 2014;11:2135–2136. [Google Scholar]
- [75].Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mirsadeghi L, Haji Hosseini R, Banaei-Moghaddam AM, et al. EARN: an ensemble machine learning algorithm to predict driver genes in metastatic breast cancer. BMC Med Genomics. 2021;14(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jimenez-Jaimez J, Palomino Doza J, Ortega A, et al. Calmodulin 2 mutation N98S is associated with unexplained cardiac arrest in infants due to low clinical penetrance electrical disorders. PLoS One. 2016;11(4):e0153851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Brohus M, Arsov T, Wallace DA, et al. Infanticide vs. inherited cardiac arrhythmias. Europace. 2021;23(3):441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Pipilas DC, Johnson CN, Webster G, et al. Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Heart Rhythm. 2016;13(10):2012–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zahavich L, Tarnopolsky M, Yao R, et al. Novel association of a de novo CALM2 mutation with long QT syndrome and hypertrophic cardiomyopathy. Circ Genom Precis Med. 2018;11(10):e002255. [DOI] [PubMed] [Google Scholar]
- [82].Reed GJ, Boczek NJ, Etheridge S, et al. CALM3 Mutation Associated with Long QT Syndrome. Heart Rhythm. 2014;12(2):419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chaix MA, Koopmann TT, Goyette P, et al. Novel CALM3 mutations in pediatric long QT syndrome patients support a CALM3-specific calmodulinopathy. HeartRhythm Case Rep. 2016;2(3):250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gomez-Hurtado N, Boczek NJ, Kryshtal DO, et al. Novel CPVT-associated calmodulin mutation in CALM3 (CALM3-A103V) activates arrhythmogenic ca Waves and Sparks. Circ Arrhythm Electrophysiol. 2016;9(8):e004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Marsman RF, Barc J, Beekman L, et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2013;63(3):259–66. [DOI] [PubMed] [Google Scholar]
- [86].Hwang HS, Nitu FR, Yang Y, et al. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114(7):1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Limpitikul WB, Dick IE, Joshi-Mukherjee R, et al. Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol. 2014;74:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Limpitikul WB, Dick IE, Tester DJ, et al. A precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndrome. Circ Res. 2017;120(1):39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yin G, Hassan F, Haroun AR, et al. Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ Ca 2+ regulation by distinct mechanisms. J Am Heart Assoc. 2014;3(3):e000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yu CC, Ko JS, Ai T, et al. Arrhythmogenic calmodulin mutations impede activation of small-conductance calcium-activated potassium current. Heart Rhythm. 2016;13(8):1716–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kato K, Isbell HM, Fressart V, et al. Novel CALM3 variant causing calmodulinopathy with variable expressivity in a 4-generation family. Circ Arrhythm Electrophysiol. 2022;15(3):e010572. [DOI] [PubMed] [Google Scholar]
- [92].Knollmann BC, Chopra N, Hlaing T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116(9):2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nakamura Y, Yamamoto T, Kobayashi S, et al. Ryanodine receptor-bound calmodulin is essential to protect against catecholaminergic polymorphic ventricular tachycardia. JCI Insight. 2019;4(11):e126112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Prakash O, Held M, McCormick LF, et al. CPVT-associated calmodulin variants N53I and A102V dysregulate Ca2+ signalling via different mechanisms. J Cell Sci. 2022;135(2):jcs258796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Limpitikul WB, Greenstein JL, Yue DT, et al. A bilobal model of Ca(2+)-dependent inactivation to probe the physiology of L-type Ca(2+) channels. J Gen Physiol. 2018;150(12):1688–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wang K, Holt C, Lu J, et al. Arrhythmia mutations in calmodulin cause conformational changes that affect interactions with the cardiac voltage-gated calcium channel. Proc Natl Acad Sci U S A. 2018;115(45):E10556–E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang K, Brohus M, Holt C, et al. Arrhythmia mutations in calmodulin can disrupt cooperativity of Ca(2+) Ca 2+ binding and cause misfolding. J Physiol. 2020;598(6):1169–1186. [DOI] [PubMed] [Google Scholar]
- [98].Ikeda S, He A, Kong SW, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29(8):2193–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chambers JS, Thomas D, Saland L, et al. Growth-associated protein 43 (GAP-43) and synaptophysin alterations in the dentate gyrus of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):283–290. [DOI] [PubMed] [Google Scholar]
- [100].Bazzazi H, Ben Johny M, Adams PJ, et al. Continuously tunable Ca(2+) regulation of RNA-edited CaV1.3 channels. Cell Rep. 2013;5:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chan CS, Guzman JN, Ilijic E, et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447(7148):1081–1086. [DOI] [PubMed] [Google Scholar]
- [102].Lee A, Westenbroek RE, Haeseleer F, et al. Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5(3):210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287(5456):1265–1269. [DOI] [PubMed] [Google Scholar]
- [104].Takahashi K, Ishikawa T, Makita N, et al. A novel de novo calmodulin mutation in a 6-year-old boy who experienced an aborted cardiac arrest. HeartRhythm Case Rep. 2017;3(1):69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. [DOI] [PubMed] [Google Scholar]
- [106].Webster G, Schoppen ZJ, George AL Jr. Treatment of calmodulinopathy with verapamil. BMJ Case Rep. 2017;2017. DOI: 10.1136/bcr-2017-220568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kanaya S, Arlock P, Katzung BG, et al. Diltiazem and verapamil preferentially block inactivated cardiac calcium channels. J Mol Cell Cardiol. 1983;15(2):145–148. [DOI] [PubMed] [Google Scholar]
- [108].Kamp TJ, Sanguinetti MC, Miller RJ. Voltage- and use-dependent modulation of cardiac calcium channels by the dihydropyridine (+)-202-791. Circ Res. 1989;64(2):338–351. [DOI] [PubMed] [Google Scholar]
- [109].Bamgboye MA, Trafficante MK, Owoyemi J, et al. Impaired CaV1.2 inactivation reduces the efficacy of calcium channel blockers in the treatment of LQT8. J Mol Cell Cardiol. 2022;173:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hwang HS, Baldo MP, Rodriguez JP, et al. Efficacy of flecainide in catecholaminergic polymorphic ventricular tachycardia is mutation-independent but reduced by calcium overload. Front Physiol. 2019;10:992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kaneko N, Matsuda R, Hata Y, et al. Pharmacological characteristics and clinical applications of K201. Curr Clin Pharmacol. 2009;4(2):126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Szentandrassy N, Magyar ZE, Hevesi J, et al. Therapeutic approaches of ryanodine receptor-associated heart diseases. Int J Mol Sci. 2022;23(8):ijms23084435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lenhardt ML, Shulman A, Goldstein BA. The role of the insula cortex in the final common pathway for tinnitus: experience using ultra-high-frequency therapy. Int Tinnitus J. 2008;14(1):13–16. [PubMed] [Google Scholar]
- [114].Shan J, Betzenhauser MJ, Kushnir A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120(12):4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Fauconnier J, Meli AC, Thireau J, et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc Natl Acad Sci U S A. 2011;108(32):13258–13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Jensen HH, Brohus M, Nyegaard M, et al. Human Calmodulin Mutations. Front Mol Neurosci. 2018;11:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yamamoto Y, Makiyama T, Harita T, et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum Mol Genet. 2017;26(9):1670–1677. [DOI] [PubMed] [Google Scholar]