

Abstract
Background:
We studied the pharmacokinetics (PK) and safety of 100mg doravirine and doravirine/lamivudine/tenofovir disoproxil fumarate fixed dose combination (100/300/300mg- DOR FDC) treatment in adolescents with HIV-1.
Methods:
Adolescents ages 12- <18 years enrolled in two sequential cohorts. Cohort 1 evaluated intensive PK and short-term safety of 100mg single-dose doravirine in adolescents ≥ 35 kg. Cohort 2 participants either initiated treatment with DOR FDC (antiretroviral (ARV)-naïve) or switched to DOR FDC from a previous ARV regimen (virologically-suppressed). The first 10 Cohort 2 participants had intensive PK evaluations, and safety, sparse PK and HIV RNA were assessed through week 24.
Results:
Fifty-five adolescents, median age 15.0 years and baseline weight 51.5 kg, enrolled. Nine participants completed Cohort 1 PK assessments (8 of the 9 participants weighed ≥45 kg) and 45 initiated study drug in Cohort 2. Doravirine geometric mean (GM) AUC0–∞ was 34.8 μM∙h and GM C24 was 514 nM following a single dose, with a predicted steady state GM C24,ss,pred of 690 nM. Cohort 2 enrolled adolescents weighing ≥ 45 kg. Plasma concentrations of doravirine, tenofovir and lamivudine achieved by Cohort 2 participants were similar to those reported in adults. No drug-related serious or grade 3 or 4 adverse events occurred. Forty-two of 45 participants (93.3%; 95% CI [81.7, 98.6]) achieved or maintained HIV-1 RNA <40 copies/mL.
Conclusions:
Doravirine and DOR FDC achieved target PK in adolescents with HIV-1. DOR FDC was well tolerated and maintained excellent virologic efficacy through 24 weeks, offering a favorable option for adolescents.
Keywords: doravirine, MK-1439, MK-1439A, HIV-1, adolescents
INTRODUCTION
The introduction of effective combination antiretroviral (ARV) therapy for treatment of HIV infection has led to significant declines in mortality in children and adolescents with HIV-11–3. The need for life-long therapy requires the availability of potent ARV regimens that are well tolerated, as virologic failure and drug resistance is unfortunately common among children and adolescents4–8. In addition, significant concerns remain regarding toxicities associated with widely-used ARVs, including neuropsychiatric toxicities with efavirenz, gastrointestinal toxicities such as diarrhea with multiple protease inhibitors (PIs), weight gain with integrase strand transfer inhibitors (INSTIs) and serum lipid abnormalities associated with multiple ARV classes. Thus, potent treatment regimens that have excellent safety and tolerability profiles and are convenient are still highly desirable. Doravirine (DOR) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) recently approved for use in ARV-naïve adults. Doravirine is a potent inhibitor of HIV-1 replication in vitro, is active against both wild type virus and the most common NNRTI-resistant variants at concentrations achieved with once daily dosing9 and has shown excellent efficacy and safety in adult trials10,11.
International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) 2014 investigated the safety, tolerability and pharmacokinetics of the adult doravirine tablet and adult doravine/lamiduvine/tenofovir disoproxil fumarate (DOR/3TC/TDF) fixed dose combination tablet (DOR FDC) in youth with HIV-1. This report presents the results of the single-dose pharmacokinetics of the 100mg doravirine tablet and the 24-week treatment outcomes for the DOR FDC (100/300/300 mg DOR/3TC/TDF) in adolescents aged 12- <18 years with HIV-1.
METHODS
Study Design and Participants:
IMPAACT 2014 is a phase I/II open-label, non-randomized, multi-center study of doravirine and DOR FDC in children and adolescents with HIV-1. Participants were enrolled into one of two cohorts. Cohort 1 (single dose of doravirine) enrolled youth ages 12 - <18 years of age who weighed ≥ 35kg and who had virologic suppression on a regimen of dolutegravir or raltegravir plus two nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs). Originally at least 4 participants in the Cohort 1 single-dose PK analysis were to weigh between 35 and 45 kg. However, eligible adolescents at least 12 years of age with weight less than 45 kg were difficult to identify, therefore Cohort 2 opened for adolescents of ≥ 45 kg while Cohort 1 remained open for continued enrollment. Cohort 2 fully enrolled before additional Cohort 1 participants were identified and enrollment into both cohorts was closed.
Cohort 2 (daily dosing of DOR FDC) enrolled youth ages 12 - <18 years of age who weighed at least 45 kg and who were either ARV-naïve or virologically suppressed with no history of documented resistance mutations to DOR/3TC/TDF. To meet criteria for virologic suppression, participants must have had one or more HIV RNA PCR results below the lower level of quantification (BLLQ) within 6 months prior to enrollment, all results BLLQ within 3 months of enrollment, and an HIV RNA PCR <40 copies/mL at screening12. The study was conducted at IMPAACT Network sites following approvals from local institutional review boards and in-country ethics committees responsible for study oversight. Parents/legal guardians provided informed consent and all youth provided assent for participation.
Cohort 1 evaluated intensive pharmacokinetics (PK) and short-term safety of doravirine. Doravirine was provided as a single 100mg tablet, with intensive PK evaluation conducted around this single dose along with a follow-up safety evaluation conducted at week 2. The PK objective was to determine the weight for which the geometric AUC0–∞ of a 100mg doravirine dose did not exceed 64μM·hr, the geometric AUC0–24hr at steady-state in adults associated with taking 200mg daily.
Cohort 2 evaluated safety, tolerability and PK of the DOR FDC tablet (100 mg DOR, 300 mg 3TC, 300 mg TDF) following once daily administration. Participants in Cohort 2 discontinued their ARVs at study entry if they were on a previous regimen and both the ARV-naïve and experienced participants initiated DOR FDC at study entry. The first ten participants had intensive PK evaluation at week 1 and all cohort 2 participants had study visits at entry, weeks 2, 4, 8, 12, 16 and 24 weeks.
Study evaluations:
Routine safety labs (chemistries, complete blood count), pregnancy tests and HIV-1 RNA were obtained at each study visit. Urinalysis, CD4 cell count and lipid profiles were collected at several visits. Plasma HIV-1 RNA concentrations were determined by the RealTime HIV-1 (Abbott Molecular, Des Plaines, IL). Plasma samples were assayed for genotypic resistance at baseline for ARV-naïve participants in Cohort 2 and all participants at virologic failure. Phenotypic resistance (Monogram Biosciences, San Francisco, CA) was also assessed if a participant experienced virologic failure. Adherence for Cohort 2 participants was determined using a self-report questionnaire13 at each visit starting at week 4 for a potential of 225 responses.
Pharmacokinetics:
In Cohort 1, intensive PK samples to determine doravirine plasma concentrations were taken pre-dose and 1, 2, 4, 8, 12, 24, 48, and 72 hours post-dose. Doravirine, tenofovir, and lamivudine plasma concentrations were also determined for the first 10 participants in Cohort 2 at week 1 at the following timepoints: pre-dose and 2, 4, 12 and 24 hours post-dose (DOR) and pre-dose and 1, 2, 4, 8, 12 and 24 hours post-dose (lamivudine/tenofovir). All Cohort 2 participants had sparse PK evaluations for all three components: pre-dose at entry and week 4, random timing at weeks 8 and 12 and pre-dose plus 0.5 – 2 hours post-dose at week 24. Doravirine, lamivudine, and tenofovir plasma concentrations were quantified by liquid chromatography with tandem mass spectrometry as described14. The lower limits of quantification were 1 ng/mL for doravirine, and 5 ng/mL and 2 ng/mL for lamivudine and tenofovir, respectively. Doravirine plasma concentrations were quantified by Syneos (Quebec, QC, Canada); lamivudine and tenofovir were quantified by Pharma Medica Research Inc. (Ontario, Canada).
Study monitoring:
Sites reported grade ≥ 3 adverse events, serious adverse events (SAE), malignancies, immune reconstitution inflammatory syndrome events and pregnancies in an expedited manner to the IMPAACT 2014 study team and to the study sponsor, National Institute of Allergy and Infectious Diseases, Division of AIDS. The study team reviewed all adverse events monthly. Overall study conduct and safety were reviewed annually by the IMPAACT safety monitoring committee. For Cohort 2 participants, virologic failure was defined as two consecutive plasma HIV-1 RNA test results ≥200 copies/mL at any time after the date of enrollment for those who entered the study with viral suppression or at or after week 24 for participants who were ARV-naïve at entry. Participants with confirmed virologic failure could remain on study drug if the reason for failure was a remediable cause such as non-adherence and there were no resistance mutations to the study agents determined by genotypic resistance testing.
Statistical analyses:
Doravirine, tenofovir, and lamivudine PK parameters for Cohort 1 and week 1 of Cohort 2 and corresponding summary statistics were calculated using WinNonLin v6.3 (Certara USA, Inc., St. Louis, MO). The primary pharmacokinetic outcomes were AUC0–∞ (Cohort 1), steady state AUC0–24 (Cohort 2), Cmax, and C24hr for 100 mg doravirine in children and adolescents infected with HIV-1. Steady state C24 and Cmax were predicted for Cohort 1 participants using the non-parametric superposition function in WinNonLin.
The primary safety and tolerability outcomes were all adverse events, regardless of severity grade, and grade 3 or higher treatment related adverse events, respectively, for cohort 1 (single dose DOR) participants at week 2 and for cohort 2 (DOR FDC) participants at week 24.
Primary safety analyses included all participants who were exposed to the study drug with data restricted through Week 2 for Cohort 1 and Week 24 for Cohort 2. Descriptive statistics were used to summarize the participant characteristics and tolerability. Unless otherwise stated, median and range were used to summarize continuous variables and proportion and 95% confidence intervals were used for categorical variables.
Primary virologic efficacy analyses were performed for Cohort 2: the virologic outcomes based on the plasma HIV-1 RNA levels ≤200 copies/mL, ≤50 copies/mL, and ≤40 copies/mL respectively were assessed at week 24 using FDA snapshot algorithm (https://www.fda.gov/media/86284/download). The proportion of participants with plasma HIV-1 RNA <200 copies/mL, <50 copies/mL, and <40 copies/mL, bounded by 95% confidence intervals, were presented.
Immunologic response measured by mean changes in CD4 count and percent from baseline to Week 24 were presented with 95% confidence intervals, both in the aggregate and broken down by ARV treatment status at entry.
RESULTS
Study population:
Between July 2, 2018 and February 26, 2020, 55 participants enrolled in the study: 10 participants into cohort 1 and 45 into cohort 2 (Figure S1). All participants, except one from cohort 1, received at least one dose of study drug resulting in an analysis population of 54 participants. Participant characteristics are summarized in Table 1. Twenty-nine (53%) participants were female and 35 (65%) were Asian. At baseline, the median age was 15 years (range, 12–17 years), weight was 51.5 kg (40.3–90.8 kg), CD4 count was 716 cells/mm3 (84–1397) and HIV-1 RNA was 1.6 log10 copies/mL (1.6–5.9). As per protocol, all Cohort 1 participants had HIV-1 RNA levels <40 copies/mL at entry on a stable ARV regimen of INSTI plus 2NRTIs. Two participants entered Cohort 2 as ARV-naïve. Duration of previous ARV treatment prior to enrollment for the 43 virologically-suppressed participants entering Cohort 2 was 2.8 years (0.3 – 14.9). Over half (51%) switched from an efavirenz-based regimen at entry. One participant from Cohort 2 became pregnant and discontinued the study early.
Table 1:
Baseline Characteristics
| Cohort 1a | Cohort 2b | Total | |||
|---|---|---|---|---|---|
| (N=9) n (%) |
Treatment-Naïve (N=2) n (%) |
Virologically-Suppressed (N=43) n (%) |
All Cohort 2 (N=45) n (%) |
(N=54) n (%) |
|
| Sex | |||||
| Male | 7 (77.8) | 1 (50.0) | 18 (41.9) | 19 (42.2) | 26 (48.1) |
| Age (years) median (min, max) | 15 (12, 16) | 15.5 (14, 17) | 15 (12, 17) | 15 (12, 17) | 15 (12, 17) |
| Race (n (%)) | |||||
| Black or African American | 7 (77.8) | 0 (0) | 10 (23.3) | 10 (22.2) | 17 (31.5) |
| White | 2 (22.2) | 0 (0) | 0 (0) | 0 (0) | 2 (3.7) |
| Asian | 0 (0) | 2 (100.0) | 33 (76.7) | 35 (77.8) | 35 (64.8) |
| Ethnicity | |||||
| Hispanic or Latino | 0 (0) | 0 (0) | 1 (2.3) | 1 (2.2) | 1 (1.9) |
| Not Hispanic or Latino | 9 (100.0) | 2 (100.0) | 42 (97.7) | 44 (97.8) | 53 (98.1) |
| Weight at Baseline (kg) | |||||
| 35 - <45 | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 1 (1.9) |
| ≥ 45 | 8 (88.9) | 2 (100.0) | 43 (100.0) | 45 (100.0) | 53 (98.1) |
| Weight at Baseline (kg) median (min,max) | 55.9 (40.3, 90.8) | 59.3 (53.3, 65.2) | 51.5 (45.1, 79.8) | 51.6 (45.1, 79.8) | 51.5 (40.3, 90.8) |
| Geography | |||||
| Africa | 0 (0) | 0 (0) | 9 (20.9) | 9 (20.0) | 9 (16.7) |
| Asia/Pacific | 0 (0) | 2 (100.0) | 33 (76.7) | 35 (77.8) | 35 (64.8) |
| North America | 9 (100.0) | 0 (0) | 1 (2.3) | 1 (2.2) | 10 (18.5) |
| Duration of Prior ART (days) | |||||
| Median (min, max) | 613 (97, 1805) | 1018 (98, 5423) | 1018 (98, 5423) | 938 (97, 5423) | |
| Class of Prior ART | |||||
| NRTI | 9 (100.0) | 0 (0) | 43 (100.0) | 43 (95.6) | 52 (96.3) |
| NNRTI | 0 (0) | 0 (0) | 32 (74.4) | 32 (71.1) | 32 (59.3) |
| INSTI | 9 (100.0) | 0 (0) | 1 (2.3) | 1 (2.2) | 10 (18.5) |
| PI | 0 (0) | 0 (0) | 10 (23.3) | 10 (22.2) | 10 (18.5) |
| Not applicable | 0 (0) | 2 (100.0) | 0 (0) | 2 (4.4) | 2 (3.7) |
N = Number of participants in each group; n (%) = Number (percent) of participants in each subcategory; N/A = Not applicable. ART = antiretroviral therapy
NRTI = nucleoside reverse transcriptase inhibitor, NNRTI = non-nucleoside reverse transcriptase inhibitor, INSTI = integrase strand transfer inhibitor, PI = protease inhibitor
100mg single-dose doravirine
Daily doravirine/lamivudine/tenofovir disoproxil fumarate fixed dose combination
Pharmacokinetics:
Following a single 100mg dose of doravirine, median peak doravirine concentrations were observed at approximately 4 hours post-dose. Doravirine plasma concentrations for the adolescents in Cohort 1 were similar to those seen in adults following single dose administration of 100mg doravirine tablets (Figure 1)15. The doravirine geometric mean single dose AUC0–∞ was 34.8 μM∙h and geometric mean C24 was 514 nM. Based on the single dose data, the predicted steady state geometric mean C24,ss,pred was 690 nM (Table 2), exceeding the lower bound for efficacy based on Phase 3 adult studies in ARV-naïve participants with HIV-1 infection16. The estimated value of the accumulation ratio was 1.3. There was no indication of trend or association between pharmacokinetic parameters and weight in the Cohort 1 participants (Table S1).
Figure 1:
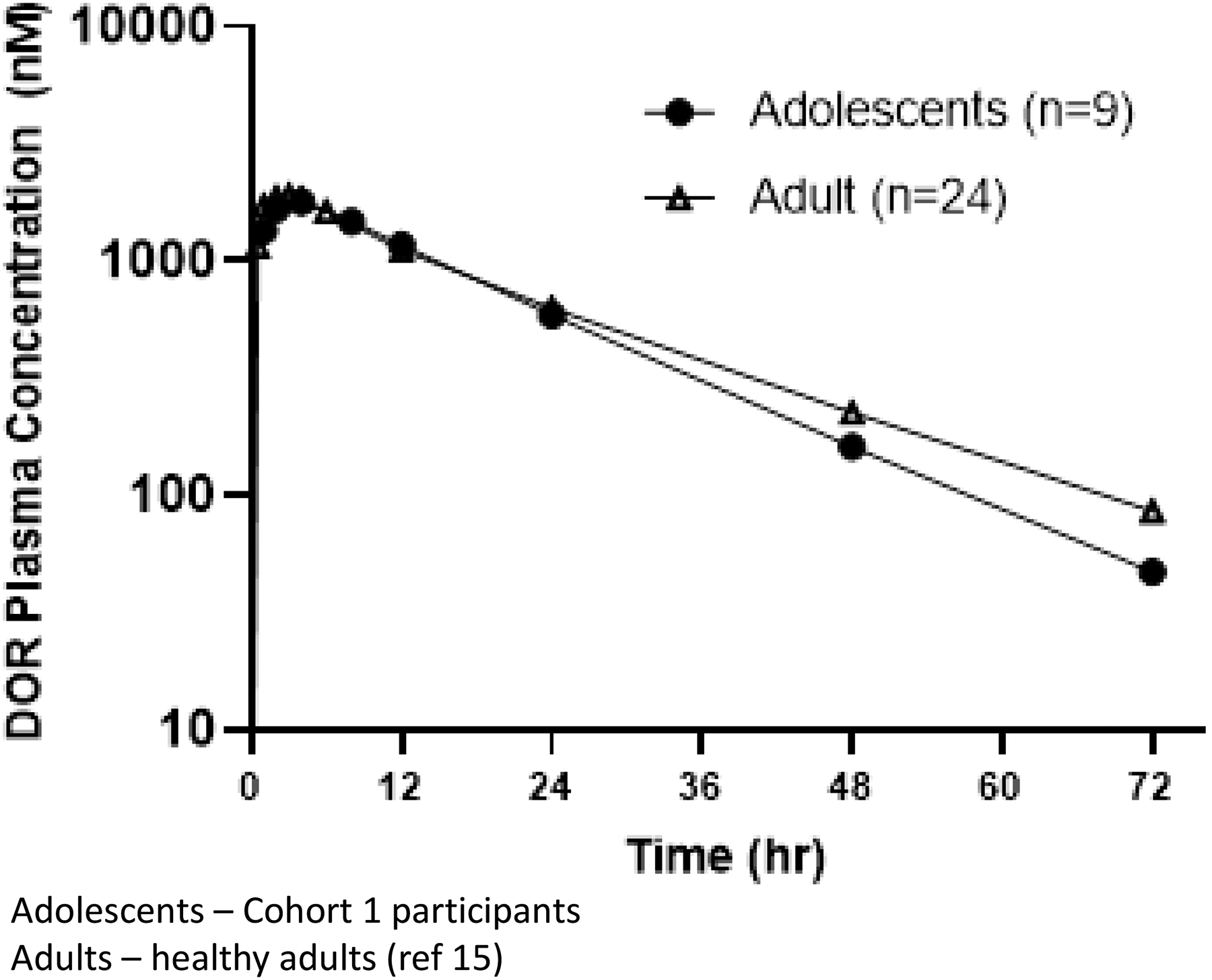
Plasma Concentrations in Adolescents and Adults Following Single Dose Administration of 100 mg Doravirine Tablets
Table 2:
Summary of Pharmacokinetic Parameter Values for Doravirine
| Geometric Mean (Geometric CV%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| AUCa (μM∙h) | C24,ssb (nM) | Cmax,ssb (μM) | Tmaxc (h) | Lz (1l/hr) | T1/2 (h) | Cl/F (L/h) | Vz/F (L) | |
| Cohort 1 (n=9) | 34.8 (43) | 690 (66) | 2.76 (28) | 3.78 (1.00–7.75) | 0.0589 (25) | 11.8 (25) | 6.75 (43) | 115 (33) |
| Cohort 2d (Week 1, n=10) | 22.9 (47) | 282 (74) | 2.13 (43) | 1.95 (1.92–3.95) | - | 10.3 (47) | - | |
Single dose AUC(0–∞) reported for Cohort 1 (100 mg single oral dose); steady state AUC(0–24) is reported for Cohort 2 (100 mg daily oral dose)
Predicted values reported for Cohort 1; observed values reported for Cohort 2
Median (range) reported for Tmax
8/10 subjects in Cohort 2 switched from an efavirenz-based regimen, resulting in decreased DOR concentrations at Week 1. Due to the sampling over 0–24 hour only at Week 1 in Cohort 2, there was insufficient data to characterize the terminal half-life; therefore, terminal half-life and Vz/F (apparent volume of distribution), could not be estimated.
Doravirine plasma concentrations at Week 1 following multiple-dose administration of DOR/3TC/TDF daily for the 10 participants in Cohort 2 with semi-intensive PK sampling peaked at approximately 2 hours post-dose. Doravirine plasma concentrations were lower than expected (geometric mean C24,ss = 282 nM) based on the PK data from Cohort 1 (Table 2). The lower concentrations were likely due to CYP3A induction as 8 of the 10 participants switched from efavirenz, a moderate CYP3A inducer known to decrease doravirine exposure17. By Week 4 and beyond, doravirine plasma concentrations in participants who switched from efavirenz were generally comparable to those who did not switch from efavirenz (Figure S2), consistent with the washout of efavirenz and CYP3A induction. At Week 4 through Week 24, the geometric mean steady state doravirine concentrations for all participants were >560 nM, exceeding the lower bound for efficacy (Table 3 and Figure S2).
Table 3:
Descriptive Statistics for DOR Plasma Concentrations by Intended Time Following Oral Administration of DOR/3TC/TDF Once Daily for All Participants: All Treated Population, Cohort 2
Based on intensive PK sampling for tenofovir and lamivudine at week 1 following oral administration of DOR/3TC/TDF, Cmax occurred at approximately 1 hour and 2 hours, respectively. Steady state AUC0–24 and Cmax were 2550 ng/mL·hr and 293 ng/mL for tenofovir and 11300 ng/mL·hr and 2100 ng/mL for lamivudine, which were consistent with those reported in adults and adolescents18,19 (Table S2).
Safety:
Overall, doravirine was well tolerated. Among 9 treated participants in Cohort 1, four participants (44.4%, 95% CI [13.7, 78.8]) had at least one Grade 1 or Grade 2 adverse event, none of which were assessed as related to the study drug (Tables 4 and S3). There were no Grade 3 or 4 adverse events in Cohort 1 participants.
Table 4:
Summary of Adverse Events according to Treatment Cohort
| Cohort 1b (N=9) n (%) |
[95 % CI] | Cohort 2c (N=45) n (%) |
[95% CI] | |
|---|---|---|---|---|
| With one or more adverse events | 4 (44.4) | [13.7, 78.8] | 45 (100.0) | [92.1, 100] |
| With no adverse events | 5 (55.6) | [21.2, 86.3] | 0 (0) | [0, 7.9] |
| With one or more drug-relateda adverse events | 0 (0) | [0, 33.6] | 1 (2.2) | [0.1, 11.8] |
| With one or more serious adverse events | 0 (0) | [0, 33.6] | 1 (2.2) | [0.1, 11.8] |
| With one or more serious drug-relateda adverse events | 0 (0) | [0, 33.6] | 0 (0) | [0, 7.9] |
| Who died due to adverse event | 0 (0) | [0, 33.6] | 0 (0) | [0, 7.9] |
| With one or more Grade 3 or greater adverse events | 0 (0) | [0, 33.6] | 9 (20.0) | [9.6, 34.6] |
| With one or more Grade 3 or greater drug-relateda adverse events | 0 (0) | [0, 33.6] | 0 (0) | [0, 7.9] |
N = number of participants in each group; n (%) = number (percent) of participants in each subcategory; 95% CI = exact 95% confidence interval
Serious adverse events include International Council for Harmonisation-defined serious adverse events and malignancies.
Relatedness to study drug is determined by the site.
100mg single-dose doravirine
Daily doravirine/lamivudine/tenofovir disoproxil fumarate fixed dose combination
In Cohort 2, all 45 participants experienced at least one adverse event (Table 4), and one participant (2.2%, 95% CI [0.1, 11.8]) experienced Grade 1 dizziness, which was assessed as related to the study drug. One participant (2.2%, 95% CI [0.1, 11.8]) experienced two SAEs, these were Grade 3 gastroenteritis and Grade 2 lip injury. Both of these were assessed as not related to the study drug. Nine (20%, 95% CI [9.6, 34.6) Cohort 2 participants experienced one or more grade 3 adverse events, none of which were attributed to the study drug by the site investigators. Three participants had grade 3 decreased estimated glomerular filtration rate (eGFR), 2 with concomitant grade 3 increased creatinine. However, for each of these participants, the grade 3 criterion was met due to an increase in creatinine or decrease in eGFR from baseline and the actual values of creatinine and eGFR remained normal for age. No participant met the safety endpoint of an eGFR <60 ml/min/1.73m2. Additional events included: increased ALT (one participant), diarrhea and gastroenteritis (one participant), and increased blood pressure (two participants). There were no Grade 4 adverse events, no deaths, nor discontinuations owing to adverse events.
Virologic efficacy:
According to the FDA Snapshot Algorithm, 42 out of 45 participants in Cohort 2 (93.3%; 95% CI [81.7, 98.6]) achieved or maintained HIV-1 RNA <50 and <40 copies/mL and, 43 out of 45 participants (95.6%; 95% CI [84.9, 99.5]) achieved or maintained HIV-1 RNA <200 copies/mL (Table S4). Based on the observed failure approach, 42/43 (97.7%) had virologic suppression to <40 copies/mL, as one participant withdrew from the study due to pregnancy prior to 24 weeks (viral load at last study visit was <40 copies/mL) and for another, the result was provided as <200 copies/mL only as the sample required dilution secondary to low volume. One of the ARV-naïve participants experienced protocol-defined virologic failure at Week 24, but as this was due to non-adherence, the participant remained on study and on study drug. For the two Cohort 2 ARV-naïve participants, the mean (95% CI) change from baseline in log10 plasma HIV-1 RNA at week 24 was −2.6 (−5.6, 19.0).
Immunologic response:
Amongst the 43 participants in Cohort 2 who had CD4 count and percent data both at baseline and at Week 24, mean (95% CI) change from baseline to week 24 in CD4 absolute counts was 84.8 (21.1, 148.4) cells/mm3 and mean (95% CI) change in CD4 percent was −1.5 (−2.8, −0.2). Mean (95% CI) change from baseline to week 24 in CD4 absolute counts was 203.5 (−99, 578.3) cells/mm3 (range: 174–233 cells/mm3) for ARV-naïve participants and 79 (12.7, 145.3) cells/mm3 for ART-experienced participants. Mean (95% CI) change from baseline to week 24 in CD4 percent was 7.3 (−7.6, 23.1) (range: 6.0–8.5) for ARV-naïve participants and −1.9 (−3.2, −0.7) for ART-experienced participants (Table S4).
Changes in clinical parameters:
Changes from baseline to week 24 in measures of liver and kidney function, lipid levels and growth for Cohort 2 participants are included in Table 5. There were no significant changes during the 24 weeks in any measure of liver or kidney function. Triglyceride levels remained stable, but cholesterol levels decreased. As expected, given the age of the participants both height and weight increased, however, BMI remained stable.
Table 5.
Summary of Clinical Parameters for Cohort 2 Participants at Baseline, change from baseline 24
| Measurements | Study Week | n | Baseline Mean | Mean Change [95% CI] | SD |
|---|---|---|---|---|---|
| Height (cm) | Baseline | 45 | 160.5 | ||
| Week 24 | 44 | 160.4 | 1.1 [0.6, 1.6] | 1.6 | |
| Weight (kg) | Baseline | 45 | 53.8 | ||
| Week 24 | 44 | 53.6 | 3.5 [2.4, 4.6] | 3.5 | |
| Body Mass Index (kg/m2) | Baseline | 45 | 20.9 | ||
| Week 24 | 44 | 20.9 | 1.1 [0.7, 1.5] | 1.3 | |
| Alanine Aminotransferase (IU/L) | Baseline | 45 | 27.6 | ||
| Week 24 | 44 | 28.0 | 1.2 [−7.3, 9.6] | 27.7 | |
| Aspartate Aminotransferase (IU/L) | Baseline | 45 | 28.0 | ||
| Week 24 | 44 | 28.3 | 0.8 [−5.1, 6.7] | 19.5 | |
| Alkaline Phosphatase (IU/L) | Baseline | 45 | 173.9 | ||
| Week 24 | 44 | 175.0 | −20.7 [−35.1, −6.3] | 47.4 | |
| Bilirubin (mg/dL) | Baseline | 45 | 0.5 | ||
| Week 24 | 44 | 0.5 | −0.0 [−0.2, 0.1] | 0.5 | |
| Creatinine (mg/dL) | Baseline | 45 | 0.6 | ||
| Week 24 | 44 | 0.6 | 0.0 [0.0, 0.1] | 0.1 | |
| eGFR from Creatinine Adjusted for BSAa (mL/min/1.73m2) | Baseline | 45 | 158.8 | ||
| Week 24 | 44 | 158.5 | −6.5 [−15.2, 2.2] | 28.6 | |
| Cholesterol (mg/dL) | Baseline | 45 | 168.7 | ||
| Week 24 | 40 | 171.7 | −28.1 [−38.8, −17.4] | 33.5 | |
| HDL Cholesterol (mg/dL) | Baseline | 45 | 57.8 | ||
| Week 24 | 40 | 58.2 | −11.3 [−15.9, −6.6] | 14.7 | |
| LDL Cholesterol (mg/dL) | Baseline | 45 | 89.2 | ||
| Week 24 | 40 | 91.6 | −12.5 [−20.7, −4.2] | 25.8 | |
| Triglycerides (mg/dL)b | Baseline | 17 | 101.0 | ||
| Week 24 | 17 | 101.0 | 0.6 [−26.6, 27.7] | 52.8 |
n = Number of participants with a result for that time point; SD = Standard deviation.
eGFR = Estimated Glomerular Filtration Rate
BSA = Body Surface Area.
Calculations of baseline mean and change from baseline statistics are based on participants with measurements both at baseline and the time point assessed.
Calculated using Modified Schwartz equation27,28
Fasting values only
Medication adherence:
The adherence summary (Table S5) is based on the total completed questionnaires: out of 216 completed questionnaires, >90% were answered that no ARV doses had been missed in the previous 30 days. The adolescents’ assessment of how well they were taking their ARVs was less, as 80% of the questionnaires were answered that their ARVs were always taken as often as they were supposed to and only 65% were answered that they did an excellent job at taking their ARVs the way they were directed.
DISCUSSION
Doravirine concentrations for the 100 mg tablet in adolescents are comparable to adult levels and the doravirine adult FDC tablet achieves high efficacy and safety in the adolescent population.
Cohort 1 established that a single 100 mg dose of doravirine given to adolescents with HIV-1 ages 12–18 years and weighing at least 35kg achieves similar PK to that seen in adults treated with 100mg, though intersubject variability for AUC in adolescents was slightly higher than that observed in adults20. In previous adult studies,21 doravirine multiple dose pharmacokinetics were consistent with predictions from single-dose data and there was no evidence of time-dependent changes in PK after multiple dosing. Thus, AUC0–∞ following a single dose is equivalent to a projected steady state AUC0–24hr and steady state C24 can be predicted from single dose C24. The projected steady state PK from the results of Cohort 1 supported opening Cohort 2 for DOR FDC treatment in adolescents with HIV-1.
Results of DRIVE-SHIFT22, an investigation of 670 virologically suppressed adults with HIV-1 who switched from their current ARV regimen to DOR/3TC/TDF, were available at the time IMPAACT 2014 Cohort 2 opened. As DRIVE-SHIFT demonstrated that DOR/3TC/TDF was not inferior to the baseline suppressive ART regimen, cohort 2 opened to adolescents with virologic suppression for at least 6 months on a previous ARV regimen as well as to ARV-naïve adolescents. All but 2 of the cohort 2 participants switched to DOR FDC from a previous regimen. Virologic efficacy was excellent as 42/45 participants had viral loads <40 copies/mL at 24 weeks (>93%).
Since the TDF and 3TC components in the DOR FDC are new generic formulations, intensive pharmacokinetic sampling for tenofovir and lamivudine was conducted on a subset of Cohort 2 participants at study week 1 to verify appropriate drug levels in adolescents. The AUC and Cmax of both the lamivudine and tenofovir were comparable to previously reported levels in adults18,19, as would be expected given that the doses of lamivudine and tenofovir in the adult FDC are also the recommended doses of these components for this age and weight range18,19. Semi-intensive PK sampling for doravirine was also done at study week 1 in the same subset of Cohort 2 participants. Unlike the results from the single dose PK in Cohort 1 which demonstrated comparable drug levels to adults, while Cmax was similar to that seen in adults23, the AUC and C24 were lower at week 1 in the Cohort 2 participants. Eight of the ten Cohort 2 participants undergoing the week 1 PK evaluation transitioned off an efavirenz-containing regimen, which likely led to the decreased doravirine concentrations due to CYP3A induction. The effects of CYP3A induction waned by week 4, resulting in improved doravirine trough levels at Week 4 and beyond. The initial lower levels of doravirine in these participants transitioning off efavirenz did not affect virologic efficacy, confirming the safety of switching from an efavirenz-based regimen to a doravirine-based regimen without a washout period17,22.
In this adolescent population, doravirine, provided as DOR/3TC/TDF fixed dose combination, was well tolerated over 24 weeks, with no drug-related serious adverse events and no toxicity-related treatment discontinuations. Although the site investigators did not attribute the few (<10%) increased creatinine/decreased eGFR values to study drug, it is possible that these changes were related to the inclusion of tenofovir disoproxil fumarate in the DOR FDC. However, as all the absolute values remained in the normal range, the effect on renal function was minimal. Similar to the results in the doravirine trials in adults10,11, rare neuropsychiatric side effects were detected among the adolescents in this study with one drug-related report of dizziness which resolved without discontinuation of DOR FDC. The low number of adverse events compares favorably with that seen in dolutegravir treatment studies in adolescents24,25. Importantly, no increase in lipid levels or unexpected weight gain was noted after switching to or initiating DOR/3TC/TDF.
Doravirine has been studied in several large adult trials. In DRIVE-FORWARD, a randomized, double-blind, phase 3 non-inferiority trial, 769 adults naïve to antiretroviral therapy were treated with either doravirine or darunavir/ritonavir plus 2 NRTIs. At week 48, doravirine was well tolerated with <1% discontinuations due to adverse events and was not inferior to darunavir/ritonavir in virologic response11. Participants receiving doravirine had significantly improved lipid levels compared to those receiving darunavir/ritonavir. DOR/3TC/TDF was also shown to be non-inferior to EFV/3TC/TDF in the 734 ARV-naïve adults participating in the DRIVE-AHEAD trial, with significantly fewer neuropsychiatric adverse events in the participants receiving doravirine10,26. In DRIVE-SHIFT, an open-label, active-controlled noninferiority trial, over 600 adults with virologically suppressed HIV-1 were randomized to switch to DOR/3TC/TDF vs. remaining on their baseline regimen. As in the previous trials, DOR/3TC/TDF was well tolerated with <3% discontinuations due to adverse events and was not inferior to the participants’ previous regimens22.
This study has limitations. First, pharmacokinetic modeling based on adult data suggests the 100mg doravirine dose would provide appropriate drug levels for adolescents of at least 35kg, however, unfortunately, as too few adolescents of weight <45 kg were identified for participation, intensive PK data were limited to only one subject in the 35 to < 45 kg weight range. Second, only two antiretroviral naïve participants were identified for inclusion in this study limiting the interpretation of results in this subset of adolescents. Furthermore, one of the two ARV-naïve adolescents had periods of non-adherence that likely falsely lowered virologic efficacy in this subset. Finally, as most of the adolescents enrolled in the study were virologically suppressed on their current ARV regimen, the study population overall may be biased towards better adherence than the general adolescent population. Despite these limitations, there are several characteristics of doravirine that support its use in adolescents with HIV-1.
In addition to excellent virologic efficacy, doravirine has fewer drug-drug interactions compared with other NNRTIs and lacks evidence of reproductive toxicity20, inappropriate weight gain27 or lipid abnormalities11,22; common side effects associated with other antiretroviral classes. Doravirine and DOR FDC can also be dosed once-daily without regard for food. The excellent medication adherence demonstrated by the adolescent participants in this study is consistent with ease of administration and lack of significant toxicities. The results of this study confirm that DOR FDC is effective, well-tolerated, achieves appropriate drug levels of all three ARV components in adolescents with HIV-1 and could be considered for a first-line regimen for the treatment of HIV-1 in adolescents.
Supplementary Material
Figure S1: Consort Diagram
Figure S2: Individual Doravirine Plasma Concentrations Relative to Actual Time Since Last Dose by Week, Cohort 2
Acknowledgements:
The IMPAACT 2014 study team gratefully acknowledges the dedication and commitment of the adolescents and young adults and site study teams without whom this study would not have been possible. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, USAID or the United States government. Financial support and antiretrovirals (DOR and DOR/3TC/TDF) were provided by Merck.
Additional Study Team Members: NIAID/DAIDS: Justine Beck, PharmD, and Thucuma Sise, PharmD; NICHD: Bill G. Kapogiannis, MD; FHI 360: Kathleen George, MPH, and Patricia Morgan, MS; Frontier Science Foundation: Yvonne Woolwine-Cunningham, BA, Rebecca Leblanc, MS, and Kathleen Trabert; Merck: Jeanne Mendell, PhD; Harvard T.H. Chan School of Public Health: Carmelita Alvero, MS, Mona Farhad, MS, Sarah Pasyar, MSc, and Petronella Muresan, MS
UNITED STATES. St. Jude CRS, TN: Nehali Patel, MD, Adrienne English, RN, Ryan Heince, MBA, Sandra Jones, MSN; Boston Medical Center CRS, MA: Debra McLaud, RN; University of Colorado, CO: Shane Curran Hays, MS, Jennifer Dunn, MS, FNP-BC, Kacey Navarro, MS, FNP-BC; Seattle Children’s Research Institute, Seattle, WA: Amanda Robson, BS.
SOUTH AFRICA. Perinatal HIV Research Unit, University of the Witwatersrand: Hilda Ndiwani, RN, Ruth Mathiba, MBChB, Avy Violari, FCPaed, Nastassja Ramsagar, BMedSc.
THAILAND. Research Institute for Health Sciences, Chiang Mai University: Linda Aurpibul, MD, MPH, Nuntisa Chotirosniramit, MD, Chintana Khamrong, MSc, Jiraporn Jantong, RN, BSN; AMS-CMU & IRD Research Collaboration, Chaing Rai: Tim R. Cressy, PhD, Pra-ornsuda Sukrakanchana, BSN, Yupawan Thaweesombat, BPharm; Chiangrai Prachanukroh Hospital: Kanyanee Kaewmamuang, BSN; Siriraj Hospital, Mahidol University, Bangkok: Nirun Vanprapar, MD, Kulkanya Chokephaibulkit, MD, Nantaka Kongstan, MSc (Toxicology), Watcharee Lermankul, PhD (Clinical Pharmacology)
Conflicts of Interest and Source of Funding:
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH), all components of the National Institutes of Health (NIH), under Award Numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC), and by NICHD contract number HHSN275201800001I. Financial support and antiretrovirals (DOR and DOR/3TC/TDF) were provided by Merck & Co., Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
KLY, HW, HT (now retired), and HC hold stock and are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD) [license holder Pifeltro® and Delstrigo®]. PF participates on a Merck safety monitoring committee for non-HIV drugs. For the remaining authors, none were declared.
Footnotes
Meetings at which parts of the data were presented:
11th International Workshop on HIV & Pediatrics 2019, Mexico City, Mexico, July 19–20. Poster# 39 Conference on Retrovirology and Opportunistic Infections, 2021, Virtual Conference March 6–10
References:
- 1.Brady MT, Oleske JM, Williams PL, et al. Declines in mortality rates and changes in causes of death in HIV-1-infected children during the HAART era. J Acquir Immune Defic Syndr. 2010;53(1):86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiappini E, Larotonda F, Lisi C, et al. Real-World Analysis of Survival and Clinical Events in a Cohort of Italian Perinatally HIV-1 Infected Children From 2001 to 2018. Front Pediatr. 2021;9:665764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapogiannis BG, Soe MM, Nesheim SR, et al. Mortality trends in the US Perinatal AIDS Collaborative Transmission Study (1986–2004). Clin Infect Dis. 2011;53(10):1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bratholm C, Johannessen A, Naman E, et al. Drug resistance is widespread among children who receive long-term antiretroviral treatment at a rural Tanzanian hospital. J Antimicrob Chemother. 2010;65(9):1996–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salou M, Dagnra AY, Butel C, et al. High rates of virological failure and drug resistance in perinatally HIV-1-infected children and adolescents receiving lifelong antiretroviral therapy in routine clinics in Togo. Journal of the International AIDS Society. 2016;19(1):20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kibalama Ssemambo P, Nalubega-Mboowa MG, Owora A, et al. Virologic response of treatment experienced HIV-infected Ugandan children and adolescents on NNRTI based first-line regimen, previously monitored without viral load. BMC Pediatr. 2021;21(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briand C, Dollfus C, Faye A, et al. Efficacy and tolerance of dolutegravir-based combined ART in perinatally HIV-1-infected adolescents: a French multicentre retrospective study. J Antimicrob Chemother. 2017;72(3):837–843. [DOI] [PubMed] [Google Scholar]
- 8.Frange P, Blanche S, Veber F, Avettand-Fenoel V. Dolutegravir in the long term in children and adolescents: frequent virological failure but rare acquired genotypic resistance. HIV Med. 2021;22(10):958–964. [DOI] [PubMed] [Google Scholar]
- 9.Lai MT, Feng M, Falgueyret JP, et al. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2014;58(3):1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orkin C, Squires KE, Molina JM, et al. Doravirine/Lamivudine/Tenofovir Disoproxil Fumarate is Non-inferior to Efavirenz/Emtricitabine/Tenofovir Disoproxil Fumarate in Treatment-naive Adults With Human Immunodeficiency Virus-1 Infection: Week 48 Results of the DRIVE-AHEAD Trial. Clin Infect Dis. 2019;68(4):535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molina JM, Squires K, Sax PE, et al. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV. 2018;5(5):e211–e220. [DOI] [PubMed] [Google Scholar]
- 12.IMPAACT 2014. protocol https://www.impaactnetwork.org/studies/impaact2014. Accessed February 25, 2022.
- 13.Wilson IB, Lee Y, Michaud J, Fowler FJ Jr., Rogers WH. Validation of a New Three-Item Self-Report Measure for Medication Adherence. AIDS Behav. 2016;20(11):2700–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yee KL, DiBenedetto A, Fan L, et al. Comparative Bioavailability of Oral Granule Formulations of the HIV Antiretroviral Drugs Doravirine, Lamivudine, and Tenofovir Disoproxil Fumarate. AAPS PharmSciTech. 2020;21(3):91. [DOI] [PubMed] [Google Scholar]
- 15.Yee KL, Cabalu TD, Kuo Y, et al. Physiologically Based Pharmacokinetic Modeling of Doravirine and Its Major Metabolite to Support Dose Adjustment With Rifabutin. J Clin Pharmacol. 2021;61(3):394–405. [DOI] [PubMed] [Google Scholar]
- 16.Khalilieh S, Yee KL, Sanchez R, Stoch SA, Wenning L, Iwamoto M. Clinical Pharmacokinetics of the Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitor Doravirine: An Assessment of the Effect of Patient Characteristics and Drug-Drug Interactions. Clin Drug Investig. 2020;40(10):927–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yee KL, Sanchez RI, Auger P, et al. Evaluation of Doravirine Pharmacokinetics When Switching from Efavirenz to Doravirine in Healthy Subjects. Antimicrob Agents Chemother. 2017;61(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epivir labeling-package insert supplement-39. In: Administration USFaD, ed 2019. [Google Scholar]
- 19.Viread labeling-package insert Supplement-58. In: Administration USFaD, ed 2019. [Google Scholar]
- 20.Pifeltro labeling-package insert Supplment-3. In: Administration USFaD, ed 2019. [Google Scholar]
- 21.Anderson MS, Gilmartin J, Cilissen C, et al. Safety, tolerability and pharmacokinetics of doravirine, a novel HIV non-nucleoside reverse transcriptase inhibitor, after single and multiple doses in healthy subjects. Antivir Ther. 2015;20(4):397–405. [DOI] [PubMed] [Google Scholar]
- 22.Johnson M, Kumar P, Molina JM, et al. Switching to Doravirine/Lamivudine/Tenofovir Disoproxil Fumarate (DOR/3TC/TDF) Maintains HIV-1 Virologic Suppression Through 48 Weeks: Results of the DRIVE-SHIFT Trial. J Acquir Immune Defic Syndr. 2019;81(4):463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yee KL, Ouerdani A, Claussen A, de Greef R, Wenning L. Population Pharmacokinetics of Doravirine and Exposure-Response Analysis in Individuals with HIV-1. Antimicrob Agents Chemother. 2019;63(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viani RM, Alvero C, Fenton T, et al. Safety, Pharmacokinetics and Efficacy of Dolutegravir in Treatment-experienced HIV-1 Infected Adolescents: Forty-eight-week Results from IMPAACT P1093. Pediatr Infect Dis J. 2015;34(11):1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viani RM, Ruel T, Alvero C, et al. Long-Term Safety and Efficacy of Dolutegravir in Treatment-Experienced Adolescents With Human Immunodeficiency Virus Infection: Results of the IMPAACT P1093 Study. J Pediatric Infect Dis Soc. 2020;9(2):159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orkin C, Squires KE, Molina JM, et al. Doravirine/Lamivudine/Tenofovir Disoproxil Fumarate (TDF) Versus Efavirenz/Emtricitabine/TDF in Treatment-naive Adults With Human Immunodeficiency Virus Type 1 Infection: Week 96 Results of the Randomized, Double-blind, Phase 3 DRIVE-AHEAD Noninferiority Trial. Clin Infect Dis. 2021;73(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orkin C, Elion R, Thompson M, et al. Changes in weight and BMI with first-line doravirine-based therapy. AIDS. 2021;35(1):91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Consort Diagram
Figure S2: Individual Doravirine Plasma Concentrations Relative to Actual Time Since Last Dose by Week, Cohort 2