

Summary
Engineered T cell therapy has shown remarkable efficacy in hematologic malignancies and has the potential for application to common epithelial cancers. Diverse T cell therapy strategies including adoptive transfer of tumor infiltrating lymphocytes, chimeric antigen receptor (CAR)-T cells, and T cell receptor (TCR)-T cells have been studied in clinical trials. Recent research has established treatment of HPV-associated cancers with TCR-T cells as a model for proof of principle studies in epithelial cancers. These studies and others have provided critical insight into mechanisms of tumor regression, therapeutic targets, treatment safety, treatment design, and barriers to curative cell therapies for common types of cancer. This Perspective will review and consolidate understanding gained from clinical trials to treat viral and non-viral epithelial cancers with cell and gene therapy and will examine how past experience may guide future strategy in treatment and biomarker discovery.
Introduction
Engineered T cell therapy has demonstrated high response rates and frequent complete responses in B cell malignancies and holds promise for the treatment of wide-ranging cancers. Five chimeric antigen receptor (CAR)-T cell therapies targeting CD19 for B cell malignancies and two CAR-T cell therapies targeting B-cell maturation antigen (BCMA) for multiple myeloma have been approved by the United States Food and Drug Administration (FDA).1–7 Relative to hematologic cancers, progress in solid tumors, and especially in common epithelial cancers, has been more measured. However, clinical trials targeting the human papillomavirus (HPV) oncoproteins in HPV-associated cancers establish clear proof of principle for the approach.8,9 Targeting of the E7 antigen with T cell receptor (TCR)-T cells resulted in robust tumor regression in treatment-refractory cancers including cervical, vulvar, anal, and oropharyngeal cancer.8 Responses included elimination of many tumors including programmed cell death protein 1 (PD-1)- and PD-1 ligand (PD-L1)- refractory lesions. In addition, Claudin18.2 (CLDN18.2)- CAR-T cells recently have been reported to show manageable toxicity as well as clinical activity in gastric cancer.10 Lessons from these and other studies are guiding expanded application of engineered T cell therapy and the development of next-generation technologies. In this Perspective we will discuss recent advances and future directions in cell therapy for epithelial cancers with an emphasis on gene-engineered T cells and principles elucidated by clinical research.
CARs and TCRs
CARs are perhaps the best-known type of engineered T cell antigen receptor due to the success of CD19 CAR-T cell therapy. However, T cells can also be engineered to target tumors through tumor-antigen-specific TCRs, and it is TCR-based approaches that thus far have demonstrated clinical activity in the greatest range of solid tumors. CARs and TCRs differ in important ways that determine their respective range of target antigens, toxicity profiles, and potential escape mechanisms. CARs directly engage target antigen through an antibody single-chain variable fragment (scFv) domain and, like antibodies, can bind to extracellular but not to intracellular antigens (Figure 1). This target range is important because many of the most attractive therapeutic targets such as driver mutations, viral oncoproteins, and cancer germline antigens, localize to the intracellular compartment. Different than CARs, TCRs engage tumor cells through heterodimeric alpha-beta chain receptors that bind a peptide derived from the target antigen complexed with human leukocyte antigen (HLA) (Figure 1). The TCR antigen recognition system has the advantage that it can target peptides generated from either intracellular or extracellular proteins (Figure 1) (Box 1). It also has the advantage that mutations that occur outside of the short target peptide, which might prevent surface expression of a CAR target, do not prevent recognition by a TCR and therefore do not offer a potential mechanism for tumor escape.11 It has the disadvantage that only patients with the target peptide-HLA complex can be treated with a given TCR (known as HLA restriction). It also has the disadvantage that tumor escape may occur through loss of the HLA restriction element or components of the antigen processing machinery that create HLA-peptide complexes. New technologies that blur the lines between CAR and TCR targeting, including CARs that recognize peptide-HLA complexes and TCRs that recognize cell surface antigens are emerging. These crossover antigen targeting systems may enhance targeting of certain low-density antigens; however, in principle they remain vulnerable to the same tumor escape mechanisms as the receptors they mimic. Clinical data with these systems will be informative.12,13
Figure 1. Differences in antigen recognition and intracellular signaling between CAR-T and TCR-T cells.
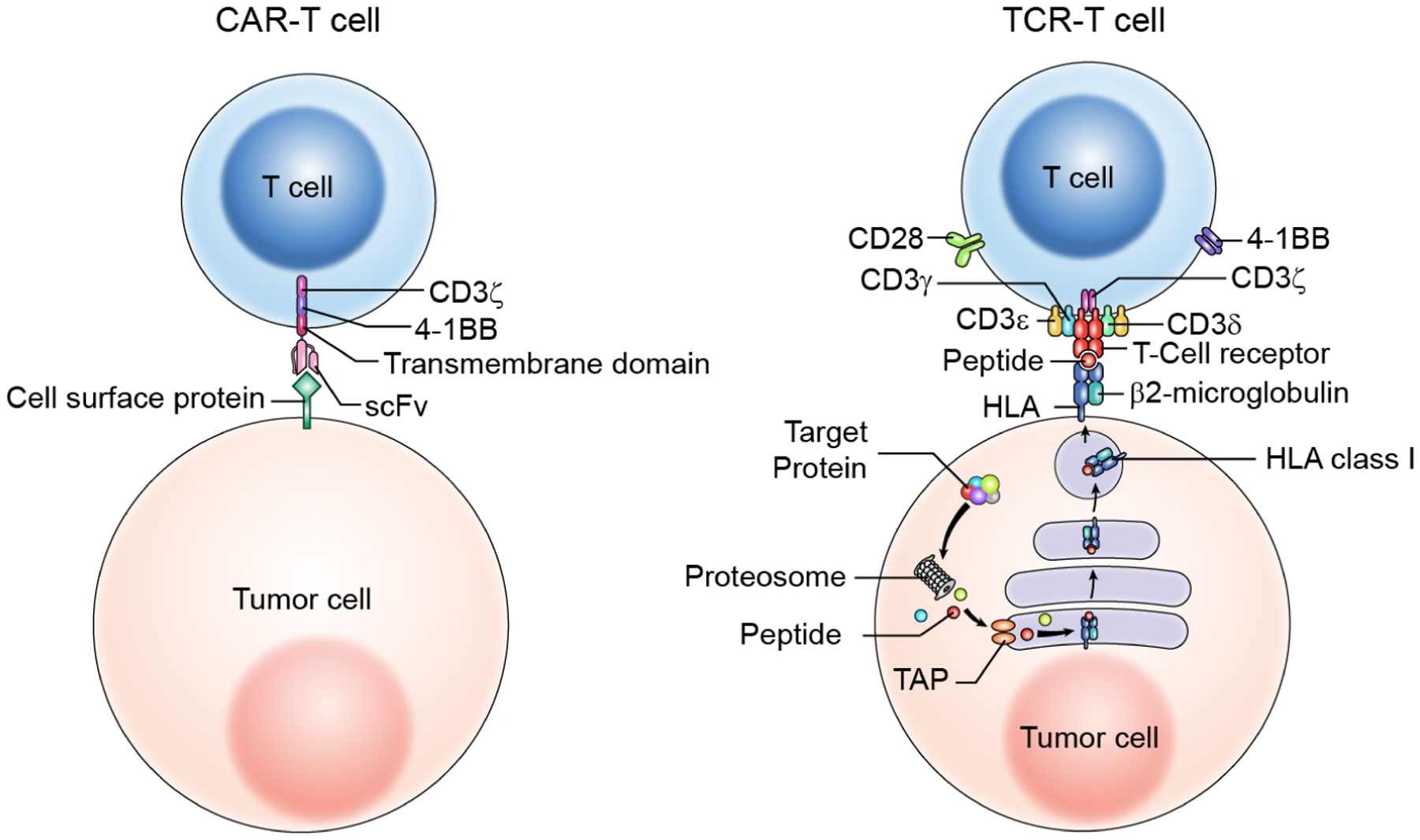
CARs are single chain synthetic molecules that target a cell surface antigen through an antibody-based single-chain variable fragment (scFv). CARs generally signal through combinations of domains from the TCR CD3 complex (e.g., CD3 zeta chain) and one or more T cell costimulatory molecules (e.g., CD28 or 4-1BB). In contrast, TCRs target a peptide-HLA complex through a heterodimeric alpha- and beta-chain. The peptide in the target complex is generated by intracellular antigen processing machinery and may be derived from an intracellular or cell surface protein. Genetically engineered TCRs signal through the natural CD3 complex, which is composed of gamma, delta, zeta, and epsilon chains. These differences in target recognition and in signaling result in a distinct range of target antigens and distinct toxicity profiles for T cells engineered with each receptor.
Box 1. Advantages and disadvantages of different adoptive T cell therapies.
| Chimeric antigen receptor | T cell receptor | Tumor infiltrating lymphocyte |
|---|---|---|
| Advantages | Advantages | Advantages |
|
|
|
| Disadvantages | Disadvantages | Disadvantages |
|
|
|
CARs and TCRs also differ in how they transduce activating signals to T cells. CARs are single-chain designer proteins that signal through synthetic activating domains most often borrowed from T cell stimulatory and costimulatory molecules. Typically, one domain derives from the TCR CD3 complex (e.g., CD3 zeta chain) and another from a costimulatory receptor or combination of costimulatory receptors (e.g., 4-1BB or CD28) (Figure 1). Engineered TCRs are heterodimeric receptors that form complexes with natural CD3 molecules and signal through the natural CD3 complex using the same mechanism as wild-type TCRs (Figure 1). These different signaling domains used in CARs versus TCRs may greatly impact T cell function and toxicity profiles. However, study of the impact is confounded by other variables such as different targeting domains (CAR scFv versus TCR alpha- and beta-chain pair) and targets (CAR epitope versus TCR peptide-HLA complex, and self-antigens versus tumor-restricted antigens), as well as variable signaling characteristics of different CAR constructs.14 Nonetheless, the differences between the CAR versus TCR receptor system signals likely result in differences in anti-tumor function and toxicity including cytokine release syndrome.15–17 This concept is supported by the observation that different signaling domains have a clinically significant impact on the characteristics of CAR-T cells.18
Engineered TCR-T cells versus TIL
Therapeutic T cells for adoptive cell transfer can be generated from tumors, peripheral blood or bone marrow, and they can target cancer antigens through endogenous TCRs or through engineered TCRs. Tumor infiltrating lymphocytes (TIL) and engineered TCR-T cells have distinct advantages and disadvantages. TIL have the advantage that they are generally composed of multiple T cell clonotypes and therefore can attack a tumor through multiple antigens presented by different HLA molecules (Figure 2A) (Box 1). However, some responses to TIL therapy in epithelial cancers have been with nearly-clonal or oligoclonal cell products that were generated to target specific tumor antigens such as HPV oncoproteins or somatic mutations.19,20 Another advantage of TIL is the potential to target varied tumor antigens including antigens that are undefined.19 TIL have the disadvantage that the cell product composition including the specificity and avidity of the T cells is highly variable. TIL often do not demonstrate reactivity against autologous tumors or tumor antigens, and clear clinical activity has been observed only in limited, immunogenic cancers such as melanoma and HPV-associated cancers.19,21–24 Another disadvantage of TIL therapy is that generation of TIL requires surgical resection of a tumor and the cell product manufacturing time is prolonged.
Figure 2. TIL and TCR-T cell therapy cell product generation, patient treatment, and mechanisms of tumor engagement.
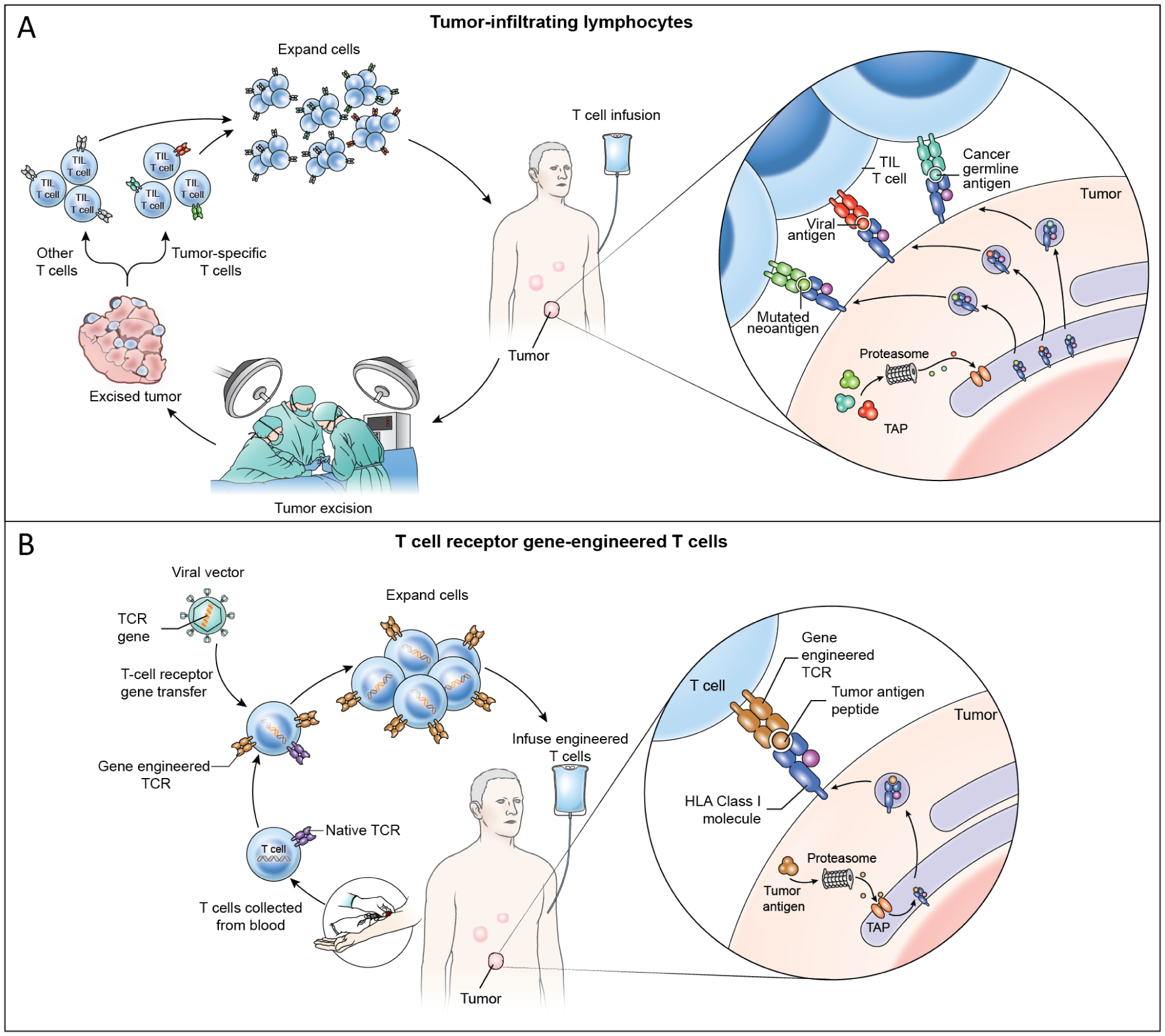
(A) In TIL therapy, a metastatic tumor is surgically resected and T cells are grown from the tumor ex vivo. The T cells are oligoclonal and may or may not target tumor antigens and may be composed of a mixture of tumor-specific T cells and other T cells. The T cells are expanded ex vivo and administered by intravenous infusion. TIL treatments resulting in durable, complete regression of cervical cancer were mediated by targeting of HPV oncoproteins as well as non-viral proteins such as mutated neoantigens and cancer germline antigens (cutout).19 Case reports have shown partial responses to TIL therapy solely targeting mutated neoantigens in non-viral epithelial cancers.20,28 (B) In TCR-T cell therapy, T cells are collected from peripheral blood and gene-engineered ex vivo to express a defined tumor-antigen-targeting TCR. TCR gene engineering may be conducted with a viral vector (shown) or with other gene transfer methods such as CRISPR-CAS9 gene editing or transposon systems. Engineered TCR-T cells are expanded ex vivo and administered intravenously. TCR-T cells engage tumors through recognition of peptide HLA complex composed of a defined peptide from a tumor antigen (cutout).
In contrast to TIL therapy, engineered TCR-T cells have defined antigen specificity and functional avidity, no requirement for surgery, and shorter manufacturing time (Figure 2B) (Box 1). The importance of having defined specificity and functional avidity should be emphasized as it permits controlled targeting of rationally chosen antigens using optimal TCRs – an advantage distinct from other immunotherapeutic approaches including TIL therapy, therapeutic cancer vaccines, and checkpoint blockade. Notably, this advantage is mostly lost when engineered TCR-T cells are used to target private neoantigens on an individualized patient-by-patient basis. Another advantage of TCR-T cells is the ability to generate the cell product from a selected subset of peripheral blood T cells with enhanced proliferative and therapeutic potential such as naïve cells, memory stem cells, or central memory cells. In addition, the in vitro differentiation of these cells might be directed toward optimal in vivo function by cytokines, signaling pathway modulators, and other culture conditions.25 The disadvantages of engineered TCR-T cells are that antigen targeting is monoclonal unless multiple different engineered TCR-T cells are combined. In addition, treatment is restricted to individuals with the combination of a particular HLA allele and target antigen, which narrows the population that can be treated. Intrapatient tumor heterogeneity is another potential challenge for TCR-T cell therapy as few target antigens display uniform tumor expression. Although not well studied, heterogeneity may also complicate TIL therapy as the immunodominant antigens may vary between tumors of the same patient such that cells that target one tumor may not optimally target another tumor.
Lessons from TIL therapy – clinical research
Clinical trials with TIL therapy have demonstrated that cell therapy can mediate clinical activity in epithelial cancers, including durable, complete tumor responses as observed in the treatment of HPV-associated cancers. A single-center study of TIL therapy in metastatic cervical cancer showed responses in 3 of 9 patients, including 2 complete responses that are ongoing more than 8 years after a single treatment.22 Expanded experience showed responses in 7 of 29 (24%) patients with varied HPV-associated malignancies including cervical, oropharyngeal, and anal cancer.21 Industry sponsored trials of TIL for head and neck, and cervical cancer are active with TIL therapy for cervical cancer having been granted FDA Breakthrough Therapy Designation.26 Emerging data also suggest that TIL may have activity in lung cancer, with a report of responses in 3 of 15 patients who were treated with TIL plus PD-1 blockade after progression on PD-1 blockade alone.27 Furthermore, case reports have described partial responses to TIL in cholangiocarcinoma and colon cancer, and a small series has described responses in breast cancer in combination with PD-1 blockade.20,28,29 These studies support proof of principle for cell therapy in epithelial cancers and highlight the need for further research to improve on the results achieved thus far with TIL-based approaches.
Clinical and translational research from TIL studies continues to be highly informative for the development of immunotherapy and cell therapy. An important lesson that is emerging from study of TIL for metastatic melanoma is the negative impact of prior treatment on cell therapy response rates. A single-center study with 101 patients, performed before PD-1 checkpoint blockade was in common use, reported an overall response rate (ORR) of 56% (36% partial response (PR) rate and 24% complete response (CR) rate).24 However, a more recent industry-sponsored, multicenter, phase II trial with 66 patients previously treated with PD-1 checkpoint blockade was less encouraging and reported an ORR of 36%, with a CR rate of 3%.23 These findings are consistent with a recent retrospective study showing ORR rates of 56% in checkpoint-naïve patients, 24% in PD-1-refractory patients (1 of 34 CRs), and 21% for targeted therapy-refractory patients (0 of 19 CRs).30 Interestingly, patients with BRAF V600E/K-mutated melanoma who were previously treated with a targeted agent demonstrated a lower response rate (21% versus 60%) and worse survival (9.3 vs. 50.7 months) than patients who were not previously treated with a targeted agent. These data comport with the oncology therapeutics principle of increasing tumor resistance with increasing line of therapy (especially within the same class of therapy, such as immunotherapy). The broader implication of these data is that adoptive T cell therapy may need to be studied in earlier lines of therapy to ascertain its true clinical activity and potential for durable, complete responses, which is arguably the most important consideration. Addition of immune checkpoint blockade to cell therapy is in principle an attractive approach for early line treatment; however, the available preclinical and clinical data thus far suggest a surprisingly limited effect for this approach.31–36 Additional clinical trials are ongoing and further data are anticipated.
Lessons from TIL therapy – translational research
Recent TIL therapy studies also have provided insight into mechanisms of action of immunotherapy. TIL treatment was initially reported to induce regression of melanomas through targeting of melanocyte differentiation antigens, which are self-antigens expressed by melanocytes and melanoma tumor cells.37 Newer research, facilitated by advances in next-generation sequencing, instead pointed to the primary importance of products of somatic gene mutations expressed by cancers (i.e., neoantigens) as tumor regression antigens.38,39 The role of neoantigens in TIL therapy for non-melanoma cancers was described in two case reports, one a partial response driven by T cell targeting of mutated ERBB2 in cholangiocarcinoma and the other a partial response driven by T cell targeting of mutated KRAS in colon cancer.20,28 These findings have expanded our understanding of antigen targeting in immunotherapy and have supported the development of new platforms to target neoantigens with therapeutic cancer vaccines and TCR-T cells.
Insight into tumor antigen targeting in immunotherapy also has been provided by the study of TIL therapy for HPV-associated cancers. HPV-associated cancers consistently express the HPV E6 and E7 oncoproteins, which are uniformly expressed, immunologically foreign, and tumor restricted, making them attractive therapeutic targets. In patients treated with TIL for HPV-associated cancers, the frequency of administered TIL cells with E6 or E7 reactivity and the persistence of these cells in vivo correlated with clinical response.21,22 However, the contribution of oncoprotein-targeting TIL to tumor response remains unclear as, in patients with complete responses, HPV-specific TIL were a subdominant tumor-antigen-reactive population.19 Indeed, the dominant tumor antigen targeting was directed against non-viral antigens such as neoantigens or cancer germline (CG) antigens (Figure 2A).19 It is important to note, however, that direct targeting of HPV antigens with TCR-T cells can mediate tumor responses indicating the potential of these antigens as therapeutic targets.8,9 Interestingly, peripheral blood T cells targeting both viral and non-viral antigens were found predominantly to express PD-1, suggesting that PD-1 blockade in viral malignancies may act to target tumors through diverse antigens.19
Proof of principle for engineered T cells in epithelial cancers
A tremendous range of CAR-T cell and TCR-T cell approaches have been described, but few strategies have resulted in manageable safety profiles and reproducible, objective (i.e., Response Evaluation Criteria in Solid Tumors) tumor responses. Data from a range of targets in diverse solid tumor indications are reviewed later in this paper. The clearest results demonstrating clinical activity in epithelial cancers come from the treatment of HPV-associated cancers with E6 or E7 oncoprotein-targeting TCR-T cells. An initial phase I/II trial targeting E6 showed responses in only 2 of 12 patients.9 However, a subsequent phase I trial targeting E7 with higher functional avidity cells demonstrated responses in 6 of 12 patients including complete regression of many lesions and marked responses in PD-1- and PD-L1-resistant tumors.8 Although many tumors were durably and completed eliminated, no patient had a complete response. In contrast to CAR-T cell therapy for hematologic cancers, cell dose was not limited by toxicity, and toxicity did not increase with dose level. Severe adverse events from neurotoxicity and cytokine release syndrome were not a major consideration in clinical management. Doses more than 500-fold higher than standard CAR-T cell doses were given safely, and a maximum dose of 100 billion cells was established (based on manufacturing considerations rather than toxicity). Safe administration of cells at this relatively high dose as compared to CAR-T cells may be possible due to the targeting of a tumor-restricted antigen and the use of the endogenous CD3 complex for TCR signaling. These data support the concept that TCR-T cell therapy can be administered safely and can mediate tumor regression in epithelial cancers, and they provide a foundation for next-generation approaches and broader application.
CD4+ T cells and other TCR considerations
Clinical trial data also indicate the potential for CD4+ T cells to mediate anti-tumor responses in cell therapy for epithelial cancers. CD4+ T cells recognize antigen epitopes presented by HLA class II molecules and have pleiotropic function in anti-tumor responses. The class II pathway utilizes distinct antigen processing and presentation machinery from the class I pathway with presentation independent of molecules such as B2M, TAP1, TAP2 and TAPBP.40–42 Direct evidence of clinical activity of CD4+ T cells comes from a case report in which administration of a highly enriched population of CD4+ TIL targeting mutant ERBB2 caused a partial response in a patient with metastatic cholangiocarcinoma.28 Indirect evidence for CD4+ T cells comes from the TIL studies finding that in complete responses to TIL therapy for HPV-associated cancers substantial fractions of the administered cell product (5% and 14% for each of 2 patients) were CD4+ T cells targeting the HPV E6 or E7 oncoprotein.19 Finally, the potential for therapeutic CD4+ T cells to mediate regression of epithelial cancers is supported by a phase I study of TCR-T cells targeting a class II restricted epitope of MAGE-A3. Responses were observed in 4 of 17 patients including 3 patients with epithelial cancers.43 These data support the clinical activity of class II restricted TCRs in cell therapy for epithelial cancers.
Targeting of tumor antigens with multiple TCRs that utilize multiple HLA molecules, and especially a combination of class I and class II HLA molecules, may overcome certain escape mechanisms related to antigen presentation. However, some defects such as HLA loss of heterozygosity (LOH) (which results in copy loss of multiple HLA and antigen presentation molecules) or loss of non-redundant interferon response pathway molecules may be more difficult to surmount. The clinical manufacturing platforms for real-time generation of multiple cell products with different TCRs for same patient, while technically achievable, are costly and clinical data on successful use are needed. In addition, the regulatory and clinical pathways for development of treatments with multiple TCR-T cell products administered together present practical challenges. Notwithstanding these challenges, combined targeting of tumors through multiple antigens and HLA alleles represents a rational strategy to overcome common resistance mechanisms and potentially increase complete tumor responses.
The contribution of T cell functional avidity to anti-tumor activity is unknown and may vary between tumors, each of which have unique characteristics that affect target (i.e., peptide-HLA) density. Since insufficient avidity results in no target engagement, and since tumor responses in epithelial cancers have been with relatively high avidity T cells, higher avidity is generally considered favorable.8,9,19,44
Practical clinical considerations for TCR-T cell therapy
The clinical trial protocols and methods for generation of engineered T cells for human administration vary, but the studies that have shown the strongest activity in solid tumors have common themes as discussed below and summarized in Table 1. Other approaches have advantages and may be effective but are less proven.
Table 1.
Characteristics of engineered T cell trials that have demonstrated clinical activity in solid cancersa
| Antigen-targeting receptor | Conditioning regimen | Maximum cell dose | Transduction efficiency (median/range) | Systemic cytokine therapy | Tumor responses (responses/N) |
|---|---|---|---|---|---|
| MART1 TCR52 | Cyclophosphamide 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
107 × 109 | 71 | Aldesleukin 720,000 IU/kg q8hrs | 6/20 |
| gp100 TCR52 | Cyclophosphamide 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
110 × 109 | 82 | Aldesleukin 720,000 IU/kg q8hrs | 3/16 |
| NY-ESO-1 TCR50 | Cyclophosphamide 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
130 × 109 | 78, 62b | Aldesleukin 720,000 IU/kg q8hrs | 22/38 |
| NY-ESO-1 TCR57 | Cyclophosphamide 1,800 mg/m2 × 2 days Fludarabine 30 mg/m2 × 4 days |
14 × 109 | N/Ac | None | 6/12 |
| NY-ESO-1 TCR58 | Cyclophosphamide 600–1,800 mg/m2 × 2–3 days Fludarabine 30 mg/m2 × 3–4 days |
N/Ac | N/Ac | None | 9/30 |
| MAGE-A3 TCR43 | Cyclophosphamide 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
120 × 109 | 90 | Aldesleukin 720,000 IU/kg q8hrs | 4/17 |
| MAGE-A3/A9/A12 TCR64 | Cyclophosphamide 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
79 × 109 | 85 | Aldesleukin 720,000 IU/kg q8hrs | 5/9 |
| E6 TCR9 | Cyclophosphamide 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
134 × 109 | 60 | Aldesleukin 720,000 IU/kg q8hrs | 2/12 |
| E7 TCR8 | Cyclophosphamide 30 or 60 mg/kg × 2 days Fludarabine 25 mg/m2 × 5 days |
120 × 109 | 96 | Aldesleukin 720,000 IU/kg q8hrs | 6/12 |
| CLDN18.2 CAR10 | Cyclophosphamide 250 mg/m2 × 3 days Fludarabine 25 mg/m2 × 2 days Nab-paclitaxel 100 mg or Gemcitabine 1,000 mg × 1 day |
5 × 108 | N/Ac | None | 18/37 |
Clinical trials with ≥2 objective responses by RECIST criteria.
CD8+ and CD4+ T cells, respectively.
Not available.
Conditioning regimen.
The use of a conditioning regimen is supported by indirect evidence including the results of sequential clinical trials, data from animal models, and translational research findings. The optimal regimen is unknown. Data from TIL therapy for melanoma suggest that there is a ceiling to how much benefit can be gained from more aggressive conditioning.24 Cyclophosphamide and fludarabine are the most employed agents, and the most common cyclophosphamide dose is 60 mg/kg. Tumor responses and high levels of engineered T cell persistence in vivo were observed with either cyclophosphamide 30 mg/kg or 60 mg/kg in the E7 TCR-T cell clinical trial.8 Given the greater toxicity of the higher dose, the lower dose may be preferable, particularly in patients with comorbidities from prior therapy.
Systemic cytokine support.
High dose aldesleukin (i.e., modified, recombinant interleukin-2) has been administered with cell therapy for solid tumors dating back to early studies for metastatic melanoma. Its use in cell therapy is supported by its single-agent clinical activity in melanoma and renal cell carcinoma, and by data from mouse models.45–48 It has been consistently included in studies that demonstrated clinical activity of TCR-T cells in solid tumors; however, its contribution to efficacy remains unknown. Objective clinical responses have been observed with as few as 1 to 3 doses of aldesleukin including responses to TIL therapy and TCR-T cell therapy in epithelial cancers.8,9,21,43,49 Despite serious toxicity when administered to maximum tolerated dosing, the first doses of aldesleukin generally cause manageable toxicity and do not require Intensive Care Unit transfer. Hence, our practice is to include high dose aldesleukin but to limit the number of doses with the aim to provide cytokine support but avoid toxicity that is not quickly reversible.
Engineered T cell dose.
The optimal dose of engineered TCR-T cells for epithelial cancers is unknown and there is a paucity of data to guide clinical trial design. The dose of cells at which responses have been observed is variable, but trials with the strongest tumor response data have used high numbers of cells (more than 1 billion) (Table 1). Reponses to E7 TCR-T cell therapy were observed at all dose levels, which ranged from 1 billion to more than 100 billion cells.8 The sample size at each dose level was small (n=3), and a correlation between dose and response was not observed, but a correlation between dose and persistence of engineered T cells was observed. These data leave unanswered the question of how many cells is “enough,” but it is notable that high doses of cells can be generated and administered and can result in high in vivo persistence and in tumor regression. It is also important to distinguish between TCR-T cells targeting tumor-restricted antigens, which have not demonstrated dose-limiting toxicity versus CAR-T cells targeting self-antigens, which have demonstrated dose-limiting toxicity in both hematologic cancer and solid tumors.8,10,21,50,51
Gene transfer efficiency and cell manufacturing.
Gene transfer platforms based on lentivirus, gamma retrovirus, transposon, CRISPR, and other technologies can be used to generate gene engineered cell products for human administration. Thus far, clinical trials that have shown clear objective responses by Response Evaluation Criteria in Solid Tumors (RECIST) criteria have used relatively efficient, retroviral or lentiviral gene transfer technologies incorporated into simple manufacturing processes that do not require T cell isolation or engineered T cell enrichment steps. Gene transfer efficiency has been high, and in vivo persistence of gene-engineered T cells has been high. In the E7 TCR T-cell trial, transduction efficiency was 93% to 99% and at the highest dose level in vivo persistence was 85% to 94% of peripheral blood T cells approximately 6 weeks after infusion.8 E7 TCR-T cells were generated by in vitro CD3- and IL-2-driven stimulation and demonstrated mostly a highly differentiated effector or effector memory phenotype yet were capable of in vivo expansion, long-term persistence, and tumor regression. Other methods of manufacturing cell products that incorporate non-viral gene transfer have distinct advantages including lower cost and faster vector generation; clinical data with these systems are anticipated.
Target antigen considerations
Clinical experience with engineered T cell therapy in solid tumors has reinforced the critical impact of the therapeutic target on the safety and efficacy of the treatment (Table 2). Here we will review from a target antigen perspective the clinical trial data with lessons for target selection. Studies that have resulted in either objective tumor regression, on-target toxicity, or both (Table 2) will be emphasized as studies that have resulted in neither tumor regression nor toxicity are difficult to interpret.
Table 2.
Target antigens for engineered T cell therapy in solid cancers
| Class | Examples | Normal tissue expression | Clinical trial outcomesa | |||
|---|---|---|---|---|---|---|
| Antigen-targeting receptor | Cancer type | Tumor responses (responses/N) | On-target toxicity | |||
| Shared tumor/self | MART1, gp100, CEA, CA9, ERBB2, ROR1, GD2, GPC3, CLDN18.2 | Variable | MART1 TCR52 | Melanoma | 6/20 | Skin, eye and ear |
| Cancer germline | NY-ESO-1, select MAGE antigens, KK-LC-1 | Germ cells | NY-ESO-1 TCR50 | Synovial cell sarcoma, melanoma | 22/38 | None |
| Neoantigen (mutation, frameshift, splice variant, etc.) | Mutant RAS, mutant BRAF, EGFRvIII | None | N/Ac | N/Ac | N/Ac | N/Ac |
| Viral | HPV, HBV, EBV | None | E6 TCR9 | HPV-associated cancers | 2/12 | None |
Only clinical trials in which systemic administration of engineered T cell therapy resulted in on-target toxicity or ≥2 objective tumor responses as measured by RECIST criteria are included.
Toxicity interpretation is confounded by the targeting of an epitope shared by multiple MAGE family members and uncertainty about the expression of each MAGE antigen in the brain. Targeting of MAGE-A12 was via an epitope that differed from the MAGE-A3 epitope by one residue, at an HLA-A*02:01 anchor site (position 2).
Not applicable. Awaiting publication.
Shared tumor-self antigens.
Shared tumor-self antigens are expressed by both tumors and normal tissues, and some are particularly attractive targets due to their uniform expression by cancers. This class of antigens is the poster child for cell therapy success due to the efficacy of treatments targeting CD19 and BCMA in hematologic malignancies. Unfortunately, in solid tumors, targeting of tumor-self antigens thus far has resulted in prohibitive toxicity from on-target autoimmunity. Examples include skin, eye, and ear toxicity from targeting MART or gp100 with TCR-T cells; gastrointestinal toxicity from targeting carcinoembryonic (CEA) with TCR-T cells; cardiopulmonary toxicity from targeting ERBB2 with CAR-T cells; and hepatotoxicity from targeting carbonic anhydrase 9 (CA9) with CAR-T cells.52–55 These results highlight the constraints of on-target toxicity as well as the need to parse the antigens that can be targeted safely and/or to develop antigen targeting systems that distinguish between normal tissues and tumors. In addition, other thoughtful targeting strategies may be possible as suggested by a report of improvement in symptoms and in some radiographic findings in a small series of patients treated with anti-GD2 CAR-T cells for pediatric brain tumors.56 Similarly, decreases in prostate-specific antigen were observed with CAR-T therapy targeting prostate-specific membrane antigen (PSMA), although toxicity was sometimes severe (a dominant-negative TGF-beta receptor was also employed).51 Finally, CAR-T cells directed against the tight junction protein CLDN18.2 showed clinical activity with objective partial responses in 16 of 28 patients with gastric cancer and 2 of 9 patients with other gastrointestinal cancers, though results should be interpreted cautiously given the use of repeated conditioning chemotherapy with repeated cell dosing as well as the relatively short-lived tumor responses and weak persistence of CAR-T cells.10
Cancer germline antigens.
CG antigens are subclassified by their expression in healthy tissues. The tumor-restricted subclass of CG antigens consists of proteins expressed exclusively by tumors and germ cells (which are not recognized by T cells due to absence of HLA expression). Targeting of the CG antigen CTAG1B (NY-ESO-1) with TCR-T cells has shown promising response rates in melanoma with responses in 11 of 20 patients and synovial cell sarcoma with responses 11 of 18 patients.50 Encouraging results were also reported in an industry-sponsored trial testing NY-ESO-1 TCR-T cells for synovial cell sarcoma with responses seen in 15 of 42 patients.57,58 Tumor responses have also been reported for targeting MAGE-A3 in a basket trial with a variety of tumors (each response was in a different type of cancer) and MAGE-A10 in non-small cell lung cancer.43,59 Unfortunately, responses in cancers other than melanoma rarely have been complete, which may be related to the heterogeneous antigen expression. In addition, the proportion of common tumors with a high tumor fraction expressing these antigens is low. One CG antigen that may be expressed in a high tumor fraction in a subset of lung, triple-negative breast, cervix, and gastric malignancies is Kita-Kyushu lung cancer antigen 1 (KK-LC-1, encoded by CT83).60–63 Treatment with TIL therapy predominantly targeting KK-LC-1 mediated a durable, complete response in a patient with metastatic cervical cancer.19,22 A clinical trial treating patients with KK-LC-1 TCR-T cells (expressing the TCR from this patient) recently opened (NCT05035407).
The importance of preclinical testing of TCRs for cross-reactivity was demonstrated with affinity-enhanced TCRs targeting the cancer germline antigen, MAGE-A3. An HLA-A*02:01-restricted TCR that was generated from HLA-A*02:01-transgenic mice and affinity enhanced by an amino acid substitution in the complementarity-determining region 2-alpha caused fatal neurotoxicity due to cross-reactivity against a nonidentical epitope of MAGE-A12, which is expressed in the brain.64 Similarly, an HLA-A*01:01-restricted TCR generated from a human and affinity enhanced by four amino acid substitutions to the complementarity-determining region 2-alpha caused fatal cardiotoxicity due to cross-reactivity against a non-identical epitope of titin, a protein expressed in myocardium.65,66 These results highlight the potential risk of autoimmunity inherent to TCRs that have not been subjected to thymic selection. They underscore the importance of preclinical testing of TCRs for cross-reactivity against peptides originating from human proteins.
Recurrent driver mutations.
Recurrent driver mutations are public neoantigens that are shared between the tumors of different patients and not expressed by healthy tissues. They represent attractive targets for engineered T cell therapy due to their homogeneous tumor expression and importance in cell transformation and survival. Case reports have described partial tumor responses following treatment with TIL or TCR-T cells targeting mutated RAS.20,67 Another case report described a partial tumor response to TCR-T cell therapy targeting mutant p53 in a patient with metastatic breast cancer.49 Larger studies that define the response rate for this type of treatment have not been reported. Sophisticated systems for the use of engineered TCR-T cells to target private neoantigens that are relatively unique to each patient’s tumor have been described.68 This type of approach has the advantage of potential broad clinical application but the disadvantages of complicated manufacturing, slow product generation, variability in the target antigens, and variability in the therapeutic TCRs. Clinical data are not yet available.
Viral antigens.
Viral antigens can be attractive targets for engineered T cells due to their restricted expression in infected or transformed tissues. Cancers that harbor viral antigens include malignancies induced by HPV, hepatitis B virus (HBV), Epstein-Barr virus, and Merkel cell polyomavirus. Clinical data showing activity with engineered antigen receptors are available only for the targeting of HPV-associated cancer through the HPV16 E6 or E7 oncoproteins.8,9 These oncoproteins are particularly well characterized and appear to be constitutively expressed and to have highly conserved sequences between isolates (although there is some variation, particularly in E6). Limited virally derived cancer antigens share these characteristics. For example, hepatocellular carcinoma can be caused by several hepatitis B virus genotypes that each have additional subtypes with varying sequence and limited shared potential epitopes.69 Early stage clinical trials are testing the safety of TCR-T cell therapy targeting HBV viral antigens.70,71 Other viruses such as Epstein-Barr virus display manifold mechanisms of T cell evasion and variable viral gene expression programs that complicate target epitope selection.72 Merkel cell polyomavirus antigens are attractive targets in Merkel cell carcinoma although published data thus far are from cell therapy in combination with other immunotherapy.73,74 Research to target a range of viral antigens is ongoing, but it is not known if the clinical activity from targeting HPV oncoproteins can be reproduced with other viral targets in other cancers.75–77
Limitations in TCR-T cell therapy
Investigation in animal models has highlighted the potential to improve T cell therapy by enhancement of T cell function (by control of T cell differentiation, transgene insertion or deletion) and/or by manipulation of the tumor microenvironment.78 However, in contrast to data from CAR-T cells, it is not clear from the limited available clinical trial data of cell therapy in epithelial cancers that T cell function and the tumor microenvironment limit treatment response. Available data from clinical trials targeting cancer germline antigens and HPV antigens demonstrate robust persistence of functional engineered TCR-T cells in the peripheral blood.8,9,43,50 Study of TCR-T cell therapy in HPV-associated cancers points to conspicuous tumor-intrinsic gene defects chiefly related to genes critical to antigen processing and interferon response as a constraint (pathways shown in Figure 3).8,9 Although the dataset is small, defects in genes with non-redundant roles in immune-related pathways have been observed consistently in cell therapy for epithelial cancers. Acquired resistance due to HLA loss was described in a case report of a patient with a response to TIL targeting RAS G12D for colorectal cancer and in a single patient with response to TCR-T cell therapy targeting mutant p53 for breast cancer.20,49 Primary resistance due to loss of HLA was identified in 1 of 2 patients treated with E6 TCR-T cells who showed treatment resistance and had tumor biopsies.9 The other patient with treatment resistance displayed a mutation in IFNGR1.9 Similarly, 4 of 4 tumors with primary or acquired resistance to E7-TCR T cell therapy demonstrated immune-related genetic defects with findings of HLA mutation, B2M mutation, B2M copy loss, and complex copy loss abnormalities involving multiple genes.8 These findings mirror those from study of immune checkpoint inhibitors showing resistance due to genetic defects in identical pathways and genes.79–82 New findings point also to the importance of tumor-intrinsic interferon-gamma signaling in solid tumor response to CAR-T cell therapy.83 Although observed in several of the few cases that have been studied, the prevalence of tumor-intrinsic gene defects that mediate cell therapy resistance in various types of cancer and at different stages of disease is not well defined. Study of the landscape and heterogeneity of immune resistance gene defects in tumors is an important future direction for defining the pervasiveness and uniformity of resistance mechanisms. This information is critical to the rational development of next-generation treatment strategies and of predictive biomarkers.
Figure 3. Critical pathways for T cell recognition and killing of tumor cells that are disrupted in tumors resistant to TCR-T cell therapy and checkpoint blockade therapy.
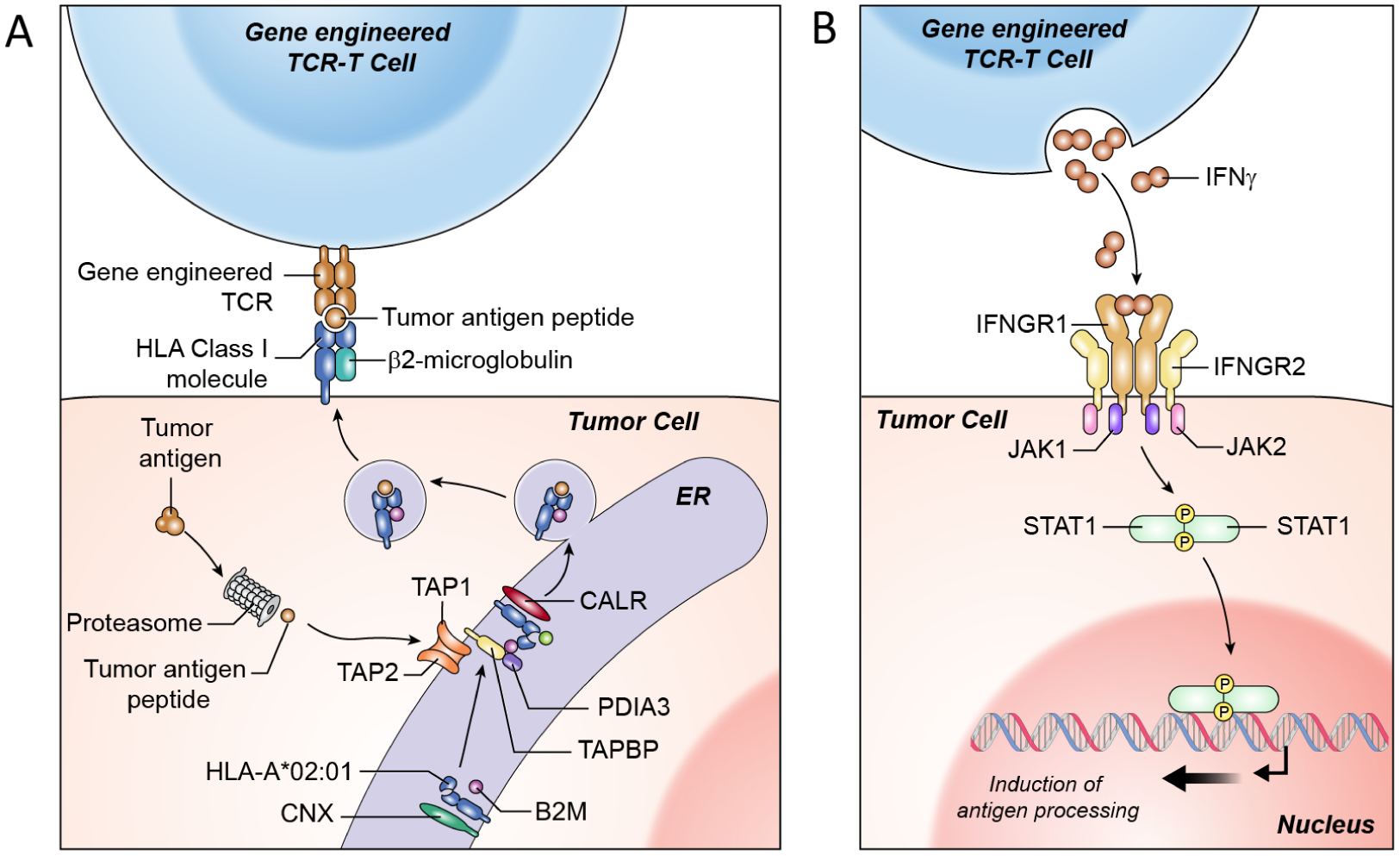
(A) T cell recognition of tumors is mediated by TCR engagement of peptide-HLA complexes on the cell surface. The generation of these target peptide-HLA complexes requires intact antigen processing and presentation machinery including proteolytic enzymes (proteosome subunits), peptide transporters (TAP molecules), HLA loading proteins (CNX, CALR, PDIA3), and HLA component molecules (HLA alpha-chain and B2M). (B) T cells mediate effector function by production of IFNγ which binds to the IFNγ receptor complex mediating complex downstream effects that are important for T cell recognition and killing of tumors cells including upregulation of antigen processing as shown in A. Damaging mutations and/or copy loss involving components of the antigen processing pathway and IFN response pathway have been implicated in tumor resistant to immune checkpoint blockade and TCR-T cell therapies.8,9,20,80–83
Future Directions
Engineered T cell therapy for epithelial cancers has gained a foothold with safety and clinical activity in HPV-associated cancers and other malignancies. However, some of the same studies demonstrating proof of principle have revealed limitations due to immune editing and tumor-intrinsic evasion mechanisms. Enhanced efficacy may rely on earlier treatment with deployment of the approach ahead of other immune-based treatments that induce acquired resistance to T cell recognition and killing. Some mechanisms of resistance such as HLA mutation or loss may be overcome by polyclonal targeting through multiple HLA restriction elements. Others such as perturbations in interferon response may require new technologies to enhance T cell potency, or combination treatment. Development of predictive biomarkers based on detection of damaging alterations in genes required for tumor recognition and killing by T cells seems to be a notable strategy to enhance response rates. The ideal treatment setting may be targeting of an oncologic driver in a relatively non-immunogenic tumor that is less subjected to immunologic pressure and adapted to immune evasion. Extension to a wider range of cancers is likely to pivot considerably on identification of rational targets that can be attacked safely and with intent to achieve complete tumor regression. Alternatively, sophisticated targeting systems that distinguish tumor from normal tissue may bring more tumor-associated antigens into range.84–86
Conclusions
Engineered T cell therapy holds promise for the treatment of wide-ranging cancers. A grounded assessment of clinical trial results reveals that we have only just begun to crack the surface of effective therapy for epithelial cancers. There is a flood of innovation aiming to enhance the efficacy and extend the range of cancers that can be treated. Lessons from clinical experience are informative in considering current treatment limitations and rational strategies to advance the field.
Acknowledgements
This work was funded by the Center for Cancer Research, National Cancer Institute, Cancer Moonshot Grant ID# BC011871-02.
Declarations of Interest
Christian S. Hinrichs serves as a consultant or advisory board member for PACT Pharma, GlaxoSmithKline, Neogene Therapeutics and Capstan Therapeutics. He is an inventor and may receive blinded royalties related to the following NIH patent applications: Methods of Preparing Anti-human Papillomavirus Antigen T Cells, PCT/US2014/046478; Anti-Human Papillomavirus 16 E6 T Cell Receptors, PCT/US2014/046480; Anti-Human Papillomavirus 16 E7 T Cell Receptors, PCT/US2015/033129; Anti-KK-LC-1 T Cell Receptors, PCT/US2017/027865; Combination PD-L1 And TGF Beta Blockade in Patients with HPV+ Malignancies, PCT/US2018/031501; Tethered Interleukin-15 and Interleukin-21, PCT/US2019/016975; HLA Class I-Restricted T Cell Receptors against CD20, PCT/US2021/38649; Enhanced Antigen Reactivity of Immune Cells Expressing a Mutant Non-signaling CD3 Zeta Chain, PCT/US2021/059109; HLA Class I-Restricted T Cell Receptors Against CD22. He receives research funding from T-Cure Biosciences and Neogene Therapeutics. Scott M. Norberg declares no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Munshi NC et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med 384, 705–716 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Abramson JS et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet Lond. Engl 396, 839–852 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Maude SL et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah BD et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet Lond. Engl 398, 491–502 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Neelapu SS et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med 382, 1331–1342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berdeja JG et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet Lond. Engl 398, 314–324 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Nagarsheth NB et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat. Med 1–7 (2021) doi: 10.1038/s41591-020-01225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doran SL et al. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J. Clin. Oncol 37, 2759–2768 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi C et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med 1–10 (2022) doi: 10.1038/s41591-022-01800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson HJ & Brentjens RJ Overcoming Antigen Escape with CAR T-cell Therapy. Cancer Discov. 5, 1238–1240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mansilla-Soto J et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med (2022) doi: 10.1038/s41591-021-01621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarmarkovich M et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 599, 477–484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Cappell KM & Kochenderfer JN A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol 18, 715–727 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Walker AJ et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. J. Am. Soc. Gene Ther 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe K et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. Baltim. Md 1950 194, 911–920 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Huang J et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity 39, 846–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cappell KM & Kochenderfer JN A comparison of chimeric antigen receptors containing CD28 versus 4–1BB costimulatory domains. Nat. Rev. Clin. Oncol 18, 715–727 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Stevanović S et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356, 200–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran E et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375, 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stevanović S et al. A phase II study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin. Cancer Res 25, 1486–1493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevanović S et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol 33, 1543–1550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarnaik AA et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol 39, 2656–2666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goff SL et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 34, 2389–2397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kishton RJ, Vodnala SK, Vizcardo R & Restifo NP Next generation immunotherapy: enhancing stemness of polyclonal T cells to improve anti-tumor activity. Curr. Opin. Immunol 74, 39–45 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Iovance Biotherapeutics. https://www.globenewswire.com/news-release/2019/05/22/1841148/0/en/Iovance-Biotherapeutics-Announces-Breakthrough-Therapy-Designation-for-LN-145-for-Treatment-of-Advanced-Cervical-Cancer-Patients-Who-Have-Progressed-on-or-After-Chemotherapy.html.
- 27.Creelan BC et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med 27, 1410–1418 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran E et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zacharakis N et al. Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. J. Clin. Oncol JCO.21.02170 (2022) doi: 10.1200/JCO.21.02170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seitter SJ et al. Impact of Prior Treatment on the Efficacy of Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res (2021) doi: 10.1158/1078-0432.CCR-21-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies JS et al. Non-synergy of PD-1 blockade with T-cell therapy in solid tumors. J. Immunother. Cancer 10, e004906 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 76, 1578–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moon EK et al. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 22, 436–447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adusumilli PS et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 11, 2748–2763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heczey A et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. J. Am. Soc. Gene Ther 25, 2214–2224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudley ME et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes. Science (2002) doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robbins PF et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med 19, 747–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Y-C et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J. Immunol. Baltim. Md 1950 190, 6034–6042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tay RE, Richardson EK & Toh HC Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 28, 5–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muranski P & Restifo NP Adoptive immunotherapy of cancer using CD4(+) T cells. Curr. Opin. Immunol 21, 200–208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poncette L, Bluhm J & Blankenstein T The role of CD4 T cells in rejection of solid tumors. Curr. Opin. Immunol 74, 18–24 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Y-C et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J. Clin. Oncol 35, 3322–3329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sim MJW et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. U. S. A 117, 12826–12835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkins MB et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 17, 2105–2116 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Rosenberg SA Interleukin 2 for patients with renal cancer. Nat. Clin. Pract. Oncol 4, 497 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin BY et al. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klebanoff CA et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 17, 5343–5352 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim SP et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor-Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res 10, 932–946 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robbins PF et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 21, 1019–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narayan V et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat. Med 1–11 (2022) doi: 10.1038/s41591-022-01726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson LA et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parkhurst MR et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. J. Am. Soc. Gene Ther 19, 620–626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamers CH et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther. J. Am. Soc. Gene Ther 21, 904–912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan RA et al. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Majzner RG et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 1–10 (2022) doi: 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.D’Angelo SP et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov. 8, 944–957 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran I et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 7, 276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blumenschein GR et al. Phase I clinical trial evaluating the safety and efficacy of ADP-A2M10 SPEAR T cells in patients with MAGE-A10+ advanced non-small cell lung cancer. J. Immunother. Cancer 10, e003581 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marcinkowski B et al. Cancer targeting by TCR gene-engineered T cells directed against Kita-Kyushu Lung Cancer Antigen-1. J. Immunother. Cancer 7, 229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen C et al. Multiomics analysis reveals CT83 is the most specific gene for triple negative breast cancer and its hypomethylation is oncogenic in breast cancer. Sci. Rep 11, 12172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fukuyama T et al. Identification of a New Cancer/Germline Gene, KK-LC-1, Encoding an Antigen Recognized by Autologous CTL Induced on Human Lung Adenocarcinoma. Cancer Res. 66, 4922–4928 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Paret C et al. CXorf61 is a target for T cell based immunotherapy of triple-negative breast cancer. Oncotarget 6, 25356–25367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morgan RA et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. Hagerstown Md 1997 36, 133–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Linette GP et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cameron BJ et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med 5, 197ra103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leidner R et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med 386, 2112–2119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tran E, Robbins PF & Rosenberg SA ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat. Immunol 18, 255–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McNaughton AL et al. Insights From Deep Sequencing of the HBV Genome—Unique, Tiny, and Misunderstood. Gastroenterology 156, 384–399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan AT et al. Use of Expression Profiles of HBV-DNA Integrated Into Genomes of Hepatocellular Carcinoma Cells to Select T Cells for Immunotherapy. Gastroenterology 156, 1862–1876.e9 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Meng F et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol. Int 15, 1402–1412 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farrell PJ Epstein- Barr Virus and Cancer. in Annual Review of Pathology: Mechanisms of Disease, Vol 14 (eds. Abbas AK, Aster JC & Feany MB) vol. 14 29–53 (Annual Reviews, 2019). [DOI] [PubMed] [Google Scholar]
- 73.Chapuis AG et al. Regression of metastatic Merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-I. Cancer Immunol. Res 2, 27–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paulson KG et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun 9, 3868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gavvovidis I et al. Targeting Merkel Cell Carcinoma by Engineered T Cells Specific to T-Antigens of Merkel Cell Polyomavirus. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 24, 3644–3655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dudaniec K, Westendorf K, Nössner E & Uckert W Generation of Epstein-Barr Virus Antigen-Specific T Cell Receptors Recognizing Immunodominant Epitopes of LMP1, LMP2A, and EBNA3C for Immunotherapy. Hum. Gene Ther 32, 919–935 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Liu Q et al. In vivo therapeutic effects of affinity-improved-TCR engineered T-cells on HBV-related hepatocellular carcinoma. J. Immunother. Cancer 8, e001748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Milone MC et al. Engineering enhanced CAR T-cells for improved cancer therapy. Nat. Cancer 2, 780–793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gettinger S et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 7, 1420–1435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zaretsky JM et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med 375, 819–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shin DS et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 7, 188–201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sade-Feldman M et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun 8, 1136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Larson RC et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature 604, 563–570 (2022). [DOI] [PubMed] [Google Scholar]
- 84.Hyrenius-Wittsten A et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med 13, eabd8836 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morsut L et al. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roybal KT et al. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432.e16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]