

Abstract
Nitric oxide can interact with a wide range of proteins including many that are involved in metabolism. In this review we have summarized the effects of NO on glycolysis, fatty acid metabolism, the TCA cycle, and oxidative phosphorylation with reference to skeletal muscle. Low to moderate NO concentrations upregulate glucose and fatty acid oxidation, while higher NO concentrations shift cellular reliance toward a fully glycolytic phenotype. Moderate NO production directly inhibits pyruvate dehydrogenase activity, reducing glucose-derived carbon entry into the TCA cycle and subsequently increasing anaploretic reactions. NO directly inhibits aconitase activity, increasing reliance on glutamine for continued energy production. At higher or prolonged NO exposure, citrate accumulation can inhibit multiple ATP-producing pathways. Reduced TCA flux slows NADH/FADH entry into the ETC. NO can also inhibit the ETC directly, further limiting oxidative phosphorylation. Moderate NO production improves mitochondrial efficiency while improving O2 utilization increasing whole-body energy production. Long-term bioenergetic capacity may be increased because of NO-derived ROS, which participate in adaptive cellular redox signaling through AMPK, PCG1-α, HIF-1, and NF-κB. However, prolonged exposure or high concentrations of NO can result in membrane depolarization and opening of the MPT. In this way NO may serve as a biochemical rheostat matching energy supply with demand for optimal respiratory function.
Keywords: nitric oxide, mitochondria, metabolism, exercise, glycolysis, cellular respiration
Introduction
A crucial aspect of metabolic homeostasis is matching adenosine 5’-triphosphate (ATP) production with demand. This process necessitates matching both in terms of total amount but also in terms of rate. Briefly, the most efficient metabolic pathways are also the slowest, so during high-intensity exercise, these pathways are unable to supply ATP at the required rate. Mitochondrial oxidative phosphorylation is required for the efficient conversion of reducing equivalents, such as nicotinamide adenine dinucleotide (NADH), to cellular energy. However, this process is considerably slower than the rates of glycolysis or the hexose monophosphate shunt. The flip side is that such high-rate energy sources are ultimately limited by the buildup of unresolved reduction equivalents that results in acidification. While this may seem like a simple supply and demand problem, it is vital that the rates and routes of ATP production and use are well matched for maximum physiological performance. This represents a complex signaling problem that centers upon the mitochondrion as the key to overall metabolic function and specific rates of oxidative phosphorylation.
Multiple cellular signals serve to regulate metabolic flux broadly. In the cytosol, increased amounts of adenosine monophosphate (AMP), adenosine diphosphate (ADP), and inorganic phosphate (Pi) signify an energy decrement, increasing glycolytic flux and the production of pyruvate and lactate1–3. Within the mitochondria, acetyl-CoA (primarily from the breakdown of carbohydrates and fats), calcium ion concentrations, O2 tension, NADH+H+, flavin adenine dinucleotide (FADH2), ADP, and Pi concentrations influence tricarboxylic acid (TCA) cycle and electron transport chain (ETC) flux respectively4,5. A multitude of other signals such as hormones (epinephrine, norepinephrine, insulin, glucagon), blood flow, and body temperature can also regulate metabolic rate. Working together, these pathways act to maintain ATP supply despite rapid increases in ATP demand during muscle contraction. However, an acute regulator of mitochondrial function, whose function can be significantly altered by available O2 tension is nitric oxide (NO). The role of NO as a fine tuner in mitochondrial function is summarized in Figure 1.
Figure 1. Nitric Oxide Operates to Balance Mitochondrial Flux and Optimize Cellular ATP Production.
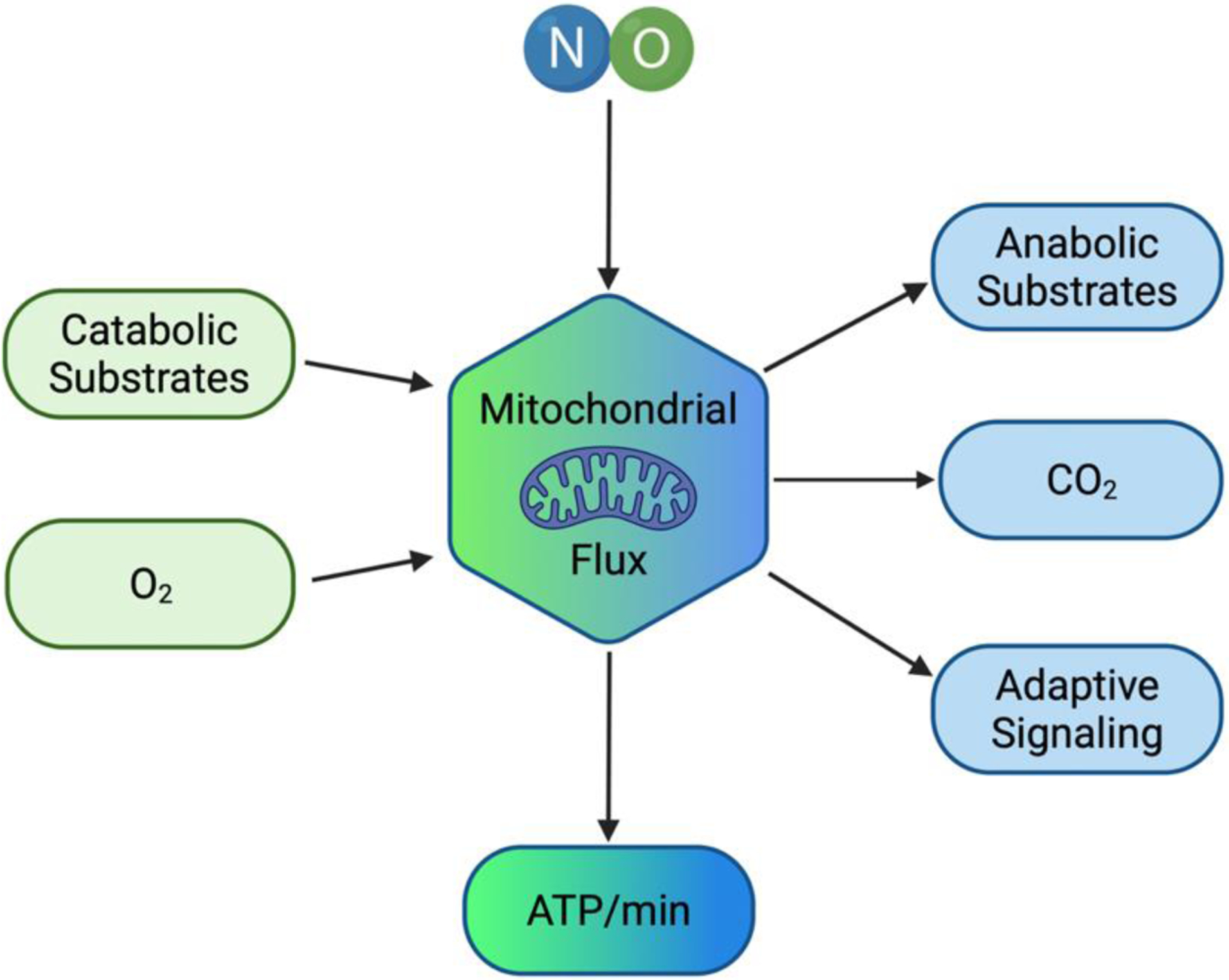
NO can regulate both catabolic and anabolic substrate flux through the mitochondrion so that O2 usage can be optimized such that the ATP produced per unit time matches the energy demand of the cell. Ultimately long-term activation of NO signaling can lead to adaptive signaling such that the cell can operate with the highest efficiency.
Nitric oxide is a powerful, short-lasting signaling molecule capable of regulating a wide range of physiological functions, including vascular homeostasis, immune response, cognitive function, and metabolism. Nitric oxide can readily participate in one electron reduction and oxidation reactions leading to the formation of a range of reactive nitrogen species, the production of which is dependent upon the cellular environment6. These reactive species can react with lipids and various proteins, including enzymes and transport proteins essential to glucose and fatty acid metabolism, the TCA cycle, and the respiratory chain7,8. Endogenously generated by several nitric oxide synthase (NOS) enzymes, NO is present both in the systemic vasculature and within mitochondrial space9. It has a short half-life (<2 seconds) that varies with O2 concentration within the tissue, and NO exerts its effects in a dose-dependent manner10. Recently, the reduction of nitrogen oxides like nitrite and nitrate has been evidenced as an important source of NO chemistry11. This review will discuss how NO regulates metabolic flux within the mitochondria by its interactions with specific proteins. In this way, NO modifies cellular utilization of metabolic substrates, the availability of O2, and the production of extramitochondrial signals to match the energy needs to their supply in the cell, which may be critical for optimal respiration.
1. Production and Molecular Targets
NO is endogenously generated by several NOS enzymes, which catalyze the oxidation of L-arginine to NO12. Regular NO production from the constitutive expression of NOS1 (neuronal; nNOS) and NOS3 (endothelial; eNOS) produces low nanomolar amounts of NO necessary to maintain vascular homeostasis13–15. During inflammatory events, NO production is rapidly increased via upregulated NOS2 (inducible; iNOS) expression12,16. NOS enzymes are not tissue specific and expression of NOS isoforms have been found within the mitochondria (termed mtNOS)17. In addition, it should be remembered, especially within mitochondria, that nitrite can be reduced back to NO by a range of metalloproteins, including xanthine oxidoreductase (XOR) and myoglobin18. NO can interact with a wide variety of biological molecules with potential signaling consequences19–21. Thus, a critical factor in NO regulation of physiological systems is its flux rate of production and the relative concentrations of target molecules.
NO will bind to metal centers, such as heme-iron or iron-sulfur centers, to produce nitrosyl-metal species or to cysteine thiols (S-nitrosylation) to produce S-nitrosothiols (SNOs)21,22. Additionally, nitrosation reactions result in the direct or indirect addition of a nitrosonium ion (NO+), which may also form nitrosothiols21. Furthermore, nitration reactions lead to the addition of a nitronium ion (NO2+), forming a nitro functional group (R-NO2)21. Finally, NO may react with reactive oxygen species (ROS), leading to the production of higher oxides of nitrogen such as peroxynitrite. The topic of NO reactivity has been discussed more thoroughly elsewhere20,21 and here we will focus on potential targets within metabolic control.
2. TCA Cycle Flux and the Fate of Carbon Substrates
3.1. Pyruvate dehydrogenase (PDH)
Part of the larger pyruvate dehydrogenase complex (PDC), PDH catalyzes the oxidative decarboxylation of glucose-derived pyruvate into acetyl-COA for entry in the TCA23–25. The downregulation of PDH reduces cellular usage of glucose, subsequently increasing the use of fatty acids for energy26,27.Several mechanisms have been proposed for how NO or NO derivates can directly inhibit PDH activity, including S-nitrosylation of PDC subunit dihydrolipoamide dehydrogenase28. PDH inhibition increases with NO production, reducing glucose-derived carbon entry into the TCA and increasing anaploretic pyruvate carboxylation and glutamonolysis29. This allows continued production of TCA intermediates such as α-ketoglutarate, citrate, or itaconate, for continued energy production (glutamine-derived NADH and FADH2 for the ETC) or for the synthesis of lipids30,31. While decreased PDH activity may blunt TCA cycle flux and downstream oxidative phosphorylation and ATP output, it also allows for the conservation of valuable metabolic substrates, such as pyruvate and lactate, that can be used for gluconeogenesis or for supporting cellular growth and other anabolic pathways32–34. Reduced PDH activity post-exercise may play a role in glycogen resynthesis post-intensive exercise by sparring carbohydrates from cellular oxidation35,36.
3.2. Aconitase
Mitochondrial aconitase, which catalyzes the isomerization of citrate to isocitrate in the TCA cycle37, is an iron–sulfur-containing dehydratase, which is a target of oxidation and inhibition by ROS, NO, and NO derivates37. Under normal conditions, high TCA cycle turnover and subsequent ETC respiration leads to increased production of ATP and mitochondrial ROS. These ROS can regulate TCA flux, inhibiting aconitase activity, reducing glucose-derived carbon oxidation and sparing glucose for use in other metabolic pathways, and activating alternative pathways for ATP production. NO and NO derivates decrease both mitochondrial and cytosolic aconitase activity in a dose-dependent manner, with a more significant effect on mitochondrial aconitase38–43. High levels of NO production lead to complete reduction in aconitase activity, preventing citrate isomerization and subsequent formation of α-ketoglutarate. In turn, this promotes increased glutaminolysis30.The inhibition of aconitase results in the accumulation of mitochondrial citrate, which has numerous effects on cellular energy production by inhibiting ATP-producing pathways44. This includes inhibition of glycolytic enzymes phosphofructokinase, pyruvate kinase, and PDH, as well as decreased carnitine palmitoyltransferase (CPT-1) mediated fatty acid transport44. Citrate accumulation upregulates ATP-consuming pathways and supports lipid synthesis45.
3.3. Glucose and Fatty Acid Metabolism
Physiological levels of NO alter blood glucose homeostasis via upregulation of glucose transporter (GLUT) expression, although the exact mechanism of action varies by cell type46–53. In skeletal muscle, insulin-independent GLUT translocation requires activation of the primary energy sensor, AMP-activated protein kinase (AMPK). Increased muscle activity will reduce ATP concentrations, activate AMPK, and stimulate NO production. This signals cGMP-dependent GLUT-4 translocation to the cell membrane50,54–57. nNOS-derived NO mediates both insulin-independent (AMPK mediated) and insulin-dependent GLUT transport in skeletal muscle58. AMPK itself is directly regulated by NO, creating a mechanism for self-regulation of glucose homeostasis in resting and exercising skeletal muscle55,59. In addition to altering glucose transport, both exogenous NO donors and eNOS derived NO reduce gluconeogenesis and glycogenesis in liver cells, i.e., reduce anabolism60–62. Not surprisingly, increased glucose uptake coupled with decreased gluconeogenesis, glycogenesis, fatty acid oxidation, and cellular respiration leads to an upregulation of glycolysis as the main source of energy production63–65.
Both endogenously and exogenously produced NO alter lipolysis, fatty acid uptake, and fatty acid oxidation in a dose-dependent manner. NO inhibits fatty acid synthesis in rat liver while decreasing acetyl-CoA carboxylase (ACC) activity, reducing malonyl-CoA concentration, and leading to upregulation of CPT166. This is further supported by studies demonstrating that the use of NOS inhibitors decreased fatty acid oxidation via decreased CPT activity67. Arginine (a precursor to NO) and low-dose nitrate supplementation increase lipolysis and fatty acid oxidation, with the latter increasing cGMP activity and CPT1 expression in mice65,68. Higher nitrate doses also upregulate PGC-1α, resulting in mitochondrial biogenesis and increased fatty acid oxidative capacity68. Post-translational modification via S-nitrosylation of acyl-CoA dehydrogenase improves the enzyme’s efficiency in mouse liver, which may increase fatty acid metabolism (although this has not been directly tested)7. Leptin alters lipolysis via several NO-mediated pathways69,70. Acute leptin exposure increases NO production and lipolysis in rat white adipose tissue, an effect that is inhibited with NOS inhibitors71. However, prolonged leptin exposure decreases lipolysis via NO-mediated signaling, potentially serving as a self-regulator for energy storage71. High flux NO decreases leptin-mediated lypolysis72; while iNOS-derived NO in stimulated macrophages decreases lipolysis and β-oxidation via inhibition of the respiratory chain73. Finally, NO reduces catecholamine-mediated lipolysis through inhibition of β-adrenergic signaling74–76.
3. Cellular Respiratory Chain
3.1. Flow of Electrons and the Utilization of Oxygen
The heme-containing enzymes of the ETC (complexes I-IV) located on the inner mitochondrial membrane are well-studied targets of NO. Electrons are transferred from one complex to the next through a sequence of redox reactions, while complexes I, III, and IV pump protons into the intermembrane space. The resulting proton gradient is used to phosphorylate ADP to ATP via ATP synthase. Oxygen serves as the final electron acceptor binding at complex IV to produce ATP, CO2, and H2O. Inhibition at any point in the ETC slows the flow of electrons, altering the rate of O2 consumption, ATP production, and the generation of ROS. Functionally, a build-up of reducing equivalents NADH and FADH2 will signal energy demands are met and slow upstream TCA cycle flux. Decreased respiratory flux allows O2 to be diverted toward other, non-ATP producing pathways.
3.2. Complex I and Complex III
Detailed mechanisms underlying the inhibition of additional complexes in the ETC have been covered elsewhere77. NO can reversibly inhibit the NADH: ubiquinone oxidoreductase (complex I). Unlike at complex IV, inhibition at complex I is likely not due to the direct binding of NO but occurs via the S-nitrosation of specific thiol residues or oxidative stress via interaction with peroxynitrite78–82. Similar inhibition has been observed at ubiquinol: cytochrome c oxidoreductase (complex III) from mtNOS and NO donor S-nitrosoglutathione (GSNO), leading to increased ROS production83,84.
3.3. Cytochrome C Oxidase
Cytochrome c oxidase (complex IV) catalyzes the final step in electron transport, oxidizing cytochrome c and reducing O2 to produce H2O. The enzyme’s O2 binding site is a binuclear heme iron/copper center, which constantly cycles between oxidized and reduced states85. The relative amounts of oxidized to reduced cytochrome c oxidase present at any time are dependent on the O2 levels in the mitochondrial environment, with oxidation increasing with high O2 tension (PO2)86. NO can bind to cytochrome c oxidase in both its reduced (competitive with O2) and oxidized (non-completive) states. This competitive and non-completive binding allows NO to regulate mitochondrial respiration over the range of normal to hypoxic cellular environments. At high PO2, NO non-competitively binds the Cu center in fully oxidized cytochrome c oxidase, reducing cytochrome c oxidase and forming nitrite87–89. At low PO2, NO reversibly binds to reduced cytochrome c oxidase, directly competing with O2. This leads to inhibition of individual mitochondrial respiration, the diversion of O2 toward other cellular targets, and alters the production of reactive oxygen and nitrogen species (RONS)90–93.
3.4. Cellular Respiration and Mitochondrial O2 Gradients
The relative amount of cytochrome c oxidase in the reduced state is increased under hypoxic conditions (i.e., contracting skeletal muscle), increasing the affinity for NO. However, in the tissue environment, concentrations of NO and O2 are not uniform94. Cells most proximal to the sources of endogenously produced NO (i.e., the vascular endothelium) will experience greater inhibition of respiration. The sparing of O2 from consumption in these proximal cells extends the half-life of O2, allowing O2 to travel further along its gradient toward deeper, more hypoxic tissue10,94–96. Additionally, the half-life of NO increases under hypoxic conditions, allowing for constant diffusion into tissue further from the blood vessel. This establishes a feed-forward mechanism that perpetuates smoothing of the concentration gradient for O210,94,96. Finally, because the maximal capacity of mitochondrial respiration exceeds O2 delivery, sites near the endothelium (or other sources of endogenously produced NO) still retain adequate energy supply, despite modest NO reductions to ETC function. The result is that NO supports increased cellular and/or whole-body energy production through an increased number of respiring mitochondria.
4. Mitochondrial Efficiency
4.1. Matching ATP Production with O2 Consumption
O2 consumption is tightly coupled to proton pumping and ATP synthesis during mitochondrial respiration. However, several other pathways exist through which either protons may re-enter the mitochondrial matrix (proton leak) or in which O2 is consumed. This uncoupling reduces mitochondrial efficiency. Under normal conditions, base levels of proton leak occur unregulated (basal leak), accounting for approximately 5% of total mitochondrial proton leak. NO can alter the electrochemical proton gradient used to synthesize ATP through several mechanisms, including changing inner mitochondrial membrane-bound protein expression or function. High concentrations of NO cause inner membrane depolarization through the formation of ONOO- and the opening of the mitochondrial permeability transition pore (MPT)97,98. Low NO flux protects against decreases in membrane potential, potentially via S-nitrosylation of membrane-bound proteins98–100. Nitrate supplementation has also been shown to decrease the expression of two mitochondrial membrane proteins, adenine nucleotide translocase (ANT) and uncoupling protein (UCP), which may be important mediators of proton leak18,101–103. In a recent study performed by Wynne and colleagues, it was observed that nitrite can stimulate the glycolytic ATP supply without the loss of the oxidative ATP supply in skeletal muscle cells104. This indicates that nitrates increase the overall rates of myocellular ATP production104. Additionally, nitrates result in an overall shift away from non-mitochondrial respiration104. Taken together, this indicates that nitrate supplementation has the potential to lower the oxygen cost of ATP production104.
4.2. Mitochondrial Efficiency and Exercise Capacity
There is strong evidence that NO can enhance mitochondrial efficiency during exercise by improving the coupling of ATP synthesis with the dissipation of the proton gradient, thus improving the mitochondrial oxidative efficiency18,105,106. These effects are likely mediated by NO control over the mitochondrial membrane potential. Several studies have shown that oral nitrate supplementation reduces whole-body O2 consumption (VO2) at various submaximal workloads18,105–109. Importantly, this observed decrease in VO2 is not accompanied by any significant changes in heart rate, respiratory exchange ratio, lactate accumulation, or maximal work capacity achieved. This suggests either a reduced O2 requirement for a given workload or an increased aerobic efficiency at submaximal exercise105–108. Additionally, nitrate supplementation during high-intensity exercise has been observed to increase time to task failure without a change in VO2 max achieved107,108. Numerous studies have replicated that nitrate supplementation reduces O2 demand and increases exercise tolerance across multiple exercise modalities, including cycling, running, and walking109–117. This effect is dose dependent, and has sustained benefits, with results persisting up to 15 days110,112. Nitrate supplementation has been observed to provide greater benefit at submaximal workloads for low-to-moderate aerobically fit individuals or individuals with reduced exercise capacity due to disease, but not for higher-level athletes. Concurrently, there have been studies that show that nitrite supplementation fails to improve mitochondrial function and efficiency, indicating that improvements seen with nitrate supplementation may occur through a mechanism other than oxygen consumption118,119. Additionally, several studies have indicated that nitrate supplementation resulted in no mitochondrial improvements or reduced oxygen cost during exercise, indicating a need for more research in this field to determine the effects of nitrate supplementation and the mechanisms by which they may occur120,121.
5. Adaptive Signaling
5.1. Indirect Effects of NO Signaling
NO regulates the rate of mitochondrial ROS production, especially superoxide (O2−)11,122,123. The fate of O2− is determined by the relative balance of other oxidants (i.e., NO, ONOO, NO2) and antioxidant defense mechanisms124–126. Superoxide can form peroxynitrite (ONOO−) by interactions with free NO127,128, and H2O2 and H2O by mitochondrial superoxide dismutase (mnSOD)129. mnSOD is abundantly present in the inner mitochondrial matrix and is essential for the removal of mitochondrially-produced free radicals, which arise during anaerobic metabolism129–131. There are several pathways through which mitochondrial ROS cross the mitochondrial membrane and participate in redox signaling within the cell132. ROS can act on several downstream bioenergetic mediators, including crucial energy regulators, AMPK and PGC-1α, as well as transcription factors such as NF-κB and HIF-1133–136.
AMPK can be activated by various stressors, including low nutrient levels and prolonged exercise137. When ATP levels are decreased, AMPK becomes activated138,139 leading to increased ATP-generation138,140. AMPK regulates glucose uptake in muscle and fat cells by increasing GLUT4 trafficking to the cell membrane and regulates the rate-limiting enzymes of lipid metabolism Acetyl-CoA carboxylase and HMG-CoA reductase138,141–143. AMPK can also increase mitochondrial biogenesis through activation of signaling pathways such as NRF-1 and PGC-1144–147. AMPK contributes to muscle fiber type remodeling, angiogenesis, and regulates autophagy and mitophagy3,140,148–150. AMPK signaling also directly affects NOS signaling. AMPK activation has been shown to inhibit iNOS expression151,152. Additionally, AMPK has been observed to phosphorylate eNOS and nNOS, leading to their activation152–154, while eNOS has been implicated in the induction of AMPK152,154. PGC1-α is a transcriptional co-activator that regulates cellular metabolism in muscle tissue147,155,156. PGC1-α responds to several stimuli, including ROS, ATP demand, and cellular stress, to initiate transcription of downstream genes155,157 encoding mitochondrial proteins, leading to mitochondrial biogenesis155,158–161. Activation of PGC1-α regulates skeletal muscle fiber type and contraction, lipid uptake, and glucose transport155,156,162. Additionally, PCG1-α activation leads to the upregulation of SOD155,156,163,164. PCG1-α is directly activated via exercise, low cellular energy155, and its expression is regulated by NO in skeletal muscle59. Short-term exposure to NO in endothelial cells downregulates PGC1-α ,however, the opposite effects occur with long-term exposure165.
HIF-1, a hypoxia sensitive transcription factor, serves to preserve O2, while maximizing ATP production by stimulating glycolysis, erythropoiesis and angiogeneis166,167. Exercise can generate micro-regions of acute hypoxia, and thus induce HIF-1 signaling166,168. NO and other reactive nitrogen species, can directly and indirectly affect HIF-1 activation169. At low NO levels promote HIF-1α degradation, whereas high concentrations promote its function, stabilizing it even in normoxic conditions169,170. This upregulation increases transcription of proteins related to mitochondrial function, glycolysis, erythropoiesis, and angiogenesis171–173. In addition, the hypoxic conditions that occur in muscle tissue during exercise lead to increased NO production, further contributing to HIF-1 activity and its downstream effects171.
5.2. Direct NO Signaling and Metabolic Adaptations
Low levels of NO, that do not significantly inhibit cellular respiration, decrease superoxide production174. Similarly, inhibition of mitochondrial respiration during hypoxic conditions (i.e., contracting skeletal muscle) temporarily decreases mitochondrial-derived ROS production via NO reduction of ETC flux175,176. However, mitochondria are the primary source for ROS following exercise, which may be important in adaptation to training177–180. Conversely, moderate and high levels of NO, result in inhibition of complex I & IV, decreasing O2 consumption and inducing a dose dependent increase in ROS and a buildup of reducing equivalents80,181.
6. Conclusions
Although often overlooked in models of metabolic regulation, NO plays a vital role in fine-tuning the catabolic and anabolic pathways of the cell. The potential interactions of NO in metabolic regulation of working tissue, such as contracting muscle, are summarized in Figure 2. The overall effect of NO on metabolic output is driven by NO concentration, rate of production, and the localization of NO within the cell or tissue environment10,98. The mobilization and subsequent oxidation of glucose and fatty acids is upregulated by low, potentially eNOS-derived, NO production. During times of increased energy demands, such as in contracting skeletal muscle, NO activates AMPK-mediated pathways for energy procurement49,54,140. Physical activity directly increases NO flux, as increased blood flow and sheer stress stimulate vascular eNOS expression and NO production182–184. Minor inhibition at the respiratory chain coupled with increased glucose transport, decreased fatty acid oxidation, and an increased need for ATP serve to increase glycolytic flux during intensive exercise. At submaximal intensities, moderate NO production improves the utilization of O2 for ATP production via decreased uncoupling across the inner membrane and increased recruitment of respiring mitochondria105. Post-physical activity recovery or long-term bioenergetic capacity may be increased because of NO-derived ROS, AMPK, PCG1-α, HIF-1, and NF-κB signaling3,175,177,185,186. Increased mitochondrial biogenesis and transcription of mitochondrial proteins increase the overall oxidative capacity of the cell, leading to long-term training adaptions and improvements.
Figure 2: NO Regulation of Mitochondrial Metabolism.
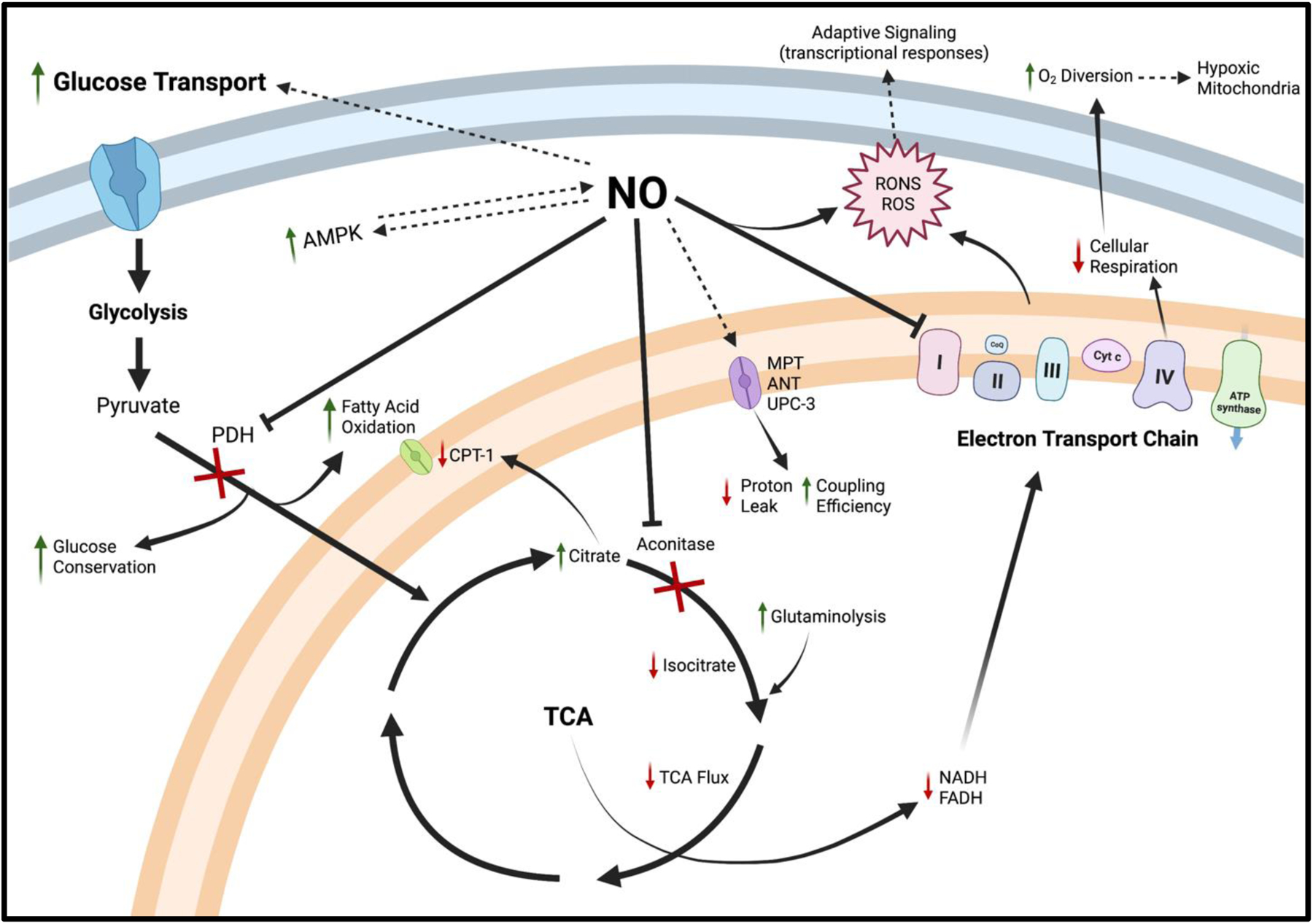
Low to moderate NO concentrations upregulate glucose and fatty acid oxidation via multiple pathways, while higher NO concentrations serve to shift cellular reliance away from fatty acid oxidation and cellular respiration toward a fully glycolytic phenotype. NO-induced AMPK activation mediates GLUT4 expression on the cell surface membrane and increased glycolytic flux. AMPK enhances NO production via NOS phosphorylation. Moderate NO production within the cytosol directly inhibits PDH, reducing glucose-derived carbon entry into the TCA, the use of fatty acids for energy while conserving lactate and pyruvate for utilization in other anabolic pathways. Within the mitochondria, NO directly inhibits aconitase, increasing TCA reliance on glutamine for continued energy production. At higher or prolonged NO exposure, subsequent citrate accumulation can inhibit multiple ATP-producing pathways, including CPT-1 and several glycolytic enzymes (not shown). Reduced TCA flux slows NADH/FADH entry into the respiratory chain, which also experiences direct NO inhibition at complex IV. Minor inhibition of the ETC, with increased glucose transport and decreased fatty acid oxidation, serves to increase glycolytic flux during high energy demand (i.e., exercise). Moderate NO production can also improve mitochondrial efficiency via decreased expression of ANT and UPC-3 across the inner membrane while improving O2 utilization by diverting O2 toward more hypoxic tissue, increasing whole-body energy production. Long-term bioenergetic capacity may be increased because of NO-derived ROS, which are able to cross the mitochondrial membrane and participate in adaptive cellular redox signaling primarily through AMPK, PCG1-α, HIF-1, and NF-κB. However, prolonged exposure or high concentrations of NO results in membrane depolarization and opening of the MPT, excessive and potentially damaging ROS production, and the shift from oxidative phosphorylation toward total reliance on less efficient glycolysis for energy production.
NO is known for its diverse roles in multiple biological processes ranging from vascular health to cognition and metabolic function. NO is capable of inhibiting or enhancing metabolic function, dependent on its own production rate and localization within the cell. Metabolic output is tightly controlled to balance ATP producing and ATP consuming processes. While there are many regulators of metabolic flux, there is clear evidence that NO is central to cellular energy control, serving as both a sensor and director of metabolic output. In this way, it appears that NO operates like a metabolic rheostat fine tuning energy production to match demand. For this function it is necessary that the right amount of NO be produced at the right time for efficient regulation, not that more is always better.
Highlights.
The mechanisms by which NO can regulate cellular metabolism are discussed.
The intersection between NO and mitochondrial function is considered especially with consideration of how metabolic rate can alter these processes.
Mechanisms that may explain how NO can improve mitochondrial efficiency are presented.
NO as a regulator of long-term bioenergetics is also considered as the concept of NO as a biological rheostat is presented.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The study received support from National Institute of Environmental Health Sciences (NIEHSP30ES005022, T32-ES007148) and National Heart and Lung Institute (NIH-HL086621).
Abbreviations:
- ATP
Adenosine 5’-triphosphate
- NADH
nicotinamide adenine dinucleotide
- AMP
adenosine monophosphate
- ADP
adenosine diphosphate
- Pi
inorganic phosphate
- FADH2
flavin adenine dinucleotide
- TCA
tricarboxylic acid
- ETC
electron transport chain
- NO
nitric oxide
- NOS
nitric oxide synthase
- XOR
xanthine oxidoreductase
- SNOs
S-nitrosothiols
- ROS
reactive oxygen species
- PDH
pyruvate dehydrogenase
- PDC
pyruvate dehydrogenase complex
- CPT-1
carnitine palmitoyltransferase
- GLUT
glucose transporter
- AMPK
AMP-activated protein kinase
- ACC
acetyl-CoA carboxylase
- GSNO
S-nitrosoglutathione
- PO2
O2 tension
- RONS
reactive oxygen and nitrogen species
- MPT
mitochondrial permeability transition pore
- ANT
adenine nucleotide translocase
- UCP
uncoupling proteins
- VO2
whole-body O2 consumption
- O2−
superoxide
- ONOO−
peroxynitrite
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- HIF-1
hypoxia-inducible factor-1
- NRF-1
nuclear respiratory factor-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Veech RL, Lawson JW, Cornell NW, Krebs HA. Cytosolic phosphorylation potential. J Biol Chem. Jul 25 1979;254(14):6538–47. [PubMed] [Google Scholar]
- 2.Wu C, Khan SA, Peng LJ, Lange AJ. Roles for fructose-2,6-bisphosphate in the control of fuel metabolism: beyond its allosteric effects on glycolytic and gluconeogenic enzymes. Adv Enzyme Regul. 2006;46:72–88. doi: 10.1016/j.advenzreg.2006.01.010 [DOI] [PubMed] [Google Scholar]
- 3.Tanaka T, Nishimura A, Nishiyama K, Goto T, Numaga-Tomita T, Nishida M. Mitochondrial dynamics in exercise physiology. Pflügers Archiv - European Journal of Physiology. 2020/02/01 2020;472(2):137–153. doi: 10.1007/s00424-019-02258-3 [DOI] [PubMed] [Google Scholar]
- 4.Lee SH, Davis EJ. Carboxylation and decarboxylation reactions. Anaplerotic flux and removal of citrate cycle intermediates in skeletal muscle. J Biol Chem. Jan 25 1979;254(2):420–30. [PubMed] [Google Scholar]
- 5.Hargreaves M, Spriet LL. Skeletal muscle energy metabolism during exercise. Nature Metabolism. 2020/09/01 2020;2(9):817–828. doi: 10.1038/s42255-020-0251-4 [DOI] [PubMed] [Google Scholar]
- 6.Fukuto JM. Chemistry of nitric oxide: biologically relevant aspects. Adv Pharmacol. 1995;34:1–15. [DOI] [PubMed] [Google Scholar]
- 7.Doulias P-T, Tenopoulou M, Greene Jennifer L, Raju K, Ischiropoulos H. Nitric Oxide Regulates Mitochondrial Fatty Acid Metabolism Through Reversible Protein S-Nitrosylation. Science Signaling. 2013/01/01 2013;6(256):rs1–rs1. doi: 10.1126/scisignal.2003252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochimica et biophysica acta. 2012;1820(6):712–721. doi: 10.1016/j.bbagen.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elfering SL, Haynes VL, Traaseth NJ, Ettl A, Giulivi C. Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. AmJPhysiol Heart CircPhysiol. 01 2004;286(1):H22–H29. [DOI] [PubMed] [Google Scholar]
- 10.Thomas DD, Liu X, Kantrow SP, Lancaster JR Jr. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc Natl Acad Sci U S A. Jan 2 2001;98(1):355–60. doi: 10.1073/pnas.011379598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tejero J, Shiva S, Gladwin MT. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol Rev. Jan 1 2019;99(1):311–379. doi: 10.1152/physrev.00036.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829–837d. doi: 10.1093/eurheartj/ehr304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costa ED, Rezende BA, Cortes SF, Lemos VS. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Mini Review. Frontiers in Physiology. 2016-June-02 2016;7(206)doi: 10.3389/fphys.2016.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Förstermann U, Münzel T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation. 2006/04/04 2006;113(13):1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532 [DOI] [PubMed] [Google Scholar]
- 15.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2011;33(7):829–837. doi: 10.1093/eurheartj/ehr304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathan CF, Hibbs JB Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Opin Immunol. Feb 1991;3(1):65–70. doi: 10.1016/0952-7915(91)90079-g [DOI] [PubMed] [Google Scholar]
- 17.Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends in Pharmacological Sciences. 2005/04/01/ 2005;26(4):190–195. doi: 10.1016/j.tips.2005.02.005 [DOI] [PubMed] [Google Scholar]
- 18.Larsen FJ, Schiffer TA, Borniquel S, et al. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. Feb 2 2011;13(2):149–59. doi: 10.1016/j.cmet.2011.01.004 [DOI] [PubMed] [Google Scholar]
- 19.Massa CM, Liu Z, Taylor S, et al. Biological Mechanisms of S - Nitrosothiol Formation and Degradation: How is Specificity of S -Nitrosylation Achieved? Antioxidants (Basel). Jul 12 2021;10(7)doi: 10.3390/antiox10071111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gow AJ, Ischiropoulos H. Nitric oxide chemistry and cellular signaling. JCell Physiol. 06 2001;187(3):277–282. [DOI] [PubMed] [Google Scholar]
- 21.Heinrich TA, da Silva RS, Miranda KM, Switzer CH, Wink DA, Fukuto JM. Biological nitric oxide signalling: chemistry and terminology. Br J Pharmacol. 2013;169(7):1417–1429. doi: 10.1111/bph.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernando V, Zheng X, Walia Y, Sharma V, Letson J, Furuta S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants (Basel). 2019;8(9):404. doi: 10.3390/antiox8090404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. Faseb j. Nov 1990;4(14):3224–33. doi: 10.1096/fasebj.4.14.2227213 [DOI] [PubMed] [Google Scholar]
- 24.Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289(24):16615–16623. doi: 10.1074/jbc.R114.563148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J, Baiesc FL, Hiromasa Y, et al. Atomic Structure of the E2 Inner Core of Human Pyruvate Dehydrogenase Complex. Biochemistry. 2018;57(16):2325–2334. doi: 10.1021/acs.biochem.8b00357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeoung Nam H, Wu P, Joshi Mandar A, et al. Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochemical Journal. 2006;397(3):417–425. doi: 10.1042/BJ20060125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crewe C, Kinter M, Szweda LI. Rapid inhibition of pyruvate dehydrogenase: an initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. PLoS One. 2013;8(10):e77280. doi: 10.1371/journal.pone.0077280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan L-J, Liu L, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli’s salt. Sheng Wu Wu Li Hsueh Bao. 2012;28(4):341–350. [PMC free article] [PubMed] [Google Scholar]
- 29.Palmieri EM, Gonzalez-Cotto M, Baseler WA, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun. 2020;11(1):698–698. doi: 10.1038/s41467-020-14433-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Experimental & Molecular Medicine. 2020/09/01 2020;52(9):1496–1516. doi: 10.1038/s12276-020-00504-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan J, Kamphorst JJ, Mathew R, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. Dec 3 2013;9:712. doi: 10.1038/msb.2013.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sradhanjali S, Reddy MM. Inhibition of Pyruvate Dehydrogenase Kinase as a Therapeutic Strategy against Cancer. Curr Top Med Chem. 2018;18(6):444–453. doi: 10.2174/1568026618666180523105756 [DOI] [PubMed] [Google Scholar]
- 33.Woolbright BL, Rajendran G, Harris RA, Taylor JA. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Molecular Cancer Therapeutics. 2019;18(10):1673. doi: 10.1158/1535-7163.MCT-19-0079 [DOI] [PubMed] [Google Scholar]
- 34.Park S, Jeon JH, Min BK, et al. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. Diabetes Metab J. 2018;42(4):270–281. doi: 10.4093/dmj.2018.0101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herbst EA, MacPherson RE, LeBlanc PJ, et al. Pyruvate dehydrogenase kinase-4 contributes to the recirculation of gluconeogenic precursors during postexercise glycogen recovery. Am J Physiol Regul Integr Comp Physiol. Jan 15 2014;306(2):R102–7. doi: 10.1152/ajpregu.00150.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pilegaard H, Neufer PD. Transcriptional regulation of pyruvate dehydrogenase kinase 4 in skeletal muscle during and after exercise. Proc Nutr Soc. May 2004;63(2):221–6. doi: 10.1079/pns2004345 [DOI] [PubMed] [Google Scholar]
- 37.Lushchak OV, Piroddi M, Galli F, Lushchak VI. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014;19(1):8–15. doi: 10.1179/1351000213Y.0000000073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stadler J, Billiar TR, Curran RD, Stuehr DJ, Ochoa JB, Simmons RL. Effect of exogenous and endogenous nitric oxide on mitochondrial respiration of rat hepatocytes. American Journal of Physiology-Cell Physiology. 1991/05/01 1991;260(5):C910–C916. doi: 10.1152/ajpcell.1991.260.5.C910 [DOI] [PubMed] [Google Scholar]
- 39.Drapier J-C. Interplay between NO and [Fe-S] Clusters: Relevance to Biological Systems. Methods. 1997/03/01/ 1997;11(3):319–329. doi: 10.1006/meth.1996.0426 [DOI] [PubMed] [Google Scholar]
- 40.Castro LA, Robalinho RL, Cayota A, Meneghini R, Radi R. Nitric oxide and peroxynitrite-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblasts. Arch Biochem Biophys. Nov 15 1998;359(2):215–24. doi: 10.1006/abbi.1998.0898 [DOI] [PubMed] [Google Scholar]
- 41.Tórtora V, Quijano C, Freeman B, Radi R, Castro L. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic Biol Med. Apr 1 2007;42(7):1075–88. doi: 10.1016/j.freeradbiomed.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 42.Cairo G, Ronchi R, Recalcati S, Campanella A, Minotti G. Nitric oxide and peroxynitrite activate the iron regulatory protein-1 of J774A.1 macrophages by direct disassembly of the Fe-S cluster of cytoplasmic aconitase. Biochemistry. Jun 11 2002;41(23):7435–42. doi: 10.1021/bi025756k [DOI] [PubMed] [Google Scholar]
- 43.Castro L, Tórtora V, Mansilla S, Radi R. Aconitases: Non-redox Iron–Sulfur Proteins Sensitive to Reactive Species. Accounts of Chemical Research. 2019/09/17 2019;52(9):2609–2619. doi: 10.1021/acs.accounts.9b00150 [DOI] [PubMed] [Google Scholar]
- 44.Williams NC, O’Neill LAJ. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front Immunol. 2018;9:141. doi: 10.3389/fimmu.2018.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim Biophys Acta. Nov 2014;1839(11):1217–1225. doi: 10.1016/j.bbagrm.2014.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balon TW, Nadler JL. Evidence that nitric oxide increases glucose transport in skeletal muscle. Journal of Applied Physiology. 1997/01/01 1997;82(1):359–363. doi: 10.1152/jappl.1997.82.1.359 [DOI] [PubMed] [Google Scholar]
- 47.Tanaka T, Nakatani K, Morioka K, et al. Nitric oxide stimulates glucose transport through insulin-independent GLUT4 translocation in 3T3-L1 adipocytes. European Journal of Endocrinology Eur J Endocrinol. 01 Jul. 2003 2003;149(1):61–67. [DOI] [PubMed] [Google Scholar]
- 48.Bergandi L, Silvagno F, Russo I, et al. Insulin Stimulates Glucose Transport Via Nitric Oxide/Cyclic GMP Pathway in Human Vascular Smooth Muscle Cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003/12/01 2003;23(12):2215–2221. doi: 10.1161/01.ATV.0000107028.20478.8e [DOI] [PubMed] [Google Scholar]
- 49.Higaki Y, Hirshman MF, Fujii N, Goodyear LJ. Nitric Oxide Increases Glucose Uptake Through a Mechanism That Is Distinct From the Insulin and Contraction Pathways in Rat Skeletal Muscle. Diabetes. 2001;50(2):241. doi: 10.2337/diabetes.50.2.241 [DOI] [PubMed] [Google Scholar]
- 50.Deshmukh AS, Long YC, de Castro Barbosa T, et al. Nitric oxide increases cyclic GMP levels, AMP-activated protein kinase (AMPK)alpha1-specific activity and glucose transport in human skeletal muscle. Diabetologia. 2010;53(6):1142–1150. doi: 10.1007/s00125-010-1716-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts CK, Barnard RJ, Scheck SH, Balon TW. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. American Journal of Physiology-Endocrinology and Metabolism. 1997/07/01 1997;273(1):E220–E225. doi: 10.1152/ajpendo.1997.273.1.E220 [DOI] [PubMed] [Google Scholar]
- 52.Tian Y, Ding Y, Liu J, et al. Nitric Oxide–Mediated Regulation of GLUT by T3 and Follicle-Stimulating Hormone in Rat Granulosa Cells. Endocrinology. 2017;158(6):1898–1915. doi: 10.1210/en.2016-1864 [DOI] [PubMed] [Google Scholar]
- 53.Li J, Hu X, Selvakumar P, et al. Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab. Nov 2004;287(5):E834–41. doi: 10.1152/ajpendo.00234.2004 [DOI] [PubMed] [Google Scholar]
- 54.Fryer LG, Hajduch E, Rencurel F, et al. Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes. 2000;49(12):1978. doi: 10.2337/diabetes.49.12.1978 [DOI] [PubMed] [Google Scholar]
- 55.Lira VA, Soltow QA, Long JHD, Betters JL, Sellman JE, Criswell DS. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. American Journal of Physiology-Endocrinology and Metabolism. 2007/10/01 2007;293(4):E1062–E1068. doi: 10.1152/ajpendo.00045.2007 [DOI] [PubMed] [Google Scholar]
- 56.Shearer J, Fueger PT, Vorndick B, et al. AMP Kinase-Induced Skeletal Muscle Glucose But Not Long-Chain Fatty Acid Uptake Is Dependent on Nitric Oxide. Diabetes. 2004;53(6):1429. doi: 10.2337/diabetes.53.6.1429 [DOI] [PubMed] [Google Scholar]
- 57.Chen Z-P, Mitchelhill KI, Michell BJ, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Letters. 1999/01/29/ 1999;443(3):285–289. doi: 10.1016/S0014-5793(98)01705-0 [DOI] [PubMed] [Google Scholar]
- 58.Kellogg DL 3rd, McCammon KM, Hinchee-Rodriguez KS, Adamo ML, Roman LJ. Neuronal nitric oxide synthase mediates insulin- and oxidative stress-induced glucose uptake in skeletal muscle myotubes. Free Radic Biol Med. 2017;110:261–269. doi: 10.1016/j.freeradbiomed.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lira VA, Brown DL, Lira AK, et al. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. The Journal of physiology. 2010;588(Pt 18):3551–3566. doi: 10.1113/jphysiol.2010.194035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horton RA, Ceppi ED, Knowles RG, Titheradge MA. Inhibition of hepatic gluconeogenesis by nitric oxide: a comparison with endotoxic shock. Biochemical Journal. 1994;299(3):735–739. doi: 10.1042/bj2990735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sprangers F, Sauerwein PH, Romijn AJ, van Woerkom MG, Meijer JA. Nitric oxide inhibits glycogen synthesis in isolated rat hepatocytes. Biochemical Journal. 1998;330(2):1045–1049. doi: 10.1042/bj3301045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abudukadier A, Fujita Y, Obara A, et al. Tetrahydrobiopterin has a glucose-lowering effect by suppressing hepatic gluconeogenesis in an endothelial nitric oxide synthase-dependent manner in diabetic mice. Diabetes. 2013;62(9):3033–3043. doi: 10.2337/db12-1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. Jan 2004;6(1):45–51. doi: 10.1038/ncb1080 [DOI] [PubMed] [Google Scholar]
- 64.Young ME, Radda GK, Leighton B. Nitric oxide stimulates glucose transport and metabolism in rat skeletal muscle in vitro. Biochemical Journal. 1997;322(1):223–228. doi: 10.1042/bj3220223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu WJ, Haynes TE, Kohli R, et al. Dietary L-Arginine Supplementation Reduces Fat Mass in Zucker Diabetic Fatty Rats. The Journal of Nutrition. 2005;135(4):714–721. doi: 10.1093/jn/135.4.714 [DOI] [PubMed] [Google Scholar]
- 66.García-Villafranca J, Guillén A, Castro J. Involvement of nitric oxide/cyclic GMP signaling pathway in the regulation of fatty acid metabolism in rat hepatocytes. Biochemical Pharmacology. 2003/03/01/ 2003;65(5):807–812. doi: 10.1016/S0006-2952(02)01623-4 [DOI] [PubMed] [Google Scholar]
- 67.Khedara A, Kawai Y, Kayashita J, Kato N. Feeding Rats the Nitric Oxide Synthase Inhibitor, L-NωNitroarginine, Elevates Serum Triglyceride and Cholesterol and Lowers Hepatic Fatty Acid Oxidation. The Journal of Nutrition. 1996;126(10):2563–2567. doi: 10.1093/jn/126.10.2563 [DOI] [PubMed] [Google Scholar]
- 68.Ashmore T, Roberts LD, Morash AJ, et al. Nitrate enhances skeletal muscle fatty acid oxidation via a nitric oxide-cGMP-PPAR-mediated mechanism. BMC Biol. Dec 22 2015;13:110. doi: 10.1186/s12915-015-0221-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frühbeck G, Gómez-Ambrosi J. Modulation of the leptin-induced white adipose tissue lipolysis by nitric oxide. Cellular Signalling. 2001/11/01/ 2001;13(11):827–833. doi: 10.1016/S0898-6568(01)00211-X [DOI] [PubMed] [Google Scholar]
- 70.Mehebik N, Jaubert AM, Sabourault D, Giudicelli Y, Ribière C. Leptin-induced nitric oxide production in white adipocytes is mediated through PKA and MAP kinase activation. Am J Physiol Cell Physiol. Aug 2005;289(2):C379–87. doi: 10.1152/ajpcell.00320.2004 [DOI] [PubMed] [Google Scholar]
- 71.Niang F, Benelli C, Ribière C, et al. Leptin Induces Nitric Oxide-Mediated Inhibition of Lipolysis and Glyceroneogenesis in Rat White Adipose Tissue. The Journal of Nutrition. 2011;141(1):4–9. doi: 10.3945/jn.110.125765 [DOI] [PubMed] [Google Scholar]
- 72.Penfornis P, Marette A. Inducible nitric oxide synthase modulates lipolysis in adipocytes. J Lipid Res. Jan 2005;46(1):135–42. doi: 10.1194/jlr.M400344-JLR200 [DOI] [PubMed] [Google Scholar]
- 73.Rosas-Ballina M, Guan XL, Schmidt A, Bumann D. Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Frontiers in immunology. 2020;11:131–131. doi: 10.3389/fimmu.2020.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klatt P, Cacho J, Crespo MD, Herrera E, Ramos P. Nitric oxide inhibits isoproterenol-stimulated adipocyte lipolysis through oxidative inactivation of the beta-agonist. Biochem J. 2000;351 Pt 2(Pt 2):485–493. [PMC free article] [PubMed] [Google Scholar]
- 75.Adam L, Bouvier M, Jones TLZ. Nitric Oxide Modulates Adrenergic Receptor Palmitoylation and Signaling *. Journal of Biological Chemistry. 1999;274(37):26337–26343. doi: 10.1074/jbc.274.37.26337 [DOI] [PubMed] [Google Scholar]
- 76.Mullins GR, Wang L, Raje V, et al. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proceedings of the National Academy of Sciences. 2014;111(49):17450. doi: 10.1073/pnas.1410530111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poderoso JJ, Helfenberger K, Poderoso C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide. 2019/07/01/ 2019;88:61–72. doi: 10.1016/j.niox.2019.04.005 [DOI] [PubMed] [Google Scholar]
- 78.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. Jun 23 1998;95(13):7631–6. doi: 10.1073/pnas.95.13.7631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Borutaite V, Budriunaite A, Brown GC. Reversal of nitric oxide-, peroxynitrite- and S-nitrosothiol-induced inhibition of mitochondrial respiration or complex I activity by light and thiols. Biochim Biophys Acta. Aug 15 2000;1459(2–3):405–12. doi: 10.1016/s0005-2728(00)00178-x [DOI] [PubMed] [Google Scholar]
- 80.Riobó NA, Clementi E, Melani M, et al. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem J. Oct 1 2001;359(Pt 1):139–45. doi: 10.1042/0264-6021:3590139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. Apr 14 2006;281(15):10056–65. doi: 10.1074/jbc.M512203200 [DOI] [PubMed] [Google Scholar]
- 82.Galkin A, Moncada S. Nitrosation of Mitochondrial Complex I Depends on Its Structural Conformation Journal of Biological Chemistry. 2007;282(52):37448–37453. doi: 10.1074/jbc.M707543200 [DOI] [PubMed] [Google Scholar]
- 83.Iglesias DE, Bombicino SS, Valdez LB, Boveris A. Nitric oxide interacts with mitochondrial complex III producing antimycin-like effects. Free Radical Biology and Medicine. 2015/12/01/ 2015;89:602–613. doi: 10.1016/j.freeradbiomed.2015.08.024 [DOI] [PubMed] [Google Scholar]
- 84.Poderoso JJ, Carreras MaC, Lisdero C, Riobó N, Schöpfer F, Boveris A. Nitric Oxide Inhibits Electron Transfer and Increases Superoxide Radical Production in Rat Heart Mitochondria and Submitochondrial Particles. Archives of Biochemistry and Biophysics. 1996/04/01/ 1996;328(1):85–92. doi: 10.1006/abbi.1996.0146 [DOI] [PubMed] [Google Scholar]
- 85.Sarti P, Forte E, Mastronicola D, Giuffrè A, Arese M. Cytochrome c oxidase and nitric oxide in action: Molecular mechanisms and pathophysiological implications. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2012/04/01/ 2012;1817(4):610–619. doi: 10.1016/j.bbabio.2011.09.002 [DOI] [PubMed] [Google Scholar]
- 86.Taylor CT, Moncada S. Nitric Oxide, Cytochrome C Oxidase, and the Cellular Response to Hypoxia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010/04/01 2010;30(4):643–647. doi: 10.1161/ATVBAHA.108.181628 [DOI] [PubMed] [Google Scholar]
- 87.Mason MG, Nicholls P, Wilson MT, Cooper CE. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(3):708. doi: 10.1073/pnas.0506562103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cooper CE, Torres J, Sharpe MA, Wilson MT. Nitric oxide ejects electrons from the binuclear centre of cytochrome c oxidase by reacting with oxidised copper: a general mechanism for the interaction of copper proteins with nitric oxide? FEBS Lett. Sep 8 1997;414(2):281–4. doi: 10.1016/s0014-5793(97)01009-0 [DOI] [PubMed] [Google Scholar]
- 89.Sarti P, Giuffré A, Forte E, Mastronicola D, Barone MC, Brunori M. Nitric oxide and cytochrome c oxidase: mechanisms of inhibition and NO degradation. Biochem Biophys Res Commun. Jul 21 2000;274(1):183–7. doi: 10.1006/bbrc.2000.3117 [DOI] [PubMed] [Google Scholar]
- 90.Sarti P, Forte E, Giuffrè A, Mastronicola D, Magnifico MC, Arese M. The Chemical Interplay between Nitric Oxide and Mitochondrial Cytochrome c Oxidase: Reactions, Effectors and Pathophysiology. International Journal of Cell Biology. 2012/07/01 2012;2012:571067. doi: 10.1155/2012/571067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. https://doi.org/10.1016/0014-5793(94)01290-3. FEBS Letters. 1994/12/19 1994;356(2–3):295–298. doi: 10.1016/0014-5793(94)01290-3 [DOI] [PubMed] [Google Scholar]
- 92.Cleeter MWJ, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AHV. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. https://doi.org/10.1016/0014-5793(94)00424-2. FEBS Letters. 1994/05/23 1994;345(1):50–54. doi: 10.1016/0014-5793(94)00424-2 [DOI] [PubMed] [Google Scholar]
- 93.Schweizer M, Richter C. Nitric Oxide Potently and Reversibly Deenergizes Mitochondria at Low Oxygen Tension. Biochemical and Biophysical Research Communications. 1994/10/15/ 1994;204(1):169–175. doi: 10.1006/bbrc.1994.2441 [DOI] [PubMed] [Google Scholar]
- 94.Shiva S, Oh J-Y, Landar AL, et al. Nitroxia: The pathological consequence of dysfunction in the nitric oxide–cytochrome c oxidase signaling pathway. Free Radical Biology and Medicine. 2005/02/01/ 2005;38(3):297–306. doi: 10.1016/j.freeradbiomed.2004.10.037 [DOI] [PubMed] [Google Scholar]
- 95.Gladwin MT, Shiva S. The Ligand Binding Battle at Cytochrome c Oxidase. Circulation Research. 2009/05/22 2009;104(10):1136–1138. doi: 10.1161/CIRCRESAHA.109.198911 [DOI] [PubMed] [Google Scholar]
- 96.Thomas DD. Breathing new life into nitric oxide signaling: A brief overview of the interplay between oxygen and nitric oxide. Redox biology. 2015;5:225–233. doi: 10.1016/j.redox.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Borutaite V, Morkuniene R, Brown GC. Release of cytochrome c from heart mitochondria is induced by high Ca2+ and peroxynitrite and is responsible for Ca2+-induced inhibition of substrate oxidation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1999/01/06/ 1999;1453(1):41–48. doi: 10.1016/S0925-4439(98)00082-9 [DOI] [PubMed] [Google Scholar]
- 98.Brookes PS, Salinas EP, Darley-Usmar K, et al. Concentration-dependent Effects of Nitric Oxide on Mitochondrial Permeability Transition and Cytochrome c Release *. Journal of Biological Chemistry. 2000;275(27):20474–20479. doi: 10.1074/jbc.M001077200 [DOI] [PubMed] [Google Scholar]
- 99.Leite ACR, Oliveira HCF, Utino FL, et al. Mitochondria generated nitric oxide protects against permeability transition via formation of membrane protein S-nitrosothiols. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2010/06/01/ 2010;1797(6):1210–1216. doi: 10.1016/j.bbabio.2010.01.034 [DOI] [PubMed] [Google Scholar]
- 100.Whiteman M, Chua YL, Zhang D, Duan W, Liou Y-C, Armstrong JS. Nitric oxide protects against mitochondrial permeabilization induced by glutathione depletion: Role of S-nitrosylation? Biochemical and Biophysical Research Communications. 2006/01/06/ 2006;339(1):255–262. doi: 10.1016/j.bbrc.2005.10.200 [DOI] [PubMed] [Google Scholar]
- 101.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brand MD, Pakay JL, Ocloo A, et al. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392(Pt 2):353–362. doi: 10.1042/BJ20050890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parker N, Vidal-Puig A, Brand MD. Stimulation of mitochondrial proton conductance by hydroxynonenal requires a high membrane potential. Biosci Rep. 2008;28(2):83–88. doi: 10.1042/BSR20080002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wynne AG, Affourtit C. Nitrite lowers the oxygen cost of ATP supply in cultured skeletal muscle cells by stimulating the rate of glycolytic ATP synthesis. PLoS One. 2022;17(8):e0266905. doi: 10.1371/journal.pone.0266905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Larsen FJ, Weitzberg E, Lundberg JO, Ekblom B. Effects of dietary nitrate on oxygen cost during exercise. Acta Physiol (Oxf). Sep 2007;191(1):59–66. doi: 10.1111/j.1748-1716.2007.01713.x [DOI] [PubMed] [Google Scholar]
- 106.Larsen FJ, Weitzberg E, Lundberg JO, Ekblom B. Dietary nitrate reduces maximal oxygen consumption while maintaining work performance in maximal exercise. Free Radical Biology and Medicine. 2010/01/15/ 2010;48(2):342–347. doi: 10.1016/j.freeradbiomed.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 107.Bailey SJ, Winyard P, Vanhatalo A, et al. Dietary nitrate supplementation reduces the O2 cost of low-intensity exercise and enhances tolerance to high-intensity exercise in humans. J Appl Physiol (1985). Oct 2009;107(4):1144–55. doi: 10.1152/japplphysiol.00722.2009 [DOI] [PubMed] [Google Scholar]
- 108.Bailey SJ, Fulford J, Vanhatalo A, et al. Dietary nitrate supplementation enhances muscle contractile efficiency during knee-extensor exercise in humans. J Appl Physiol (1985). Jul 2010;109(1):135–48. doi: 10.1152/japplphysiol.00046.2010 [DOI] [PubMed] [Google Scholar]
- 109.Lansley KE, Winyard PG, Bailey SJ, et al. Acute Dietary Nitrate Supplementation Improves Cycling Time Trial Performance. Medicine & Science in Sports & Exercise. 2011;43(6) [DOI] [PubMed] [Google Scholar]
- 110.Wylie LJ, Kelly J, Bailey SJ, et al. Beetroot juice and exercise: pharmacodynamic and dose-response relationships. Journal of Applied Physiology. 2013/08/01 2013;115(3):325–336. doi: 10.1152/japplphysiol.00372.2013 [DOI] [PubMed] [Google Scholar]
- 111.Kelly J, Vanhatalo A, Wilkerson DP, Wylie LJ, Jones AM. Effects of nitrate on the power-duration relationship for severe-intensity exercise. Med Sci Sports Exerc. Sep 2013;45(9):1798–806. doi: 10.1249/MSS.0b013e31828e885c [DOI] [PubMed] [Google Scholar]
- 112.Vanhatalo A, Bailey SJ, Blackwell JR, et al. Acute and chronic effects of dietary nitrate supplementation on blood pressure and the physiological responses to moderate-intensity and incremental exercise. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2010/10/01 2010;299(4):R1121–R1131. doi: 10.1152/ajpregu.00206.2010 [DOI] [PubMed] [Google Scholar]
- 113.Cermak NM, Gibala MJ, van Loon LJ. Nitrate supplementation’s improvement of 10-km time-trial performance in trained cyclists. Int J Sport Nutr Exerc Metab. Feb 2012;22(1):64–71. doi: 10.1123/ijsnem.22.1.64 [DOI] [PubMed] [Google Scholar]
- 114.Porcelli S, Ramaglia M, Bellistri G, et al. Aerobic Fitness Affects the Exercise Performance Responses to Nitrate Supplementation. Medicine & Science in Sports & Exercise. 2015;47(8) [DOI] [PubMed] [Google Scholar]
- 115.Muggeridge DJ, Howe CC, Spendiff O, Pedlar C, James PE, Easton C. The effects of a single dose of concentrated beetroot juice on performance in trained flatwater kayakers. Int J Sport Nutr Exerc Metab. Oct 2013;23(5):498–506. doi: 10.1123/ijsnem.23.5.498 [DOI] [PubMed] [Google Scholar]
- 116.Peeling P, Cox GR, Bullock N, Burke LM. Beetroot Juice Improves On-Water 500 M Time-Trial Performance, and Laboratory-Based Paddling Economy in National and International-Level Kayak Athletes. Int J Sport Nutr Exerc Metab. Jun 2015;25(3):278–84. doi: 10.1123/ijsnem.2014-0110 [DOI] [PubMed] [Google Scholar]
- 117.Bond H, Morton L, Braakhuis AJ. Dietary nitrate supplementation improves rowing performance in well-trained rowers. Int J Sport Nutr Exerc Metab. Aug 2012;22(4):251–6. doi: 10.1123/ijsnem.22.4.251 [DOI] [PubMed] [Google Scholar]
- 118.Whitfield J, Ludzki A, Heigenhauser GJ, et al. Beetroot juice supplementation reduces whole body oxygen consumption but does not improve indices of mitochondrial efficiency in human skeletal muscle. J Physiol. Jan 15 2016;594(2):421–35. doi: 10.1113/JP270844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ntessalen M, Procter NEK, Schwarz K, et al. Inorganic nitrate and nitrite supplementation fails to improve skeletal muscle mitochondrial efficiency in mice and humans. Am J Clin Nutr. Jan 1 2020;111(1):79–89. doi: 10.1093/ajcn/nqz245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nabben M, Schmitz JPJ, Ciapaite J, et al. Dietary nitrate does not reduce oxygen cost of exercise or improve muscle mitochondrial function in patients with mitochondrial myopathy. Am J Physiol Regul Integr Comp Physiol. May 1 2017;312(5):R689–R701. doi: 10.1152/ajpregu.00264.2016 [DOI] [PubMed] [Google Scholar]
- 121.Woessner MN, Neil C, Saner NJ, et al. Effect of inorganic nitrate on exercise capacity, mitochondria respiration, and vascular function in heart failure with reduced ejection fraction. J Appl Physiol (1985). May 1 2020;128(5):1355–1364. doi: 10.1152/japplphysiol.00850.2019 [DOI] [PubMed] [Google Scholar]
- 122.Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. Sep 15 2011;15(6):1517–30. doi: 10.1089/ars.2010.3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Buetler TM, Krauskopf A, Ruegg UT. Role of superoxide as a signaling molecule. News Physiol Sci. Jun 2004;19:120–3. doi: 10.1152/nips.01514.2003 [DOI] [PubMed] [Google Scholar]
- 124.Wang Y, Branicky R, Noe A, Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. Jun 4 2018;217(6):1915–1928. doi: 10.1083/jcb.201708007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. Jan 2012;5(1):9–19. doi: 10.1097/WOX.0b013e3182439613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. Jul 25 2016;15(1):71. doi: 10.1186/s12937-016-0186-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hayyan M, Hashim MA, AlNashef IM. Superoxide Ion: Generation and Chemical Implications. Chem Rev. Mar 9 2016;116(5):3029–85. doi: 10.1021/acs.chemrev.5b00407 [DOI] [PubMed] [Google Scholar]
- 128.Radi R Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc Natl Acad Sci U S A. Jun 5 2018;115(23):5839–5848. doi: 10.1073/pnas.1804932115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miriyala S, Holley AK, St Clair DK. Mitochondrial superoxide dismutase--signals of distinction. Anticancer Agents Med Chem. Feb 2011;11(2):181–90. doi: 10.2174/187152011795255920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. Aug 1 2002;33(3):337–49. doi: 10.1016/s0891-5849(02)00905-x [DOI] [PubMed] [Google Scholar]
- 131.McCord JM, Keele BB Jr., Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci U S A. May 1971;68(5):1024–7. doi: 10.1073/pnas.68.5.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569. doi: 10.1016/j.cell.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao Y, Hu X, Liu Y, et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer. Apr 13 2017;16(1):79. doi: 10.1186/s12943-017-0648-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am J Physiol Cell Physiol. Jan 2009;296(1):C116–23. doi: 10.1152/ajpcell.00267.2007 [DOI] [PubMed] [Google Scholar]
- 135.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. Jan 2011;21(1):103–15. doi: 10.1038/cr.2010.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem. May 2015;116(5):696–703. doi: 10.1002/jcb.25074 [DOI] [PubMed] [Google Scholar]
- 137.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. Mar 1 2009;418(2):261–75. doi: 10.1042/BJ20082055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. Sep 2 2011;13(9):1016–23. doi: 10.1038/ncb2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oakhill JS, Steel R, Chen ZP, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. Jun 17 2011;332(6036):1433–5. doi: 10.1126/science.1200094 [DOI] [PubMed] [Google Scholar]
- 140.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nature Reviews Molecular Cell Biology. 2018/02/01 2018;19(2):121–135. doi: 10.1038/nrm.2017.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. Nov 2 1987;223(2):217–22. doi: 10.1016/0014-5793(87)80292-2 [DOI] [PubMed] [Google Scholar]
- 142.Sato R, Goldstein JL, Brown MS. Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. Proc Natl Acad Sci U S A. Oct 15 1993;90(20):9261–5. doi: 10.1073/pnas.90.20.9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. Jul 2008;295(1):E29–37. doi: 10.1152/ajpendo.90331.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. Feb 2018;19(2):121–135. doi: 10.1038/nrm.2017.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bergeron R, Ren JM, Cadman KS, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. Dec 2001;281(6):E1340–6. doi: 10.1152/ajpendo.2001.281.6.E1340 [DOI] [PubMed] [Google Scholar]
- 146.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. Mar 20 1998;92(6):829–39. doi: 10.1016/s0092-8674(00)81410-5 [DOI] [PubMed] [Google Scholar]
- 147.Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. Jul 9 1999;98(1):115–24. doi: 10.1016/S0092-8674(00)80611-X [DOI] [PubMed] [Google Scholar]
- 148.Choi HC, Song P, Xie Z, et al. Reactive Nitrogen Species Is Required for the Activation of the AMP-activated Protein Kinase by Statin in Vivo*. Journal of Biological Chemistry. 2008;283(29):20186–20197. doi: 10.1074/jbc.M803020200 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 149.Liang H, Ward WF. PGC-1α: a key regulator of energy metabolism. Advances in Physiology Education. 2006/12/01 2006;30(4):145–151. doi: 10.1152/advan.00052.2006 [DOI] [PubMed] [Google Scholar]
- 150.Gureev AP, Shaforostova EA, Popov VN. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Review. Frontiers in Genetics. 2019-May-14 2019;10(435)doi: 10.3389/fgene.2019.00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pilon G, Dallaire P, Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem. May 14 2004;279(20):20767–74. doi: 10.1074/jbc.M401390200 [DOI] [PubMed] [Google Scholar]
- 152.Lira VA, Soltow QA, Long JH, Betters JL, Sellman JE, Criswell DS. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am J Physiol Endocrinol Metab. Oct 2007;293(4):E1062–8. doi: 10.1152/ajpendo.00045.2007 [DOI] [PubMed] [Google Scholar]
- 153.Chen ZP, Mitchelhill KI, Michell BJ, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. Jan 29 1999;443(3):285–9. doi: 10.1016/s0014-5793(98)01705-0 [DOI] [PubMed] [Google Scholar]
- 154.Chen ZP, Stephens TJ, Murthy S, et al. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes. Sep 2003;52(9):2205–12. doi: 10.2337/diabetes.52.9.2205 [DOI] [PubMed] [Google Scholar]
- 155.Kang C, Li Ji L. Role of PGC-1alpha signaling in skeletal muscle health and disease. Ann N Y Acad Sci. Oct 2012;1271:110–7. doi: 10.1111/j.1749-6632.2012.06738.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. Aug 15 2002;418(6899):797–801. doi: 10.1038/nature00904 [DOI] [PubMed] [Google Scholar]
- 157.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. Feb 2003;24(1):78–90. doi: 10.1210/er.2002-0012 [DOI] [PubMed] [Google Scholar]
- 158.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. Jul 2003;34(3):267–73. doi: 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- 159.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. Apr 2008;88(2):611–38. doi: 10.1152/physrev.00025.2007 [DOI] [PubMed] [Google Scholar]
- 160.Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol Metab. Oct 2001;12(8):360–5. doi: 10.1016/s1043-2760(01)00457-x [DOI] [PubMed] [Google Scholar]
- 161.Russell AP. PGC-1alpha and exercise: important partners in combating insulin resistance. Curr Diabetes Rev. May 2005;1(2):175–81. doi: 10.2174/1573399054022811 [DOI] [PubMed] [Google Scholar]
- 162.Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care. May 2001;4(3):183–90. doi: 10.1097/00075197-200105000-00003 [DOI] [PubMed] [Google Scholar]
- 163.Geng T, Li P, Okutsu M, et al. PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol. Mar 2010;298(3):C572–9. doi: 10.1152/ajpcell.00481.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Baar K, Wende AR, Jones TE, et al. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. Dec 2002;16(14):1879–86. doi: 10.1096/fj.02-0367com [DOI] [PubMed] [Google Scholar]
- 165.Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. Sep 2006;20(11):1889–91. doi: 10.1096/fj.05-5189fje [DOI] [PubMed] [Google Scholar]
- 166.Lindholm ME, Rundqvist H. Skeletal muscle hypoxia-inducible factor-1 and exercise. Exp Physiol. Jan 2016;101(1):28–32. doi: 10.1113/EP085318 [DOI] [PubMed] [Google Scholar]
- 167.Stroka DM, Burkhardt T, Desbaillets I, et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. Nov 2001;15(13):2445–53. doi: 10.1096/fj.01-0125com [DOI] [PubMed] [Google Scholar]
- 168.Tang K, Breen EC, Wagner H, Brutsaert TD, Gassmann M, Wagner PD. HIF and VEGF relationships in response to hypoxia and sciatic nerve stimulation in rat gastrocnemius. Respir Physiol Neurobiol. Nov 30 2004;144(1):71–80. doi: 10.1016/j.resp.2004.04.009 [DOI] [PubMed] [Google Scholar]
- 169.Olson N, van der Vliet A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide. Aug 1 2011;25(2):125–37. doi: 10.1016/j.niox.2010.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mateo J, Garcia-Lecea M, Cadenas S, Hernandez C, Moncada S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J. Dec 1 2003;376(Pt 2):537–44. doi: 10.1042/BJ20031155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Muangritdech N, Hamlin MJ, Sawanyawisuth K, et al. Hypoxic training improves blood pressure, nitric oxide and hypoxia-inducible factor-1 alpha in hypertensive patients. Eur J Appl Physiol. Aug 2020;120(8):1815–1826. doi: 10.1007/s00421-020-04410-9 [DOI] [PubMed] [Google Scholar]
- 172.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. Sep 23 1994;269(38):23757–63. [PubMed] [Google Scholar]
- 173.Semenza GL, Agani F, Booth G, et al. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. Feb 1997;51(2):553–5. doi: 10.1038/ki.1997.77 [DOI] [PubMed] [Google Scholar]
- 174.Dikalov SI, Mayorov VI, Panov AV. Physiological Levels of Nitric Oxide Diminish Mitochondrial Superoxide. Potential Role of Mitochondrial Dinitrosyl Iron Complexes and Nitrosothiols. Frontiers in physiology. 2017;8:907–907. doi: 10.3389/fphys.2017.00907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bouviere J, Fortunato RS, Dupuy C, Werneck-de-Castro JP, Carvalho DP, Louzada RA. Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants. 2021;10(4)doi: 10.3390/antiox10040537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.He F, Li J, Liu Z, Chuang C-C, Yang W, Zuo L. Redox Mechanism of Reactive Oxygen Species in Exercise. Frontiers in physiology. 2016;7:486–486. doi: 10.3389/fphys.2016.00486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Trewin AJ, Berry BJ, Wojtovich AP. Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants (Basel). 2018;7(1):7. doi: 10.3390/antiox7010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Goncalves RLS, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem. 2015;290(1):209–227. doi: 10.1074/jbc.M114.619072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sahlin K, Shabalina IG, Mattsson CM, et al. Ultraendurance exercise increases the production of reactive oxygen species in isolated mitochondria from human skeletal muscle. Journal of Applied Physiology. 2010/04/01 2010;108(4):780–787. doi: 10.1152/japplphysiol.00966.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pearson T, Kabayo T, Ng R, Chamberlain J, McArdle A, Jackson MJ. Skeletal muscle contractions induce acute changes in cytosolic superoxide, but slower responses in mitochondrial superoxide and cellular hydrogen peroxide. PLoS One. 2014;9(5):e96378. doi: 10.1371/journal.pone.0096378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Poderoso JJ, Peralta JG, Lisdero CL, et al. Nitric oxide regulates oxygen uptake and hydrogen peroxide release by the isolated beating rat heart. American Journal of Physiology-Cell Physiology. 1998/01/01 1998;274(1):C112–C119. doi: 10.1152/ajpcell.1998.274.1.C112 [DOI] [PubMed] [Google Scholar]
- 182.Whyte JJ, Laughlin MH. The effects of acute and chronic exercise on the vasculature. Acta Physiol (Oxf). Aug 2010;199(4):441–50. doi: 10.1111/j.1748-1716.2010.02127.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nosarev AV, Smagliy LV, Anfinogenova Y, Popov SV, Kapilevich LV. Exercise and NO production: relevance and implications in the cardiopulmonary system. Front Cell Dev Biol. 2015;2:73–73. doi: 10.3389/fcell.2014.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Guizoni DM, Dorighello GG, Oliveira HCF, Delbin MA, Krieger MH, Davel AP. Aerobic exercise training protects against endothelial dysfunction by increasing nitric oxide and hydrogen peroxide production in LDL receptor-deficient mice. Journal of Translational Medicine. 2016/07/19 2016;14(1):213. doi: 10.1186/s12967-016-0972-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Calbet JAL, Martín-Rodríguez S, Martin-Rincon M, Morales-Alamo D. An integrative approach to the regulation of mitochondrial respiration during exercise: Focus on high-intensity exercise. Redox Biology. 2020/08/01/ 2020;35:101478. doi: 10.1016/j.redox.2020.101478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Powers SK, Deminice R, Ozdemir M, Yoshihara T, Bomkamp MP, Hyatt H. Exercise-induced oxidative stress: Friend or foe? Journal of Sport and Health Science. 2020/09/01/ 2020;9(5):415–425. doi: 10.1016/j.jshs.2020.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]