

Abstract
Bruton tyrosine kinase inhibitors (BTKi) have transformed the therapeutic landscape of chronic lymphocytic leukemia (CLL) and Non-Hodgkin lymphoma (NHL). However, primary and acquired resistance to BTKi can be seen due to a variety of mechanisms including tumor intrinsic and extrinsic mechanisms such as gene mutations, activation of bypass signaling pathways, and tumor microenvironment. Herein, we provide an updated review of the key clinical data of BTKi treatment in CLL, mantle cell lymphoma (MCL), and diffuse large B-cell lymphoma (DLBCL). We incorporate the most recent findings regarding mechanisms of resistance to covalent and non-covalent inhibitors, including ibrutinib, acalabrutinib, zanubrutinib and pirtobrutinib. We also cover the clinical sensitivity of certain molecular subtypes of DLBCL to an ibrutinib-containing regimen. Lastly, we summarize ongoing clinical investigations aimed at overcoming resistance via use of BTKi-containing combination therapies or the novel non-covalent BTK inhibitors. The review article targets an audience of clinical practitioners, clinical investigators, and translational researchers.
Keywords: B-cell receptor, BTK, ibrutinib, CLL, non-Hodgkin lymphoma
Short Summary
BTK targeted therapy in B cell lymphomas is a fast-moving field. Despite the remarkable efficacy of BTKi, several mechanisms of resistance have been identified that lead to lack of or shortened duration of response. In this review, we will summarize key clinical trials of BTKi in CLL, MCL, and DLBCL as well as the unique mechanisms of resistance within these histological subtypes. We also discuss efforts to overcome resistance via combination targeted therapies and use of novel non-covalent BTKi therapy.
Introduction
B cell non-Hodgkin lymphomas and leukemias make up ~3.5% of all cancer diagnoses in the United States and consist of a heterogeneous group of malignancies. Historically, the backbone of treatment has been cytotoxic chemotherapy, but in recent decades, targeted therapies have been incorporated into earlier line settings. The B-cell receptor (BCR) signaling pathway is a key driver of B cell malignancies (Fig. 1) and agents disrupting this pathway have changed the landscape of management in both the frontline and relapsed/refractory (R/R) settings. A key target is the Bruton tyrosine kinase (BTK), a component of early BCR signaling pathway. In normal B-cells, BTK activation, reflected by its phosphorylation, triggers downstream events and ultimately, the activation of nuclear factor kappa B (NFκB) pathways enabling increased B-cell survival, proliferation, differentiation into plasma cells, and subsequent antibody production [1, 2] (Fig. 1). In this review, we will discuss the use of Bruton tyrosine kinase inhibitors in B cell malignancies and cover the mechanisms of resistance. These understandings not only help improve care and survival of patients treated with BTK inhibitors but also help direct BCR-targeted therapeutic strategies for future clinical trial design.
Fig. 1. Resistance-Relevant mutations and signaling pathways in CLL, MCL and DLBCL.
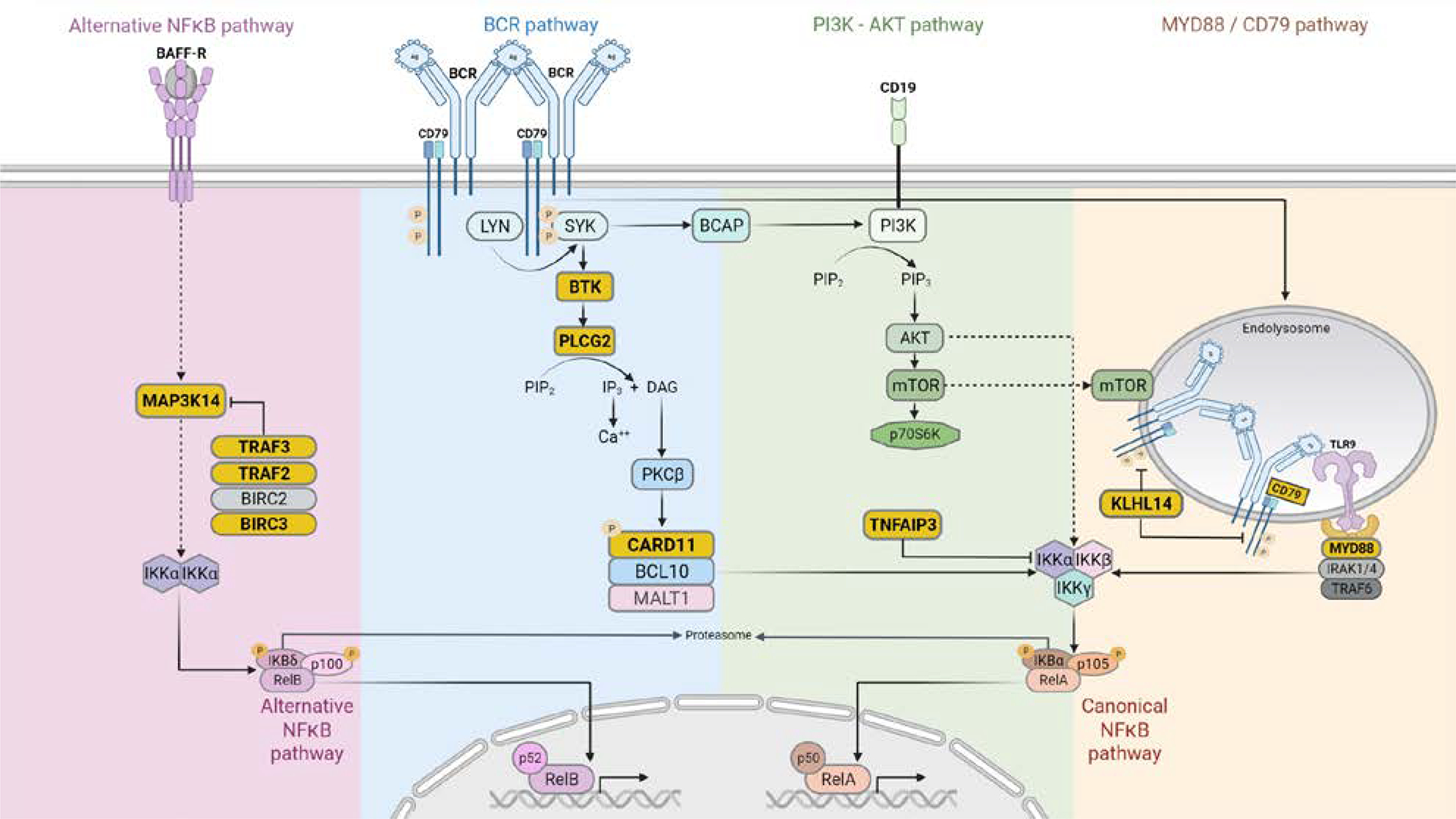
Key components of the signaling pathways including BCR, PI3K-AKT, MYD88/CD79, canonical NFκB and alternative NFκB are depicted. Mutated genes along these pathways associated with BTKi resistance are highlighted in yellow. Also see Table 4 for their relationship with the disease and resistance setting. Positive interactions are indicated by arrows, indirect interactions by dashed arrows, and inhibitory interactions by T-bars. The graph was generated with BioRender.
The first in class BTK inhibitor is ibrutinib, an orally available small molecular inhibitor of the kinase. At the molecular level, the drug binds covalently to the cysteine 481 at the ATP binding site of BTK (Fig. 2) to inhibit its activity and downstream signaling cascade [3, 4]. At the cellular level, the consequence of BCR inhibition is primarily cell proliferation deceleration rather than direct cell killing [5, 6]. Aside from cell proliferation, ibrutinib has demonstrated inhibitory effects on cell adhesion and migration of malignant cells as well as on the tumor microenvironment including T cells and mesenchymal stromal cells [7–11]. On the systemic level, inhibition of cell adhesion results in dislodging of CLL cells from the lymph node and their relocation (or release) to the periphery. Inhibition of cell migration, on the other hand, prevents homing of the circulating CLL cells back into the lymph nodes leading to lymphocytosis [12].
Fig.2. Map of clinically documented BTK mutations.
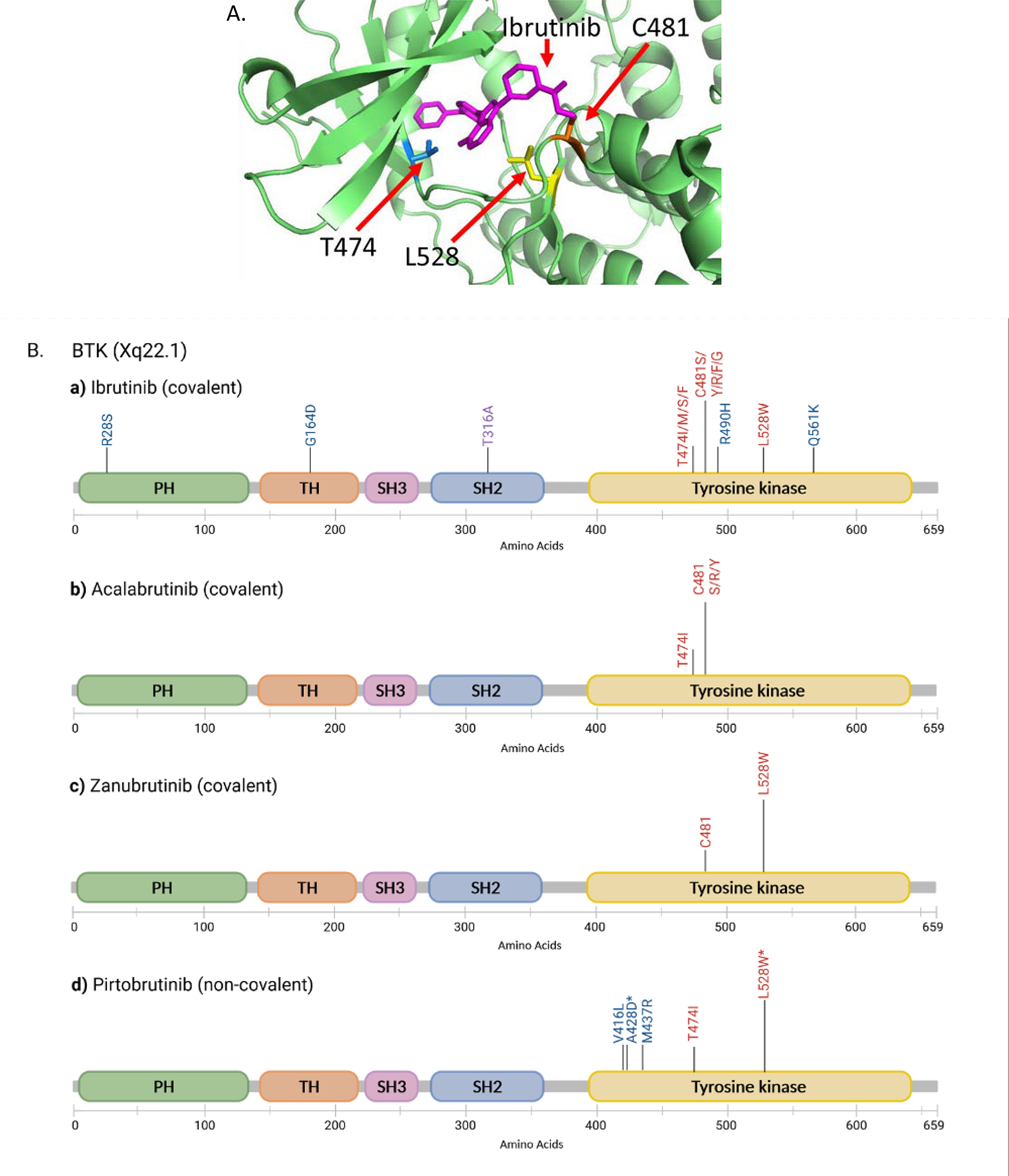
A) 3D mapping of common BTK missense mutations associated with resistance to multiple BTK inhibitors (previously published in Sharma et al, PMID: 27626698). B) Clinically reported BTK mutations in patients treated with ibrutinib, acalabrutinib, zanubrutinib and pirtobrutinib. Note that not all mapped mutations have been functionally validated. PH, pleckstrin homology domain; TH, Tec homology domain; SH, Src homology domain; Tyrosine kinase, tyrosine kinase domain. The height of the vertical bars represents the relevant abundance of the particular variants. *Experimentally predicted to be resistant to multiple BTK inhibitors. The graph was generated with BioRender.
Ibrutinib has been FDA approved for use in various B cell malignancies including MCL, CLL, lymphoplasmacytic lymphoma/ Waldenström’s macroglobulinemia and marginal zone lymphoma (Table 1 summarizes the trials that led to the approval). Ibrutinib was first approved in MCL based on phase II data revealing an overall response rate (ORR) of 68% in the relapsed/refractory setting. This response rate is close to that of highly myelosuppressive salvage chemotherapy but with a much more favorable toxicity profile [13, 14]. In CLL, ibrutinib demonstrated an ORR of 71% in a phase I/II study of heavily pre-treated patients. Phase III data comparing ibrutinib to chemotherapy confirmed this superior rate and durability of response, particularly for those patients with del(17p) and other high-risk genetic features such as presence of del(11q) and unmutated immunoglobulin heavy chain (UM-IGHV) [15, 16]. Notably, ibrutinib does not produce high rates of complete response (CR), probably related to its inability to produce significant apoptotic activity as a monotherapy [5, 7].
Table 1.
FDA Approved Indications for BTKi Therapy
| Clinical Trial that led to FDA Approval | BTKi | Date of approval | Disease Setting |
|---|---|---|---|
| Chronic Lymphocytic Leukemia | |||
| Byrd 2013 [114] | Ibrutinib | 2/12/14 | Relapsed/Refractory disease |
| Byrd 2014 [15] | Ibrutinib | 7/28/14 | Del(17p) disease |
| Burger 2020 [16] | Ibrutinib | 3/4/16 | Treatment Naive |
| Sharman 2020 [57] Ghia 2020 [115] Byrd 2021 [48] |
Acalabrutinib | 11/21/19 | Treatment Naïve and Relapsed/Refractory disease |
| Tam 2019 [116] | Zanubrutinib | Not FDA approved Included in NCCN guidelines | Treatment Naïve and Relapsed/Refractory disease |
| Mantle Cell Lymphoma | |||
| Wang 2012 [13] | Ibrutinib | 11/13/13 | Relapsed/Refractory disease |
| Wang 2018 [68] | Acalabrutinib | 10/31/17 | Relapsed/Refractory disease |
| Tam 2021 [117] | Zanubrutinib | 11/14/19 | Relapsed/Refractory disease |
| DLBCL | |||
| Wilson 2015 [118] | Ibrutinib | Not FDA approved Included in NCCN guidelines | Relapsed/Refractory non-GCB DLBCL |
| Lymphoplasmacytic Lymphoma | |||
| Treon 2015 [119] | Ibrutinib | 1/29/15 | Relapsed/Refractory disease |
| Dimopoulos 2017 [120] | Ibrutinib | 8/27/18 | Treatment Naïve and Relapsed/Refractory disease, with rituximab |
| Tam 2020 [121] | Zanubrutinib | 9/1/21 | Treatment Naïve and Relapsed/Refractory disease |
| Marginal Zone Lymphoma | |||
| Noy 2020 [122] | Ibrutinib | 1/19/17 | Relapsed/Refractory Disease |
| Opat 2020 [123] | Zanubrutinib | 9/15/21 | Relapsed/Refractory disease |
Despite its remarkable clinical efficacy, resistance to ibrutinib does develop. Conceptually, resistance is classified as primary resistance and secondary (acquired resistance) from a clinical perspective. Primary resistance is seen in patients who fail to respond to ibrutinib upfront whereas secondary resistance is seen in patients who initially respond but then relapse. Meanwhile, predisposition to resistance refers to pathological parameters present at baseline. Although they are not associated with upfront failure to respond, rather, they increase the risk of later disease progression while patients are on treatment. From the biological point of view, the mechanisms of resistance can be intrinsic or extrinsic to the tumor cells. Intrinsic mechanisms include mutations and activation of bypass pathways leading to restoration of tumor cell survival and proliferation. Extrinsic factors include protection of tumor cells through cytokines or cell-cell contact with the tumor microenvironment. In this article, we primarily focus on intrinsic mechanisms.
Resistance to ibrutinib may be overcome by other newer BTK inhibitors. As a prototype, ibrutinib is followed by an expanding class of BTK inhibitors with either irreversible (covalent) or reversible (non-covalent) BTK binding mechanism [17]. Second generation covalent BTKi, acalabrutinib and zanubrutinib, have generally more favorable toxicity profiles than ibrutinib but are susceptible to similar resistance mechanisms as ibrutinib [18, 19]. Meanwhile, novel noncovalent BTKi including pirtobrutinib (LOXO-305) and ARQ-531 (MK-1026) do not form a covalent bond with BTK molecule and have demonstrated activity in ibrutinib resistant disease [20, 21].
In this article, we will cover B-cell malignancies including CLL, MCL and DLBCL. We will first review ibrutinib clinical data, then the primary and secondary resistance mechanism. We will summarize results of the clinical trials involving combination therapies and newer BTK inhibitors. The most recent developments in the understanding of resistance to newer BTK inhibitors will be discussed.
Chronic Lymphocytic Leukemia
CLL and small lymphocytic lymphoma (SLL) are a spectrum of a single mature B cell malignancy presenting with predominantly lymphocytosis and/or nodal disease, respectively. Constitutive activation of the BCR signaling pathway and upregulation of the BCL2 anti-apoptotic protein leads to increased cell survival and proliferation. The advent of ibrutinib, has drastically changed the landscape of CLL management in recent years. In addition to inhibiting the BCR pathway, the drug also promotes mobilization of malignant cells out of the protective nodal niche of nodal or bone marrow microenvironment into circulation, leading to decreased lymphadenopathy [22].
Clinical data with ibrutinib
Historically, chemoimmunotherapy (CIT) had been the backbone of treatment in symptomatic patients with CLL. While fixed duration CIT demonstrates a 90% response rate, more than half of patients have disease progression at 5 years on this regimen and treatment carries significant toxicity risk [23].Ibrutinib has demonstrated superior duration of response over CIT, particularly in those with the high-risk features such as del(17p)/TP53, in addition to a more tolerable side effect profile [24, 25] (Table 2). Additionally, high rates of response are seen with BTKi even in patients with multiple prior lines of chemotherapy [25, 26]. However, ibrutinib monotherapy fails to achieve deep responses in 71–90% of patients with CLL due to absence of direct killing [27].
Table 2.
Ibrutinib based Regimens in CLL in the Frontline Setting
| Study | Regimen | N | Phase | PFS (median follow up in months) | uMRD in peripheral blood |
|---|---|---|---|---|---|
| Woyach, 2018 [124] | BR vs Ibr vs Ibr-R | 208 | II | 74% vs 87% vs 88% (24) | 8% vs 1% vs 4% |
| Moreno 2019 [125] | Ibr + O vs Chl + O | 229 | III | 90% vs 31% (30) | 35% vs 25% |
| Shanafelt 2019 [126] | Ibr+R vs FCR | 529 | III | PFS 89.4 vs 72.9% (36) | 8.3 vs 59.2% |
| Tam 2019 [58] Wierda 2021 [60] |
Ibr + Ven | 164 | II | 95% (12)* | 75% |
| Burger 2020 [16] | Ibr vs Chl | 269 | III | 70% vs 12% (60) | N/A |
| Kater 2021 [59] | Ibr + Ven vs Chl-O | 211 | III | NR vs 21 months** | 54.7% vs 39% |
end point of disease free survival
median progression free survival
Ibr: Ibrutinib; R: Rituximab; FCR: Fludarabine/Cyclophosphamide/Rituximab; BR: Bendamustine/Rituximab; O: Obinutuzumab, Chl: Chlorambucil; Acal: acalabrutinib; Ven: Venetoclax; NR: not reached; uMRD: undetectable MRD
Predisposition to future relapse
Primary resistance to ibrutinib develop in 10–16% of cases and the mechanisms are mostly unknown. Predominant baseline CLL mutations in treatment naïve patients are mostly unrelated to the BCR pathway, including mutations in ATM, BIRC3, NOTCH, SF3B1, and TP53 [28, 29]. However, more is known about what genetic characteristics predispose to future relapse. Baseline molecular and cytogenetic features including del(17p)/TP53 and complex karyotype (≥3 chromosomal abnormalities) increase the risk of disease progression in CLL patients treated with ibrutinib [30–32]. TP53 abnormality, in particular, is the only independent molecular factor that was included in a four-factor model that predicts inferior overall survival (OS) and progression-free survival (PFS) on ibrutinib treatment [33]. Thus, patients with del(17p)/TP53 aberrations continue to have worse prognosis even in the era of targeted therapies. In addition, del(18p), occurs in 2–7% of cases in untreated population, was found at a particularly high frequency in 56% (5 of 9 patients) of ibrutinib-relapsed patients and is also associated with the development of BTK mutation [30].
Secondary resistance to ibrutinib
Disease relapse/progression develop in 10–18% of CLL patients [32, 34–36]. In ~70 % of cases, this is due to mutations at C481 residue of BTK which disrupts the covalent binding of ibrutinib to the BTK kinase domain [30, 37–41] (Fig. 2A and major mutation mechanisms summarized in Table 4). The cysteine residue is most commonly mutated to serine (C481S), but mutations to other amino acid residues are also seen in practice, including C481Y/R/F/G (tyrosine arginine/phenylalanine/glycine, approximately in the order of decreasing frequencies) [32, 36, 42, 43].
Table 4.
Major Mutation Mechanisms of Ibrutinib Resistance
| Primary Sensitivity | Primary Resistance/Reduced Sensitivity | Predisposition for Later Progression | Secondary Resistance | |
|---|---|---|---|---|
| CLL | UM-IGHV * | TP53 del/mut, complex cytogenetics Del 18p |
BTK PLCG2 Del 8p/TRAIL-R |
|
| MCL | CARD11 CCND1 TRAF2, TRAF3 & BIRC3 and MAP3K14 |
BTK CCND1 CDKN2A/MTAP |
||
| DLBCL | MCD or N1 molecular subtypes | CARD11 TNFAIP3 KLHL14 PIM1 |
C481 and other mutations are clustered in the tyrosine kinase domain of the BTK protein (Fig. 2B). T316A, however, is located at the Src homology 2 domain. Cells carrying BTK T316A showed resistance to ibrutinib at both cellular and molecular levels to a similar extent as BTK C481S [44]. The functional impacts of other mutations have not yet been demonstrated (Fig. 2B).
The next most common mechanism is gain of function PLCG2 missense mutations, seen in 11% of ibrutinib resistant cases (Fig. 1 and Kadri S et al 2017). PLCG2 is the enzymatic substrate of BTK, its activation enables CLL cell proliferation independent of BTK control [30, 36, 38, 41]. Interestingly, PLCG2 mutations often co-exist with the BTK mutations [30, 36, 38, 41, 45]. In the remaining ~20% of relapsed/progression cases, del (8p) leading to loss of TRAIL-R was reported in 3 of 5 ibrutinib resistant patients [46]. Besides the genetic mechanisms, epigenetic changes have been shown to play a role in conferring the CLL resistance [47].
Secondary resistance to newer covalent BTK inhibitors
Second generation covalent BTK inhibitors have been developed including acalabrutinib and zanubrutinib. These newer BTKi demonstrated similar or improved efficacy and superior toxicity profiles in phase III comparison to ibrutinib in the R/R setting [48, 49]. Resistance to these BTKi is also driven by mutations in the BTK and, to a much less degree, in the downstream enzyme PLCG2. For patients treated with acalabrutinib, the most frequent mutation is BTK C481 [19] (Fig.2B). However, different from ibrutinib, in cases of zanubrutinib, BTK L528W seems to be the more predominant mutation that occurs in more cases and at a much higher variant allele frequency than the C481 mutation [18, 50] (Fig.2 A&B). Interestingly, in this small cohort, L528W may be present in the same cells as the C481 substitutions [50]. Finding multiple BTK mutations in the same patient samples has been reported previously in ibrutinib-relapsed cases as well [30].
Non-covalent BTKi therapies
As mentioned in the Introduction, one way of overcoming resistance mediated by BTK C481 mutation is the use of reversible, noncovalent BTKi such as Pirtobrutinib [20, 21]. Pirtobrutinib is highly selective for BTK and significantly inhibits BTK phosphorylation, cell proliferation and tumor growth in mice [51, 52]. It binds to BTK but does not depend on C481. Therefore, this agent is predicted to overcome ibrutinib resistance. Both pirtobrutinib and ARQ531, another non-covalent BTK inhibitor, are active in CLL/SLL with either C481 mutated or unmutated BTK enzyme [20, 21]. The ORR in multiple relapsed/refractory CLL is 63%, of which the majority had prior BTKi exposure [21]. Ongoing trials evaluating this agent in the front line setting and as part of combination regimens are underway (NCT05023980 and NCT04965493).
Secondary resistance to non-covalent BTKi
Acquired resistance to pirtobrutinib has emerged. Several non-C481mutations were newly acquired in patients with progressive disease. These include V416L, A428D, M437R, T474I, and L528W that are clustered in the tyrosine kinase domain (Fig.2B). Of note, L528W was commonly found in patients resistant to the covalent inhibitor ibrutinib, zanubrutinib, and non-covalent pirtobrutinib. Together with A428D, L528W is predicted by the in vitro assay to be universally resistant to covalent and non-covalent BTK inhibitors as well [53]. Apparently, positions of BTK mutations may be different for different BTK inhibitors. Therefore, sequencing the entire BTK gene rather than hot spot locations may help reveal less frequent mutations, accumulate data for further understanding and foster ongoing research in addressing next line treatment.
Overcoming resistance with combination therapies
Deepening response by achieving undetectable minimal residual disease (uMRD) would perhaps reduce the emergence of resistance. uMRD correlates with improved survival in CLL treated with CIT and venetoclax-based regimens, particularly in patients with high-risk disease such as those with del(17p)/TP53 or del(11q) [54, 55]. While BTKi monotherapy fails to achieve high rates of deep response, this can be achieved with BTKi combination therapies.
Addition of anti-CD20 monoclonal antibody to ibrutinib induces higher rates of undetectable MRD, although it did not demonstrate statistically significant improvements in response or PFS [56]. Similarly, addition of obinutuzumab achieved slightly higher rates of complete response in post hoc analysis of acalabrutinib based treatments which correlated with improvement in PFS [57]. BCL2 inhibitor venetoclax delivers direct killing by enhancing rate of apoptosis of the resting subpopulation of CLL cells [6]. When combined with ibrutinib, the regimen improved rate of uMRD from <10% to 55–75% in the frontline setting [58–60]. While long term survival data is not yet available, patients who achieved uMRD in this study showed a promising 30 month PFS of ≥ 95% [60]. However, it remains to be seen if combination therapies may overcome resistance in patients who progress on BTKi monotherapy. Clinical efficacy of BTKi containing combination therapies is summarized in Table 2 which bring promises for long-term disease control and a potential cure [58–60].
Mantle Cell Lymphoma
MCL is a mature B cell neoplasm characterized by a t(11;14) translocation involving the cyclin D1 (CCND1) cell cycle regulator gene and immunoglobulin heavy chain locus (IGH) leading to overexpression of cyclin D1 [61, 62]. However, presence of other mutations are frequently seen in treatment naïve MCL contributing to the clonal proliferation and resistance mechanisms of this disease [62]. Activation of BCR signaling and overexpression of BTK has been observed in MCL cells that served as the main rationale for use of Ibrutinib [63, 64].
Clinical data
Patients with R/R MCL after standard of care frontline intensive chemotherapy therapy historically have had poor prognosis. Those with TP53-mutated disease have particularly poor outcomes, with a median overall survival (OS) of less than 2 years compared to 10.2 years in those with unmutated wild-type TP53 [62, 65]. While ibrutinib monotherapy has shown efficacy in the R/R setting, 30% of patients fail to respond and 60% achieve only a partial response [66]. Moreover, outcomes after ibrutinib failure are dismal [67]. The second generation covalent irreversible BTKi did not seem to improve outcomes which can be predicted from the chemical binding mechanism [68].
Primary resistance
Primary resistance to ibrutinib has been reported in 32% of patients with MCL [13]. Compared to CLL, patients with MCL have higher rates of high-risk mutations at baseline that predispose to treatment resistance and relapse with CIT. These include the ATM and TP53 gene mutations, seen in 44% and 27% of patients, respectively, according to a meta-analysis of 2045 samples [62]. Several gene mutations are associated with primary ibrutinib resistance in cell lines including CARD11 and CCND1 [69, 70] (Fig.1).
Primary resistance is also mediated by upregulation of other oncogenic pathways, including activation of the alternative NFκB pathway with associated mutations in MAP3K14, TRAF2, TRAF3, BIRC3 [71] (Fig. 1). Other bypass pathways including PI3K/AKT, MEK/ERK, canonical NFκB activation have also been described as a mechanism of primary resistance, as well as ROR1 overexpression and MYC activation mainly based on cell line studies [72–74]. Resistance may also be mediated by BCR-induced upregulation of RAC2, a master control of the cell adhesion program, counteracting ibrutinib’s ability to dislodge malignant cells from nodal stroma [75].
Secondary resistance
With single agent ibrutinib in the R/R setting, 69% of patients progress within 2 years of therapy [14]. Unlike CLL, mutations in BTK are rarely detected at disease progression after ibrutinib [67, 76, 77]. Instead, newly acquired recurrent mutations were found in the cyclin D1 (CCND1) gene and CDKN2A/MTAP genes, closely located on 9p, in a study of small number of ibrutinib resistant patients.
Secondary resistance, similar to primary resistance, mainly involve bypass pathways of the B cell receptor signaling. Activation of PI3K/AKT/mTOR pathway (Fig.1) was consistently identified by several studies in cell line models [64, 78, 79] as well as in a patient-derived xenograft model [78]. In the xenograft model, it is further demonstrated that adding PI3K or mTOR inhibitors to ibrutinib significantly slows down the growth of the ibrutinib-resistant tumor [78]. Moreover, in clinical samples collected from patients, in addition to pathway changes (including mTOR), downstream metabolic reprogramming towards oxidative phosphorylation and glutaminolysis was identified as one of the main changes in ibr-resistant samples versus ibr-sensitive ones [77].
Overcoming ibrutinib resistance with non-covalent BTK inhibitors and combination therapies
Despite the rare occurrence of acquired BTK mutations in MCL, non-covalent BTKi pirtobrutinib has also demonstrated activity in ibrutinib-resistant MCL with an ORR of 52% in R/R cases [21, 80]. This may have to do with a higher target binding and a longer exposure of BTK to the drug [80]. Head-to-head to comparison of this agent versus early generation BTKi in BTK-naïve patients with MCL is ongoing (NCT04662255) (Table 3).
Table 3.
Ibrutinib Based Regimens in MCL in R/R and Frontline Settings
| Study | Regimen | N | Phase | PFS (median follow-up in month) | ORR (CR) |
|---|---|---|---|---|---|
| R/R MCL Single Agent BTKi | |||||
| Wang 2013 [13, 14] | Ibr | 111 | II | 21% (24) | 67% (23%) |
| Dreyling 2016 [127] | Ibr vs. temsirolimus | 280 | III | 14.6 vs. 6.2 m (20)* | 81% (40%) |
| R/R MCL Combination Regimens | |||||
| Wang 2016 [84] | Ibr + R | 50 | II | 69% (15) 75% (12) |
88% (44%) |
| Tam 2018 [83] | Ibr + Ven | 24 | II | 43 m (48) | 71% (71%) |
| Jerkeman 2020 [82] | Ibr + Len + R | 50 | II | 18 m (40) 56.9% (12) |
76% (56%) |
| Lee 2021 [85] | Ibr + cirmtuzumab | 20 | I/II | NR (25) | 90% (35%) |
| Frontline MCL | |||||
| Wang 2019 [87] | Ibr + R induction prior to CIT | 131 | II | 82% (36) | 100% (88%) |
| Jain 2021 [88] | Ibr + R in elderly | 50 | II | NR* | 98% (60%) |
| Le Gouill 2021 [89] | Ibr + Ven + O | 15 | I | 74.5% (12) | 75% (67%) |
| Wang 2022 [90] | BR +/− Ibr | 523 | III | 80.6 vs. 52.9 (85)* | |
BR: Bendamustine/rituximab Ibr: Ibrutinib; R: rituximab; Ven: Venetoclax; Len: Lenalidomide; O: Obinutuzumab; NR: not reached; m: months
median progression free survival
Much like CLL, combination therapies can deepen response with ibrutinib. Combination with venetoclax or rituximab +/− lenalidomide have demonstrated deeper responses than single agent BTKi, with higher CR rate of 44–71% compared to 27% with single agent ibrutinib. U-MRD is as high as 67–68% [66, 80–82]. Median PFS was not reached in the dose finding cohort of venetoclax/ibrutinib combination and phase III investigation is ongoing [83] (Table 3). Combination of ibrutinib with rituximab +/− lenalidomide demonstrated 12-month PFS to 57–75% in the R/R setting [82, 84]. Combination of ibrutinib with novel anti-ROR1 monoclonal antibody, cirmtuzumab is also promising in R/R MCL with ORR of 80% and CR of 35% based on preliminary phase I/II data [85]. CDK4/6 inhibitor palbociclib, which prolongs the G1 cell cycle arrest has also been investigated in combination with ibrutinib and demonstrated an ORR of 67% and CR of 37% in patients with R/R MCL [86] (Summarized in Table 3).
Lastly, incorporating BTKi into the frontline setting has shown promising activity as either part of a chemotherapy-free regimen for elderly/frail patients or induction therapy prior to consolidative CIT in medically fit patients [87, 88] (Table 3). In a single center phase II study of older patients with newly diagnosed classic MCL (excluding histologically aggressive variants, Ki67<50%), first line treatment with ibrutinib and rituximab demonstrated a 3 year PFS of 87% [88]. In a dose finding phase I/II study evaluating frontline ibrutinib combined with obinutuzumab and venetoclax, 87% and 100% patients achieved CR and uMRD, respectively, with a 1 year PFS of 93% [89]. In a large phase III study of 523 patients aged ≥65 years old, ibrutinib was combined with lower intensity chemotherapy regimen bendamustine/rituximab in the upfront setting. Median PFS was improved by a remarkable 2.3 years with fewer patients requiring subsequent therapy (20% vs. 41%, respectively) at 7-year follow up [90]. These and other ongoing studies of novel BTK agents, combination therapies, and incorporation of targeted therapies in earlier line settings will likely change the treatment paradigm of MCL and lower occurrence of BTK resistance.
Diffuse Large B-cell Lymphoma
Molecular subclassification of DLBCL
Diffuse large B cell lymphoma represents a heterogeneous disease derived from germinal or post germinal center B cells. Gene expression profiling has been used to separate these into distinct subgroups based on the cell of origin, those related to germinal center B cell (GCB) and those related to activated B cell (ABC) [91, 92]. ABC-subtype defined by gene expression profiling is closely related to, but not equivalent to the non-GCB subtype defined by immunohistochemistry [93]. The ABC subtype is characterized by chronically active NFκB signaling downstream of the BCR pathway (Fig. 1) which has served as the rationale for investigational BTKi therapy in this subtype [94–96]. In contrast, GCB-DLBCL is more dependent on PI3K and BCL2 signaling pathways.
In the era of emerging precision medicine, many efforts have been made to sub-classify DLBCL by genetic and biological features which are potentially amenable to targeted therapeutic agents. Hundreds of DLBCL tumors have been characterized using multi-omic technologies interrogating point mutations, indels, chromosomal structural alterations and gene expression profiles [97–99]. In the recent LymphGen algorithm developed by Staudt’s group, DLBCL is subclassified into 7 groups with 37% of tumors unclassifiable [100]. Among molecular subgroups is MCD, which is enriched with gain of function of MYD88 L265P and/or CD79B mutations (Fig. 1). MYD88 is a key molecule mediating Toll-like receptor signaling while CD79B is part of the B-cell receptor complex that plays a role in maintaining the cell surface expression of the receptor [95]. Other subgroups include BN2 defined by BCL6 fusion and NOTCH2 mutations, EZB enriched for EZH2 mutations and BCL2 translocations and N1 characterized by NOTCH1 mutations.
In a separate study from Shipp’s group, the consensus clustering algorithm subclassified the tumors into C1-C5 clusters [98]. There are substantial molecular and biological similarity and overlaps between LymphGen and the consensus clusters categories. For example, MCD is related with C5, BN2 with C1, and EZB with C3.
Finally, to reduce the molecular classification into routine clinical practice, a UK network subclassified DLBCL into 6 molecular groups based mostly on mutations detected in FFPE tissues by targeted sequencing of ~300 genes instead of the multiomic whole genome sequencing. The subgroups are named after the major genetic features. Grossly, the MYD88 group is the counterpart of MCD/C5 group, NOTCH2 group corresponds to BN2/C1 while BCL2 group is similar to EZB/C3.
While these various molecular subgroup classifications are not yet widely available for routine clinical practice, use of NGS testing as a correlative in clinical trials is rapidly increasing in order to determine which particular subtypes benefit from therapies targeted to the underlying molecular pathology (Also see below).
Clinical data
Patients with ABC DLBCL have significantly inferior survival with standard R-CHOP based treatment in comparison to those with GCB subtype [96]. Since ABC lymphomas are characterized by chronic active BCR signaling, clinical investigations were started with correlative subtype determination of DLBCL. In the initial trial, ibrutinib has demonstrated activity predominantly in the ABC subtype with ORR of 37% versus only 5% in those with GCB subtype [101].
Encouraged by these initial results, the phase III PHOENIX trial evaluated ibrutinib vs. placebo in combination with frontline R-CHOP in patients with non-GCB subtype disease and failed to show an overall survival benefit of ibrutinib plus R-CHOP. However, it did demonstrate event free survival (EFS), PFS, and OS benefit in the <60-year-old population [102]. This lack of benefit in older patient population was attributed in part to increased toxicity with combination therapy thereby limiting optimal CIT dosing. Later, a more in-depth subgroup analysis in the younger patients revealed that ABC lymphomas of MCD and N1 subtype had a 3-year EFS and OS of 100% with ibrutinib plus R-CHOP compared to 42.9 and 50% in the R-CHOP alone arm. While this study was not powered to assess differences in response or survival among these subgroups, it is hypothesis generating and warrants larger studies to identify which genetic subgroups will have better outcomes on BTKi-containing therapy [103].
Primary sensitivity and resistance
Notably, the MCD and N1 subgroups only make up 14% and 2.8% of the total of 574 DLBCL tumors, respectively [97, 98, 100]. Therefore, only about 1/5 of patients seem to benefit from ibrutinib-containing CIT regimen and a significant number of ABC tumors did not respond well to ibrutinib. In the GCB subtype, ibrutinib is not as active as in ABC DLBCL as demonstrated in vitro and in clinical studies [101, 104] (107, 110, 109). GCB DLBCL is featured by activation of the PI3K/AKT pathway enabling cell survival and proliferation [105, 106] (Fig.1).
Regarding molecular mechanisms of primary resistance, activating CARD11 and inactivating mutations in TNFAIP3 (aka A20), a negative regulator of NFκB, are associated with primary ibrutinib resistance in a phase I/II clinical trial [95, 107, 108] (Fig.1). These genes act downstream of BTK, promoting NFκB activity with no regards to the upstream BTK activity. Mutations in KLHL14 gene is also associated with primary resistance [109] (Fig.1). KLHL14 is a negative regulator of the BCR signaling. It promotes the ubiquitination and degradation of the BCR subunits IgM, CD79A, and CD79B. KLHL14 inactivating mutations is present in ~11% of DLBCL tumors, especially in the MYD88/CD79B double mutant (MCD) genetic subtype of ABC DLBCL.
Regarding sensitivity to other BTK inhibitors, in a phase II clinical trial evaluating zanubrutinib in R/R DLBCL, the ORR to zanubrutinib was 46.2% (6/13) and 28.6% (6/21) in patients with or without CD79B mutations, and 40% (4/10) and 33.3% (8/24) in patients with or without MYD88 L265P mutations. Notably, these differences in ORR was not statistically significant [110]. Larger studies will be needed to investigate the association of genetic biomarkers with response to zanubrutinib and other BTK inhibitors.
Secondary resistance
Clinical experience of DLBCL treated with BTKi is limited, and long-term follow-up is still lacking. Thus, secondary resistance to ibrutinib in DLBCL is mostly studied in cell line models. In DLBCL cell lines cultivated to become resistant to ibrutinib, BTK and PLCG2 mutations were not identified, however, PI3K/AKT/mTOR signaling is upregulated leading to increased tumor cell survival [111, 112] (Fig. 1). In addition to pathway alterations, the role of epigenetic mechanisms was recently revealed by a study using ABC DLBCL cell lines [47]. Interestingly, RAC2, the small GTPase, was identified as the mediator of the epigenetic ibrutinib resistance. As mentioned above, RAC2 is also involved in primary ibrutinib resistance in MCL, but through enhanced cell adhesion [75]. This molecular commonality between DLBCL and MCL may worth further investigation. Long term follow-up on patients treated with BTKi-containing regimens is still in progress. Mechanisms of secondary resistance in patients who relapse after initial BTKi response remain to be seen.
Trials with newer BTK inhibitors and other targeted therapies
While certain DLBCL genetic subgroups appear to have increased ibrutinib sensitivity, the role of BTKi in the frontline and R/R setting remains unclear. Investigation of 2nd generation covalent BTKi combined with CIT, such as the ESCALADE study (NCT04529772) is still ongoing.
Additionally, reversible non-covalent BTKi provide an alternative to patients who are intolerant to covalent BTKi or who develop progressive diseases during therapies [21]. Compared to covalent BTKi, pirtobrutinib has demonstrated higher BTK selectivity and more durable target inhibition over 24 hours on pharmacokinetic studies [51]. This suggests a potential role for this agent in more proliferative B cell malignancies like DLBCL where earlier generation BTKi have been less efficacious. In a phase I/II evaluation, 24% of patients with R/R DLBCL responded to pirtobrutinib [21].
As mentioned above, upregulation of AKT/PI3K serves as a bypass pathway for tumor survival. PI3Ki demonstrated in vitro activity in BTKi resistant cells lines [112]. However, in an early clinical trial investigation, durability of response to umbralisib was poor and the study was prematurely closed [113]. It remains to be seen if other PI3Ki would have clinical activities in the BTKi-resistant patient population.
Conclusion
Targeting the B cell receptor signaling pathway via BTK inhibition has played a pivotal role in treatment of B cell malignancies. In this review, we summarized the clinical data on using ibrutinib in CLL, MCL and DLBCL. The mutational mechanisms of primary and secondary resistance to ibrutinib and novel covalent and non-covalent BTK inhibitors are summarized in Table 4 and Fig. 1 and 2. We also reviewed the current clinical investigations on overcoming such resistance with either newer BTK inhibitors or combination therapies. Due to its success as a class, several more BTK inhibitors as well as BTK degraders are being developed. Since single agent therapy is unlikely to completely eliminate the malignant B-cell clones, the combination therapies either with other class of targeted therapies or with antibody-based therapies would stand a better chance for a durable remission.
Additional clinical assessments of non-covalent BTKi in upfront setting and as part of combination regimens are still underway. Optimal sequencing of covalent and non-covalent BTKi and combination regimens has yet to be determined. Looking forward, understanding the resistance mechanisms and sensitive detection of emergent resistant clones during therapies would help guide new therapeutic development, therapeutic sequencing, and rational drug combination in the future.
Acknowledgement
This work is supported by NCI R21CA263415 to YLW. We apologize that we have to leave out some important publications to meet the limitations of number of cited references.
Footnotes
Competing Interest
The authors do not have any financial conflicts of interest to disclose.
References
- 1.Bajpai UD, et al. , Bruton’s tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J Exp Med, 2000. 191(10): p. 1735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petro JB, et al. , Bruton’s Tyrosine Kinase Is Required for Activation of Iκb Kinase and Nuclear Factor κb in Response to B Cell Receptor Engagement. Journal of Experimental Medicine, 2000. 191(10): p. 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan Z, et al. , Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem, 2007. 2(1): p. 58–61. [DOI] [PubMed] [Google Scholar]
- 4.Honigberg LA, et al. , The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences, 2010. 107(29): p. 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng S, et al. , BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia, 2014. 28(3): p. 649–57. [DOI] [PubMed] [Google Scholar]
- 6.Lu P, et al. , Ibrutinib and venetoclax target distinct subpopulations of CLL cells: implication for residual disease eradication. Blood Cancer Journal, 2021. 11(2): p. 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponader S, et al. , The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood, 2012. 119(5): p. 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Rooij MF, et al. , The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood, 2012. 119(11): p. 2590–4. [DOI] [PubMed] [Google Scholar]
- 9.Dubovsky JA, et al. , Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood, 2013. 122(15): p. 2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herman SE, et al. , Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin Cancer Res, 2015. 21(20): p. 4642–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niemann CU, et al. , Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib--Findings from an Investigator-Initiated Phase II Study. Clin Cancer Res, 2016. 22(7): p. 1572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman SE, et al. , Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia, 2014. 28(11): p. 2188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang ML, et al. , Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med, 2013. 369(6): p. 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang ML, et al. , Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood, 2015. 126(6): p. 739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrd JC, et al. , Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med, 2014. 371(3): p. 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burger JA, et al. , Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia, 2020. 34(3): p. 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zain R and Vihinen M, Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Frontiers in Immunology, 2021. 12(2675). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y, et al. , A two-part, single-arm, multicentre, phase I study of zanubrutinib, a selective Bruton tyrosine kinase inhibitor, in Chinese patients with relapsed/refractory B-cell malignancies. British Journal of Haematology, 2022. 198(1): p. 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woyach J, et al. , Resistance to Acalabrutinib in CLL Is Mediated Primarily By BTK Mutations. Blood, 2019. 134(Supplement_1): p. 504–504. [Google Scholar]
- 20.Woyach J, et al. , Final Results of Phase 1, Dose Escalation Study Evaluating ARQ 531 in Patients with Relapsed or Refractory B-Cell Lymphoid Malignancies. Blood, 2019. 134(Supplement_1): p. 4298–4298. [Google Scholar]
- 21.Mato AR, et al. , Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. The Lancet, 2021. 397(10277): p. 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman SEM, et al. , Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood, 2011. 117(23): p. 6287–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer K, et al. , Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood, 2016. 127(2): p. 208–15. [DOI] [PubMed] [Google Scholar]
- 24.Farooqui MZ, et al. , Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol, 2015. 16(2): p. 169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munir T, et al. , Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol, 2019. 94(12): p. 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Brien SM, et al. , Outcomes with ibrutinib by line of therapy and post-ibrutinib discontinuation in patients with chronic lymphocytic leukemia: Phase 3 analysis. Am J Hematol, 2019. 94(5): p. 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Brien S, et al. , Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood, 2018. 131(17): p. 1910–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J and Wang YL, Prognostic and Predictive Molecular Biomarkers in Chronic Lymphocytic Leukemia. The Journal of Molecular Diagnostics, 2020. 22(9): p. 1114–1125. [DOI] [PubMed] [Google Scholar]
- 29.Smith CIE and Burger JA, Resistance Mutations to BTK Inhibitors Originate From the NF-κB but Not From the PI3K-RAS-MAPK Arm of the B Cell Receptor Signaling Pathway. Front Immunol, 2021. 12: p. 689472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadri S, et al. , Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Advances, 2017. 1(12): p. 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campo E, et al. , TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica, 2018. 103(12): p. 1956–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maddocks KJ, et al. , Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol, 2015. 1(1): p. 80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahn IE, et al. , Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia Treated With Ibrutinib: Development and Validation of a Four-Factor Prognostic Model. Journal of Clinical Oncology, 2020. 39(6): p. 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain P, et al. , Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood, 2015. 125(13): p. 2062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mato AR, et al. , Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood, 2016. 128(18): p. 2199–2205. [DOI] [PubMed] [Google Scholar]
- 36.Ahn IE, et al. , Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood, 2017. 129(11): p. 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furman RR, et al. , Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med, 2014. 370(24): p. 2352–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woyach JA, et al. , Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med, 2014. 370(24): p. 2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng S, et al. , Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia, 2015. 29(4): p. 895–900. [DOI] [PubMed] [Google Scholar]
- 40.Woyach JA, et al. , BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J Clin Oncol, 2017. 35(13): p. 1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinquenel A, et al. , Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood, 2019. 134(7): p. 641–644. [DOI] [PubMed] [Google Scholar]
- 42.Hamasy A, et al. , Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia, 2017. 31(1): p. 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinquenel A, et al. , Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood, 2019. 134(7): p. 641–644. [DOI] [PubMed] [Google Scholar]
- 44.Sharma S, et al. , Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget, 2016. 7(42): p. 68833–68841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu TM, et al. , Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood, 2015. 126(1): p. 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burger JA, et al. , Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nature Communications, 2016. 7(1): p. 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaffer AL III, et al. , Overcoming Acquired Epigenetic Resistance to BTK Inhibitors. Blood Cancer Discovery, 2021. 2(6): p. 630–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Byrd JC, et al. , First results of a head-to-head trial of acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia. Journal of Clinical Oncology, 2021. 39(15_suppl): p. 7500–7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hillmen P, et al. First Interim Analysis of ALPINE Study: Results of a Phase 3 Randomized Study of Zanubrutinib vs. Ibrutinib in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. in European Hematology Association Virtual Congress. 2021. Vienna, Austria. [Google Scholar]
- 50.Handunnetti SM, et al. , BTK Leu528Trp - a Potential Secondary Resistance Mechanism Specific for Patients with Chronic Lymphocytic Leukemia Treated with the Next Generation BTK Inhibitor Zanubrutinib. Blood, 2019. 134(Supplement_1): p. 170–170. [Google Scholar]
- 51.Brandhuber B, et al. , LOXO-305, A Next Generation Reversible BTK Inhibitor, for Overcoming Acquired Resistance to Irreversible BTK Inhibitors. Clinical Lymphoma Myeloma and Leukemia, 2018. 18: p. S216. [Google Scholar]
- 52.Liu Y, et al. Pirtobrutinib Overcomes Ibrutinib and Venetoclax Resistance in Mantle Cell Lymphoma. in American Society of Hematology Annual Meeting & Exposition. 2021. Atlanta, GA. [Google Scholar]
- 53.Wang E, et al. , Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N Engl J Med, 2022. 386(8): p. 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dimier N, et al. , A model for predicting effect of treatment on progression-free survival using MRD as a surrogate end point in CLL. Blood, 2018. 131(9): p. 955–962. [DOI] [PubMed] [Google Scholar]
- 55.Seymour JF, et al. , Enduring undetectable MRD and updated outcomes in relapsed/refractory CLL after fixed-duration venetoclax-rituximab. Blood, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burger JA, et al. , Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood, 2019. 133(10): p. 1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharman JP, et al. , Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet, 2020. 395(10232): p. 1278–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tam CS, et al. , Ibrutinib (Ibr) Plus Venetoclax (Ven) for First-Line Treatment of Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL): Results from the MRD Cohort of the Phase 2 CAPTIVATE Study. Blood, 2019. 134(Supplement_1): p. 35–35. [Google Scholar]
- 59.Kater A, et al. Fixed-Duration Ibruitnib and Venetoclax (I+V) versus Chlorambucil Plus Obinutuzumab (Clb + O) for First-Line (1L) Chronic Lymphocytic Leukemia (CLL): Primary Analysis of the Phase 3 GlOW Study. in European Hematology Association Virtual Congress. 2021. Vienna, Austria. [Google Scholar]
- 60.Wierda WG, et al. , Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J Clin Oncol, 2021. 39(34): p. 3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pérez-Galán P, Dreyling M, and Wiestner A, Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood, 2011. 117(1): p. 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill HA, et al. , Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv, 2020. 4(13): p. 2927–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pighi C, et al. , Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling. Cell Oncol (Dordr), 2011. 34(2): p. 141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma J, et al. , Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol, 2014. 166(6): p. 849–61. [DOI] [PubMed] [Google Scholar]
- 65.Eskelund CW, et al. , TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood, 2017. 130(17): p. 1903–1910. [DOI] [PubMed] [Google Scholar]
- 66.Rule S, et al. , Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: extended 3.5-year follow up from a pooled analysis. Haematologica, 2019. 104(5): p. e211–e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin P, et al. , Postibrutinib outcomes in patients with mantle cell lymphoma. Blood, 2016. 127(12): p. 1559–63. [DOI] [PubMed] [Google Scholar]
- 68.Wang M, et al. , Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. The Lancet, 2018. 391(10121): p. 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu C, et al. , Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget, 2016. 7(25): p. 38180–38190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mohanty A, et al. , CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget, 2016. 7(45): p. 73558–73572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rahal R, et al. , Pharmacological and genomic profiling identifies NF-κB–targeted treatment strategies for mantle cell lymphoma. Nature Medicine, 2014. 20(1): p. 87–92. [DOI] [PubMed] [Google Scholar]
- 72.Lee J, et al. , Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv, 2018. 2(16): p. 2039–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Q, et al. , Cutting Edge: ROR1/CD19 Receptor Complex Promotes Growth of Mantle Cell Lymphoma Cells Independently of the B Cell Receptor–BTK Signaling Pathway. 2019. 203(8): p. 2043–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ming M, et al. , XPO1 Inhibitor Selinexor Overcomes Intrinsic Ibrutinib Resistance in Mantle Cell Lymphoma via Nuclear Retention of IκB. Molecular Cancer Therapeutics, 2018. 17(12): p. 2564–2574. [DOI] [PubMed] [Google Scholar]
- 75.Wu W, et al. , Inhibition of B-cell receptor signaling disrupts cell adhesion in mantle cell lymphoma via RAC2. Blood Advances, 2021. 5(1): p. 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jain P, et al. , Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol, 2018. 183(4): p. 578–587. [DOI] [PubMed] [Google Scholar]
- 77.Zhang L, et al. , Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med, 2019. 11(491). [DOI] [PubMed] [Google Scholar]
- 78.Zhao X, et al. , Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nature Communications, 2017. 8(1): p. 14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang SQ, et al. , Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br J Haematol, 2015. 170(4): p. 445–56. [DOI] [PubMed] [Google Scholar]
- 80.Wang M, et al. Pirtobrutinib, A Next Generation, Highly Selective, Non-Covalent BTK Inhibitor in Previously Treated Mantle Cell Lymphoma: Updated Results from the Phase 1/2 BRUIN Study. in American Society of Hematology Annual Meeting & Exposition. 2021. Atlanta, GA. [Google Scholar]
- 81.Jain P, et al. , Four-year follow-up of a single arm, phase II clinical trial of ibrutinib with rituximab (IR) in patients with relapsed/refractory mantle cell lymphoma (MCL). Br J Haematol, 2018. 182(3): p. 404–411. [DOI] [PubMed] [Google Scholar]
- 82.Jerkeman M, et al. , Ibrutinib-Lenalidomide-Rituximab in Patients with Relapsed/Refractory Mantle Cell Lymphoma: Final Results from the Nordic Lymphoma Group MCL6 (PHILEMON) Phase II Trial. Blood, 2020. 136: p. 36.32430502 [Google Scholar]
- 83.Tam CS, et al. , Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. New England Journal of Medicine, 2018. 378(13): p. 1211–1223. [DOI] [PubMed] [Google Scholar]
- 84.Wang ML, et al. , Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol, 2016. 17(1): p. 48–56. [DOI] [PubMed] [Google Scholar]
- 85.Lee HJ, et al. Phase 1b/2 Study of Cirmtuzumab and Ibrutinib in Mantle Cell Lymphoma (MCL) or Chronic Lymphocytic Leukemia (CLL). in American Society of Hematology Annual Meeting & Exposition. 2021. Atlanta, GA. [Google Scholar]
- 86.Martin P, et al. , A phase 1 trial of ibrutinib plus palbociclib in previously treated mantle cell lymphoma. Blood, 2019. 133(11): p. 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang ML, et al. , Frontline Treatment with Ibrutinib Plus Rituximab (IR) Followed By Short Course R-Hypercvad/MTX Is Extremely Potent and Safe in Patients (age ≤ 65 years) with Mantle Cell Lymphoma (MCL) - Results of Phase-II Window-1 Clinical Trial. Blood, 2019. 134(Supplement_1): p. 3987–3987. [Google Scholar]
- 88.Jain P, et al. , Ibrutinib With Rituximab in First-Line Treatment of Older Patients With Mantle Cell Lymphoma. Journal of Clinical Oncology, 2021: p. JCO.21.01797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Le Gouill S, et al. , Ibrutinib, obinutuzumab, and venetoclax in relapsed and untreated patients with mantle cell lymphoma: a phase 1/2 trial. Blood, 2021. 137(7): p. 877–887. [DOI] [PubMed] [Google Scholar]
- 90.Wang ML, et al. , Ibrutinib plus Bendamustine and Rituximab in Untreated Mantle-Cell Lymphoma. N Engl J Med, 2022. 386(26): p. 2482–2494. [DOI] [PubMed] [Google Scholar]
- 91.Alizadeh AA, et al. , Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 2000. 403(6769): p. 503–11. [DOI] [PubMed] [Google Scholar]
- 92.Rosenwald A, et al. , The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. 2002. 346(25): p. 1937–1947. [DOI] [PubMed] [Google Scholar]
- 93.Hans CP, et al. , Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood, 2004. 103(1): p. 275–82. [DOI] [PubMed] [Google Scholar]
- 94.Davis RE, et al. , Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med, 2001. 194(12): p. 1861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davis RE, et al. , Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature, 2010. 463(7277): p. 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lenz G, et al. , Stromal gene signatures in large-B-cell lymphomas. New England Journal of Medicine, 2008. 359(22): p. 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmitz R, et al. , Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med, 2018. 378(15): p. 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chapuy B, et al. , Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nature medicine, 2018. 24(5): p. 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reddy A, et al. , Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell, 2017. 171(2): p. 481–494.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wright GW, et al. , A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell, 2020. 37(4): p. 551–568.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wilson WH, et al. , Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med, 2015. 21(8): p. 922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Younes A, et al. , Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol, 2019. 37(15): p. 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wilson WH, et al. , Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell, 2021. 39(12): p. 1643–1653.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goy A, et al. , Ibrutinib plus lenalidomide and rituximab has promising activity in relapsed/refractory non–germinal center B-cell–like DLBCL. Blood, 2019. 134(13): p. 1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lenz G, et al. , Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A, 2008. 105(36): p. 13520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frick M, Dörken B, and Lenz G, The molecular biology of diffuse large B-cell lymphoma. Ther Adv Hematol, 2011. 2(6): p. 369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lenz G, et al. , Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science, 2008. 319(5870): p. 1676–9. [DOI] [PubMed] [Google Scholar]
- 108.Compagno M, et al. , Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature, 2009. 459(7247): p. 717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Choi J, et al. , Regulation of B cell receptor-dependent NF-κB signaling by the tumor suppressor KLHL14. Proc Natl Acad Sci U S A, 2020. 117(11): p. 6092–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang H, et al. , Zanubrutinib Monotherapy for Relapsed or Refractory Non-Germinal Center Diffuse Large B-Cell Lymphoma. Blood Advances, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kapoor I, et al. , Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis, 2019. 10(12): p. 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jain N, et al. , Targeting phosphatidylinositol 3 kinase-β and -δ for Bruton tyrosine kinase resistance in diffuse large B-cell lymphoma. Blood Advances, 2020. 4(18): p. 4382–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lunning M, et al. , A Phase IIA Study Evaluating the Safety and Tolerability of Umbralisib and Ibrutinib in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. 2019. 37(S2): p. 519–519. [Google Scholar]
- 114.Byrd JC, et al. , Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med, 2013. 369(1): p. 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ghia P, et al. , ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. Journal of Clinical Oncology, 2020. 38(25): p. 2849–2861. [DOI] [PubMed] [Google Scholar]
- 116.Tam CS, et al. , Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood, 2019. 134(11): p. 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tam CS, et al. , Zanubrutinib for the treatment of relapsed or refractory mantle cell lymphoma. Blood Advances, 2021. 5(12): p. 2577–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilson WH, et al. , Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nature Medicine, 2015. 21(8): p. 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Treon SP, et al. , Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. New England Journal of Medicine, 2015. 372(15): p. 1430–1440. [DOI] [PubMed] [Google Scholar]
- 120.Dimopoulos MA, et al. , Ibrutinib for patients with rituximab-refractory Waldenström’s macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol, 2017. 18(2): p. 241–250. [DOI] [PubMed] [Google Scholar]
- 121.Tam CS, et al. , A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood, 2020. 136(18): p. 2038–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Noy A, et al. , Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv, 2020. 4(22): p. 5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Opat S, et al. , Efficacy and Safety of Zanubrutinib in Patients with Relapsed/Refractory Marginal Zone Lymphoma: Initial Results of the MAGNOLIA (BGB-3111–214) Trial. Blood, 2020. 136(Supplement 1): p. 28–30. [Google Scholar]
- 124.Woyach JA, et al. , Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N Engl J Med, 2018. 379(26): p. 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moreno C, et al. , Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. The Lancet Oncology, 2019. 20(1): p. 43–56. [DOI] [PubMed] [Google Scholar]
- 126.Shanafelt TD, et al. , Ibrutinib–Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. New England Journal of Medicine, 2019. 381(5): p. 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dreyling M, et al. , Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet, 2016. 387(10020): p. 770–8. [DOI] [PubMed] [Google Scholar]
- 128.Guo A, et al. , Heightened BTK-dependent cell proliferation in unmutated chronic lymphocytic leukemia confers increased sensitivity to ibrutinib. Oncotarget, 2016. 7(4): p. 4598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shanafelt TD, et al. , Long-term outcomes for ibrutinib–rituximab and chemoimmunotherapy in CLL: updated results of the E1912 trial. Blood, 2022. 140(2): p. 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]