

Abstract
Single cell combinatorial indexing RNA sequencing (sci-RNA-seq) is a powerful method for recovering gene expression data from an exponentially scalable number of individual cells or nuclei. However, sci-RNA-seq is a complex protocol that has historically exhibited variable performance on different tissues, as well as lower sensitivity than alternative methods. Here we report a simplified, optimized version of the three-level sci-RNA-seq protocol that is faster, higher yield, more robust, and more sensitive, than the original sci-RNA-seq3 protocol, with reagent costs on the order of 1 cent per cell or less. The total hands-on time from nuclei isolation to final library preparation takes 2 to 3 days, depending on the number of samples sharing the experiment. The improvements also allow RNA profiling from tissues rich in RNases like older mouse embryos or adult tissues that were problematic for the original method. We showcase the optimized protocol via whole organism analysis of an E16.5 mouse embryo, profiling ~380,000 nuclei in a single experiment. Finally, we introduce a “Tiny-Sci” protocol for experiments where input material is very limited.
Introduction
Single cell combinatorial indexing (sci-*) combines in situ molecular indexing and a “split-pool” framework in order to uniquely label an exponentially scalable number of cells or nuclei with a unique combination of nucleic acid barcodes. Following its demonstration in the context of chromatin accessibility1,2 in 2015, we and others have additionally developed sci-* methods for profiling gene expression3–7, genome sequence8,9, genome architecture10, genome-wide methylation11, co-assays of mRNA and chromatin accessibility12, transcriptional dynamics13, transcription factor occupancy14,15, surface proteins14, small molecule exposures16 and spatial locations17, all at single cell resolution.
A split-pool method for profiling gene expression, sci-RNA-seq, was first reported in 20173, and an improved three-level version, sci-RNA-seq33,4, in 2019. Among other applications, sci-RNA-seq3 has been applied to generate the largest atlases of single cell gene expression for both mouse3 (~2 million cells) and human18 (~4 million cells) to date. Both of these datasets were largely generated within a single lab, each within a few weeks and by one or two individuals. Nonetheless, the underlying protocol remains cumbersome. Here, we describe the culmination of extensive efforts to simplify and optimize the protocol, enabling the transcriptional profiling of previously difficult tissues. In addition, we describe a modified version of the protocol, called “Tiny-Sci”, for experiments where input material is very limited. For example, we recently applied Tiny-Sci to a series of individual embryonic day 8.5 (E8.5) mouse embryos to generate a sci-RNA-seq3 dataset with somite-level resolution19. These embryos were only a few millimeters in length, such that Tiny-Sci extends the range of sci-RNA-seq3 to samples for which only small amounts of tissue are available.
Briefly, the sci-RNA-seq3 protocol (Figure 1) starts by allocating fixed cells or nuclei to the wells of one or more 96-well plates. The first index is introduced during reverse transcription with barcoded oligo-dT primers. Cells or nuclei are then pooled and split to a new set of one or more 96-well plates. The second index is ligated onto the end of the first index, and then the cells or nuclei are pooled and split again. In the third set of plates, second strand synthesis occurs and the double-stranded product is then tagmented with Tn5 transposase. PCR amplification adds the third index and, finally, the library is purified and sequenced. A detailed schematic of primers and adaptor sequences is shown in Figure 2.
Figure 1. Summary of optimized sci-RNA-seq3 method.

Colors in plate wells represent 96 unique index sequences for each of the 3 rounds of indexing. RNA transcript in red, DNA in dark blue. a, Nuclei (in green) are isolated in lysis buffer with DEPC, then fixed with DSP and methanol. b, Nuclei are then distributed to a 96-well plate for reverse transcription, where the first index (in purple well) is introduced. If desired, nuclei from different samples can be deposited to different wells during this first round of indexing, facilitating multi-sample processing while minimizing batch effects. c, After indexed reverse transcription, the nuclei are pooled and split into a new plate to add a second index (in orange well) via ligation. d, The nuclei are pooled and split again. After second-strand synthesis (e), protease digestion, and tagmentation (f), the third index is added by PCR (in blue well), with an optional plate index (aqua) (g)., Finally, the library is purified and sequenced (h).
Figure 2. Detailed schematic of the sci-RNA-seq3 combinatorial indexing strategy.

a, The indexed primer sets used. “#” indicates the index sequence, “N” are random bases incorporated into the primer for the unique molecular identifier (UMI). There are 96–384 indexed reverse transcription (RT) primers, 96–384 indexed ligation primers, 96–384 indexed PCR P7 primers, and indexed PCR P5 primers. The P5 primer can be used as a plate index for multiple plates, or optionally a full set of 96–384 for fully dual indexed PCRs. b, In the first round of indexing, the oligodT of the RT primer binds to the polyA tail of mRNA, extending to make the complementary cDNA strand. c, After pooling and redistribution of the nuclei, the ligation primer, which can form a hairpin, anneals to the 6bp linker on the RT primer allowing ligation to the phosphorylated 5’ end of the RT primer. d, Nuclei are pooled and redistributed into the third and final plate, and during second strand synthesis the RNA is nicked and used as primers to create the second strand with DNA Polymerase I. e, Tn5 Tagmentation fragments the now double-stranded DNA and adds adaptors for the PCR primers. f, PCR adds the final indexes and sequencing adaptors. g, The final product can be sequenced on Illumina platforms with their standard primers.
Development of the protocol
Here we describe the optimized sci-RNA-seq3 protocol, primarily focusing on nuclei (although to apply the protocol to cells, one need only omit the lysis step; see “Limitations” section below). In order to expand the range of tissues that can be processed with sci-RNA-seq3, a first set of changes were directed at better neutralizing endogenous RNases found in older embryonic and adult tissues. A secondary consequence of these changes has been an increase in the number of unique molecular identifiers (UMIs) obtained per nucleus. Specifically, diethyl pyrocarbonate (DEPC) is now used to inactivate RNases during the lysis step. DEPC has commonly been used to deactivate RNases in water and solutions used for RNA work by modifying the RNase protein, but it has been unclear whether it negatively impacts RNA itself20. We compared the effectiveness of DEPC versus commercial RNase inhibitors on RNase activity in the lysate of an E13.5 day mouse embryo. The differences were stark (Supplementary Figures 1–2). Using an RNase Alert kit (IDT), we quickly realized why older embryos often failed the sci-RNA-seq3 protocol. The expensive SuperaseIn RNase Inhibitor that was used in the original sci-RNA-seq3 protocol was ineffective at inactivating the RNases in older tissues, and the change to DEPC alone has had the biggest impact on sci-RNA-seq3 success. Moreover, the resulting data indicates that the DEPC is not detrimental to the transcriptional readout, at least not for sci-RNA-seq3. Of note, DEPC can phase-separate at the concentration used here, but the detergent in the lysis buffer seems to help keep it emulsified and effective.
The RNase Alert checkpoint step is now included to ensure no RNase activity still exists before proceeding with the bulk of the protocol, preventing wasted time and reagents. The lysis buffer had to be changed from a Tris-based buffer to a hypotonic phosphate buffer to accommodate DEPC. Another new buffer, 0.3M SPBSTM, replaces the original nuclei suspension buffer, and enables better nuclei recovery during washes and spins. It contains sucrose for osmolarity and cushioning the nuclei during spins, PBS to keep the buffer isotonic, Triton-X 100 which makes the nuclei pellet more cleanly, and Magnesium Chloride is necessary for nuclei integrity.
DSP/methanol fixation replaces the need for a separate permeabilization step and results in more UMIs per cell compared to paraformaldehyde fixation in the original protocol. DSP is an amine-crosslinker that doesn’t alter RNA, but unfortunately precipitates in aqueous solutions. Methanol both fixes and permeablizes nuclei well for access to the transcripts, but often leaves nuclei too fragile to tolerate the complete protocol. When methanol is used on its own, this results in large, unusable clumps of hundreds of nuclei at the end of the protocol. However, the two fixatives work well together–the DSP is easily dissolved in the methanol and confers integrity to the fixed cells. The resulting nuclei are stable, accessible, and less prone to clumping compared with the paraformaldehyde fixation in the original protocol.
We have eliminated the USER step from the original protocol, and deoxyU is no longer needed in the ligation primer. We realized that the second strand synthesis step can take care of the hairpin and so an additional step is not necessary to open it up.
After the nuclei are in their third and final plate, and have undergone second strand synthesis, we have found that extracting the nuclei before tagmentation allows better access for the Tn5. However, we have simplified this extraction with a simple digestion with a heat-inactivatable protease, thereby removing any need for a whole-plate ampure/spri bead cleanup, significantly reducing the hands-on time and cost for the final library preparation steps.
These changes result in a streamlined protocol that allows transcripts to be recovered from RNase-rich tissues that were previously problematic for sci-RNA-seq3, while also mitigating nuclei losses for precious samples, as more nuclei are able to tolerate the entire process. Additionally, we have drastically reduced the cost of conducting these experiments as compared with the original protocol (Table 1), which was already considerably less expensive than commercial alternatives.
Table 1. Cost comparison between original4 vs. optimized sci-RNA-seq3 protocols.
We focus on the most expensive reagents here (all enzymes) as other reagent costs are comparatively negligible.
| Enzyme | Cost/μl | Original amount/plate | Original cost/plate | Optimized amount/plate | Optimized cost/plate |
|---|---|---|---|---|---|
| Superscript IV | $7.11 | 205 μl | $1458 | 55 μl | $391 |
| RNaseOUT | $1.56 | 205 μl | $320 | not used | $0 |
| SuperaseIN | $1.00 | 250 μl | $250 | not used | $0 |
| Quick Ligase | $2.59 | 215 μl | $557 | not used | $0 |
| T4 DNA Ligase | $1.04 | not used | $0 | 65 μl | $68 |
| USER | $1.21 | 110 μl | $133 | not used | $0 |
| Second Strand Synthesis | $2.95 | 73.3 μl | $216 | 35 μl | $103 |
| AmpureXP | $0.014 | 3840 μl | $53 | 230 μl | $3 |
| Tn5 | $24.50 | 0.92 μl | $22 | 4.6 μl | $112 |
| NEBNext 2X PCR mix | $0.058 | 1920 μl | $111 | 2200 μl | $127 |
| Total enzyme cost per plate (96×96×96) | $3,120 | $804 | |||
| Total enzyme cost per 4 plates (384×384×384) | $12,480 | $3,207 |
Application of the Method
Other single cell techniques may benefit from the nuclei preparation and fixation method presented here, particularly with older embryos and tissues, which are rendered challenging by the abundant presence of RNases. Using DEPC as an RNase inhibitor has been very successful in our hands, but it has a short half life in water and is broken down by Tris-containing buffers. It is only needed until the nuclei are fixed, so it might be useful in other methods to replace more expensive inhibitors if you can substitute buffers until the RNase inactivation is complete. Additional modifications to streamline or even remove the tagmentation step may further improve the protocol, and the method can also potentially be combined with oligo-based hashing techniques16,17. Using DSP as a fixative might prohibit some single cell techniques because of the ability of DTT (commonly found in reaction buffers) to reverse the crosslinks. For such protocols, one might try using DSS (disuccinimidyl suberate) instead, which acts similarly to DSP but with cross-links that are irreversible.
Comparison to other single cell methods
Key contrasts between sci-RNA-seq3 and widely used commercial solutions such as that of 10X Genomics include that sci-RNA-seq3 incurs a considerably lower cost per cell or nucleus when used at scale, does not require a kit, is open source, and is exponentially scalable. The optimized sci-RNA-seq3 protocol presented here is written here as the basic “1-plate version”, with 96 reverse transcription indexes, 96 ligation indexes, and 96 PCR indexes. In our hands, a 96 × 96 × 96 experiment typically nets ~100,000 nuclei. However, there are usually enough nuclei to fill an additional 3 plates or more for the final round of indexing, such that it is straightforward to boost the number of nuclei profiled to ~400,000 (i.e. 96 × 96 × 384). Further scaling to 384 × 384 × (384 to 768) enables further scaling to one or several million or more nuclei per experiment. As with all sci-* protocols, cells or nuclei from different samples can be deposited to different wells during the first round of indexing, facilitating multi-sample processing while minimizing batch effects3.
Limitations
In our hands, “organism-scale” transcriptional profiling is much more straightforward and less biased when performed on nuclei, rather than cells, because whereas each tissue might require a different protocol to dissociate cells, cell lysis is a “universal” means of freeing nuclei. Of course, a drawback is that one loses cytoplasmic transcripts. However, we note that it is straightforward to apply the protocol presented here to whole cells, simply by omitting the nuclear lysis step. We have done this successfully with mammalian cell lines, achieving similar performance, although we note that we have not characterized how the subsequent buffers impact cell integrity.
Overview of the Protocol
There are three main sections to the protocol that can be split over multiple days. If there is only one sample, you can combine Days 1 and 2 into a single day.
Day 1 - Nuclei preparation and fixing. Fixed samples can be stored at −80°C.
Day 2 - Reverse Transcription, Ligation, and Second Strand Synthesis (overnight).
Day 3 - Protease digestion, tagmentation, PCR, final library cleanup and load on sequencer.
Experimental Design
General Notes
This protocol is written for a small experiment with one plate of primers at each indexing round (which typically yields ~100,000 single cell profiles). It can be scaled up easily to use as many as 4 plates of primers at each round (which would be expected to yield over ~1 million single cell profiles) - just multiply each step by 4, combine all 4 plates when pooling, and put more nuclei (4000 per well) into the final plates.
Everything is to be kept cold at all times. Have lots of ice ready, pre-cool centrifuges to 4°C. Pre-cool all tubes on ice before you put nuclei in them.
Clean workspace and fume hood area ahead of time - wipe pipettes, racks, centrifuges down with RNAseZap, change gloves often.
Spin the primer plates down before opening, but don’t spin plates with nuclei until the second strand synthesis stage.
Use lo-bind (DNA/RNA) microcentrifuge tubes for the nuclei if possible.
Samples
Samples can be cell lines, tissues, or whole embryos. Ideally, samples (e.g. embryos or other) are isolated and frozen individually in 1.5 ml microfuge tubes with very little extra fluid with them. For a 96 × 96 × 96 sci- experiment, you’ll need 2M fixed cells to distribute into the first plate. If you’re using a cell line, count out at least 4–6M cells to start with, and wash cells in PBS before lysis. For tissues, a good size is 0.5–0.75cm3 or about 200mg. Bigger chunks of tissue will necessitate more lysis buffer. For instance, a E13.5 mouse embryo, which is about ≤1cm long, can be lysed in 5ml buffer with one filter, but an E18.5 mouse at about 3cm long will need to be split into 4 tubes of 20ml lysis buffer each and 4 filters since there is so much more debris involved. Alternatively you can powder a large sample, keeping it frozen and save the extra powder for lysing later if needed. If you don’t have enough cells for 2M in the first plate, share with another sample to fill out the plate for 2M nuclei total. The RT indexes in each well will allow you to sort the different samples later in the analysis.
If you are working with extremely small samples a few millimeters in size (such as E8.5 mouse embryos), see the “Tiny-Sci” section for alternative nuclei isolation and fixing steps. This is the same sci- protocol, but with the lysis and fixation scaled significantly down, and with less transfers that will contribute to nuclei loss. We have used this method for E8.5 single embryos, which are about 2–3mm in size, but it can more generally be used for instances where starting material is very limited.
Counting Nuclei
Nuclei prepared from frozen tissue are not simple to count, as debris can interfere with interpretation. Staining with yoyo-1 dye helps to discern nuclei from debris and is visualized on the GFP channel. Mix 10 μl of diluted yoyo-1 (see reagent setup) with 10 μl of nuclei (2 fold dilution) or do a 10–20-fold dilution if the nuclei are concentrated.
For the example experiment described below, nuclei were counted manually on a Countess Cell counter, as the automatic cell counting was unreliable for nuclei (Figure 3). On the GFP channel, with the view zoomed all the way out, yoyo-1-stained nuclei were counted in a 6cm × 6cm square. This count was multiplied by the dilution factor x10,000 to get an approximate number of nuclei/ml.
Figure 3. Yoyo-1 stained nuclei from an E16.5 mouse embryo visualized on a Countess Cell Counter.

Nuclei are counted by hand in a 6cm × 6cm square. The method is perhaps inelegant, but in our hands fast and remarkably consistent.
Cell Lysis
Tissue is dissociated by simply smashing it on dry ice (Figure 4, Supplementary Video 1). At this point, RNA is going to be especially vulnerable to RNases that are released by the cells, so a sufficient volume of DEPC-containing lysis buffer is necessary to inactivate them. This is the step that will make or break your experiment, so be sure to do an RNaseAlert check before proceeding to fixation.
Figure 4. Smashing tissue in a foil packet on a slab of dry ice with a hammer.
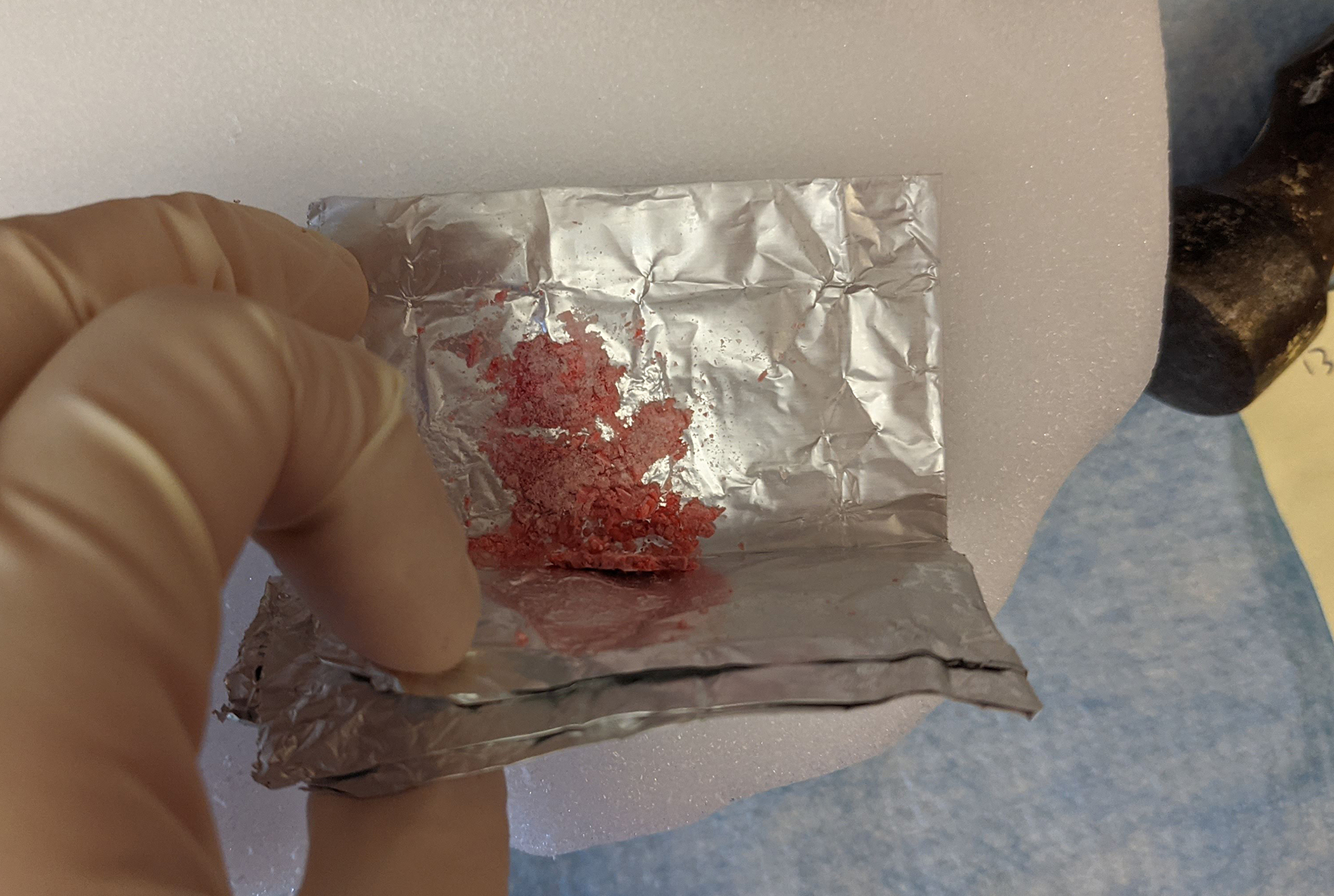
Foil packet with sample must stay on the dry ice until the powdered tissue is added to the lysis buffer.
Lysis Buffer Selection
There are two lysis buffers to choose from, depending on the tissues involved. Older mouse embryos perform better with buffer A, which is sucrose-based and lacks BSA. Most other tissues, like younger embryos, isolated adult tissues (including mouse and human), and cell lines should use buffer B with BSA. We’re not exactly sure why the older mouse embryos benefit from a lack of BSA, it could be that they have so much protein of their own already and that adding more somehow limits the DEPC’s effectiveness.
Reverse Transcription (RT)
This step will take you longer than you think because you will need to get the right amount of nuclei loaded into the first plate, and especially if you are including multiple samples. Allow plenty of time for counting and resuspending samples at the correct concentrations. For a single sample, you will need 2M nuclei to fill out a plate.
Data Analysis
The pipeline for generating single cell digital expression matrix is posted on Github (https://github.com/JunyueC/sci-RNA-seq3_pipeline). The processing is slightly different from the pipeline used for analyzing data which are generated by other technologies (e.g. 10X Genomics): base calls are first converted to fastq format followed by demultiplexing based on PCR i5 and i7 barcodes. Reads are filtered for legitimate RT and ligation indexes and these indexes are added to the read information. After mapping to the reference genome, reads are split into constituent cellular indices by further demultiplexing reads using the RT index and ligation index, and deduplicated by the UMI. The pipeline is based on implementing qsub command on a Linux system (e.g. Centos 7), and we recommend running it on a high performance computing cluster. Of note, it will create some intermediate profiles which are space consuming (around 3–4 times bigger than the original data).
We tested the pipeline on a sample data (created from a mouse embryo at E16.5) with around 7 billion reads. To speed up the processing, we split the whole data set into seven batches after demultiplexing by PCR barcodes, and then run the pipeline on each batch in parallel. Each batch utilized 10 cores (Intel Xeon Gold 6238) with 20G RAM for each core.
Materials
Reagents
Dithiobis (succinimidyl propionate) (DSP, Lomant’s Reagent; Thermo Fisher, cat. no. 22586 or PG82081 ) CRITICAL DSP is sensitive to water and should be used immediately after dissolving in DMSO.
Methanol (Millipore Sigma, cat. no. 494437–2L)
DMSO (Millipore Sigma, cat. no. D2438–5X10ML) CRITICAL DMSO used for dissolving DSP should be new and unopened so that water is not introduced. These smaller bottles are useful for this reason.
Sodium Phosphate Dibasic (Millipore Sigma, cat. no. S3264–250G)
Sodium Phosphate Monobasic Monohydrate (Millipore Sigma, cat. no. 71507–250G)
Potassium Phosphate Monobasic (Millipore Sigma, cat. no. P9791–100G)
Sodium Chloride (Millipore Sigma, cat. no. S3014–500G)
Potassium Chloride (Millipore Sigma, cat. no. P9541–500G)
Magnesium Chloride solution 2M (Millipore Sigma, cat. no. 68475–100ML-F)
Igepal CA-630 (Millipore Sigma, cat. no. I8896–50ML)
Bovine Serum Albumin 20mg/ml (New England Biolabs, cat. no. B9000S)
DEPC (Diethyl Pyrocarbonate) (Millipore Sigma, cat. no. D5758–25ML) CAUTION handle DEPC, and samples containing it, in a fume hood
Sucrose (VWR, cat. no. 97061–428)
TritonX-100 (Millipore Sigma, cat. no. T8787–100ML)
Tween 20 (Thermo Fisher BP-337–100)
10X Dulbecco’s Phosphate Buffered Saline (10XDPBS; Thermo Fisher, cat. no. 14200075)
Superscript IV Reverse Transcriptase (Thermo Fisher, cat. no. 18090200) CRITICAL This protocol has not been tested with other reverse transcriptases.
T4 DNA Ligase (New England Biolabs, cat. no. M0202L) CRITICAL The previous version of this protocol used Quick Ligase, but the buffer that is included with that enzyme interferes with the pelleting of the nuclei during centrifugation.
Tagmentase (Tn5 transposase) - unloaded (Diagenode Cat# C01070010–20) CRITICAL The tuning of the tagmentase activity is written for this particular enzyme.
Tn5-N7 oligo (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′, Eurofins, High-Purity Salt-Free)
Mosaic End (ME) oligo (5′-/5Phos/CTGTCTCTTATACACATCT-3′, Eurofins, High-Purity Salt-Free)
NEBNext® mRNA Second Strand Synthesis Module (New England Biolabs, cat. no. E6111L)
NEBNext high fidelity 2x PCR master mix (New England Biolabs, cat. no. M0541L)
dNTP mix (New England Biolabs, cat. no. N0447L)
Agencourt AMPure XP (Beckman Coulter, cat. no. A63882)
YoYo dye (Thermo Fisher, cat. no. Y3601)
RNaseAlert kit (IDT, cat. no. 11–02-01–02)
RNaseZap (Thermo Fisher, cat. no. AM9780)
Elution buffer (EB, 10mM Tris pH8.5, Qiagen 19086)
Protease (Qiagen, cat. no. 19157) CRITICAL Do not use any other protease/proteinase. This one can be heat inactivated at the temperature and time listed in the protocol.
Qubit dsDNA HS quantitation kit (Thermo Q32851)
sci-RNA-seq3 indexed primer plates at 10μM dilution (standard desalting for purification, random bases do NOT need hand-mixing) The complete list of primers is found in Supplementary Table 1.
Plate(s) of indexed oligo-dT RT primers (5′-/5Phos/CAGAGCNNNNNNNN[10bpRTindex]TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′, where “N” is any base; IDT)
Plate(s) of indexed ligation primers (5’- GCTCTG[9bp or 10bp ligation index]TACGACGCTCTTCCGATCT[reverse complement of ligation index]-3’)
Plate of Indexed PCR P7 primers (5′-CAAGCAGAAGACGGCATACGAGAT[PCR P7 index]GTCTCGTGGGCTCGG-3′, IDT)
PCR P5 primers (this primer doesn’t need to be indexed if you only do one plate of pcr) (5′-AATGATACGGCGACCACCGAGATCTACAC[PCR P5 index]ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′, IDT)
Equipment
Hammer
Lo-bind tubes DNA/RNA (Eppendorf, cat. no. 022431021)
Refrigerated centrifuges that hold 1.5 ml microfuge tubes, microwell plates, 15 ml and 50 ml conical tubes
Chemical fume hood
Multichannel pipettes and tips
FloMi filter 40μm (VWR, cat. no. 10032–802)
Falcon cell strainer 40μm (VWR, cat. no. 21008–949)
Pestle for cell strainer (Midsci, cat. no. SG-PEST)
96-well plates (Eppendorf, cat. no. 951020401)
96-well LoBind plates (Eppendorf, cat. no. 30129512) or Thomas Scientific (cat. no. 1149V59) if LoBind are not available
Thermomixer
Sonicator (Diagenode Bioruptor Plus)
Cell counter with GFP channel, or a hemocytometer that allows visualization with GFP
Biological Materials
E16.5 or E18.5 C57BL/6J embryos
C57BL/6J (WT) mice (Mus musculus) were maintained in a specific pathogen-free environment in the Animal Research and Care Facility (ARCF) at the University of Washington. Mice were kept on a 12-h light/dark cycle with ad libitum access to food and water. Mice were originally bought from the Jackson Laboratory and bred in our facility. Male WT mice at least 6 weeks of age were bred to female WT mice at least seven weeks of age; the first day of plug positive was marked as embryonic day (E)0.5. On specific gestational days (E16.5 or E18.5), dams were euthanized and embryos were collected. Embryos were rinsed in 1xPBS buffer, dabbed dry, and immediately flash frozen in liquid nitrogen. Male or female embryos went into the sci-RNA-seq workflows. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
E8.5 Embryos
E8.5 mouse embryos were harvested as recently described19, dissected free of extraembryonic membranes. Embryos were transferred from the dissecting dish within a minimal volume of PBS (~5μl), placed on the wall of individual cryovials and snap frozen in liquid nitrogen. Samples were stored at −80 °C until further processing.
Reagent Setup
10X-PBS-hypotonic stock solution
Mix 5.45g Na2HPO4 (dibasic), 3.1g NaH2PO4-H20, 1.2g KH2PO4, 1g KCl, 3g NaCl in nuclease-free water and bring to a final volume of 500 ml. This stock solution will be ~pH 6.8, but when diluted to 1X should end up at pH 7.0–7.4. The buffer can be stored at room temperature (20–23°C) for 6 months.
Hypotonic Lysis buffer solution A
Used for whole mouse embryos E16.5 and older
Mix 5ml of the 10X-PBS-hypotonic stock solution, 5.7g sucrose, 75 μl of 2M MgCl2, and nuclease-free water to a final volume of 50 ml to make the lysis base solution. Right before lysis, for every 1 ml of lysis buffer needed, add 2.5 μl 10% igepal (vol/vol) and 10 μl DEPC, then vortex solution to disperse the DEPC throughout. Example: If a sample needs 5 ml of lysis buffer, take a 5 ml aliquot of lysis buffer stock solution, add 12.5 μl 10% igepal (vol/vol) and 50 μl DEPC. Keep buffer on ice. Make fresh for each experiment.
CAUTION DEPC needs to be used in a fume hood.
CRITICAL DEPC has a short half life in aqueous solutions, so it’s important to add it to the buffer just before the cells are added.
Hypotonic Lysis buffer solution B
Used for Tiny-Sci, cell lines, mouse embryos under E16.5, isolated tissues.
Mix 5ml of the 10X-PBS-hypotonic stock solution, 75 μl of 2M MgCl2, and nuclease-free water to a final volume of 50 ml to make the lysis base solution. Right before lysis, for every 1 ml of lysis buffer needed, add 40μl BSA (20mg/ml), 2.5 μl 10% igepal (vol/vol) and 10 μl DEPC, then vortex solution to disperse the DEPC throughout. Example: If a sample needs 5 ml of lysis buffer, take a 5 ml aliquot of lysis buffer base solution, add 200μl BSA, 12.5 μl 10% igepal (vol/vol), 50 μl DEPC. Keep buffer on ice. Make fresh for each experiment.
CAUTION DEPC needs to be used in a fume hood.
CRITICAL DEPC has a short half life in aqueous solutions, so it’s important to add it to the buffer just before the cells are added.
0.3M SPBSTM (Sucrose PBS TritonX MgCl2)
This is the main buffer used throughout the protocol for washing and diluting nuclei. Dissolve 28.5g sucrose in 25ml 10X DPBS (regular DPBS, not the hypotonic version) and 125ml nuclease-free water (about half the volume of water you’ll need). Once the sucrose has dissolved, add 2.5ml 10% TritonX-100 (vol/vol), 375 μl of 2M MgCl2, and more water to the final volume of 250ml. Store this buffer at 4°C for up to 3 months.
DSP 50 mg/ml stock
Dissolve a 50 mg vial of DSP in 1 ml of anhydrous DMSO (use a new vial of DMSO), as DSP will precipitate in aqueous solutions. Dissolved DSP should be used immediately.
Yoyo-1 dye for counting
Dilute 1 μl of Yoyo-1 dye in 1 ml of 0.3M SPBSTM in a dark or amber microfuge tube, and store the reagent at 4°C for up to 3 months. This will be used to dilute nuclei for counting.
Annealed N7 oligos
Tn5-N7 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′
Mosaic End (ME) 5′-[phos]CTGTCTCTTATACACATCT-3′
Resuspend both oligos to 100μM in annealing buffer (50 mM NaCl, 40 mM Tris-HCl pH8.0).
Mix one volume of Tn5-N7 with one volume of ME. This creates a working stock at 50 μM. Anneal them with the following PCR program: 95°C 5min, cool to 65°C (0.1°C/sec), 65°C 5min, cool to 4°C (0.1°C/sec). Store annealed oligos at 4°C for 6 months or aliquot and freeze at −20°C.
N7-loaded Tn5
Tagmentase (Tn5 transposase) - unloaded (Diagenode Cat# C01070010–20).
To 20 μl of Tn5, add 20 μl of annealed N7 oligos. Place in a thermomixer and shake at 350 rpm, 23°C, 30 minutes.
Add 20 μl of glycerol. Store at −20°C for up to 6 months.
Tagment DNA (TD) buffer (2X)
To 38.75 ml of nuclease-free water, add 1 ml 1M Tris pH7.6, 250 μl of 2M MgCl2, 10 ml dimethylformamide. Final volume is 50 ml. Make 550 μl aliquots and store at −20°C for up to 6 months.
Indexed Primer Plates
Primers for reverse transcription, ligation, and PCR indexing steps are ordered at 100 μM. Working dilutions are made to 10 μM in EB, and kept at 4°C up to six months.
10% Igepal (vol/vol)
Dilute 5 ml Igepal in 45 ml nuclease-free water. Store at room temperature up to 6 months.
10% TritonX-100 (vol/vol)
Dilute 5 ml TritonX-100 in 45 ml nuclease-free water. Store at room temperature up to 6 months.
10% Tween20 (vol/vol)
Dilute 5 ml Tween20 in 45 ml nuclease-free water. Store at room temperature up to 6 months.
Protease
Add 7ml water to a bottle of lyophilized Qiagen protease (Qiagen #19157). Make 200μl aliquots and store −20C up to 6 months. Don’t freeze/thaw. CRITICAL: This must be protease, NOT proteinaseK.
Protocol
Nuclei Isolation
Timing: 2 hours
-
1
Remember everything is to be kept cold all the time. Prepare two ice buckets with wet ice, a bucket with crushed dry ice to hold your frozen tissues, and a thick, flat slab of dry ice for smashing tissues. Precool a centrifuge that will hold 50ml tubes, and a microfuge, to 4°C.
-
2
Determine how much lysis buffer you will need for the tissue you will be processing. (For extremely small samples, skip to Box 1: Tiny-Sci). A E13.5 mouse embryo (~200 mg) works with 5 ml of lysis buffer. A E16.5 embryo (~500 mg) will need 20 ml. An adult mouse heart needs 5 ml. Adult mouse kidneys need 5 ml per kidney. Adult mouse liver needs 20 ml. Adult mouse pancreas needs 15ml. Adult tissues and tissues high in RNases will necessitate a bigger lysis volume. The buffer is inexpensive to make so don’t worry about using too much.
CAUTION DEPC is flammable and toxic. Avoid breathing vapors. The following steps should be performed in the chemical hood from this point until the DEPC is washed from the sample (step 17).
-
3
For every 1 ml of lysis buffer needed, add 2.5 μl 10% igepal (vol/vol), 10 μl DEPC to the hypotonic lysis buffer solution (and 40μl BSA if using lysis buffer B), then vortex solution to disperse the DEPC throughout. Have complete lysis buffer in a 50 ml tube for each sample on ice ready to go.
-
4
Fold a piece of aluminum foil 4 times so that you have a small pouch with 8 layers of foil on each side. Place this on a slab of dry ice to chill.
-
5
Place your frozen tissue inside this foil and hold it firmly closed on the dry ice and smash it with a hammer (Figure 4, Supplementary Video 1). You want to be gentle enough not to tear the foil, but thorough enough to make a powder of the tissue. Do not let the tissue thaw.
-
6
Use the foil to guide your powdered tissue into the tube of the lysis buffer. It will stick a bit, pipet some of the lysis buffer from the tube to rinse the sample from the foil into the tube. Try to make sure that the sample is only thawing if it is in lysis buffer.
-
7
Cap the 50 ml tube and shake to disperse the chunks in the buffer. Let sit on ice for 10 min. Triturate the chunks with a 1 ml pipette tip to help tease them apart a bit.
-
8
Set up another 50 ml tube on ice with a 40 μm cell strainer on top. Pour your lysate through that – there will still be a lot of chunks. Use a disposable pestle to coax the tissue through the filter. Don’t worry about getting all of it through.
-
9
Take 45 μl sample of the filtered lysate and check for Rnase activity with the IDT RnaseAlert kit. The RNaseAlert test will guide you on whether to proceed or not. There should not be any RNase detected, and if there is, you will have to restart with a new sample and adjust either the sample size or the volume of lysis buffer, so that there is enough DEPC to inactivate the RNases. You cannot continue with a sample that has RNases detected at this point, the damage is already done. TROUBLESHOOTING
-
10
While the RNaseAlert sample is incubating, spin down the remainder of the lysate (500×g, 3 min, 4°C). Keep the nuclei in the 50ml tube. Resuspend the nuclei in 1 ml 0.3M SPBSTM with 10μl DEPC added (or more buffer if there are a lot of nuclei - roughly 1ml buffer per 200mg-500mg starting material at minimum). TROUBLESHOOTING
Box 1. Tiny-Sci.
Timing: 1 hour
Procedure
Samples should be no more than 2–3mm in size, frozen individually in a microfuge tube and kept on dry ice until lysis. The key to these small samples is to limit pipetting and tube transfers that will leave behind precious nuclei.
You will need 100 μl lysis buffer B per embryo, but make up enough for several samples.
-
Right before lysis, for every 1 ml of lysis buffer needed, add 2.5 μl 10% igepal (vol/vol), 40 μl BSA (20 mg/ml), 10 μl DEPC, then vortex solution to disperse the DEPC throughout.
CAUTION you must work in the hood because the DEPC is toxic.
Add 100 μl of complete lysis buffer (with BSA/DEPC/igepal) to the tube with the frozen embryo, and make sure the embryo is actually in the buffer. Let sit a couple of minutes on ice, then triturate the embryo slowly and gently with a pipette set to 50 μl with a yellow tip. You shouldn’t see any chunks left. Lysis time is only about 5 min.
Optional: You can take 1 μl of this and mix with 9 μl of diluted yoyo dye to quickly check under a microscope with a GFP filter to make sure things are looking good at this point.
Mix fixative: 400 μl ice-cold methanol + 10 μl DSP stock solution. Add 400 μl to the embryo on ice, dripwise over a minute or two. Do not pipette up and down, instead flick gently to mix and occasionally over 5–10 min. You may see some clumping happening now.
Add 1 ml 0.3M SPBSTM, dripwise, slowly and mixing gently by flicking. Do not pipette up and down.
Spin 500g, 3 min, 4°C. You should see a very tiny pellet. Remove all but about 50 μl of the supernatant, without disturbing the pellet.
Add 500 μl 0.3M SPBSTM. Set 1 ml pipette to 100μl and resuspend by pipetting gently.
If you got clumps from fixing, sonicate the tube for 12s on low.
Spin again 500×g, 3 min, 4°C. Carefully remove supernatant and resuspend gently in SPBSTM so that the volume is 42.5 μl. Add 2.5 μl 10mM dNTPs and put 5 μl into each well of 1 column on a plate, noting which wells the nuclei went into.
Fill up the plate with more embryos or some other nuclei that you have a lot of, and continue with the sci protocol at the reverse transcription step 24 as normal.
Nuclei Fixation
Timing: 1 hour
-
11
For each sample, prepare fixative: In a 5 ml tube, add 100 μl of 50 mg/ml DSP stock solution to 4 ml of ice cold methanol for every 1 ml of nuclei that you are starting with.
-
12
Add the prepared fixative to the 50ml tube containing the nuclei gradually, swirling as you are adding it.
-
13
Fix on ice for 15 min, swirling occasionally.
-
14
Add 2 volumes 0.3 SPBSTM gradually, 2–3 milliliters at a time, swirling in-between additions to rehydrate the nuclei. For instance, with 1 ml of nuclei and 4 mls of fixative, you would need 10ml of buffer to rehydrate.
-
15
Spin down the nuclei at 500×g 3min 4°C.
-
16
Carefully remove supernatant and dispose properly. The nuclei pellet is at the bottom and should look a little white-ish from the DSP.
-
17
Resuspend the nuclei in 1 ml (or more) 0.3M SPBSTM. Triturate gently with a pipette tip to separate nuclei.
-
18
OPTIONAL: If there are obvious clumps at this point that won’t tease apart, you will need to sonicate them to break them up. Sonicate (Diagenode Bioruptor Plus) on low intensity for 12s only at 4°C. Spin and resuspend the nuclei in 1 ml 0.3M SPBSTM. TROUBLESHOOTING
-
19
Divide fixed nuclei into aliquots in microfuge tubes. Spin 500×g 3 min 4°C and remove supernatant. Snap freeze tubes in LN2 and store at −80°C. PAUSE POINT - Fixed samples may be stored at −80C up to 6 months.
Reverse Transcription (RT)
Timing: 2–3 hours
-
20Follow the chart below to determine how many starting nuclei you need and their volume. If you are only doing sci on one sample (filling all 12 columns of the 96-well plate with that sample), then follow the volumes outlined by the box. Figure 5 shows the approximate size of a pellet with 2M nuclei needed for a whole plate. If you are dividing the plate among multiple samples, then the chart will outline the cell number you need depending on how many columns in the plate each sample will occupy. For example, if you will have two samples on one plate, each one will take 6 columns and you will need 1M cells of each sample in a tube to start with in a volume of 250μl each, and you will add 28μl of dNTPs to each before distributing to their RT plate wells.
cell number: 2M 1M 800K 500K 400K 200K number of columns: 12 6 4 3 2 1 nuclei volume 500 μl 250 μl 170 μl 125 μl 85 μl 42.5 μl 10mM dNTP 56 μl 28 μl 19 μl 14 μl 9.5 μl 4.75 μl -
21
Resuspend an aliquot of frozen nuclei in 500 μl of 0.3M SPBSTM to start. Count. Dilute nuclei if necessary to get an accurate count. If nuclei are clumpy, even after sonicating, and can’t be teased apart with pipetting, then put them over a 40μm Flow-mi pipette tip filter before counting. Flow-mi filter is a last resort as it results in nuclei loss, but is helpful if you have an excess of nuclei.
-
22
Pull out the desired amount of nuclei into a new tube and spin. Remove supernatant and resuspend nuclei in the necessary volume determined by the chart and add the appropriate amount of dNTPs.
-
23
Aliquot 5 μl nuclei+dNTP mix to each well of the plate on ice.
-
24
Quickly spin the plate of 3-level RT primers (10μM).
(5′-/5Phos/CAGAGCNNNNNNNN[10bpRTindex]TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′, where “N” is any base)
-
25
Add 2 μl of primer to each well. Don’t pipet up and down to mix, just stir gently with the pipet tips.
-
26
Incubate plate at 55°C for 5 min (heated lid set to 65°C) and then immediately place on ice.
-
27While this is incubating, make the reaction mix. Note: we are not including DTT in this mix, as it will undo the DSP crosslinks (it is not necessary for the RT to work).
RT mix per plate: each X120 5X Superscript IV buffer 2 μl 240 μl Superscript IV (200 u/μl) 0.5 μl 60 μl water 0.5 μl 60 μl -
28
Put 3μl of reaction mix into each well (45 μl mix x8 in strip tube for multichannel), stirring gently with tips. 10 μl total now.
-
29
Incubate 55°C 10 min (heated lid at 65°C) and then immediately place on ice.
-
30
Ice plates until they are cold (10–15min). Add 5 μl cold 0.3M SPBSTM per well. To maximize recovery, pool wells by using a 12-multichannel with 200μl tips to pipet gently up and down (the pipetting up and down is important to dislodge the nuclei, but try to avoid creating excessive bubbles), and combine each row of the plate into the bottom row. You can use the same tips for the whole plate. Then collect these wells into 2 cold microfuge tubes. (It will be bubbly so it’s difficult to squeeze into 1 tube).
-
31
Spin 500×g, 3min, 4°C. Pellet will be small but you should be able to see it. Remove supernatant.
-
32
Combine tubes and wash once more in 1 ml cold 0.3 SPBSTM. Spin 500×g, 3min, 4°C. Remove supernatant.
Figure 5. Nuclear pellet size.
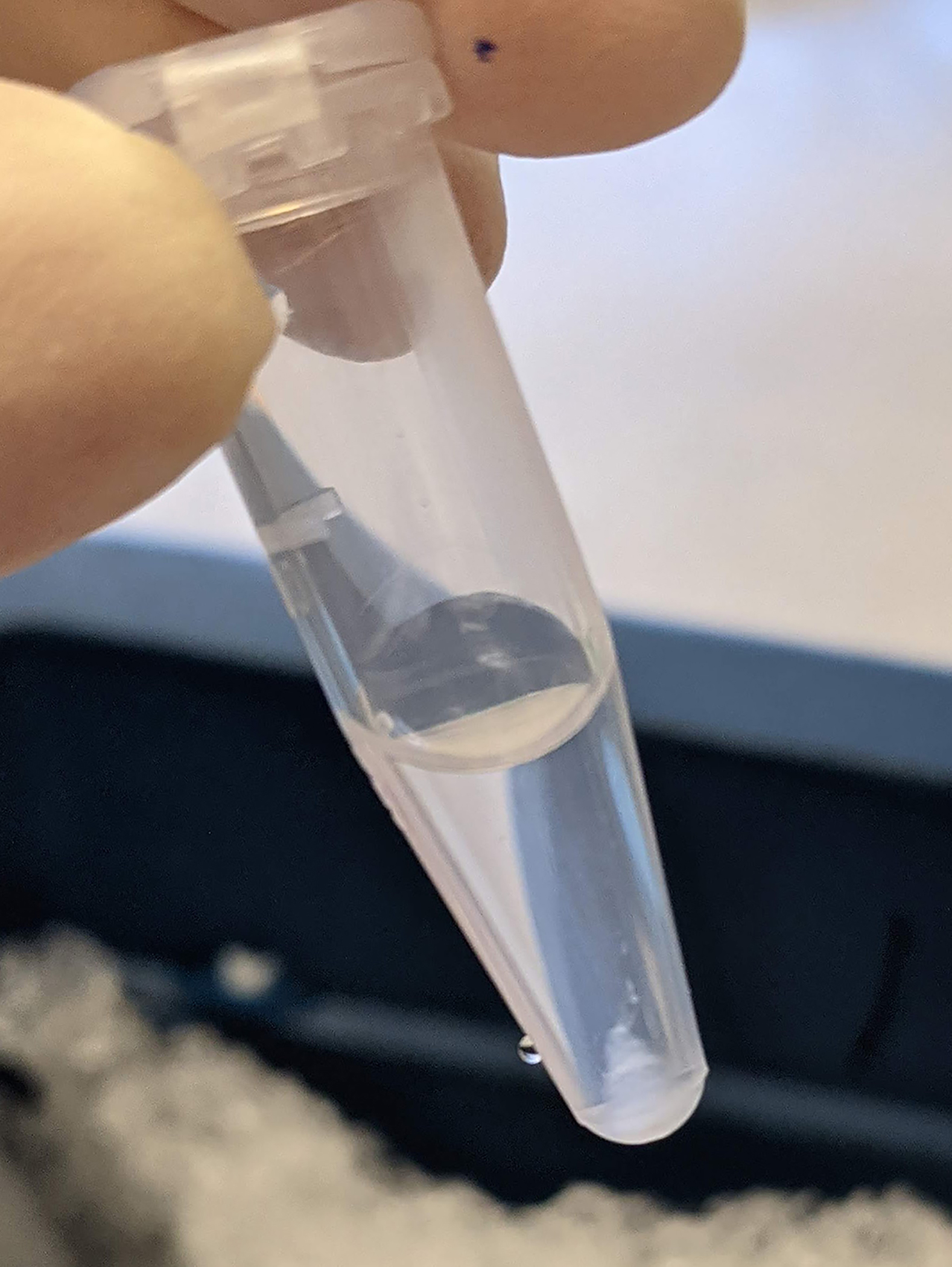
This is approximately the size of the nuclei pellet (~2 million nuclei) needed for 1 plate of reverse transcription. Extra fixed nuclei can be aliquoted and snap-frozen.
Ligation
Timing: 1.5 hours
-
33
Resuspend nuclei in 1200 μl 0.3M SPBSTM.
-
34
Distribute 11 μl to each well of a new plate on ice.
-
35
Quick spin the plate of 3-level ligation primers (10 μM)
(5’- GCTCTG[9bp or 10bp ligation index]TACGACGCTCTTCCGATCT[reverse complement of ligation index]-3’)
-
36
Add 2 μl of primer to each well. Don’t pipet up and down.
-
37
Make a 3:1 mix of 10X T4 ligation buffer and T4 DNA ligase. (195 μl10X buffer + 65 μlT4 DNA Ligase)
-
38
Add 2 μl ligase mix to each well. (32 μl x8 in strip tube for multichannel).15 μl total now.
-
39
Incubate 20 min at room temperature.
-
40
Ice plates until cold.
-
41
Add 10μl cold SPBSTM to each well. This helps keep the cells from clumping and allows for more cell recovery.
-
42
Pool wells by using a 12-multichannel to pipet gently up and down (the pipetting up and down is important to dislodge the nuclei), and combine each row of the plate into the bottom row. Then collect these wells into 2 cold microfuge tubes.
-
43
Spin 500×g, 3min, 4°C. Remove supernatant.
-
44
Combine the two tubes and wash twice more with 1 ml 0.3M SPBSTM per wash.
-
45
Resuspend in 1 ml 0.3M SPBSTM to count. If they are clumpy and can’t be teased apart with gentle pipetting, put through a tip filter and recount. TROUBLESHOOTING
Final Distribution
Timing: 1 hour
In the final plate you will want 1000 nuclei/well (or 4000/well if you’ve scaled up the experiment to 384×384×384). You should have enough nuclei to freeze multiple plates if you like.
-
46
Make 400 μl 1X Second Strand Synthesis buffer for each plate in the final distribution: Dilute 40 μl 10X Second Strand buffer in 360 μl water to get 1X concentration.
-
47
Spin down 100K nuclei for each plate desired for the final distribution. (400K per plate if this is a 384×384×384 experiment). For each plate/100K, resuspend in 400 μl 1X Second Strand Synthesis buffer.
-
48
Put 4 μl nuclei into each well of a regular, not lo-bind, plate on ice.
-
49
Cover with foil seals and freeze plates at −80°C or proceed with second strand synthesis. PAUSE POINT - Plates may be kept frozen at −80°C for up to 6 months.
Second Strand Synthesis
Timing: 3 hours (or overnight)
-
50
Thaw plate on ice.
-
51Make second strand synthesis mix on ice as follows:
reaction mix per plate: each X140 water 0.675 μl 94.5 μl second strand buffer (10X) 0.075 μl 10.5 μl second strand enzyme (20X) 0.25 μl 35 μl -
52
Put 1 μl of second strand synthesis mix into each well (17 μl mix x8 in strip tube for multichannel). 5 μl total now.
-
53
Incubate 16°C 2.5hours. (No heated lid) PAUSE POINT - Keep the plate at 4°C up to 24 hours.
Protease Digestion
Timing: 2 hours
-
54
Add 1 μl protease to each well. NOTE: This is NOT proteinaseK. It’s important to use Qiagen protease (#19157) because it can be heat-inactivated. See reagent preparation section.
-
55
Incubate 37°C for 30min (47°C heated lid). Check 1μl on microscope: mix 1μl sample with 2μl of diluted yo-yo1 dye and put this on a slide and check on the GFP channel. You should see whisps of DNA instead of intact nuclei. (Figure 6)
-
56
Heat-inactivate the protease 75°C 20min (85°C heated lid). CRITICAL Do not lower this temperature, do not shorten this time. Qiagen lists different conditions for heat-inactivating their protease, but it is not sufficient. Put plate on ice after inactivating. PAUSE POINT - Store plate at 4°C for up to a week.
Figure 6. Visualizing protease digestion of the nuclei.

a, Nuclei after about 10min of protease digestion, swelling and starting to lose integrity. b, Nuclei after 30min of protease digestion; DNA has been released and now the protease can be heat-inactivated.
Tagmentation
Timing: 1 hour
-
57On ice, make tagmentation mix as follows:
reaction mix per plate: each X110 TD buffer 5 μl 550 μl N7-loaded Tn5 0.125 μl 13.75 μl -
58
On ice, add 5 μl tagmentation mix to each well. ~10–11 μl total now.
-
59
Incubate 55°C 5min, do not put on ice afterwards, just keep on the bench at room temperature as you add the next reaction mix, or else the SDS will come out of solution at the next step.
-
60Remove the transposases with this buffer (keep at room temperature):
reaction mix per plate: each X120 1% SDS (wt/vol) 0.4 μl 48 μl BSA 0.4 μl 48 μl water 1.8 μl 216 μl 2.6 μl -
61
Add 2.6 μl to each well and mix (39 μl x8 into a strip tube for multichannel).
-
62
Incubate 55°C 15min.
-
63
Quench SDS by adding 2 μl 10% Tween 20 (vol/vol) to each well (bolded this because it is a very easy step to forget)
PCR amplification
Timing: 1 hour for PCR, 2–3 hours for gel purification
-
64
Assemble the PCR master mix. PCR is done with 96 indexed P7 primers and 1 P5 primer. You can also add an optional index sequence on the P5 primer for multiplexing multiple plates.
Plate of 96 Indexed PCR P7 primers (5′-CAAGCAGAAGACGGCATACGAGAT[PCR P7 index]GTCTCGTGGGCTCGG-3′)
P5 primer(s) (5′-AATGATACGGCGACCACCGAGATCTACAC[PCR P5 index]ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′)reaction mix per plate: each X110 2X NEBNext 20 μl 2200 μl TruSeqP5 primer (100μM) 0.2 μl 22 μl water 3.2 μl 352 μl total 23.4 μl 2574 μl -
65
Add 2 μl of indexed P7 primers (10μM) to each well.
-
66
Add 23.4 μl of PCR master mix to each well.
-
67Amplify 16 cycles with a pre-extension step in the following program:
1 70°C 3 min 2 98°C 30s 3 98°C 10s 4 63°C 30s 5 72°C 1 min 6 go to step 3, 15 more times 7 72°C 5 min -
68
Run 1.5 μl of a few wells on a 6% PAGE gel to check. You should see a smear of products with primer-dimers underneath (Figure 7a). We will be isolating a section of the smear, centered on 400bp. TROUBLESHOOTING
-
69
Concentration of library and agarose gel purification: Pool 3 μl of each well and do a 0.8X ampureXP cleanup (230 μl beads). (Save the remaining plate in case you need to redo cleanup or if you anticipate needing more library for a large NovaSeq run). Wash the ampure bead pellet twice gently with 70% EtOH (vol/vol) and elute pool in 50 μl. Load this into a single 1 cm well on a 1% agarose gel (wt/vol). (Figure 7c). Cut out the smear between about 250–600 bp and use the NEB gel extraction kit, using extra dissolving buffer since it will be bigger than a normal slice and run it all through the same purification column. Wash twice with 200 μl NEB wash buffer, elute in 20 μl EB. Quantitate library with Qubit dsDNA HS.
-
70
Run the library on NextSeq (or NovaSeq depending on final cell numbers or sequencing depth desired), using standard primers. Read1 34 cycles, Index 10 cycles, Read2 48 cycles. If you’ve also used a P5 index for pcr, then add a second Index read of 10bp.
Figure 7. Evaluating the libraries after PCR.

Yellow bars indicate the size range to look for. a, A good quality library - sampling of 8 wells from the pcr plate, 1.5 μl of each well is run on a 6% PAGE gel, 200V for 30 min. A bright, long smear of products above the 200 bp marker indicates a robust library. This one is from a 384×384 experiment with 4000 nuclei plated in each well of the last plate. b, A medium quality library with fainter smears. This was from a 96×96 experiment that underperformed - quality of the data was good and the number of UMIs per cell was as expected, but had less total cells overall, most likely due to counting estimates during the last round of plating. c, Gel size-selection. Libraries have been concentrated via ampure purification and run on a 1% agarose gel at 100V for about 1 hour. Most of the primer dimers are gone, and the libraries are cut out of the gel at the. This is a 384×384 experiment so these smears are very intense. d, A failed experiment. The smaller number of primer dimer bands suggests that the failure resulted from incomplete protease digestion. e,f, The final, gel-purified library fragment distribution as measured on an Agilent tapestation. Primer dimers have been removed and you should be left with a library ranging from 300–700bp in size.
Troubleshooting Guide
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 9 | RNase check is positive for RNase | Sample:lysis buffer volume ratio is too large, tissue is especially rich in RNases. | Redo with fresh sample, and add more lysis buffer, or decrease sample size, until you find a ratio that shows that all RNase has been deactivated. More lysis buffer is always better. |
| 10 | Nuclei are clumping when they are lysed, | Some cells are more delicate and clumping indicates that DNA is leaking out of the nuclei. | Try a shorter lysis, less igepal in the lysis buffer, or more BSA in the lysis buffer. Make sure that you included MgCl2 in the lysis buffer |
| 18 | Nuclei looked fine after lysis, but clumped after fixation. | Some cells are more delicate and clumping indicates that DNA is leaking out of the nuclei. | If sonicating the nuclei doesn’t break up the clumps, try a shorter lysis, less igepal in the lysis buffer, and/or more BSA in the lysis buffer. Try fixing and rehydrating more gradually. |
| 45 | Losing too many nuclei when washing | It’s not unexpected to lose half the nuclei from the 2M that started in the RT plate. Recovery can be maximized by being mindful that every transfer of the nuclei will lose some to the walls of the tubes and pipettes. The supernatant doesn’t always have to be completely removed for the washes if there is a chance to disturb the pellet. The biggest losses seem to happen when pooling wells, so at those steps make sure to gently pipet up/down a few times to dislodge settled nuclei before pulling them out of the well. | |
| 46 | Cells are sticking after ligation | DSP not fixing | Make a fresh batch of DSP, be sure to use anhydrous DMSO for a solvent. Some lots of DSP have been problematic. Some little clumps are expected. If you have a lot of cells, put them over a flowmi filter before continuing, to remove the clumps. |
| 68 | No smear of library on gel | Sample quality is the biggest factor, bad reagents. | If you’ve saved extra fixed nuclei, or extra final plates, you can retry from RT at step 20 or second strand synthesis at step 48 with fresh reagents. Optionally, take an aliquot of frozen nuclei and bulk RNA extract to make sure you are seeing RNA at all. Retry experiment with less tissue in the lysis. |
| 68 | Spotty wells, or missing smears | Spotty wells can indicate that the protease was incompletely inactivated. | |
| 68 | Smear is mostly too high or mostly too low | You might see issues here with under- or over-tagmenting. If you have extra plates, you might try increasing or decreasing your Tn5 2-fold. But don’t worry if it’s not perfectly sized, with all the different length transcripts this is difficult to tune. Any smear centered at about 400–600 is great. | |
Timing
Steps 1–10, nuclei isolation: 2 h
Steps 11–19, nuclei fixation: 1 h
Steps 20–32, reverse transcription: 2–3 h
Steps 33–45, ligation: 1.5 h
Steps 46–49, final distribution: 1 h
Steps 50–53, second-strand synthesis: 3 h (or overnight)
Steps 54–56, protease digestion: 2 h
Steps 57–63, tagmentation: 1 h
Steps 64–69, PCR and gel purification: 3–4 hours
Step 70, Loading the sequencer 1 h
Sequencing 13–20 h
Bioinformatic Analysis 8–10 h
Anticipated Results
The success of the experiment can be evaluated at the end of library generation after the third-round PCR step. You should see a smear of products on 6% TBE polyacrylamide gel above 200bp. (Figure 7a). The intensity of the smear is a good indicator of the cell/UMI number that you will see in the final data (compare Figures 7a and 7b). An absence of a smear (Figure 7d) can result from poor quality tissue where the RNA has been degraded, or a failure somewhere in the protocol. Primer dimers are expected, and after gel purification should be completely removed, as visualized on the Tapestation (Figure 7e–f).
In bioinformatic analysis, during the UMI Attach step where the reads are filtered for the expected indexes, you should see a filter rate of about 75–80% or higher. Aligning reads with STAR should see a mapping rate over 70% (okay) to 80% (better). For a single NextSeq 550 High sequencing run of one plate of PCR at 1000 cells/well, you should expect 300,000–400,000 reads, with less than 30% PCR duplicates. At 4000 cells/well, you should see a duplication rate less than 20%.
To evaluate the performance of the improved protocol, we utilized the sci-RNA-seq3 method on an E16.5 mouse in a 192×192 experiment (2 plates of RT indexes, 2 plates of Ligation indexes) with 3.5 plates of PCR indexes. There were 2000 nuclei/well placed in the final PCR plates, for potentially 700,000 nuclei in total. The pooled library was sequenced on a NovaSeq and after filtering resulted in 381,888 nuclei with high quality (for these, the median UMI count per cell is 2,648; the median gene count detected per cell is 1,493). The resulting 2D UMAP visualization of cells with their cell-type annotation labels is shown in Figure 8. The details of data analysis and more results can be found in Supplementary Information.
Figure 8. High-quality data of E16.5 mouse embryo generated by application of the optimized sci-RNA-seq3 protocol.

2D UMAP visualization of the new E16.5 dataset. All nuclei colored by each of the 20 cell trajectories are shown on the left. Subview of global 2D UMAP visualization highlighting subpopulations of the white blood cells trajectory is shown on the right.
SOURCE DATA/DATA AVAILABILITY
Raw data from the E16.5 mouse embryo is available for download from the NCBI Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE186824.
Original photos and gels have been deposited at Figshare (10.6084/m9.figshare.c.5915834)
Supplementary Material
Acknowledgements
We thank Bridget Kulesakara for the sucrose buffer advice, Jase Gehring for fixative expertise and Riza Daza for helpful feedback. We thank Diana O’Day, Mai Le, Roshella Gomes, Saskia Ilcisin, Dana Jackson for protocol testing, and the entire Shendure Lab for support and encouragement. This work was funded in part by NIH R01HG010632 to JS and CT, UM1HG011586 to JS and BB, R35GM137916 to BB and T32HG000035 to EN. JS is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Ethics declarations
Competing interests
J.S. is a scientific advisory board member, consultant and/or cofounder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies and Scale Biosciences. C.T. is a founder of Scale Biosciences. All other authors have no competing interests.
References
- 1.Domcke S et al. A human cell atlas of fetal chromatin accessibility. Science 370, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao J et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg AB et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datlinger P et al. Ultra-high throughput single-cell RNA sequencing by combinatorial fluidic indexing. doi: 10.1101/2019.12.17.879304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vitak SA et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302–308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin Y et al. High-Throughput Single-Cell Sequencing with Linear Amplification. Mol. Cell 76, 676–690.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramani V et al. Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulqueen RM et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 36, 428–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao J, Zhou W, Steemers F, Trapnell C & Shendure J Sci-fate characterizes the dynamics of gene expression in single cells. Nat. Biotechnol. 38, 980–988 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang B et al. SCITO-seq: single-cell combinatorial indexed cytometry sequencing. Nat. Methods 18, 903–911 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q et al. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 76, 206–216.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Srivatsan SR et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 367, 45–51 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivatsan SR et al. Embryo-scale, single-cell spatial transcriptomics. Science 373, 111–117 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao J et al. A human cell atlas of fetal gene expression. Science 370, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu C et al. Systematic reconstruction of cellular trajectories across mouse embryogenesis. Nat. Genet. 54, 328–341 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrenberg L, Fedorcsak I & Solymosy F Diethyl pyrocarbonate in nucleic acid research. Prog. Nucleic Acid Res. Mol. Biol. 16, 189–262 (1976). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.