

Summary
T cells are at the centerstage of cancer immunology due to their ability to recognize mutations within tumor cells and directly mediate cancer cell killing. Immunotherapies to rejuvenate exhausted T cell responses have transformed the clinical management of several malignancies. In parallel, the development of novel multidimensional analysis platforms such as single-cell RNA-sequencing and high-dimensional flow cytometry has yielded unprecedented insights into immune cell biology. This convergence has revealed substantial heterogeneity of tumor-infiltrating immune cells, both within single tumors, across tumor types, and among cancer patients. Here, we discuss the opportunities and challenges of studying the complex tumor microenvironment with -omics technologies that generate vast amounts of data, highlighting the opportunities and limitations of these technologies with a particular focus on interpreting high-dimensional studies of CD8+ T cells in the tumor microenvironment.
The promise of immunotherapy
Cancer immunotherapy – the augmentation of anti-tumor immunity – has revolutionized the treatment of many cancers. Dozens of immunotherapies have received FDA approval, and thousands of immuno-oncology agents are under development (Upadhaya et al., 2020). Current types of cancer immunotherapy include cytokines, depleting antibodies, adoptive cell therapy, oncolytic viruses, cancer vaccines, and immune checkpoint inhibitors (ICIs) (Esfahani et al., 2020). As key mediators of anti-tumor immunity, T cells are the target of most of these therapeutic strategies. ICIs, in particular, are the most widely used cancer immunotherapy and modulate signaling pathways of exhausted CD8+ T cells in order to enhance the anti-tumor response (Robert, 2020). The unprecedented success of ICIs in the clinical management of cancer combined with the transience or complete lack of responses in most patients has spurred significant efforts over the past decade to gain a better understanding of CD8+ T cell differentiation and dysfunction in cancer to ultimately improve immunotherapy response rates.
Studies of T cell differentiation were revolutionized by the use of peptide-MHC (pMHC) I multimers, allowing the tracking of antigen-specific T cells in a manner independent of their functionality (Altman et al., 1996). Much early work on antigen-specific T cell differentiation was performed in mouse models, especially the lymphocytic choriomeningitis virus (LCMV) system that enables the investigation of T cells specific for identical epitopes under acute and chronic antigen stimulation settings (Wherry et al., 2007; Zajac et al., 1998). Until recently, studies of antigen-specific T cell differentiation in humans had focused almost exclusively on viral infections, including yellow fever virus (YFV) (Akondy et al., 2017; Akondy et al., 2009; Miller et al., 2008), Epstein-Barr Virus (EBV) (Amyes et al., 2003; Long et al., 2019; Murray et al., 1992), Cytomegalovirus (CMV) (Khan et al., 2002a; Klenerman and Oxenius, 2016; Pourgheysari et al., 2007), and influenza (Gotch et al., 1987; Hufford et al., 2014; Koutsakos et al., 2019).
These studies showed that, following antigen exposure, naïve CD8+ T cells differentiate into effector cells and express high amounts of cytotoxic proteins, cytokines, and markers of division (Fig. 1). Antigen clearance results in de-differentiation of a subset (Kaech et al., 2003) of effector cells into memory cells, which lack effector molecules but are primed for rapid recall of cytotoxic function (Youngblood et al., 2017). In cases of antigen persistence, such as cancer, CD8+ T cells exist in several differentiation states (Eberhardt et al., 2021; Hudson et al., 2019a; Im et al., 2016; Philip and Schietinger, 2019). One such subset, PD-1+ stem-like cells, has been of immense interest in the field of cancer immunology as this subset of CD8+ T cells serves as a reservoir maintaining a relatively stable pool of antigen-specific T cells during antigen persistence (Siddiqui et al., 2019). Most importantly, these stem-like cells mediate the proliferative burst of antigen-specific CD8+ T cells observed after PD-1 pathway blockade, a finding first described in the murine LCMV model (Im et al., 2016).
Figure 1:
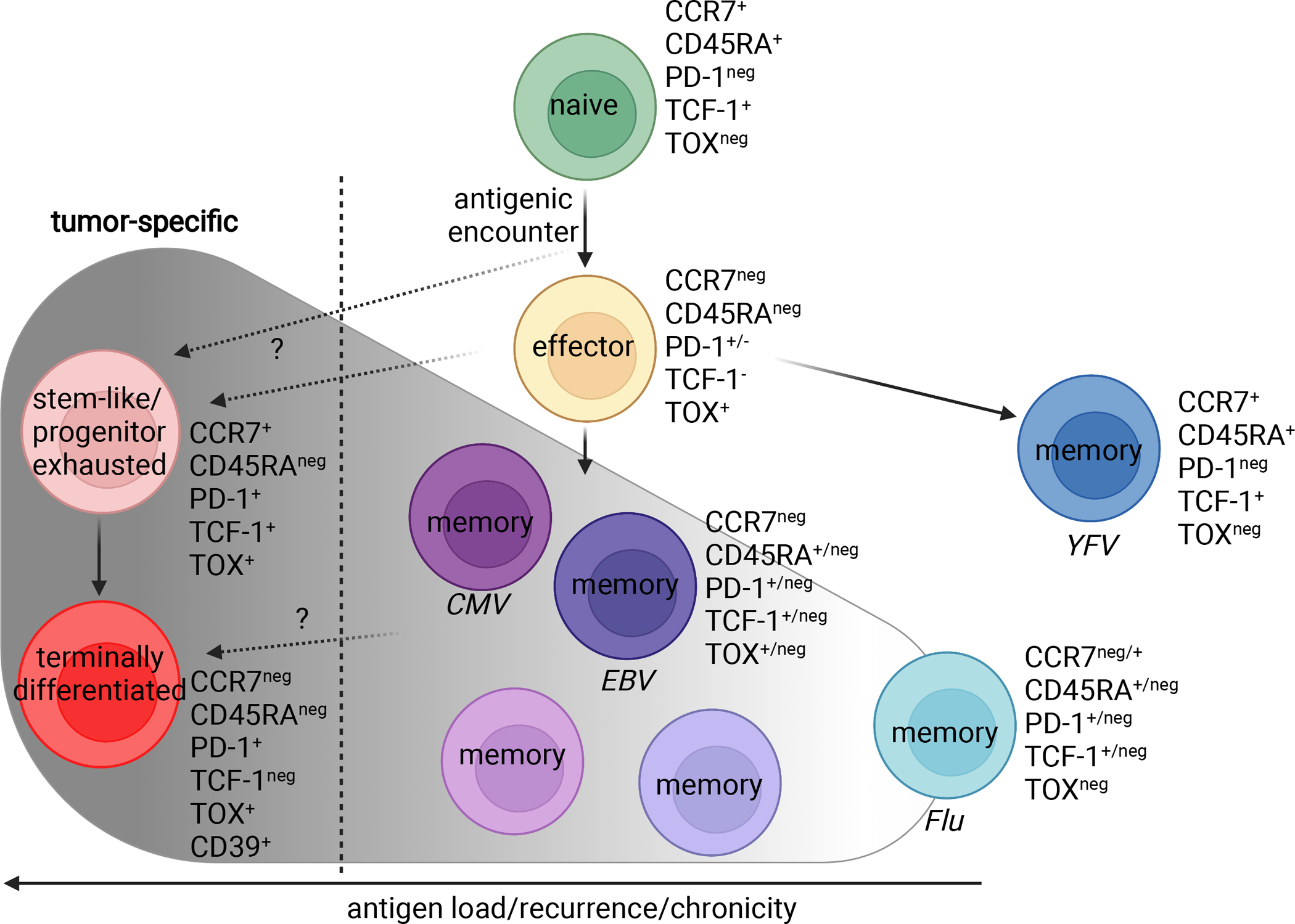
Human CD8+ T cell differentiation and associated phenotypic markers. Tumor-specific phenotypes are shown in red. Upon antigenic encounter, naïve CD8+ T cells differentiate into effector cells and further differentiate into different states depending on antigen clearance and chronic/recurrent antigen loads. Memory T cells specific for yellow fever virus (YFV) and the common human pathogens Influenza virus (Flu), Epstein-Barr virus (EBV) and cytomegalovirus (CMV), as well as tumor-specific cells (stem-like/progenitor exhausted and terminally differentiated) are shown along a schematic continuum of antigen load/recurrence. Of note, CMV- and EBV-specific memory CD8+ T cells can adopt various phenotypes and even resemble the phenotype of tumor-specific stem-like CD8+ T cells. Figure created with BioRender.
The heterogeneity of the tumor microenvironment
In contrast to the early focus on viral infections, hundreds of studies have been performed in recent years to characterize T cell responses to cancer. This increased interest in anti-tumor T cell responses has coincided with a revolution in biological techniques that has advanced tumor immunology. In particular, next-generation sequencing has provided the ability to measure the expression of thousands of genes in pools of cells, in situ within tissue sections, or in single cells. Other technologies, including flow cytometry and gene editing, have greatly increased their capabilities and adoption by researchers in the cancer immunotherapy era.
These advances in methodology have revealed many important features of tumor immunology; most notably, the diversity of tumor-infiltrating immune cells. This diversity includes differences in anti-tumor immunity between cancer patients as well as the variety of immune cell types within a single tumor microenvironment (TME). Further, not only are many cell types present within tumors, but each of these cell types can also be present in multiple states within the same TME, a feature true not only of T cells, but also of B cells (Wei et al., 2021), myeloid cells (Veglia et al., 2021), fibroblasts (Helms et al., 2020), as well as tumor cells themselves (Patel et al., 2014). In this Perspective, we will discuss the opportunities and challenges that have arisen as modern immunology techniques have encountered the complexity of the TME, with a particular focus on anti-tumor CD8+ T cell immunity.
Specificity is key: bystanders within the TME
Heterogeneity in clinical responses was an early feature of ICI use in cancer (Brahmer et al., 2010; Topalian et al., 2012), prompting the search for clinical correlates of immunotherapy response. One of the first variables tested for correlation with clinical response to PD-1 pathway blockade was pre-existing infiltration of tumors by CD8+ T cells (Herbst et al., 2014; Tumeh et al., 2014). In many cases, a higher level of CD8+ T cell infiltration predicts response to ICIs (Galon and Bruni, 2019; Tumeh et al., 2014), but this correlation has not been found in all studies (Braun et al., 2020; Herbst et al., 2014). Studies in patients with various cancers have shown that a substantial proportion - in some cases even the majority - of intratumoral CD8+ T cells are bystander cells; that is, they are not specific for tumor antigens and are either predominantly reactive to common human pathogens such as EBV, CMV, and influenza (Cheng et al., 2021; Oliveira et al., 2021; Rosato et al., 2019; Scheper et al., 2018; Simoni et al., 2018; Sudmeier et al., 2022) or have unknown antigen specificity.
Thus, the composition of the intratumoral CD8+ T cell compartment is not only the result of available and recognized tumor antigens but also of the immunological history of the patient. Without knowledge of their antigen specificity, this infiltration by microbe-specific CD8+ T cells has the potential to seriously confound studies assessing the number, phenotype, and differentiation of tumor-specific CD8+ TILs. This presence of bystander cells is not only restricted to CD8+ TILs, as CMV-specific CD4+ T cells have also been reported with in the TME of various cancers (Li et al., 2022; Oliveira et al., 2022). However, less is known about bystander infiltration of CD4+ T cells due to inherent difficulties of their detection with pMHC multimers when compared to CD8+ T cells (Christophersen, 2020).
In some tumors, even the most abundant CD8+ clones may not be tumor-specific. Elderly patients – in which cancer incidence is high - can accumulate oligoclonal CD8+ T cell repertoires in a process termed memory inflation (Klenerman, 2018). Many of these clones are CMV-specific (Khan et al., 2002b; Wieland et al., 2018). In a nonagenarian melanoma patient we recently found that the ten most abundant CD8+ T cell clones in circulation accounted for nearly 60% of the entire blood CD8+ T cell repertoire, with the most dominant clone comprising 30% (Wieland et al., 2018). Importantly, while these peripherally expanded clones were less prevalent in tumor and did not expand in response to anti-PD-1 therapy (thus likely representing virus-specific bystanders), they still ranked among the top tumor-infiltrating CD8+ T cell clones due to their vast abundance in the periphery.
While these and other data suggest that a negative enrichment of CD8+ T cell clonotype frequency in tumor versus blood can identify bystander cells, bystander cells can also be found at similar or even slightly higher frequencies than in blood (Ning et al., 2022; Rosato et al., 2019; Scheper et al., 2018). This is likely due to the relative exclusion of naïve T cells from tumors (Egelston et al., 2018; Sudmeier et al., 2022) and highlights the impact of the respective denominator for such comparisons. While techniques allowing for the direct determination of antigen specificity such as tetramer staining, MANAFEST and TCR sequencing of cells with confirmed antigen/tumor specificity are tedious (Caushi et al., 2021; Danilova et al., 2018; Oliveira et al., 2022; Oliveira et al., 2021; Rosato et al., 2019; Simoni et al., 2018), they are necessary to accurately distinguish bystanders from tumor-specific T cells.
This presence of bystander CD8+ T cells in the absence of their cognate antigen has also been recapitulated in mouse tumor models, demonstrating that pre-existing virus-specific CD8+ T cells can infiltrate tumors and acquire phenotypic traits such as PD-1 expression (Erkes et al., 2017; Rosato et al., 2019; Sudmeier et al., 2022) which are often erroneously associated with T cell dysfunction or even equated with tumor specificity (Danahy et al., 2020). Importantly, bystander recruitment seems to be mostly restricted to antigen-experienced cells (Erkes et al., 2017; Ning et al., 2022; Rosato et al., 2019; Sudmeier et al., 2022) and thus likely results in substantially higher frequencies of tumor-specific CD8+ T cells in standard mouse models due to the relative absence of microbe-specific memory CD8+ T cells in mice kept under standard pathogen-free conditions.
Bystander infiltration into tumors has two major implications for the study of tumor-infiltrating CD8+ T cells. First, since tumor-specific CD8+ T cells are required for efficient tumor killing, the presence of bystanders may complicate the correlation of CD8+ T cell infiltration with response to ICIs. A potential contributor to this phenomenon might be the variable fraction of bystanders compared to bona fide tumor-specific cells in different tumors and among individuals. Another contributor may be the potentially beneficial, antigen-independent role of bystander cells for the maintenance of a permissive local immune milieu by secretion of various cytokines and chemokines. This too may be variable: cells that have experienced multiple sequential stimulations by their cognate antigen exhibit increased antigen-independent effector functions in the TME (Danahy et al., 2020). These data suggest that functional bystander cells are not equal from an anti-tumor perspective and that their antigen specificity and thus antigenic history (i.e. time since last antigen encounter and number of antigenic encounters throughout their lifetime) will significantly impact their anti-tumor functions. Experiments in mice exposed to various pathogens in a controlled manner prior to tumor inoculation (Danahy et al., 2020; Erkes et al., 2017; Ning et al., 2022; Rosato et al., 2019) or the use of “dirty” mice (Beura et al., 2016) will continue to improve our understanding of the role of bystander CD8+ T cells and their potential to be specifically harnessed to promote anti-tumor immune responses.
Looks can be deceiving
A second implication of bystander cell infiltration is their potential to confound the study of tumor-specific T cells within the TME. As discussed above, tumor specificity of T cells cannot be inferred from their simple presence within the TME. As a corollary, the phenotype and number of tumor-infiltrating cells should not be confused with the phenotype and number of tumor-specific cells.
The marked heterogeneity of antigen specificities within the tumor raises the important question to what degree the observed phenotypic heterogeneity of CD8+ TILs can be attributed to divergent antigen specificities and thus differential histories of antigenic stimulation versus differentiation of the tumor-specific cells themselves. As noted, CD8+ T cell differentiation has been extensively studied in the context of both acute and chronic antigen exposure in both preclinical mouse models as well as humans (Akondy et al., 2017; Kaech et al., 2003; Miller et al., 2008). While acute antigen exposure results in the ultimate development of polyfunctional memory cells, chronic antigen stimulation leads to the alternate T cell differentiation state of exhaustion. Although the ultimate products of acute and chronic antigen exposure (i.e., memory and exhausted CD8+ T cells) differ in their functional capacity, they still share a substantial number of expressed molecules at various stages of their differentiation trajectories, while simultaneously exhibiting significant heterogeneity along each trajectory (Figure 1).
Hence, multiple markers are required to accurately describe the differentiation state of a given CD8+ T cell. In settings in which the number of employed markers is small, such as in low-dimensional flow cytometry or immunohistochemistry, determining distinct differentiation states may mislead data interpretation. One misleading marker is PD-1 itself, whose presence on CD8+ T cells is commonly equated to an exhausted phenotype; however, PD-1 is more accurately described as an activation marker reflecting TCR stimulation. Indeed, PD-1 expression is not limited to exhausted T cells but also observed on antigen-specific cells early after antigenic stimulation (Ahn et al., 2018), with memory precursors - fated to become long-lived memory CD8+ T cells - expressing higher levels of PD-1 than terminal effector cells (Hudson et al., 2019b).
The above observations were made in a mouse model of acute viral infection, which provides detailed information about the antigen specificity of the analyzed CD8+ T cells and when a cell first/last encountered its cognate antigen. With some notable exceptions of experimental studies in viral infections (Akondy et al., 2017; Akondy et al., 2009; Fuertes Marraco et al., 2015; Miller et al., 2008), temporal information is typically not available to study human CD8+ T cell differentiation. This is particularly true in tumor studies, where a snapshot of the TME is generated at the time of resection or biopsy.
Pitfalls of trajectory analysis
The resulting difficulty in determining distinct lineage relationships between complex human T cell populations in vivo has spurred the use of differentiation trajectory algorithms to develop a biological framework for tumor-infiltrating lymphocyte differentiation. Key methods of this kind include trajectory analysis using pseudotime and RNA velocity to infer differentiation pathways among cells analyzed by scRNA-seq (La Manno et al., 2018; Trapnell et al., 2014). However, while these methods can be useful for hypothesis generation, they are not substitutes for experimental determination of lymphocyte differentiation trajectories and may lead to erroneous results even in relatively simple experiments.
As an example, to determine the differentiation pathways of virus-specific CD8+ T cells, Kurd et al. (Kurd et al., 2020) transferred P14 cells – TCR-transgenic CD8+ T cells specific for a LCMV epitope – to wild type mice. After infection of recipient mice with the acute strain of LCMV Armstrong, the authors isolated donor P14 cells at various time points and performed scRNA-sequencing (Fig. 2A–B). Since the stage of differentiation and time after antigen exposure are known for every cell in this experiment, this is an excellent dataset to compare the ground truth (real time) with inferred pseudotime. We re-analyzed their data set, performing dimensionality reduction of sequenced cells with Uniform Manifold Approximation and Projection (UMAP) and trajectory inference with Slingshot (McInnes et al., 2018; Street et al., 2018). The actual, real-time trajectory of CD8+ T cell differentiation followed a clockwise pattern in UMAP space (Fig. 2C) whereas trajectory inference predicted the opposite differentiation pathway as determined by biological experimentation: naïve cells were predicted to differentiate into memory cells first, followed by subsequent transition into late and early effectors (Fig. 2D).
Figure 2:
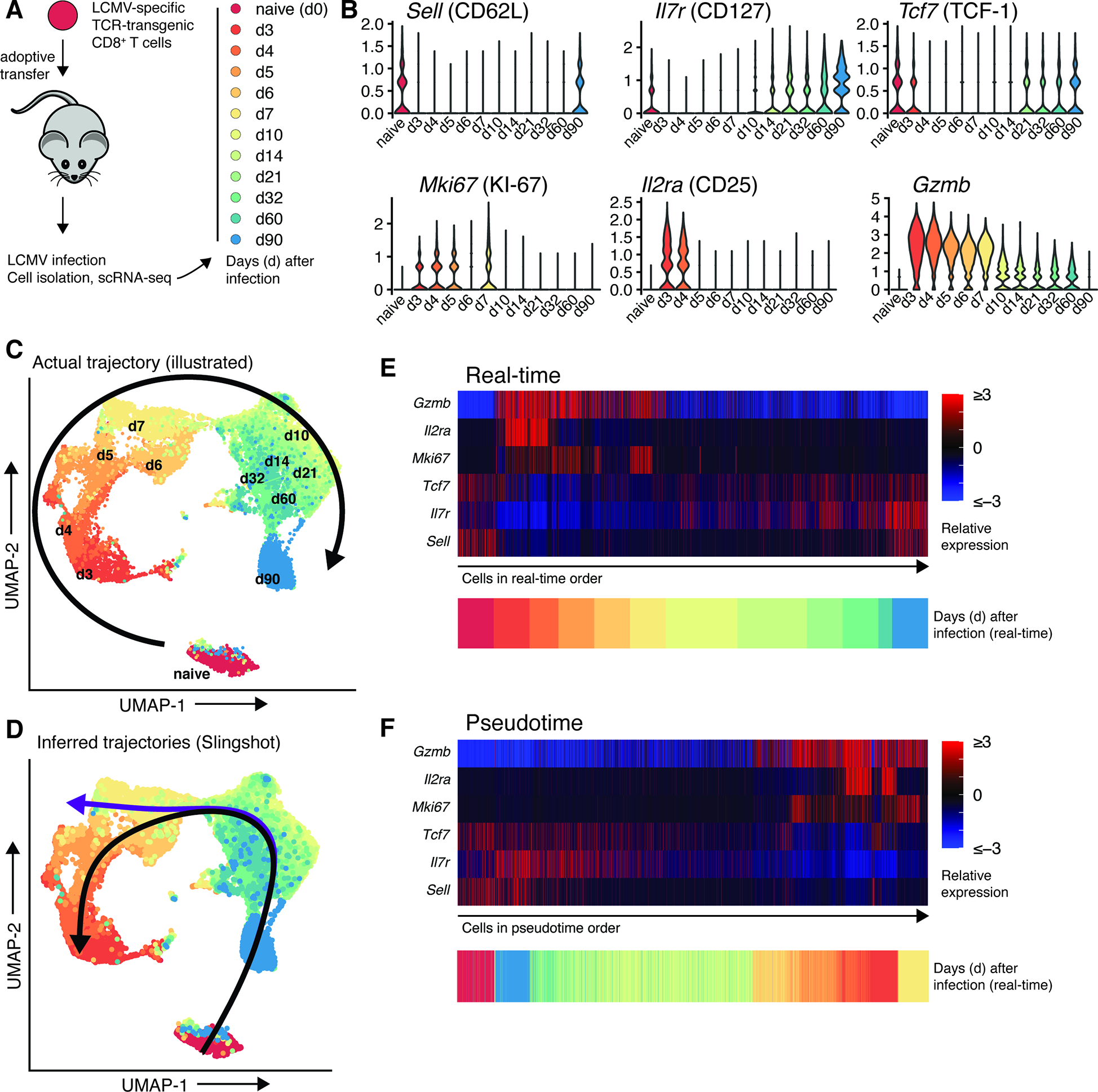
Inference of a reverse differentiation trajectory from scRNA-seq data. A: TCR-transgenic CD8+ T cells were transferred to mice which were subsequently infected with LCMV Armstrong. At illustrated timepoints, cells were sorted and subjected to scRNA-seq. B: Expression of T cell differentiation markers at indicated time points. C: UMAP projection of sequenced cells, with an illustrated arrow showing actual cell differentiation trajectory. D: Identical UMAP projection, with inferred trajectories. E: Heatmap of T cell differentiation marker expression, with cells in real-time order. F: Heatmap of T cell differentiation marker expression, with cells in pseudotime order. Raw data are from (Kurd et al., 2020). Code and data to reproduce the scRNA-seq analysis are available on Mendeley Data, doi: 10.17632/3dvt79c7yt.1.
Even in this exceptionally “clean” example, where CD8+ T cells with an identical antigen receptor of known specificity are transferred to a congenically distinct host and subsequently isolated, trajectory analysis from scRNA-seq data infers a wildly incorrect differentiation pathway compared to the known ground truth. In human tumors, such analyses are often performed on scRNA-seq data from heterogenous pools of tumor-infiltrating T cells comprised of cells with multiple TCR clonotypes, multiple antigen specificities and thus distinct differentiation histories, often from multiple patients, each isolated at a single time point. Any trajectory inference from such a heterogeneous population of cells should thus be treated with skepticism. In many cases, it appears that bystander and tumor-specific T cells are found in mutually exclusive differentiation states (Oliveira et al., 2021; Sudmeier et al., 2022). Given the inability of exhausted CD8+ T cells to form central memory cells after antigen removal (Abdel-Hakeem et al., 2021; Hensel et al., 2021; Tonnerre et al., 2021; Yates et al., 2021), the poor response of memory CD8+ T cells to sustained antigenic load (West et al., 2011), and the epigenetically-enforced irreversibility of the exhausted T cell state (Pauken et al., 2016), two distinct tumor-infiltrating T cell populations may not share a common differentiation pathway and any attempt to infer trajectories between such states would be incorrect. An extreme example is a recent study (Wilk et al., 2020) that used RNA velocity analysis from scRNA-seq to propose a “differentiation bridge from plasmablasts to developing neutrophils”, a conclusion seemingly at odds with basic immunological principles (Alquicira-Hernandez et al., 2021).
In our example (Fig. 2), the discrepancy between real-time and pseudotime trajectories is due to a conflict between inherent biological properties of CD8+ T cell differentiation and the design of differentiation trajectory algorithms. Upon activation, naïve T cells undergo extensive proliferation and acquire an effector gene expression pattern including high levels of MKI67, GZMB, PRF1, IL2RA (encoding CD25), and PDCD1 (encoding PD-1) (Fig. 2B,E). This is accompanied by downregulation of memory/stemness-associated genes such as SELL (encoding CD62L), IL7R, TCF7, and CCR7 (Fig. 2B,E). After antigen clearance, a subset of effector cells will ultimately de-differentiate into memory CD8+ T cells that re-acquire a more naïve and quiescent-like state but simultaneously maintain epigenetic marks of an effector phase such as demethylation of loci encoding for effector molecules such as granzyme B (Akondy et al., 2017; Youngblood et al., 2017). Similar reversals of gene expression are also observed in the differentiation of exhausted CD8+ T cells (with CX3CR1 and CD69, for example (Beltra et al., 2020; Hudson et al., 2019a)). Unfortunately, this gene expression pattern – inherent to T cell biology - violates “a key assumption that enables pseudotemporal ordering,” namely “that genes do not change direction very often, and thus samples with similar transcriptional profiles should be close in order” (Bacher and Kendziorski, 2016). The result in our example is that naïve and memory cells, which are the most transcriptionally similar yet temporally distant, are projected to be adjacent in differentiation state (Fig. 2F). Similar issues likely exist with CD4+ T cells, and inference of their differentiation trajectories may be further complicated by their potential to differentiate into distinct helper T cell subsets, compared to the (relatively) simplistic lifestyle of cytotoxic CD8+ T cells. Thus, although it is tempting to use trajectory inference techniques to infer differentiation relationships between cell subsets, experimental validation is required before making firm conclusions about immune cell differentiation pathways.
Another method commonly used to infer immune cell phenotype or differentiation state is gene set enrichment analysis (GSEA) (Subramanian et al., 2005). GSEA determines whether differences in expression levels of a pre-defined set of genes are statistically significant between two populations. When using GSEA or gene ontology data for analysis, it is critical to properly frame the comparison between the gene set(s) and populations being analyzed and to also consider confounding biological processes that may be present between two otherwise-different cell types. For example, despite their distinct differentiation states, both effector and exhausted CD8+ T cells will express high levels of cytotoxicity-related genes compared to naïve or memory cells. In our analysis of CD8+ T cells following acute LCMV infection, expression of an exhaustion-associated gene set is high in effector cells isolated 5–7 days following infection (Fig. 3A,B). However, no exhausted cells are present in this dataset; the CD8+ T cells expressing these genes are not exhausted but are effectors that share an overlapping gene expression program with exhausted T cells. It is also worth noting that any errors or omissions in annotation or construction of gene sets will be carried forward in the analysis. For example, the gene set in our example was constructed from microarray data (Doering et al., 2012), likely omitting more recently discovered or annotated genes associated with T cell exhaustion. However, careful use of gene sets does have several advantages, including the availability of thousands of gene sets for analysis (Liberzon et al., 2011) and less sensitivity to scRNA-seq dropout.
Figure 3:
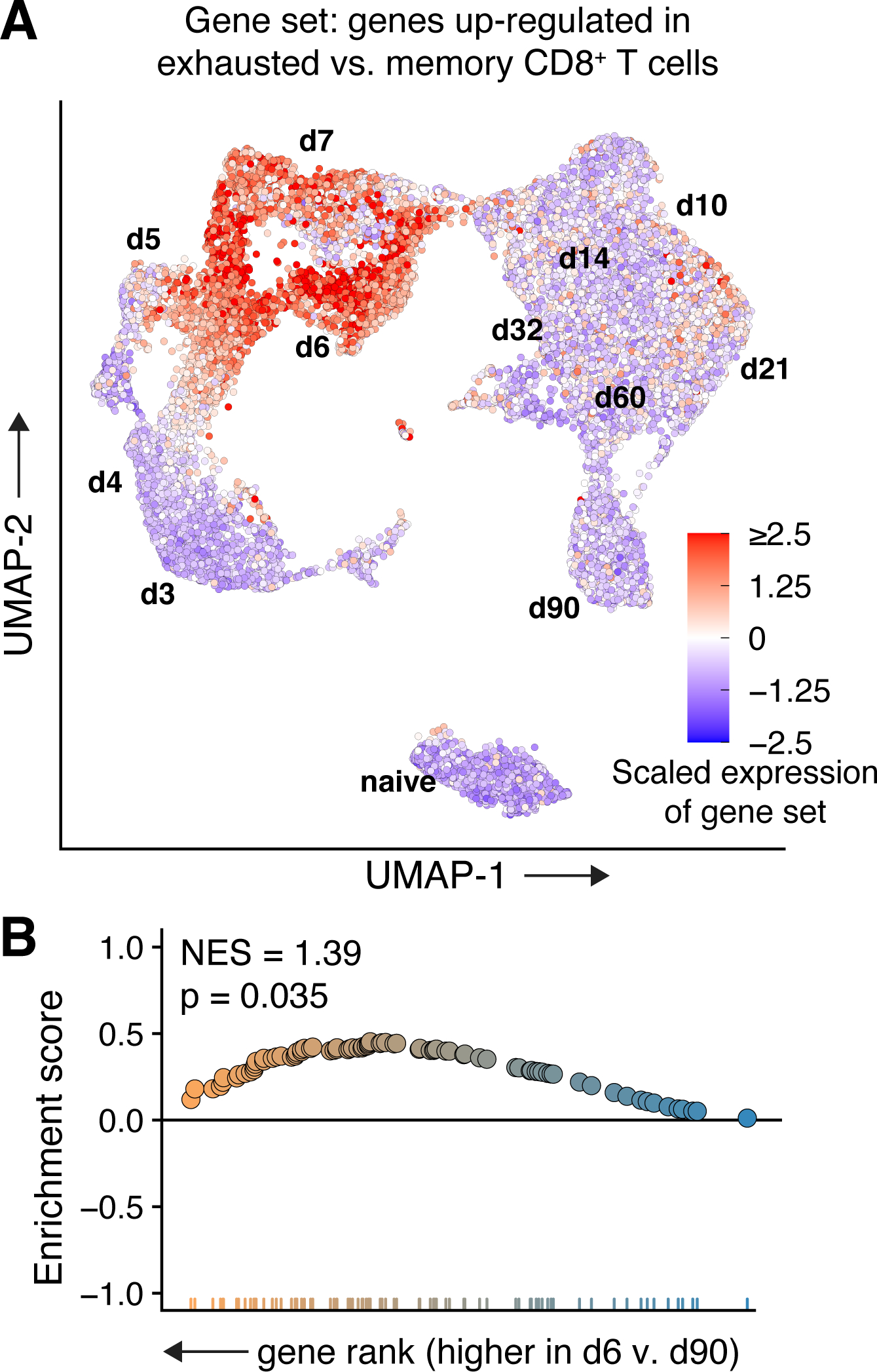
Expression of an exhaustion-associated gene set in non-exhausted CD8+ T cells. A: Relative expression of the GSE41867_MEMORY_VS_EXHAUSTED_CD8_TCELL_DAY30_LCMV_DN gene set (Godec et al., 2016) in CD8+ T cells following acute infection. B: Exhaustion-associated genes are significantly enriched in day 6 vs day 90 cells, despite neither subset being biologically exhausted.
One key advantage to studying lymphocytes with scRNA-seq is the ability to determine antigen receptor sequences (B cell receptors/BCRs and T cell receptors/TCRs) (Morgan and Tergaonkar, 2022; Pai and Satpathy, 2021; Pauken et al., 2022). At a broad level, measuring TCR diversity yields important information on the clonality and breadth of T cell responses. On the single-cell level, since lymphocytes with the same antigen receptor sequence are clonally related and share antigen specificity, BCR/TCR sequencing can deliver profound biological insight into lymphocyte differentiation and is a necessary addition for scRNA-seq studies of lymphocytes. When sequenced to an appropriate depth, analysis of cells with the same BCR/TCR clonotype will reveal differentiation states available to lymphocytes with a given antigen receptor sequence within a particular tissue or tumor. This insight is strongest when paired with experimental determination of antigen specificity; several recent, elegant studies have used peptide-MHC I multimers and/or in vitro stimulation assays to determine bystander and/or tumor-specific TCRs which are then linked with scRNA-seq gene expression data (Caushi et al., 2021; Eberhardt et al., 2021; Oliveira et al., 2022; Oliveira et al., 2021; Sudmeier et al., 2022). Even without experimental determination of antigen specificity, careful use of databases (Bagaev et al., 2020) containing TCR sequences with known specificity can assist in the identification of bystander, self-reactive, or tumor-associated antigen-specific cells (Park et al., 2020a; Sudmeier et al., 2022). Although determination of their antigen specificity is more difficult due to the recognition of both linear and conformational epitopes, studies of tumor-specific B cells are also possible (Mazor et al., 2022; Wieland et al., 2020).
Markers of tumor-specific T cells
Unfortunately, the cost of scRNA-seq makes it prohibitive for many sample sets, and in all cases the number of cells analyzed is fewer than in other methods, particularly flow cytometry. Thus, protein markers capable of distinguishing tumor-specific from bystander T cells have become a topic of particular interest. Memory CD8+ T cells to common human pathogens exist in a continuum of differentiation states that likely are imparted by differences in their antigenic history and strength of antigenic stimulation. Human memory T cells can exhibit a wide range of phenotypes with YFV-specific cells ultimately acquiring a CCR7+CD45RA+PD-1negTCF-1+ T stem cell memory (TSCM) phenotype (Gattinoni et al., 2011) and cells specific for other common human pathogens such as Influenza (Flu), EBV, and CMV expressing a mix of the aforementioned markers (Fig. 1) (Debes et al., 2004; Sekine et al., 2020).
However, both YFV- and Influenza-specific memory cells lack expression of TOX, a transcription factor heavily associated with CD8+ T cell exhaustion and tumor-specificity (Alfei et al., 2019; Khan et al., 2019; Oliveira et al., 2021; Scott et al., 2019). Unfortunately, TOX expression can also be induced in mouse and human memory T cells by cytokine signaling, independent of TCR stimulation (Maurice et al., 2021). CD8+ T cells reactive to EBV and CMV, two common human herpesviruses that periodically reactivate and can thus repeatedly stimulate cognate CD8+ T cells, show a marked heterogeneity in terms of the aforementioned memory markers and can express significant levels of PD-1 as well as TOX (Sekine et al., 2020). Despite the expression of these “exhaustion” markers, these cells are highly cytolytic and polyfunctional (Sekine et al., 2020), highlighting that the exclusive reliance on these markers for differentiation state determination can falsely label cells as tumor-specific or exhausted. Of note, EBV- and CMV-specific CD8+ T cells can co-express TCF-1 and TOX, making them particularly resemble the antigen-specific stem-like CD8+ T cells found in chronic viral infection and some tumors (Eberhardt et al., 2021; Im et al., 2016; Siddiqui et al., 2019).
Stem-like CD8+ T cells are characterized by co-expression of the transcription factors TOX and TCF-1 and possess substantial proliferative capacity and the potential to give rise to more differentiated cytotoxic progeny (Im et al., 2016; Leong et al., 2016; Utzschneider et al., 2016). The proliferative burst observed after immune checkpoint blockade depends on the presence of these cells, which makes them an intense topic of study (Im et al., 2016). In the steady state, stem-like CD8+ T cells serve as a resource to maintain antigen-specific T cell responses in the face of chronic antigen stimulation through slow self-renewal and differentiation (Im et al., 2016; Leong et al., 2016; Utzschneider et al., 2016). These cells are also referred to as progenitor exhausted cells and have been found in various settings of persistent antigen such as chronic viral infections, cancer and most recently in autoimmune settings such as diabetes (Gearty et al., 2021). Note that despite potentially confusing similarities in nomenclature, human stem-like CD8+ T cells are highly distinct from T memory stem cells (TSCM) that have been described based on the expression of the CD45RA isoform and form a distinct and quite rare subset of memory T cells (Gattinoni et al., 2011) emerging slowly after antigen clearance (Akondy et al., 2017; Fuertes Marraco et al., 2015). In contrast to TSCM, human stem-like CD8+ T cells express the CD45RO isoform and high levels of PD-1 (Eberhardt et al., 2021) (Fig. 1).
In contrast to the somewhat promiscuous expression of PD-1 and TOX on exhausted and polyfunctional cells, CD39 has been identified and confirmed in several studies to faithfully demarcate exhausted tumor-specific CD8+ T cells (Duhen et al., 2018; Hanada et al., 2022; Simoni et al., 2018). CD39 is also expressed on tumor-specific conventional CD4+ (Kortekaas et al., 2020; Oliveira et al., 2022) TILs, although its constitutive expression on regulatory T cells precludes its use to capture tumor-specific Tregs (Borsellino et al., 2007). Furthermore, CXCL13 transcript expression has been reported to be enriched among exhausted tumor-specific CD4+ and CD8+ T cells (Hanada et al., 2022; Thommen et al., 2018; Veatch et al., 2022; Zheng et al., 2022). While CD39 appears to be a specific marker to enrich for tumor-specific CD8+ TILs without requiring cognate tumor antigen identification, CD39 expression alone does not necessarily capture all tumor-specific CD8+ T cells in the TME, as tumor-specific stem-like cells do not express CD39 (Eberhardt et al., 2021; Oliveira et al., 2021). Rather, CD39 marks an exhausted, dysfunctional differentiation state that is available primarily to tumor-specific CD8+ T cells within the TME.
Studying T cell differentiation states
In most studies, the majority of intratumoral tumor-specific CD8+ T cells have been found in the CD39+ terminally exhausted state (Caushi et al., 2021; Li et al., 2019; Oliveira et al., 2021; Philip et al., 2017; Simoni et al., 2018), while stem-like cells are present at higher frequencies in tumor-draining lymph nodes (Buchwald et al., 2020; Connolly et al., 2021). Combined with the high frequency of TCF-1+ bystanders, it has remained unclear if intratumoral TCF-1+ cells are truly tumor-reactive (Philip and Schietinger, 2019). The answer likely depends on the tumor type and nature of the surrounding tissue. Using tetramer staining for tumor-associated viral antigens, we recently identified intratumoral TCF-1+ tumor-specific cells in human papilloma virus (HPV)+ head and neck cancer (HNC) (Eberhardt et al., 2021), which arises mostly in lymphoid tissues such as tonsils. Other studies examining tumors in non-lymphoid tissues have found very few TCF-1+ tumor-specific cells (Caushi et al., 2021; Li et al., 2019; Oliveira et al., 2021; Philip et al., 2017). Importantly, while HPV-specific T cells were readily detectable in the HPV+ HNC tumors, these cells were undetectable in the peripheral blood directly ex vivo (Eberhardt et al., 2021), a finding consistent with other studies (Caushi et al., 2021; Oliveira et al., 2021). Overall, these data demonstrate a differential distribution of tumor-specific CD8+ T cells and subsets not only macroscopically between blood and tissue but also between lymphoid and non-lymphoid tissues. This suggests that tumor-specific stem-like cells mostly reside in lymphoid tissues, with the possible exception of the cells residing in tertiary lymphoid structures. This distribution aligns nicely with data from the murine LCMV model, where viral antigen is present in all tissues, but stem-like cells are resident in lymphoid organs (Im et al., 2020).
This divergent localization presents challenges to the faithful detection of different CD8+ T cell subsets and their associated markers by flow cytometry. Without cells available from tumor and lymphoid tissues (or blood), accurate quantification of cell- or tissue-restricted proteins can be difficult. One such example is TIM-3, a key marker and co-inhibitory molecule co-expressed with CD39 on differentiated tumor-infiltrating CD8+ T cells (Sade-Feldman et al., 2018). Since TIM-3+ CD39+ CD8+ T cells are rarely found in circulation (Eberhardt et al., 2021; Hudson et al., 2019a; Im et al., 2020; Oliveira et al., 2021; Sudmeier et al., 2022) (Fig. 4A), it is difficult to assess the quality of TIM-3 staining and true fraction of TIM-3+ cells using circulating T cells alone. However, simultaneous staining of PBMCs and TILs (the latter acting as a positive control) facilitates assessment of the TIM-3+ population (Fig. 4B–C). Unfortunately, such samples are not readily available in all studies, and measurements of the TIM-3+ fraction in circulating T cells of healthy adults has thus varied wildly, from <1% (Bachmann et al., 2012; Kawashima et al., 2020; Wu et al., 2013) to more than 40% (Bonifacius et al., 2021) and various points in between (Cai et al., 2015; Liu et al., 2010; Sada-Ovalle et al., 2015; Wu et al., 2011; Xu et al., 2015) (Fig. 4D). Such variability highlights the importance of sample diversity, biological controls, and careful downstream analysis of studies using cytometry-based approaches.
Figure 4:
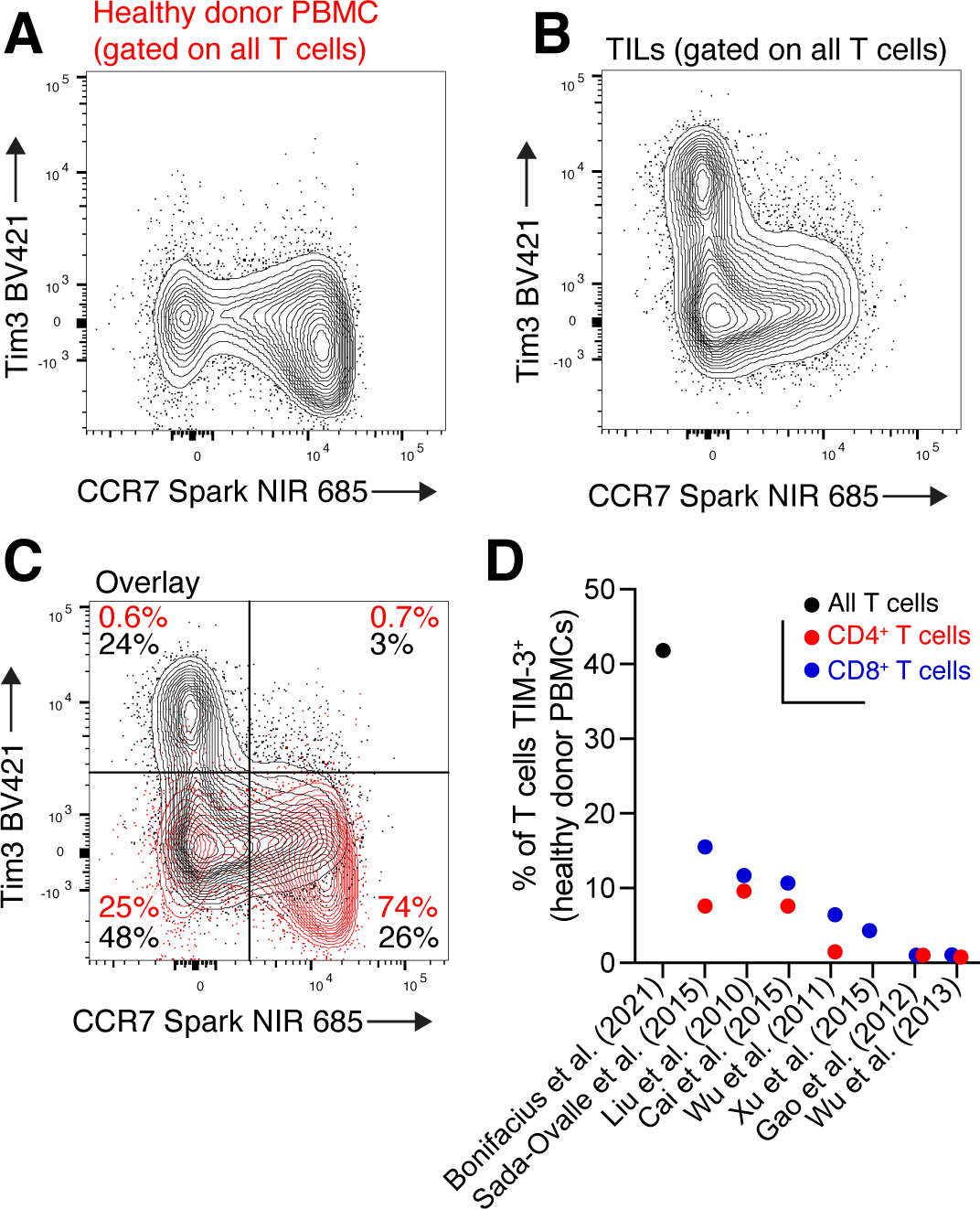
Quantification of TIM-3 on human T cells. A: CCR7 and TIM-3 staining on circulating T cells of a healthy donor. With this tissue only, it is unclear if TIM-3 staining was successful, and if so, where TIM-3+ cells should be gated. B: CCR7 and TIM-3 expression on tumor-infiltrating T cells, stained at the same time as PBMCs in (A). C: Overlay of CCR7 and TIM-3 staining in healthy donor PBMCs (red) and TILs (black). D: Estimates of TIM-3 expression on circulating T cells of healthy donors from various studies. Flow data in panels A-C were previously reported (Sudmeier et al., 2022).
Another challenge for cytometry-based approaches is the analysis, visualization, and reporting of high dimensional data. This challenge dates to the introduction of mass cytometry (or CyTOF, cytometry by time-of-flight), which at its introduction greatly increased the number of measured parameters per cell (Bandura et al., 2009). CyTOF measures the cellular abundance of isotope-labeled antibodies, theoretically enabling collection of up to 120 parameters, although this number is limited in practice due to current conjugation chemistries and the availability of sufficiently purified heavy metal isotopes to currently about 50 parameters (Bandura et al., 2009; Gadalla et al., 2019; Iyer et al., 2022).
Recent years have seen the proliferation of advanced fluorescence flow cytometers with five or more lasers and equipped with 64 or more detectors, allowing the full-spectrum assessment of each fluorophore rather than limiting its detection to a narrow bandwidth, a technique known as spectral flow cytometry. Coupled with advances in fluorophore chemistry, it is now possible to analyze expression of 40 or more proteins on single cells using fluorescence flow cytometry (Park et al., 2020b), thus nearly closing the gap with mass cytometry in terms of measurable parameters per cell. Compared to CyTOF or scRNA-seq, modern fluorescence-based flow cytometry offers an outstanding balance between the number of parameters acquired per cell, data acquisition rates, reagent and instrumentation cost, widespread availability of required equipment, and ease of downstream analysis (Bonilla et al., 2021).
Unfortunately, these properties of fluorescence flow cytometry create a tradeoff: while a higher number of measured parameters increases the potential biological insight gathered from an experiment, the complexity of data acquisition, analysis, and presentation increase in parallel (Fig. 5A). Compensation – correction for spectral overlap in fluorescence flow cytometry experiments – is both more crucial and complicated in high-parameter experiments. Several excellent articles have been written on the technical aspects of high-parameter flow cytometry data collection and analysis (Ferrer-Font et al., 2020; Fox et al., 2020; Liechti et al., 2021), but we will provide an example demonstrating how small inaccuracies collection or analysis can lead to erroneous conclusions from cytometry data.
Figure 5:
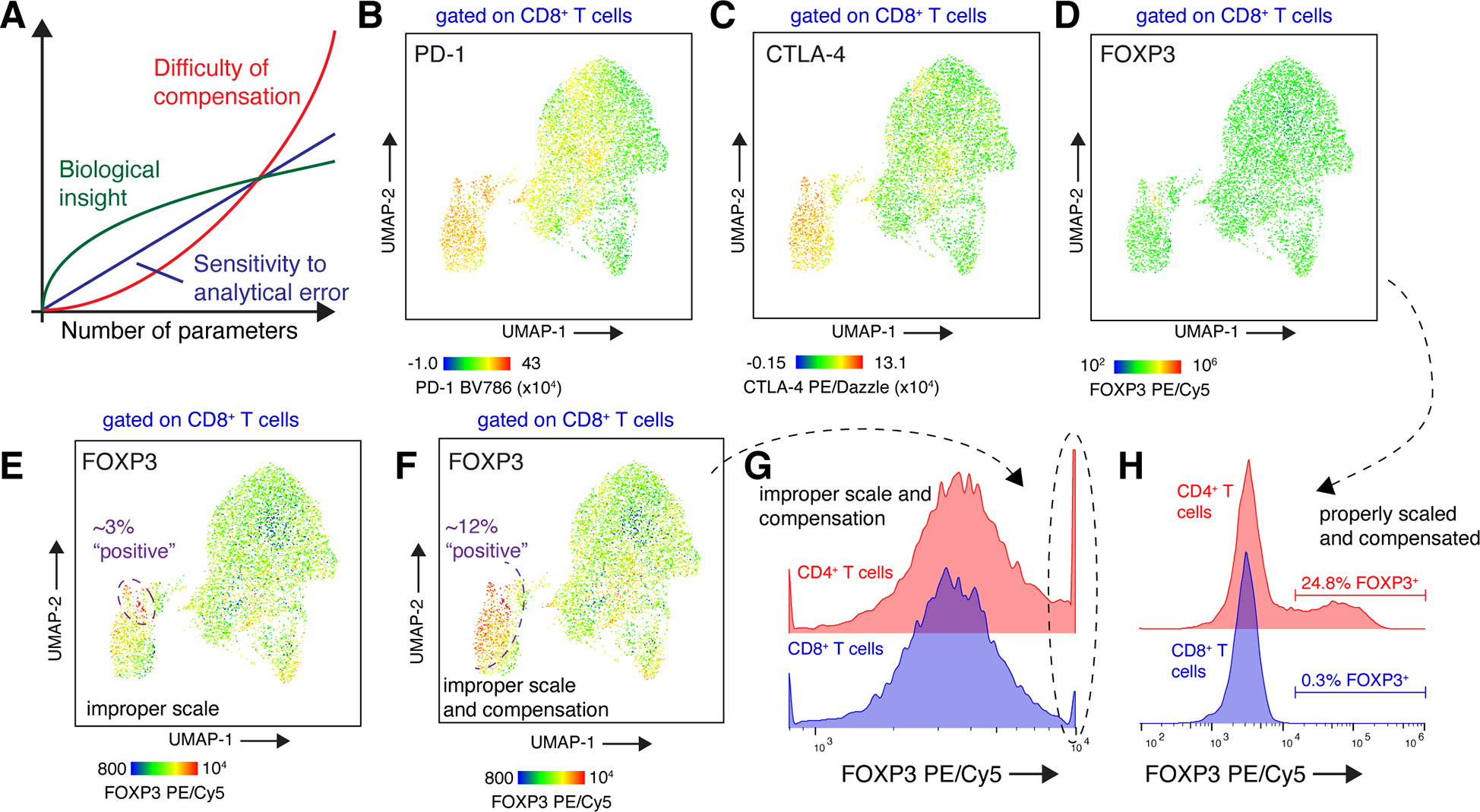
Perils of data visualization and analysis in high-parameter flow cytometry. A: As more parameters are collected in a flow cytometry experiment, the potential biological insight increases. Unfortunately, both the difficulty of analysis and sensitivity of the experiment to such error also increase. An example is shown in panels B-H, where tumor-infiltrating lymphocytes from a brain metastasis patient were analyzed with a T cell-focused, 22-color flow cytometry panel. B-D: UMAP projection of CD8+ T cells, with cells colored by expression of PD-1 (B), CTLA-4 (C), and FOXP3 (D). Data shown are properly compensated and scaled. E: UMAP projection of CD8+ T cells, with cells colored by expression of FOXP3. The color intensity scale has been modified compared to panel (D); data are properly compensated. F: UMAP projection of CD8+ T cells, with cells colored by expression of FOXP3 (same scale as panel E). Fluorescence spillover of CTLA-4 PE/Dazzle into FOXP3 PE/Cy5 has been undercompensated by 10%. (G) An alternate display method, such as a histogram, using an internal positive control for FOXP3 expression would reveal improper scaling of FOXP3 fluorescence intensities (circled population). H: The true percentage of FOXP3+ CD8+ T cells in this sample is <0.5%. Data to reproduce the flow cytometry analysis are available on Mendeley Data, doi: 10.17632/3dvt79c7yt.1.
We recently collected 22-color flow cytometry data of brain metastasis-infiltrating CD8+ T cells on a four-laser spectral flow cytometer (Sudmeier et al., 2022). All antibodies in the staining panel were expected to bind to at least some proportion of T cells; some cells had 16 or more bound fluorophores. Thus, both the difficulty of compensation and sensitivity to compensation error in this experiment were high. Given the high number of parameters, we used UMAP to reduce data collected from CD8+ T cells to two dimensions for visualization (McInnes et al., 2018). This analysis showed an exhausted CD8+ T cell population with high expression of immune checkpoint molecules PD-1 and CTLA-4 (Fig. 5B–C).
As expected, nearly all CD8+ T cells were negative for expression of FOXP3, a transcription factor expressed in regulatory CD4+ T cells (Rudensky, 2011) (Fig. 5D). However, a false positive population of FOXP3+ CD8+ T cells can be generated through errors in analysis – or even choices in data visualization. Compressing the scale of FOXP3 fluorescence intensity produces the appearance of a small population of FOXP3hi CD8+ T cells, even when data are properly compensated (Fig. 5E). When compounded by an error in compensation, the result is a striking - and false - FOXP3+ population that represents 12% of CD8+ T cells (Fig. 5F).
The experimental details needed to decipher such an erroneous result are often not reported alongside the data; unlike scRNA-seq experiments, deposition of raw cytometry data is not yet (Kozlov, 2022) required for most publications. While dimensionality reduction methods such as UMAP (McInnes et al., 2018) or tSNE (Van der Maaten and Hinton, 2008) are frequently used to visualize and analyze high-dimensional data (Keyes et al., 2020), their use often results in limited inspection and presentation of direct fluorescence measurements. In our example, “old-fashioned” examination of the individual parameter channels in low-dimensional space (Fig. 5G–H) is crucial to revealing these issues. Further, the inclusion of scales is critical to assess antibody staining, particularly when using color scales in dimensionality reduction projections. Finally, when in doubt, positive control samples or populations should be used to validate expression of key markers. In this example, FOXP3 expression on CD8+ T cells can be compared to a known positive population (CD4+ Tregs) to quantify the true level of FOXP3+ cells in this sample and to guide gating strategies (Fig. 5H). However, as described above, these controls may not always be available if the expression of the marker in question is cell-type or spatially restricted.
Induced sources of heterogeneity
Experimental uncertainty is also introduced into flow cytometry and scRNA-seq experiments through artifacts from sample processing. Batch effects result from the separate sample collection, processing, and/or data acquisition of different samples, and can introduce artificial heterogeneity. When data are collected from different patients, cell types, or tissues at different times, technical variation may also obscure true biological differences (Hicks et al., 2018). This is a particular problem in samples like tumors where significant biological heterogeneity is already present. Preclinical studies often allow the simultaneous processing of samples from different treatment groups or separate animals, but batch effects remain a particular challenge in human studies, where patient samples are often collected over many weeks or months. In such cases, batch effects can be quantified and controlled by including a reference cell population in each technical sequencing run (Stuart et al., 2019). For single-cell studies, an additional strategy is to prepare samples and acquire data from different individuals and/or experimental conditions simultaneously. In flow cytometry experiments, this can be done by barcoding individual samples with unique fluorophores (Krutzik and Nolan, 2006), but this technique is more relevant in single-cell sequencing studies where cells from each analyzed sample can be barcoded with a unique, DNA-labeled sequencing antibody specific for a ubiquitously-expressed antigen (Stoeckius et al., 2018). This technique, Cellular Indexing of Transcriptomes and Epitopes by Sequencing (Stoeckius et al., 2017), or CITE-seq, is also a powerful method for detecting hundreds of proteins simultaneously on single cells. Panels of over 250 antibodies have been used to characterize immune tumor infiltrate with this method (Pombo Antunes et al., 2021), and large CITE-seq antibody cocktails are now available commercially, providing unprecedented insights into the heterogeneity at the protein level. As fluorescence spillover is not an issue in CITE-seq experiments, it is an excellent complementary and confirmatory technique to flow cytometry.
In human studies, T cell phenotype may also be affected by the current and previous medical and pharmacological history of the patient cohort. Importantly, immunotherapies may directly change T cell phenotype, abundance, and clonality within the tumor, and this likely varies by treatment type. PD-1 pathway blockade does not markedly change exhausted T cell phenotype, which is limited by stability of a dysfunctional epigenetic state (Pauken et al., 2016). Instead, in both preclinical mouse models and human tumors, PD-1 pathway blockade results in a higher abundance of classically-exhausted CD8+ T cells (Bassez et al., 2021; Hudson et al., 2019a; Zhang et al., 2021) caused by division and differentiation of the TCF-1+ stem-like population (Im et al., 2016). Most – but not all – human tumor studies show responding cells originate from pre-existing clones found in the pre-treatment tumor (Bassez et al., 2021; Chang et al., 2020; Liu et al., 2021; Yost et al., 2019). As with differentiation trajectory analysis, the interplay of methodology with T cell biology in such studies is critical to interpretation of these results. In the chronic LCMV infection model, the stem-like population is spatially restricted to lymphoid tissues and contains a more diverse TCR repertoire (Chang et al., 2020; Im et al., 2020). In human cancer, the TCR repertoire varies spatially throughout the tumor microenvironment (Joshi et al., 2019; Sudmeier et al., 2022). Thus, appropriate sequencing depth and spatial sampling are critical to accurately define changes in T cell phenotype and TCR repertoire in response to immunotherapy.
In contrast to PD-1 pathway blockade monotherapy, its combination with IL-2 administration results in drastic changes in antigen-specific CD8+ T cell phenotype (Hashimoto et al., In press; West et al., 2013). Immunologic effects of other medications should also be considered when assessing TIL phenotype and function in patients. Glucocorticoids, for example, are used both to directly treat various hematological malignancies and also to manage sequelae of other cancers (Keith, 2008) but may promote T cell dysfunction in the tumor microenvironment (Acharya et al., 2020). Systemic chemotherapy agents may also induce changes in T cell phenotype within the tumor microenvironment (Zhang et al., 2021).
Tissue disruption and cell isolation is an additional processing step with the potential to confound interpretation of cell states. Enzymatic digestion with various proteases is commonly used to generate viable single-cell suspensions from tumors and other tissues (Carney and Malmgren, 1967; Corgnac et al., 2021; Reichard and Asosingh, 2018; Rodriguez de la Fuente et al., 2021). While this method is employed to obtain the highest possible number of viable cells, these digestion steps can cleave or otherwise alter extracellular proteins, creating false negatives in flow cytometry or CITE-sequencing experiments (Autengruber et al., 2012; Hines et al., 2014). A more insidious effect of enzymatic digestion is its effects on cellular transcriptomes. Incubation of immune cells at room temperature for as little as two hours can impact gene expression (Massoni-Badosa et al., 2020). Collagenase digestion at 37 °C causes large changes in cellular transcriptomes, including upregulation of immune-associated genes such FOS, JUN, and NR4A1 (O’Flanagan et al., 2019). Even though these changes may only be present in a subset of digested cells, the magnitude of these changes can be high, requiring careful inspection of clusters in single-cell experiments in addition to careful analysis of stressrelated genes in bulk RNA-sequencing experiments (van den Brink et al., 2017). As a potential remedy, some groups and commercial suppliers have investigated fixation to “lock-in” cell states before subsequent downstream processing (Alles et al., 2017). While this method may introduce transcript length and GC content-dependent bias (Wang et al., 2021), it also enables cell isolation based on fluorescence sorting with intracellular markers (Thomsen et al., 2015).
Location, location, location
Generation of single-cell suspensions from tumors or other tissue may result in the underestimation of target cell abundance (Stark et al., 2018; Steinert et al., 2015) and causes the loss of spatial information. Recent studies have shown that spatial restriction of CD8+ T cell subsets is not limited to differences among tissues, but T cell phenotype is also closely linked to position within the TME. For example, TCR repertoires vary across space in tumors (Joshi et al., 2019), and TCF-1+ cells are more likely to be situated in tertiary lymphoid structures or stromal antigen-presenting “niches” (Jansen et al., 2019). Spatial biology has advanced significantly in recent years, allowing the detection of many proteins and gene expression signatures in situ in tissue sections. While low-parameter immunohistochemistry has been a mainstay of cancer pathology for decades, new methods have been developed to measure multiple proteins by microscopy, with multiplexing ranging from a handful to 40 or more detected markers (Tan et al., 2020). When combined with the ability to image whole tumor sections, the spatial architecture of cell types within the TME can be defined and even correlated with clinical outcomes (Berry et al., 2021; Jansen et al., 2019).
Interrogating the spatial architecture of the TME has already provided significant insights, such as demonstrating the importance of immune cell co-localization - spanning from small lymphoid aggregates to fully-fledged tertiary lymphoid structures (TLS) - for anti-tumor immunity and response to ICIs (Berry et al., 2021; Helmink et al., 2020; Jansen et al., 2019; Schumacher and Thommen, 2022). As mentioned, tumor-specific T cells express the B cell chemoattractant CXCL13 and are thus equipped to potentially remodel their microenvironment, with CXCL13+ cells being found in close proximity to tumor-infiltrating B cells and within TLS (Gu-Trantien et al., 2017; Ukita et al., 2022). TLS and smaller immune structures have been suggested to provide a protective niche for the local maintenance of stem-like CD8+ T cells (Jansen et al., 2019; Pagliarulo et al., 2022). However, most protein-based multiplex imaging approaches do not provide antigen-specificity information of interrogated cells, hindering discrimination of bystander and tumor-specific cells. One notable exception is in situ tetramer staining, which has shown the presence of EBV- and influenza-specific CD8+ T cells in glioblastoma (Ning et al., 2022).
A potential solution to this problem lies in spatial transcriptomics, a new technique that permits the measurement of RNA levels – often on a whole-transcriptome basis – in situ within tissue sections (Rao et al., 2021). While spatial transcriptomics currently provides neither the resolution nor the sequencing depth of scRNA-seq, integration of the two modalities can elucidate significant biological insight (Longo et al., 2021). Spatially-resolved methods that can capture complete transcriptomes may be a particular boon for tumor immunology. Targeted amplification with these techniques can recover lymphocyte antigen receptor sequences; we recently used targeted TCR amplification combined with spatial transcriptomics to show that clones with a CD39+ phenotype were preferentially enriched within tumor beds, whereas clones expressing TCF-1 were found in the surrounding stroma (Hudson and Sudmeier, 2022; Sudmeier et al., 2022). Similar techniques have also been used to identify B cell receptor sequences in tumors (Meylan et al., 2022). Long-read sequencing could also be used to detect mutated mRNAs and copy number variations in cancer, aiding in localization of neoantigen-expressing tumor cells (Erickson et al., 2022; Lebrigand et al., 2022). The combination of these modalities will allow extremely detailed studies of the TME, such as determining how the distribution and gene expression patterns of neo-antigen-specific T cell clones are influenced by expression of their cognate antigens.
Putting it all together
The emergence of new multidimensional technologies such as scRNA-seq, high-parameter flow cytometry, and spatial transcriptomics have transformed our view of anti-tumor immune responses. In the future, as these technologies expand in capability and are adopted more widely, they will provide deep insights into immune cell behavior within the TME. Such knowledge has the potential to form the foundation of rationally-designed and more effective immunotherapies. To achieve this goal, however, the sheer volume of data generated by these technologies must be matched by focused experimental questions, accurate analysis, clear interpretation, and supportive mechanistic studies.
Recent years have witnessed a slowdown in FDA approval of ICIs, and the vast majority of approvals have targeted the PD-1/PD-L1 inhibitory pathway (de Miguel and Calvo, 2020). To accurately identify targets for next-generation ICIs, determination of antigen specificity within the TME must be a crucial component of future immuno-oncology studies. Studies identifying tumor-specific T cells (Eberhardt et al., 2021; Oliveira et al., 2022; Oliveira et al., 2021; Simoni et al., 2018) have already provided important insights into their differentiation states; studying the signaling pathways within and in spatial proximity of tumor-specific cells is an important task to identify effective and specific ICI targets. Understanding tumor-specific T cells will also inform strategies for engineering improved function of CAR and TCR transgenic T cells, which have been widely used for hematologic malignancies and are now seeing use in other tumor types (Leidner et al., 2022; Majzner et al., 2022; Singh and McGuirk, 2020). While infiltration of bystander T cells may complicate the study of tumor-specific cells, they also represent an important therapeutic target, particularly to increase infiltration of “cold” tumors or promote inflammation and antigen processing in patients with poor pre-existing anti-tumor T cell responses (Lapteva et al., 2014; Rosato et al., 2019; Wang et al., 2020). Thus, the study of antiviral immune responses in the infectious disease and basic immunology fields also directly benefits immuno-oncology research efforts.
Generating therapies and therapeutic strategies – or even working biological hypotheses – from this deluge of -omics data may represent the greatest current challenge in immunology and most other fields of biology (Anderson, 2008; Denecker and Lelandais, 2022; Krassowski et al., 2020). As we have discussed here, it is crucial to be aware of the intrinsic limitations and potential sources of error in modern immunology techniques. As ever more raw data are collected, processed data attract more of our attention, often obscuring experimental error and true biological complexity in the process (Chari et al., 2021; Gorin et al., 2022). To combat this, the use of complementary techniques can test and control for both biological, experimental, and analytical sources of error. For example, CITE-seq and scRNA-seq respectively measure protein and transcript levels in single cells, providing two independent measurements of gene and gene product expression. CITE-seq and flow cytometry both measure single-cell protein expression but have different cell throughput and sources of experimental error. However, while these complementary and multidimensional techniques provide unprecedented insights into human anti-tumor immune responses, they are intrinsically descriptive, should be mostly used for hypothesis generation and ultimately require experimental validation for establishing causal relationships.
While recent advances, such as an ex vivo tumor platform to assess patient response to ICIs (Voabil et al., 2021), have permitted more mechanistic study of human tumors, preclinical tumor immunology models remain an irreplaceable cornerstone to evaluate findings in the - omics era. These models allow the normalization of many variables across treatment groups, most notably antigen specificity, tumor burden, age, and genetic background. Much work has been performed to make these models better recapitulate human cancer, including varied processes for tumor initiation and the use of different tumor antigens (Connolly et al., 2022). In these models, the use of technologies such as scRNA-seq, high-parameter flow cytometry, and spatial transcriptomics can deliver unparalleled phenotypic data that can be directly attributed to an experimental treatment or condition. Such studies combining hypothesis-driven science with emerging immunological techniques have the potential to create accurate models of immune function, provide invaluable insights into anti-tumor immune responses, and ultimately to improve patient outcomes.
Acknowledgements
We thank Andres Chang, Andrew Gunderson, and Daniel McManus for critical reading of the manuscript. W.H.H. is supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number K99AI153736. A.W. is supported by the Pelotonia Institute for Immuno-Oncology (PIIO). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the PIIO.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Hakeem MS, Manne S, Beltra J-C, Stelekati E, Chen Z, Nzingha K, Ali M-A, Johnson JL, Giles JR, Mathew D, et al. (2021). Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nature Immunology 22, 1008–1019. 10.1038/s41590-021-00975-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya N, Madi A, Zhang H, Klapholz M, Escobar G, Dulberg S, Christian E, Ferreira M, Dixon KO, Fell G, et al. (2020). Endogenous Glucocorticoid Signaling Regulates CD8+ T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity 53, 658–671.e656. 10.1016/j.immuni.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA, and Ahmed R (2018). Role of PD-1 during effector CD8 T cell differentiation. Proceedings of the National Academy of Sciences 115, 4749–4754. 10.1073/pnas.1718217115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, et al. (2017). Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367. 10.1038/nature24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, Quyyumi F, Garg S, Altman JD, Del Rio C, et al. (2009). The Yellow Fever Virus Vaccine Induces a Broad and Polyfunctional Human Memory CD8+ T Cell Response. The Journal of Immunology 183, 7919–7930. 10.4049/jimmunol.0803903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, et al. (2019). TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269. 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault P-L, Ayoub S, Schreyer L, Boltengagen A, Birchmeier C, et al. (2017). Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biology 15. 10.1186/s12915-017-0383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alquicira-Hernandez J, Powell JE, and Phan TG (2021). No evidence that plasmablasts transdifferentiate into developing neutrophils in severe COVID-19 disease. Clinical & Translational Immunology 10. 10.1002/cti2.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, and Davis MM (1996). Phenotypic Analysis of Antigen-Specific T Lymphocytes. Science 274, 94–96. 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- Amyes E, Hatton C, Montamat-Sicotte D, Gudgeon N, Rickinson AB, McMichael AJ, and Callan MFC (2003). Characterization of the CD4+ T Cell Response to Epstein-Barr Virus during Primary and Persistent Infection. Journal of Experimental Medicine 198, 903–911. 10.1084/jem.20022058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C (2008). The End of Theory: The Data Deluge Makes the Scientific Method Obsolete. Wired Magazine. [Google Scholar]
- Autengruber A, Gereke M, Hansen G, Hennig C, and Bruder D (2012). Impact of enzymatic tissue disintegration on the level of surface molecule expression and immune cell function. European Journal of Microbiology and Immunology 2, 112–120. 10.1556/EuJMI.2.2012.2.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher R, and Kendziorski C (2016). Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biology 17. 10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MP, Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, Sun J, Yang Q, Zhang X, and Lu B (2012). TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 7. 10.1371/journal.pone.0030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagaev DV, Vroomans RMA, Samir J, Stervbo U, Rius C, Dolton G, Greenshields-Watson A, Attaf M, Egorov ES, Zvyagin IV, et al. (2020). VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Research 48, D1057–D1062. 10.1093/nar/gkz874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, and Tanner SD (2009). Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Analytical Chemistry 81, 6813–6822. 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, Boeckx B, Vanden Bempt M, Nevelsteen I, Lambein K, et al. (2021). A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nature Medicine 27, 820–832. 10.1038/s41591-021-01323-8. [DOI] [PubMed] [Google Scholar]
- Beltra J-C, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841.e828. 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry S, Giraldo NA, Green BF, Cottrell TR, Stein JE, Engle EL, Xu H, Ogurtsova A, Roberts C, Wang D, et al. (2021). Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockade. Science 372. 10.1126/science.aba2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacius A, Tischer-Zimmermann S, Dragon AC, Gussarow D, Vogel A, Krettek U, Gödecke N, Yilmaz M, Kraft ARM, Hoeper MM, et al. (2021). COVID-19 immune signatures reveal stable antiviral T cell function despite declining humoral responses. Immunity 54, 340–354.e346. 10.1016/j.immuni.2021.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla DL, Reinin G, and Chua E (2021). Full Spectrum Flow Cytometry as a Powerful Technology for Cancer Immunotherapy Research. Frontiers in Molecular Biosciences 7. 10.3389/fmolb.2020.612801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Höpner S, Centonze D, Bernardi G, Dell’Acqua ML, et al. (2007). Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 110, 1225–1232. 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. (2010). Phase I Study of Single-Agent Anti–Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. Journal of Clinical Oncology 28, 3167–3175. 10.1200/jco.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J, Ross-Macdonald P, Berger AC, Jegede OA, Elagina L, et al. (2020). Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nature Medicine 26, 909–918. 10.1038/s41591-020-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald ZS, Nasti TH, Lee J, Eberhardt CS, Wieland A, Im SJ, Lawson D, Curran W, Ahmed R, and Khan MK (2020). Tumor-draining lymph node is important for a robust abscopal effect stimulated by radiotherapy. Journal for ImmunoTherapy of Cancer 8. 10.1136/jitc-2020-000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Xu Y-F, Wu Z-J, Dong Q, Li M-Y, Olson JC, Rabinowitz YM, Wang L-H, and Sun Y (2015). Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World Journal of Urology 34, 561–567. 10.1007/s00345-015-1656-7. [DOI] [PubMed] [Google Scholar]
- Carney PG, and Malmgren RA (1967). COMPARISON OF TECHNIQUES FOR OBTAINING SINGLE CELL SUSPENSIONS FROM TUMORS. Transplantation 5, 455–458. [DOI] [PubMed] [Google Scholar]
- Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH-C, Mog BJ, Hou W, Justesen S, Blosser R, et al. (2021). Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132. 10.1038/s41586-021-03752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YM, Wieland A, Li Z. r., Im SJ, McGuire DJ, Kissick HT, Antia R, Ahmed R, and Heise MT (2020). T Cell Receptor Diversity and Lineage Relationship between Virus-Specific CD8 T Cell Subsets during Chronic Lymphocytic Choriomeningitis Virus Infection. Journal of Virology 94. 10.1128/jvi.00935-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari T, Banerjee J, and Pachter L (2021). The Specious Art of Single-Cell Genomics. bioRxiv. 10.1101/2021.08.25.457696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Gunasegaran B, Singh HD, Dutertre C-A, Loh CY, Lim JQ, Crawford JC, Lee HK, Zhang X, Lee B, et al. (2021). Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 54, 1825–1840.e1827. 10.1016/j.immuni.2021.06.013. [DOI] [PubMed] [Google Scholar]
- Christophersen A (2020). Peptide-MHC class I and class II tetramers: From flow to mass cytometry. Hla 95, 169–178. 10.1111/tan.13789. [DOI] [PubMed] [Google Scholar]
- Connolly KA, Fitzgerald B, Damo M, and Joshi NS (2022). Novel Mouse Models for Cancer Immunology. Annual Review of Cancer Biology 6, 269–291. 10.1146/annurev-cancerbio-070620-105523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, Hornick NI, Fitzgerald BL, Damo M, Kasmani MY, et al. (2021). A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Science Immunology 6. 10.1126/sciimmunol.abg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corgnac S, Lecluse Y, and Mami-chouaib F (2021). Isolation of tumor-resident CD8+ T cells from human lung tumors. STAR Protocols 2. 10.1016/j.xpro.2020.100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danahy DB, Berton RR, and Badovinac VP (2020). Cutting Edge: Antitumor Immunity by Pathogen-Specific CD8 T Cells in the Absence of Cognate Antigen Recognition. The Journal of Immunology 204, 1431–1435. 10.4049/jimmunol.1901172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova L, Anagnostou V, Caushi JX, Sidhom J-W, Guo H, Chan HY, Suri P, Tam A, Zhang J, Asmar ME, et al. (2018). The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunology Research 6, 888–899. 10.1158/2326-6066.Cir-18-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miguel M, and Calvo E (2020). Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 38, 326–333. 10.1016/j.ccell.2020.07.004. [DOI] [PubMed] [Google Scholar]
- Debes GF, Bonhagen K, Wolff T, Kretschmer U, Krautwald S, Kamradt T, and Hamann A (2004). CC Chemokine Receptor 7 Expression by Effector/Memory CD4+ T Cells Depends on Antigen Specificity and Tissue Localization during Influenza A Virus Infection. Journal of Virology 78, 7528–7535. 10.1128/jvi.78.14.7528-7535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecker T, and Lelandais G (2022). Omics Analyses: How to Navigate Through a Constant Data Deluge. In Yeast Functional Genomics, pp. 457–471. 10.1007/978-1-0716-2257-5_25. [DOI] [PubMed] [Google Scholar]
- Doering Travis A., Crawford A, Angelosanto Jill M., Paley Michael A., Ziegler Carly G., and Wherry EJ (2012). Network Analysis Reveals Centrally Connected Genes and Pathways Involved in CD8+ T Cell Exhaustion versus Memory. Immunity 37, 1130–1144. 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. (2018). Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nature Communications 9. 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt CS, Kissick HT, Patel MR, Cardenas MA, Prokhnevska N, Obeng RC, Nasti TH, Griffith CC, Im SJ, Wang X, et al. (2021). Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nature 597, 279–284. 10.1038/s41586-021-03862-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelston CA, Avalos C, Tu TY, Simons DL, Jimenez G, Jung JY, Melstrom L, Margolin K, Yim JH, Kruper L, et al. (2018). Human breast tumor-infiltrating CD8+ T cells retain polyfunctionality despite PD-1 expression. Nature Communications 9. 10.1038/s41467-018-06653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson A, He M, Berglund E, Marklund M, Mirzazadeh R, Schultz N, Kvastad L, Andersson A, Bergenstråhle L, Bergenstråhle J, et al. (2022). Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature 608, 360–367. 10.1038/s41586-022-05023-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkes DA, Smith CJ, Wilski NA, Caldeira-Dantas S, Mohgbeli T, and Snyder CM (2017). Virus-Specific CD8+T Cells Infiltrate Melanoma Lesions and Retain Function Independently of PD-1 Expression. The Journal of Immunology 198, 2979–2988. 10.4049/jimmunol.1601064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, and Miller WH (2020). A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Current Oncology 27, 87–97. 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, and Price KM (2020). Panel Design and Optimization for High-Dimensional Immunophenotyping Assays Using Spectral Flow Cytometry. Current Protocols in Cytometry 92. 10.1002/cpcy.70. [DOI] [PubMed] [Google Scholar]
- Fox A, Dutt TS, Karger B, Obregón-Henao A, Anderson GB, and Henao-Tamayo M (2020). Acquisition of High-Quality Spectral Flow Cytometry Data. Current Protocols in Cytometry 93. 10.1002/cpcy.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Maillard SA, Montandon N, Rufer N, Waldvogel S, Delorenzi M, and Speiser DE (2015). Long-lasting stem cell–like memory CD8+ T cells with a naïve-like profile upon yellow fever vaccination. Science Translational Medicine 7. 10.1126/scitranslmed.aaa3700. [DOI] [PubMed] [Google Scholar]
- Gadalla R, Noamani B, MacLeod BL, Dickson RJ, Guo M, Xu W, Lukhele S, Elsaesser HJ, Razak ARA, Hirano N, et al. (2019). Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Frontiers in Oncology 9. 10.3389/fonc.2019.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, and Bruni D (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 18, 197–218. 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. (2011). A human memory T cell subset with stem cell–like properties. Nature Medicine 17, 1290–1297. 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearty SV, Dündar F, Zumbo P, Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, Socci ND, Trivedi P, Lowe SW, Lauer P, et al. (2021). An autoimmune stem-like CD8 T cell population drives type 1 diabetes. Nature 602, 156–161. 10.1038/s41586-021-04248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godec J, Tan Y, Liberzon A, Tamayo P, Bhattacharya S, Butte Atul J., Mesirov Jill P., and Haining WN (2016). Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation. Immunity 44, 194–206. 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorin G, Fang M, Chari T, and Pachter L (2022). RNA velocity unraveled. bioRxiv. 10.1101/2022.02.12.480214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotch F, McMichael A, Smith G, and Moss B (1987). Identification of viral molecules recognized by influenza-specific human cytotoxic T lymphocytes. Journal of Experimental Medicine 165, 408–416. 10.1084/jem.165.2.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu-Trantien C, Migliori E, Buisseret L, de Wind A, Brohée S, Garaud S, Noël G, C.V LD, Lodewyckx J-N, Naveaux C, et al. (2017). CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI Insight 2. 10.1172/jci.insight.91487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K. i., Zhao C, Gil-Hoyos R, Gartner JJ, Chow-Parmer C, Lowery FJ, Krishna S, Prickett TD, Kivitz S, Parkhurst MR, et al. (2022). A phenotypic signature that identifies neoantigen-reactive T cells in fresh human lung cancers. Cancer Cell 40, 479–493.e476. 10.1016/j.ccell.2022.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Araki K, Cardenas M, Li P, Jadhav R, Kissick H, Hudson W, McGuire D, Im SJ, Lee J, et al. (In press). PD-1 combination therapy with IL-2 modifies CD8 T cell exhaustion program. Nature. 10.1038/s41586-022-05257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms E, Onate MK, and Sherman MH (2020). Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discovery 10, 648–656. 10.1158/2159-8290.Cd-19-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel N, Gu Z, Sagar, Wieland D, Jechow K, Kemming J, Llewellyn-Lacey S, Gostick E, Sogukpinar O, Emmerich F, et al. (2021). Memory-like HCV-specific CD8+ T cells retain a molecular scar after cure of chronic HCV infection. Nature Immunology 22, 229–239. 10.1038/s41590-020-00817-w. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks SC, Townes FW, Teng M, and Irizarry RA (2018). Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 19, 562–578. 10.1093/biostatistics/kxx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines William C., Su Y, Kuhn I, Polyak K, and Bissell Mina J. (2014). Sorting Out the FACS: A Devil in the Details. Cell Reports 6, 779–781. 10.1016/j.celrep.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin J-X, Konieczny BT, Im SJ, Freeman GJ, et al. (2019a). Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity 51, 1043–1058.e1044. 10.1016/j.immuni.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Prokhnevska N, Gensheimer J, Akondy R, McGuire DJ, Ahmed R, and Kissick HT (2019b). Expression of novel long noncoding RNAs defines virus-specific effector and memory CD8+ T cells. Nature Communications 10. 10.1038/s41467-018-07956-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, and Sudmeier LJ (2022). Localization of T cell clonotypes using the Visium spatial transcriptomics platform. STAR Protocols 3. 10.1016/j.xpro.2022.101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford MM, Kim TS, Sun J, and Braciale TJ (2014). The Effector T Cell Response to Influenza Infection. In Influenza Pathogenesis and Control - Volume II, pp. 423–455. 10.1007/82_2014_397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Konieczny BT, Hudson WH, Masopust D, and Ahmed R (2020). PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. Proceedings of the National Academy of Sciences 117, 4292–4299. 10.1073/pnas.1917298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A, Hamers AAJ, and Pillai AB (2022). CyTOF® for the Masses. Frontiers in Immunology 13. 10.3389/fimmu.2022.815828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K, de Massy MR, Ismail M, Reading JL, Uddin I, Woolston A, Hatipoglu E, Oakes T, Rosenthal R, Peacock T, et al. (2019). Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nature Medicine 25, 1549–1559. 10.1038/s41591-019-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, and Ahmed R (2003). Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nature Immunology 4, 1191–1198. 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- Kawashima A, Kanazawa T, Kidani Y, Yoshida T, Hirata M, Nishida K, Nojima S, Yamamoto Y, Kato T, Hatano K, et al. (2020). Tumour grade significantly correlates with total dysfunction of tumour tissue-infiltrating lymphocytes in renal cell carcinoma. Scientific Reports 10. 10.1038/s41598-020-63060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith BD (2008). Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 8. 10.1186/1471-2407-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes TJ, Domizi P, Lo YC, Nolan GP, and Davis KL (2020). A Cancer Biologist’s Primer on Machine Learning Applications in High-Dimensional Cytometry. Cytometry Part A 97, 782–799. 10.1002/cyto.a.24158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Cobbold M, Keenan R, and Moss Paul A.H. (2002a). Comparative Analysis of CD8+T Cell Responses against Human Cytomegalovirus Proteins pp65 and Immediate Early 1 Shows Similarities in Precursor Frequency, Oligoclonality, and Phenotype. The Journal of Infectious Diseases 185, 1025–1034. 10.1086/339963. [DOI] [PubMed] [Google Scholar]
- Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, and Moss PAH (2002b). Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals. The Journal of Immunology 169, 1984–1992. 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218. 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenerman P (2018). The (gradual) rise of memory inflation. Immunol Rev 283, 99–112. 10.1111/imr.12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenerman P, and Oxenius A (2016). T cell responses to cytomegalovirus. Nature Reviews Immunology 16, 367–377. 10.1038/nri.2016.38. [DOI] [PubMed] [Google Scholar]
- Kortekaas KE, Santegoets SJ, Sturm G, Ehsan I, van Egmond SL, Finotello F, Trajanoski Z, Welters MJP, van Poelgeest MIE, and van der Burg SH (2020). CD39 Identifies the CD4+ Tumor-Specific T-cell Population in Human Cancer. Cancer Immunology Research 8, 1311–1321. 10.1158/2326-6066.Cir-20-0270. [DOI] [PubMed] [Google Scholar]
- Koutsakos M, Illing PT, Nguyen THO, Mifsud NA, Crawford JC, Rizzetto S, Eltahla AA, Clemens EB, Sant S, Chua BY, et al. (2019). Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nature Immunology 20, 613–625. 10.1038/s41590-019-0320-6. [DOI] [PubMed] [Google Scholar]
- Kozlov M (2022). NIH issues a seismic mandate: share data publicly. Nature 602, 558–559. 10.1038/d41586-022-00402-1. [DOI] [PubMed] [Google Scholar]
- Krassowski M, Das V, Sahu SK, and Misra BB (2020). State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Frontiers in Genetics 11. 10.3389/fgene.2020.610798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik PO, and Nolan GP (2006). Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nature Methods 3, 361–368. 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- Kurd NS, He Z, Louis TL, Milner JJ, Omilusik KD, Jin W, Tsai MS, Widjaja CE, Kanbar JN, Olvera JG, et al. (2020). Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Science Immunology 5. 10.1126/sciimmunol.aaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapteva N, Monjazeb AM, Tietze JK, Grossenbacher SK, Hsiao H-H, Zamora AE, Mirsoian A, Koehn B, Blazar BR, Weiss JM, et al. (2014). Bystander Activation and Anti-Tumor Effects of CD8+ T Cells Following Interleukin-2 Based Immunotherapy Is Independent of CD4+ T Cell Help. PLoS ONE 9. 10.1371/journal.pone.0102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrigand K, Bergenstråhle J, Thrane K, Mollbrink A, Meletis K, Barbry P, Waldmann R, and Lundeberg J (2022). The spatial landscape of gene expression isoforms in tissue sections. bioRxiv. 10.1101/2020.08.24.252296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih Y-P, Leung A, Payne R, Sutcliffe K, Cramer J, et al. (2022). Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. New England Journal of Medicine 386, 2112–2119. 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, et al. (2016). CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nature Immunology 17, 1187–1196. 10.1038/ni.3543. [DOI] [PubMed] [Google Scholar]
- Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen JBAG, Blank CU, et al. (2019). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789.e718. 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhuang S, Heit A, Koo S-L, Tan AC, Chow IT, Kwok WW, Tan IB, Tan DSW, Simoni Y, and Newell EW (2022). Bystander CD4+ T cells infiltrate human tumors and are phenotypically distinct. OncoImmunology 11. 10.1080/2162402x.2021.2012961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechti T, Weber LM, Ashhurst TM, Stanley N, Prlic M, Van Gassen S, and Mair F (2021). An updated guide for the perplexed: cytometry in the high-dimensional era. Nature Immunology 22, 1190–1197. 10.1038/s41590-021-01006-z. [DOI] [PubMed] [Google Scholar]
- Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, Zhang Y, Corse E, Hu Y, Han W, and Zhang Z (2021). Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nature Cancer 3, 108–121. 10.1038/s43018-021-00292-8. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shu Q, Gao L, Hou N, Zhao D, Liu X, Zhang X, Xu L, Yue X, Zhu F, et al. (2010). Increased Tim-3 expression on peripheral lymphocytes from patients with rheumatoid arthritis negatively correlates with disease activity. Clinical Immunology 137, 288–295. 10.1016/j.clim.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Long HM, Meckiff BJ, and Taylor GS (2019). The T-cell Response to Epstein-Barr Virus–New Tricks From an Old Dog. Frontiers in Immunology 10. 10.3389/fimmu.2019.02193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo SK, Guo MG, Ji AL, and Khavari PA (2021). Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nature Reviews Genetics 22, 627–644. 10.1038/s41576-021-00370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, Richards RM, Jiang L, Barsan V, Mancusi R, et al. (2022). GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603, 934–941. 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoni-Badosa R, Iacono G, Moutinho C, Kulis M, Palau N, Marchese D, Rodríguez-Ubreva J, Ballestar E, Rodriguez-Esteban G, Marsal S, et al. (2020). Sampling time-dependent artifacts in single-cell genomics studies. Genome Biology 21. 10.1186/s13059-020-02032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice NJ, Berner J, Taber AK, Zehn D, and Prlic M (2021). Inflammatory signals are sufficient to elicit TOX expression in mouse and human CD8+ T cells. JCI Insight 6. 10.1172/jci.insight.150744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor RD, Nathan N, Gilboa A, Stoler-Barak L, Moss L, Solomonov I, Hanuna A, Divinsky Y, Shmueli MD, Hezroni H, et al. (2022). Tumor-reactive antibodies evolve from non-binding and autoreactive precursors. Cell 185, 1208–1222.e1221. 10.1016/j.cell.2022.02.012. [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv. 10.48550/arXiv.1802.03426. [DOI] [Google Scholar]
- Meylan M, Petitprez F, Becht E, Bougoüin A, Pupier G, Calvez A, Giglioli I, Verkarre V, Lacroix G, Verneau J, et al. (2022). Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 55, 527–541.e525. 10.1016/j.immuni.2022.02.001. [DOI] [PubMed] [Google Scholar]
- Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, et al. (2008). Human Effector and Memory CD8+ T Cell Responses to Smallpox and Yellow Fever Vaccines. Immunity 28, 710–722. 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Morgan D, and Tergaonkar V (2022). Unraveling B cell trajectories at single cell resolution. Trends in Immunology 43, 210–229. 10.1016/j.it.2022.01.003. [DOI] [PubMed] [Google Scholar]
- Murray RJ, Kurilla MG, Brooks JM, Thomas WA, Rowe M, Kieff E, and Rickinson AB (1992). Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. Journal of Experimental Medicine 176, 157–168. 10.1084/jem.176.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning J, Gavil NV, Wu S, Wijeyesinghe S, Weyu E, Ma J, Li M, Grigore F-N, Dhawan S, Skorput AGJ, et al. (2022). Functional virus-specific memory T cells survey glioblastoma. Cancer Immunology, Immunotherapy. 10.1007/s00262-021-03125-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flanagan CH, Campbell KR, Zhang AW, Kabeer F, Lim JLP, Biele J, Eirew P, Lai D, McPherson A, Kong E, et al. (2019). Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biology 20. 10.1186/s13059-019-1830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira G, Stromhaug K, Cieri N, Iorgulescu JB, Klaeger S, Wolff JO, Rachimi S, Chea V, Krause K, Freeman SS, et al. (2022). Landscape of helper and regulatory antitumour CD4+ T cells in melanoma. Nature 605, 532–538. 10.1038/s41586-022-04682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, et al. (2021). Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 596, 119–125. 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarulo F, Cheng PF, Brugger L, van Dijk N, van den Heijden M, Levesque MP, Silina K, and van den Broek M (2022). Molecular, Immunological, and Clinical Features Associated With Lymphoid Neogenesis in Muscle Invasive Bladder Cancer. Frontiers in Immunology 12. 10.3389/fimmu.2021.793992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai JA, and Satpathy AT (2021). High-throughput and single-cell T cell receptor sequencing technologies. Nature Methods 18, 881–892. 10.1038/s41592-021-01201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IA, Rajaei H, Kim Y-A, Lee H, Lee H, Seo J-H, Heo S-H, Song IH, Gong G, and Lee HJ (2020a). T cell receptor repertoires of ex vivo–expanded tumor-infiltrating lymphocytes from breast cancer patients. Immunologic Research 68, 233–245. 10.1007/s12026-020-09150-8. [DOI] [PubMed] [Google Scholar]
- Park LM, Lannigan J, and Jaimes MC (2020b). OMIP-069: Forty-Color Full Spectrum Flow Cytometry Panel for Deep Immunophenotyping of Major Cell Subsets in Human Peripheral Blood. Cytometry Part A 97, 1044–1051. 10.1002/cyto.a.24213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Lagattuta KA, Lu BY, Lucca LE, Daud AI, Hafler DA, Kluger HM, Raychaudhuri S, and Sharpe AH (2022). TCR-sequencing in cancer and autoimmunity: barcodes and beyond. Trends in Immunology 43, 180–194. 10.1016/j.it.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, and Schietinger A (2019). Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Current Opinion in Immunology 58, 98–103. 10.1016/j.coi.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, Kancheva D, Martens L, De Vlaminck K, Van Hove H, et al. (2021). Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nature Neuroscience 24, 595–610. 10.1038/s41593-020-00789-y. [DOI] [PubMed] [Google Scholar]
- Pourgheysari B, Khan N, Best D, Bruton R, Nayak L, and Moss PAH (2007). The Cytomegalovirus-Specific CD4+ T-Cell Response Expands with Age and Markedly Alters the CD4+ T-Cell Repertoire. Journal of Virology 81, 7759–7765. 10.1128/jvi.01262-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Barkley D, França GS, and Yanai I (2021). Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220. 10.1038/s41586-021-03634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard A, and Asosingh K (2018). Best Practices for Preparing a Single Cell Suspension from Solid Tissues for Flow Cytometry. Cytometry Part A 95, 219–226. 10.1002/cyto.a.23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C (2020). A decade of immune-checkpoint inhibitors in cancer therapy. Nature Communications 11. 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de la Fuente L, Law AMK, Gallego-Ortega D, and Valdes-Mora F (2021). Tumor dissociation of highly viable cell suspensions for single-cell omic analyses in mouse models of breast cancer. STAR Protocols 2. 10.1016/j.xpro.2021.100841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, Pennell CA, Blazar BR, Chen CC, Geller MA, et al. (2019). Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nature Communications 10. 10.1038/s41467-019-08534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensky AY (2011). Regulatory T cells and Foxp3. Immunological Reviews 241, 260–268. 10.1111/j.1600-065X.2011.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sada-Ovalle I, Ocaña-Guzman R, Pérez-Patrigeón S, Chávez-Galán L, Sierra-Madero J, Torre-Bouscoulet L, and Addo MM (2015). Tim-3 blocking rescue macrophage and T cell function againstMycobacterium tuberculosisinfection in HIV+ patients. Journal of the International AIDS Society 18. 10.7448/ias.18.1.20078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e1020. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M, Dijkstra K, et al. (2018). Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nature Medicine 25, 89–94. 10.1038/s41591-018-0266-5. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, and Thommen DS (2022). Tertiary lymphoid structures in cancer. Science 375. 10.1126/science.abf9419. [DOI] [PubMed] [Google Scholar]
- Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T, Perez-Potti A, Nguyen S, Gorin J-B, Wu VH, Gostick E, Llewellyn-Lacey S, Hammer Q, Falck-Jones S, Vangeti S, et al. (2020). TOX is expressed by exhausted and polyfunctional human effector memory CD8+T cells. Science Immunology 5. 10.1126/sciimmunol.aba7918. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e110. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- Singh AK, and McGuirk JP (2020). CAR T cells: continuation in a revolution of immunotherapy. The Lancet Oncology 21, e168–e178. 10.1016/s1470-2045(19)30823-x. [DOI] [PubMed] [Google Scholar]
- Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, van Gisbergen KPJM, and van Lier RAW (2018). TRM maintenance is regulated by tissue damage via P2RX7. Science Immunology 3. 10.1126/sciimmunol.aau1022. [DOI] [PubMed] [Google Scholar]
- Steinert Elizabeth M., Schenkel Jason M., Fraser Kathryn A., Beura Lalit K., Manlove Luke S., Igyártó Botond Z., Southern Peter J., and Masopust D (2015). Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749. 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nature Methods 14, 865–868. 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biology 19. 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19. 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmeier LJ, Hoang KB, Nduom EK, Wieland A, Neill SG, Schniederjan MJ, Ramalingam SS, Olson JJ, Ahmed R, and Hudson WH (2022). Distinct phenotypic states and spatial distribution of CD8+ T cell clonotypes in human brain metastases. Cell Reports Medicine 3, 100620. 10.1016/j.xcrm.2022.100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WCC, Nerurkar SN, Cai HY, Ng HHM, Wu D, Wee YTF, Lim JCT, Yeong J, and Lim TKH (2020). Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Communications 40, 135–153. 10.1002/cac2.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. (2018). A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nature Medicine 24, 994–1004. 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ER, Mich JK, Yao Z, Hodge RD, Doyle AM, Jang S, Shehata SI, Nelson AM, Shapovalova NV, Levi BP, and Ramanathan S (2015). Fixed single-cell transcriptomic characterization of human radial glial diversity. Nature Methods 13, 87–93. 10.1038/nmeth.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnerre P, Wolski D, Subudhi S, Aljabban J, Hoogeveen RC, Damasio M, Drescher HK, Bartsch LM, Tully DC, Sen DR, et al. (2021). Differentiation of exhausted CD8+ T cells after termination of chronic antigen stimulation stops short of achieving functional T cell memory. Nature Immunology 22, 1030–1041. 10.1038/s41590-021-00982-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. New England Journal of Medicine 366, 2443–2454. 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukita M, Hamanishi J, Yoshitomi H, Yamanoi K, Takamatsu S, Ueda A, Suzuki H, Hosoe Y, Furutake Y, Taki M, et al. (2022). CXCL13-producing CD4+ T cells accumulate in the early phase of tertiary lymphoid structures in ovarian cancer. JCI Insight 7. 10.1172/jci.insight.157215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhaya S, Hubbard-Lucey VM, and Yu JX (2020). Immuno-oncology drug development forges on despite COVID-19. Nature Reviews Drug Discovery 19, 751–752. 10.1038/d41573-020-00166-1. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods 14, 935–936. 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- Van der Maaten L, and Hinton G (2008). Visualizing data using t-SNE. Journal of machine learning research 9. [Google Scholar]
- Veatch JR, Lee SM, Shasha C, Singhi N, Szeto JL, Moshiri AS, Kim TS, Smythe K, Kong P, Fitzgibbon M, et al. (2022). Neoantigen-specific CD4+ T cells in human melanoma have diverse differentiation states and correlate with CD8+ T cell, macrophage, and B cell function. Cancer Cell 40, 393–409.e399. 10.1016/j.ccell.2022.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nature Reviews Immunology 21, 485–498. 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, Broeks A, Sanders J, Herzig P, Zippelius A, et al. (2021). An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nature Medicine 27, 1250–1261. 10.1038/s41591-021-01398-3. [DOI] [PubMed] [Google Scholar]
- Wang X, Waschke BC, Woolaver RA, Chen SMY, Chen Z, and Wang JH (2020). MHC class I-independent activation of virtual memory CD8 T cells induced by chemotherapeutic agent-treated cancer cells. Cellular & Molecular Immunology 18, 723–734. 10.1038/s41423-020-0463-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yu L, and Wu AR (2021). The effect of methanol fixation on single-cell RNA sequencing data. BMC Genomics 22. 10.1186/s12864-021-07744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Huang C-X, Xiao X, Chen D-P, Shan H, He H, and Kuang D-M (2021). B cell heterogeneity, plasticity, and functional diversity in cancer microenvironments. Oncogene 40, 4737–4745. 10.1038/s41388-021-01918-y. [DOI] [PubMed] [Google Scholar]
- West EE, Jin H-T, Rasheed A-U, Penaloza-MacMaster P, Ha S-J, Tan WG, Youngblood B, Freeman GJ, Smith KA, and Ahmed R (2013). PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. Journal of Clinical Investigation 123, 2604–2615. 10.1172/jci67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West Erin E., Youngblood B, Tan Wendy G., Jin H-T, Araki K, Alexe G, Konieczny Bogumila T., Calpe S, Freeman Gordon J., Terhorst C, et al. (2011). Tight Regulation of Memory CD8+ T Cells Limits Their Effectiveness during Sustained High Viral Load. Immunity 35, 285–298. 10.1016/j.immuni.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 27, 670–684. 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Wieland A, Kamphorst AO, Adsay NV, Masor JJ, Sarmiento J, Nasti TH, Darko S, Douek DC, Xue Y, Curran WJ, et al. (2018). T cell receptor sequencing of activated CD8 T cells in the blood identifies tumor-infiltrating clones that expand after PD-1 therapy and radiation in a melanoma patient. Cancer Immunology, Immunotherapy 67, 1767–1776. 10.1007/s00262-018-2228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland A, Patel MR, Cardenas MA, Eberhardt CS, Hudson WH, Obeng RC, Griffith CC, Wang X, Chen ZG, Kissick HT, et al. (2020). Defining HPV-specific B cell responses in patients with head and neck cancer. Nature 597, 274–278. 10.1038/s41586-020-2931-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, Ivison GT, Ranganath T, Vergara R, Hollis T, et al. (2020). A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nature Medicine 26, 1070–1076. 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Liu C, Qian S, and Hou H (2013). The Expression of Tim-3 in Peripheral Blood of Ovarian Cancer. DNA and Cell Biology 32, 648–653. 10.1089/dna.2013.2116. [DOI] [PubMed] [Google Scholar]
- Wu W, Shi Y, Li J, Chen F, Chen Z, and Zheng M (2011). Tim-3 expression on peripheral T cell subsets correlates with disease progression in hepatitis B infection. Virology Journal 8. 10.1186/1743-422x-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Yuan L, Gao Q, Yuan P, Zhao P, Yuan H, Fan H, Li T, Qin P, Han L, et al. (2015). Circulating and tumor-infiltrating Tim-3 in patients with colorectal cancer. Oncotarget 6, 20592–20603. 10.18632/oncotarget.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates KB, Tonnerre P, Martin GE, Gerdemann U, Al Abosy R, Comstock DE, Weiss SA, Wolski D, Tully DC, Chung RT, et al. (2021). Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nature Immunology 22, 1020–1029. 10.1038/s41590-021-00979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nature Medicine 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, Araki K, West EE, Ghoneim HE, Fan Y, et al. (2017). Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552, 404–409. 10.1038/nature25144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, and Ahmed R (1998). Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. Journal of Experimental Medicine 188, 2205–2213. 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, Liu B, Niu L, Sun X, Yu X, et al. (2021). Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593.e1578. 10.1016/j.ccell.2021.09.010. [DOI] [PubMed] [Google Scholar]
- Zheng C, Fass JN, Shih Y-P, Gunderson AJ, Sanjuan Silva N, Huang H, Bernard BM, Rajamanickam V, Slagel J, Bifulco CB, et al. (2022). Transcriptomic profiles of neoantigen-reactive T cells in human gastrointestinal cancers. Cancer Cell 40, 410–423.e417. 10.1016/j.ccell.2022.03.005. [DOI] [PubMed] [Google Scholar]