

SUMMARY
Therapy resistance is a major challenge in the treatment of cancer. Here, we performed CRISPR-Cas9 screens across a broad range of therapies used in acute myeloid leukemia to identify genomic determinants of drug response. Our screens uncover a selective dependency on RNA splicing factors whose loss preferentially enhances response to the BCL2 inhibitor venetoclax. Loss of the splicing factor RBM10 augments response to venetoclax in leukemia yet is completely dispensable for normal hematopoiesis. Combined RBM10 and BCL2 inhibition leads to mis-splicing and inactivation of the inhibitor of apoptosis XIAP and downregulation of BCL2A1, an anti-apoptotic protein implicated in venetoclax resistance. Inhibition of splicing kinase families CLKs and DYRKs leads to aberrant splicing of key splicing and apoptotic factors that synergize with venetoclax and overcomes resistance to BCL2 inhibition. Our findings underscore the importance of splicing in modulating response to therapies and provide a strategy to improve venetoclax-based treatments.
Keywords: Acute myeloid leukemia, BCL2, CLK, DYRK, RBM10, RNA splicing, venetoclax, XIAP
eTOC Blurbs
Wang et al. perform genetic screens to identify mediators of sensitization and resistance to acute myeloid leukemia therapies. This reveals a unique relationship between expression of RNA splicing factors and response to the BCL2 inhibitor venetoclax as well as a inhibitor of splicing-dependent kinases which overcomes venetoclax resistance.
Graphical Abstract
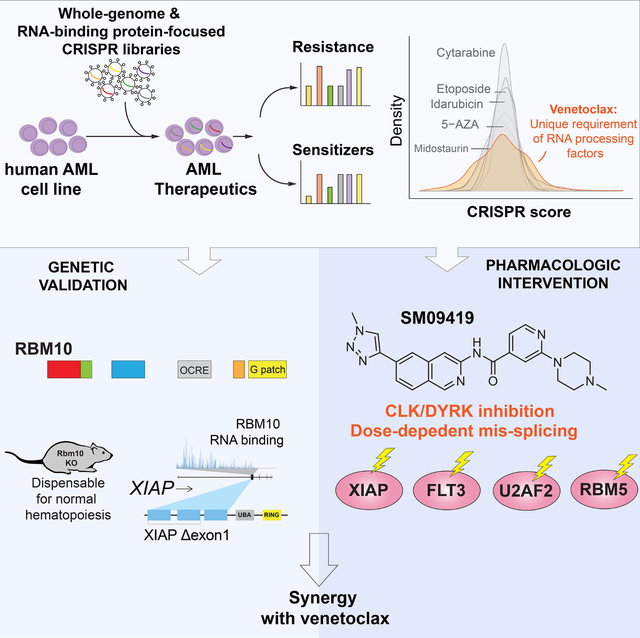
INTRODUCTION
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy marked by a dismal prognosis 1. For decades, the standard therapy for newly diagnosed AML has been intensive cytotoxic chemotherapy. Recently, several targeted therapies have been approved for AML, including inhibitors of IDH1/2, FLT3, and BCL2 2. Despite the introduction of these agents, most patients ultimately relapse and acquire resistance to long-term, continuous drug exposure 3,4. Genetic mutations, such as in TP53, have been shown to contribute to poor prognosis in patients treated with chemotherapy or the BCL2 inhibitor venetoclax 5–7. More recently, acquired BAX mutations have been shown to confer resistance to venetoclax in a subset of AML patients 8. However, in the majority of cases, genetic lesions are not known to be the main underlying mechanism of AML relapse 9,10, possibly implicating non-genetic mechanisms that allow persistent survival of leukemia cells upon exposure to drug therapy 11. For instance, upregulation of anti-apoptotic proteins 12,13 and dysregulated mitochondrial metabolism 14–16 can alter responsiveness to venetoclax. Such findings have demonstrated that epigenetic plasticity and transcriptional variability can act as critical evolutionary drivers of clonal fitness and drug resistance in leukemia 17,18.
The use of combinatorial therapies has been widely used to circumvent acquired drug resistance and is an approach with proven clinical efficacy for the treatment of several cancer types 19,20. In AML, the combination of venetoclax with hypomethylating agents is now widely used and has significantly improved the response and survival rates of patients 21,22. However, despite the success of venetoclax and hypomethylating agent combination therapy, this regimen is not curative. Furthermore, the majority of patients are unable to undergo curative allogeneic stem cell transplantation and ultimately become resistant to therapy 22. As such, identifying and targeting drug resistance mechanisms in AML with combinatorial treatment regimens is of critical importance.
Here, we utilized unbiased genetic screens to map drug/gene interactions for a variety of clinically approved therapies used in the treatment of AML. This effort highlighted a unique genetic relationship between response to venetoclax and the function of specific RNA splicing factors. While there is a well-established role for RNA splicing in the regulation of apoptosis 23, clinically viable means to manipulate splicing to enhance cell death in cancer have been limited to date. As genetic proof of concept, we identified a number of splicing factors whose loss promotes cell death in the setting of venetoclax and are dispensable for normal hematopoiesis, suggesting a therapeutic index for augmenting venetoclax response by modulating RNA splicing. Moreover, we present a compound to modulate RNA splicing and enhance venetoclax response via inhibition of the splicing kinase families known as CLKs (CDC-like kinases) and DYRKs (dual-specificity tyrosine-regulated kinases).
RESULTS
Mapping genomic determinants of AML drug response
To explore drug-gene interactions that underpin response to AML therapies, we performed a genome-wide Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) screen containing 77,441 single guide RNA (sgRNA) targeting 19,115 genes 24. We transduced this library into the human AML cell line, MOLM-13 (an MLL-AF9 translocated cell line bearing a concomitant FLT3ITD mutation) and after 8 days post-transduction, cells were treated with a broad range of clinically approved AML drugs (venetoclax, 5-azacytidine, cytarabine, etoposide, midostaurin, and idarubicin) (Figure 1A). Changes in sgRNA abundance were assessed at day 20 post-transduction by measuring the average fold change (drug/DMSO) of all sgRNAs targeting a given gene and top scoring candidates were classified as genes that sensitize (negative CRISPR score) or confer resistance (positive CRISPR score) to individual drugs (Table S1).
Figure 1. Mapping genomic determinants of AML drug response and synthetic lethal relationship between RNA splicing factors and venetoclax sensitivity.
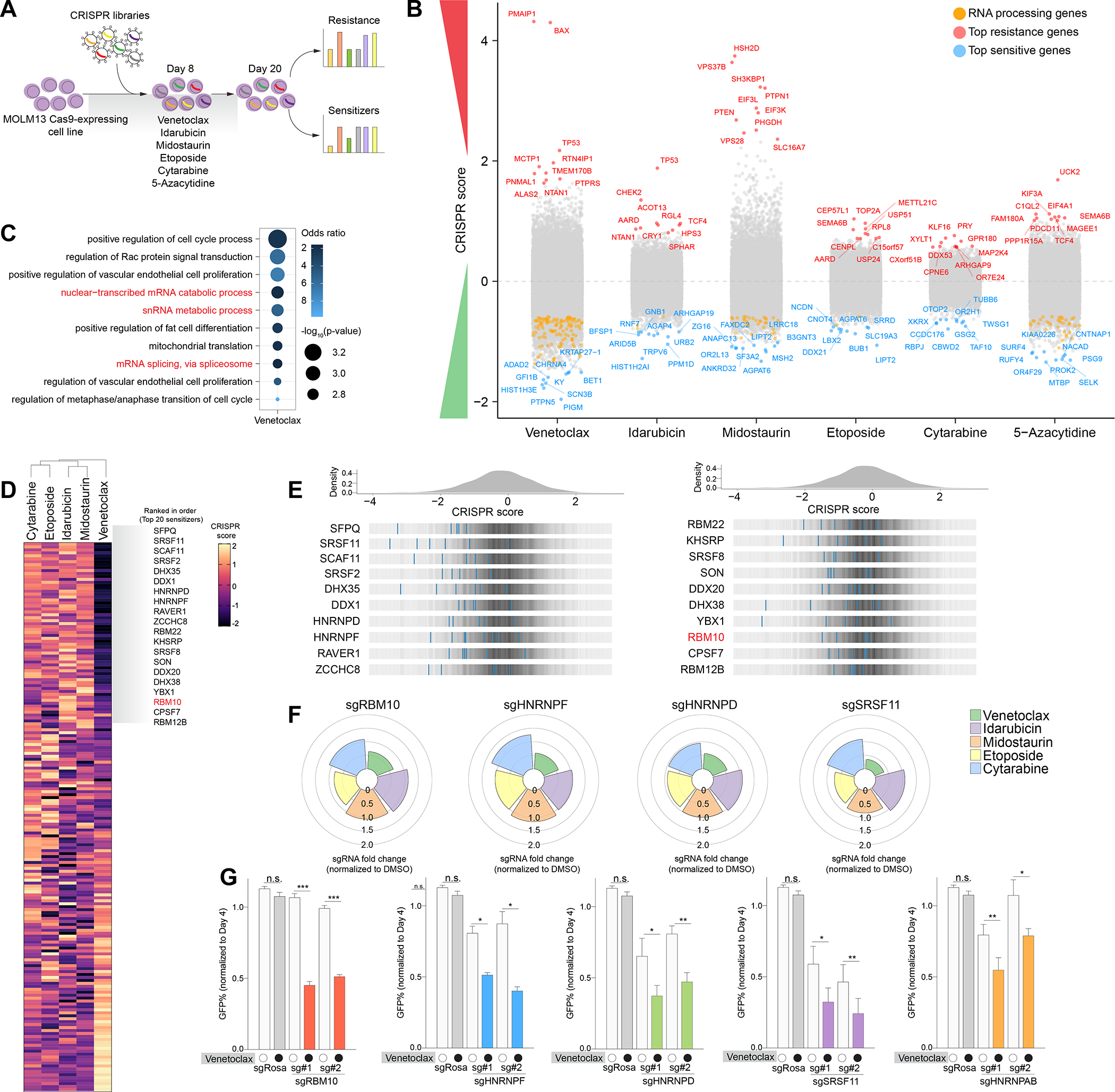
(A) Schematic of genome-wide CRISPR screens in MOLM-13 AML cells treated with a panel of clinically approved AML drugs. (B) Manhattan plot depicting top 10 genes that sensitizes (blue) or confer resistance (red) in individual CRISPR drug screens. Orange dots represent RNA processing genes. CRISPR score represents the log2 (fold-change) values of sgRNAs normalized to DMSO. (C) Gene ontology (GO) enrichment analysis of top sensitizers in the venetoclax screen. (D) Clustered heatmap of results of the RNA-binding protein-focused CRISPR drug screens in MOLM-13 AML cells treated with drugs. CRISPR score represents log2 fold change of sgRNAs normalized to DMSO. (E) Histogram of CRISPR scores for all sgRNAs in the venetoclax screen in (D). Values represent the log2 (fold-change) values of sgRNAs normalized to DMSO. The blue lines represent individual sgRNAs targeting the indicated genes among the top splicing factor candidates. (F) Polar plots of top synergistic splicing factors identified in (D) treated with various AML drugs. The height of the wedge corresponds to the sgRNA fold change normalized to DMSO. (G) Competition-based assay in MOLM-13 cells 10 days post-transduction with top 2 sgRNAs targeting each splicing factor or non-targeting sgRosa control (n=3 per condition, mean+SEM) treated with 50 nM venetoclax. Statistical analysis was performed using unpaired Student’s t test by Prism GraphPad (*p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant). See also Figure S1 and Table S1.
We identified previously characterized genes shown to mediate resistance to these compounds, including sgRNAs targeting the pro-apoptotic factors, BAX and PMAIP (also known as NOXA), as well as TP53 to confer venetoclax resistance 5 (Figure 1B). We also confirmed that inactivation of TOP2A, a target of etoposide, promoted survival of AML cells against etoposide exposure. Of note, sgRNAs targeting the uridine-cytidine kinase UCK2 scored as the top positive hit in our 5-azacytidine screen and UCK has been previously implicated to confer resistance to hypomethylating agent 25,26.
To identify combinatorial strategies that enhance existing AML therapies, we explored genes whose sgRNAs were significantly depleted upon drug exposure. We performed Gene Ontology (GO) enrichment analysis on the top scoring negative hits from each CRISPR screen and uncovered significant terms associated with RNA splicing and regulation of mRNAs, linked to venetoclax sensitization (Figure 1C). Consistently, we observed a significantly wider distribution (higher variance) of CRISPR scores for sgRNAs targeting RNA processing genes in the setting of venetoclax treatment compared to other drugs (Figure S1A). These data suggest a unique relationship between perturbation of RNA processing and response to venetoclax compared to other commonly used AML therapies. Previous reports have indicated the importance of leukemia cells exploiting alternative splicing and post-transcriptional mechanisms to promote tumor growth and therapy resistance 27–31. Moreover, clinical observations in AML patients have also demonstrated correlations between spliceosome mutations and alterations in response to venetoclax 6,32.
To further investigate the functional impact of RNA splicing factors in modulating drug response, we applied a previously developed CRISPR library targeting functional domains of 492 RNA-binding proteins (RBPs) consisting of 2,855 sgRNAs 27 to enhance CRISPR-Cas9 negative selection by targeting functional protein domains 33 (Figure 1D). Consistent with our initial findings from the genome-wide screen, we identified that loss-of-function of several RNA splicing factors enhanced sensitivity or resistance to venetoclax treatment (Figure 1E–F). We further validated the top scoring sensitizers such as RBM10, SRSF11, SRSF8, HNRNPD, HNRNPAB, and HNRNPF whose inactivation led to preferential sensitivity in AML cells treated with venetoclax, which was not seen with other tested therapeutics (Figure 1G and Figure S1B–D).
Loss of RBM10 sensitizes leukemia cells to venetoclax
Among the top gene candidates whose loss sensitized cells to venetoclax was RBM10, whose loss-of-function exclusively enhanced venetoclax efficacy in AML amongst other drugs screened (Figure 1F–G and Figure S2A). We further explored publicly available genome-wide CRISPR screens performed in a broad range of human cancer cell lines which revealed that RBM10 loss is least essential in leukemia cell lines compared other cancer subtypes (Figure S2B). However, in the presence of venetoclax, RBM10 deletion strikingly conferred preferential lethality and anti-leukemic effects in human AML cell lines, across a variety of molecular subtypes (Figure 2A–C and Figure S2C). Of note, RBM10 deletion even augmented BCL2 inhibition in TP53-mutated AML cell lines (THP-1, and U937) 34, which have been previously described as venetoclax resistant 5,6 (Figure S2D).
Figure 2. RBM10 loss enhances BCL2 inhibition in AML cells but is dispensable for normal hematopoiesis.
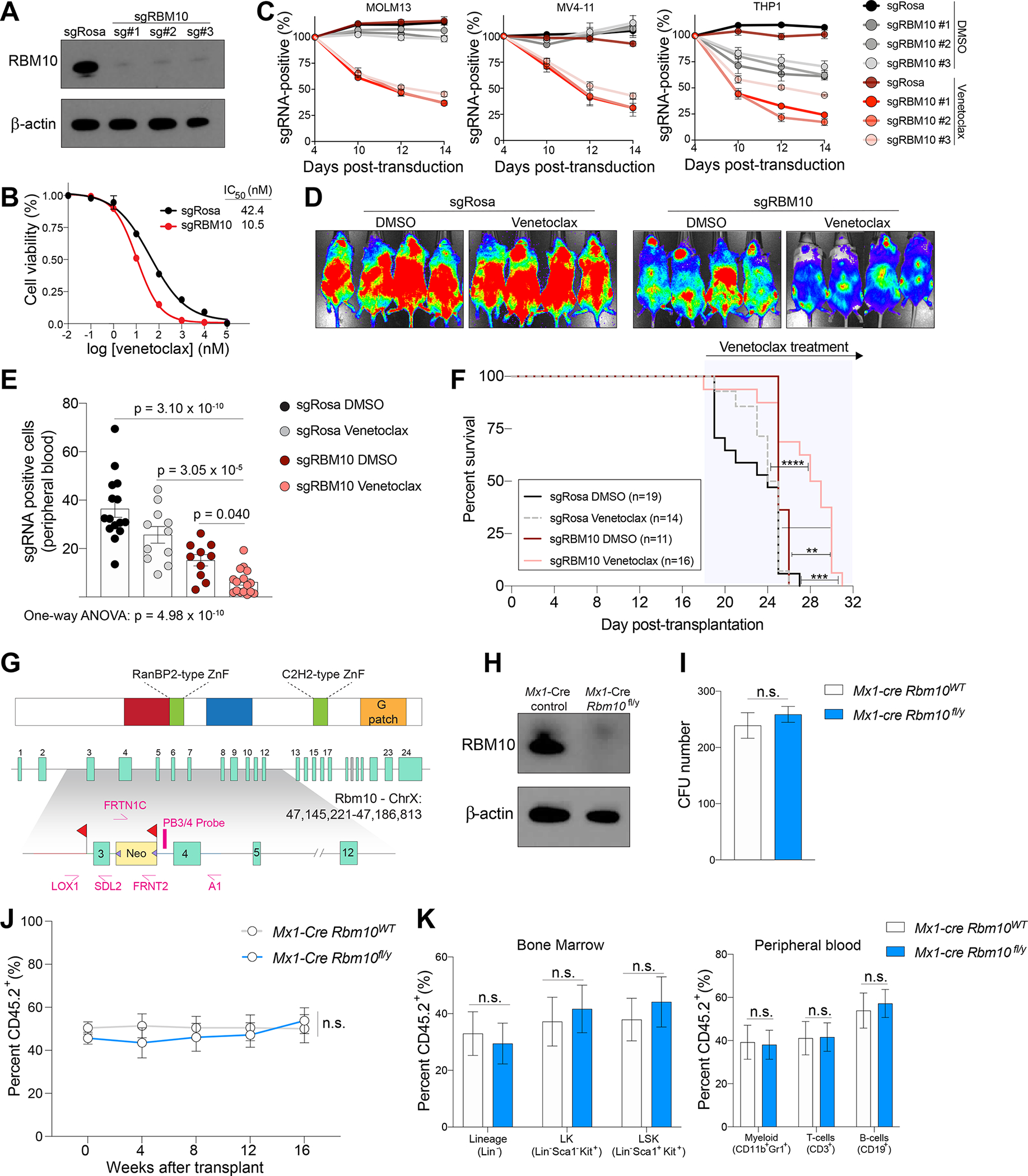
(A) Western blot of CRISPR-mediated knockout of RBM10 in MOLM-13 cells. (B) Dose-response curves of sgRBM10 or sgRosa treated with indicated venetoclax concentrations on the x-axis and cell viability on the y-axis at 48 hours. IC50 values were calculated from technical triplicates per experiment, error bars represent SEM. (C) Competition proliferation assays of sgRBM10 or non-targeting sgRosa in human AML cell line expressing Cas9 and treated with 50 nM of venetoclax or DMSO (n=3 biological replicates per time point and condition, mean+SEM). (D) Bioluminescent imaging of mice transplanted with MOLM-13 cells transduced with sgRBM10 or sgRosa and treated daily with venetoclax (100 mg/kg) or vehicle control. Representative images of 4 mice per condition is shown. Images were taken 4 days post-treatment. (E) Flow cytometry analysis of GFP-positive sgRNA-expressing indicated by y-axis MOLM-13 cells in peripheral blood at day 6 post-treatment. Statistical analysis was performed using One-way ANOVA with post-hoc testing as indicated (number of mice used in each group is indicated in F, mean+SEM). (F) Kaplan-Meier survival curves of mice transplanted with MOLM-13 cells transduced with sgRBM10 or sgRosa and treated daily with venetoclax (100 mg/kg) or vehicle. The p values were determined using a log-rank Mantel-Cox test (**p < 0.01, ***p < 0.001, n.s., not significant). (G) Schematic depiction of the targeting strategy to generate Rbm10 cKO mice. The Rbm10 allele was deleted by targeting exon 3 that resulted in a frameshift following excision. Two LoxP sites flanking exon 3 and an Frt-flanked neomycin selection cassette were inserted in the downstream intron. (H) Western blot of Rbm10 in bone marrow mononuclear cells from Mx1-cre Rbm10fl/y (Rbm10 cKO) or Mx1-cre control 7 days after polyinosinic-polycytidylic acid (pIpC) treatment. (I) Total number of colony-forming units (CFU) from bone marrow cells of Mx1-cre Rbm10fl/y (Rbm10 cKO) or Mx1-cre control mice following 7 days of culture (n=6, mean+SEM). The p values were determined by unpaired student t test. n.s., not significant. (J) Percentage of CD45.2+ cells in peripheral blood over the course of 4 months competitive transplantation (n=6 for Mx1-cre control and n=7 mice for Rbm10 cKO, mean+SEM). (K) Percentage of CD45.2+ of hematopoietic stem and progenitor cells in the bone marrow (left) and mature immune cells in the peripheral blood (right) (n=6 for Mx1-cre control and n=7 mice for Rbm10 cKO, mean+SEM). See also Figure S2.
We next assessed the impact of RBM10 deletion on the response of human AML cells to venetoclax in vivo. To achieve this, we transplanted MOLM-13 cells stably expressing firefly luciferase and anti-RBM10 sgRNAs or the non-targeting control (sgRosa) into (NOD)/severe combined immunodeficiency (SCID) IL2Rgammanull (NSG) mice. Upon disease onset, mice were treated with venetoclax (100 mg/kg/day) or vehicle control (Figure 2D). Consistent with our in vitro findings, RBM10 deletion reduced leukemia burden and extended survival in the setting of venetoclax treatment (Figure 2E–F and Figure S2E). Indel analysis of prolonged RBM10 sgRNA editing by next-generation sequencing showed an outgrowth of cells containing in-frame RBM10 mutations, implicating that mice succumb to an outgrowth of sgRNA-expressing cells that retain RBM10 functionality (Figure S2F). Overall, these findings provide genetic evidence that loss of RBM10 has a synthetic lethal relationship with BCL2 inhibition in AML.
Many RNA splicing factors are known to be pan-essential for cell survival35. To evaluate the therapeutic potential of RBM10 modulation as a therapeutic candidate for venetoclax-based therapies, we generated an Rbm10 conditional knockout (cKO) mouse by inserting loxP sites flanking exon 3 of Rbm10 (Rbm10fl/fl; Figure 2G) and crossing with interferon-induced Mx1-driven Cre recombinase mice. Following intraperitoneal polyinosinic;polycytidylic acid (pIpC) injections, Rbm10 cKO mice were confirmed to excise exon 3 of Rbm10 leading to an early frameshift and loss of Rbm10 protein in bone marrow cells (Figure 2H and Figure S2G–I). We next assessed stem cell functionality using in vitro colony-replating assays which demonstrated that Rbm10 deletion in hematopoietic precursors did not impair colony formation (Figure 2I). In parallel, bone marrow-derived cells from CD45.2+ Rbm10 floxed mice were transplanted in a competitive manner along with competitor Rbm10 wild-type CD45.1+ cells and treated with pIpC after stable reconstitution of hematopoiesis. There was no significant effect of Rbm10 deletion on absolute numbers or frequency of peripheral blood and bone marrow cells (Figure 2J–K and Figure S2J–K). These data demonstrate that Rbm10 is dispensable for normal hematopoiesis.
Dual inhibition of RBM10 and BCL2 promotes XIAP mis-splicing
We next sought to understand the mechanistic basis for the relationship between RBM10 loss and enhanced response to venetoclax. The effects of RBM10 knockout (KO) on venetoclax response were rescued by expressing an RBM10 cDNA impervious to anti-RBM10 sgRNAs (due to mismatches between cDNA sequence and the RBM10 sgRNAs; Figure 3A). However, expression of RBM10 lacking its second RNA recognition motif 2 (RRM2) or C2H2-type zinc finger (C2H2 ZnF) 36 failed to rescue response to venetoclax (Figure 3A).
Figure 3. Impact of RBM10 on RNA binding, RNA splicing, and response to venetoclax.
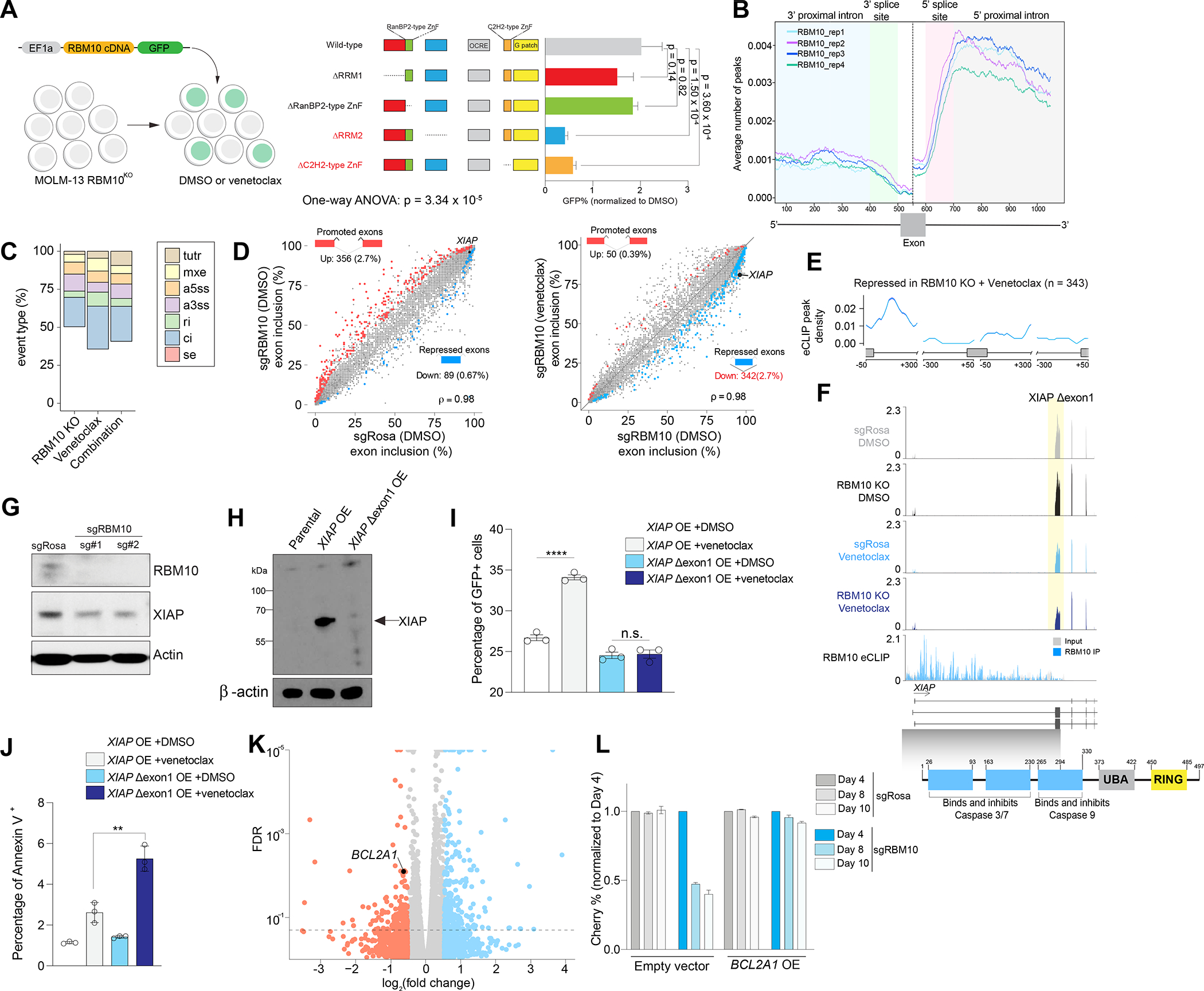
(A) Competition-based assay of RBM10 KO in MOLM-13 cells and transduced with RBM10 cDNA wild-type (WT) or individual mutant (lacking RNA-binding domains) RBM10 cDNA and treated with venetoclax (50 nM) or DMSO at 48 hours (n=3, mean+SEM). The p-values were determined by One-way ANOVA with post-hoc testing. (B) Metaintron plots of average number indicated by y-axis of RBM10 peaks mapped to intronic regions flanking exons in MOLM-13 cells (n=4 eCLIP replicates). This plot is exon-centered (500–600 bp) on the x-axis. Enhanced crosslinking and immunoprecipitation (eCLIP) was performed in 4 replicates. (C) Percentage of treatment-responsive (RBM10 KO, venetoclax, or RBM10 KO and venetoclax) differentially spliced event types: cassette exons (SE), alternative 5’ ss exon (A5E), alternative 3’ ss exon (A3E), mutually exclusive exons (MXE), retained intron (RI), constitutive intron (CI), and tandem 3’ UTR (TUTR) from RNA-seq (n=3 per condition). (D) Scatter plot of cassette exons (SE) promoted (red circles) or repressed (blue circles) in MOLM-13 cells transduced with sgRosa (y-axis) or sgRBM10 (x-axis) treated with DMSO or venetoclax RNA-seq (n=3 per condition). ρ denotes Spearman’s rank correlation coefficient. (E) RBM10 splicing map generated by integrating RBM10 KO splicing changes from RNA-seq and RBM10 eCLIP binding sites. (F) RNA-seq and eCLIP (bottom) coverage plots of XIAP Δexon1 in MOLM-13 cells with RBM10 KO or non-targeting sgRNAs treated with DMSO or venetoclax. Yellow shadow depicts exon exclusion event in RBM10KO venetoclax-treated MOLM-13 cells overlapped with functional protein domains of XIAP. (G) Western blotting of XIAP after 50 nM venetoclax treatment of MOLM-13 cells with sgRosa or sgRBM10 for 48 hrs. (H) Western blotting of XIAP protein levels after ectopic overexpression of XIAP full-length (FL) or XIAP Δexon1. (I) Competition-based assay of XIAP full-length (FL) or XIAP Δexon1 linked to GFP reporter after 24 hrs of venetoclax treatment (n=3, mean+SEM). Y-axis denotes GFP positive cells. (J) Annexin V staining of XIAP full-length (FL) or XIAP Δexon1 after 24 hrs of venetoclax treatment (n=3, mean+SEM). Y-axis denotes Annexin V positive cells. Statistical analysis was performed using unpaired Student’s t test by Prism GraphPad (*p < 0.05, **p < 0.01, n.s., not significant). (K) Volcano plot of differentially expressed genes (DEGs) upon RBM10KO venetoclax-treated MOLM-13 cells compared to DMSO RNA-seq (n=3 per condition). (L) Competition-based assay measuring Cherry-expressing sgRBM10 or sgRosa cells transduced with overexpression (OE) of BCL2A1 cDNA or empty vector GFP-positive cells in MOLM-13 cells treated with 50 nM venetoclax for 48 hours (n=3, mean+SEM). Y-axis denotes mCherry positive cells. See also Figure S3 and Tables S2–4.
The above data indicate the importance of RBM10’s RNA binding domains on venetoclax response. We therefore further assessed the direct impact of RBM10-RNA interactions on pre-mRNA binding and splicing, which have not been explored in hematopoietic cells previously. We performed anti-RBM10 enhanced UV cross-linking immunoprecipitation (eCLIP) 37 in MOLM-13 AML cells (Figure S3A). This approach identified approximately 29,000 significant sequence clusters bound by RBM10, which corresponded to ~5,000 annotated transcripts (Table S2). Approximately 90% of RBM10 binding sites mapped to intronic sites, with a preferential occupancy of distal (further than 500 nucleotides (nt) from the splice site region) (77.1%) and proximal (within 500 nt of splice site region) intronic (8%) sequences near 5’ and 3’ splice sites throughout the transcriptome (Figure 3B and Figure S3B).
Next, we evaluated the transcriptional and splicing changes in RBM10-deleted AML cells treated with venetoclax or DMSO, compared to non-targeting sgRosa, by RNA sequencing (RNA-seq) (Table S3–4). We measured isoform usage frequencies across seven main types of alternative splicing events [skipped (or retained) cassette exons (SE), alternative 5’ splice sites (A5SS), alternative 3’ splice sites (A3SS), mutually exclusive exons (MXE), tandem 3’ UTRs (TUTR), and retained (RI) and constitutive introns (CI)] to quantify splicing changes across treatments (Figure S3C–D). RBM10 KO primarily led to changes in cassette exon splicing (Figure 3C), suggesting that RBM10 most commonly regulates exon usage in AML cells. In comparison, RBM10 deletion in the presence of venetoclax amplified the degree of aberrant splicing involving constitutive introns and cassette exons. Most notably, we observed an increase in exon exclusion events in the combination treatment versus RBM10 deletion alone (n=342) (Figure 3D).
We further investigated the link between RBM10 binding and differential splicing observed in combined RBM10 KO and venetoclax. These analyses revealed RBM10 binding signal in the 5′ region of the upstream intron of repressed cassette exons following combination treatment, suggestive of a role of RBM10 binding in this region in promoting exon exclusion (Figure 3E). Interestingly, we found that the inhibitor of apoptosis protein (IAP) family member, XIAP, displayed increased exclusion of the first coding exon in venetoclax-treated RBM10 KO AML cells, which also had significant RBM10 binding at this region (Figure 3F and Figure S3E). XIAP, also known as BIRC4, binds and sequesters pro-apoptotic caspases through direct protein-protein interactions with its BIR domains to prevent caspase homodimerization thereby inactivating apoptosis 38–41. Based on our findings, we hypothesize that activation of apoptosis is a consequence of skipping the first coding exon of XIAP. The resulting mRNA lacks XIAP’s canonical start codon as well as the sequence encoding the majority of its BIR1-3 domains, strongly suggesting that this splicing change results in loss of functional XIAP production (Figure 3G). We also functionally evaluated the mis-spliced isoform of XIAP event induced by RBM10 KO and venetoclax treatment (which we refer to as XIAP Δexon 1) by ectopically expressing full-length XIAP (FL) or XIAP Δexon 1 linked to a GFP reporter in MOLM-13 cells (Figure 3H). Consistent with the function of IAP proteins, we found XIAP FL overexpression allowed survival of AML cells after venetoclax treatment, whereas XIAP Δexon 1 resulted in increased apoptosis (Figure 3I–J and Figure S3F). Overall, these results demonstrate that XIAP Δexon 1 cannot rescue cell death induced by venetoclax treatment and RBM10 deletion (Figure S3G). Importantly, prior work has demonstrated that inhibition of XIAP synergized with venetoclax 42, highlighting the importance of XIAP levels in BCL2 inhibitor sensitivity.
Gene expression analysis of venetoclax-treated RBM10 KO AML cells revealed downregulated expression of BCL2A1, which encodes an anti-apoptotic factor whose expression is correlated with venetoclax resistance in AML patients 6,43 (Figure 3K and Figure S3H–I). Consistent with these data, overexpression of BCL2A1 cDNA was able to fully rescue the anti-leukemic effects seen with the combined loss of RBM10 and BCL2 inhibition (Figure 3L). Moreover, we did not observe significant RBM10 eCLIP peaks or splicing alteration of BCL2A1 mRNA which suggests that upstream factors may regulate BCL2A1 transcript. Overall, these data provide mechanistic evidence that the combined loss of RBM10 and BCL2 leads to altered splicing and expression of mRNAs encoding key apoptotic genes.
Pharmacologic inhibition of splicing kinases synergizes with venetoclax
Utilizing our CRISPR screens to identify pharmacologically intervenable splicing factors to augment venetoclax response, we found that inactivation of several serine/arginine (SR)-rich proteins (SRSF2, SRSF3, SRSF8, and SRSF11) sensitized AML cells to venetoclax (Figure 1D and Table S1). The family of SR splicing factors are essential for alternative pre-mRNA splicing and their activity is tightly regulated by post-translational modifications placed by serine/threonine kinases 44–46. For example, CLKs phosphorylate Arginine-Serine (RS) domains in SR proteins and regulate pre-mRNA splicing 45,47. Moreover, DYRK1A has been reported to regulate alternative splicing via phosphorylation of SF3B148–50. In addition, analysis of publicly available genome-wide CRISPR screens from DepMap51 revealed BCL2 as one of the top co-dependencies with DYRK1A loss (Figure S4A).
These findings support the rationale to inhibit splicing-dependent kinases as a combinatorial strategy with venetoclax treatment. To pursue therapeutic inhibition of splicing-dependent kinases, SM09419, a pan-CLK pan-DYRK inhibitor was developed via rational design and iterative medicinal chemistry to achieve drug-like and favorable pharmacokinetic profiles (Figure 4A–D). We confirmed the selectivity of SM09419 to target CLK kinases CLK1-4 as well as DYRK1A-B and DYRK2 using in cell NanoBRET target engagement assays (Figure 4C and Figure S4B). Accordingly, SM09419 treatment resulted in dose-dependent reduction of CLK activity and SR protein phosphorylation in AML cells (Figure 4E and Figure S4C). Next, we assessed the combinatorial effects of SM09419 with a panel of drugs (venetoclax, 5-azacytidine, cytarabine, and midostaurin) in human AML cell lines. We observed a synergistic effect exclusively when combining SM09419 and venetoclax in MOLM-13 parental and venetoclax-resistant cells but not with other drugs (Figure 4F–I and Figure S4D–E). Despite robust anti-leukemic effects of SM09419 in vitro, SM09419 (25 mg/kg) treatment in wild-type C57BL/6 mice was well tolerated in vivo with no signs of hematologic toxicities (based on serial blood counts, in vitro hematopoietic progenitor cell assays, and detailed analysis of hematopoietic cell composition in blood and bone marrow) or liver or kidney dysfunction, thus providing a rationale for pharmacologic inhibition of CLK/DYRK in combination with venetoclax in AML (Figure S5A–H).
Figure 4. Pharmacologic inhibition of splicing-dependent kinases synergizes with venetoclax. (A) Structure of SM09419 selectivity.
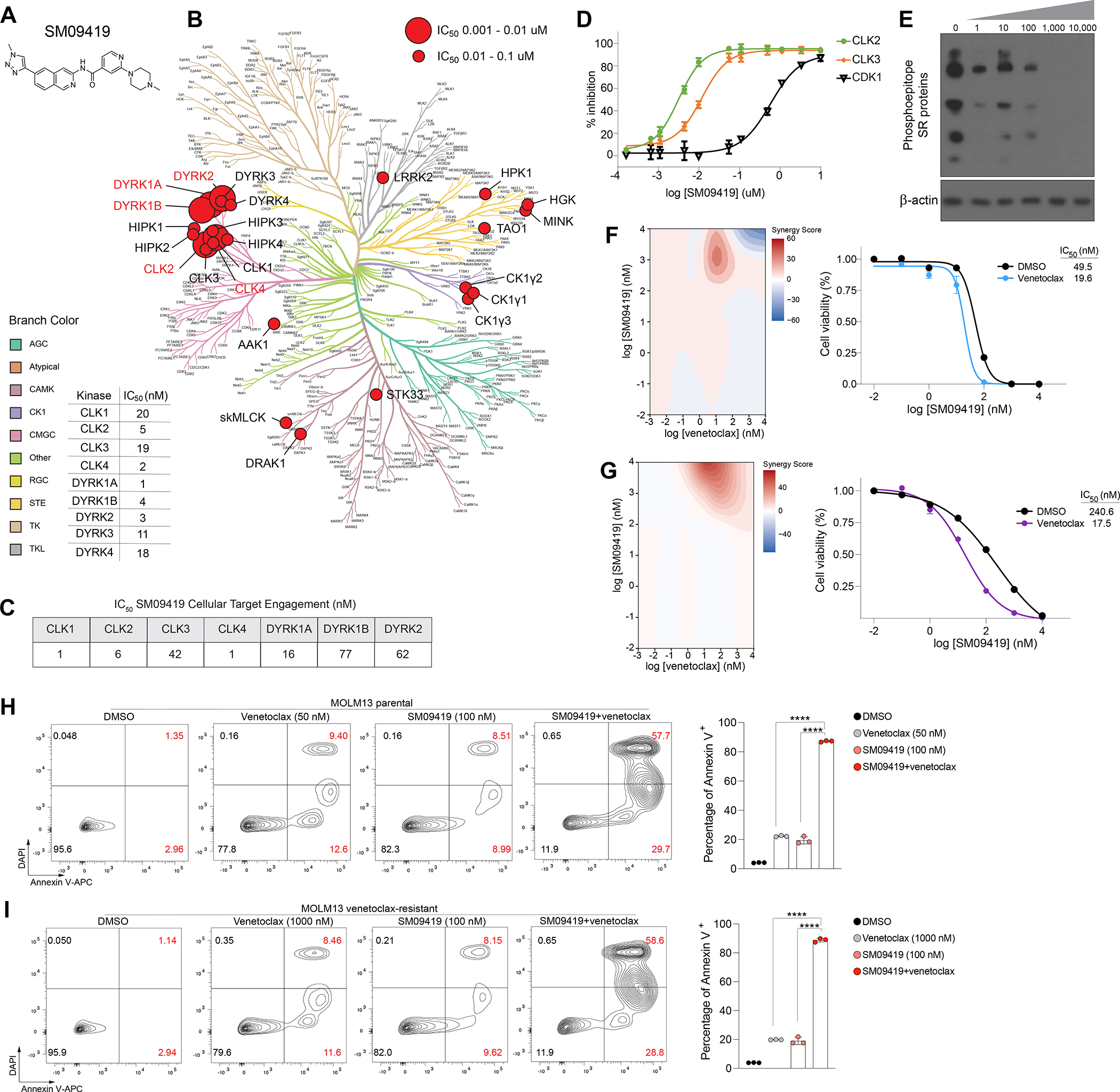
(B) Kinase dendrogram of SM09419. Kinases with IC50 values of 0.01 to 0.1 μM are indicated by small red circle, whereas larger red circles represent more potent IC50 values with 0.001 to 0.01 μM. (C) NanoBRET target engagement assay of CLK1-4, DYRK1A/B, and DYRK2 upon 24 hrs of SM09419 treatment. (D) Inhibition of CLK kinases (CLK2 and CLK3) and CDK1 kinase (n=3). IC50 values were determined from dose response curves. Y-axis denotes the percent inhibition for CLK2, CLK3, and CDK1 (n=3, mean+SEM). (E) Western blot of phosphorylated SR proteins treated with increasing concentration of SM09419 for 48 hrs in MOLM-13 cells. (F) 2D synergy plots using Zero interaction potency (ZIP) model (left) and dose-response curves (right) of SM09419 and venetoclax combination at various concentration treated for 48 hrs in MOLM-13 (n=3, mean+SEM) and (G) KG-1 cells (n=3, mean+SEM). The presence of synergy was determined using the SynergyFinder computational package and the ZIP synergy index in which red signifies synergism and blue is antagonism. A positive synergy score is the percent more cell death than expected. IC50 values were calculated from technical triplicates per experiment. (H) Annexin V staining (left) and quantification (right) of MOLM-13 parental and (I) venetoclax-resistant cell lines treated with SM09419, venetoclax, or the combination at 48 hrs post-treatment (n=3, mean+SEM). Y-axis denotes percent of Annexin V positive cells. Statistical analysis was performed using unpaired Student’s t test by Prism GraphPad (****p < 0.0001). See also Figure S4–5.
To understand the mechanistic basis for the synergy of SM09419 and venetoclax combination, we performed RNA-seq on MOLM-13 human AML cells treated with SM09419 alone or in combination with venetoclax (Table S5–6). Splicing analyses showed that SM09419 alone, or in combination with venetoclax, mainly resulted in changes in the processing of constitutive/retained introns and cassette exons (Figure 5A). CLK/DYRK inhibition affects cassette exon recognition in a sequence-specific manner, as evidenced by the enrichment of pyrimidines in exons preferentially excluded upon SM09419 treatment (Figure 5B). While venetoclax monotherapy had no significant effects on RNA splicing, treatment with SM09419 or the combination resulted in striking reductions in RNA splicing efficiency as manifested by cassette exon skipping and intron retention (Figure 5A, Figure 5C, and Table S6). Of note, these splicing shifts resulted in substantial increases in levels of mRNAs that contain premature termination codons and are therefore predicted substrates for degradation by nonsense-mediated decay (NMD).
Figure 5. SM09419 promotes mis-splicing of key oncogenic pathways in AML.
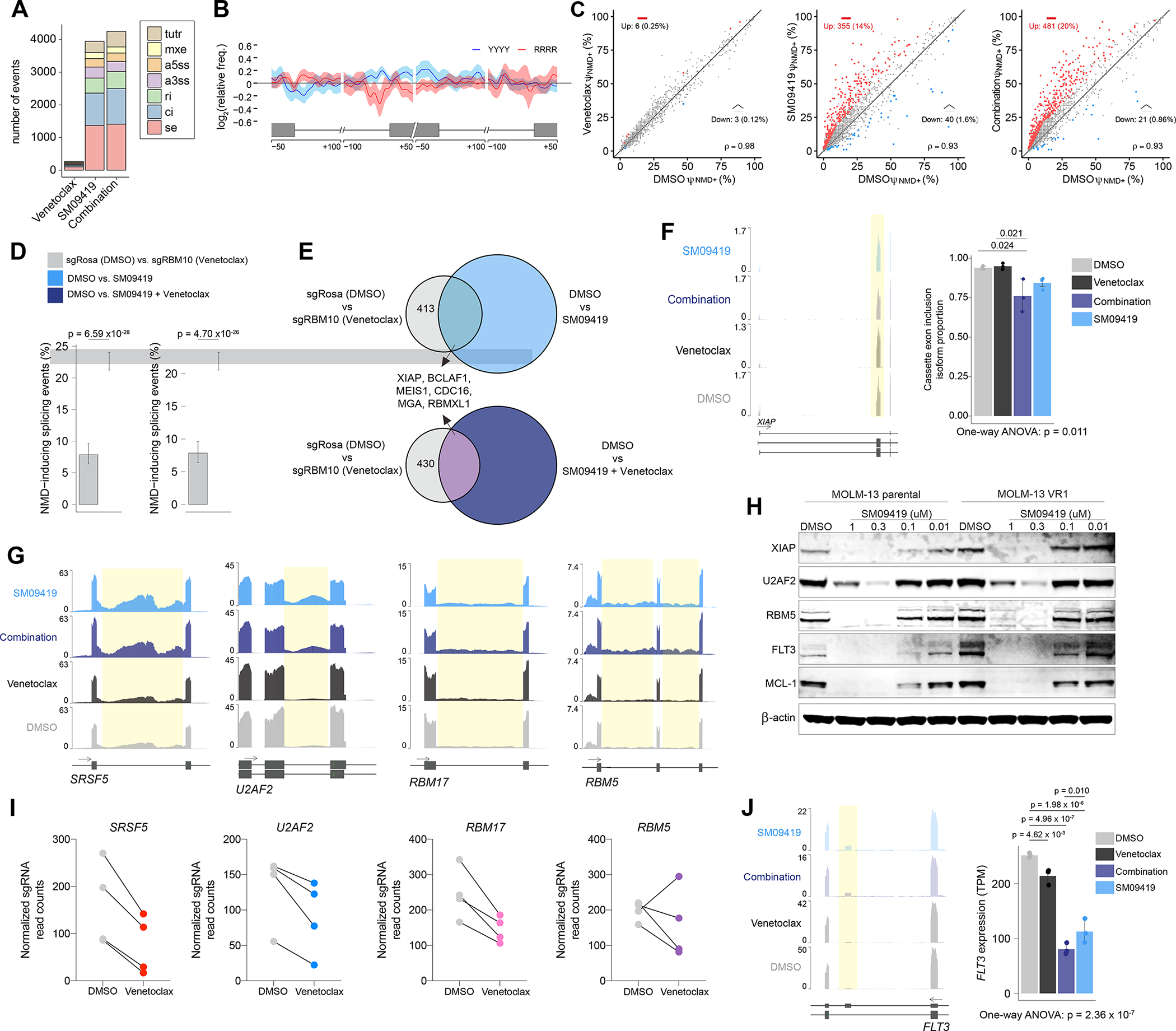
(A) Total number of splicing changes observed after SM09419 (100 nM), venetoclax (10 nM), or combination of SM09419 (100 nM) and venetoclax (10 nM) treatment for 48 hours RNA-seq (n=3 per condition). Cassette exons (SE), alternative 5’ ss exon (A5E), alternative 3’ ss exon (A3E), mutually exclusive exons (MXE), retained intron (RI), constitutive intron (CI), and tandem 3’ UTR (TUTR). (B) Spatial distribution of pyrimidines-rich (YYYY) and purine-rich (RRRR) motifs comparing sequence enrichment of excluded exons (n = 674) against included exons (n = 370) in SM0419-treated (100 nM) MOLM-13 cells. (C) Scatter plot of NMD-inducing retained intron (RI) events (red circles) in MOLM-13 cells treated with venetoclax (left), SM09419 (middle) or the combination of venetoclax and SM09419 (right) RNA-seq in triplicates for each condition. (D) Percentage of NMD-inducing events indicated on the y-axis in RBM10 KO venetoclax (compared to non-targeting sgRosa) and SM09419, or SM09419+venetoclax (compared to DMSO) RNA-seq in triplicates for each condition (mean+SEM). (E) Venn diagram of NMD-inducing events in RBM10 KO venetoclax (compared to non-targeting sgRosa) and SM09419, or SM09419+venetoclax (compared to DMSO). (F) RNA-seq coverage plot (left) and mean PSI of XIAP cassette exon inclusion isoform (n=3 per condition, mean+SEM). (G) RNA-seq coverage plots of the splicing factors SRSF5, U2AF2, RBM17, and RBM5 in MOLM-13 cells. Yellow regions represent retained intron events in each of the genes. (H) Western blotting of XIAP, U2AF2, RBM5, FLT3, MCL-1, and actin in MOLM-13 parental or venetoclax-resistant (VR1) cells treated with varying concentration of SM09419 for 24 hrs. (I) Normalized sgRNA counts of top splicing factors from RNA-binding protein CRISPR screen that synergized with venetoclax treatment in MOLM-13 cells. (J) RNA-seq coverage plots (left) and gene expression (right) plots for FLT3 mRNA (n=3 per condition, mean+SEM). p-values were determined by One-way ANOVA with post-hoc testing as indicated. See also Figure S6 and Tables S5–6.
In order to understand how the effects of SM09419 relates to deletion of RBM10, we next performed a systematic comparison of the splicing changes and gene expression across both conditions in the same MOLM-13 cells. Both RBM10 deletion and SM09419 treatment cause splicing changes which promote nonsense-mediated decay (NMD)-inducing transcripts. However, the magnitude of NMD-inducing splicing events is greater with SM09419 treatment (a result consistent with the fact that RBM10 deletion in the absence of any drug treatment is well-tolerated in MOLM-13 cells) (Figure 5D). Nonetheless, a number of mRNA isoforms were shared across RBM10 deletion versus SM09419 treatment in the absence and presence of concomitant venetoclax treatment. Interestingly, one concordant effect was the same mis-splicing event in XIAP seen with RBM10 deletion (Figure 5E–F). Finally, both RBM10 deletion and SM09419 treatment prominently downregulate TNFAIP3 (also known as A20) (Figure S6A). TNFAIP3 is a well described regulator of NF-κB signaling 52 and its inhibition may explain the BCL2A1 downregulation in RBM10 KO cells exposed to venetoclax. Transcriptomic analysis of SM09419-treated AML cells also demonstrated downregulation of MYB and MYC mRNA levels, which are essential oncogenic factors in AML (Figure S6B) 53,54.
SM09419 induces splicing alterations of key survival genes in AML
Further characterization of SM09419-associated splicing changes revealed increased intron retention within the transcripts of a number of RNA splicing factors (SRSF5, U2AF2, RBM17, and RBM5) which also led to decreased protein expression in two independent human AML cell lines (Figure 5G–H and Figure S6C–E). Interestingly, several of these same splicing factors were also identified by our CRISPR screens as genes whose inactivation enhanced venetoclax efficacy and therefore explains the synergistic effects when combining SM09419 and venetoclax (Figure 5E and Figure 5I). Furthermore, we found that SM09419-treated AML cells led to downregulation of several key apoptotic proteins, such as MCL-1, which have been shown to be upregulated in hematologic neoplasms and confers resistance to BCL2 inhibitors 12 (Figure 5H and Figure S6E). Finally, SM09419 treatment promoted inclusion of an exon with an in-frame stop codon (a “poison exon,” whose inclusion renders the transcript NMD-sensitive) in the receptor tyrosine kinase FLT3 (Figure 5J). As such, there was reduced FLT3 mRNA and protein expression in SM09419 treated cells (which is especially pertinent given the known dependence of this FLT3 mutant AML cell line on FLT3 expression) (Figure 5H and Figure S6E). Importantly, the above results on the impact of SM09419 treatment on XIAP, FLT3, and MCL-1 levels were confirmed in an additional AML cell line. Both venetoclax resistant and sensitive KG-1a cells were similarly susceptible to SM09419 treatment and experienced comparable dose-dependent reductions in XIAP, FLT3, and MCL-1 (Figure S6D–E).
Additional predicted NMD-inducing splicing events upon SM09419 treatment with commensurate reduction in mRNA expression in SMYD2 (a lysine methyltransferase recognized as a therapeutic target in AML 53, DHODH (a metabolic enzyme and recent AML therapeutic target) 55, ATAD3A (a metabolic enzyme whose expression has been included in leukemia stem cell signatures) 56, the MYC target gene CDC16 57, and the additional RNA processing genes SRPK3, TRA2A, and DDX51 (Figure S6F). Overall, these data identify that SM09419 downregulates expression of key RNA splicing factors as well as important apoptotic factors and FLT3 via impaired splicing to enhance response to venetoclax in AML while having minimal impact on normal hematopoiesis.
SM09419 overcomes venetoclax-based therapy resistance
We next tested the efficacy of SM09419 across a spectrum of human AML cell lines. SM09419 treatment resulted in broad anti-leukemic effects with potent inhibitory activity across AML subtypes, including cell lines that were highly resistant to venetoclax treatment (Figure 6A). Based on these data, we evaluated the ability of SM09419 to overcome venetoclax resistance. We developed three independent venetoclax-resistant MOLM-13 cell lines following continuous exposure to venetoclax for 3 weeks. Dose-response curves after drug selection confirmed that venetoclax-resistant cell lines displayed a high inhibitory effect concentration (IC50 > 99 nM) approximately six times greater than parental cells (IC50 = ~15 nM) (Figure 6B). Whole-exome sequencing (WES) and targeted capture sequencing (MSKCC-IMPACT) did not reveal any known genomic alterations that may cause venetoclax resistance. SM09419 as single agent led to approximately equally potent inhibitory activity against venetoclax-resistant AML cells as parental, venetoclax sensitive cells (Figure 6B). Moreover, addition of venetoclax and SM09419 led to synergistic effects in venetoclax-resistant MOLM-13 cells (Figure 4I and Figure S4D). Consistent with our previous findings in MOLM-13 parental cells, we observed downregulation of essential apoptotic proteins (XIAP, MCL-1), splicing factors (RBM5, U2AF2), and the tyrosine kinase FLT3 in venetoclax-resistant MOLM-13 and KG-1a cells (Figure 5H and Figure S6E). We further extended these findings to patient-derived xenograft (PDX) models of AML from patients with de novo resistance to venetoclax combination regimens (5-azacytidine or low-dose cytarabine) (Figure 6C–D). Following xenotransplantation from two individual venetoclax-resistant patients into NSGS mice, we detected disease engraftment with ≥10% human hCD45+ hCD34+ hCD38+ cells and exposed mice to SM09419 (25 mg/kg) or vehicle administered orally and daily for 3 weeks. SM09419 resulted in significant reduction of hCD45 AML cells in the peripheral blood and bone marrow of mice treated with SM09419 when compared to vehicle control (Figure 6D–F). Moreover, ex vivo culturing of these AML patient samples demonstrated single-agent potency of SM09419 as well as synergistic effects when combined with venetoclax (Figure 6G–H). Collectively, these findings demonstrate the in vivo efficacy of SM09419 to overcome resistance to venetoclax-based therapies.
Figure 6. SM09419 circumvents therapeutic resistance to venetoclax.
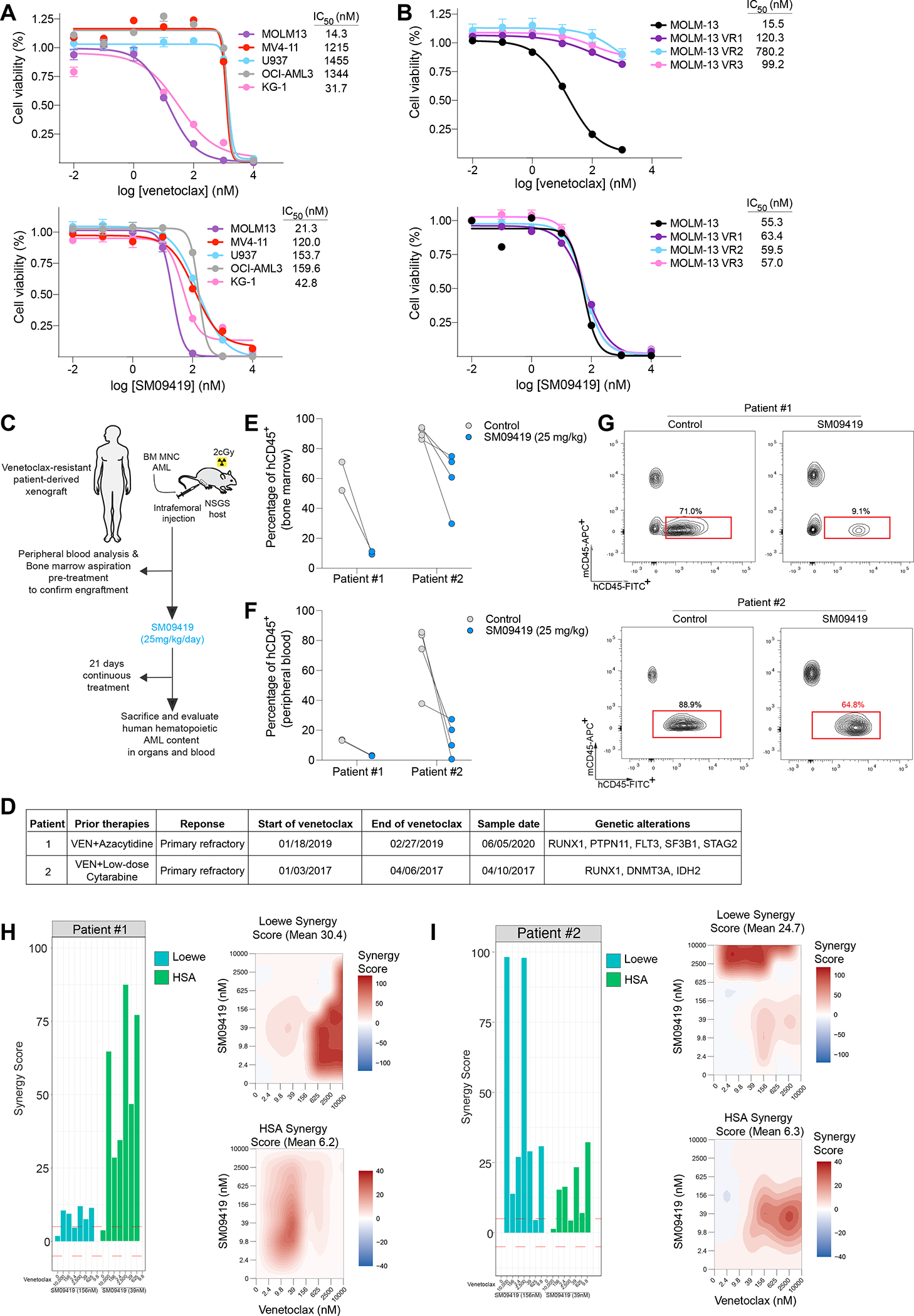
(A) Dose-response curves of human AML cell lines treated with various concentrations of venetoclax (top) or SM09419 (bottom). IC50 values were calculated from technical triplicates per experiment, error bars represent SEM. (B) Dose-response curves of venetoclax-resistant MOLM-13 cells treated with different concentrations of venetoclax (top) and SM09419 (bottom) as indicated by x-axis (n=3, mean+SEM). Cell viability is denoted on the y-axis. (C) Schematic of patient-derived xenograft (PDXs) generation and treated daily with SM09419 (25 mg/kg, QD, PO) or vehicle. (D) Diagnosis, treatment regimen and genetic characteristics of AML patient-derived xenograft samples. (E) Percentage of human CD45+ (hCD45+) cells in bone marrow and (F) peripheral blood of PDXs following 3-weeks of SM09419 treatment. (G) Representative flow-cytometry plots of hCD45+ and mouse CD45+ (mCD45+) in bone marrow from PDXs treated daily with 25 mg/kg SM09419 after 3-weeks. (H) Synergy scores (Loewe and HSA) (left) and 2D synergy plots (right) from ex vivo cultured patient #1 and (I) patient #2 samples treated with venetoclax, SM09419 or the combination after 48 hours.
DISCUSSION
Here, we performed comprehensive mapping of drug-gene interactions that dictate response to a broad range of AML therapies. This effort uncovered genetic strategies which enhance the effects of these existing AML drugs which may ultimately lead to combinatorial strategies to improve patient outcomes. We focused on those genetic events which augment response to venetoclax given the clinical need to develop venetoclax-based combinatorial treatment regimens for AML. Overall, our findings establish a functional link between splicing modulation and therapeutic efficacy of BCL2 inhibition in AML.
Given the established role of RNA splicing in regulating the expression and function of key proteins involved in cell death signaling and apoptosis, a number of prior studies have attempted to pharmacologically perturb splicing to promote response to BCL2 inhibition. For example, one prior study demonstrated that E7107, a potent SF3b inhibitor, can synergize with venetoclax in B-cell malignancies and solid tumors 58,59. However, toxicities associated with E7107 have led to its suspension from clinical use 60,61, and the clinical efficacy of more recent SF3b inhibitors (such as the drug H3B-8800) 62 for high-risk myeloid neoplasms remain unclear. Here, we provide rationale for a clinical modality to modulate RNA splicing through pharmacological inhibition of splicing-dependent kinases. Specifically, our data suggest that inhibition of CLKs and DYRKs in combination with venetoclax or as a single agent represent a therapeutic strategy to circumvent resistance to venetoclax.
CLK and DYRK kinases are two highly related families of kinases within the CMGC (cyclin-dependent kinase [CDK], mitogen-activated protein kinase [MAPK], glycogen synthase kinase [GSK3], CLK) group of the eukaryotic kinome. Each CLK and DYRK are dual specificity kinases which phosphorylate serine/threonine and tyrosine residues63,64. A variety of structurally diverse CLK inhibitors have been developed and most of these also inhibit DYRK kinases to varying degrees 64. Despite the fact that CLKs and DYRKs perform multi-site phosphorylation of a number of substrates, it is clear that perturbing the activity of CLK1-4 or DYRK1A/B globally impacts RNA splicing via altering splicing factor protein phosphorylation. DYRK1A resides in nuclear speckles and overexpression of DYRK1A induces redistribution of SR proteins from nuclear speckles to active sites of transcription/splicing in a manner that depends on its kinase activity 65. Similarly, CLK1 has been shown to regulate the cellular localization and splicing impact of SRSF1 via phosphorylation of Serine-Proline dipeptides at multiple sites on SRSF1 66.
Interestingly, prior data have identified that dephosphorylation of RNA splicing factors occurs during apoptosis and that the ensuing change in splicing may be necessary for cells to execute apoptosis 67. For example, SR proteins are targets for a number of apoptosis agonists and splicing factor kinases are inactivated during cell death by caspase-mediated proteolysis 67. This includes caspases 8, 9, and 3/6 cleaving SRPK1, SPRK2, and topoisomerase respectively, thereby altering splicing during apoptosis. Moreover, FAS activation results in dephosphorylation of SR proteins via induction of PP1 phosphatase 68. Thus, pharmacologic inhibition of phosphorylation of splicing factors may enhance response to venetoclax by mimicking the impact of apoptosis signaling cascade on RNA splicing.
SM09419 has pharmacologic properties that are very similar to Cirtuvivint, one of the first CLK/DYRK ATP-competitive inhibitors that has entered first-in-human and phase 1b clinical trials in patients with advanced solid tumors (NCT03355066 and NCT05084859)69,70. In the Cirtuvivint first-in-human study, pharmacodynamic evidence for proof of mechanism in human whole blood was reported at well tolerated doses. Importantly, infrequent grade 3 adverse hematologic events have been observed with a grade 3 anemia rate of <15% and even lower frequency for neutropenia or thrombocytopenia. In the dose escalation portion of this trial and in the combination study, Cirtuvivint has shown early evidence of anti-tumor activity with declines in PSA in prostate cancer subjects, tumor shrinkage in several tumor types, and prolonged stable disease (treatment reaching cycle 6 and beyond). Similarly, in the phase I trial of the pan-CLK inhibitor CTX-712 that evaluated subjects with solid tumors and hematologic malignancies, two complete remissions in refractory AML patients along with two partial responses in ovarian cancers subjects were reported with single agent therapy 71. While it is too early to make conclusions about efficacy from these early data, hematologic recovery is required for CR in AML which indicates that CLK/DYRK inhibition is feasible with manageable hematologic toxicity. Using the SM09419 compound herein, we provide preclinical data demonstrating the utility of CLK/DYRK inhibition as a single agent to overcome resistance to venetoclax-based therapies.
Beyond chemical modulation of RNA splicing, our genetic studies also highlighted a number of additional therapeutic targets to rationally enhance response to venetoclax and/or overcome venetoclax resistance. For instance, we demonstrated that loss of RBM10 enhances efficacy of venetoclax. RBM10 is a known splicing factor that promotes exon inclusion 72. While loss-of-function RBM10 mutations have been described in certain solid tumors 73–75 and in the genetic disease TARP syndrome 76, we found that loss of Rbm10 does not alter hematopoiesis. These data suggest context-specific roles for RBM10 and nominate RBM10 as a therapeutic vulnerability in combination with BCL2 inhibitors.
Importantly, loss of RBM10 was associated with reduced expression of the anti-apoptotic BCL2 homologue BCL2A1 as well as alternative splicing of XIAP, the most well-characterized IAP protein. Amongst BCL2 family members, expression of BCL2A1 has been most consistently associated with venetoclax resistance in a variety of leukemias. Upregulation of BCL2A1 has been reported to be associated with resistance to venetoclax in both the BeatAML as well as Leucegene cohorts of AML patients as well as AML preclinical models 6,77. In addition, BCL2A1 mRNA is heavily expressed (>10-fold more than BCL2) in monocytes, which is thought to at least partially explain the relationship between monocytic leukemia differentiation and impaired response to venetoclax 6. Consistent with these findings, genetic downregulation of BCL2A1 restores venetoclax sensitivity in AML models with venetoclax resistance 6. While there is no clinical means to chemically inhibit RBM10, our data underscores the need to develop potent small molecule inhibitors or peptide aptamers of BCL2A1 in the treatment of AML.
Lastly, our data demonstrates that co-targeting of RBM10 and BCL2 or pharmacologic inhibition of CLK/DYRK converge on the mis-splicing of the IAP-antagonist protein, XIAP as a mechanism to enhance venetoclax efficacy. These findings suggest the potential benefit of chemical campaigns to develop mimetics of IAP-antagonist proteins to negatively regulate XIAP as seen with RBM10 loss.
LIMITATIONS OF THE STUDY
Here we describe the therapeutic rationale for combination of venetoclax and SM09419, a multi-kinase inhibitor targeting CLK1-4 as well as DYRK1A-B and DYRK2 kinases. A limitation of this study its currently unknown which of these specific kinase targets inhibited by SM09419 is responsible for exerting its function to sensitize AML cells to venetoclax therapy. This is a very important point given the development of other CLK and DYRK inhibitor agents which may be more selective for individual CLK or DYRK kinases. Another limitation of this study is the evaluation of SM09419 across diverse genetic subtypes of AML. Given prior literature on the preferential sensitivity of splicing factor mutant myeloid leukemias via targeting of the spliceosome27,78,79, it will be important to test if certain genetic alterations are more susceptible to SM09419.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Omar Abdel-Wahab (abdelwao@mskcc.org).
Material availability
All unique reagents generated in this study are available from the lead contact without restriction.
Data and code availability
All bulk RNA-seq and eCLIP as well as whole-exome sequencing of venetoclax resistant MOLM-13 cells have been deposited at GEO (GSE199161) and are publicly available as of the date of publication which is listed in the key resources table. The Tyner et al., 2018 data used for this study can be found with the Accession number (dbGaP:phs001657).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TruStain Fcblock | BioLegends | 422302 |
| Anti-RBM10 rabbit polyclonal | Bethyl Laboratories | A301-006A |
| B-actin HRP | Sigma Aldrich | A3854 |
| Anti-Phosphoepitope SR proteins Antibody, clone 1H4 | Millipore Sigma | MABE50 |
| Anti-total SR Antibody (1H4) | Santa Cruz | sc-13509 |
| FITC anti-human CD45 | BioLegend | 368507 |
| APC anti-mouse CD45 | BioLegend | 103111 |
| APC anti-human CD19 | BioLegend | 302211 |
| APC anti-human CD3 | BioLegend | 300411 |
| PE anti-human CD34 | BioLegend | 343605 |
| APC/Cyanine7 anti-human CD38 | BioLegend | 303533 |
| Anti-XIAP | Cell Signaling | 2042S |
| Anti-U2AF2 | Abcam | ab37530 |
| Anti-RBM5 | Abcam | ab245646 |
| Anti-MCL-1 | SantaCruz | sc-12756 |
| Anti-FLT3 | Cell Signaling | 3462S |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Fisher | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-Luciferin, Potassium Salt | Gold-Bio Technology | Cat#LUCK-500 |
| G418 | ThermoFisher Scientific | 10131027 |
| Blasticidin | Fisher | Cat#A1113903 |
| Polybrene | Sigma Aldrich | TR-1003-G |
| Polyethylenimine (PEI) | Polysciences, Inc | Cat#23966 |
| Venetoclax | Fisher | NC1235948 |
| SM09419 | BioSplice | N/A |
| Cytarabine | Sigma | BP383 |
| Etoposide | Sigma | E1383-25MG |
| Idarubicin hydrocholoride | Sigma | I1656-10MG |
| Midostaurin | SelleckChem | S8064 |
| 5-Azacytidine | SelleckChem | S1782 |
| Critical Commercial Assays | ||
| RNeasy Plus Mini Kit | Qiagen | Cat#74136 |
| MethoCult GF M3434 | StemCell Technologies | Cat#03434 |
| CellTiter-Glo Luminescent Cell Viability | Promega | G7572 |
| NucleoSpin Blood XL | Takara | 740950.1 |
| Verso cDNA synthesis kit | ThermoFisher Scientific | AB1453B |
| Nano-Glo Dual Luciferase Reporter Assay | Promega | N1610 |
| Z’-LYTE Kinase assay | ThermoFisher Scientific | PV3190 |
| Phusion Flash High Fidelity PCR Master Mix | Fisher | F548L |
| KAPA Hyper Library Preparation Kit | Roche | KK8500 |
| Deposited Data | ||
| RNA-seq Raw data | This paper | GSE199161 |
| RBM10 eCLIP data | This paper | GSE199161 |
| Whole exome sequencing data on venetoclax sensitive and resistant MOLM-13 cells | This paper | GSE199161 |
| CRISPR screen data | This paper | Table S1 |
| List of differentially expressed genes in RBM10-deleted AML cells treated with venetoclax or DMSO | This paper | Table S3 |
| List of differentially spliced events in RBM10-deleted AML cells treated with venetoclax or DMSO | This paper | Table S4 |
| List of differentially expressed genes in SM09419-treated AML cells treated with venetoclax or DMSO | This paper | Table S5 |
| List of differentially spliced events in SM09419-treated AML cells treated with venetoclax or DMSO | This paper | Table S6 |
| BeatAML database | Tyner et al. 43 | https://www.nature.com/articles/s41586-018-0623-z |
| Experimental Models: Cell Lines | ||
| Human: MOLM-13 | DSMZ | ACC 554 |
| Human: NKM-1 | JCRB | IFO50476 |
| Human: MV4-11 | ATCC | CRL-9591 |
| Human: THP-1 | ATCC | TIB-202 |
| Human: U937 | ATCC | CRL-1593.2 |
| Human: TF-1 | ATCC | CRL-2003 |
| Human: HEK293T | ATCC | CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| NOD scid gamma | JAX | Cat#005557 |
| NSG-S | JAX | Cat#013062 |
| C57BL/6J | JAX | Cat#000664 |
| Mx-1 cre transgenic mice | JAX | Cat#005673 |
| Rbm10 conditional knockout | This paper | N/A |
| Biological Samples | ||
| Patient 1 PDX (bone marrow) | This paper | N/A |
| Patient 2 PDX (PBMCs) | This paper | N/A |
| Oligonucleotides | ||
| sgRNAs and primers, see Table S1 | This paper | N/A |
| Human pooled genome-wide sgRNA library (Brunello) sequences | Doench et al. 83 | Addgene: 73179 |
| Recombinant DNA | ||
| Lenti-Cas9 Blasticidin | Sanjana et al. 80 | Addgene: 52962 |
| psPAX2 | Gift from Didier Trono | Addgene: 12260 |
| pVSVG | Gift from Didier Trono | Addgene: 12259 |
| LRG2.1-GFP sgRNA vector | Tarumoto et al. 84 | Addgene: 108098 |
| Software and Algorithms | ||
| FlowJo V8.7 | TreeStar (BD Biosciences) | https://www.flowjo.com/ |
| Prism 9.0 | GraphPad | https://www.graphpad.com |
| Living Image Software | Perkin Elmer | http://www.perkinelmer.com/product/li-software-for-spectrum-1-seat-add-on-128113 |
| GSEA | Broad Institute | http://software.broadinstitute.org/gsea/ |
| IGV | Broad Institute | http://software.broadinstitute.org/software/igv/ |
| CRISPResso | Pinello et al. 85 | http://crispresso.pinellolab.partners.org/) |
| Trim_galore v0.6.4 | Martin et al.88 | https://github.com/FelixKrueger/TrimGalore |
| STAR v2.7.5 | Dobin et al.89 | https://github.com/alexdobin/STAR |
| samtools v1.9 | Danecek et al.90 | http://www.htslib.org/ |
| deeptools v3.3.1 | Ramírez, et al.91 | https://github.com/deeptools/deepTools |
| subread v1.5.0 | Liao et al.92 | https://subread.sourceforge.net/ |
| RSEM v1.2.4 | Li and Dewey.96 | https://github.com/deweylab/RSEM |
| Bowtie | Langmead et al.97 | https://github.com/BenLangmead/bowtie |
| edgeR | Robinson et al.98 | Bioconductor |
| TopHat v2.0.8b | Trapnell et al.99 | https://ccb.jhu.edu/software/tophat/index.shtml |
| MISO v2.0 | Katz et al.95 | http://hollywood.mit.edu/burgelab/miso/ |
| tidyverse | Wickham et al.101 | Bioconductor |
| UpSetR | Conway et al.102 | Bioconductor |
| GenomicRanges | Lawrence et al.106 | Bioconductor |
| picard v2.6.0 | Broad Institute | https://github.com/broadinstitute/picard |
| bamutils v0.5.7 | Breese et al.103 | https://ngsutils.org/modules/bamutils/ |
| DEWSeq | Huppertz et al.105 | Bioconductor |
| htseq-clip | Anders et al.104 | https://github.com/EMBL-Hentze-group/htseq-clip/ |
| IHW | Ignatiadis et al.107 | Bioconductor |
| fgsea | Korotkevich, et al.108 | Bioconductor |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cell Culture
All human leukemia cell lines were cultured in recommended media, typically RPMI medium with 20% FBS and 1% penicillin/streptomycin. TF-1 human AML cell line was cultured in RPMI 20% FBS, 1% penicillin/streptomycin and 2 ng/ml GM-CSF. HEK293T cells were grown in DMEM medium with 10% FBS and 1% penicillin streptomycin. Cell lines transduced with lentiviral Cas9 blasticidin (Addgene plasmid no. 52962)80 were selected with blasticidin (Fisher) 48 hours after transduction. All transfections were performed in HEK293T cells using Polyethylenimine (PEI) reagent at 4:2:3 ratios of plasmid: pVSVG: pPax2 in OPTI-MEM solution. Viral supernatant was collected 48 hrs and 72 hrs post-transfection. Spin infections were performed at room temperature at 1,800 RPM for 30 mins with polybrene reagent (1:2000 dilution) (Fisher Scientific). All cell lines were authenticated in-house by our Integrated Genomics Operation (IGO) core based on fragment and STR analysis.
Animals
8–10 weeks-old female and male C57BL/6 and Mx1-Cre mice were purchased from Jackson Laboratory. 8 weeks-old NOD scid gamma and NSG-S female mice were obtained from Jackson Laboratory. Mice were bred and maintained in individual ventilated cages and fed with autoclaved food and water at Memorial Sloan Kettering Animal Facility. All animal procedures were completed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at MSKCC. All mouse experiments were performed in accordance with a protocol approved by the MSKCC Institutional Animal Care and Use Committees (13-04-003).
Human patient samples
Studies were approved by the Institutional Review Boards of Memorial Sloan Kettering Cancer Center and conducted in accordance with the Declaration of Helsinki protocol. Primary human de-identified AML samples derived from whole peripheral blood or BM mononuclear cells were utilized. Mutational genotyping of each sample was performed by the MSKCC IMPACT assay as described previously 81,82. Cord blood was acquired from NY Blood Bank. Informed consent was obtained from all subjects to obtain the patient specimens used in the studies described. Patient 1 is a 62 year old male and patient 2 is a 85 year old male. Specimens were obtained as part of the Memorial Sloan-Kettering Cancer Center Institutional Review Board approved clinical protocol #16-171 to which all subjects consented. O.A-W is a participating investigator on this protocol.
METHOD DETAILS
CRISPR Screen
250 million MOLM13 Cas9-expressing cells were transduced with the Brunello sgRNA library 83 at a low multiplicity of infection (~0.3) to obtain at least 500 cells per sgRNA (500X). Spin infections were performed at room temperature at 1,500 RCF for 90 mins with polybrene reagent (1:2000 dilution) (Fisher Scientific). On Day 4 post-transduction, GFP percentage was assessed to determine infection efficiency and sgRNA coverage (~300–500X). Remaining 300–500X cells were placed back into culture after each passage until 20 days post-transduction. At day 8 post-transduction, pooled sgRNA cells were treated with either DMSO (1%), cytarabine (50 nM), 5-azacytidine (3 uM), etoposide (400 nM), idarubicin (5 nM), midostaurin (25 nM) or venetoclax (25 nM). Genomic DNA (gDNA) extraction using NucleoSpin Blood XL, Maxi kit for DNA from blood (Takara) according to manufacturer’s protocol. For pooled CRISPR screen analysis, sgRNAs were normalized using the formula (sgRNA read count/total read count) × CPM+1. Subsequently, normalized reads were then used to calculate log2 fold change (normalized read count drug treatment/normalized read count DMSO). CRISPR library amplifications were performed according to published study 83. Competition assays were performed using MOLM-13 cells transduced with sgRNA or cDNA constructs and mixed with parental cells at fixed ratios followed by 4 days of treatment with either vehicle (DMSO) or venetoclax, and GFP percentages were analyzed using BD LSR Fortessa FlowCytometer. The RNA processing factor (genes in the “RNA processing” gene ontology term, GO:0006396) sgRNA log2 fold change distributions in cytarabine, 5-azacytidine, etoposide, idarubicin, and midostaurin were compared to venetoclax. Specifically, a two-sided F-test for equality of variances was used to assess if the drug:venetoclax ratios significantly deviated from 1. Variance ratios and the 95% confidence intervals were estimated using the stats R package. For individual sgRNA validations, sgRNAs were cloned into LRG2.1 vector84.
CRISPR indel analysis
To quantify the spectrum of indel mutations with RBM10 sgRNAs, we transduced MOLM-13 cells with sgRBM10 or sgRosa (non-targeting), followed by cell sorting of GFP+/sgRNA+ populations at day 4 and day 28 post-infection. Cells were then harvested for gDNA and PCR amplicon (~200 bp) was designed to flank the sgRNA recognition sequence. 200 ng of gDNA was amplified using 2x Phusion Master Mix. Sequencing libraries were prepared from amplicons with an average size of 200 bp. The reported concentration was 3–7 ng/μL, and 50 μL were used as input for the KAPA Hyper Library Preparation Kit (Kapa Biosystems KK8504) according to the manufacturer’s instructions with 8 cycles of PCR. Barcoded libraries were pooled in equal volumes and run on MiSeq in a PE150 run, using the MiSeq Reagent Micro Kit v2 (300 Cycles) (Illumina). The average number of read pairs per sample was 203,000.
Indel analysis was performed using CRISPResso (http://crispresso.pinellolab.partners.org/)85.
RNA-sequencing library preparation and sequencing
For cell line RNA sequencing (RNA-seq), RNA was extracted from MOLM13 cells using the Qiagen RNeasy extraction kit, according to the manufacturer’s instructions. A minimum of 500 ng of high-quality RNA (as determined by Agilent Bioanalyzer) per replicate was used as input for library preparation. Poly(A)-selected, strand-specific (dUTP method) Illumina libraries were prepared by the Integrated Genomics Operation (IGO) at Memorial Sloan Kettering with a modified TruSeq protocol and sequenced on the Illumina HiSeq 2000 to obtain ~50–60M 2×101 bp paired-end reads per sample.
eCLIP library preparation
eCLIP studies were performed in duplicates by Eclipse Bioinnovations Inc (San Diego, www.eclipsebio.com) according to the published single-end enhanced CLIP protocol with the following modifications. For Rbm10 immunoprecipitation, 10% of IP samples and 1% of input samples were run on NuPAGE 4–12% Bis-Tris protein gels, transferred to PVDF membrane, probed with 1:1,000 of RBM10 antibody and 1:10,000 TrueBlot Anti Rabbit IgG (HRP) and imaged with C300 Imager for 1 minute on normal settings using Azure Radiance ECL. Only the region from ~100 kDa to 180 kDa (protein size to 80 kDa above) was isolated during eCLIP. For RNA visualization, 10% of IP samples were run on NuPAGE 4–12% Bis-Tris protein gels, transferred to nitrocellulose membrane, visualized using the Chemiluminescent Nucleic Acid Detection Kit (cat. no. 89880) from Thermo Fisher Scientific and imaged with C300 Imager for 30 seconds on normal settings. For eCLIP preparation, 10 million MOLM-13 cells were UV crosslinked at 400 mJoules/cm2 with 254 nm radiation, and snap frozen. Cells were then lysed and treated with RNase I to fragment RNA as previously described. RBM10 antibody (A301-006A, Bethyl) was then pre-coupled to Protein G Dynabeads (Thermo Fisher), added to lysate, and incubated overnight at 4 deg C. Prior to immunoprecipitation, 2% of the sample was taken as the paired input sample, with the remainder magnetically separated and washed with lysis buffer only (as the standard high-salt eCLIP wash buffer gave poor immunoprecipitation yield). eCLIP was performed by excising the area from ~100 kDa to ~180 kDa. RNA adapter ligation, IP-western, reverse transcription, DNA adapter ligation, and PCR amplification were performed as previously described.
Whole-exome sequencing and targeted capture sequencing
For MSKCC-IMPACT, after PicoGreen quantification and quality control by Agilent BioAnalyzer, 100 ng of DNA were used to prepare libraries using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with 8 cycles of PCR. 80–190 ng of each barcoded library were captured by hybridization in pools of 6–14 samples using the IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets) assay 81 (Nimblegen SeqCap), designed to capture all protein-coding exons and select introns of 505 commonly implicated oncogenes, tumor suppressor genes, and members of pathways deemed actionable by targeted therapies. Captured pools were sequenced on a NovaSeq 6000 in a PE100 run using the NovaSeq 6000 S4 Reagent Kit (200 Cycles) (Illumina) producing an average of 540X coverage per sample. For exome capture and sequencing, after PicoGreen quantification and quality control by Agilent BioAnalyzer, 100 ng of DNA were used to prepare libraries using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with 8 cycles of PCR. After sample barcoding, 500 ng of library were captured by hybridization using the xGen Exome Research Panel v2.0 (IDT) according to the manufacturer’s protocol. PCR amplification of the post-capture libraries was carried out for 12 cycles. Samples were run on a NovaSeq 6000 in a PE100 run, using the NovaSeq 6000 S4 Reagent Kit (200 Cycles) (Illumina). Samples were covered to an average of 251X.
Western blotting
MOLM-13 Cas9-expressing cells were transduced with sgRNAs and harvested for protein on day 6 post-transduction. For SM09419, MOLM-13 cells were treated with varying concentrations of SM09419, and protein was harvested 48 hours post-treatment. Lysate protein concentration was measured with the BCA reagent and 10–30 mcg was loaded per lane onto 4–12% NuPAGE™ Bis-Tris protein gels. After transfer, PVDF membranes were probed with anti-RBM10 (Bethyl Laboratories), anti-Phosphoepitope SR proteins Antibody (clone 1H4, Millipore Sigma), total SR protein (Santa Cruz), anti-XIAP (Cell signaling), anti-MCL-1 (Cell signaling), anti-RBM5 (Abcam), anti-FLT3 clone 8F2 (Cell signaling), anti-U2AF2/U2AF65 (Abcam) and anti-BCL-2 (Abcam) at 1:1,000 and visualized by standard methods.
Colony-forming assays
Total bone marrow from Mx1-Cre WT and Mx1-Cre Rbm10fl/y mice were harvested and seeded at a density of 20,000 cells per replicate into cytokine-supplemented methylcellulose medium (MethoCult M3434, Stemcell Technologies). For SM09419 experiments, total bone marrow from C57BL/6 treated with 25 mg/kg SM09419 for 3 weeks were harvested and seeded as described above. Colonies propagated in culture were scored at day 7.
Annexin V assay
Apoptotic analysis was determined using APC Annexin V (BD Bioscience) and performed according to manufacturer’s specifications and co-stained with 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) for DNA content. Cells were analyzed by flow cytometry and FlowJo software.
Generation of Rbm10 conditional knockout mice
The Rbm10 allele was deleted by targeting exon 4. Two LoxP sites flanking exon 3 and a Frt flanked neomycin selection cassette were inserted in the downstream intron. Ten micrograms of the targeting vector were linearized and then transfected by electroporation of HF4 (129/SvEv × C57Bl/6) (FLP Hybrid) embryonic stem cells. After selection with G418 antibiotic, surviving clones were expanded for PCR analysis to identify recombinant ES clones. The Neo cassette in targeting vector has been removed during ES clone expansion. Screening primer A1 was designed downstream of the short homology arm (SA) outside the 3’ region used to generate the targeting construct. Clones 182, 184, 211, 212, and 284 were expanded and reconfirmed for SA integration. A PCR was performed on clones 182, 184, 211, 212, and 284 to detect presence of the distal LoxP site using the LOX1 and SDL2 primers. This reaction amplifies a wild-type product 472 bp in size. The presence of a second PCR product 48 bp greater than the wild-type product indicates a positive LoxP PCR. Confirmation of distal LoxP retention was performed by PCR using the LOX1 and FRTN2C primers. This reaction produces a product 1.05 kb in size. Sequencing was performed on purified PCR DNA to confirm presence of the distal LoxP cassette using the SDL2 primer. Secondary confirmation of positive clones identified by PCR was performed by Southern Blotting analysis. DNA was digested with Apa I, and electrophoretically separated on a 0.8% agarose gel. After transfer to a nylon membrane, the digested DNA was hybridized with a probe targeted against the 5’ external region. DNA from HF4 mouse ES cells was used as a wild-type control. Positive clones were further confirmed by Southern Blotting analysis using an internal probe. DNA was digested with BamH I, and electrophoretically separated on a 0.8% agarose gel. After transfer to a nylon membrane, the digested DNA was hybridized with a probe targeted against the 3’ internal region. DNA from HF4 mouse ES cells was used as a wild-type control. Primer set NDEL1 and NDEL2 was used to screen mice for the deletion of the Neo cassette. The PCR product for the wild-type is 322 bp. After Neo deletion, one set of LoxP-FRT sites remains (147 bp). A second band with a size of 469 bp indicates Neo deletion. A PCR was performed to detect presence of the distal LoxP site using the LOX1 and SDL2 primers. This reaction amplifies a wild-type product 473 bp in size. The presence of a second PCR product 48 bp greater than the wild-type product indicates a positive LoxP PCR. Tail DNA samples from positive mice were amplified with primers NEOGT and A1. NEOGT is located inside the Neo cassette and A1 is located downstream of the short homology arm, outside the region used to create the targeting construct. NEOGT / A1 amplifies a fragment of 2.32 kb in length.
Bone marrow (BM) transplantation
Freshly dissected femora and tibiae were isolated from Mx1-cre WT and Mx1-cre Rbm10fl/y, CD45.2+ mice. BM was flushed with a 3-cc insulin syringe into PBS supplemented with 3% fetal bovine serum. The BM was spun at 0.5 g by centrifugation and RBCs were lysed in ammonium chloride-potassium bicarbonate lysis buffer for 5 min. After centrifugation, cells were resuspended in PBS plus 3 % FBS, passed through a cell strainer, and counted. Finally, 0.5 million total BM cells of Mx1-cre WT and Mx1-cre Rbm10fl/y CD45.2+ mice were mixed with 0.5 million WT CD45.1+ support BM and transplanted via tail vein injection into lethally irradiated (two times 450 cGy) CD45.1+ recipient mice. Chimerism was measured by FACS from the peripheral blood 4 weeks after transplant. Chimerism was followed via FACS in the peripheral blood every 4 weeks (week 0, 4, 6, 8,12, and 16 after polyI:polyC injection). For noncompetitive transplantation experiments, 1 million total BM cells of Mx1-cre WT and Mx1-cre Rbm10fl/y CD45.2+ mice were injected into lethally irradiated (two times 450 cGy) CD45.1+ recipient mice.
Drug treatment IC50 measurements
Cell lines were plated in 96 well plates and exposed to the indicated compounds at various concentration ranges with a minimum of three technical replicates per concentration per cell line. Cell viability was measured with the CellTiter Glo reagent (Promega) as per manufacturer’s instructions. Absolute viability values were converted to percentage viability versus DMSO control treatment, and then non-linear fit of log(inhibitor) versus response (three parameters) was performed in GraphPad Prism v7.0 to obtain an IC50 values. Two-dimensional heatmaps of Synergy Scores from Bliss synergy models were generated based on Demidenko et al., 2019 86.
QPCR measurement of BCL2A1 gene expression
RNA was extracted from the indicated cell lines and reverse transcribed into cDNA using the Verso cDNA synthesis Kit (ThermoFisher Scientific). Measurement of BCL2A1 gene expression was performed using primers amplifying BCL2A1 CDS region and designed by primer3 (https://bioinfo.ut.ee/primer3-0.4.0/) with ACTB as the housekeeping gene. Relative expression levels across cell lines were calculated using the Delta-delta Ct method as per standard procedures.
cDNA overexpression
BCL2A1, RBM10 wild-type and RBM10 domain mutants as well as XIAP full-length and Δexon 1 were codon optimized and synthesized as gene blocks by Integrated DNA Technologies (IDT) and was sublconed cloned into lentiviral Puro-IRES-GFP construct using NEBuilder Hifi DNA assembly. MOLM-13 RBM10-KO cells were transduced with either BCL2A1, RBM10 wild-type, or RBM10 mutant constructs and treated with venetoclax.
Animal experiments
For in vivo Cas9 experiments, MOLM-13 Cas9-expressing cells were transduced with sgRosa (negative control) or sgRBM10 constructs. At day 2 post-transduction, sgRNA positive cells (GFP+) were sorted by FACS. 100,000 leukemia-sgRNA expressing cells were intravenously injected into each sub-lethal irradiated (5.5 Gy) 8 weeks-old NOD scid gamma mice mice. For venetoclax trials, a 100 mg/ml venetoclax (Sigma Aldrich) stock was diluted in a carrier containing 10% ethanol, 30% polyethyleneglycol-400 (Sigma), and 60% phosal 50 propylene glycol (Lipoid) to obtain a final concentration of 100 mg/kg. Upon disease onset as measured by bioluminescent imaging, we performed oral gavage once daily with either 100 mg/kg venetoclax or vehicle (1% DMSO). All whole-body bioluminescent imaging was performed by intraperitoneally injection of Luciferin (Goldbio) at a 50 mg/kg concentration and imaging was performed after 5 mins using an IVIS imager. Bioluminescent signals (radiance) were quantified using Living Image software with standard regions of interests (ROI) rectangles.
Kinase assays
IC50 values for CLK2, CLK3, DYRK1A and CDK1 were determined by transferring test compounds to 1536-well plates (Echo 550, LabCyte) and by optimizing and performing Z’-LYTE™ kinase assays per the manufacturer’s instructions (Thermo Fisher). In addition, a full kinome screen (464 kinases) with 1 μM SM09419 was performed by Thermo Fisher Select Screen service. The IC50 for each kinase demonstrating >80% inhibition was then determined. Kinase tree dendrogram was generated using Coral 87.
NanoBRET target engagement assay
Cellular target engagement assays were performed using NanoBRET in 293T cells expressing CLK1, CLK2, CLK3, CLK4, DYRK1A, DYRK1B, and DYRK2 in-frame with a nanoluciferase (NanoLuc) tag. A cell permeable NanoBRET fluorescent tracer was then added to the cells which reversibly binds the target-NanoLuc Fusion protein in live cells to result in a BRET signal. SM09419 or vehicle were then added to each cell over a dose range and the degree of drug-target protein binding was assessed via loss of NanoBRET signal. An IC50 value indicating SM09419-protein binding was then identified via 10-point dose response curves.
Patient-derived xenograft experiments
Frozen human peripheral blood mononuclear cells (PBMCs) from two individual PDX models were rapidly thawed and transferred into 50 ml conical tubes. 20 mL pre-warmed RPMI 1640 (Corning) was added dropwise to tubes. After centrifuging at 300 × g at 4 degrees Celsius, cell pellet was resuspended in PBS (Corning). 4 million cells were intrafemorally injected per mouse. Blood was collected by retro-orbital bleeding using heparinized microhematocrit capillary tubes (Thermo Fisher Scientific) and a flow cytometry panel consisting of mCD45/hCD45/hCD3/hCD11b/hB220 were used to discriminate human from mouse cells and human myeloid vs T-cell engraftment. Upon disease onset as measured by hCD45-positive cells by flow cytometry, we performed oral gavage once daily with either 25 mg/kg SM09419 or vehicle (5% polyvinylpyrrolidone).
QUANTIFICATION AND STATISTICAL ANALYSIS
Genome-wide differential gene expression analysis
FASTQ files were first trimmed using Trim_galore (v0.6.4)88 to remove sequencing adapters and low quality (Q<15) reads. Trimmed sequencing reads were aligned to the human Hg19 reference genome (GENCODE, GRCh37.p13) using STAR (v2.7.5) 89. SAM files were subsequently converted to BAM files, sorted, and indexed using samtools (v1.9)90. BAM files were used to generate bigwig files using bamCoverage (part of the Deeptools package; v3.3.1)91. Read counting across genomic features was performed using featureCounts (part of the subread package; v1.5.0) 92.
Gene expression estimation and alternative splicing analysis
Annotations from UCSC knownGene 93, Ensembl 71 94, and MISO v2.095 were combined to create a genome annotation for the human UCSC hg19 (GRCh37) assembly. We mapped all reads to the transcriptome via RSEM v1.2.496, using the Bowtie alignment option “-v 2”97. RSEM produces gene-level estimates of expression in units of transcripts per million (TPM). All gene expression estimates were normalized via the trimmed mean of M values (TMM) method 98. Reads which failed to align were mapped to the genome with TopHat v2.0.8b 99, as well to an expanded annotation created by computing all possible combinations of annotated 5′ and 3′ splice sites per gene. We quantified isoform expression with MISO v2.095, using the combined RSEM and TopHat alignments as input. We used the two-sided t-test to test differential isoform expression between sample groups. Differentially spliced events were defined as those with at least 20 isoform-identifying reads in each sample, a minimum absolute difference of 10% in isoform expression, and a p-value < 0.05. All analyses were conducted within the R Programming environment with tools from Bioconductor100. The visualizations were created using the dplyr, ggplot2, tidyverse101, and UpSetR 102 packages.
Purine/Pyrimidine Motif Enrichment Analysis
Differentially spliced cassette exon events following SM09419 treatment were identified. The enrichment of purines/pyrimidines in excluded, relative to included, cassette exons was measured within exonic regions and immediately adjacent intronic sequences. The 95% confidence interval was estimated with bootstrapping (1000 resampling iterations). The motif enrichment analysis was conducted within the R Programming environment with GenomicRanges from Bioconductor 100.
eCLIP data analysis
The eCLIP data was processed similarly as described previously 37 and is outlined shortly in the following. First, adapter sequences were trimmed from both reads of all read-pairs using cutadapt version 1.14. Then, all remaining reads longer than 16 bases were aligned against the human reference genome sequence hg19/GRCh37 using STAR version 2.5.0c. Only uniquely mapped reads were kept. Read-pair duplicates by position were removed using picard tools version 2.6.0. To identify binding sites, we first ran a custom script to identify clusters of overlapping reads that had a read-depth of at least 10 reads. Then, we calculated significant enrichments for all such identified clusters by comparing IP-samples versus input-samples using edgeR. More specifically, we ran bamutils count version 0.5.7103 to counted stranded reads within all identified clusters for all samples. Using this output, we calculated differential coverage between IP-vs-input for each cluster with edgeR after normalizing for total sequencing depth per replicate (resulting in counts per million/CPM per cluster). Final binding sites were called by applying logFC > 2 and FDR < 0.05 thresholds between IP-vs-input. Identification of RBM10 binding positions in events alternatively spliced following RBM10 KO relied on the htseq-clip suite (https://htseq-clip.readthedocs.io)104, and the DEWSeq 105 and GenomicRanges Bioconductor packages 106. In brief, the GRCh38.v40 GENCODE annotation was processed into 50 nucleotide (nt) genomic sliding windows, with step size of 20 nt, using htseq-clip. From the STAR-aligned eCLIP BAM files, htseq-clip was used to identify crosslink positions and count their abundance in each window. The htseq-clip counts matrix was used as input to DEWSeq for normalization and identification of IP-vs-input significantly enriched windows (adjusted p-value < 0.05, logFC > 2) in protein-coding regions. The p-values were FDR-adjusted via Independent Hypothesis Weighting 107 and overlapping significantly enriched windows were combined. The positions of enriched windows in alternatively spliced regions identified from our RNA-seq analyses were determined using the GenomicRanges package.
Gene Ontology analysis
Gene set enrichment was performed using the fgsea R package (1.4.0)108 using the KEGG, GO and MsigDB specific signatures according to the manual.
Statistical analysis
Kaplan-Meier survival curve p-values were performed using Log rank Mantel-COX test. For statistical comparison, we performed unpaired Student’s t test. Statistical analyses were performed using Prism 7 software (GraphPad). Data with statistical significance are as indicated, *p< 0.05, **p< 0.01, ***p< 0.001. Kaplan-Meier survival curves were compared using the Wilcoxon Rank-Sum test via GraphPad Prism. Information on replicates, independent experiments and statistical test can be found in the Figure Legends.
Supplementary Material
Table S1. CRISPR-Cas9 drug screens in AML cells, Related to Figure 1.
Table S2. eCLIP identification of RBM10 interacting RNAs, Related to Figure 3.
Table S3. Differential gene expression in dual inhibition of RBM10 and BCL2, Related to Figure 3.
Table S4. Differential splicing changes in dual inhibition of RBM10 and BCL2, Related to Figure 3.
Table S5. Differential gene expression upon SM09419 treatment in AML cells, Related to Figure 5.
Table S6. Differential splicing changes upon SM09419 treatment in AML cells, Related to Figure 5.
Highlights.
Genome-wide screens identify genomic determinants of drug response in AML.
RBM10 loss enhances venetoclax efficacy and is dispensable for hematopoiesis.
Inhibition of splicing-dependent kinases overcomes venetoclax resistance.
SM09419 synergizes with venetoclax by impairing XIAP and splicing factors.
ACKNOWLEDGMENTS
O.A.-W. and R.K.B. were supported in part by the Edward P. Evans Foundation, NIH/NCI (R01 CA251138), and NIH/NHLBI (R01 HL128239). O.A.-W. was supported in part by the NIH/NCI (R01 CA242020; P50 CA254838-01), and The Leukemia & Lymphoma Society. R.K.B. was supported in part by NIH/NHLBI (R01 HL151651) and the Blood Cancer Discoveries Grant program through the Leukemia & Lymphoma Society, Mark Foundation for Cancer Research, and Paul G. Allen Frontiers Group (8023-20). R.K.B is a Scholar of The Leukemia & Lymphoma Society (1344-18) and holds the McIlwain Family Endowed Chair in Data Science. W.J.K. was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number: T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. Computational studies were supported in part by FHCRC’s Scientific Computing Infrastructure (ORIP S10 OD028685). We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Footnotes
DECLARATION OF INTERESTS
E.M., E.C., M.J., C.B., C.-C.M., and D.M.B. are employees of Biosplice Therapeutics. O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc., Merck, Prelude Therapeutics, and Janssen, and is on the Scientific Advisory Board of Envisagenics Inc., AIChemy, Harmonic Discovery Inc., and Pfizer Boulder; O.A.-W. has received prior research funding from H3B Biomedicine, Nurix Therapeutics, and LOXO Oncology unrelated to the current manuscript. The remaining authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ferrara F, and Schiffer CA (2013). Acute myeloid leukaemia in adults. Lancet 381, 484–495. 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- 2.Short NJ, Konopleva M, Kadia TM, Borthakur G, Ravandi F, DiNardo CD, and Daver N (2020). Advances in the Treatment of Acute Myeloid Leukemia: New Drugs and New Challenges. Cancer Discov 10, 506–525. 10.1158/2159-8290.CD-19-1011. [DOI] [PubMed] [Google Scholar]
- 3.Ganzel C, Sun Z, Cripe LD, Fernandez HF, Douer D, Rowe JM, Paietta EM, Ketterling R, O’Connell MJ, Wiernik PH, et al. (2018). Very poor long-term survival in past and more recent studies for relapsed AML patients: The ECOG-ACRIN experience. Am J Hematol 93, 1074–1081. 10.1002/ajh.25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breems DA, Van Putten WL, Huijgens PC, Ossenkoppele GJ, Verhoef GE, Verdonck LF, Vellenga E, De Greef GE, Jacky E, Van der Lelie J, et al. (2005). Prognostic index for adult patients with acute myeloid leukemia in first relapse. J Clin Oncol 23, 1969–1978. 10.1200/JCO.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 5.Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, Culp-Hill R, d’Almeida A, Joshi SK, Rosenberg M, et al. (2019). The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov 9, 910–925. 10.1158/2159-8290.CD-19-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Nakauchi Y, Kohnke T, Stafford M, Bottomly D, Thomas R, Wilmot B, McWeeney SK, Majeti R, and Tyner JW (2020). Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer 1, 826–839. 10.1038/s43018-020-0103-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuber J, Radtke I, Pardee TS, Zhao Z, Rappaport AR, Luo W, McCurrach ME, Yang MM, Dolan ME, Kogan SC, et al. (2009). Mouse models of human AML accurately predict chemotherapy response. Genes Dev 23, 877–889. 10.1101/gad.1771409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blombery P, Lew TE, Dengler MA, Thompson ER, Lin VS, Chen X, Nguyen T, Panigrahi A, Handunnetti SM, Carney DA, et al. (2022). Clonal hematopoiesis, myeloid disorders and BAX-mutated myelopoiesis in patients receiving venetoclax for CLL. Blood 139, 1198–1207. 10.1182/blood.2021012775. [DOI] [PubMed] [Google Scholar]
- 9.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown AL, et al. (2016). Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 22, 792–799. 10.1038/nm.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fennell KA, Vassiliadis D, Lam EYN, Martelotto LG, Balic JJ, Hollizeck S, Weber TS, Semple T, Wang Q, Miles DC, et al. (2021). Non-genetic determinants of malignant clonal fitness at single-cell resolution. Nature. 10.1038/s41586-021-04206-7. [DOI] [PubMed] [Google Scholar]
- 12.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, et al. (2016). Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov 6, 1106–1117. 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minn AJ, Rudin CM, Boise LH, and Thompson CB (1995). Expression of bcl-xL can confer a multidrug resistance phenotype. Blood 86, 1903–1910. [PubMed] [Google Scholar]
- 14.Jones CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, Nemkov T, Gehrke S, Gamboni F, Krug A, Winters A, et al. (2020). Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 27, 748–764 e744. 10.1016/j.stem.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, et al. (2018). Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 34, 724–740 e724. 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, Sakellaropoulos T, Gong Y, Kloetgen A, Yap YS, et al. (2019). Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov 9, 890–909. 10.1158/2159-8290.CD-19-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, et al. (2015). BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542. 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. (2015). Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, Pouliot F, Alekseev B, Soulieres D, Melichar B, et al. (2019). Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 380, 1116–1127. 10.1056/NEJMoa1816714. [DOI] [PubMed] [Google Scholar]
- 20.Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, et al. (2012). Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 366, 109–119. 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, Arellano M, Frattini MG, Kantarjian H, Popovic R, et al. (2018). Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol 19, 216–228. 10.1016/S1470-2045(18)30010-X. [DOI] [PubMed] [Google Scholar]
- 22.DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, Konopleva M, Dohner H, Letai A, Fenaux P, et al. (2020). Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med 383, 617–629. 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 23.Schwerk C, and Schulze-Osthoff K (2005). Regulation of apoptosis by alternative pre-mRNA splicing. Mol Cell 19, 1–13. 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, and Doench JG (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9, 5416. 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu X, Tohme R, Tomlinson B, Sakre N, Hasipek M, Durkin L, Schuerger C, Grabowski D, Zidan AM, Radivoyevitch T, et al. (2021). Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia 35, 1023–1036. 10.1038/s41375-020-1003-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sripayap P, Nagai T, Uesawa M, Kobayashi H, Tsukahara T, Ohmine K, Muroi K, and Ozawa K (2014). Mechanisms of resistance to azacitidine in human leukemia cell lines. Exp Hematol 42, 294–306 e292. 10.1016/j.exphem.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, Hockemeyer K, Ghebrechristos YE, Yoshimi A, Inoue D, et al. (2019). Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 35, 369–384 e367. 10.1016/j.ccell.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Han C, Wang E, Lorch AH, Serafin V, Cho BK, Guttierrez Diaz BT, Calvo J, Fang C, Khodadadi-Jamayran A, et al. (2020). Posttranslational regulation of the exon skipping machinery controls aberrant splicing in leukemia. Cancer Discov. 10.1158/2159-8290.CD-19-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang E, Zhou H, Nadorp B, Cayanan G, Chen X, Yeaton AH, Nomikou S, Witkowski MT, Narang S, Kloetgen A, et al. (2021). Surface antigen-guided CRISPR screens identify regulators of myeloid leukemia differentiation. Cell Stem Cell 28, 718–731 e716. 10.1016/j.stem.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witkowski MT, Lee S, Wang E, Lee AK, Talbot A, Ma C, Tsopoulidis N, Brumbaugh J, Zhao Y, Roberts KG, et al. (2022). NUDT21 limits CD19 levels through alternative mRNA polyadenylation in B cell acute lymphoblastic leukemia. Nat Immunol. 10.1038/s41590-022-01314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han C, Khodadadi-Jamayran A, Lorch AH, Jin Q, Serafin V, Zhu P, Politanska Y, Sun L, Gutierrez-Diaz BT, Pryzhkova MV, et al. (2022). SF3B1 homeostasis is critical for survival and therapeutic response in T cell leukemia. Sci Adv 8, eabj8357. 10.1126/sciadv.abj8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lachowiez CA, Loghavi S, Furudate K, Montalban-Bravo G, Maiti A, Kadia T, Daver N, Borthakur G, Pemmaraju N, Sasaki K, et al. (2021). Impact of splicing mutations in acute myeloid leukemia treated with hypomethylating agents combined with venetoclax. Blood Adv 5, 2173–2183. 10.1182/bloodadvances.2020004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, and Vakoc CR (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol 33, 661–667. 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto K, Toyoshima H, Sakai R, Miyagawa K, Hagiwara K, Ishikawa F, Takaku F, Yazaki Y, and Hirai H (1992). Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood 79, 2378–2383. [PubMed] [Google Scholar]
- 35.Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Collins KM, Kainov YA, Christodolou E, Ray D, Morris Q, Hughes T, Taylor IA, Makeyev EV, and Ramos A (2017). An RRM-ZnF RNA recognition module targets RBM10 to exonic sequences to promote exon exclusion. Nucleic Acids Res 45, 6761–6774. 10.1093/nar/gkx225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514. 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, and Shi Y (2003). Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 11, 519–527. 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 39.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, and Alnemri ES (2001). A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410, 112–116. 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, and Wu H (2001). Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104, 781–790. [PubMed] [Google Scholar]
- 41.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, and Salvesen GS (2001). Structural basis for the inhibition of caspase-3 by XIAP. Cell 104, 791–800. 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 42.Hashimoto M, Saito Y, Nakagawa R, Ogahara I, Takagi S, Takata S, Amitani H, Endo M, Yuki H, Ramilowski JA, et al. (2021). Combined inhibition of XIAP and BCL2 drives maximal therapeutic efficacy in genetically diverse aggressive acute myeloid leukemia. Nature Cancer 2, 340–356. 10.1038/s43018-021-00177-w. [DOI] [PubMed] [Google Scholar]
- 43.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. (2018). Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531. 10.1038/s41586-018-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gui JF, Tronchere H, Chandler SD, and Fu XD (1994). Purification and characterization of a kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc Natl Acad Sci U S A 91, 10824–10828. 10.1073/pnas.91.23.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ, Fu XD, and Adams JA (2016). Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-mRNA Splicing. Mol Cell 63, 218–228. 10.1016/j.molcel.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, and Fu XD (1996). SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J Biol Chem 271, 24569–24575. 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- 47.Prasad J, Colwill K, Pawson T, and Manley JL (1999). The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol 19, 6991–7000. 10.1128/MCB.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian W, Liang H, Shi J, Jin N, Grundke-Iqbal I, Iqbal K, Gong CX, and Liu F (2011). Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res 39, 6161–6171. 10.1093/nar/gkr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, Zhou J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. (2008). Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem 283, 28660–28669. 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Graaf K, Czajkowska H, Rottmann S, Packman LC, Lilischkis R, Luscher B, and Becker W (2006). The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochem 7, 7. 10.1186/1471-2091-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P, and Krappmann D (2009). A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol 182, 7718–7728. 10.4049/jimmunol.0803313. [DOI] [PubMed] [Google Scholar]
- 53.Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, et al. (2011). An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev 25, 1628–1640. 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, Lewis TA, Schajnovitz A, Jain E, Lee D, et al. (2016). Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 167, 171–186 e115. 10.1016/j.cell.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, and Orkin SH (2010). A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143, 313–324. 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM, Chang HY, Shurtleff SA, Downing JR, and Cleary ML (2009). Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 4, 129–140. 10.1016/j.stem.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aird D, Teng T, Huang CL, Pazolli E, Banka D, Cheung-Ong K, Eifert C, Furman C, Wu ZJ, Seiler M, et al. (2019). Sensitivity to splicing modulation of BCL2 family genes defines cancer therapeutic strategies for splicing modulators. Nat Commun 10, 137. 10.1038/s41467-018-08150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ten Hacken E, Valentin R, Regis FFD, Sun J, Yin S, Werner L, Deng J, Gruber M, Wong J, Zheng M, et al. (2018). Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight 3. 10.1172/jci.insight.121438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O’Brien J, and LoRusso P (2014). A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs 32, 436–444. 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- 61.Eskens FA, Ramos FJ, Burger H, O’Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, and Tabernero J (2013). Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res 19, 6296–6304. 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- 62.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24, 497–504. 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindberg MF, and Meijer L (2021). Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int J Mol Sci 22. 10.3390/ijms22116047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin Moyano P, Nemec V, and Paruch K (2020). Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. Int J Mol Sci 21. 10.3390/ijms21207549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alvarez M, Estivill X, and de la Luna S (2003). DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J Cell Sci 116, 3099–3107. 10.1242/jcs.00618. [DOI] [PubMed] [Google Scholar]
- 66.Aubol BE, Plocinik RM, Hagopian JC, Ma CT, McGlone ML, Bandyopadhyay R, Fu XD, and Adams JA (2013). Partitioning RS domain phosphorylation in an SR protein through the CLK and SRPK protein kinases. J Mol Biol 425, 2894–2909. 10.1016/j.jmb.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamachi M, Le TM, Kim SJ, Geiger ME, Anderson P, and Utz PJ (2002). Human autoimmune sera as molecular probes for the identification of an autoantigen kinase signaling pathway. J Exp Med 196, 1213–1225. 10.1084/jem.20021167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chalfant CE, Ogretmen B, Galadari S, Kroesen BJ, Pettus BJ, and Hannun YA (2001). FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem 276, 44848–44855. 10.1074/jbc.M106291200. [DOI] [PubMed] [Google Scholar]
- 69.Tolcher A, Babiker HM, Chung V, Kim E, Moser J, Karim R, Vandross A, Sommerhalder D, Scott AJ, Fakih M, et al. (2021). Abstract CT112: Initial results from a Phase 1 trial of a first-in-class pan-CDC-like kinase inhibitor (SM08502) with proof of mechanism in subjects with advanced solid tumors. Cancer Research 81, CT112–CT112. 10.1158/1538-7445.Am2021-ct112. [DOI] [Google Scholar]
- 70.Scott A, Call JA, Chandana S, Borazanci E, Falchook GS, Bordoni R, Richey S, Starodub A, Chung V, Lakhani NJ, et al. (2022). 451O Preliminary evidence of clinical activity from phase I and Ib trials of the CLK/DYRK inhibitor cirtuvivint (CIRT) in subjects with advanced solid tumors. Annals of Oncology 33, S742–S743. 10.1016/j.annonc.2022.07.580. [DOI] [Google Scholar]
- 71.Shimizu T, Yonemori K, Koyama T, Katsuya Y, Sato J, Fukuhara N, Yokoyama H, Iida H, Ando K, Fukuhara S, et al. (2022). A first-in-human phase I study of CTX-712 in patients with advanced, relapsed or refractory malignant tumors. Journal of Clinical Oncology 40, 3080–3080. 10.1200/JCO.2022.40.16_suppl.3080. [DOI] [Google Scholar]
- 72.Wang Y, Gogol-Doring A, Hu H, Frohler S, Ma Y, Jens M, Maaskola J, Murakawa Y, Quedenau C, Landthaler M, et al. (2013). Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med 5, 1431–1442. 10.1002/emmm.201302663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. (2015). Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, 6744. 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cancer Genome Atlas Research, N. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. (2016). Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep 15, 857–865. 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, and Biesecker LG (2011). Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am J Med Genet A 155A, 2516–2520. 10.1002/ajmg.a.34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bisaillon R, Moison C, Thiollier C, Krosl J, Bordeleau ME, Lehnertz B, Lavallee VP, MacRae T, Mayotte N, Labelle C, et al. (2020). Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 34, 63–74. 10.1038/s41375-019-0485-x. [DOI] [PubMed] [Google Scholar]
- 78.Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. (2016). Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 30, 404–417. 10.1016/j.ccell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee SC, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nat Med 22, 976–986. 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17, 251–264. 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23, 703–713. 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tarumoto Y, Lu B, Somerville TDD, Huang YH, Milazzo JP, Wu XS, Klingbeil O, El Demerdash O, Shi J, and Vakoc CR (2018). LKB1, Salt-Inducible Kinases, and MEF2C Are Linked Dependencies in Acute Myeloid Leukemia. Mol Cell 69, 1017–1027 e1016. 10.1016/j.molcel.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan GC (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34, 695–697. 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Demidenko E, and Miller TW (2019). Statistical determination of synergy based on Bliss definition of drugs independence. PLoS One 14, e0224137. 10.1371/journal.pone.0224137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Metz KS, Deoudes EM, Berginski ME, Jimenez-Ruiz I, Aksoy BA, Hammerbacher J, Gomez SM, and Phanstiel DH (2018). Coral: Clear and Customizable Visualization of Human Kinome Data. Cell Syst 7, 347–350 e341. 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 89.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, and Li H (2021). Twelve years of SAMtools and BCFtools. Gigascience 10. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 93.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. (2013). The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41, D64–69. 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. (2013). Ensembl 2013. Nucleic Acids Res 41, D48–55. 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7, 1009–1015. 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25. 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wickham H, Averick M, Bryan J, Chang W, McGowan LDA, François R, Grolemund G, Hayes A, Henry L, and Hester J (2019). Welcome to the Tidyverse. Journal of open source software 4, 1686. [Google Scholar]
- 102.Conway JR, Lex A, and Gehlenborg N (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Breese MR, and Liu Y (2013). NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29, 494–496. 10.1093/bioinformatics/bts731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huppertz I, Perez-Perri JI, Mantas P, Sekaran T, Schwarzl T, Russo F, Ferring-Appel D, Koskova Z, Dimitrova-Paternoga L, Kafkia E, et al. (2022). Riboregulation of Enolase 1 activity controls glycolysis and embryonic stem cell differentiation. Mol Cell 82, 2666–2680 e2611. 10.1016/j.molcel.2022.05.019. [DOI] [PubMed] [Google Scholar]
- 106.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ignatiadis N, Klaus B, Zaugg JB, and Huber W (2016). Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat Methods 13, 577–580. 10.1038/nmeth.3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. bioRxiv, 060012. 10.1101/060012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. CRISPR-Cas9 drug screens in AML cells, Related to Figure 1.
Table S2. eCLIP identification of RBM10 interacting RNAs, Related to Figure 3.
Table S3. Differential gene expression in dual inhibition of RBM10 and BCL2, Related to Figure 3.
Table S4. Differential splicing changes in dual inhibition of RBM10 and BCL2, Related to Figure 3.
Table S5. Differential gene expression upon SM09419 treatment in AML cells, Related to Figure 5.
Table S6. Differential splicing changes upon SM09419 treatment in AML cells, Related to Figure 5.
Data Availability Statement
All bulk RNA-seq and eCLIP as well as whole-exome sequencing of venetoclax resistant MOLM-13 cells have been deposited at GEO (GSE199161) and are publicly available as of the date of publication which is listed in the key resources table. The Tyner et al., 2018 data used for this study can be found with the Accession number (dbGaP:phs001657).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TruStain Fcblock | BioLegends | 422302 |
| Anti-RBM10 rabbit polyclonal | Bethyl Laboratories | A301-006A |
| B-actin HRP | Sigma Aldrich | A3854 |
| Anti-Phosphoepitope SR proteins Antibody, clone 1H4 | Millipore Sigma | MABE50 |
| Anti-total SR Antibody (1H4) | Santa Cruz | sc-13509 |
| FITC anti-human CD45 | BioLegend | 368507 |
| APC anti-mouse CD45 | BioLegend | 103111 |
| APC anti-human CD19 | BioLegend | 302211 |
| APC anti-human CD3 | BioLegend | 300411 |
| PE anti-human CD34 | BioLegend | 343605 |
| APC/Cyanine7 anti-human CD38 | BioLegend | 303533 |
| Anti-XIAP | Cell Signaling | 2042S |
| Anti-U2AF2 | Abcam | ab37530 |
| Anti-RBM5 | Abcam | ab245646 |
| Anti-MCL-1 | SantaCruz | sc-12756 |
| Anti-FLT3 | Cell Signaling | 3462S |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Fisher | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-Luciferin, Potassium Salt | Gold-Bio Technology | Cat#LUCK-500 |
| G418 | ThermoFisher Scientific | 10131027 |
| Blasticidin | Fisher | Cat#A1113903 |
| Polybrene | Sigma Aldrich | TR-1003-G |
| Polyethylenimine (PEI) | Polysciences, Inc | Cat#23966 |
| Venetoclax | Fisher | NC1235948 |
| SM09419 | BioSplice | N/A |
| Cytarabine | Sigma | BP383 |
| Etoposide | Sigma | E1383-25MG |
| Idarubicin hydrocholoride | Sigma | I1656-10MG |
| Midostaurin | SelleckChem | S8064 |
| 5-Azacytidine | SelleckChem | S1782 |
| Critical Commercial Assays | ||
| RNeasy Plus Mini Kit | Qiagen | Cat#74136 |
| MethoCult GF M3434 | StemCell Technologies | Cat#03434 |
| CellTiter-Glo Luminescent Cell Viability | Promega | G7572 |
| NucleoSpin Blood XL | Takara | 740950.1 |
| Verso cDNA synthesis kit | ThermoFisher Scientific | AB1453B |
| Nano-Glo Dual Luciferase Reporter Assay | Promega | N1610 |
| Z’-LYTE Kinase assay | ThermoFisher Scientific | PV3190 |
| Phusion Flash High Fidelity PCR Master Mix | Fisher | F548L |
| KAPA Hyper Library Preparation Kit | Roche | KK8500 |
| Deposited Data | ||
| RNA-seq Raw data | This paper | GSE199161 |
| RBM10 eCLIP data | This paper | GSE199161 |
| Whole exome sequencing data on venetoclax sensitive and resistant MOLM-13 cells | This paper | GSE199161 |
| CRISPR screen data | This paper | Table S1 |
| List of differentially expressed genes in RBM10-deleted AML cells treated with venetoclax or DMSO | This paper | Table S3 |
| List of differentially spliced events in RBM10-deleted AML cells treated with venetoclax or DMSO | This paper | Table S4 |
| List of differentially expressed genes in SM09419-treated AML cells treated with venetoclax or DMSO | This paper | Table S5 |
| List of differentially spliced events in SM09419-treated AML cells treated with venetoclax or DMSO | This paper | Table S6 |
| BeatAML database | Tyner et al. 43 | https://www.nature.com/articles/s41586-018-0623-z |
| Experimental Models: Cell Lines | ||
| Human: MOLM-13 | DSMZ | ACC 554 |
| Human: NKM-1 | JCRB | IFO50476 |
| Human: MV4-11 | ATCC | CRL-9591 |
| Human: THP-1 | ATCC | TIB-202 |
| Human: U937 | ATCC | CRL-1593.2 |
| Human: TF-1 | ATCC | CRL-2003 |
| Human: HEK293T | ATCC | CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| NOD scid gamma | JAX | Cat#005557 |
| NSG-S | JAX | Cat#013062 |
| C57BL/6J | JAX | Cat#000664 |
| Mx-1 cre transgenic mice | JAX | Cat#005673 |
| Rbm10 conditional knockout | This paper | N/A |
| Biological Samples | ||
| Patient 1 PDX (bone marrow) | This paper | N/A |
| Patient 2 PDX (PBMCs) | This paper | N/A |
| Oligonucleotides | ||
| sgRNAs and primers, see Table S1 | This paper | N/A |
| Human pooled genome-wide sgRNA library (Brunello) sequences | Doench et al. 83 | Addgene: 73179 |
| Recombinant DNA | ||
| Lenti-Cas9 Blasticidin | Sanjana et al. 80 | Addgene: 52962 |
| psPAX2 | Gift from Didier Trono | Addgene: 12260 |
| pVSVG | Gift from Didier Trono | Addgene: 12259 |
| LRG2.1-GFP sgRNA vector | Tarumoto et al. 84 | Addgene: 108098 |
| Software and Algorithms | ||
| FlowJo V8.7 | TreeStar (BD Biosciences) | https://www.flowjo.com/ |
| Prism 9.0 | GraphPad | https://www.graphpad.com |
| Living Image Software | Perkin Elmer | http://www.perkinelmer.com/product/li-software-for-spectrum-1-seat-add-on-128113 |
| GSEA | Broad Institute | http://software.broadinstitute.org/gsea/ |
| IGV | Broad Institute | http://software.broadinstitute.org/software/igv/ |
| CRISPResso | Pinello et al. 85 | http://crispresso.pinellolab.partners.org/) |
| Trim_galore v0.6.4 | Martin et al.88 | https://github.com/FelixKrueger/TrimGalore |
| STAR v2.7.5 | Dobin et al.89 | https://github.com/alexdobin/STAR |
| samtools v1.9 | Danecek et al.90 | http://www.htslib.org/ |
| deeptools v3.3.1 | Ramírez, et al.91 | https://github.com/deeptools/deepTools |
| subread v1.5.0 | Liao et al.92 | https://subread.sourceforge.net/ |
| RSEM v1.2.4 | Li and Dewey.96 | https://github.com/deweylab/RSEM |
| Bowtie | Langmead et al.97 | https://github.com/BenLangmead/bowtie |
| edgeR | Robinson et al.98 | Bioconductor |
| TopHat v2.0.8b | Trapnell et al.99 | https://ccb.jhu.edu/software/tophat/index.shtml |
| MISO v2.0 | Katz et al.95 | http://hollywood.mit.edu/burgelab/miso/ |
| tidyverse | Wickham et al.101 | Bioconductor |
| UpSetR | Conway et al.102 | Bioconductor |
| GenomicRanges | Lawrence et al.106 | Bioconductor |
| picard v2.6.0 | Broad Institute | https://github.com/broadinstitute/picard |
| bamutils v0.5.7 | Breese et al.103 | https://ngsutils.org/modules/bamutils/ |
| DEWSeq | Huppertz et al.105 | Bioconductor |
| htseq-clip | Anders et al.104 | https://github.com/EMBL-Hentze-group/htseq-clip/ |
| IHW | Ignatiadis et al.107 | Bioconductor |
| fgsea | Korotkevich, et al.108 | Bioconductor |