

Abstract
A third of diffuse large B-cell lymphoma (DLBCL) patients present with extranodal dissemination, associated with inferior clinical outcome. MYD88L265P is a hallmark extranodal DLBCL mutation that supports lymphoma proliferation. Yet, extranodal lymphomagenesis and the role of MYD88L265P in transformation remain mostly unknown. Here, we show that B-cells expressing Myd88L252P (MYD88L265P murine equivalent) activate, proliferate, and differentiate, with minimal T-cell co-stimulation. Additionally, Myd88L252P skewed B-cells towards the memory fate. Unexpectedly, the transcriptional and phenotypic profiles of B-cells expressing Myd88L252P, or other extranodal lymphoma founder mutations, resembled those of CD11c+T-BET+ aged/autoimmune memory B-cells (AiBC). AiBC-like cells progressively accumulated in animals prone to develop lymphomas, and ablation of T-BET, the AiBC master regulator, stripped mouse and human mutant B-cells of their competitive fitness. By identifying a phenotypically-defined prospective lymphoma precursor population and its dependencies, our findings pave the way for the early detection of premalignant states and targeted prophylactic interventions in high-risk patients.
Keywords: B-cell lymphoma, Extranodal disease, MYD88-L265P, Aged B-cells, Autoimmune B-cells
INTRODUCTION
Diffuse large B-cell lymphomas (DLBCL) are biological and clinically heterogeneous diseases that develop from mature activated B-cells (1). Under normal circumstances, B-cell activation requires stimulation of the B-cell receptor (BCR) by a cognate antigen, and co-stimulation from specialized T-cells (2). Fully activated B-cells can form transient structures called germinal centers (GC), where they undergo somatic hypermutation (SHM) and compete for T-cell help to promptly expand and improve the affinity of their BCR against foreign antigens, before differentiating into effector cells, namely plasma cells (PC, antibody secreting cells) or memory B-cells (MBC, long-lived cells that activate upon antigen re-encounter, enabling faster and enhanced responses) (3). These fine-tuned processes require B-cells to be transiently endowed with tumorigenic-like features (heightened proliferation, genomic instability, tolerance to DNA damage, dysregulated metabolism, cell death escape, etc.), which are hijacked and perpetuated by mutations in lymphomas (4). These mutations are believed to largely arise as aberrant byproducts of SHM, a process catalyzed by the enzyme activation-induced cytidine deaminase (AID/AICDA) (3).
While DLBCLs frequently initiate from, and localize to, the lymph nodes, one third of patients present with extranodal tumors in non-lymphoid organs, including immune privileged sites, with an often-fatal outcome (5–7). Extranodal DLBCLs manifest in advanced stages of the nodal disease or occur as primary events. The localization of these aggressive tumors and their elevated relapse rates (5) create significant clinical challenges. Standard DLBCL treatments (1) (chemotherapy, anti-B-cell antibodies, and radiotherapy) are insufficient to cure most patients, and carry significant toxicity. Moreover, there is no way to identify their onset at early stages, where they may be more susceptible to targeted therapies.
Recent large-scale profiling efforts have yielded a genetically-defined classification for DLBCLs, including the identification of a highly aggressive subtype called “C5”/“MCD” (hereafter MCD), with the highest frequency of extranodal dissemination (8,9). MCD and primary extranodal tumors in immune privileged sites share founder mutations targeting MYD88 [core Toll-like-receptor (TLR) signaling mediator], CD79B (BCR complex component), TBL1XR1 (transcriptional repression complexes component) and others (8,9). Approximately 70% of these carry a MYD88L265P gain-of-function missense mutation (9) that facilitates proliferation and survival through constitutive NF-κB signaling (10). This same mutation occurs in other B-cell malignancies, including Waldenstrom macroglobulinemia (WM) and chronic lymphocytic leukemia (11,12), highlighting its widespread biological relevance. Mice expressing Myd88L252P (murine equivalent to MYD88L265P) in all B-cells develop lymphadenopathy and occasional lymphomas with old age (13). Combination of Myd88L252P with BCL2 overexpression, a common feature in MCD-DLBCLs (8,9), results in higher incidence of lymphomas that closely resemble their human counterparts (14). However, the mechanisms through which these tumors arise from the immune system and progress to an overt and advanced disease remain unknown.
MCD tumors show the highest AID activity footprint, suggesting that these have transited through the GC during transformation (8). Still, AICDA is expressed in activated B-cells before entering the GC (15), and even in proliferating B-cells outside of lymphoid follicles (16), suggesting that MCD precursors could resort to alternative activated states to accumulate mutations (17). Since the immune system keeps a tight control on B-cell activation through selective co-stimulation, we hypothesized that MCD founder mutations altered these requirements, enabling the progressive transformation and expansion of lymphoma precursors. Here, we explore the evolutionary advantage conferred to mature B-cells by the hallmark mutation MYD88L265P, as well as the pathogenic trajectories delineated by this mutation, to provide critical insight into extranodal lymphoma transformation and the precursor populations involved.
RESULTS
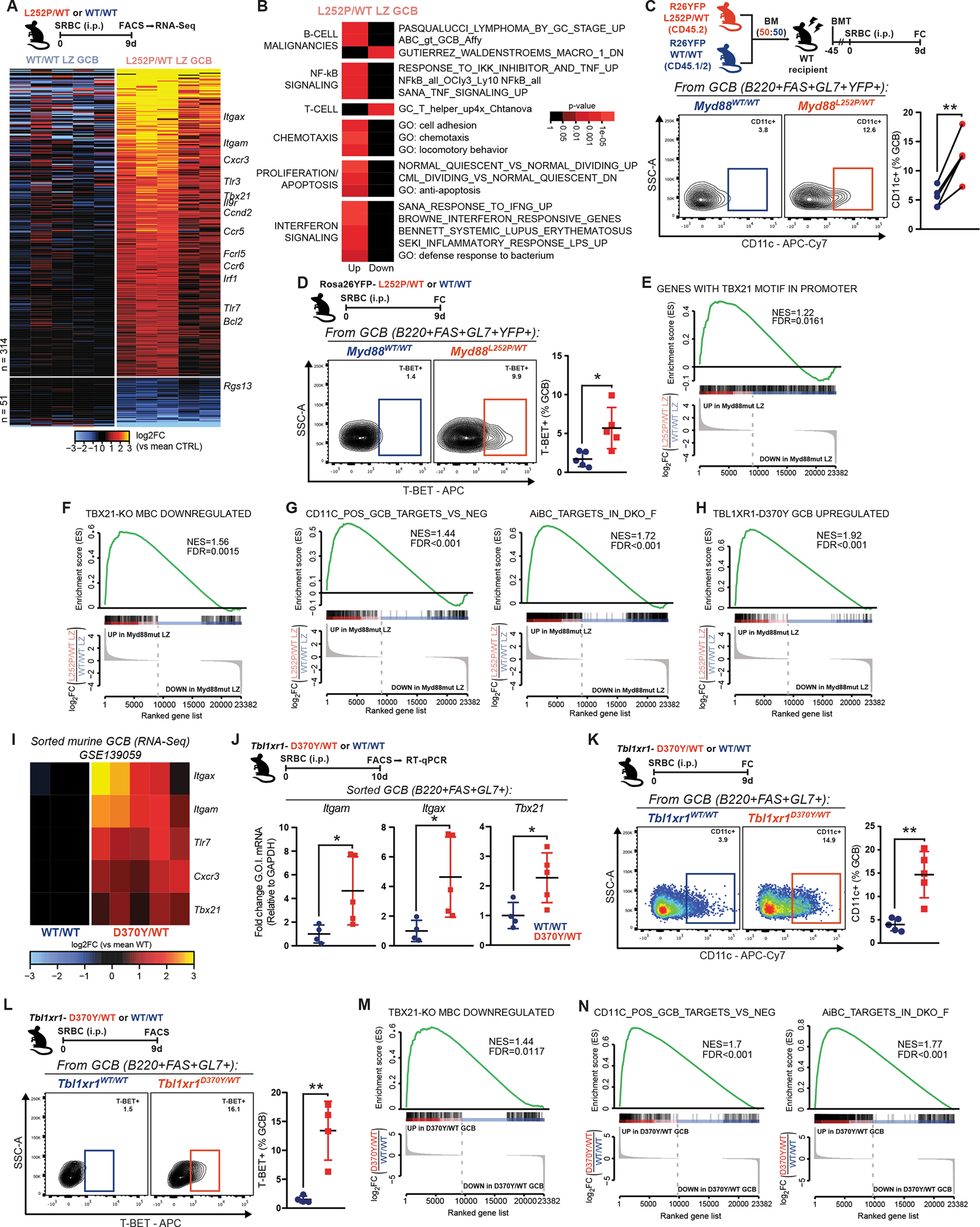
Myd88 mutations confer a competitive advantage to GCB cells
To dissect how MCD founder mutations shape pathogenic trajectories, we first explored the impact of the class-defining lesion MYD88L265P on the GC. We crossed an existing Cre-inducible Myd88L252P mouse model (13) to the Cγ1Cre strain (18), restricting MYD88-mutant expression to (pre-)germinal center B-cells (GCB) and GC-derived cells. We immunized young Cγ1Cre;Myd88L252P/WT or Cγ1Cre;Myd88WT/WT (WT) mice with the T-cell-dependent antigen sheep red blood cells (SRBC) and profiled spleens at the peak of the GC. While B-cell levels were unaltered (Fig. 1A; Supplementary Fig. S1A), Myd88L252P/WT mice showed a significant increase in the fraction (Fig. 1B) and absolute number (Supplementary Fig. S1B) of FAS+GL7+ GCB. This expansion extended to GCB with a mature phenotype (FAS+CD38−; Supplementary Fig. S1C). An alternative T-cell-dependent antigen yielded similar results (Supplementary Fig. S1D). In line with these findings, immunohistochemistry (IHC) staining for B-cells (B220) and GCB (peanut agglutinin, PNA) revealed intact follicles harboring an increase in GC numbers and size in Myd88L252P/WT mice (Fig. 1C–E).
Figure 1. Myd88 mutations increase the competitive fitness of GCB.
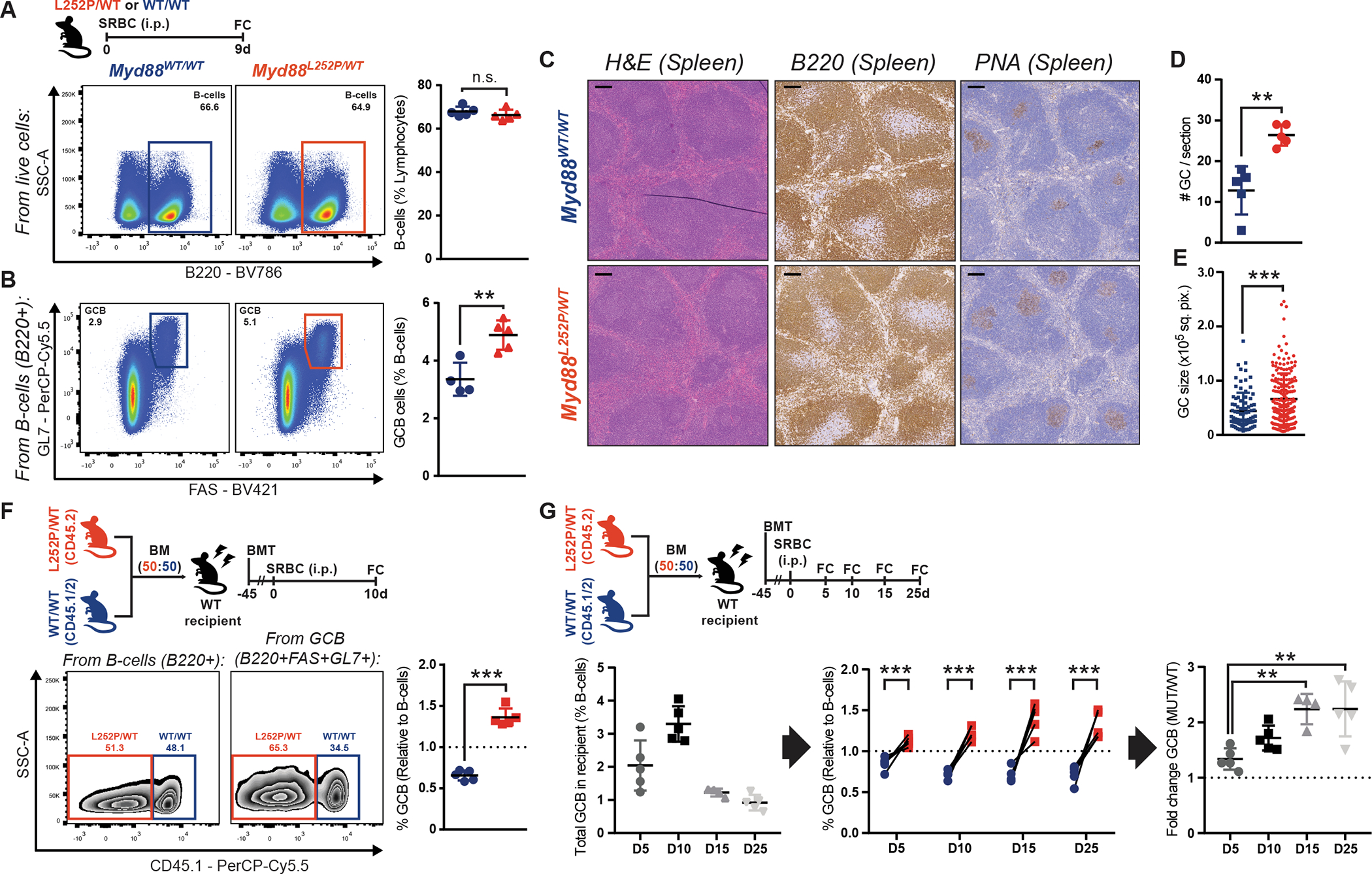
A-B, Flow cytometry (FC) analysis of splenic (A) B-cells or (B) GCB.
C, H&E, B220 IHC and PNA IHC in consecutive splenic sections from animals treated as in (A). Scale = 100μm.
D-E, GC (D) numbers or (E) individual area, based on PNA staining. Dots represent individual (D) animals or (E) GCs. Results for 5 animals per genotype.
F, FC analysis of Myd88L252P/WT and Myd88WT/WT relative contribution to B-cells and GCB, based on CD45 allele frequencies.
G, FC analysis of splenic GCB.
Values represent mean ± SEM. Data reproducible with two repeats. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001, using unpaired (A,B) or paired (F,G) two-tailed Student’s t-test with the two-stage step-up method of Benjamini, Krieger and Yekutieli where applicable; or Mann-Whitney U-test (D,E).
To test whether Myd88L252P enhanced the competitive fitness of GCB, we conducted mixed chimera experiments where equal numbers of Cd45.2;Myd88L252P/WT or Cd45.1/2;Myd88WT/WT bone marrow (BM) cells were transplanted into irradiated WT recipients. Following immunization, Myd88L252P GCB manifested significant competitive advantage, whereas the proportions of CD45.2+ and CD45.1/2+ total B-cells were comparable (Fig. 1F). To dissect the kinetics of this advantage, we investigated additional timepoints following immunization. While the GC response in chimeric mice showed the expected profile and duration (Fig. 1G, left), Myd88L252P GCB were consistently expanded, and developed cumulative dominance over time (Fig. 1G, center and right). These results indicate that GCB harboring MYD88 mutations are endowed with a significant competitive advantage.
Myd88 mutant GCB exhibit increased proliferative capacity
The size of the GC is determined through a fine-tuned balance between proliferation and cell death (3). Notably, the fraction of apoptotic GCBs was comparable in WT and Myd88L252P/WT mice, as assessed by cleaved Caspase (Supplementary Fig. S2A) or AnnexinV/DAPI (Supplementary Fig. S2B) staining, suggesting aberrant survival does not explain Myd88L252P GCB fitness. On the other hand, Myd88L252P GCB showed increased levels of the proliferation marker Ki67, as compared to WT (Fig. 2A). Similarly, mutant GCB exhibited higher incorporation of 5-ethynyl-2-deoxyuridine (EdU) (Fig. 2B), indicative of increased DNA synthesis and proliferation. Notably, while the fraction of proliferating WT GCB progressively decreased over the GC, as expected (19), the proportion of Ki67+ Myd88L252P GCB remained elevated and fairly constant (Fig. 2C). These differences align with the observed cumulative expansion of Myd88L252P/WT GCB (Fig. 1G).
Figure 2. Myd88 mutations confer a proliferative advantage to GCB.
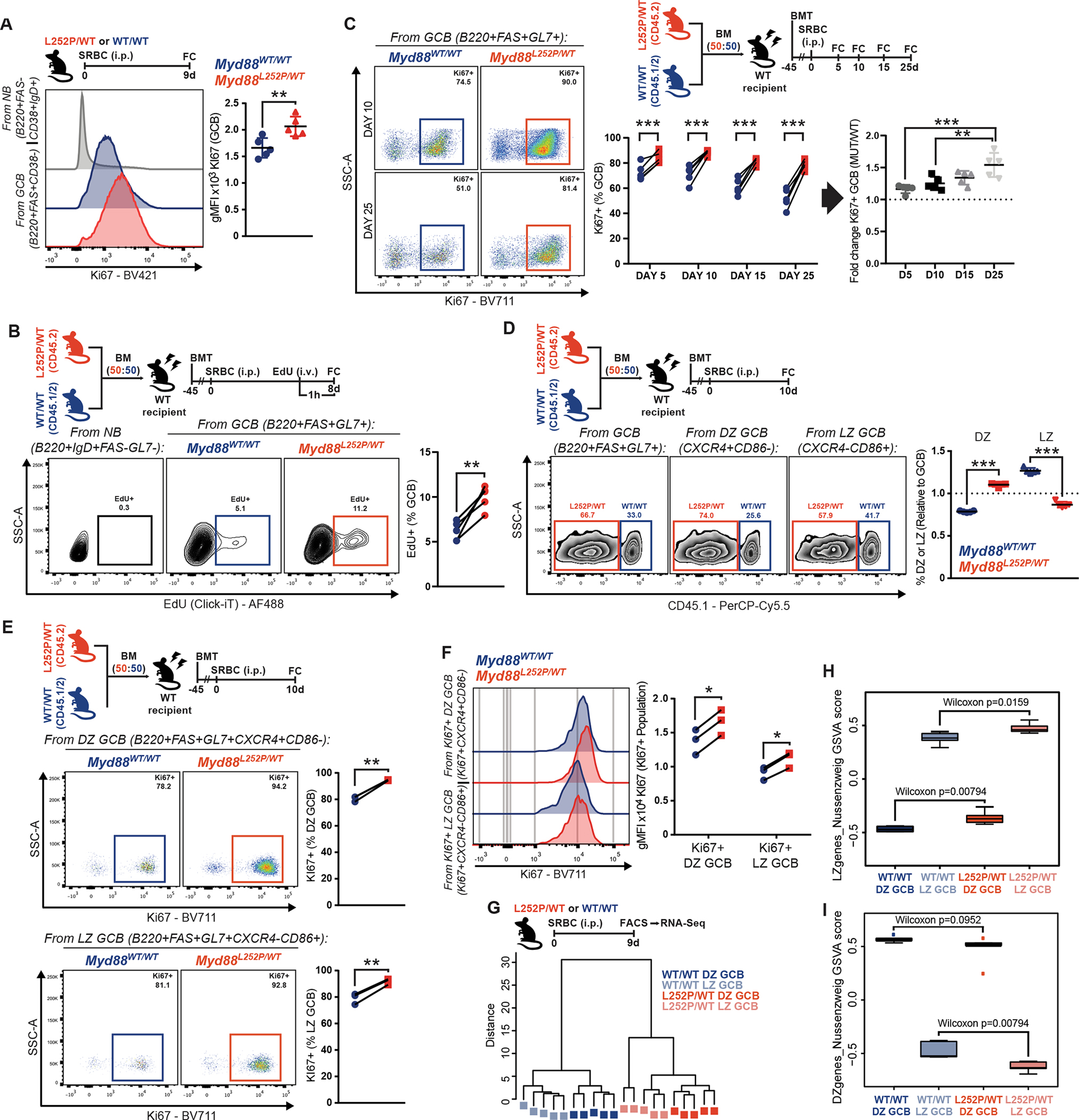
A, Geometric mean fluorescence intensity (gMFI) of Ki67 in splenic GCB.
B, FC analysis of EdU incorporation by GCB. NB illustrate non-proliferating cells.
C, FC analysis of Ki67+ GCB.
D, FC analysis of Myd88L252P/WT and Myd88WT/WT relative contribution to total, DZ or LZ GCB.
E, FC analysis of Ki67+ DZ and LZ GCB from (D).
F, FC analysis of Ki67 expression in Ki67+ DZ and LZ GCB from (E).
G, Hierarchical clustering, based on Euclidean distance, for RNA-Seq samples from sorted GCB.
H-I, GSVA analysis for samples in (G), relative to canonical (H) LZ or (I) DZ GCB signatures (GSE38696).
Values represent mean ± SEM. P-values calculated using unpaired (A,D) or paired (B,C,E,F) two-tailed Student’s t-test with the two-stage step-up method of Benjamini, Krieger and Yekutieli where applicable.
To gain insight into the proliferative capacity of Myd88L252P/WT B-cells, we used an established culture system, where naive B-cells (NB) exposed to high levels of co-stimulatory signals develop a GC-like phenotype (iGCB; (20,21)). To avoid artifactual effects related to the extent and timing of Cγ1Cre-induced recombination ex vivo, we isolated NB from CD19Cre;Myd88L252P/WT or CD19Cre;Myd88WT/WT mice. While both WT and mutant B-cells acquired an iGCB profile to similar extent (see below), Myd88L252P/WT iGCB divided faster, as revealed by accelerated proliferation dye dilution (Supplementary Fig. S2C). These results suggest that the proliferative advantage of Myd88L252P/WT GCB is supported, at least in part, through a cell-intrinsic effect.
Myd88 mutations blur the separation between GC cell compartments
The GC reaction is made up of anatomically and functionally specialized compartments, the dark zone (DZ; CXCR4+CD86−) and light zone (LZ; CXCR4-CD86+). Positively selected GCB undergo repeated rounds of division in the DZ (3). In line with the increase in proliferating GCB, Myd88L252P mice showed relative expansion of DZ GCB (Supplementary Fig. S2D), and Myd88L252P GCB preferentially acquired a DZ profile in chimeric mice (Fig. 2D). This became more pronounced at later stages in the GC reaction (Supplementary Fig. S2E). However, the fraction of proliferating cells (Fig. 2E), and the relative expression of KI67 among them (Fig. 2F), was higher in both compartments for Myd88L252P GCB than for WT. Thus, the increase in proliferating mutant GCB does not simply reflect an abnormal distribution across zones, but rather appears as a core feature conferred by Myd88L252P to all GCB, that obscures canonical compartmentalization.
To explore this, we sorted Myd88L252P/WT or WT GCB from each compartment, and conducted RNA sequencing (RNA-Seq). Hierarchical clustering and principal component analysis revealed segregation by both Myd88 status and cell type (Fig. 2G; Supplementary Fig. S2F). Interestingly, despite the phenotypic DZ/LZ asymmetry observed by flow cytometry, Myd88L252P/WT DZ and LZ GCB were transcriptionally closer to prototypical LZ GCBs (Fig. 2H; Supplementary Fig. S2G) and further away from prototypical DZ GCBs (Fig. 2I; Supplementary Fig. S2G) than their WT counterparts. Hence, while Myd88L252P DZ GCB portray canonical features, such as their proliferative status, their transcriptional identity appears more ambiguous.
Myd88 mutations reduce the threshold for T-cell-derived co-stimulatory signals
B-cells require co-stimulation to fully activate, escape cell death, and differentiate (2). In the GC, specialized follicular helper T-cells (TFH) provide membrane-bound (e.g., CD40L) and soluble (e.g., IL-4) signals to B-cells (3). Interestingly, the transcriptional profile of MCD tumors is depleted of GC TFH signatures (9), which may reflect their immune evasive nature, but could also suggest lower reliance on T-cell help. Myd88L252P and WT mice showed comparable levels of CD4+ cells (Fig. 3A). Myd88L252P mice presented an expansion of GC TFH (Fig. 3B), proportional to that of GCB (Fig. 3C), suggesting that TFH dosage in Myd88L252P GCs is adequate.
Figure 3. Myd88 mutations lower the requirement for T-cell-derived co-stimulatory signals.
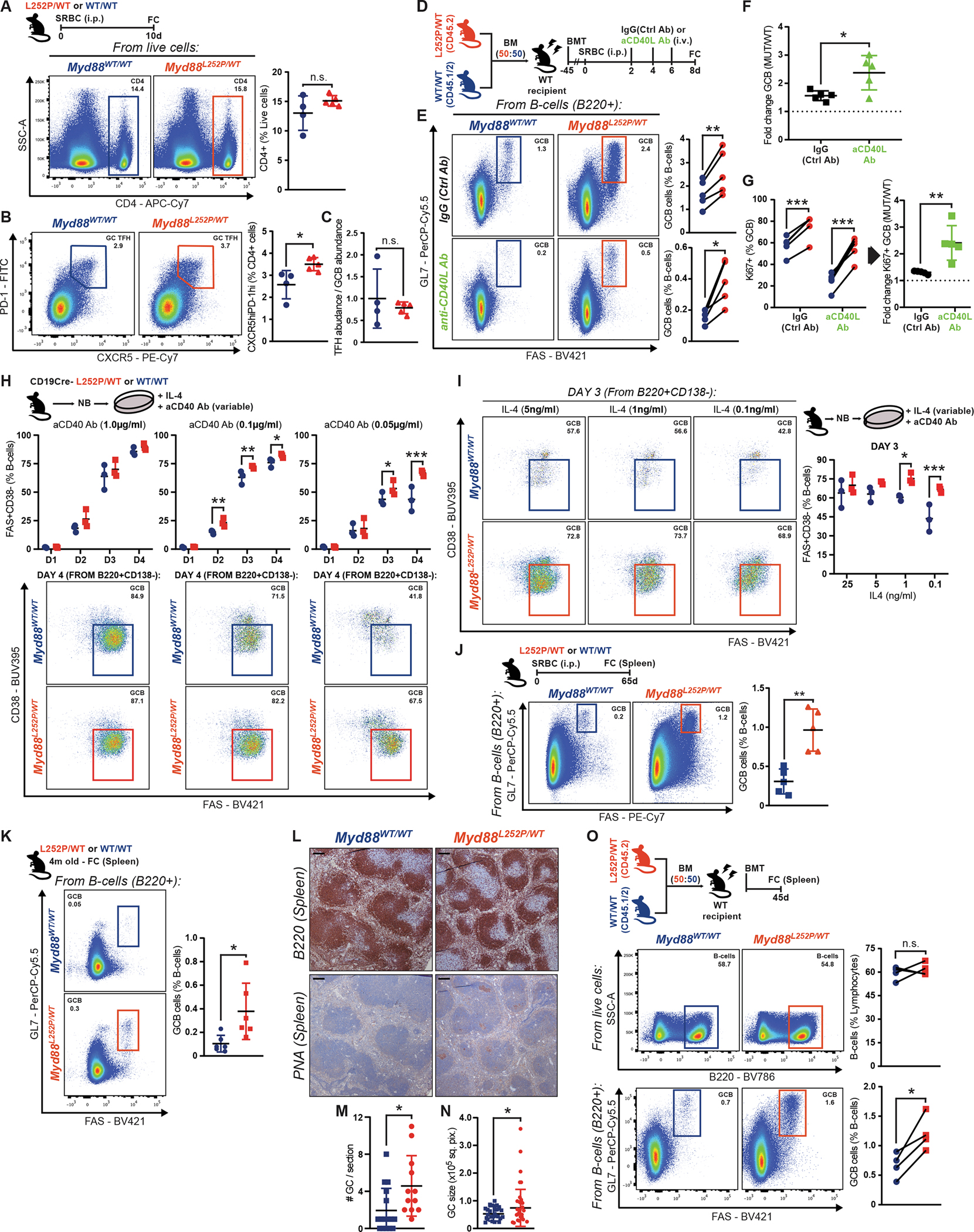
A-C, FC analysis of (A) CD4+ or (B) GC TFH cells. (C) GC TFH abundance relative to GCB in the same animals.
D, Experimental scheme for E-G.
E-F, FC analysis of GCB as (E) percentage of B-cells or (F) change between conditions.
G, FC analysis of Ki67 expression in GCB from (E).
H-I, FC analysis of iGCB with variable (H) CD40 or (I) IL-4 stimulation.
J, FC analysis of splenic GCB.
K, FC analysis of GCB in non-immunized mice.
L, B220 and PNA IHC in consecutive sections from animals treated as in K. Scale = 100μm.
M-N, GC (M) numbers or (N) individual area in naive mice. Dots represent individual (M) animals or (N) GCs. Results for 12–18 animals per genotype, from 2 experiments.
O, FC analysis of Myd88L252P/WT and Myd88WT/WT relative contribution to B-cells and GCB.
Values represent mean ± SEM. P-values calculated using unpaired (A-C,F,G,J,K) or paired (E,G,O) two-tailed Student’s t-test; or Mann-Whitney U-test (M,N); or two-way ANOVA with Tukey’s post-test (H,I).
On the other hand, NF-κB priming by MYD88L265P (13) could lower signaling thresholds in B-cells, enabling heightened responses with minimal T-cell help. To test whether GCB-TFH crosstalk was essential for MYD88-mutant B-cells, as is for WT, we administered CD40L-blocking or control antibodies to SRBC-immunized chimeric mice (Fig. 3D). In IgG-control-treated mice, Myd88L252P GCB showed the expected advantage (Fig. 3E, top row). Strikingly, whereas anti-CD40L treatment almost completely abrogated WT GCB, there was significantly less reduction of Myd88L252P GCB (Fig. 3E, bottom row). Myd88L252P GCBs resisting CD40L blockage still grouped in clusters within follicles (Supplementary Fig. S3A). Similar results were obtained using a strictly T-cell dependent antigen (Supplementary Fig. S3B). These results suggest that, while Myd88L252P B-cells benefit from T-cell help, their lower signaling threshold allows them to fully activate, become GCB, and expand with minimal co-stimulation. In fact, the relative Myd88L252P GCB expansion was more pronounced after blocking CD40L (Fig. 3F; Supplementary Fig. S3C), suggesting that normal T-cell help partially masks the advantage conferred by Myd88L252P. Differences in proliferation were also magnified after blocking CD40L, with a large proportion of Myd88L252P GCB retaining their Ki67+ status (Fig. 3G; Supplementary Fig. S3D).
To validate our findings, we assessed the impact of limited stimulation on iGCB formation. When exposed to high concentrations of a CD40-activating antibody, WT and Myd88L252P/WT activated and differentiated into mature iGCB at similar rates (Fig. 3H, left column). However, when dosage was reduced by 10- or 20-fold, a larger fraction of Myd88L252P/WT B-cells acquired an iGCB phenotype, compared to WT (Fig. 3H, center and right columns), supporting that the activation threshold for mutant cells is significantly lower. Accordingly, Myd88L252P/WT B-cells showed higher relative IкBα phosphorylation levels, and reduced total IкBα levels, compared to WT, upon acute CD40 stimulation (Supplementary Fig. S3E–G).
ABC(activated B-cell)-DLBCL tumors carrying MYD88L265P harbor increased p-STAT3 levels, and MYD88 silencing in MCD cell lines impairs STAT3 phosphorylation (10). Such an increase in p-STAT could enhance the response to IL-4. Thus, we conducted additional ex vivo stimulation assays limiting IL-4 concentration. Again, Myd88L252P/WT B-cells managed to proliferate and acquire a fully mature iGCB phenotype even with highly-restrictive IL-4 concentrations that were detrimental to WT B-cell expansion/activation (Fig. 3I). In all, our data shows that MYD88-mutant B-cells can thrive with minimal T-cell-derived co-stimulation.
Myd88 mutations favor spontaneous GC reactions
The lower reliance of Myd88L252P B-cells on T-cell help could facilitate their recurring or persistent activation, a requirement for MCD pathogenesis (22) and tumor cell survival (23). To explore this, we profiled Myd88L252P/WT and WT mice two months after SRBC immunization, a time point at which GCs should be fully resolved. Akin to the GC peak, Myd88L252P mice exhibited significantly more GCB than WT, which only presented a residual population (Fig. 3J). Detected Myd88L252P GCB could represent aberrantly long-lasting cells derived from the immunization, and/or originate from spontaneous GCs (24). To explore whether Myd88L252P exacerbated spontaneous GC formation/expansion, we profiled adult naive mice housed in our specific-pathogen free facility. Indeed, Myd88L252P animals presented significantly more spontaneous splenic GCB by FC (Fig. 3K; Supplementary Fig. S3H) or IHC (Fig. 3L–N). A similar phenotype was observed in inguinal lymph nodes (Supplementary Fig. S3I). To exclude potential artifactual environmental variables -despite animals with different genotypes being housed as cagemates-, we profiled naive chimeric mice, and observed comparable phenotypes (Fig. 3O; Supplementary Fig. S3J). This lower threshold to form GCB shown by MYD88-mutant B-cells could facilitate the aberrant reactivation of MCD precursor populations.
Myd88 mutations enable an aberrantly increased and pervasive GC output
TBL1XR1 mutations in GCB bias them towards MBC and away from PC (22), which prompted us to explore whether this was a common theme among MCD founder mutations. First, we conducted a supervised analysis of the Myd88L252P/WT LZ GCB transcriptome (Fig. 2G) and, indeed, found strong enrichment for GCB-to-MBC transition signatures (Fig. 4A; (25)), and a depletion for genes associated with the PC fate (Fig. 4B). Differentially upregulated genes in mutant LZ GCB included MBC-associated markers (26,27), including Bcl2, Il9r and Ccr6 (Supplementary Table S1). We then aimed to validate whether the transcriptional deviation from the PC fate resulted in constrained plasmacytic differentiation, using our ex vivo system (Supplementary Fig. S2C). The frequency (Fig. 4C) and absolute number (Fig. 4D) of CD138+ cells produced by Myd88L252P/WT B-cells was moderately but significantly lower than for WT. Interestingly, while Myd88L252P/WT B-cells lost IgD surface expression (reflective of activation) before WT did (Supplementary Fig. S4A), and underwent more division rounds per unit of time (Supplementary Fig. S4B), the relative CD138+ cell output was consistently inferior after each cell cycle (Fig. 4E).
Figure 4. Myd88 mutations enable an aberrantly increased and pervasive GC output.
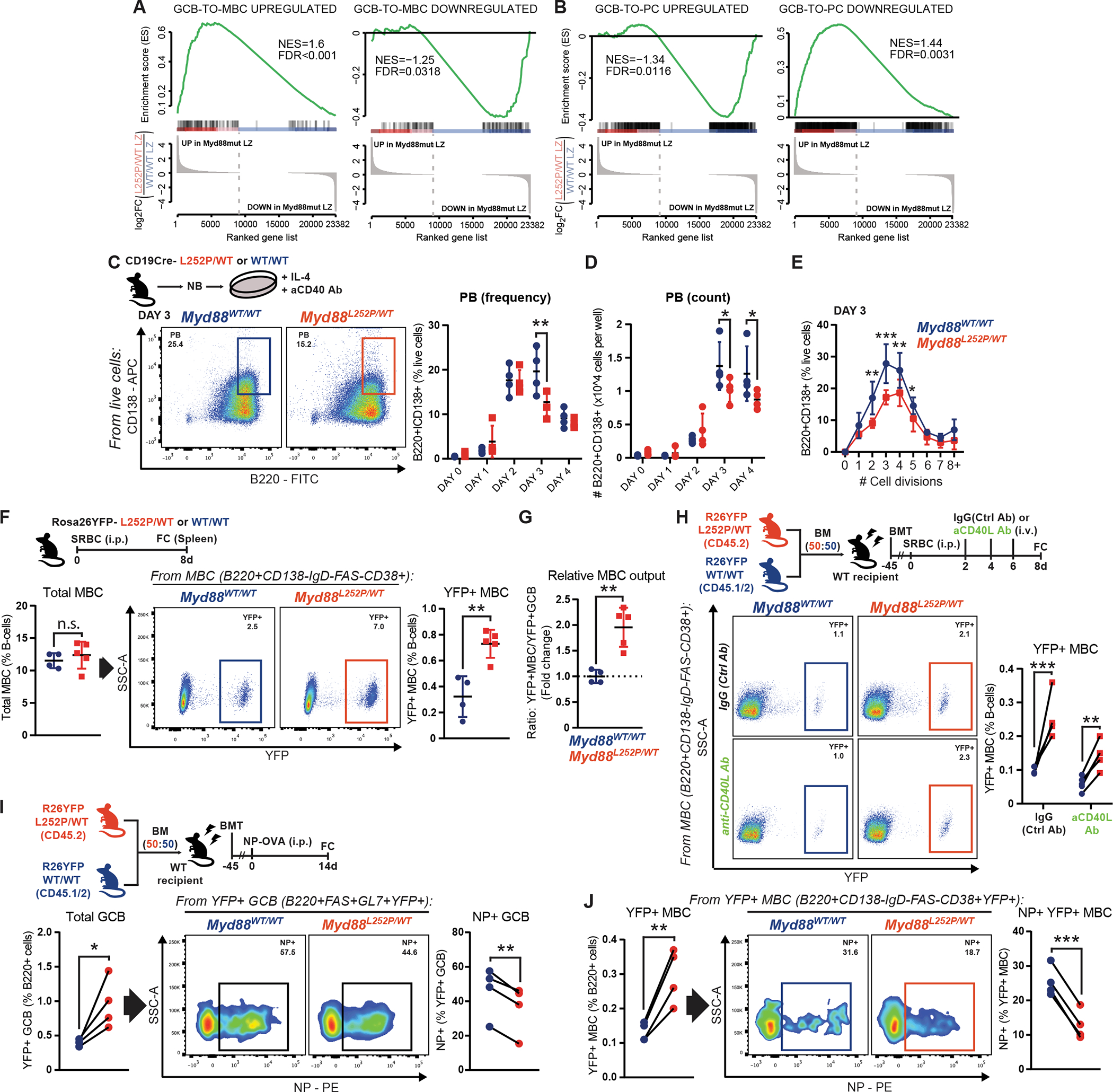
A-B, GSEA of (A) MBC or (B) PC signatures (GSE4142), against Myd88L252P/WT LZ GCB.
C-D, FC analysis of CD138+ cells (C) relative or (D) absolute abundance.
E, FC analysis of the relative fraction of CD138+ cells per cell division, determined by proliferation dye dilution, in cells from (C). Results for 3 animals per genotype.
F, FC analysis of (left) total or (right) YFP+ MBC.
G, YFP+ MBC abundance relative to YFP+ GCB in the same animals.
H, FC analysis of YFP+ MBC.
I-J, FC analysis of (left) total or (right) NP-specific (I) YFP+ GCB or (J) YFP+ MBC.
Values represent mean ± SEM. P-values calculated using unpaired (F,G) or paired (H-J) two-tailed Student’s t-test; or two-way ANOVA with Tukey’s post-test (C-E).
Currently, there are no equivalent systems to induce and compare MBC formation. Thus, to assess whether the transcriptional bias translated into MBC expansion, we used a Cre-inducible fluorescent reporter (Rosa26YFP; (28)) to track (pre)GC-derived MBC in vivo (Supplementary Fig. S4C). Following immunization, Rosa26YFP;Cγ1Cre;Myd88L252P/WT mice exhibited a significant increase in GC-derived (YFP+) MBC, compared to controls (Fig. 4F; Supplementary Fig. S4D). This expansion was disproportionately higher than that of GCBs (Fig. 4G), suggesting that MYD88L265P/WT GCs generate significantly greater abundance of MBC. This exacerbated output could produce an abnormal “drainage” of LZ GCBs, and relative DZ GCB overrepresentation (Fig. 2D). Next, to determine whether the MBC expansion depended on T-cell co-stimulation, we generated Rosa26YFP-harboring chimeric animals and administered CD40L-blocking antibodies following immunization (Fig. 4H). Similar to GCBs (Fig. 3E), mutant YFP+ MBC were significantly expanded in control conditions and upon impairment of GCB-TFH crosstalk (Fig. 4H). Thus, the aberrant phenotypes caused by MYD88 mutation appear hardwired into B-cells and show little dependence on co-stimulation.
TFH fulfill a gatekeeper function ensuring that GCB that develop high affinity for the antigen are preferentially selected to survive, expand, and exit as long-lived effectors (2,29). Given the observations above, we assessed antigen specificity in our Rosa26YFP-harboring models. NP-OVA-immunized chimeric mice showed the expected overrepresentation of total mutant GCB (Fig. 4I, left). However, the relative frequency of cells with high affinity for the antigen (NP+) was significantly lower among mutant GCB than for WT (Fig. 4I, right). More importantly, among the expanded GC-derived mutant MBC pool, there was a dramatically lower fraction of cells with detectable NP affinity (Fig. 4J). These data suggest that MYD88 mutations enable highly permissive GC transit and exit, which could facilitate the spurious expansion and persistence of B-cell clones.
Myd88 mutations trigger an aged/autoimmune-like program in GCB
To gain mechanistic insight into the observed phenotypes, we conducted further analysis of RNA-Seq profiles (Fig. 2G). Differential expression analysis identified Myd88L252P-driven signatures in DZ and LZ GCB (|FC|>1.5; FDR<0.05), skewed towards gene activation (Fig. 5A; Supplementary Fig. S5A; Supplementary Table S1). Pathway analysis revealed enrichment for ABC-DLBCL, advanced-stage DLBCL, and WM signatures, supporting that our models recapitulate MYD88L265P effects in human lymphomas (Fig. 5B; Supplementary Fig. S5B; Supplementary Table S1). Accordingly, we saw enrichment for NF-kB-regulated genes, the central cascade activated by MYD88L265P (Fig. 5B; Supplementary Fig. S5B; (30)). We further saw depletion of TFH signatures (Fig. 5B; Supplementary Fig. S5B), partly driven by downregulation of the transcription factors Jun and Fos. We also observed enrichment for cell migration and chemotaxis signatures (Fig. 5B; Supplementary Fig. S5B), which could relate to the MBC-like program in GCB (Fig. 4A), but also to the highly-prevalent extranodal presentation of MYD88L265P tumors. Related to cell motility, Myd88L252P LZ GCB downregulated Rgs13 (Fig. 5A), a negative modulator of CXCR4 function (31), which could contribute to the observed DZ skew (Fig. 2D). Myd88L252P LZ GCB also upregulated Il13ra1 (Supplementary Table S1), which pairs with the IL-4 receptor and signals through overlapping effectors (32), providing a plausible mechanic explanation to the enhanced response to IL-4 by Myd88L252P B-cells (Fig. 3I).
Figure 5. MCD mutations trigger an aged/autoimmune-like program in GCBs.
A, Differentially expressed genes in Myd88L252P/WT LZ GCB.
B, Pathway analysis for genes in (A).
C-D, FC analysis of (C) CD11c+ or (D) T-BET+ GCB.
E, GSEA of genes with a T-BET binding motif in their promoter (HOCOMOCO v11; >90% match within −5kb:TSS:+2kb), against Myd88L252P/WT LZ GCB.
F, GSEA of T-BET-KO MBC (GSE81189), against Myd88L252P/WT LZ GCB.
G, GSEA of canonical murine AiBC signatures (GSE175365), against Myd88L252P/WT LZ GCB.
H, GSEA of Tbl1xr1 mutant GCB (GSE139059), against Myd88L252P/WT LZ GCB.
I, RNA-Seq-based expression of genes of interest (G.O.I.) in Tbl1xr1 mutant (D370Y/WT) or WT GCB (GSE139059).
J, RT-qPCR validation of selected genes from (I), on independent animals.
K-L, FC analysis of (K) CD11c+ or (L) T-BET+ GCB in Tbl1xr1MUT mice.
M, GSEA of T-BET-KO MBC (GSE81189), against Tbl1xr1MUT GCB.
N, GSEA of canonical murine AiBC signatures, against Tbl1xr1MUT GCB.
Values represent mean ± SEM. P-values calculated using unpaired (D,J-L) or paired (C) two-tailed Student’s t-test.
Beyond the aforementioned profiles, the most prominent signal in Myd88L252P GCB related to interferon (IFN) signaling, and IFN gamma (IFNγ) in particular (Fig. 5B; Supplementary Fig. S5B). IFNγ instructs B-cell function against infections, but also drives autoimmune phenotypes (33,34). Indeed, our analysis showed enrichment for signatures related to immune responses and the autoimmune disorder Systemic Lupus Erythematosus (Fig. 5B). IFNγ induces the expression of the transcription factor T-BET in B-cells, and drives the formation of “aged/autoimmune” B-cells (AiBC; (35–37)). AiBC are a MBC subpopulation, phenotypically distinguishable as T-BET+ and/or CD11c+ (38), that expand and differentiate into PCs responsible for pathogenic auto-antibody production (36). B-cells with similar phenotypes accumulate during viral/parasitic infections and in aged healthy females (38). Interestingly, Myd88L252P GCB showed Tbx21 (T-BET) and Itgax (CD11c) upregulation (Fig. 5A; Supplementary Fig. S5A) by RNA-Seq, a finding confirmed by RT-qPCR in independent mice (Supplementary Fig. S5C). Enhanced CD11c (Fig. 5C) and T-BET (Fig. 5D) protein expression was further observed in Myd88L252P GCB driven by SRBC, or alternative antigens (Supplementary Fig. S5D). In line with T-BET induction, genes with T-BET binding motifs in their promoter were overrepresented among upregulated genes in Myd88L252P LZ GCB (Fig. 5E). Similarly, these genes matched those downregulated in T-BET knockout MBC (Fig. 5F; (39)). These findings suggest that aberrant T-BET expression induces transcriptional rewiring of Myd88L252P GCB. Myd88L252P GCB showed upregulation of other AiBC-related genes, including Itgam (CD11b), Fcrl5, Tlr7, and Cxcr3 (Fig. 5A; Supplementary Fig. S5E; (38–40)). Beyond individual markers, Myd88L252P GCB showed significant enrichment for canonical AiBC signatures (Fig. 5G; Supplementary Fig. S5F; (41)). In all, Myd88L252P induced a program in GCB that closely resembles that of pathogenic B-cells driving autoimmune disorders.
Induction of an AiBC program is a common feature among MCD founder mutations
In addition to IFNγ, Toll like receptor (TLR) activation is typically required to generate AiBCs (40). Since MYD88 is a critical signaling hub for most TLRs, the AiBC-related phenotype could pertain to MYD88L265P, but not MCDs as a class. Hence, we explored whether TBL1XR1 mutations would have similar effects. TBL1XR1 is a structural component of the SMRT/HDAC3 corepressor complex (22), with no evident functional link to MYD88. However, the transcriptome of Myd88L252P GCB showed significant enrichment for that of Tbl1xr1MUT GCB (Fig. 5H; Supplementary Fig. S5G). Further analysis of RNA-Seq data from Tbl1xr1MUT GCB (22) revealed differential upregulation of AiBC markers, including Tbx21, Itgax and Itgam (Fig. 5I), a finding confirmed by RT-qPCR in independent mice (Fig. 5J). Accordingly, Tbl1xr1MUT GCB showed protein-level upregulation of T-BET (Fig. 5K), CD11c (Fig. 5L), and TLR7 (Supplementary Fig. S5H). Furthermore, as observed for Myd88L252P, Tbl1xr1MUT GCB exhibited transcriptional enrichment for genes downregulated upon T-BET knockout (Fig. 5M), and for canonical AiBC signatures (Fig. 5N). These data suggest that the acquisition of an AiBC-like program is a common prevalent feature in MCD transformation, triggered by mechanistically distinct founder mutations.
MCD mutations cause a cumulative expansion of AiBC-like MBC
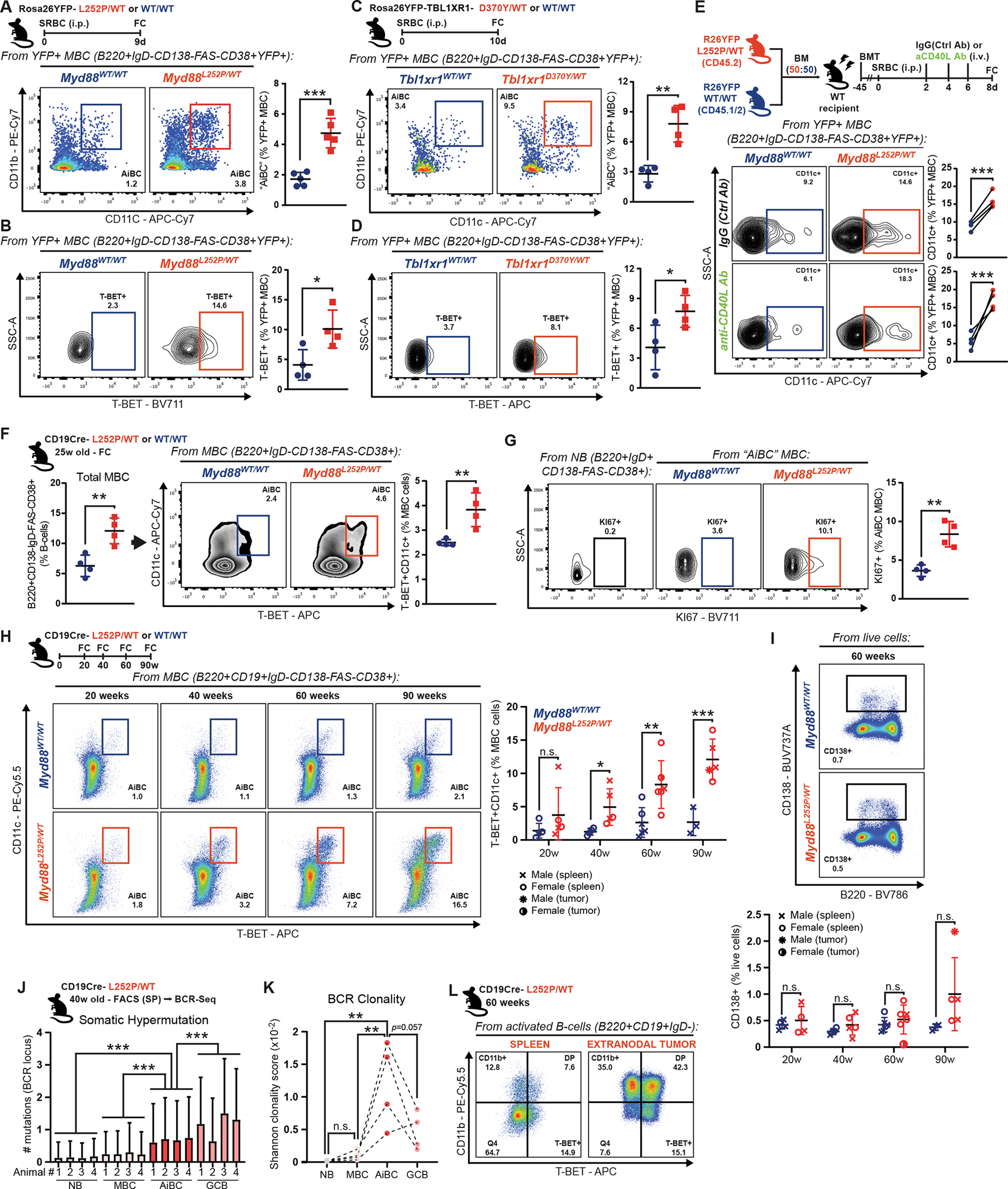
Given the transcriptional imprint exerted by T-BET in GCB harboring MCD founder mutations, we explored whether this resulted in the generation of AiBC-like MBCs, something of particular relevance considering that these mutations aberrantly increase MBC output ((22) and results herein). Notably, a significantly higher fraction of GC-derived MBC co-expressed the canonical AiBC markers CD11b/CD11c in Myd88L252P mice (Fig. 6A). Accordingly, a higher proportion of Myd88L252P MBC expressed T-BET (Fig. 6B). Studies in Rosa26YFP;Cγ1Cre;Tbl1xr1MUT animals revealed a similar expansion of CD11b+CD11c+ (Fig. 6C) and T-BET+ (Fig. 6D) MBC.
Figure 6. MCD mutations cause a cumulative expansion of AiBC-like MBCs.
A-D, FC analysis of (A, C) CD11b+CD11c+ or (B, D) T-BET+ YFP+ MBC.
E, FC analysis of CD11c+ YFP+ MBC.
F, FC analysis of (left) total or (right) T-BET+CD11c+ MBC in non-immunized young animals.
G, FC analysis of Ki67+ AiBC-like MBC in mice from (F). NB illustrate non-proliferating cells.
H, FC analysis of splenic T-BET+CD11c+ MBC in non-immunized mice. Tumor samples are plotted but not considered for statistical analysis.
I, FC analysis of CD138+ cells in animals from (H).
J, BCR mutation burden in splenic B-cells. Values = mean ± SD.
K, Clonality based on productive VDJ combinations. Datasets were down-sampled to a common minimum, to prevent size-based bias.
L, FC analysis of T-BET and CD11b expression in activated B-cells from the spleen or extranodal tumor from an animal in (H).
Values represent mean ± SEM. P-values calculated using unpaired (A-D,F,G-J) or paired (E) two-tailed Student’s t-test with the two-stage step-up method of Benjamini, Krieger and Yekutieli where applicable; or a Kruskal-Wallis test (J,K).
Although AiBC generation was first described as absolutely dependent on CD40 signaling (42), recent studies suggest this could be context-dependent (43). Given the observed lower dependency on CD40 stimulation among Myd88L252P B-cells, we asked whether the AiBC-like MBC expansion would occur under poor co-stimulation. CD40L-blocking antibodies reduced WT CD11c+ MBC by ~50%, whereas mutant CD11c+ MBC abundance remained elevated and comparable to that in IgG-control-treated mice (Fig. 6E; Supplementary Fig. S6A), suggesting little-to-no co-stimulation is needed for mutant B-cells to follow this cell fate.
CD19Cre;Myd88L252P mice, which express the mutation in all B-cells, develop lymphadenopathy and occasional lymphomas with old age (median survival ~70 weeks; (13)). We explored whether AiBC-like MBC would also expand in this model. At 25 weeks of age, a time point at which CD19Cre;Myd88L252P mice do not evidence lymphoproliferative disease (13), total MBC were expanded, and, within this population, there was a significant overrepresentation of AiBC-like (T-BET+CD11c+) phenotypes (Fig. 6F; Supplementary Fig. S6B). NB did not express these markers, suggesting the phenotype was only acquired post activation (Supplementary Fig. S6C). Notably, mutant AiBC-like MBC manifested a more active proliferative phenotype than WT counterparts, as per Ki67 staining (Fig. 6G). Such differences were absent in non-AiBC-like MBC (Supplementary Fig. S6D). This prompted us to explore whether AiBC-like MBC progressively accumulated during malignant transformation. Hence, we conducted longitudinal tracking of B-cells in Myd88L252P ageing mice (20–90 weeks). Strikingly, mutant mice showed increasing accumulation of T-BET+CD11c+ MBC (Fig. 6H), whereas there was little change in sex- and age-matched WT animals. Similar results were obtained when defining AiBC-like MBC as CD11b+CD11c+ or CD11b+T-BET+ (Supplementary Fig. S6E–F). Conversely, we found little splenic plasmacytic differentiation (CD138+ cells), even at old age (Fig. 6I), suggesting these cells do not play a leading role in the longitudinal transformation.
To gain insights into the origin and trajectory of AiBC-like MBC, we isolated this and other B-cell subsets from CD19Cre;Myd88L252P/WT mice at 40 weeks of age (Supplementary Fig. S6G), and performed targeted BCR sequencing. Despite reports that Aicda can activate in developing B-cells under BCR/TLR co-stimulation (44), NB from mutant mice showed largely undetectable SHM levels (Fig. 6J). On the other hand, AiBC exhibited elevated SHM levels (Fig. 6J), suggesting these transited through the GC, or acquired a GCB-like phenotype during their formation/expansion. SHM in AiBC was significantly higher than that in other MBC, but still lower than in (spontaneous) GCBs in the same animals (Fig. 6K). Notably, AiBC showed the highest clonality across all studied populations, as would be expected from prospective lymphoma precursors. Two animals in our longitudinal study presented overt B-cell lymphomas at the time of necropsy, one of which had extranodal peritoneal localization (Supplementary Fig. S6H). While a fraction of all splenic activated (IgD-) B-cells expressed CD11b and/or T-BET in this animal, more than 90% of tumor cells expressed one or both of these (Fig. 6L). In all, these observations support a role for AiBC-like MBC as prospective precursor populations for MCD lymphomas.
T-BET supports the fitness of MYD88-mutant B-cells
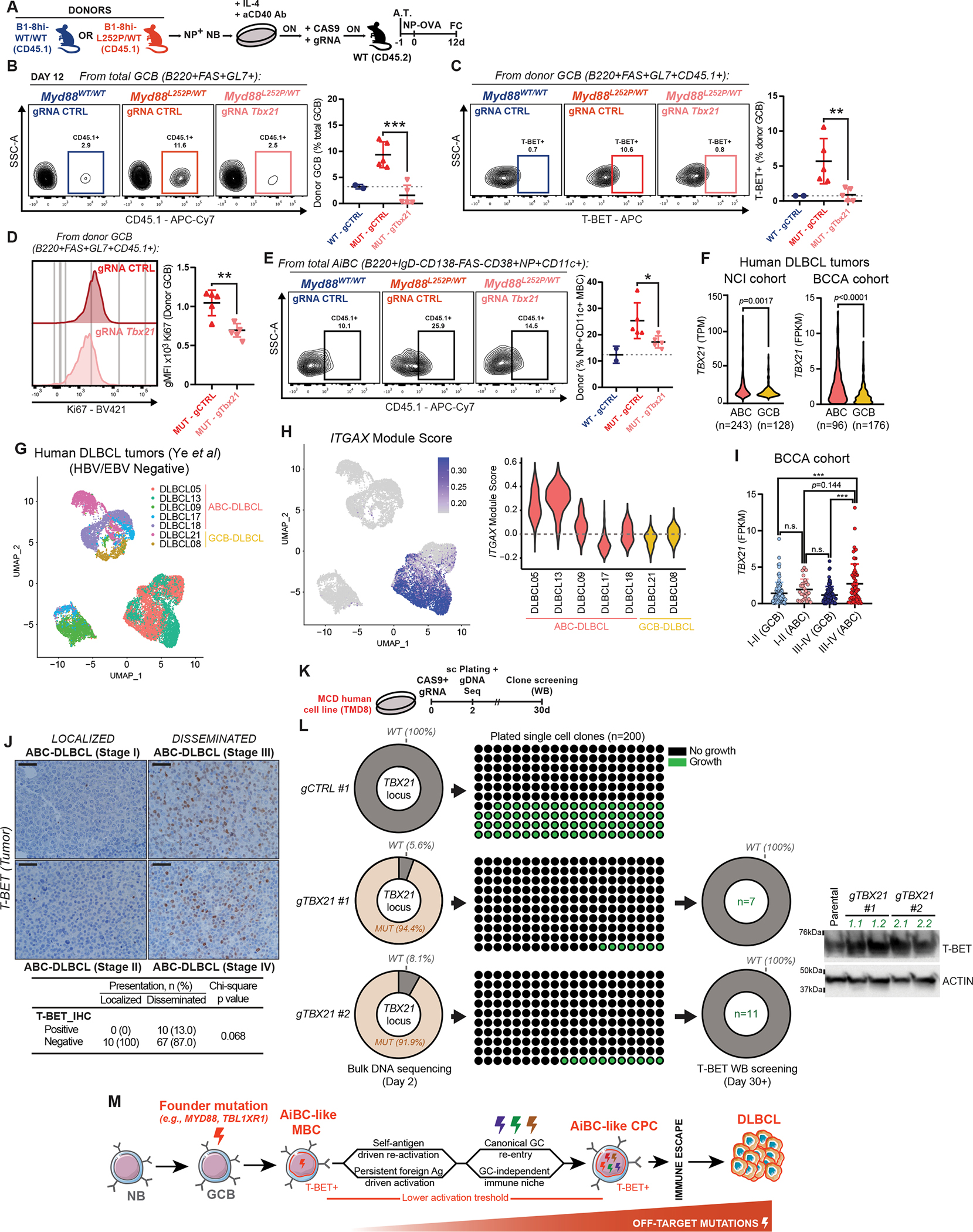
As the master regulator of canonical AiBC, T-BET ablation results in loss of this population and amelioration of autoimmune disease severity in animal models (36). The observed T-BET-driven GCB transcriptional program and AiBC-like MBC expansion in our models suggests a similar dependency may exist in MCD precursors. To test this, we ablated T-BET in non-transformed primary Myd88L252P B-cells using CRISPR/CAS9, and assessed their fitness to form GCBs after transplantation into immunocompetent recipients. Only a minimal fraction of NB from donor mice would be expected to recognize a given antigen, greatly limiting our capacity to study transferred cells. To overcome this, we crossed our models to incorporate an engineered B-cell receptor with defined specificity for NP (B1–8hi strain; (45)). Briefly, NP-specific NB from B1–8hi;Cd45.1;Myd88L252P or B1–8hi;Cd45.1;Myd88WT mice were first primed to enable editing (46), nucleofected with CAS9 complexes carrying control or Tbx21 guide RNAs (gRNA), and adoptively transferred into WT recipients (Fig. 7A). A fluorescently-labeled trans-activating RNA was used to track effective nucleofection with CAS9 complexes (Supplementary Fig. S7A). Targeted sequencing 24h after nucleofection confirmed the presence of disruptive lesions at the intended loci (Supplementary Fig. S7B). Recipient animals were immunized with an NP-protein conjugate the day after cell transfer, and profiled at the peak of the NP-induced GC. Approximately 10% of GCB cells in recipients derived from gCtrl-treated B1–8hi;Cd45.1;Myd88L252P transferred B-cells (Fig. 7B; Supplementary Fig. S7C). Strikingly, gTbx21-treated mutant donors manifested significantly impaired expansion, similar to that of B1–8hi;Cd45.1;Myd88WT donor B-cells (Fig. 7B). As expected, Myd88L252P GCBs featured higher T-BET expression compared to WT (Fig. 7C). However, GCB derived from gTbx21-treated B1–8hi;Cd45.1;Myd88L252P cells showed minimal T-BET expression, confirming efficient editing (Fig. 7C). In line with the observed loss of competitive fitness, gTbx21-treated cells showed significant reduction in Ki67 expression, reflecting their impaired proliferative status (Fig. 7D). T-BET ablation also significantly impaired the expansion of AiBC-like (CD11c+) MBC derived from transferred cells (Fig. 7E). Conversely, WT B-cell fitness was unaltered by Tbx21 knockout (Supplementary Fig. S7D–G). In all, these results show that T-BET supports the competitive fitness of B-cells harboring founder MCD mutations.
Figure 7. T-BET supports the fitness of MYD88-mutant B-cells.
A, Experimental scheme for B-D. ON = Overnight; A.T. = Adoptive transfer.
B, FC profiling of donor B-cell contribution to total GCB.
C, FC analysis of T-BET+ donor-derived GCB.
D, FC analysis of KI67 expression in donor-derived GCB.
E, FC analysis of donor B-cell contribution to AiBC-like MBC.
F, RNA-Seq-based TBX21 expression in primary specimens from (left) NCI (47) or (right) BCCA (48,49) cohorts.
G, uMAP depiction of single-cell RNA-Seq data from EBV/HBV-negative DLBCL tumors (50).
H, Relative expression of the ITGAX module among specimens in (G).
I, RNA-Seq-based TBX21 expression in primary human DLBCL specimens.
J, Representative images and quantification of T-BET IHC in specimens from the BCCA cohort (52). Scale = 20μm.
K, Experimental scheme for (L).
L, (Left) Penetrance of targeted genomic alterations, at time of cell plating. (Center) Number of detectable clonal outgrows 30 days after plating. (Right) WB-based T-BET expression in clonal outgrows. Representative blots for 2 clones per gRNA.
M, Schematic representation of the proposed transformation model.
Values represent mean ± SEM. P-values calculated using unpaired two-tailed Student’s t-test (B-F), or one-way ANOVA with Tukey’s post-test (I).
Human ABC-DLBCL tumors exhibit AiBC-like features
In the context of autoimmune disorders, some AiBC retain their transcriptional and phenotypic profiles despite T-BET downregulation following disease onset (41). To assess whether T-BET expression could be detected in human DLBCL, we first analyzed transcriptional profiles of primary tumors from the NCI (47) and BCCA (48,49) cohorts, and found that ABC-DLBCLs showed significantly higher TBX21 expression than GCB-DLBCLs (Fig. 7F). When narrowing down to MCD-DLBCL, the significant difference was upheld (Supplementary Fig. S7H), despite the limited number of MCD cases. MCD showed the highest levels of TBX21 among ABC-DLBCL, only after N1 tumors (another subtype thought to originate from MBC (9)). RT-qPCR and WB profiling of TBX21/T-BET in human cell lines supported these findings (Supplementary Fig. S7I–K). These observations in clinical specimens are consistent with the accumulation of T-BET+ B-cells in our preclinical models (Fig. 6H).
To provide greater evidence that AiBC-like phenotypes exist in human ABC-DLBCL, beyond TBX21, we analyzed published scRNA-Seq profiles from primary tumors (50). Most cases in that study (n=10/17) were EBV/HBV+, which could drive the expansion of AiBC (38). After excluding these to avoid confounding effects (Fig. 7G), most ABC-DLBCLs (n=5/7) showed a high proportion of cells with elevated expression of ITGAX and other canonical AiBC markers (e.g., CD22, FCRL5 (51)), whereas only a minor fraction of cells expressed these in GCB-DLBCLs (n=2) (Fig. 7H; Supplementary Fig. S7L–M).
Our models strongly suggest that the AiBC phenotype is linked to dissemination-prone DLBCLs. Disseminated ABC-DLBCL (stages III-IV; n=60) showed a trend towards higher TBX21 expression than localized tumors (stages I-II; n=30) (Fig. 7I). Given the small number of cases in the latter category, this analysis was likely underpowered to reach statistical significance. Still, disseminated ABC-DLBCL (but not localized cases) showed significantly higher TBX21 levels than localized (n=100) or disseminated (n=80) GCB-DLBCL (Fig. 7I). To complement this, we conducted T-BET IHC staining of ABC-DLBCL tissue microarrays (52). T-BET expression in tumor cells was variable, and generally lower than in T-cells in the same sections (Supplementary Fig. S7N). Still, in line with the mRNA data, we found that a fraction of disseminated ABC-DLBCL harbored T-BET+ tumor cells, whereas these were not detected in limited-stage cases (Fig. 7J).
T-BET supports the self-renewal capacity of MCD tumor cells
To test whether T-BET influences the fitness of fully-transformed MCD-DLBCL, we ablated T-BET in cell lines using CRISPR/CAS9, and assessed their ability to outgrow from single cells, as a measure of self-renewal capacity (Fig. 7K). Bulk sequencing 48h after delivery of TBX21-directed CAS9 complexes revealed extremely high penetrance of targeted disruptive mutations (Fig. 7L, left column). Only a minor fraction of plated cells (7/200 and 11/200, for two independent gRNAs) underwent clonal expansion, as compared to gCTRL-treated cells (78/200 outgrows; Fig. 7L, center column). Importantly, the few gTBX21-treated clones that did expand, showed T-BET levels comparable to parental, suggesting these derived from cells that escaped TBX21 editing (Fig. 7L, right column). Conversely, TBX21 ablation in a GCB-DLBCL model only had a mild non-significant effect on clonal fitness (Supplementary Fig. S7O–R). These findings suggest that T-BET retains an important role beyond lymphomagenesis, by supporting the clonogenic potential of fully transformed MCD tumor cells.
DISCUSSION
During an adaptive immune response, lack of co-stimulation results in B-cell death, preventing the misfiring and inappropriate engagement of B-cells in inflammatory responses (2,29). Here, we found that MYD88L256P alleviates this requirement, enabling B-cells to activate, proliferate, and differentiate into long-lived MBC, even under poor co-stimulation. TBL1XR1 mutations also imprint pathogenic trajectories on B-cells despite limited co-stimulation (22), highlighting a common theme in MCD lymphomagenesis. Such autonomous programs illustrate how B-cells normally attuned with their niche and under strict surveillance, may initiate and sustain transformation. Moreover, while T-cell help may be readily available to B-cells undergoing transformation in lymphoid tissues, this may not be the case at extranodal sites. Immune-privileged sites, which are targeted by MYD88L256P lymphomas (53), represent an extreme scenario, in that they offer a significantly different stromal landscape and greatly restrict lymphocytic infiltration, so they are likely ill-equipped to support transformation programs reliant on extensive B/T-cell crosstalk. Our results suggest that MYD88 mutations would still enable lymphomagenesis in such contexts. This is different from lymphomas such as classical Hodgkin’s, where transformation and tumor maintenance rely on strong co-stimulation (54).
Our results show that MYD88 mutations endow B-cells with a lower threshold and even semi-autonomous capacity to engage in proliferative responses to external signals, consistent with studies showing increased proliferation of MYD88MUT B-cells (55,56). We find that this property leads to increased competitive fitness against WT B-cells. Along these lines, we observed that mutant GCB formation and expansion occurred even in the absence of immunization. Age-associated spontaneous GC formation can be supported by TLR signaling (24). Beyond the direct effects of the MYD88 mutation downstream of TLRs, Myd88L252P GCB exhibited upregulation of Tlr3 and Tlr7, which could further contribute to these phenomena. TLRs activate by sensing conserved molecular patterns in pathogens, but can also become aberrantly stimulated by self-antigens (57). Notably, MCD and primary extranodal DLBCLs distinctively harbor BCRs that solely or promiscuously recognize self-antigens (9,58). Accordingly, MCD cell lines depend on auto-reactive BCR signaling for their survival in vitro (23). Self-reactivity could also serve as a source of continuous or repeated activation for lymphoma precursors. Self-reactive antibodies have been detected in aged mice expressing Myd88L252P in all B-cells (14). While the early introduction of the mutation could artifactually alter central tolerance mechanisms in the bone marrow, recent findings showed that spontaneous GCBs from animals where Myd88L252P was expressed in mature B-cells, harbored BCRs with similar properties to the self-reactive receptors in MCD cell lines (59). Here, we largely limited Myd88L252P expression to activated mature B-cells, in line with the current understanding that founder mutations most likely arise as aberrant SHM byproducts (8). Normally, lack of co-stimulation further prevents the expansion of self-reactive B-cells that escaped central tolerance checkpoints (2), and even anergic self-reactive B-cells awakened by antigen mimicry need to mutate their BCRs away from self to “redeem” themselves and be positively selected (29). Here, we show that MYD88-mutant GCs are significantly more permissive in terms of antigen specificity, which could set the stage for the aberrant activation and selection of clonal precursors driven by self-reactivity.
Previous studies on the effects of MYD88 mutations focused exclusively on plasmacytic fates, reporting abnormally elevated IgM antibodies and increased plasmablast proliferation in aged animal models (55,60). While informative, these did not delineate transformation trajectories, or elucidate the selective advantage conferred by MYD88L265P as a founder mutation. We and others recently showed that MBCs are the most likely MCD precursor (22,61), challenging the long-held belief that these arise from plasmablasts (17). Similarly, WM, a disease where >90% of tumors carry MYD88L265P and tumor cells exhibit plasmacytic features, is thought to derive from MBC (62). In line with this revised framework, we now show that MYD88 mutation not only provides a competitive advantage to B-cells, but also shepherds them towards the MBC fate. The fact that early mutations in two functionally unrelated genes, namely TBL1XR1 and MYD88, harness overlapping pathogenic trajectories, is a bona fide example of convergent evolution during lymphomagenesis.
MBC represent a heterogeneous and largely uncharted population. Here, we found that MCD founder mutations introduce a T-BET-driven transcriptional program in GCB, and cause the cumulative expansion of AiBC-like MBC. Unlike other MBCs, AiBC reactivation can be supported not only through BCR engagement and canonical co-stimulation (63), but also through TLR activation (35). TLR stimulation ex vivo is enough to induce T-BET expression in B-cells, and increased TLR7 dosage promotes AiBCs expansion (41). In turn, T-BET induces AICDA expression in MBC (39), which may favor the accumulation of mutations outside of a canonical GC context. Such plasticity and increased autonomy make AiBC ideal vectors for driving pathogenic processes, as first described for autoimmune disorders (35–37). Notably, mutations similar to those in ABC-DLBCLs were identified in self-reactive pathogenic MBC in patients with Sjogren’s Syndrome (SS; (64)), a rheumatic disease. Ours is the first demonstration that lymphoma mutations directly promote the generation of AiBC-like MBC, in response to foreign antigens or even in the absence of immunization. A relationship between autoimmune disorders and lymphomas has been extensively reported (65), whereby patients with Lupus, SS, or others, show increased risk of developing aggressive lymphomas. One study on Lupus patients that developed DLBCL found most tumors corresponded to the non-GCB subtype (66). Also, a third of patients had only extranodal involvement at DLBCL diagnosis, and their onset was significantly shorter than for nodal disease alone. Given that MCD fall into the non-GCB subclass (9), show significant association with extranodal disease (8,47), and share a mutational landscape with primary extranodal tumors (9,53), it is tempting to speculate that MCD-DLBCL may be disproportional prevalent among patients with autoimmune disorders. Our finding that lymphomas and autoimmune disorders harness similar pathogenic populations provides a rationale for these clinical observations, and prompts us to think of them as a spectrum, rather than separate entities. While our findings more directly support such conceptual framework for MCD and primary extranodal DLBCLs, BN2- and A53-DLBCLs also harbor self-reactive-prone BCRs (9), suggesting this may be a common theme among ABC-DLBCLs. Alternative stimuli, such as chronic infections, could similarly drive the production and re-stimulation of AiBC-like MBC involved in lymphomagenesis. A clinical association between chronic viral infections and DLBCL has been made (67–69), although this appears specific to BN2-DLBCLs (70).
DLBCLs are most frequently diagnosed among people in their seventh decade, and the incidence of ABC-type tumors increases with age (71). It has been hypothesized that this skew reflects a change in normal B-cell populations during aging (71). Clonal expansion of B-cells, overall reduction in B-cell diversity (72), and increased usage of the self-reactive-prone VH4–34 BCR (73) have been observed in aged individuals with no evident hematological malignancies. This further supports a model where a clonal and potentially auto-reactive B-cell population, such as AiBC, is directly involved in lymphomagenesis. Our findings indicate that MCD founder mutations instruct these phenotypes. The incidence of MYD88L265P was also significantly higher in tumors from older individuals (74). Mutations targeting MYD88 have also been recently reported in lymphoid clonal hematopoiesis of indeterminate potential (L-CHIP, (75)), and normal precursors and mature B-cells from patients with lymphoma (76), supporting the idea that MCD transformation involves a longitudinal process (Fig. 7M). While these observations also indicate that MCD founder mutations can be acquired early in the B-cell lineage, our findings suggest that AiBC phenotypes manifest at the mature B-cell stage, and support the idea that these cells acquire a GC-like state at some point during transformation.
The identification of precursor populations for aggressive and potentially incurable lymphomas remains a long sought-after but elusive goal. Here, we show that founder mutations associated with extranodal DLBCL give rise to a phenotypically distinguishable MBC subpopulation that differentially accumulates in time. In line with their prospective role as precursor cells, canonical AiBC disseminate and can be found in circulation and distributed to many tissues (77). These observations raise the provocative idea that an accumulation of AiBC-like MBC may be detectable in lymphoma patients preceding primary tumor onset and/or ensuing relapses, paving the way for risk-based stratification and prophylactic interventions. This could further attain premalignant conditions known to harbor MYD88L265P, such as monoclonal gammopathy of undetermined significance (78) or L-CHIP (75). Given their dismal outcome, patients at risk of developing central nervous system relapses routinely receive prophylactic treatment with methotrexate, a broad immunosuppressant, but recent studies have shown this to be largely ineffective (79). In this regard, we have unveiled a new axis involved in extranodal DLBCL pathogenesis, amenable to therapeutic targeting. Despite advances in direct pharmacological inhibition (80,81) or proteolysis targeting chimeras (82,83) against transcription factors, blocking of signaling cascades upstream of T-BET, including the use of TLR7/9 inhibitors (84), appears as an alternative that could be more readily implemented. In addition to exploring these aspects, future studies should determine the exact mechanisms of actions of T-BET and other AiBC features in the context of extranodal lymphomagenesis, and explore their potential value as predictive biomarkers for dissemination.
MATERIALS AND METHODS
Mouse models
Animal care was in strict compliance with institutional guidelines established by Weill Cornell Medicine (WCM), the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, 1996), and the Association for Assessment and Accreditation of Laboratory Animal Care International. The Research Animal Resource Center of WCM (protocol #2011–0031) and the Landesamt für Natur, Umwelt und Verbraucherschutz NRW (LANUV; (AZ: 84–02.04.2014.A146, 84–02.04.2017.A131, 81–02.04.2019.A009) approved all mouse procedures. The following strains were obtained from The Jackson Laboratory (Ben Harbor, ME, USA): C57Bl/6J (CD45.2, stock 000664; RRID:IMSR_JAX:000664), C57BL/6-Myd88tm1.1Rein/J (Myd88-L252P, stock 029349; RRID:IMSR_JAX:029349), Cγ1-Cre (stock 010611; RRID:MMRRC_010611-UCD), CD19-Cre (stock 006785; RRID:IMSR_JAX:006785), B6.SJL-PtprcaPepcb/Boy (CD45.1, stock 002014; RRID:IMSR_JAX:002014), Rosa26-lox-stop-lox-YFP (Rosa26YFP; stock 006148; RRID:MMRRC_006148-UCD), and B1–8hi (stock 007594; RRID:IMSR_JAX:007594). The generation of the conditional Tbl1xr1-D370Y mouse model has been described (22). Animals were housed in specific-pathogen free facilities. Experiments were conducted using aged and sex-matched littermates wherever possible. Experiments were designed to include male and female specimens in all groups, and no sex-based influence/bias was detected in the observations made in this work. Unless stated otherwise in the text, animals were 8 to 12 weeks of age at the time of experimentation. Bone marrow transplantations to generate chimeric animals, immunization strategies, and use of CD40L-blocking antibodies were all conducted as described (22).
Cell lines
The DLBCL cell lines OCI-Ly1 (CVCL_1879; male origin; RRID:CVCL_1879), OCI-Ly3 (CVCL_8800; male origin; RRID:CVCL_8800), OCI-Ly8 (CVCL_8803; male origin; CVCL_8803) and OCI-Ly10 (CVCL_8795; female origin; RRID:CVCL_8795) were grown in Iscove Modified Dulbecco Media (12440061; ThermoFisher Scientific), supplemented with 10% FBS and penicillin G/streptomycin; U2932 (CVCL_1896; female origin; CVCL_1896), HBL1 (CVCL_4213; male origin; RRID:CVCL_4213), TMD8 (CVCL_A442, male origin), MD901 (CVCL_D709; male origin; RRID:CVCL_A442), RIVA (CVCL_1885; female origin; RRID:CVCL_1885), RC-K8 (CVCL_1883; male origin; RRID:CVCL_1883) and HLY-1 (CVCL_H207; RRID:CVCL_H207) were grown in Roswell Park Memorial Institute medium (10–040-CV; Corning; Corning, NY, USA), supplemented with 10% FBS, penicillin G/streptomycin, L-glutamine, and HEPES. All cells were grown in incubators at 37°C in a 5% CO2 atmosphere. Cells were maintained in culture for up to 4–6 weeks between the time of thawing and experimentation. HBL-1, HLY-1, and U2932 were obtained from Jose Martinez-Climent (Universidad de Navarra, Pamplona, Spain); OCI-Ly3 were obtained from Anas Younes (Memorial Sloan Kettering Cancer Center, New York, NY, USA); OCI-Ly1, OCI-Ly7, OCI-Ly8 and OCI-Ly10 were obtained from the Ontario Cancer Institute (OCI); TMD8 was obtained from Louis M. Staudt (National Cancer Institute, Bethesda, MA, USA); RC-K8 and RIVA were obtained from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ). Cell line authentication was performed at IDEXX BioResearch, using methods recommended by the American National Standards Institute (ANSI ASN-0002–2011). Cell lines were confirmed to be of human origin and tested for evidence of cross-species contamination (mouse, rat, Chinese hamster and African Green monkey). All cell lines were routinely tested for Mycoplasma contaminations using a MycoAlert PLUS Detection Kit (LT07–705; Lonza, Basel, Switzerland).
Flow cytometry analysis and cell sorting
Single-cell suspensions from mouse spleens, lymph nodes, bone marrows or tumors were stained using the following fluorescent-labeled anti-mouse antibodies: from eBioscience ThermoFisher Scientific (Waltham, MA, USA): PE-Cy5.5 anti-CD11c (35–0114-80, dilution 1:200; RRID:AB_11219866), PE-Cy7 anti-CD11b (25–0112-82, dilution 1:400; RRID:AB_469588), APC anti-CD38 (17–0381, dilution 1:500; RRID:AB_469381), PE anti-CXCR4 (12–9991, dilution 1:400; RRID:AB_891391), PerCP-Cy5.5 anti-CD45.1 (45–0453, dilution 1:500; RRID:AB_1107003), FITC anti-PD-1 (11–9985, dilution 1:150; RRID:AB_465472); from BD Biosciences (San Jose, CA, USA): AF647 anti-active Caspase 3 (560626, dilution 1:100; RRID:AB_1727414), BUV615 anti-CD45 (752418, dilution 1:500; RRID:AB_2917432), BV421 and BV711 anti-Ki67 (562899 and 563755, dilution 1:500; RRID:AB_2686897 and RRID:AB_2738406), FITC, PE-Cy7 and BV786 anti-B220 (553087, 552772 and 563894, dilution 1:500; RRID:AB_394617, RRID:AB_394458 and RRID:AB_2738472), BV421, BUV805 and PE-Cy7 anti-FAS (562633, 741968, and 557653, dilution 1:500; RRID:AB_2737690, RRID:AB_2871273 and RRID:AB_396768), BUV563 and BUV395 anti-CD38 (741271 and 740245, dilution 1:500; RRID:AB_2870812 and RRID:AB_2739992), PE-Cy7 anti-CXCR5 (560617, dilution 1:100; RRID:AB_1727521), PE-Cy7 anti-CD86 (560582 and 564198, dilution 1:300; RRID:AB_1727518 and RRID:AB_2738663), BV510 anti-IgD (563110, dilution 1:500; RRID:AB_2737003), APC and BUV737A anti-CD138 (558626 and 564430, dilution 1:500; RRID:AB_1645216 and RRID:AB_2738805), FITC anti-CD19 (553785, dilution 1:500; RRID:AB_395049), FITC and AF647 anti-GL7 (553666 and 561529, dilution 1:500; RRID:AB_394981 and RRID:AB_10716056); from BioLegend (San Diego, CA, USA): APC anti-CD11b (101212, dilution 1:400; RRID:AB_312795), BV785 and APC-Cy7 anti-CD11c (117336 and 117324, dilution 1:200; RRID:AB_2565268 and RRID:AB_830649) BV711 and APC anti-T-BET (644820 and 644814, dilution 1:100; RRID:AB_2715766 and RRID:AB_10901173), PercP-Cy5.5 anti-CD138 (142510, dilution 1:500; RRID:AB_2561601) Spark NIR 685 anti-CD19 (115567, dilution 1:500; RRID:AB_2819828), AF488 anti-IgD (405717, dilution 1:500; RRID:AB_10730618), APC-Cy7 anti-CD4 (100414, dilution 1:500; RRID:AB_312699), APC-Cy7 anti-CD45.1 (110716, dilution 1:500; RRID:AB_313505), PerCP-Cy5.5 anti-CD45.2 (109828, dilution 1:500; RRID:AB_893350), APC anti-CD3 (100235, dilution 1:500; RRID:AB_2561455), APC-Cy7 and PE anti-B220 (103224 and 103208, dilution 1:500; RRID:AB_313007 and RRID:AB_312993), PerCP-Cy5.5 anti-GL7 (144610, dilution 1:500; RRID:AB_2562979), PerCP-Cy5.5 anti-FAS (152610, dilution 1:500; RRID:AB_2632905), BV605 anti-CD86 (105037, dilution 1:300; RRID:AB_11204429), PE anti-TLR7 (160004, dilution 1:100; RRID:AB_2876562); from Biosearch Technologies: PE NP (N-5070–1, dilution 1:100). NIP-haptenated FITC was obtained from M.J. Shlomchik (University of Pittsburgh, PA, USA). A Zombie NIR Fixable Viability Kit (423105, Biolegend), a LIVE/DEAD Fixable Violet Dead Cell Stain Kit (L34963; ThermoFisher Scientific), or DAPI (D1306; ThermoFisher Scientific) were used for exclusion of dead cells. Intracellular stains, AnnexinV/DAPI stains, and detection of EdU incorporation were performed as described (22). Proliferation was assessed using a CellTrace CFSE Cell Proliferation Kit (C34554; ThermoFisher Scientific), according to the vendor’s protocol. Data were acquired on a BD Fortessa or BD Symphony instrument (BD Biosciences), or a Cytek Aurora spectral flow cytometry analyzer (Cytek, Bethesda, MD, USA), and analyzed using FlowJo software package (BD Biosciences; RRID:SCR_008520).
When B-cell populations were sorted, single-cell suspensions were pre-enriched in B-cells using positive selection with anti-B220 magnetic microbeads (130–049-501; Miltenyi Biotec), or negative selection with the EasySep Mouse B-Cell Isolation Kit (19854; StemCell Technologies, Vancouver, BC, Canada). The stated populations were then isolated using a BD FACSAria II or a BD Influx cell sorter (BD Biosciences).
Primary B-cell cultures
Total splenocytes were harvested from 8–16 weeks old naive mice, and NB were isolated using negative selection with CD43 magnetic beads (130–049-801; Miltenyi Biotec; Somerville, MA, USA) in accordance with manufacturer’s protocol. In some experiments, cells were stained with a CellTrace proliferation dye, as noted above. Cells were seeded at 1×10^6 cells/ml in RPMI media with 10% FBS, penicillin G/streptomycin, MEM non-essential AA (11140050, ThermoFisher Scientific), 50 μM 2-Mercaptoethanol (21985023, ThermoFisher Scientific), containing the indicated concentrations of murine recombinant IL-4 (0–25ng/ml; 404-ML; R&D Systems) and a functional grade anti-CD40 monoclonal antibody (0–1μg/ml; 16–0402-82, ThermoFisher Scientific; RRID:AB_468945). Cells were incubated at 37°C with 5% CO2 and culture medium was renewed every 2–3 days.
CRISPR editing of DLBCL cell lines and primary B-cells
Human cell lines were electroporated using an Amaxa Nucleofector and the SF Cell Line 4D-Nucleofector X Kit (PBC2–22500; Lonza) to incorporate a recombinant Cas9 nuclease (Alt-R® S.p. Cas9 Nuclease V3, #1081058), control (#1072544 or #1072545) or TBX21-targeting gRNAs (Hs.Cas9.TBX21.1.AB “#1”: 5’- GCGGUACCAGAGCGGCAAGU-3’; Hs.Cas9.TBX21.1.AC “#2”: 5’-GAUUAAACUUGGACCACAAC −3’), electroporation enhancer (#1075915), and a tracrRNA-ATTO550 (#1075927; all from Integrated DNA Technologies; Coralville, IA, USA), following vendor’s recommendations. For primary murine B-cells, the P4 Primary Cell 4D-Nucleofector X Kit L (V4XP4012; Lonza) was used, along with control or Tbx21-targeting (Mm.Cas9.TBX21.1.AA: 5’- UCCAAGGAAGCGACCCGGCG-3’; Mm.Cas9.TBX21.1.AC: 5’-GGUUGAACUUGGACCACAAC −3’; Integrated DNA Technologies) gRNAs.
B-cell adoptive transfer
For B-cell adoptive transfers, total splenocytes were harvested from 8–12 weeks old donor mice (Cd45.1/1 or Cd45.1/2), and B-cells were isolated using negative selection, as described above. To increase the number of productive (NP-binding) B-cells in the mix, cells were further subjected to Igκ light chain-based depletion. To this end, an anti-Igκ antibody (409502, BioLegend; RRID:AB_2563297) biotinylated in-house (ab201796; Abcam, Cambridge, United Kingdom), was added during magnetic B-cell isolation. Cells were grown overnight in complete media containing 25ng/ml murine recombinant IL-4 and 1μg/ml of an anti-CD40 antibody. Cells were nucleofected with an ATTO-labeled CAS9 ribonucleoprotein complexes as stated above, and were allowed to recover overnight in complete media supplemented with IL-4 and the CD40 agonist. The percentage of ATTO+ and NP-binding cells in each population was determined by FC, to allow for normalization across conditions. Cells were allowed to rest in complete media without IL-4/anti-CD40 for >2h prior to transfer. A number of mature B-cells corresponding to 1×105 ATTO+NP+ B-cells was injected i.v. into C57Bl/6J recipient mice (Cd45.2/2). Recipient animals were immunized with an NP-conjugate 16h after cell transfer, and euthanized for analysis at the stated timepoints.
Quantitative real-time PCR
Total RNA extracts and cDNA synthesis were conducted as described (22). Expression of genes of interest was detected using a Fast SYBR Green Master Mix (4385614; ThermoFisher Scientific) on a QuantStudio6 Flex Real-Time PCR System (ThermoFisher Scientific). Gene expression was normalized to Actin or GAPDH levels, using the ΔΔC(t) method. Primer sequences (5’->3’): hTBX21.F: GGTTGCGGAGACATGCTGA; hTBX21.R: GTAGGCGTAGGCTCCAAGG; mTbx21.F: AGCAAGGACGGCGAATGTT; mTbx21.R: GGGTGGACATATAAGCGGTTC; mItgax.F: CTGGATAGCCTTTCTTCTGCTG; mItgax.R: GCACACTGTGTCCGAACTCA; mItgam.F: ATGGACGCTGATGGCAATACC; mItgam.R: TCCCCATTCACGTCTCCCA.
Targeted genomic sequencing
Genomic DNA from primary B-cells or human DLBCL cell lines was extracted as described (22). DNA concentration was determined using Qubit Fluorometric Quantification, and the amount of template DNA across samples was normalized for amplification. The CRISPR-targeted Tbx21 or TBX21 loci were PCR-amplified using the following primers (5’->3’): hTBX21.AB.F: AGGATGTTTGTGGACGTGGT; hTBX21.AB.R: CAGGAAGCCAGAAACAGGAG; hTBX21.AC.F: AGGTGTCGGGGAAACTGAG; hTBX21.AC.R: CCTGTCTCCCTACGCTGAAG; mTbx21AA.F: CTC AGCTTCCCAGACACCTC; mTbx21AA.R: GACCAACAGCATCGTTTCTTC. PCR products were resolved by agarose gel electrophoresis, bands of interest were excised, and DNA was retrieved using the QIAquick Gel Extraction Kit (28706X4; QIAGEN, Hilden, Germany). Library preparation, amplicon sequencing and variant calling were performed by GENEWIZ (South Plainfield, NJ, USA) or the Massachusetts General Hospital Center for Computational & Integrative Biology DNA Core (Cambridge, MA, USA).
BCR sequencing from primary murine cells was conducted by Adaptive Biotechnologies (Seattle, WA, USA) using an ImmunoSEQ Assay (85), as described (86). BCR repertoire analysis, including clonality and mutation burden calculations, were performed using the immunoSEQ Analyzer 3.0 (Adaptive Biotechnologies).
RNA sequencing
Library preparation, sequencing and post-processing of the raw data was performed at the Genomics Core at WCM, as described (22). Paired end-sequencing (PE75×2) was performed on an Illumina NextSeq500 instrument (Illumina; San Diego, CA, USA). Hierarchical clustering was performed using Euclidean distance of log TPM+0.1 values of genes within the top 5th percentile of standard deviation across replicates and Ward’s minimum variance. Gene set enrichment analysis was performed using the GSEA algorithm, as described in (87). Pathway analysis was performed using PAGE algorithm (88).
Histology and immunohistochemistry
Murine tissue preparation and staining were conducted by the Laboratory of Comparative Pathology at Memorial Sloan Kettering Cancer Center (MSKCC; New York, NY, USA), as described (22,89). The following primary antibodies were used for IHC: biotin-conjugated anti-B220 (550286; BD Biosciences; RRID:AB_393581) and anti-PNA (B1075; Vector Laboratories; RRID:AB_2313597). IHC on human specimens was performed at the BC Cancer Agency, as described previously (49,52), using an anti-T-BET (4B10) antibody (561262; BD Biosciences; dilution 1:50; RRID:AB_10565981). T-BET staining was semi-quantitatively assessed on tumor cells using HistoScore (HS = IxP): intensity (I = [1–3]) and percentage of positive cells (p = [0–100]). Specimens with HS > 10 were defined as T-BET positive.
Cell lysis and immunoblotting
Whole cell protein lysates preparation and SDS-PAGE analysis were performed as described (22), using the following primary antibodies: ß-ACTIN (C-4) (sc-47778; Santa Cruz Biotechnology, Dallas, TX, USA; RRID:AB_626632), T-BET (D6N8B) (#13232; RRID:AB_2616022), IκBα (44D4) (#4812; RRID:AB_10694416), and Phospho-IκBα (Ser32) (14D4) (#2859S; RRID:AB_561111; Cell Signaling Technology, Danvers, MA, United States).
Data availability
RNA sequencing data from this paper has been deposited in the Gene Expression Omnibus (GEO) database (RRID:SCR_005012), under accession number GSE201058. Gene expression data from individuals with de novo DLBCL from the NCI (47), BC Cancer (49), and Sun Yat-sen University Cancer Center (50) cohorts had been previously published. Code used for analysis is available upon request.
Statistical analysis
Statistical parameters, including the exact value and definition of n, precision measures (mean ± SEM or SD), and statistical significance are reported in figures and figure legends. No statistical methods were used to pre-determine animal sample sizes, but these were decided based on reports using similar models and approaches (13,14,22,89). Statistical analysis was conducted using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA; RRID:SCR_002798), or R statistical language scripts and packages specified. Data was judged to be statistically significant when p<0.05. Asterisks in figures denote statistical significance (∗, p<0.05; ∗∗, p<0.01; ∗∗∗, p<0.001).
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Extranodal lymphomas feature very poor prognosis. The identification of phenotypically distinguishable prospective precursor cells represents a milestone in the pursuit of earlier diagnosis, patient stratification, and prophylactic interventions. Conceptually, we found that extranodal lymphomas and autoimmune disorders harness overlapping pathogenic trajectories, suggesting these B-cell disorders develop and evolve within a spectrum.
ACKNOWLEDGEMENTS
We thank all members of the Melnick Lab for thoughtful discussions and suggestions, Mohamed Moustafa for his help with FC optimization, Hao Shen for his outstanding support with mouse colonies, the Genomics Core and Flow Cytometry Core (WCM), Molecular Cytology Core Facility (MSKCC), and Center of Comparative Medicine and Pathology (WCM/MSKCC). L. Venturutti is a Michael Smith Health Research BC Scholar, and is funded by the BC Cancer Foundation, CIHR (Project Grant #180613) and LLS-TRP 6663–23. Funding M.A. Rivas: ASH Junior Faculty Scholar Award. Funding B.W. Pelzer: Deutsche Krebshilfe (Mildred Scheel Nachwuchszentrum Grant #70113307). Funding C.E. Mason: Scientific Computing Unit, XSEDE Supercomputing Resources, Starr Cancer Consortium (I7-A765, I9-A9–071, I13–0052), Vallee Foundation, WorldQuant Foundation, Pershing Square Sohn Cancer Research Alliance, NIH (R01MH117406, R01CA249054, R01AI151059, P01CA214274), and LLS (9238–16, LLS-MCL-982). Funding Q. Pan-Hammarström: Swedish Research Council, Swedish Cancer Society, CIMED, Radiumhemmets research fund, and Knut and Alice Wallenberg Foundation. Funding A. Pernis: NIH (AR064883, AR070146). Funding E. Ricker: T32 Rheumatology Research Training Grant. Funding H.C Reinhardt: Deutsche Forschungsgemeinschaft (RE2246/13–1, SFB-1430-A09, SFB-1530-A01), Deutsche Jose Carreras Leukämie Stiftung (R12/08), Else Kröner-Fresenius Stiftung (EKFS-2014-A06, 2016_Kolleg.19) and Deutsche Krebshilfe (1117240, 70113041). Funding A.M. Melnick: NCI-R35 CA220499 and LLS-SCOR 7012–16.
Footnotes
COI statement: A.M.M. reports grants from Janssen, Epizyme, and Daiichi Sankyo and consulting fees from Janssen, Epizyme, AstraZeneca, Daiichi Sankyo, Treeline Biosciences and Exo Therapeutics outside the submitted work. H.C.R. received consulting and lecture fees from Abbvie, AstraZeneca, Novartis, Vertex, BMS and Merck. H.C.R. received research funding from Gilead Pharmaceuticals and AstraZeneca. H.C.R. is a co-founder of CDL Therapeutics GmbH. C.E.M. is a co-founder of Onegevity. No COI were reported by the other authors.
REFERENCES
- 1.Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med 2021;384(9):842–58 doi 10.1056/NEJMra2027612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brink R, Phan TG. Self-Reactive B Cells in the Germinal Center Reaction. Annu Rev Immunol 2018;36:339–57 doi 10.1146/annurev-immunol-051116-052510. [DOI] [PubMed] [Google Scholar]
- 3.Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol 2022;40:413–42 doi 10.1146/annurev-immunol-120419-022408. [DOI] [PubMed] [Google Scholar]
- 4.Venturutti L, Melnick AM. The Role of Epigenetic Mechanisms in B Cell Lymphoma Pathogenesis. Annu Rev Cancer Biology 2021;5(1):311–30 doi 10.1146/annurev-cancerbio-060820-125304. [DOI] [Google Scholar]
- 5.Bobillo S, Joffe E, Lavery JA, Sermer D, Ghione P, Noy A, et al. Clinical characteristics and outcomes of extranodal stage I diffuse large B-cell lymphoma in the rituximab era. Blood 2021;137(1):39–48 doi 10.1182/blood.2020005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo JJ, Winer ES, Olszewski AJ. Sites of extranodal involvement are prognostic in patients with diffuse large B-cell lymphoma in the rituximab era: an analysis of the Surveillance, Epidemiology and End Results database. Am J Hematol 2014;89(3):310–4 doi 10.1002/ajh.23638. [DOI] [PubMed] [Google Scholar]
- 7.Lu CS, Chen JH, Huang TC, Wu YY, Chang PY, Dai MS, et al. Diffuse large B-cell lymphoma: sites of extranodal involvement are a stronger prognostic indicator than number of extranodal sites in the rituximab era. Leuk Lymphoma 2015;56(7):2047–55 doi 10.3109/10428194.2014.982636. [DOI] [PubMed] [Google Scholar]
- 8.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24(5):679–90 doi 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020;37(4):551–68 e14 doi 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011;470(7332):115–9 doi 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shuai W, Lin P, Strati P, Patel KP, Routbort MJ, Hu S, et al. Clinicopathological characterization of chronic lymphocytic leukemia with MYD88 mutations: L265P and non-L265P mutations are associated with different features. Blood Cancer J 2020;10(8):86 doi 10.1038/s41408-020-00351-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Treon SP, Xu L, Liu X, Hunter ZR, Yang G, Castillo JJ. Genomic Landscape of Waldenstrom Macroglobulinemia. Hematol Oncol Clin North Am 2018;32(5):745–52 doi 10.1016/j.hoc.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Knittel G, Liedgens P, Korovkina D, Seeger JM, Al-Baldawi Y, Al-Maarri M, et al. B-cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood 2016;127(22):2732–41 doi 10.1182/blood-2015-11-684183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flumann R, Rehkamper T, Nieper P, Pfeiffer P, Holzem A, Klein S, et al. An Autochthonous Mouse Model of Myd88- and BCL2-Driven Diffuse Large B-cell Lymphoma Reveals Actionable Molecular Vulnerabilities. Blood Cancer Discov 2021;2(1):70–91 doi 10.1158/2643-3230.BCD-19-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019;51(2):337–50 e7 doi 10.1016/j.immuni.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cattoretti G, Buttner M, Shaknovich R, Kremmer E, Alobeid B, Niedobitek G. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood 2006;107(10):3967–75 doi 10.1182/blood-2005-10-4170. [DOI] [PubMed] [Google Scholar]
- 17.Venturutti L, Melnick AM. The dangers of deja vu: memory B cells as the cells of origin of ABC-DLBCLs. Blood 2020;136(20):2263–74 doi 10.1182/blood.2020005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, et al. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci U S A 2006;103(19):7396–401 doi 10.1073/pnas.0602353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkenschlager J, Finkin S, Ramos V, Kraft J, Cipolla M, Nowosad CR, et al. Dynamic regulation of TFH selection during the germinal centre reaction. Nature 2021;591(7850):458–63 doi 10.1038/s41586-021-03187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivas MA, Meydan C, Chin CR, Challman MF, Kim D, Bhinder B, et al. Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation. Nat Immunol 2021;22(2):240–53 doi 10.1038/s41590-020-00827-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venturutti L, Teater M, Zhai A, Chadburn A, Babiker L, Kim D, et al. TBL1XR1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-tumorigenic Memory Fate. Cell 2020;182(2):297–316 e27 doi 10.1016/j.cell.2020.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A 2015;112(44):13447–54 doi 10.1073/pnas.1514944112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, et al. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol 2014;193(9):4400–14 doi 10.4049/jimmunol.1401720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, et al. Transcriptional profiling of antigen-dependent murine B cell differentiation and memory formation. J Immunol 2007;179(10):6808–19 doi 10.4049/jimmunol.179.10.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suan D, Krautler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, et al. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 2017;47(6):1142–53 e4 doi 10.1016/j.immuni.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 27.Takatsuka S, Yamada H, Haniuda K, Saruwatari H, Ichihashi M, Renauld JC, et al. IL-9 receptor signaling in memory B cells regulates humoral recall responses. Nat Immunol 2018;19(9):1025–34 doi 10.1038/s41590-018-0177-0. [DOI] [PubMed] [Google Scholar]
- 28.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 2001;1:4 doi 10.1186/1471-213x-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burnett DL, Reed JH, Christ D, Goodnow CC. Clonal redemption and clonal anergy as mechanisms to balance B cell tolerance and immunity. Immunol Rev 2019;292(1):61–75 doi 10.1111/imr.12808. [DOI] [PubMed] [Google Scholar]
- 30.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2001;194(12):1861–74 doi 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol 2002;169(5):2507–15 doi 10.4049/jimmunol.169.5.2507. [DOI] [PubMed] [Google Scholar]
- 32.Umeshita-Suyama R, Sugimoto R, Akaiwa M, Arima K, Yu B, Wada M, et al. Characterization of IL-4 and IL-13 signals dependent on the human IL-13 receptor alpha chain 1: redundancy of requirement of tyrosine residue for STAT3 activation. Int Immunol 2000;12(11):1499–509 doi 10.1093/intimm/12.11.1499. [DOI] [PubMed] [Google Scholar]
- 33.Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med 2016;213(5):733–50 doi 10.1084/jem.20151724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prabhu N, Ho AW, Wong KH, Hutchinson PE, Chua YL, Kandasamy M, et al. Gamma interferon regulates contraction of the influenza virus-specific CD8 T cell response and limits the size of the memory population. J Virol 2013;87(23):12510–22 doi 10.1128/JVI.01776-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancro MP. Age-Associated B Cells. Annu Rev Immunol 2020;38:315–40 doi 10.1146/annurev-immunol-092419-031130. [DOI] [PubMed] [Google Scholar]
- 36.Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest 2017;127(4):1392–404 doi 10.1172/JCI91250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol 2007;178(10):6624–33 doi 10.4049/jimmunol.178.10.6624. [DOI] [PubMed] [Google Scholar]
- 38.Phalke S, Marrack P. Age (autoimmunity) associated B cells (ABCs) and their relatives. Curr Opin Immunol 2018;55:75–80 doi 10.1016/j.coi.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, et al. Cutting Edge: B Cell-Intrinsic T-bet Expression Is Required To Control Chronic Viral Infection. J Immunol 2016;197(4):1017–22 doi 10.4049/jimmunol.1500368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 2011;118(5):1305–15 doi 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricker E, Manni M, Flores-Castro D, Jenkins D, Gupta S, Rivera-Correa J, et al. Altered function and differentiation of age-associated B cells contribute to the female bias in lupus mice. Nat Commun 2021;12(1):4813 doi 10.1038/s41467-021-25102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell Knode LM, Naradikian MS, Myles A, Scholz JL, Hao Y, Liu D, et al. Age-Associated B Cells Express a Diverse Repertoire of VH and Vkappa Genes with Somatic Hypermutation. J Immunol 2017;198(5):1921–7 doi 10.4049/jimmunol.1601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song W, Antao OQ, Condiff E, Sanchez GM, Chernova I, Zembrzuski K, et al. Development of Tbet- and CD11c-expressing B cells in a viral infection requires T follicular helper cells outside of germinal centers. Immunity 2022;55(2):290–307 e5 doi 10.1016/j.immuni.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity 2007;27(1):64–75 doi 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shih TA, Roederer M, Nussenzweig MC. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat Immunol 2002;3(4):399–406 doi 10.1038/ni776. [DOI] [PubMed] [Google Scholar]
- 46.Hartweger H, McGuire AT, Horning M, Taylor JJ, Dosenovic P, Yost D, et al. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J Exp Med 2019;216(6):1301–10 doi 10.1084/jem.20190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378(15):1396–407 doi 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arthur SE, Jiang A, Grande BM, Alcaide M, Cojocaru R, Rushton CK, et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun 2018;9(1):4001 doi 10.1038/s41467-018-06354-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ennishi D, Healy S, Bashashati A, Saberi S, Hother C, Mottok A, et al. TMEM30A loss-of-function mutations drive lymphomagenesis and confer therapeutically exploitable vulnerability in B-cell lymphoma. Nat Med 2020;26(4):577–88 doi 10.1038/s41591-020-0757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye X, Wang L, Nie M, Wang Y, Dong S, Ren W, et al. A single-cell atlas of diffuse large B cell lymphoma. Cell Rep 2022;39(3):110713 doi 10.1016/j.celrep.2022.110713. [DOI] [PubMed] [Google Scholar]
- 51.Sutton HJ, Aye R, Idris AH, Vistein R, Nduati E, Kai O, et al. Atypical B cells are part of an alternative lineage of B cells that participates in responses to vaccination and infection in humans. Cell Rep 2021;34(6):108684 doi 10.1016/j.celrep.2020.108684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scott DW, Mottok A, Ennishi D, Wright GW, Farinha P, Ben-Neriah S, et al. Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J Clin Oncol 2015;33(26):2848–56 doi 10.1200/JCO.2014.60.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 2016;127(7):869–81 doi 10.1182/blood-2015-10-673236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wein F, Kuppers R. The role of T cells in the microenvironment of Hodgkin lymphoma. J Leukoc Biol 2016;99(1):45–50 doi 10.1189/jlb.3MR0315-136R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmidt K, Sack U, Graf R, Winkler W, Popp O, Mertins P, et al. B-Cell-Specific Myd88 L252P Expression Causes a Premalignant Gammopathy Resembling IgM MGUS. Front Immunol 2020;11:602868 doi 10.3389/fimmu.2020.602868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang JQ, Jeelall YS, Beutler B, Horikawa K, Goodnow CC. Consequences of the recurrent MYD88(L265P) somatic mutation for B cell tolerance. J Exp Med 2014;211(3):413–26 doi 10.1084/jem.20131424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farrugia M, Baron B. The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int J Inflam 2017;2017:8391230 doi 10.1155/2017/8391230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montesinos-Rongen M, Terrao M, May C, Marcus K, Blumcke I, Hellmich M, et al. The process of somatic hypermutation increases polyreactivity for central nervous system antigens in primary central nervous system lymphoma. Haematologica 2021;106(3):708–17 doi 10.3324/haematol.2019.242701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pindzola GM, Razzaghi R, Tavory RN, Nguyen HT, Morris VM, Li M, et al. Aberrant expansion of spontaneous splenic germinal centers induced by hallmark genetic lesions of aggressive lymphoma. Blood 2022;140(10):1119–31 doi 10.1182/blood.2022015926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouk C, Roland L, Gachard N, Poulain S, Oblet C, Rizzo D, et al. Continuous MYD88 Activation Is Associated With Expansion and Then Transformation of IgM Differentiating Plasma Cells. Front Immunol 2021;12:641692 doi 10.3389/fimmu.2021.641692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. Journal of Experimental Medicine 2020;217(10) doi 10.1084/jem.20200483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garcia-Sanz R, Jimenez C, Puig N, Paiva B, Gutierrez NC, Rodriguez-Otero P, et al. Origin of Waldenstrom’s macroglobulinaemia. Best Pract Res Clin Haematol 2016;29(2):136–47 doi 10.1016/j.beha.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 63.Glass DR, Tsai AG, Oliveria JP, Hartmann FJ, Kimmey SC, Calderon AA, et al. An Integrated Multi-omic Single-Cell Atlas of Human B Cell Identity. Immunity 2020;53(1):217–32 e5 doi 10.1016/j.immuni.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh M, Jackson KJL, Wang JJ, Schofield P, Field MA, Koppstein D, et al. Lymphoma Driver Mutations in the Pathogenic Evolution of an Iconic Human Autoantibody. Cell 2020;180(5):878–94 e19 doi 10.1016/j.cell.2020.01.029. [DOI] [PubMed] [Google Scholar]
- 65.Cuttner J, Spiera H, Troy K, Wallenstein S. Autoimmune disease is a risk factor for the development of non-Hodgkin’s lymphoma. J Rheumatol 2005;32(10):1884–7. [PubMed] [Google Scholar]
- 66.Tessier-Cloutier B, Twa DD, Baecklund E, Gascoyne R, Johnson NA, Backlin C, et al. Cell of origin in diffuse large B-cell lymphoma in systemic lupus erythematosus: molecular and clinical factors associated with survival. Lupus Sci Med 2019;6(1):e000324 doi 10.1136/lupus-2019-000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rong X, Wang H, Ma J, Pan S, Wang H, Jing S, et al. Chronic hepatitis B virus infection is associated with a poorer prognosis in diffuse large B-cell lymphoma: a meta-analysis and systemic review. J Cancer 2019;10(15):3450–8 doi 10.7150/jca.31033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Visco C, Finotto S. Hepatitis C virus and diffuse large B-cell lymphoma: Pathogenesis, behavior and treatment. World J Gastroenterol 2014;20(32):11054–61 doi 10.3748/wjg.v20.i32.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren W, Ye X, Su H, Li W, Liu D, Pirmoradian M, et al. Genetic landscape of hepatitis B virus-associated diffuse large B-cell lymphoma. Blood 2018;131(24):2670–81 doi 10.1182/blood-2017-11-817601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arcaini L, Rossi D, Lucioni M, Nicola M, Bruscaggin A, Fiaccadori V, et al. The NOTCH pathway is recurrently mutated in diffuse large B-cell lymphoma associated with hepatitis C virus infection. Haematologica 2015;100(2):246–52 doi 10.3324/haematol.2014.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mareschal S, Lanic H, Ruminy P, Bastard C, Tilly H, Jardin F. The proportion of activated B-cell like subtype among de novo diffuse large B-cell lymphoma increases with age. Haematologica 2011;96(12):1888–90 doi 10.3324/haematol.2011.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gibson KL, Wu YC, Barnett Y, Duggan O, Vaughan R, Kondeatis E, et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009;8(1):18–25 doi 10.1111/j.1474-9726.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang X, Stollar BD. Immunoglobulin VH gene expression in human aging. Clin Immunol 1999;93(2):132–42 doi 10.1006/clim.1999.4781. [DOI] [PubMed] [Google Scholar]
- 74.Lee JH, Jeong H, Choi JW, Oh H, Kim YS. Clinicopathologic significance of MYD88 L265P mutation in diffuse large B-cell lymphoma: a meta-analysis. Sci Rep 2017;7(1):1785 doi 10.1038/s41598-017-01998-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niroula A, Sekar A, Murakami MA, Trinder M, Agrawal M, Wong WJ, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med 2021;27(11):1921–7 doi 10.1038/s41591-021-01521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodriguez S, Celay J, Goicoechea I, Jimenez C, Botta C, Garcia-Barchino MJ, et al. Preneoplastic somatic mutations including MYD88(L265P) in lymphoplasmacytic lymphoma. Sci Adv 2022;8(3):eabl4644 doi 10.1126/sciadv.abl4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, et al. The Transcription Factor T-bet Resolves Memory B Cell Subsets with Distinct Tissue Distributions and Antibody Specificities in Mice and Humans. Immunity 2020;52(5):842–55 e6 doi 10.1016/j.immuni.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varettoni M, Zibellini S, Arcaini L, Boveri E, Rattotti S, Pascutto C, et al. MYD88 (L265P) mutation is an independent risk factor for progression in patients with IgM monoclonal gammopathy of undetermined significance. Blood 2013;122(13):2284–5 doi 10.1182/blood-2013-07-513366. [DOI] [PubMed] [Google Scholar]
- 79.Jeong H, Cho H, Kim H, Chae H, Lee JB, Lee K, et al. Efficacy and safety of prophylactic high-dose MTX in high-risk DLBCL: a treatment intent-based analysis. Blood Adv 2021;5(8):2142–52 doi 10.1182/bloodadvances.2020003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cardenas MG, Yu W, Beguelin W, Teater MR, Geng H, Goldstein RL, et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest 2016;126(9):3351–62 doi 10.1172/JCI85795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020;368(6489):387–94 doi 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jain N, Hartert K, Tadros S, Fiskus W, Havranek O, Ma MCJ, et al. Targetable genetic alterations of TCF4 (E2–2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci Transl Med 2019;11(497) doi 10.1126/scitranslmed.aav5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCoull W, Cheung T, Anderson E, Barton P, Burgess J, Byth K, et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem Biol 2018;13(11):3131–41 doi 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- 84.An B, Zhu S, Li T, Wu J, Zang G, Lv X, et al. A Dual TLR7/TLR9 Inhibitor HJ901 Inhibits ABC-DLBCL Expressing the MyD88 L265P Mutation. Front Cell Dev Biol 2020;8:262 doi 10.3389/fcell.2020.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun 2013;4:2680 doi 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 86.Leung W, Teater M, Durmaz C, Meydan C, Chivu AG, Chadburn A, et al. SETD2 Haploinsufficiency Enhances Germinal Center-Associated AICDA Somatic Hypermutation to Drive B-cell Lymphomagenesis. Cancer Discov 2022;12(7):1782–803 doi 10.1158/2159-8290.CD-21-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goodarzi H, Elemento O, Tavazoie S. Revealing global regulatory perturbations across human cancers. Mol Cell 2009;36(5):900–11 doi 10.1016/j.molcel.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA, et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 2020;37(5):655–73 e11 doi 10.1016/j.ccell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data from this paper has been deposited in the Gene Expression Omnibus (GEO) database (RRID:SCR_005012), under accession number GSE201058. Gene expression data from individuals with de novo DLBCL from the NCI (47), BC Cancer (49), and Sun Yat-sen University Cancer Center (50) cohorts had been previously published. Code used for analysis is available upon request.