

Abstract
Human calcitonin (hCT) is a polypeptide hormone that participates in calcium-phosphorus metabolism. Irreversible aggregation of 32-amino acid hCT into β-sheet-rich amyloid fibrils impairs physiological activity and increases the risk of medullary carcinoma of the thyroid. Amyloid-resistant hCT derivatives substituting critical amyloidogenic residues are of particular interest for clinical applications as therapeutic drugs against bone-related diseases. Uncovering the aggregation mechanism of hCT at the molecular level, therefore, is important for the design of amyloid-resistant hCT analogs. Here, we investigated the aggregation dynamics of hCT, non-amyloidogenic salmon calcitonin (sCT), and two hCT analogs with reduced aggregation tendency – TL-hCT and phCT – using long timescale discrete molecular dynamics simulations. Our results showed that hCT monomers mainly adopted unstructured conformations with dynamically formed helices around the central region. hCT self-assembled into helix-rich oligomers first, followed by a conformational conversion into β-sheet-rich oligomers with β-sheets formed by residues 10–30 and stabilized by aromatic and hydrophobic interactions. Our simulations confirmed that TL-hCT and phCT oligomers featured more helices and fewer β-sheets than hCT. Substitution of central aromatic residues with leucine in TL-hCT and replacing C-terminal hydrophobic residue with hydrophilic amino acid in phCT only locally suppressed β-sheet propensities in the central region and C-terminus, respectively. Having mutations in both central and C-terminal regions, sCT monomers and dynamically-formed oligomers predominantly adopted helices, confirming that both central aromatic and C-terminal hydrophobic residues played important roles in the fibrillization of hCT. We also observed the formation of β-barrel intermediates, postulated as the toxic oligomers in amyloidosis, for hCT but not sCT. Our computational study depicts a complete picture of the aggregation dynamics of hCT and the effects of mutations. The design of next-generation amyloid-resistant hCT analogs should consider the impact on both amyloidogenic regions and also take into account the amplification of transient β-sheets population in monomers upon aggregation.
Graphical Abstract
Introduction
Human calcitonin (hCT) is a 32-residue hormone polypeptide produced by C cells of the thyroid glands1, 2. hCT biologically acts to decrease the calcium level in the blood and reduce bone resorption by suppressing the activity of osteoclast cells3, 4. Primarily, hCT hormone has been used to treat a series of bone-related diseases including osteoporosis, hypercalcemia, and Paget’s disease for decades5–8. However, due to the intrinsic self-assembly tendency of forming various aggregates with low bioactivity2, 9, the food and drug administration (FDA) has terminated the clinical application of hCT9. Besides, emerging evidence suggests the amyloid aggregation of hCT into insoluble, β-sheet rich fibrillar aggregates is associated with medullary carcinoma of the thyroid (MTC) after discovering hCT amyloid deposits from patients10, 11. The misfolding and amyloid aggregation of peptides – e.g., amyloid-β (Aβ), α-synuclein, and human amylin (hIAPP) – are also the hallmark of numerous amyloid degenerative diseases (e.g., Alzheimer’s disease, Parkinson’s disease, and type 2 diabetes)12–15. Preventing the aggregation of hCT from forming amyloid aggregates has thus become crucially important for both its clinical application of hCT and future MTC therapeutics2, 16.
The aggregation of hCT decreases its bioavailability and therapeutic efficiency, stimulates undesirable immune responses, and enhances the risk of drug-induced toxicity3, 9, 17, 18. For example, the viability of SH-SY5Y cells exposed to incubated 4μM hCT was reduced to 58.3 ± 0.7%19. hCT is thus not suitable to be used in the clinic. The calcitonin of salmon (sCT) with a much lower aggregation propensity than hCT is used in the clinic alternatively, although sCT displays a much weaker potency than hCT16. In addition, sCT must be administrated by frequent injections because the half-life is relatively short ~1h20. Furthermore, the sCT administration also causes immunogenic reactions (e.g., anorexia and vomiting) because the sequence similarity to hCT is only ~50%21–23. Possible strategies for overcoming these problems involve the design of non-amyloidogenic hCT analogs with high similarity in both physicochemical properties and sequence and/or the development of biocompatible inhibitors against hCT amyloidosis2, 16. Many small-molecule polyphenols (e.g., magnolol and EGCG) with anti-amyloid effects on other amyloidogenic proteins (e.g., Aβ, hIAPP, and tau) display the capability of inhibiting hCT aggregation24–27. However, potential side effects and the pharmacological efficacies of these phenolic inhibitors in inhibiting hCT aggregation in vivo are still elusive. Mitigating the amyloidosis of hCT through substituting few critically-important amyloidogenic residues is of particular interest due to the expected high biocompatibility, low immunogenicity, and the high chance of preserving hCT function2, 19, 28. Therefore, hCT analogs with low aggregation propensity may have promising clinical applications as therapeutic drugs against bone-related diseases.
The mechanistic insight into the fibrillization of hCT is essential for the rational design of aggregation-resistant hCT analogs. The fibrillization tendency of hCT is strong both in vivo and in vitro16, 17. The fibrillization kinetics of hCT features a typical sigmoidal curve as other pathological amyloid proteins (e.g., Aβ, hIAPP, and α-synuclein)20, 28, 29. A two-step reaction mechanism was proposed, isolated hCT monomers first nucleated β-sheet rich protofibrils via helical accumulation along with a series of complex conformation conversion, followed by fibril growth of forming mature fibrils3, 17, 30, 31. As revealed by numerous studies of other disease-related amyloid peptides, soluble low-molecular-weight, β-sheet rich oligomers formed in the early nucleation stages are likely the major cytotoxic species of hCT aggregation13–15. All the calcitonin family proteins contain a highly conserved intramolecular disulfide bond formed by residues Cys-1 and Cys-7 at the N-terminus5, 9. Reducing the disulfide bond of sCT causes the loss of helical conformation indicating that the disulfide bond may stabilize the helices of calcitonin29, 32. hCT monomer also mainly adopts α-helix, and the helical structure in hCT is more flexible than sCT2, 17, 33, 34. Residues from 15 to 19 of hCT, 15DFNKF19, are known as the amyloidogenic core of hCT28, 35, 36. A recent study showed that the nitration of tyrosine Y12 of hCT would significantly decrease the aggregation and cytotoxicity of hCT, indicating a critical role of this particular aromatic residue in the amyloid aggregation of hCT19. Two single-point mutants of F16L and F19L hCT aggregated much slower than the wild type but faster than a multi-point mutant TL-hCT with Y12L, F16L, and F19L substitutions28. Another fibrillization-resistant polar hCT analog (phCT) with five residues replaced by the corresponding sCT residues – Y12L, N17H, A26N, I27T, and A31T – displayed low aggregation propensity and similar solution structure as sCT2. However, detailed molecular mechanisms of the hCT forming β-sheet-rich aggregates and effects of known mutations are still elusive, hindering the development of next-generation hCT analogs with increased biocompatibility, reduced immunogenicity, and also preserved functionality.
In this study, we systematically investigated the self-assembly of hCT, sCT, TL-hCT, and phCT to explore the aggregation mechanics of hCT and the effects of various mutations using all-atom discrete molecular dynamics37, 38 (DMD) simulations. DMD is a fast and predictive molecular dynamics algorithm that has been widely used by our group12, 39 and others37, 40–42 to study protein folding and amyloid aggregation. Our simulation results revealed that hCT monomers mainly adopted unstructured conformations with dynamic helices around the central region. The helical structures of monomeric sCT were more stable than hCT, consistent with prior experiments2, 17, 33, 34. hCT readily self-assembled into helical oligomers followed by a helix-to-sheet conformational conversion to form β-sheet rich oligomers. The self-assembly of hCT rendered the loss of helical conformations along with the amplifications of the transient β-sheet structures in monomers. Both the central aromatic and C-terminal hydrophobic residues of hCT participated in forming β-sheets structures. Substitution of central aromatic residues with leucine in TL-hCT and replacing C-terminal hydrophobic residue with hydrophilic amino acid in phCT only locally suppressed β-sheet propensities in the central region and C-terminus, respectively. Having mutations in both central and C-terminal regions, sCT mainly formed dynamical helical oligomers. The β-barrel formations with well-defined structures postulated to be the toxic oligomer of amyloidosis were also observed in the hCT self-assemblies. The direct observation of β-barrel oligomers during the self-assembly of cytotoxic hCT rather than the nontoxic sCT also supported β-barrels as the potential toxic oligomers of amyloidosis. By recapitulating the experimentally observed self-assembly of hCT forming β-sheet rich formations and amyloid-resistant mutants of TL-hCT and phCT, our computational results not only uncovered the self-assembly mechanism of hCT at the molecular level but also offered insights to help future design of next-generation hCT analogs for clinical applications. The design of amyloid-resistant hCT derivatives for clinical application should consider the impact on the central and C-terminal regions and the conformational amplification of even transient β-sheets in monomers upon aggregation.
Materials and Methods.
Molecular Systems.
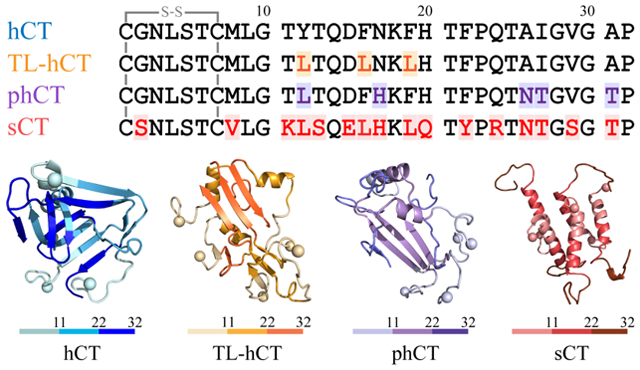
The initial structures of phCT (PDBid: 2jxz2) and sCT (PDBid:2glh43) used in our simulation are taken from the NMR structures solved in an aqueous solution. The hCT and TL-hCT are constructed using the PyMol mutagenesis based on the structure of phCT (PDBid: 2jxz2). The amino acid sequences of four calcitonin peptides are presented in Table 1. For each type of peptide, two molecular systems with the one and five peptides were performed to investigate monomer conformation and self-assembly dynamics. For each molecular system, thirty independent all-atom DMD simulations were performed starting with different initial states (i.e., coordinates, orientations, and velocities). In five-peptide simulations, the initial configurations had a minimum intermolecular distance of more than 1.5 nm between any two molecules. The duration of each DMD trajectory in one- and five-peptide systems was 500 ns and 600 ns, respectively. The details of all the simulations are summarized in Table 2. The hCT decreases the calcium level in the blood and reduce bone resorption by suppressing the activity of osteoclast cells via binding to its GPCR receptor3, 4, rather than directly interacts with calcium ion. Thus, prior experimental and computation simulation studies of calcitonin didn’t consider the calcium2, 28, 34, 43. The calcium ions were not included in our simulations.
Table. 1.
Amino acid sequences of hCT, TL-hCT, phCT, and sCT used in our simulation. All the four calcitonin peptides have a Cys1-Cys7 intra-molecular disulfide bond. For clarity, the hydrophobic, hydrophilic, negatively charged, and positively charged residues are colored black, green, red, and blue, respectively.

|
Table 2.
Details of molecular system in each DMD simulation. Including the number of simulated peptides (System), the type of calcitonin (Peptide), corresponding dimensions of the cubic simulation box (Box edge size), the number of independent DMD simulations performed (DMD run), the length of each DMD simulation (Time), and the accumulative total simulation time (Total Time).
| System | Peptide | Box edge size (nm) | DMD run | Time (μs) | Total time (μs) |
|---|---|---|---|---|---|
| 1-peptide | hCT | 6.5 | 30 | 0.5 | 15 |
| TL-hCT | 6.5 | 30 | 0.5 | 15 | |
| phCT | 6.5 | 30 | 0.5 | 15 | |
| sCT | 6.5 | 30 | 0.5 | 15 | |
| 5-peptides | hCT | 9.5 | 30 | 0.6 | 18 |
| TL-hCT | 9.5 | 30 | 0.6 | 18 | |
| phCT | 9.5 | 30 | 0.6 | 18 | |
| sCT | 9.5 | 30 | 0.6 | 18 |
Discrete Molecular Dynamics (DMD) Simulations.
All simulations are performed at 300K using the all-atom DMD algorithm with the Medusa force field38, which has been benchmarked for the accurate prediction of protein stability change upon mutation and protein–ligand binding affinity44, 45. DMD is a particular approach of molecular dynamics algorithm with significantly enhanced sampling efficiency, widely used in studying protein folding, amyloid peptides aggregation, and nanoparticle–protein interactions by our groups12, 39 and others37, 40–42. The major difference between DMD and conventional MD approaches is in the form of the interatomic interaction potential functions, where step functions in DMD replace the continuous potential functions of the MD force field. The VDW parameters of the Medusa force field are adopted from the CHARMM force field46, and the EEF1 implicit solvent model is used to model the solvation term47. The hydrogen bond interactions are explicitly modeled with a reaction-like approach48. Screened charge-charge interactions are modeled using the Debye–Hückel approximation by setting the Debye length approximately at 10 Å. The units of mass, time, length, and energy used in our all-atom DMD simulations with an implicit water model are 1 Da, ~50 fs, 1 Å, and 1 kcal/mol, respectively.
Computational Analysis.
Secondary structures are analyzed by using the dictionary secondary structure of protein (DSSP) method49. According to our prior studies, a hydrogen bond was considered to be formed if the distance between backbone N and O atoms was ≤ 3.5 Å and the angle of N-H⋯O ≥ 120°50. Two residues are in contact when the minimum heavy atoms distance between two non-sequential residues is less than 0.55 nm. The residue-pairwise contact map frequency is a two-dimensional matrix representing the propensity between each residue-pair forming contact in all the conformations used for the data analysis. If two peptides were connected by at least one inter-peptide contact, they were treated to belong to the same oligomer. Cluster analysis is performed using the Daura algorithm and a backbone root-mean-square-deviation cutoff of 0.35 nm51. A two-dimensional (2D) free energy surface was constructed using −RT ln P(x, y), where P(x, y) corresponds to the probability of selected reaction ordinates (x and y). If the β-strand segments of an oligomer could form a closed cycle with each β-strand connected by two β-strands neighbors through no less than two hydrogen bonds, this oligomer was treated as a β-barrel oligomer12, 14, 15. Only the conformations from the saturated states were used for the average secondary structure propensity and residue-pairwise contact frequency analysis. The error bars of secondary structure propensities corresponded to the standard deviations of means from 30 independent simulations. All the 600 ns simulation data from each independent DMD trajectory was considered in the β-barrel propensity analysis due to the β-barrel formations were also present in the first 300 ns simulations.
Results and discussion
Monomeric structures of calcitonin variants.
We first investigated the conformational dynamics of hCT, TL-hCT, phCT, and sCT monomers. Thirty independent DMD simulations were performed for each molecular peptide to ensure sufficient sampling. The conformational dynamics of each peptide were monitored by the time evolution of the secondary structure of each residue (one trajectory was randomly selected for each type of peptide, Figure 1). As shown in Figure 1a, the monomeric conformations of hCT were very dynamic and populated with partial helices and transient β-sheets. Dynamical helices were mainly formed by residues 5–20. Residues in the C-terminal predominantly adopted unstructured conformations (i.e., coil and bend). Transient β-sheets appeared around both the central region and C-terminus of hCT. The helical conformations of TL-hCT monomers were more stable with longer duration than hCT, indicating that the Y12L, F16L, and F19L substitutions of hCT enhanced the helix formation (Figure 1b). The C-terminal transient β-sheets observed in hCT were suppressed in phCT monomers (Figure 1c) due to residues in that region being replaced by the corresponding sCT residues (e.g., Y12L, N17H, A26N, I27T, and A31T). Differently, residues 5–20 of sCT formed stable helices without frequent folding/refolding as observed in hCT and hCT analogs (Figure 1d), consistent with prior experimental studies34, 52. The formation of β-sheets was also rare in the sCT.
Figure 1.
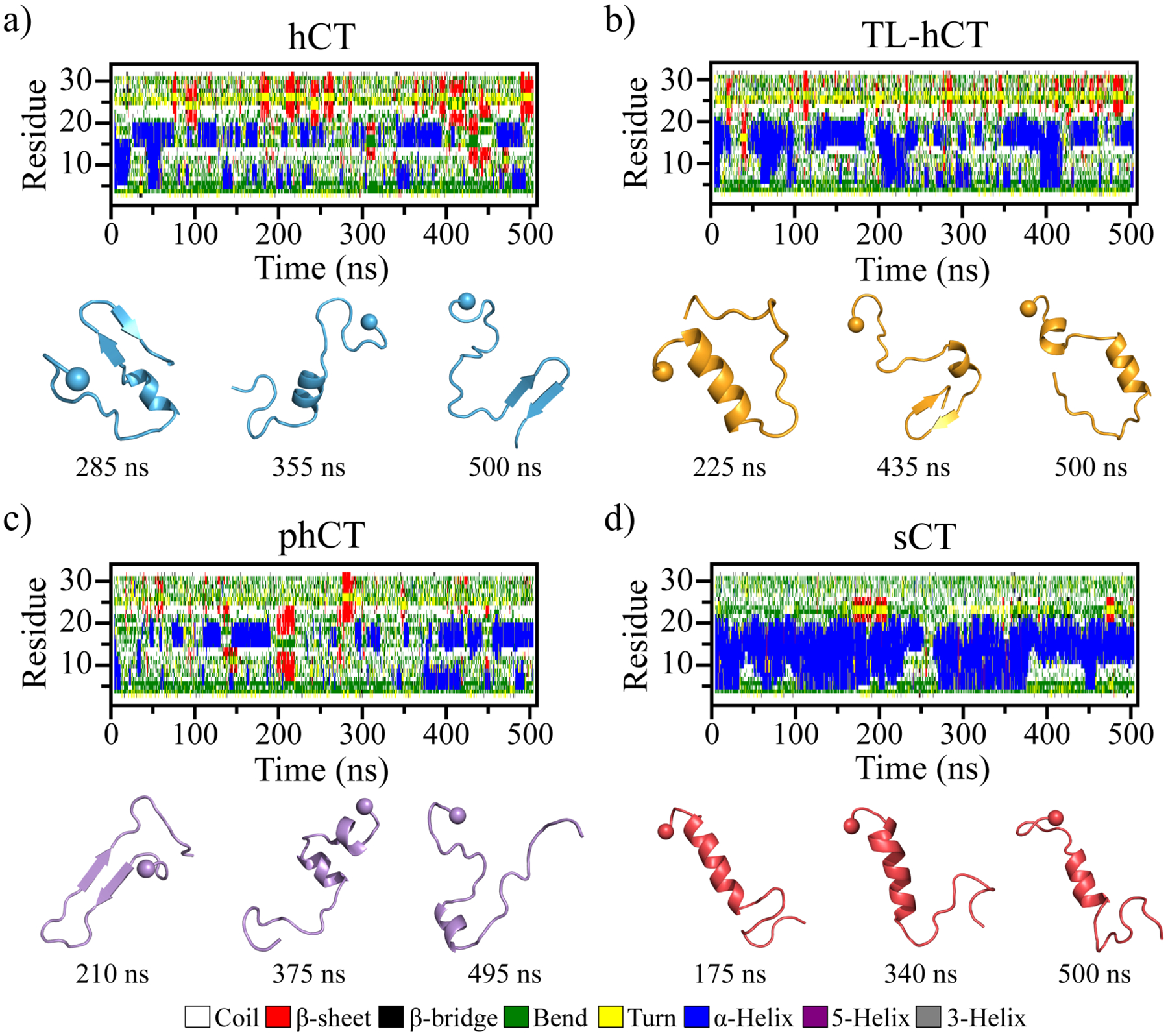
Conformational dynamics of each monomeric calcitonin peptide. Time evolution of the secondary structure of each residue from hCT (a), TL-hCT (b), phCT (c), and sCT (d) peptide in the monomeric peptide simulation is shown on the upper panel. Transient ordered conformations formed along the simulation (the time-stamped blew) are also presented. For each system, one 500 ns DMD trajectory is randomly selected out of thirty independent simulations. The N-terminal Cα atom is highlighted by a sphere for clarity.
The convergence of each molecular system was examined by the structural parameters of radius gyration (Rg) and the secondary structure probability as a function of simulation time (Figures S1&S2). The significant conformational changes in each calcitonin indicated that sufficient sampling was achieved (Figure S1). Not many changes in the time-evolution of ensemble-averaged conformational parameters (e.g., Rg and secondary structure content) over thirty independent simulations during the last 200 ns indicated that all the simulation systems were reasonably converged (Figure S2). Therefore, all calcitonin monomer conformations from the last 200 ns of 30 independent DMD trajectories were used for the equilibrium structural analysis to avoid potential biases in the single trajectory data53, 54. The calculation of average secondary structure propensities revealed that all the four types of calcitonin monomers predominately adopted unstructured conformation, with the population of the random coil and bend structures ~60%−70% (Figure 2a&b). The population of helix structures of hCT (~16.9%) was a little smaller than TL-hCT (~22.2%) and phCT (~19.8%). Helical structures of hCT were mainly formed by residues 4–10 and 14–21 (Figure 2b). Averaged helical propensity of residues 14–21 (~50%) was much stronger than residues 4–10 (~25%) in hCT. The residue-pairwise contact frequencies formed among main-chain atoms also displayed two helical patterns along the diagonal (Figures 2c&S3a). Similar helical regions were also observed by prior solid-state NMR spectra measurements31, 34, 55. The high-resolution solid-state 13C NMR spectroscopy measurement suggested that a local helical form was present around G10, whereas the regions around residues F22, A26, and A31 were dominant with the random coil in the hCT monomer at pH 3.331. An analysis of the NMR restraints violations together with the angular order parameters S(φ) and S(ψ) of hCT monomers indicated that residues 13–21 were populated with helix34. The aromatic interactions caused residues Y12, F16, and F19 of hCT aligning on the same side of the helix, resulting in an amphiphilic helix was also observed in prior experimental measurement2.
Figure 2.
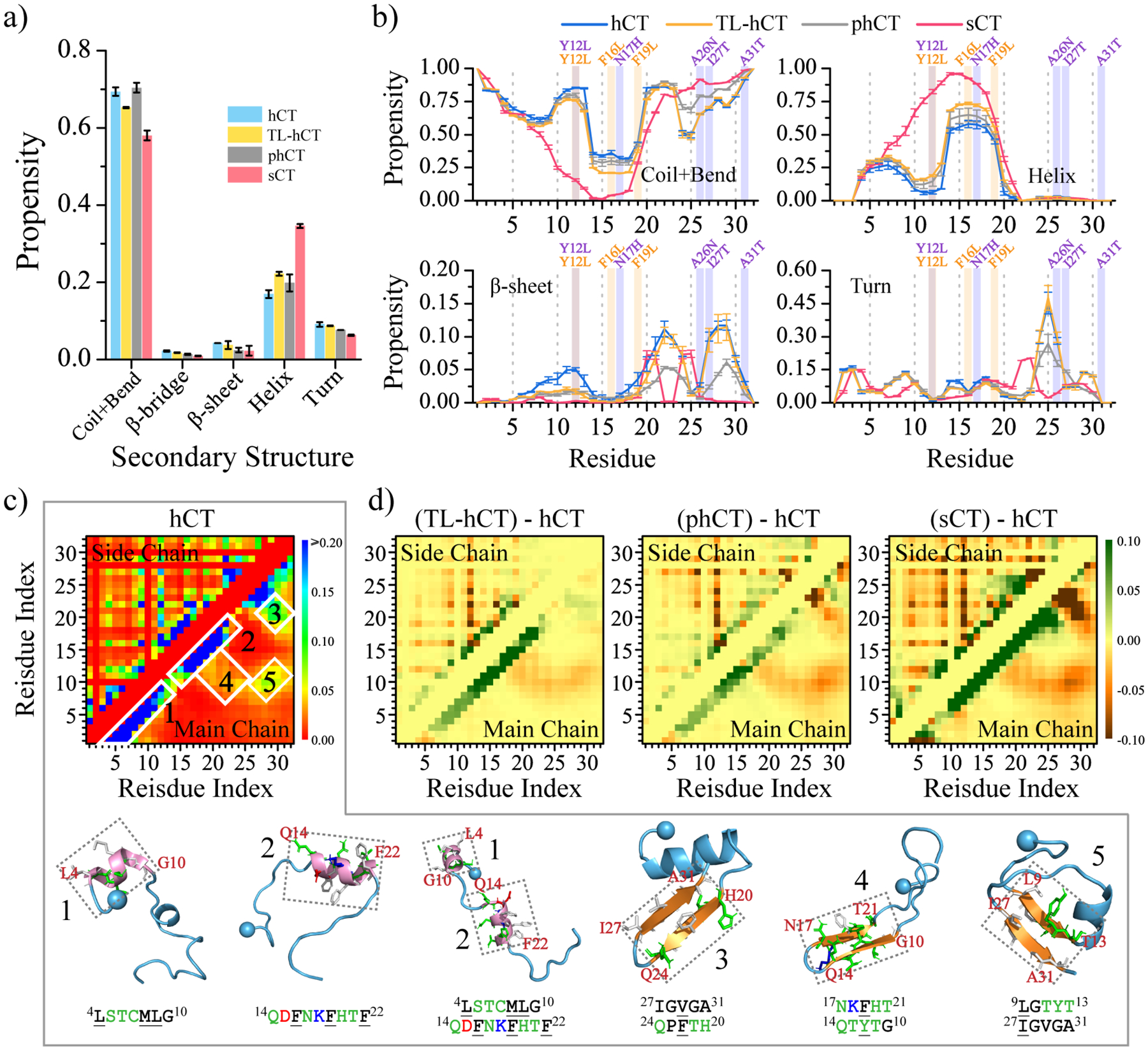
Secondary structure analysis for each calcitonin monomer. (a) Average secondary structure propensity in the monomers of each calcitonin. (b) Probability of each residue adopting different secondary structures. The mutation sites in TL-hCT and phCT are labeled in orange and purple. (c) The frequency of intra-molecular residue-pairwise contact formed between atoms from main-chain (upper diagonal) and side-chain (lower diagonal). The representative structured contact patterns and their corresponding structures (selected according to the contact frequency) are labeled and presented as 1–5. (d) The residue-pairwise contact frequency differences of TL-hCT, phCT, and sCT with respect to the wild-type hCT are calculated by subtracting each corresponding residue-pairwise contact propensity of hCT from the hCT derivatives. Only the last 200 ns data from 30 independent 500 ns DMD simulations are used for the above analysis. The error bars of secondary structure propensities correspond to the standard deviations of means from 30 independent simulations.
The aggregation-resistant mutants of TL-hCT and phCT didn’t alter the helical regions but enhanced the helical propensities of the corresponding residues of hCT (Figures 2b&S3b–c). The NMR measurement of TL-hCT monomers indicated that residue G10 was in the helical state but residues F22, A26, and A31 featured unstructured formations28. NMR spectroscopy demonstrated helical structures in phCT monomer spanned residues 4–21, and other residues mainly adopted random coil2. The residue-pairwise contact frequency difference maps of TL-hCT and phCT with respect to the wild-type hCT also showed that the helical patterns formed by residues 4–21 were enhanced (Figure 2d). The backbone Ca root-mean-square deviation (RMSD) of the helical region (residues 3–22) with respect to the NMR phCT structure (PDBid: 2jxz2) had an average value of ~0.40 nm (Figure S4a–d). Averaged helical conformations of sCT (~34.5%) were much larger than hCT (~16.9%), in agreement with prior experimental data34, 43. Residues 9–19 of sCT displaying high propensities (Figures 2b&S3d) to form helices in our simulations were also observed in previous NMR measurement2, 34. Helices of sCT (residues 3–22) were well maintained with the backbone RMSD mostly within 0.25 nm compared to the NMR structure (PDBid:2glh43) (Figure S4e–h). In addition, the phCT monomers had more helical content than hCT but smaller than sCT also agreed with the CD spectra2, 7, 56.
Transient β-sheets of hCT were mainly formed by residues 6–14, 16–24, and 26–31, where were stabilized by hydrophobic and aromatic interactions among residues 6–31 (Figures 2c&S3a). Conformational analysis showed that the Y12L, F16L, and F19L substitutions reduced the β-sheet formations in residues 6–14 and enhanced more helical formation for residues 4–10 and 14–21 (Figure 2d). The observation can be explained by the fact that leucine (L or Leu) was more favorable with helix but phenylalanine (F or Phe) preferred to form β-sheet57, 58. Residue-pairwise contact map demonstrated that the transient β-hairpin formation formed by residues 10–21 (snapshot 4 in Figure 2c) was inhibited in TL-hCT due to the enhanced formation of helical conformations (Figure 2d). The anti-aggregation mutants in the TL-hCT didn’t alter the β-hairpin formations around residues 19–31 (Figures 2d&S3b). The Y12L, N17H, A26N, I27T, and A31T replacements of hCT disrupted the interactions among hydrophobic residues from the C-terminal tail (residue 26–31) and residues 9–22, which destabilized the β-sheet conformations and increased the stability of helical formations in phCT (Figures 2d&S3c). Representative conformations of top eight most populated conformational clusters of each calcitonin peptide could also demonstrate that sCT was more helical than hCT, TL-hCT, and phCT (Figure S5).
Differential oligomerization dynamics of calcitonin variants.
To investigate the self-assembly dynamics and structures of hCT, TL-hCT, phCT, and sCT, we performed aggregation simulations of five peptides for each calcitonin variant (Methods). The time evolution of radius gyration and secondary structure contents indicated that all our molecular systems reached the corresponding steady states of oligomerization during the last 300 ns (Figures S6&S7). Hence, the simulation data from the last 300 ns of each independent DMD trajectory was used for equilibrium analyses of aggregate structures and conformational dynamics. We found that all the five isolated hCT peptides first accumulated into a single helix-rich oligomer followed by a helix-to-sheet conformation conversion (Figures 3a–c&S6a). The β-sheet conformations, very transient in the monomeric hCT (Figure 1a), became the dominant secondary structures in hCT oligomers. The nucleation of β-sheet rich aggregates via helical accumulation was also observed in many other amyloid peptides2, 30, 31, 59, 60. All the TL-hCT and phCT peptides also readily self-assembled into a single oligomer, but their aggregates displayed more helices and fewer β-sheets than hCT oligomers (Figure 3d–f&3g-i). These results suggested that amyloid-resistant mutants of hCT hindered the helix-to-sheet conformational conversion, resulting in more helical formations as observed in prior experiment studies2, 28, 61. Time evolution of the oligomer size of each peptide aggregated into revealed that the hCT, TL-hCT, and phCT oligomers were relatively stable in size without apparent association and dissociation (Figure 3a–i). The self-assembly dynamics of sCT were dramatically different from hCT and its two analogs (Figure 3j–l). Five sCT peptides were able to form helical oligomers, which undergo frequent association and dissociation with dynamics in size (Figure 3k). The oligomeric sCT predominantly adopted helical formations instead of β-sheet consistent with prior CD measurements7, 56. The association and dissociation process of sCT was much more frequent than hCT, TL-hCT, and phCT (Figure 3j–l). Time evolution of the largest oligomer size in each independent DMD simulation and oligomer size probably distribution during the last 300 ns further confirmed the oligomers of sCT were more dynamic in size (Figure S8).
Figure 3.
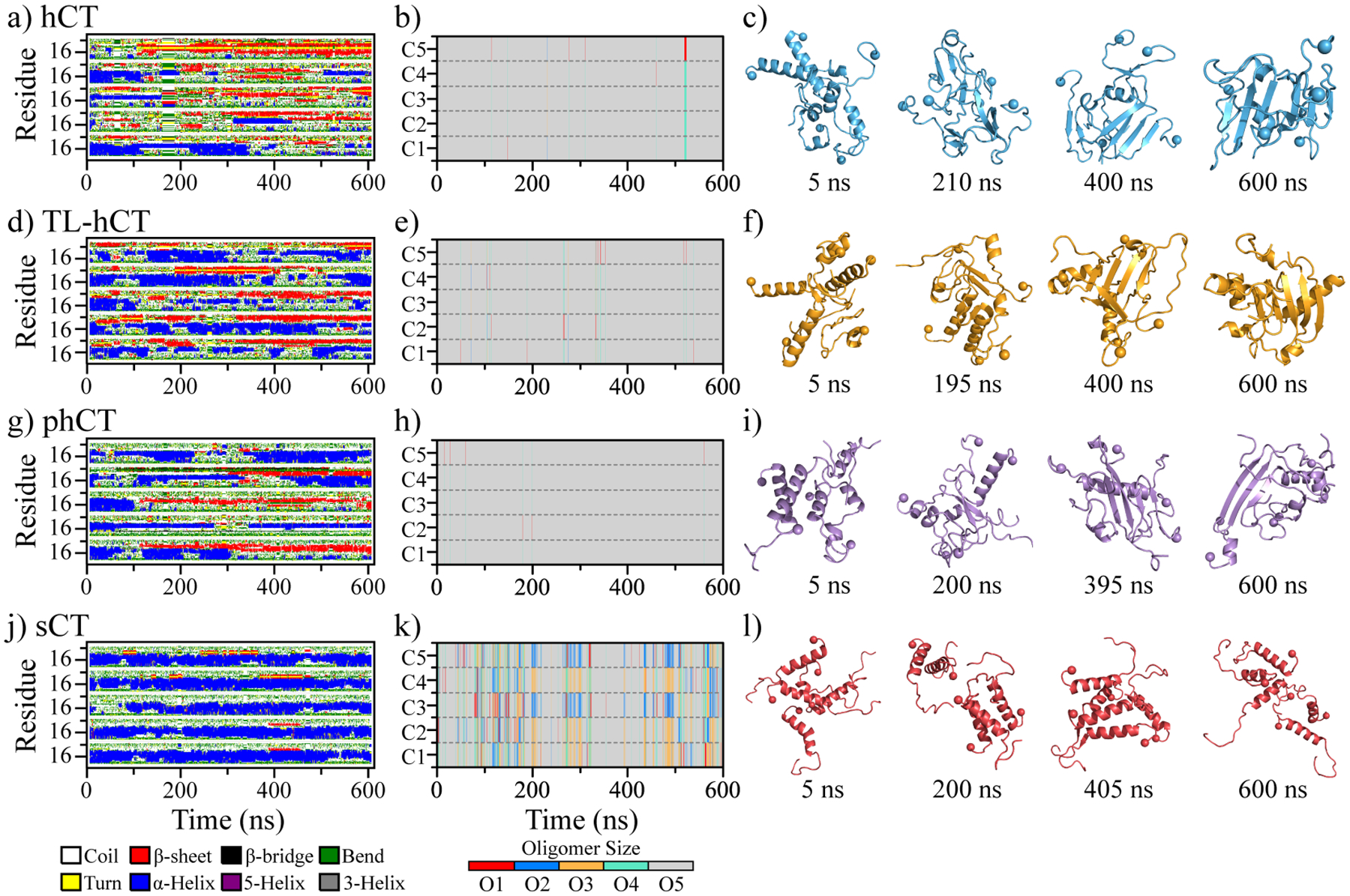
Oligomerization dynamics and conformational changes of each calcitonin variant. The time evolution of secondary structure per residue (a, d, g, j), and the oligomer size into which a peptide aggregated (b, e, h, k) are shown for representative trajectories. Four snapshots with time stamps below (c, f, i, l) are also shown to illustrate transient conformations. For each system, the trajectory is randomly selected out of 30 independent DMD simulations.
Secondary structure content revealed that compared to the monomer oligomerization of hCT enhanced the β-sheet conformations and reduced the helical structures (Figure 4a). For example, the averaged β-sheet and helix contents of oligomeric hCT were ~19.2% and ~10.2% (Figure 4a), but monomeric hCT had ~4.2% β-sheet and 16.9% helix (Figure 2a). Oligomeric hCT also featured two helical regions around residues 4–10 and 14–21 with a similar averaged propensity of ~25% (Figure 4a&b). Meanwhile, the helical propensity of residues 14–21 in the hCT monomer (~50%) was much larger than in hCT oligomers (~25%). Unlike hCT monomers with relatively weak β-sheet propensities (Figure 2a&b), the β-sheet propensities of residues 10–31 in oligomeric hCT were ~15%−45% (Figure 4b). Residues 19–31 displayed a strong tendency to form a typical β-strand-turn-β-strand motif with the β-strand propensity of ~33%, indicating C-terminal residues may adopt a β-hairpin conformation (Figure 4b). Residues Y12, F16, F19, and F22 displayed significantly strong β-sheet propensity suggesting these aromatic residues played critical roles in the fibrillization of hCT, consistent with prior numerous experimental and computational studies19, 35, 36, 62. For example, the aggregation propensity and cytotoxicity of hCT derivative with Y12 mutation were much weaker than the wild-type hCT19. Single-crystal X-ray experiment demonstrated that fragments 15–19 of hCT could independently aggregate into cross-β fibrillar structures35. Differently, the average secondary structure content of sCT oligomers was predominantly helix (~40.1%), and β-sheet formation was rare (~1.8%) (Figure 4a), which was similar to the sCT monomers (Figure 2a). Apart from residues 4–21 populated with helical conformations in the sCT aggregates, other residues mainly adopted the unstructured conformations (Figure 4b). The initially helical aggregates underwent helix-to-sheet conformational conversion in hCT, but not in sCT. The averaged β-sheet and helix content of each independent trajectory showed all the trajectories of sCT were populated with helical formations further confirmed that (Figure 4c). Indeed, the sCT as the non-amyloidogenic peptide without fibrillar propensity was also supported by numerous prior experiments2, 16. Overall, the self-assembly of hCT rendered the loss of helical conformations along with the amplifications of β-sheet structures in monomers (Figure 4d).
Figure 4.
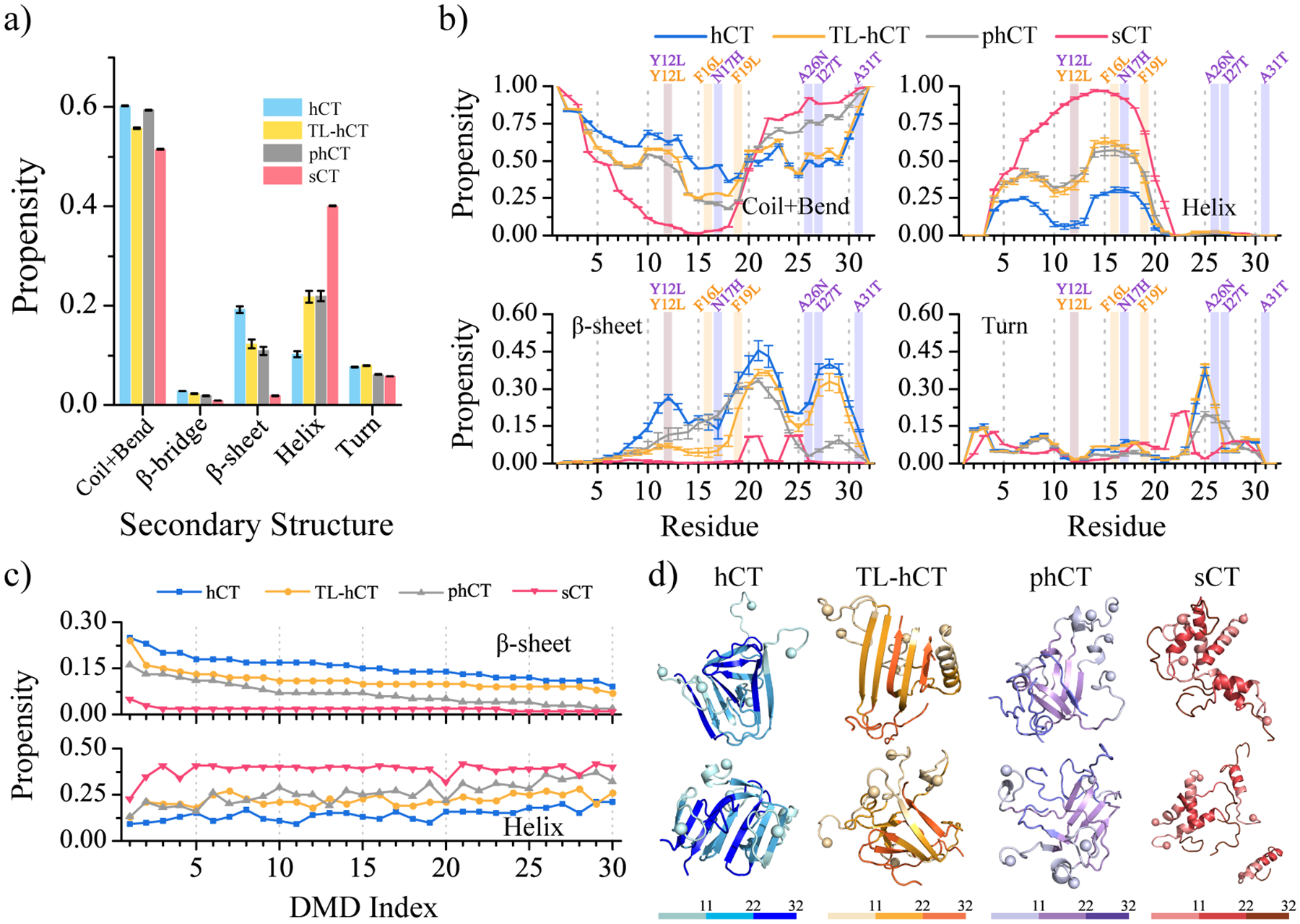
Secondary structure analysis for calcitonin oligomers. (a) The secondary structure contents in oligomers formed by hCT, TL-hCT, phCT, and sCT. (b) Probability of each residue adopting different secondary structures. The mutation sites in TL-hCT and phCT are labeled in orange and purple. The averaging was calculated over all 30 independent simulations during the last 300 ns. The error bars of secondary structure propensities correspond to the standard deviations of means from 30 independent simulations. (c) Simulation trajectories are sorted according to the β-sheet content from high to low, and the corresponding trajectories’ helical content is shown below. (d) Snapshot structures with high β-sheet contents from the top ranked trajectories are also shown.
The self-assemblies of TL-hCT and phCT were populated with more helices and fewer β-sheets than wild-type hCT (Figure 4a&c). Both residues 4–10 and 14–21 had stronger helical propensities ranging around ~35%−63% in the aggregates of TL-hCT and phCT (Figure 4b). Despite TL-hCT and phCT oligomers displaying a similar β-sheet content of ~11%, which was much smaller than hCT aggregates. The β-sheet populated regions of TL-hCT and phCT were dramatically different due to the mutating sites being different. Compared to hCT oligomers, the fibrillization-resistant mutations significantly prevented residues 10–18 of TL-hCT and residues 26–31 of phCT from forming β-sheets in oligomers (Figure 4b). However, the other β-sheet abundant regions observed in the hCT aggregates still had strong β-sheet propensities in the self-assemblies of TL-hCT and phCT, albeit a little smaller than that in hCT oligomers (Figure 4b). The Y12L, F16L, and F19L substitutions of hCT preventing the β-sheet conformation around mutant regions through stabilizing the helical formation in the TL-hCT self-assemblies were consistent with prior NMR assay28. The oligomeric structure of phCT suggested residues replacement of Y12L, N17H, A26N, I27T, and A31T rendered β-sheet decrease along with helices increase also agreed with the previous experiment2. These residue replacements leading the oligomers with more helices and fewer β-sheets suggested that residues 26–31 played a critical role in the fibrillization of hCT. The observation that amyloid-resistant mutation sites in TL-hCT and phCT resulted in low β-sheet formations around the corresponding mutation region (Figure 4b&d) suggests that there should be more than one aggregation prone region in hCT.
Direct observation of β-barrel intermediates in the self-assembly of cytotoxic hCT and absence in nontoxic sCT.
The β-barrel oligomers first discovered in the aggregation of an 11-residue fragment from the heat-shock protein αB-crystalline (residues 90–100) by X-ray crystallography were postulated as the potentially cytotoxic oligomers of amyloidosis63. A series of prior computational studies reported the observation of β-barrel intermediates in the aggregation of experimentally-determined, toxic amyloid peptides12, 50, 64, 65. The formation of β-barrel intermediate was also experimentally and computationally supported for full-length Aβ and hIAPP14, 15, 66, 67. Consistent with the toxic β-barrel intermediates hypothesis15, 67, hCT19 could also spontaneously nucleate into β-barrel oligomers (Figure 5). The averaged β-barrel tendency of TL-hCT was lower than the hCT (Figure 5a). No β-barrel oligomers were observed in the self-assemblies of phCT and sCT. The direct observation of β-barrel oligomers during the self-assembly of toxic hCT but not the nontoxic sCT also supported β-barrels as the toxic oligomers in amyloidosis. With well-ordered 3D structures, the β-barrel oligomers might serve as novel therapeutic targets against amyloid-related diseases.
Figure 5.
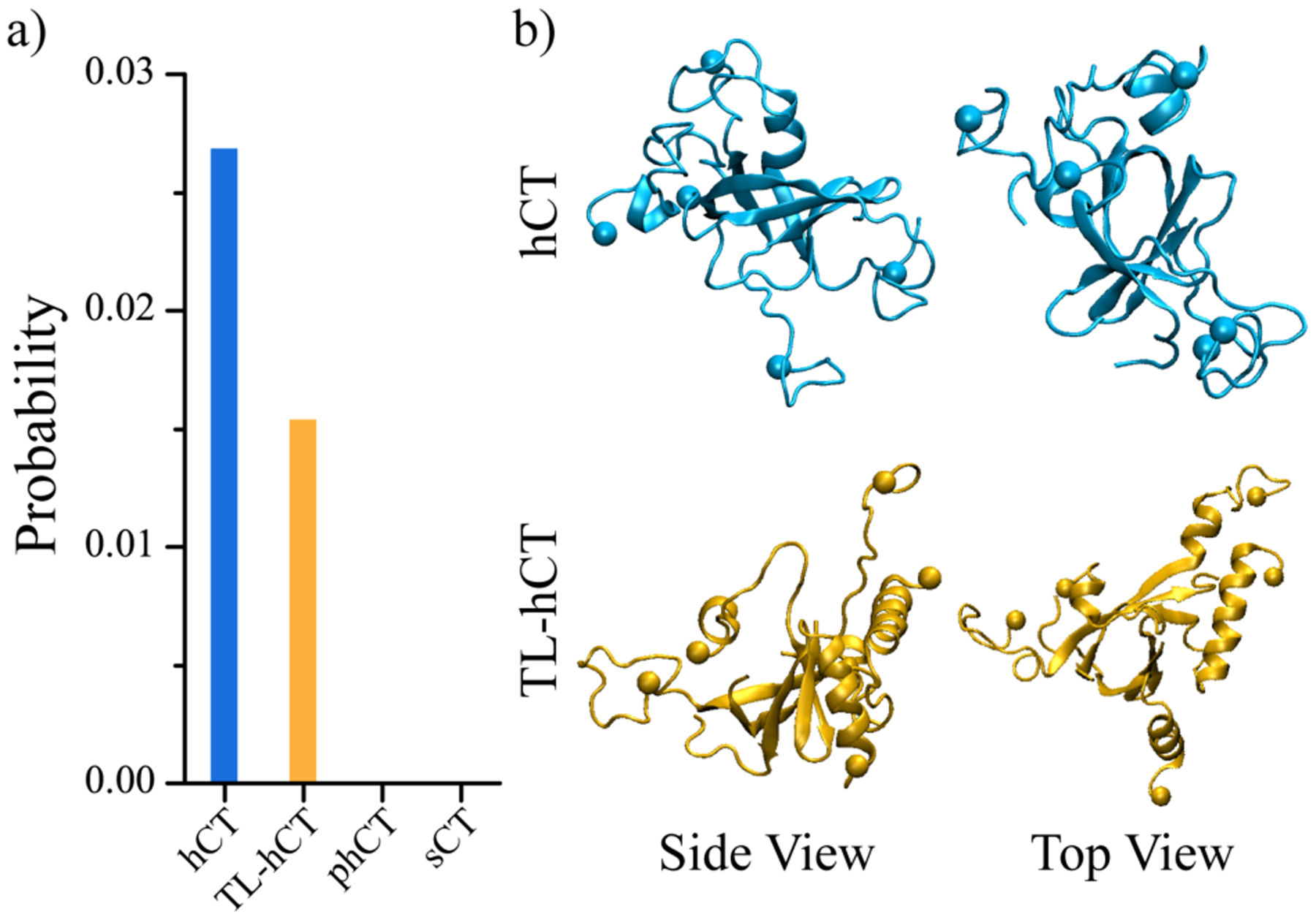
The population of β-barrel oligomers. (a) Probability of β-barrel oligomers observed in the 30 independent DMD trajectories for each calcitonin. (b) One representative β-barrel structure formed by hCT and TL-hCT is also presented in two different views (side and top).
Both the central aromatic and C-terminal hydrophobic residues played important roles in the amyloid aggregation of hCT.
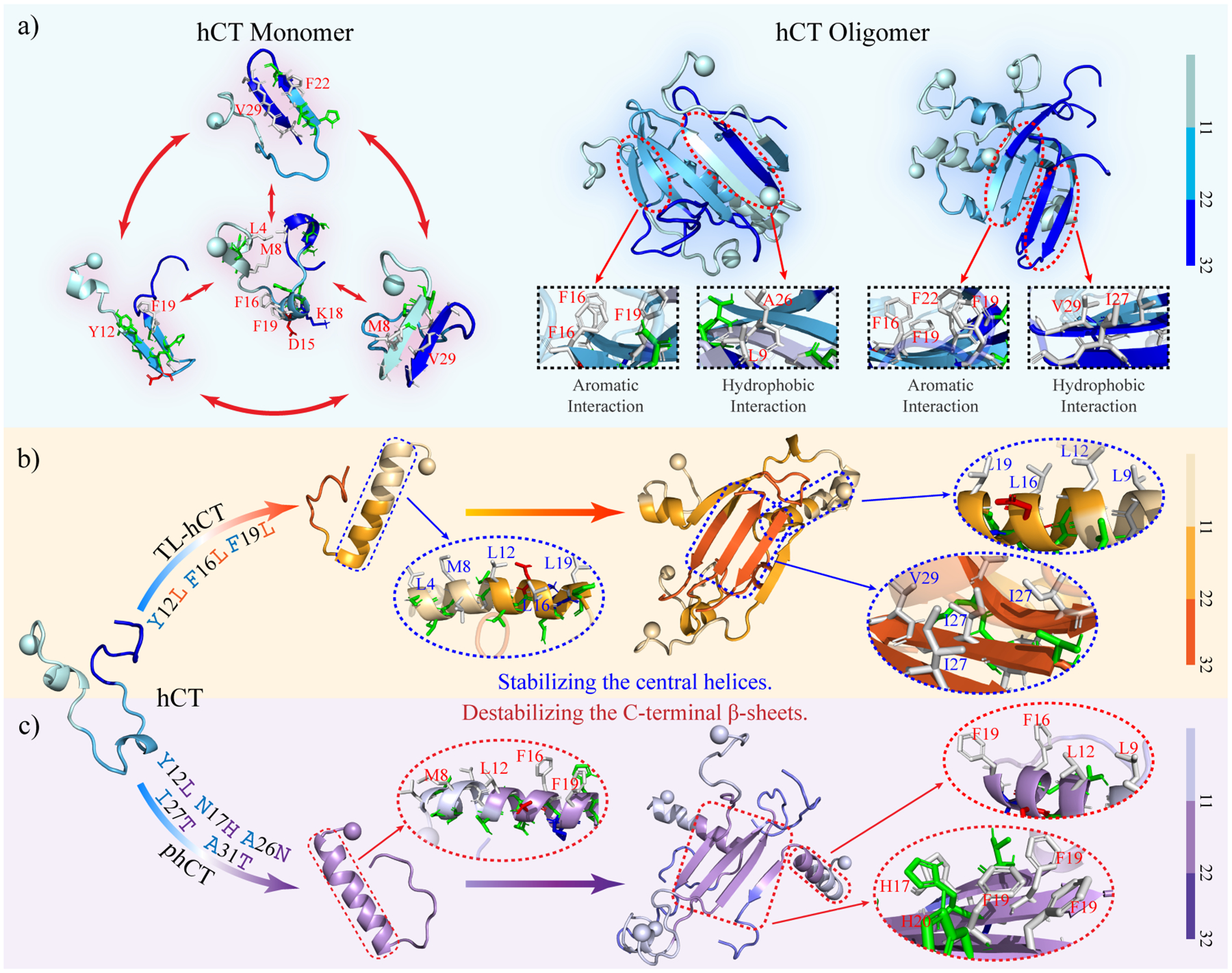
To investigate the key interactions driving the aggregation of calcitonin variants, we calculated intra- and inter-chain contact probabilities between all residue pairs formed by main-chain and side-chain atoms (Figure 6). Intra-chain contact frequency map of hCT self-assemblies featured patterns of two helixes (residues 4–10 and 15–21) and one β-hairpin (residues 19–31) (left panel, Figure 6a). There were multiple inter-chain contact patterns perpendicular to the diagonal for residues 10–31 of hCT, suggesting that residues 10–31 mainly assembled into antiparallel β-sheets. The results agreed with previous experiments30, 31. The side-chain contact frequencies and the total number of inter-chain contacts per residue (Figure 6a right panel) revealed that aromatic residues Y12, F16, F19, H20, and F22 were important in stabilizing the β-sheet patterns. Previous NMR measurements also demonstrated that interactions between these central aromatic residues – i.e., governed by electrostatics between polarized pi systems and also hydrophobic interactions68 – were the main driving force for the nucleation and fibril elongation of hCT28, 60. In addition, the C-terminal hydrophobic residues A25, I27, V29, and A31 participated in forming both intra-chain β-hairpins and inter-chain β-sheets (Figure 6a right panel). The average contact numbers of the C-terminal hydrophobic residues were lower than that of central aromatic residues. Previous HDX-MS measurements of hCT fibrillization suggested the involvement of the central 12–19 fragments was much stronger than C-terminal fragments 26–3229, 60.
Figure 6.
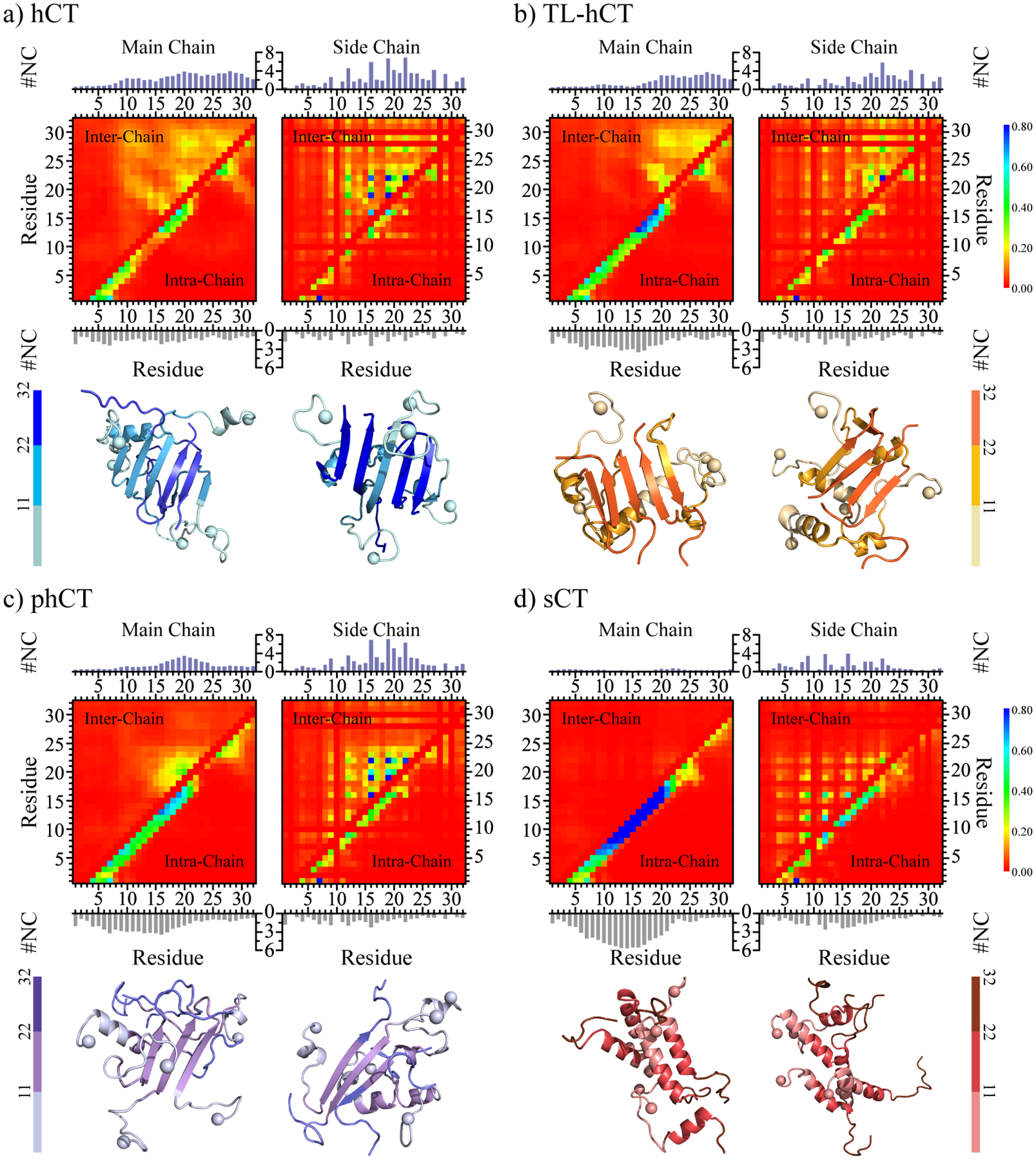
Inter-residue interactions driving the self-assembly of calcitonin variants. The intra-chain (lower diagonal) and inter-chain (upper diagonal) residue-wise contact frequencies of main-chain (left column) and side-chain (right column) atoms for the aggregates of (a) hCT, (b) TL-hCT, (c) phCT, and (d) sCT. The average number of inter-chain (topper) and intra-chain (bottom) contacts per residue formed by atoms from the main-chain (left panel) and side-chain (right panel) are also computed for each molecular system. For each molecular system, two representative aggregate structures selected according to contact frequency are also presented with N-terminal Cα atoms highlighted by spheres. For clarity, residues 1–11, 12–22, and 22–32 are colored from light, to moderate and deep, respectively.
Compared to hCT, the contact map of TL-hCT self-assemblies displayed more helical structures around residues 4–21, but the inter-peptide interaction among residues 10–18 was significantly decreased (Figure 6b). The mutations of Y12L, F16L, and F19L resulted in more helical and less β-sheet formation around the central residue region in TL-hCT aggregates, consistent with prior NMR spectra measurements28. The NMR assays revealed that the fibrillization rates of TL-hCT were significantly slower than that of hCT28, indicating the substitutions of aromatic residues delayed the fibrillization rather than complete inhibition. The fibrillar-resistant mutants didn’t alter the interactions among residues 20–31, and thus, the C-terminal residues of TL-hCT were still abundant with various β-sheets (Figure 6b left panel). Side-chain atoms of residues F22, A26, and V29 had a larger number of intermolecular contacts than other residues, indicating these were important in stabilizing the β-sheets of the TL-hCT self-assemblies (Figure 6b right panel). These results suggested that Y12L, F16L, and F19L enhanced the stability of the central helix, which suppressed the helix-to-sheet transition around the central helical region. The contact map of phCT oligomers showed that the Y12L, N17H, A26N, I27T, and A31T substitutions strongly inhibited the intermolecular interactions around the mutation sites and enhanced the intra-chain interactions around the central helical region (Figure 6c). Therefore, the β-sheet formation around residues 12 and 26–31 was inhibited. The aromatic interactions among F16, F19, and F22 drove the conversion of residues 15–23 into β-sheet structures. The phCT became more helical and had weaker intermolecular interaction than hCT, also in agreement with prior experimental studies2, 61. Mutations of A26N, I27T, and A31T significantly decreased the hydrophobicity of the C-terminus, which not only disrupted interaction among the C-terminus but also between the C-terminal and central hydrophobic residues (Figure 6c).
The main-chain contact map of sCT only displayed one long helical pattern formed by residues 10–20 without any β-sheet patterns (Figure 6d left panel). The side-chain interaction among hydrophobic residues L9, L12, and L16 was observed (Figure 6d right panel), suggesting the dynamic formation of helix-rich sCT oligomers was driven by hydrophobic interactions among the aforementioned residues. The formation of sCT oligomers didn’t induce β-sheet conformational conversion in silico, consistent with numerous prior experimental studies2, 33. Hence, the replacements of central aromatic residues (Y12, F16, and F19) by the leucine and substitutions of C-terminal hydrophobic residues by hydrophilic residues (A26N, I27T, V29S, and A31T) allowed both sCT monomers and oligomers predominantly adopted stable helixes and completely inhibited β-sheet formations. Together, the contact analysis suggested that both the central aromatic and C-terminal hydrophobic residues played critically important roles in the amyloid aggregation of hCT and future design of amyloid-resistant hCT derivatives for clinical application should consider the impact on both regions.
Conformational free energy landscape analysis.
To better understand the conformation characteristics of the oligomers aggregated by hCT, TL-hCT, phCT, and sCT, we calculated the potential of mean force (PMF, the effective free energy landscape) as a function of the helix ratio and β-sheet content. The last 300 ns simulations from 30 independent simulations were used for the PMF calculation. Conformations of hCT oligomers in the free energy basin had the average β-sheet and α-helix contents ~20–30% and ~0–15%, respectively (Figure 7a). The probability distribution of helix and β-sheet content of hCT oligomer also displayed more β-sheet and fewer helix (Figure 7e&f), with the β-sheets populated around both the central aromatic region and C-terminus (Figure 7g&h). The representative snapshots with low free energy further confirmed that both aforementioned regions participated in forming β-sheet. The Y12L, F16L, and F19L residue replacements rendered the TL-hCT oligomer more helical along with the decrease of β-sheet (Figure 7e&f). For example, the self-assemblies of TL-hCT with low free energy were most distributed around regions with β-sheet contented less than 20% and helical ratio ~20%−30% (Figure 7b). The representative TL-hCT structures (snapshots in Figure 7b) and the β-sheet propensity distribution (Figure 7g&h) suggested that β-sheets were more populated in the C-terminus than in the central region due to the substitutions enhanced the stability of the central helix. The low free energy regions of phCT aggregates were even more helical (~20%−40%) and had less β-sheet content (~0–10%) (Figure 7c). The central region of phCT had a stronger β-sheet tendency than C-terminus (Figure 7g&h). Oligomers of sCT were predominantly adopted with high helical content (~35%−45%) and relatively rare β-sheets (less than 2%) (Figure 7d). The probability distribution of inter-chain and intra-chain hydrogen bonds formed by main-chain atoms was also analyzed (Figure S9). Overall, the non-amyloidogenic sCT aggregates were more populated with intra-chain hydrogen bonds due to the abundance of the helix, but amyloidogenic hCT oligomers were more abundant with inter-chain hydrogen bonds corresponding to forming intermolecular β-sheet (Figure S9a&b). In addition, due to the oligomer of sCT being dynamic in size with frequent association and dissociation (Figure S8), the self-assemblies of sCT displayed larger radius gyration than hCT aggregates. The anti-amyloid mutants resulted in the TL-hCT and phCT oligomers with fewer inter-chain hydrogen bonds and more intra-chain hydrogen bonds than hCT assemblies.
Figure 7.
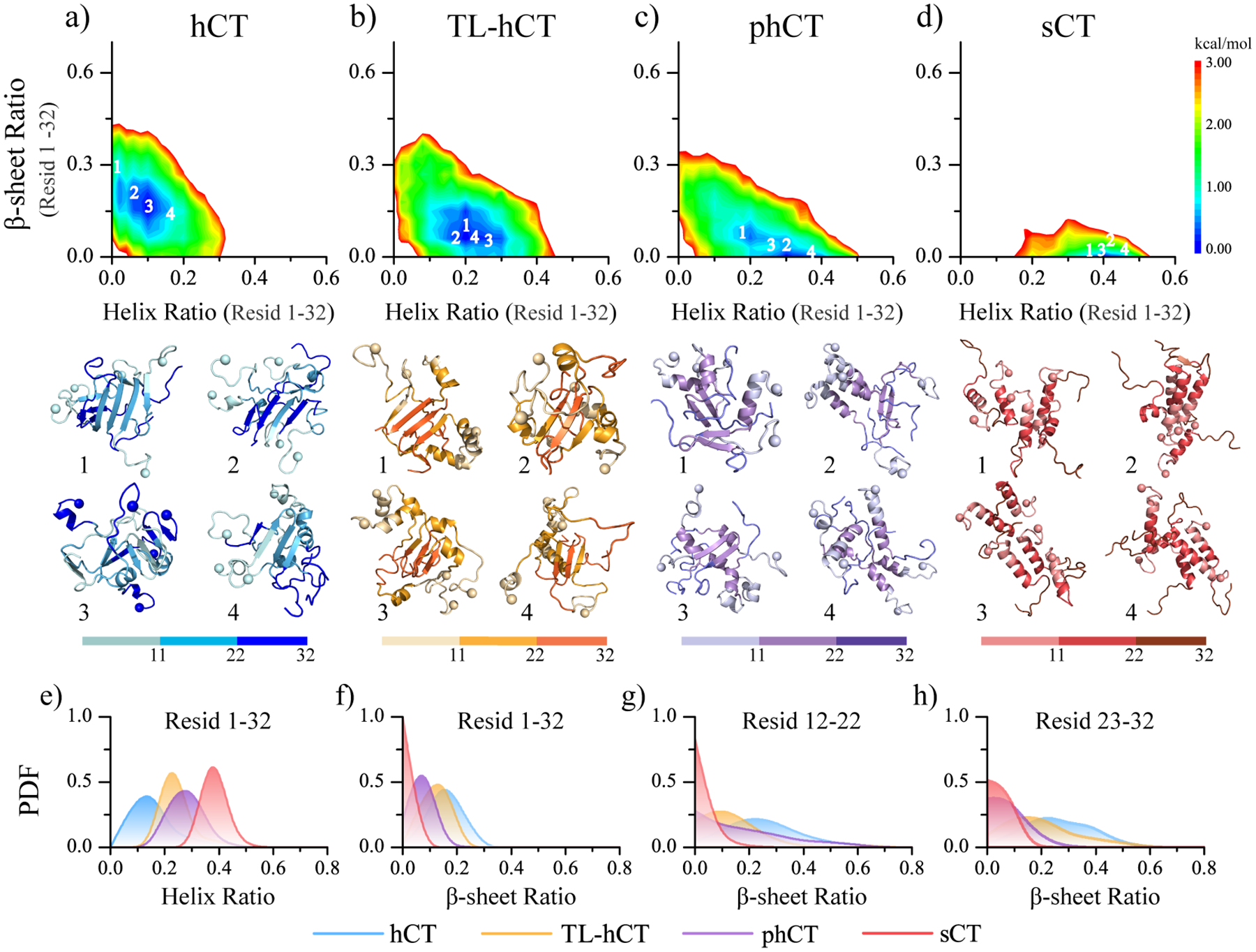
Conformational free energy landscape analysis. The conformational free energy landscape as a function of the helix and β-sheet contents for the self-assemblies aggregated by hCT (a), TL-hCT (b), phCT (c), and sCT (d) peptides. For each molecular system, four representative structures with the helix and β-sheet contents featuring low free energy (labelled as 1–4 on the free energy landscape surface) are also presented. The N-terminal Cα atoms are highlighted as a sphere for clarity. The probability distribution as a function of average helix content (e) and the β-sheet ratio of the whole peptide (f), as well as the β-sheet ratio around the central helical region (residues 12–22) (g) and C-terminus (residues 23–32) (h) in each calcitonin variants oligomer are also present.
A proposed hCT amyloid aggregation mechanism.
The hCT monomer is a typical IDP, with high conformational dynamics. It had less helical content than TL-hCT, phCT, and sCT. The central helical conformations were mainly maintained by the local aromatic interactions among residues Y12, F16, F19, H20, and F22 (Figures S3a&2c). On the other hand, aromatic residues of phenylalanine and tyrosine were more frequently present in β-sheet than a-helix in proteins,57, 58 suggesting both the phenylalanine and tyrosine preferred to form β-sheets than a-helixes. Indeed, transient β-strand was observed in the central region, and these aromatic residues also stabilized the β-sheet structures with the rest of the protein (Figure 8a). Thus, substituting the aromatic residues Y12, F16, and F19 with leucine (with a stronger helical tendency57, 58) promoted the helix propensity and weakened the β-sheet propensity in the central region. However, since replacing central aromatic residues with hydrophobic leucine did not abolish the corresponding interactions that stabilized transient β-sheets (Figure 8b), TL-hCT were able to form β-sheets in the aggregates. Mutating the hydrophobic residues to hydrophilic amino acids (including A26N, I27T, and A31T) around the C-terminus disrupted the hydrophobic interactions stabilizing the transient β-sheets between the central region and C-terminus, rendering phCT adopting more stable helical formations (Figure 8c). β-sheets in oligomers were mainly stabilized by interactions among aromatic and hydrophobic residues from the central region and C-terminus, suggesting both central aromatic and C-terminal hydrophobic residues were critically important in the fibrillization of hCT. The C-terminal tail of phCT mostly adopts unstructured formations. With the central aromatic residues mutant to the more helical leucine amino acids (Y12L, F16L, and F19L) and substitutions of the C-terminal hydrophobic residues replaced by hydrophilic residues (A26N, I27T, V29S, and A31T), the sCT predominantly adopted helix, and the β-sheet formations were nearly completely inhibited. Overall, our results suggested the strategies of the design of non-amyloidogenic hCT analogs included enhancing central helix stability and decreasing C-terminal hydrophobicity.
Figure 8.
Proposed mechanism of hCT forming β-sheet rich aggregates. (a) The hCT monomers are dynamic and mainly adopted unstructured conformations with partial helix and transient β-sheet structures. Oligomers of hCT are abundant in diverse β-sheets formed by the central region and C-terminus of hCT. The interactions among aromatic and hydrophobic residues from the central region and C-terminus stabilize β-sheets and promoted the helix-to-sheet transition. (b) The Y12L, F16L, and F19L substitutions enhance the helix propensity and decrease the helix-to-sheet conformational conversion, because the intrinsic helical tendency of leucine amino acid is stronger than aromatic tyrosine and phenylalanine. Oligomeric β-sheets of TL-hCT are mainly formed by C-terminal residues. (c) The Y12L, N17H, A26N, I27T, and A31T replacements disrupt the interaction between the central region and C-terminus that stabilize transient β-sheet in monomers and protect the central helix of phCT monomer forming conformational convert. The β-sheet formations of phCT oligomers mainly span central region stabilized by interactions among aromatic residues. The C-terminal tail of phCT mostly adopts unstructured conformation.
Conclusion
We systematically studied the self-assembly of hCT, TL-hCT, phCT, and sCT through multiple long-timescale all-atom DMD simulations. Prior computational studies have shown the secondary structure tendency of some peptides may depend on the force field69–71. Therefore, whether or not a computational study could recapitulate experimental measurements is the ultimate test of the method. The predictive power of DMD with Medusa force field has been demonstrated in our previous studies, such as ab initio protein folding38, 72 and conformational dynamics of large multi-domain proteins73, 74. Using the same method, we were able to recapitulate available experimental measurements in terms of monomer structures and aggregation propensities of different calcitonin variants. Specifically, our results demonstrated that the hCT monomer was intrinsically disordered with dynamic formation of helices. The helical structures of sCT monomers were more abundant and stable than hCT monomers, as observed in prior experimental measurements34, 52. Oligomers of hCT were populated with β-sheets conformations around residues 10–31. Self-assembly dynamics analysis revealed that hCT monomers readily self-assembled initially into helix-rich oligomers, followed by a conformational conversion into β-sheets. The sCT, on the other hand, assembled into dynamic helix-rich oligomers with frequent association and dissociation. There were no helix-to-sheet conversion in sCT, in agreement with numerous studies2, 19, 33, 35, 36, 62. Residue-wise contact frequency analysis demonstrated that interactions among central aromatic residues (Y12, F16, F19, and F22) and C-terminal hydrophobic residues (A25, I27, V29, and A31) played important roles in stabilizing transient β-sheet in monomers and also promoting helix-to-sheet conversions in aggregates of hCT. The Y12L, F16L, and F19L substitutions increased the helical propensity and weakened the β-sheet propensity of the central region, but failed to disrupt interactions with the hydrophobic residues in the C-terminus that stabilized the transient β-sheets between the two adjacent regions. In phCT, hydrophilic mutations of N17H, A26N, I27T, and A31T mainly prevented C-terminal residues from forming β-sheets, but Y12L only partially weaken the β-sheet propensity of the central regions; and thus the amplification of β-sheets was observed in phCT aggregates. Together, mutations in TL-hCT and phCT only locally suppressed β-sheet formations in central or C-terminal regions. Only in sCT, mutations in both central and C-terminus stabilized the helixes, destabilized the aggregates, prevented the helix-to-sheet conversion, and thus, inhibiting amyloid fibrillization. The aggregation of hCT limits the bioavailability and therapeutic activity of hCT peptides3, 9, 17, 18 and increases the risk of MTC disease10, 11. The β-barrel intermediates observed in the aggregation of numerous toxic amyloid disease-related peptides were postulated as the potentially cytotoxic oligomers12, 14, 15, 50, 63–67. Interestingly, the β-barrel oligomers were only observed in the self-assemblies of hCT rather than the nontoxic sCT aggregates. Overall, our theoretical insights offer a complete picture of the aggregation mechanism of full-length hCT at the molecular level and reveal the importance of both central and C-terminal regions in amyloid aggregation. Our work suggests that the design of amyloid-resistant hCT derivatives for clinical application should consider the impact on both regions and also take into account the conformational amplification of even transient β-sheets in monomers upon aggregation.
Data and Software Availability.
DMD simulation engine is available at Molecules In Action, LLC. (www.moleculesinaction.com). Initial conformations, input parameter and topology files for DMD simulation, and representative DMD output trajectories for each system are available (https://doi.org/10.5281/zenodo.7109325).
Supplementary Material
Acknowledgments.
This work was supported in part by the National Natural Science Foundation of China under the grant no. 11904189 (Y.S.), K. C. Wong Magna Fund in Ningbo University, China (Y.S.), NSF CBET-1553945 (F.D.), and NIH R35GM145409 (F.D.). Computer simulations were supported by the multi-scale computational modeling core of NIH P20GM121342. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH, and NSF.
Footnotes
The authors declare no competing financial interest.
Supporting Information: The Supporting Information is available free of charge on the website. The convergence assessments of each monomeric calcitonin simulation (Figures S1–2); residue-pairwise contact frequency of each calcitonin monomer (Figure S3); root-mean-square deviation of phCT and sCT corresponding to the NMR determined structures (Figure S4); representative structures of each calcitonin monomer (Figure S5); equilibrium analysis of each calcitonin oligomerization simulation (Figures S6–7); oligomer size analysis for each calcitonin (Figure S8); conformational analysis of each oligomer formed by each calcitonin (Figure S9) (PDF).
References.
- 1.Chambers TJ; Magnus CJ, Calcitonin alters behaviour of isolated osteoclasts. J Pathol 1982, 136, 27–39. [DOI] [PubMed] [Google Scholar]
- 2.Andreotti G; Vitale RM; Avidan-Shpalter C; Amodeo P; Gazit E; Motta A, Converting the highly amyloidogenic human calcitonin into a powerful fibril inhibitor by three-dimensional structure homology with a non-amyloidogenic analogue. J Biol Chem 2011, 286, 2707–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaidi M; Inzerillo AM; Moonga BS; Bevis PJ; Huang CL, Forty years of calcitonin--where are we now? A tribute to the work of Iain Macintyre, FRS. Bone 2002, 30, 655–663. [DOI] [PubMed] [Google Scholar]
- 4.Naot D; Musson DS; Cornish J, The Activity of Peptides of the Calcitonin Family in Bone. Physiol Rev 2019, 99, 781–805. [DOI] [PubMed] [Google Scholar]
- 5.Inzerillo AM; Zaidi M; Huang CL, Calcitonin: physiological actions and clinical applications. J Pediatr Endocrinol Metab 2004, 17, 931–940. [DOI] [PubMed] [Google Scholar]
- 6.Wootten D; Miller LJ; Koole C; Christopoulos A; Sexton PM, Allostery and Biased Agonism at Class B G Protein-Coupled Receptors. Chem Rev 2017, 117, 111–138. [DOI] [PubMed] [Google Scholar]
- 7.Gaudiano MC; Colone M; Bombelli C; Chistolini P; Valvo L; Diociaiuti M, Early stages of salmon calcitonin aggregation: effect induced by ageing and oxidation processes in water and in the presence of model membranes. Biochim Biophys Acta 2005, 1750, 134–145. [DOI] [PubMed] [Google Scholar]
- 8.Schneider D; Hofmann MT; Peterson JA, Diagnosis and treatment of Paget’s disease of bone. Am Fam Physician 2002, 65, 2069–2072. [PubMed] [Google Scholar]
- 9.Colman E; Hedin R; Swann J; Orloff D, A brief history of calcitonin. Lancet 2002, 359, 885–886. [DOI] [PubMed] [Google Scholar]
- 10.Koopman T; Niedlich-den Herder C; Stegeman CA; Links TP; Bijzet J; Hazenberg BPC; Diepstra A, Kidney Involvement in Systemic Calcitonin Amyloidosis Associated With Medullary Thyroid Carcinoma. Am J Kidney Dis 2017, 69, 546–549. [DOI] [PubMed] [Google Scholar]
- 11.Khurana R; Agarwal A; Bajpai VK; Verma N; Sharma AK; Gupta RP; Madhusudan KP, Unraveling the amyloid associated with human medullary thyroid carcinoma. Endocrinology 2004, 145, 5465–5470. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y; Kakinen A; Xing Y; Faridi P; Nandakumar A; Purcell AW; Davis TP; Ke PC; Ding F, Amyloid Self-Assembly of hIAPP8–20 via the Accumulation of Helical Oligomers, alpha-Helix to beta-Sheet Transition, and Formation of beta-Barrel Intermediates. Small 2019, 15, e1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke PC; Pilkington EH; Sun Y; Javed I; Kakinen A; Peng G; Ding F; Davis TP, Mitigation of Amyloidosis with Nanomaterials. Adv Mater 2020, 32, e1901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y; Kakinen A; Xing Y; Pilkington EH; Davis TP; Ke PC; Ding F, Nucleation of beta-rich oligomers and beta-barrels in the early aggregation of human islet amyloid polypeptide. Biochim Biophys Acta Mol Basis Dis 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y; Kakinen A; Wan X; Moriarty N; Hunt CPJ; Li Y; Andrikopoulos N; Nandakumar A; Davis TP; Parish CL; Song Y; Ke PC; Ding F, Spontaneous Formation of beta-sheet Nano-barrels during the Early Aggregation of Alzheimer’s Amyloid Beta. Nano Today 2021, 38, 101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cudd A; Arvinte T; Das RE; Chinni C; MacIntyre I, Enhanced potency of human calcitonin when fibrillation is avoided. J Pharm Sci 1995, 84, 717–719. [DOI] [PubMed] [Google Scholar]
- 17.Kamgar-Parsi K; Tolchard J; Habenstein B; Loquet A; Naito A; Ramamoorthy A, Structural Biology of Calcitonin: From Aqueous Therapeutic Properties to Amyloid Aggregation. Isr J Chem 2017, 57, 634–650. [Google Scholar]
- 18.Rastogi N; Mitra K; Kumar D; Roy R, Metal ions as cofactors for aggregation of therapeutic peptide salmon calcitonin. Inorg Chem 2012, 51, 5642–5650. [DOI] [PubMed] [Google Scholar]
- 19.Ye H; Li H; Gao Z, Y12 nitration of human calcitonin (hCT): A promising strategy to produce non-aggregation bioactive hCT. Nitric Oxide 2020, 104–105, 11–19. [DOI] [PubMed] [Google Scholar]
- 20.Shang H; Zhou A; Jiang J; Liu Y; Xie J; Li S; Chen Y; Zhu X; Tan H; Li J, Inhibition of the fibrillation of highly amyloidogenic human calcitonin by cucurbit[7]uril with improved bioactivity. Acta Biomater 2018, 78, 178–188. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto Y; Nakamuta H; Koida M; Seyler JK; Orlowski RC, Calcitonin-induced anorexia in rats: a structure-activity study by intraventricular injections. Jpn J Pharmacol 1982, 32, 1013–1017. [DOI] [PubMed] [Google Scholar]
- 22.Feletti C; Bonomini V, Effect of calcitonin on bone lesions in chronic dialysis patients. Nephron 1979, 24, 85–88. [DOI] [PubMed] [Google Scholar]
- 23.Muff R; Dambacher MA; Fischer JA, Formation of neutralizing antibodies during intranasal synthetic salmon calcitonin treatment of postmenopausal osteoporosis. Osteoporos Int 1991, 1, 72–75. [DOI] [PubMed] [Google Scholar]
- 24.Cheng B; Gong H; Xiao H; Petersen RB; Zheng L; Huang K, Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta 2013, 1830, 4860–4871. [DOI] [PubMed] [Google Scholar]
- 25.Guo C; Ma L; Zhao Y; Peng A; Cheng B; Zhou Q; Zheng L; Huang K, Inhibitory effects of magnolol and honokiol on human calcitonin aggregation. Sci Rep 2015, 5, 13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang R; Vivekanandan S; Brender JR; Abe Y; Naito A; Ramamoorthy A, NMR characterization of monomeric and oligomeric conformations of human calcitonin and its interaction with EGCG. J Mol Biol 2012, 416, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lantz R; Busbee B; Wojcikiewicz EP; Du D, Flavonoids with Vicinal Hydroxyl Groups Inhibit Human Calcitonin Amyloid Formation. Chemistry 2020, 26, 13063–13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itoh-Watanabe H; Kamihira-Ishijima M; Javkhlantugs N; Inoue R; Itoh Y; Endo H; Tuzi S; Saito H; Ueda K; Naito A, Role of aromatic residues in amyloid fibril formation of human calcitonin by solid-state 13C NMR and molecular dynamics simulation. Phys Chem Chem Phys 2013, 15, 8890–8901. [DOI] [PubMed] [Google Scholar]
- 29.Renawala HK; Chandrababu KB; Topp EM, Fibrillation of Human Calcitonin and Its Analogs: Effects of Phosphorylation and Disulfide Reduction. Biophys J 2021, 120, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naito A; Kamihira M; Inoue R; Saito H, Structural diversity of amyloid fibril formed in human calcitonin as revealed by site-directed 13C solid-state NMR spectroscopy. Magn Reson Chem 2004, 42, 247–257. [DOI] [PubMed] [Google Scholar]
- 31.Kamihira M; Naito A; Tuzi S; Nosaka AY; Saito H, Conformational transitions and fibrillation mechanism of human calcitonin as studied by high-resolution solid-state 13C NMR. Protein Sci 2000, 9, 867–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epand RM; Stahl GL; Orlowski RC, Conformational and biological properties of partial sequences of salmon calcitonin. Int J Pept Protein Res 1986, 27, 501–507. [DOI] [PubMed] [Google Scholar]
- 33.Andreotti G; Motta A, Modulating calcitonin fibrillogenesis: an antiparallel alpha-helical dimer inhibits fibrillation of salmon calcitonin. J Biol Chem 2004, 279, 6364–6370. [DOI] [PubMed] [Google Scholar]
- 34.Amodeo P; Motta A; Strazzullo G; Castiglione Morelli MA, Conformational flexibility in calcitonin: the dynamic properties of human and salmon calcitonin in solution. J Biomol NMR 1999, 13, 161–174. [DOI] [PubMed] [Google Scholar]
- 35.Bertolani A; Pizzi A; Pirrie L; Gazzera L; Morra G; Meli M; Colombo G; Genoni A; Cavallo G; Terraneo G; Metrangolo P, Crystal Structure of the DFNKF Segment of Human Calcitonin Unveils Aromatic Interactions between Phenylalanines. Chemistry 2017, 23, 2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamid AKM; Salvatore JC; Wang K; Murahari P; Guljas A; Ragyanszki A; Owen M; Jojart B; Szori M; Csizmadia IG; Viskolcz B; Fiser B, Oxidatively-mediated in silico epimerization of a highly amyloidogenic segment in the human calcitonin hormone (hCT15–19). Comput Biol Chem 2019, 80, 259–269. [DOI] [PubMed] [Google Scholar]
- 37.Proctor EA; Dokholyan NV, Applications of Discrete Molecular Dynamics in biology and medicine. Curr Opin Struct Biol 2016, 37, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding F; Tsao D; Nie H; Dokholyan NV, Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y; Liu Y; Zhao W; Sun Y, Hydroxylated single-walled carbon nanotube inhibits beta2m21–31 fibrillization and disrupts pre-formed proto-fibrils. Int J Biol Macromol 2021, 193, 1–7. [DOI] [PubMed] [Google Scholar]
- 40.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci Adv 2017, 3, e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emperador A; Orozco M, Discrete Molecular Dynamics Approach to the Study of Disordered and Aggregating Proteins. J Chem Theory Comput 2017, 13, 1454–1461. [DOI] [PubMed] [Google Scholar]
- 42.Zhu C; Beck MV; Griffith JD; Deshmukh M; Dokholyan NV, Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2018, 115, 4661–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreotti G; Mendez BL; Amodeo P; Morelli MA; Nakamuta H; Motta A, Structural determinants of salmon calcitonin bioactivity: the role of the Leu-based amphipathic alpha-helix. J Biol Chem 2006, 281, 24193–24203. [DOI] [PubMed] [Google Scholar]
- 44.Yin S; Ding F; Dokholyan NV, Eris: an automated estimator of protein stability. Nat Methods 2007, 4, 466–467. [DOI] [PubMed] [Google Scholar]
- 45.Yin S; Biedermannova L; Vondrasek J; Dokholyan NV, MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem Inf Model 2008, 48, 1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M, Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem 1983, 4, 187–217. [Google Scholar]
- 47.Lazaridis T; Karplus M, Effective energy functions for protein structure prediction. Curr Opin Struct Biol 2000, 10, 139–145. [DOI] [PubMed] [Google Scholar]
- 48.Ding F; Borreguero JM; Buldyrey SV; Stanley HE; Dokholyan NV, Mechanism for the alpha-helix to beta-hairpin transition. Proteins 2003, 53, 220–228. [DOI] [PubMed] [Google Scholar]
- 49.Kabsch W; Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y; Huang J; Duan X; Ding F, Direct Observation of beta-Barrel Intermediates in the Self-Assembly of Toxic SOD128–38 and Absence in Nontoxic Glycine Mutants. J Chem Inf Model 2021, 61, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daura X; Gademann K; Jaun B; Seebach D; van Gunsteren WF; Mark AE, Peptide folding: When simulation meets experiment. Angew Chem Int Edit 1999, 38, 236–240. [Google Scholar]
- 52.Motta A; Pastore A; Goud NA; Castiglione Morelli MA, Solution conformation of salmon calcitonin in sodium dodecyl sulfate micelles as determined by two-dimensional NMR and distance geometry calculations. Biochemistry 1991, 30, 10444–10450. [DOI] [PubMed] [Google Scholar]
- 53.Shao J; Tanner SW; Thompson N; Cheatham TE, Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J Chem Theory Comput 2007, 3, 2312–2334. [DOI] [PubMed] [Google Scholar]
- 54.Gonzalez-Aleman R; Hernandez-Castillo D; Caballero J; Montero-Cabrera LA, Quality Threshold Clustering of Molecular Dynamics: A Word of Caution. J Chem Inf Model 2020, 60, 467–472. [DOI] [PubMed] [Google Scholar]
- 55.Motta A; Andreotti G; Amodeo P; Strazzullo G; Castiglione Morelli MA, Solution structure of human calcitonin in membrane-mimetic environment: the role of the amphipathic helix. Proteins 1998, 32, 314–323. [PubMed] [Google Scholar]
- 56.Kamgar-Parsi K; Hong L; Naito A; Brooks CL 3rd; Ramamoorthy A, Growth-incompetent monomers of human calcitonin lead to a noncanonical direct relationship between peptide concentration and aggregation lag time. J Biol Chem 2017, 292, 14963–14976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swindells MB; MacArthur MW; Thornton JM, Intrinsic phi, psi propensities of amino acids, derived from the coil regions of known structures. Nat Struct Biol 1995, 2, 596–603. [DOI] [PubMed] [Google Scholar]
- 58.Munoz V; Serrano L, Intrinsic secondary structure propensities of the amino acids, using statistical phi-psi matrices: comparison with experimental scales. Proteins 1994, 20, 301–311. [DOI] [PubMed] [Google Scholar]
- 59.Bauer HH; Aebi U; Haner M; Hermann R; Muller M; Merkle HP, Architecture and polymorphism of fibrillar supramolecular assemblies produced by in vitro aggregation of human calcitonin. J Struct Biol 1995, 115, 1–15. [DOI] [PubMed] [Google Scholar]
- 60.Kanaori K; Nosaka AY, Study of human calcitonin fibrillation by proton nuclear magnetic resonance spectroscopy. Biochemistry 1995, 34, 12138–12143. [DOI] [PubMed] [Google Scholar]
- 61.Chen YT; Hu KW; Huang BJ; Lai CH; Tu LH, Inhibiting Human Calcitonin Fibril Formation with Its Most Relevant Aggregation-Resistant Analog. J Phys Chem B 2019, 123, 10171–10180. [DOI] [PubMed] [Google Scholar]
- 62.Rigoldi F; Metrangolo P; Redaelli A; Gautieri A, Nanostructure and stability of calcitonin amyloids. J Biol Chem 2017, 292, 7348–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laganowsky A; Liu C; Sawaya MR; Whitelegge JP; Park J; Zhao M; Pensalfini A; Soriaga AB; Landau M; Teng PK; Cascio D; Glabe C; Eisenberg D, Atomic view of a toxic amyloid small oligomer. Science 2012, 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun Y; Ge X; Xing Y; Wang B; Ding F, beta-barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-beta Aggregates. Sci Rep 2018, 8, 10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun Y; Kakinen A; Zhang C; Yang Y; Faridi A; Davis TP; Cao W; Ke PC; Ding F, Amphiphilic surface chemistry of fullerenols is necessary for inhibiting the amyloid aggregation of alpha-synuclein NACore. Nanoscale 2019, 11, 11933–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osterlund N; Moons R; Ilag LL; Sobott F; Graslund A, Native Ion Mobility-Mass Spectrometry Reveals the Formation of beta-Barrel Shaped Amyloid-beta Hexamers in a Membrane-Mimicking Environment. J Am Chem Soc 2019, 141, 10440–10450. [DOI] [PubMed] [Google Scholar]
- 67.Wu J; Blum TB; Farrell DP; DiMaio F; Abrahams JP; Luo J, Cryo-electron Microscopy Imaging of Alzheimer’s Amyloid-beta 42 Oligomer Displayed on a Functionally and Structurally Relevant Scaffold. Angew Chem Int Ed Engl 2021, 60, 18680–18687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martinez CR; Iverson BL, Rethinking the term “pi-stacking”. Chem Sci 2012, 3, 2191–2201. [Google Scholar]
- 69.Best RB; Buchete NV; Hummer G, Are current molecular dynamics force fields too helical? Biophys J 2008, 95, L07–L09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Somavarapu AK; Kepp KP, The Dependence of Amyloid-beta Dynamics on Protein Force Fields and Water Models. Chemphyschem 2015, 16, 3278–3289. [DOI] [PubMed] [Google Scholar]
- 71.Lindorff-Larsen K; Maragakis P; Piana S; Eastwood MP; Dror RO; Shaw DE, Systematic validation of protein force fields against experimental data. PLoS One 2012, 7, e32131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV, Discrete Molecular Dynamics: An Efficient And Versatile Simulation Method For Fine Protein Characterization. Journal of Physical Chemistry B 2012, 116, 8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orozco ISY; Mindlin FA; Ma JY; Wang B; Levesque B; Spencer M; Adariani SR; Hamilton G; Ding F; Bowen ME; Sanabria H, Identifying weak interdomain interactions that stabilize the supertertiary structure of the N-terminal tandem PDZ domains of PSD-95. Nat Commun 2018, 9, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamilton GL; Saikia N; Basak S; Welcome FS; Wu F; Kubiak J; Zhang CC; Hao Y; Seidel CA; Ding F; Sanabria H; Bowen ME, Fuzzy supertertiary interactions within PSD-95 enable ligand binding. Elife 2022, 11, e77242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.