

Abstract
A variety of cancers have been found to have chromosomal rearrangements, and the genomic abnormalities often induced expression of fusion oncogenes. To date, a pair of engineered nucleases including ZFNs, TALENs, and CRISPR-Cas9 nucleases have been used to generate chromosomal rearrangement in living cells and organisms for disease modeling. However, these methods induce unwanted indel mutations at the DNA break junctions, resulting in incomplete disease modeling. Here, we developed prime editor nuclease-mediated translocation and inversion (PETI), a method for programmable chromosomal translocation and inversion using prime editor 2 nuclease (PE2 nuclease) and paired pegRNA. Using PETI method, we successfully introduced DNA recombination in episomal fluorescence reporters as well as precise chromosomal translocations in human cells. We applied PETI to create cancer-associated translocations and inversions such as NPM1-ALK and EML4-ALK in human cells. Our findings show that PETI generated chromosomal translocation and inversion in a programmable manner with efficiencies comparable of Cas9. PETI methods, we believe, could be used to create disease models or for gene therapy.
Keywords: CRISPR-Cas9, genome editing, prime editing, genome rearrangement, chromosomal translocation, chromosomal inversion
Graphical abstract
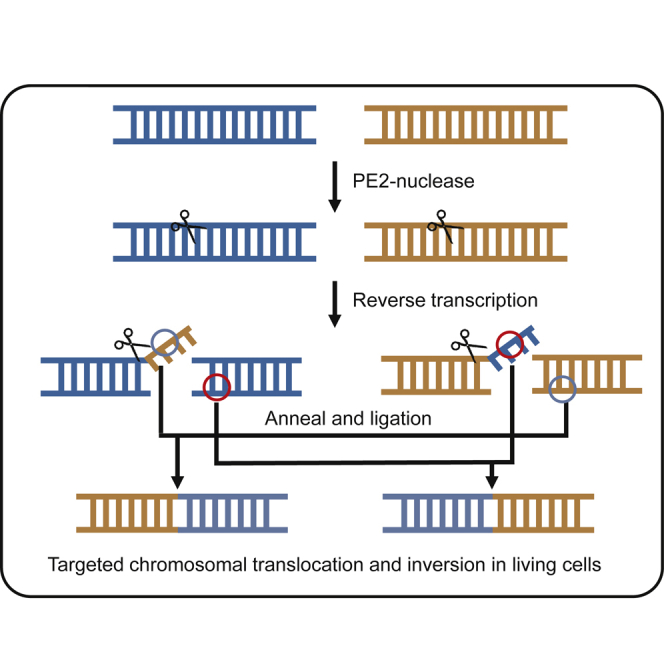
Kweon et al. developed prime editor nuclease-mediated translocation and inversion (PETI), a method for programmable chromosomal translocation and inversion using prime editor 2 nuclease (PE2 nuclease) and paired pegRNA in human cells.
Introduction
Chromosomal rearrangements, including deletions, inversions, duplications, and translocations, are chromosome abnormalities that are induced mainly by the generation of DNA double-strand breaks (DSBs) at two different locations, followed by the rejoining of the DNA ends in a new configuration.1 These structural variations are frequently identified in various types of cancers and are often the underlying cause of tumorigenesis.2,3 To elucidate the effects of such rearrangements, previous studies have introduced targeted chromosomal rearrangements in cells and organisms using a pair of programmable nucleases including zinc finger nucleases, TAL effector nucleases, and CRISPR-Cas9 nucleases.4,5,6,7,8,9,10,11 In particular, Cas9 nuclease with paired guide RNAs (gRNAs) has been used to generate cancer-associated chromosomal rearrangements, such as EML4-ALK, NPM-ALK, CD74-ROS1, KIF5B-RET, EWSR1-FLI1, and AML1-ETO.10,11,12 Two gRNAs targeting two chromosomes can lead to chromosomal translocation by generating DNA DSBs on each chromosome, However, Cas9-mediated translocations are accompanied by additional indel mutations at the fragment junctions, and it is difficult to generate precise translocation or to insert or delete desired sequences at the junction.
Prime editor 2 (PE2) enables the generation of genetic modifications, including all types of substitutions, small insertions, and deletions, as well as combinations of these mutations, at target sites without the formation of DNA double-strand breaks in cultured cells and whole organisms.13,14,15,16,17,18,19,20,21 PE2 consists of Cas9 H840A nickase fused to an engineered Moloney murine leukemia virus reverse transcriptase (RT) and a prime editing guide RNA (pegRNA) that includes a spacer sequence, a primer binding sequence (PBS), and an RT template (RTT) sequence containing the desired edit. Prime editing is composed of (1) the generation of a single-strand break in the non-target strand via Cas9 H840A nickase, (2) hybridization of the PBS in the pegRNA to the 3′ end of the nicked strand, making a DNA/RNA complex; (3) RT-mediated DNA extension of the nicked strand according to the RTT sequence, which generates a 3′-flap containing the intended edit sequence; and (4) incorporation of the 3′-flap sequence into the genome. Prime editing activities are improved by using an additional nicking guide RNA, which leads to the generation of a nick in the non-edited strand.
Recently, advanced prime editor (PE)-based methods, known as Prime-Del22 and PE-Cas9-based deletion and repair (PEDAR),23 have been developed to induce large deletions of up to 10 kb. Both methods involve a pair of pegRNAs targeting opposite strands; in Prime-Del, the pegRNAs are used together with PE2, whereas in PEDAR, they are used with a Cas9 nuclease-RT fusion, here referred to as PE2 nuclease. We reasoned that prime editing systems based on paired pegRNAs could be used to induce not only large deletions but also other chromosomal structural rearrangements like translocations and inversions. Here, we developed the PE2 nuclease-based translocation and inversion (PETI) method, which uses PE2 nuclease and paired pegRNAs to induce programmable translocations and inversions in mammalian cells with high efficiency and purity.
Results
DNA recombination induced by PETI using a fluorescent reporter system
To test the feasibility of using the PETI strategy to induce programmed DNA recombination, we designed two plasmids encoding fluorescent reporters. The first contains a promoter, a HEK4 target site, and the mCherry gene, and the second contains a stop codon, a HEK3 target site, and the eGFP gene. Because the eGFP gene lacks a promoter, reporter-transfected cells can express mCherry but not eGFP (Figure 1A). When PETI induces DNA recombination between the HEK4 and HEK3 target sites of these two plasmids in cells, the promoter moves in front of the eGFP gene, resulting in eGFP expression (Figure 1A). We constructed paired pegRNAs to induce this recombination using the PETI system. The pegRNA that targets the HEK4 site generates a 3′-flap containing the desired sequence (blue circle in Figure 1A), which is complementary to the HEK3 target site at the recombination junction; the pegRNA that targets the HEK3 site generates a 3′-flap that is complementary to the HEK4 target at the recombination junction (Figure 1A, red circle). In contrast to the low expression of eGFP observed when HEK293T cells were transfected with plasmids encoding PE2 and the paired pegRNAs, transfection of plasmids encoding PE2 nuclease and the paired pegRNAs induced higher eGFP expression (Figure 1B). Cas9 nuclease and paired single guide RNAs (sgRNAs) targeting the HEK3 and HEK4 target sites also induced eGFP expression (Figure 1B). Note that PE2, which induces extremely low indel mutations, significantly reduced mCherry expression, because the PE2 protein and the pegRNA complex that bound between the promoter and the mCherry coding sequence could block RNA polymerase elongation. The length of the RTT in the paired pegRNAs targeting the HEK4 and HEK3 sites was then changed from 0 to 35 bp in 5 bp increments to generate 3′-flaps of different lengths; plasmids encoding each of the new pegRNA pairs were transfected with plasmids encoding PE2, PE2 nuclease, or Cas9 nuclease. The length of the 3′ extension did not affect Cas9 nuclease-mediated DNA recombination activity, whereas the frequency of PE2 nuclease-mediated recombination increased as the length of the 3′-flap was gradually increased from 0 to 20 bp, but there was no significant change when it was increased from 20 to 35 bp (Figure 1C). Using the fluorescent reporter, we then investigated the synergistic effects of paired 3′-flaps (Figures 1A and 1D). Transfection of plasmids encoding PE2 nuclease and paired pegRNAs resulted in higher DNA recombination frequencies than those induced by PE2 nuclease, a single pegRNA, and sgRNA (Figure 1D), indicating that paired 3′-flaps have a synergistic effect.
Figure 1.

PETI-mediated translocation in an episomal fluorescent reporter
(A) Overview of the PETI method for generating episomal DNA recombination in human cells. The paired pegRNAs target the HEK3 and HEK4 sites that are incorporated into the two plasmids that make up the episomal fluorescent reporter system. PETI induces the formation of 3′-flaps at each of the target sites, which are complementary to sequences at the other target site, resulting in episomal translocations. (B) Representative fluorescence microscope images showing the absence or presence of eGFP expression, which results from episomal translocations potentially induced by PE2, PE2 nuclease, or Cas9 nuclease. Scale bar represents 100 μM. (C) Effects of 3′-flap length on translocation outcomes. Cells were transfected with plasmids encoding PE2, PE2 nuclease, or Cas9 nuclease and pegRNAs that generated 3′-flaps of different lengths (0–35 bp, increasing in increments of 5 bp), and the percentage of GFP-positive cells was evaluated using fluorescence-activated cell sorting. (D) Effects of combinations of pegRNA and gRNA on translocation outcomes. Cells were transfected with plasmids encoding PE2, PE2 nuclease, or Cas9 nuclease and pegRNA or gRNA pairs, and the GFP-positive cells were evaluated using fluorescence-activated cell sorting. (E) Cells were transfected with plasmids encoding Cas9 nuclease with gRNAs or PE2 nuclease with two types of pegRNA pairs (direct junction guided or 3 bp insertion guided), and the GFP-positive cells were evaluated using fluorescence-activated cell sorting. Data are presented as mean ± SEM (n = 2 biologically independent samples). (F) The relative frequency of sequence reads of amplicons generated using translocation-specific primers. Direct translocation and PETI-guided translocation (3 bp insertion) are shown in blue and red bars, respectively. (G) Results of targeted sequencing of amplicons generated using translocation-specific primers.
To examine the accuracy of the PETI strategy for DNA recombination, we further constructed paired pegRNAs inducing 3 bp (5′-ACG-3′) insertion at the recombination. We first conducted flow cytometry assays to measure the recombination efficiency and found that both paired pegRNAs (direct junction guided and 3 bp insertion guided) induced DNA recombination with comparable efficiency (Figure 1E). Targeted deep sequencing showed that PE2 nuclease with paired pegRNAs inducing direct junction induced DNA recombination with a higher accuracy than Cas9 nuclease (91.7% for PE2 nuclease, 62.8% for Cas9 nuclease) (Figures 1F and S1). Furthermore, we found that paired pegRNAs inducing 3 bp (5′-ACG-3′) insertion at the junction could induced the insertional mutation in episomal reporter plasmids (51.7%) as well as reporter integrated cell line (76.9%) (Figures 1F, 1G, and S2). Although episomal reporter assays may overestimate the efficiency of rearrangements because of the use of multiple copies of plasmids in a single cell, these results showed that PETI indeed induce DNA recombination in a precise and programmable manner.
PETI induces chromosomal translocations in genomic DNA
We next investigated whether PETI induces precise chromosomal translocation between endogenous loci in human cells. We co-transfected plasmids encoding paired pegRNAs for inducing reciprocal translocations between the HEK3 and HEK4 genomic sites and PE2 or PE2 nuclease into HEK293T cells (Figure 2A). For a positive control, we also co-transfected plasmids encoding Cas9 nuclease and sgRNAs targeting the HEK3 and HEK4 sites. We then amplified the expected translocation junctions with reciprocal translocation-specific primer pairs and found that translocations were induced in cells transfected with plasmids encoding PE2 nuclease and paired pegRNAs, as well as Cas9 nuclease and dual sgRNAs, but not in cells transfected with plasmids encoding PE2 and paired pegRNAs (Figure S3A). We analyzed these amplicons by targeted deep sequencing and found that PE2 nuclease induced more precise reciprocal translocation than Cas9 nuclease (43.9% and 23.9% with PE2 nuclease; 11.0% and 0% with Cas9 nuclease at each junction) (Figures 2B–2D). Following this experiment, we designed paired pegRNAs to induce a 3 bp deletion or a 4 or 5 bp insertion (containing an EcoRI or BamHI site) at the junction of the reciprocal translocation and transfected plasmids encoding the pegRNAs and PE2 or PE2 nuclease or Cas9 nuclease and sgRNAs. Translocation junctions were also amplified translocation-specific primer pairs in the presence of PE2 nuclease with these paired pegRNAs. The amplified junction generated by paired pegRNAs designed to insert an EcoRI or BamHI site was digested by EcoRI or BamHI, respectively but that the junction generated by Cas9 nuclease was not (Figure S3B). We then used deep sequencing of PCR-amplified translocation junctions to validate the editing accuracy. At both reciprocal translocation junctions, we revealed that PE2 nuclease was more accurate than Cas9 nuclease (Figures 2C and 2D). The translocation frequencies were also measured by dilution PCR,5,24 and we found that PE2 nuclease induced reciprocal translocation with a similar frequency to Cas9 nucleases (Table 1; Figure S4). These results indicated that PE2 nuclease with appropriate paired pegRNAs can induce reciprocal translocations with desired mutations at the junctions in human cells.
Figure 2.

PETI-mediated translocation at an endogenous genomic locus
(A) Representative schematic showing PETI-mediated translocations at endogenous target sites. PETI induces the formation of 3′-flaps at each of the target sites, which are complementary to sequences at the other target site, resulting in translocations. (B) Targeted sequencing of amplicons generated using translocation-specific primers. The bar charts represent the fraction of reads with direct translocations (blue), translocations with the intended insertion (orange) or deletion (green), and translocations with unwanted indels (gray). (C and D) Representative results of relative frequency of sequence reads in PCR amplicons amplified by translocation-specific primers, either H3F (HEK3 forward) and H4R (HEK4 reverse) or H4F and H3R. Direct translocation and PETI-induced translocation are indicated by blue and red bars, respectively. (E) Schematic overview of multiplex PCR to detect PE2-, PE2 nuclease-, or Cas9 nuclease-mediated translocations. Un-translocated genomic DNA is amplified by primers T1F and T1R or T2F and T2R, the junctions of desired translocations are amplified by T1F and T2R or T2F and T1R, and the junctions of undesired translocations are amplified by T1F and T2F or T2R and T1R. (F) Fraction of sequencing reads of multiplex PCR amplicons generated from desired and undesired PE2-, PE2 nuclease-, or Cas9 nuclease-mediated translocations. Data are presented as mean ± SEM (n = 3 biologically independent samples).
Table 1.
Estimated frequencies of Cas9- or PETI-mediated chromosomal translocations and inversions
| Target sites | Target rearrangements | Types | Junction-1 |
Junction-2 |
||||
|---|---|---|---|---|---|---|---|---|
| Lower, % | Estimated, % | Upper, % | Lower, % | Estimated, % | Upper, % | |||
| HEK3-HEK4 | Translocation | Cas9 | 0.10 | 0.18 | 0.33 | 0.22 | 0.37 | 0.61 |
| insertion guided | 0.07 | 0.13 | 0.25 | 0.31 | 0.50 | 0.80 | ||
| deletion guided | 0.04 | 0.08 | 0.18 | 0.41 | 0.66 | 1.06 | ||
| direct junction guided | 0.07 | 0.14 | 0.27 | 0.11 | 0.20 | 0.36 | ||
| HEK3-HEK4-OFF-1 | Translocation | Cas9 | 0.14 | 0.26 | 0.51 | 0.18 | 0.36 | 0.72 |
| PE2 nuclease | 0.02 | 0.05 | 0.10 | 0.05 | 0.11 | 0.24 | ||
| HEK3-HEK4-OFF-2 | Translocation | Cas9 | 0.11 | 0.20 | 0.38 | 0.14 | 0.26 | 0.48 |
| PE2 nuclease | 0.02 | 0.03 | 0.07 | 0.03 | 0.06 | 0.12 | ||
| NPM1-ALK | Translocation | Cas9 | 0.04 | 0.08 | 0.17 | 0.07 | 0.14 | 0.29 |
| PE2 nuclease | 1.00 | 2.00 | 4.00 | 0.19 | 0.37 | 0.72 | ||
| KIF5B-ALK | Translocation | Cas9 | 0.04 | 0.07 | 0.14 | 0.04 | 0.08 | 0.16 |
| PE2 nuclease | 0.07 | 0.13 | 0.26 | 0.04 | 0.08 | 0.16 | ||
| EML4-ALK-V1 | Inversion | Cas9 | 0.21 | 0.45 | 0.97 | 0.26 | 0.54 | 1.13 |
| PE2 nuclease | 0.65 | 1.26 | 2.45 | 0.87 | 1.87 | 4.02 | ||
| EML4-ALK-V2 | Inversion | Cas9 | 0.85 | 1.80 | 3.86 | 2.57 | 5.20 | 10.6 |
| PE2 nuclease | 5.24 | 10.1 | 19.6 | 5.74 | 11.6 | 23.3 | ||
| EML4-ALK-V3 | Inversion | Cas9 | 0.36 | 0.71 | 1.44 | 0.20 | 0.44 | 0.94 |
| PE2 nuclease | 0.87 | 1.87 | 4.02 | 0.33 | 0.67 | 1.36 | ||
In general, when two DSBs are induced simultaneously in different chromosomes, translocations could cause four different types of genomic rearrangements25 (Figure S5). Possible rearrangements include not only reciprocal translocations but also unusual results such as dicentric and acentric chromosomes. Cas9 and PE2 nuclease, which induce DNA DSBs in the genome, could potentially generate not only reciprocal translocations but also unusual translocations resulting in dicentric and acentric chromosomes. We used multiplex PCR and deep sequencing to detect all types of translocations, amplifying not only reciprocal translocation junctions but also junctions formed by unusual translocations as well as un-translocated DNA (Figure 2E). Although multiplex PCR-based assay cannot detect precise translocation frequencies because it depends on target-specific primers, the assay can be used to detect the relative frequency of each translocation as well as translocation accuracies at their junctions. Our multiplex PCR-based assay revealed that compared with Cas9 nuclease, PE2 nuclease increased the relative frequency of desired translocations while decreasing the frequency of unwanted translocations (Figure 2F). In addition to inducing chromosomal rearrangements, Cas9 and PE2 nucleases were also capable of inducing DSBs at their on- and off-target sites, which results in unintended off-target indel mutations. Targeted deep sequencing was used to elucidate the unwanted off-target mutation, and we found that PE2 nuclease, compared with Cas9, reduced indel frequencies at their on- and off-target sites (Figure S6). We also measured translocation frequencies between HEK3 on-target site and HEK4 off-target sites by dilution PCR and found that PETI decreased unwanted translocation compared with Cas9 (Table 1; Figure S7). These results demonstrated that PETI generate the desired translocations with higher efficiency and purity than Cas9.
Generation of a cancer-associated translocation using PETI
Anaplastic lymphoma kinase (ALK) gene fusion induced by chromosomal translocation or inversion is well-known driving mutation underlying the development of various type of cancers such as lung adenocarcinoma, non-small cell lung carcinoma, and inflammatory myofibroblastic tumor.26 We first chose two types of ALK gene fusion, NPM1-ALK and KIF5B-ALK, generated by reciprocal translocation (Figures 3A and 3B). We generated paired pegRNAs that target ALK and either NPM1 or KIF5B to induce these translocations. In case of NPM1-ALK fusion, the ALK-targeting pegRNA produces 3′-flaps that are complementary to NPM1, and the NPM1-targeting pegRNA produces 3′-flaps that are complementary to ALK. The pegRNAs were designed to insert a 5 bp (ALK-targeting pegRNA) or a 6 bp (NPM1-targeting pegRNA) insertion at the chromosomal translocation junctions. In case of KIF5B-ALK fusion, the pegRNA targeting ALK produces 3′-flaps that are complementary to KIF5B, and pegRNA targeting KIF5B generate 3′-flaps that are complementary to ALK.
Figure 3.

Use of PETI to generate a cancer-associated translocation
(A and B) Schematic of the chromosomes before and after the reciprocal translocations generating the NPM1-ALK (a) and KIF5B-ALK fusion (b). (C and D) Representative results of relative frequency of sequence reads in PCR amplicons amplified by translocation-specific primers amplifying either junction-1 or junction-2. (E) Targeted deep sequencing of amplicons generated using translocation-specific primers. The bar charts represent the fraction of reads with direct translocations (blue), PETI-guided translocations (orange), and translocations with unwanted indels (gray). (F) Fraction of sequencing reads of multiplex PCR amplicons generated from desired and undesired PE2-, Cas9 nuclease-, and PE2 nuclease-mediated translocations. Data are presented as mean ± SEM (n = 3 biologically independent samples).
We then co-transfected HEK293T cells with plasmids encoding PE2, PE2 nuclease, or positive control Cas9 nuclease, as well as the paired pegRNAs, and investigated the resulting translocations using primers that amplify the translocation junctions. Unlike PE2, both Cas9 and PE2 nuclease could cause reciprocal translocations (Figures S8A and S8B). We conducted targeted deep sequencing using translocation-specific primer pairs and found that PETI induced a significantly higher fraction of desired translocations with the intended insertion at the NPM1-ALK and KIF5B-ALK junctions (Figures 3C–3E). To further our understanding of these outcomes, we used multiplex PCR, as described above, and revealed that PE2 nuclease induced precise translocations with PETI-guided manner and reduced undesired translocation compared with Cas9 nuclease (Figure 3F).
To determine if the generated NPM1-ALK gene fusions were transcribed, we isolated mRNA from the transfected cells described above and performed reverse transcription PCR (RT-PCR) using primers amplifying the translocation junction. The presence of a PCR amplicon showed that the NPM1-ALK fusions induced by both the Cas9 and PE2 nuclease-mediated translocations were transcribed (Figure S8C). We conducted dilution PCR to measure translocation frequencies of NPM1-ALK and KIF5B-ALK and found that PE2 nuclease induced the reciprocal translocations with higher frequencies compared with Cas9 (Table 1; Figure S9). We also co-transfected plasmids encoding PE2 nuclease or positive control Cas9 nuclease, as well as the paired pegRNAs to induce NPM1-ALK and KIF5B-ALK translocation in A549 lung cancer cell line and performed targeted deep sequencing using PCR product, which amplifies the translocation junctions using translocation-specific primer pairs. Sequencing results indicated that PETI induced a PETI-guided translocation and direct translocation better than Cas9 nuclease (Figure S10A), which is similar to that performed in HEK293T cells. These findings indicate that PETI can be used to produce cancer-related chromosomal translocations with high fidelity.
Generation of cancer-associated inversions using PETI
Next, we asked whether PETI could be used to generate cancer-associated inversions in endogenous genomes. One of the most common oncogenic chromosomal rearrangements in human cancers, particularly non-small cell lung cancers, leads to the expression of EML4-ALK fusion proteins. There are several EML4-ALK fusion protein variants, which are produced as a result of paracentric inversions within the short arm of chromosome 2 (Figure 4A). EML4-ALK variants 1, 2, and 3 (V1, V2, and V3) account for more than 90% of oncogenic EML4-ALK variants (Figure 4B).27 We first designed paired pegRNAs targeting intron 19 of the ALK gene and intron 13 of the EML4 gene to induce an inversion resulting in EML4-ALK V1 and then transfected HEK293T cells with plasmids encoding PE2, PE2 nuclease, or Cas9 and the appropriate pegRNAs. When HEK293T cells were treated with plasmids encoding Cas9 or PE2 nuclease and paired pegRNAs, inversions were detected using inversion-specific PCR, however, PE2 and paired pegRNAs did not cause inversion (Figure S11A). Next, we generated pegRNAs to generate the inversions expected to result in the oncogenic fusions EML4-ALK V2 and V3. We transfected HEK293T with plasmids encoding these paired pegRNAs or sgRNAs and PE2, PE2 nuclease, or Cas9. Inversions were induced by PE2 nuclease and Cas9 nuclease but not by PE2, which was consistent with the results of the experiment involving EML4-ALK V1 (Figure S11A). Next, we performed targeted amplicon sequencing of PCR products generated by inversion-specific primers and indicated that PETI produced a higher ratio of correct inversions and inversions with the intended insertion than Cas9 (Figures 4C and 4D). We then used multiplex PCR and deep sequencing to determine the relative frequency of inversion and found that the frequencies generated by PE2 nuclease are significantly higher than those of Cas9 in three types of EML4-ALK inversion (Figure 4E). Using dilution PCR, we revealed that PETI could induce EML4-ALK V2 inversion by 10.1% and 11.6% at junction-1 and junction-2, respectively (Table 1; Figure S12). We also induced PETI-mediated inversion in A549 cells and found that PE2 nuclease could induce a higher fraction of PETI-guided inversion and direct inversion compared with Cas9 nuclease (Figure S10B).
Figure 4.

Use of PETI to generate cancer-associated inversions
(A) Schematic of chromosome 2 before and after the paracentric inversion generating the EML4-ALK fusion. (B) Schematic of EML4-ALK fusion variants. EML4-ALK variant 1 (V1), variant 2 (V2), and variant 3 (V3) are generated by fusion of EML4 exon 13 to ALK exon 20, EML4 exon 20 to ALK exon 20, and EML4 exon 6 to ALK exon 20, respectively. (C) Representative results of relative frequency of sequence reads in PCR amplicons amplified by inversion-specific primers amplifying either junction-1 or junction-2. (D) Targeted deep sequencing of amplicons generated using inversion-specific primers. The bar charts represent the fraction of reads with direct inversions (blue), PETI-guided inversions (orange), and inversions with unwanted indels (gray). (E) Fraction of sequencing reads of multiplex PCR amplicons generated from desired and undesired PE2-, Cas9 nuclease-, and PE2 nuclease-mediated inversions. (F) Sequence chromatogram of a region of the EML4-ALK fusion transcripts showing the correct EML4-ALK junctions. (G and H) Flow cytometry analysis to estimate the percentage of cells with EML4-ALK V2 rearrangement by Cas9 and PE2 nuclease. p values were derived from Student’s two-tailed t test. Data are presented as mean ± SEM (n = 3 biologically independent samples).
To investigate whether the PETI- and Cas9 nuclease-induced EML4-ALK transcripts were expressed, we isolated total mRNA from the transfected cells described above and performed RT-PCR. We confirmed that PETI and Cas9 nuclease, but not PE2, led to the expression of EML4-ALK transcripts via paracentric inversion (Figures 4F and S11B). In addition, we further conducted a flow cytometric assay to quantify ALK protein expression at the single cell level10 and observed increased ALK expression in Cas9 or PE2 nuclease treated cells (Figures 4G and 4H). These results suggested that PETI can be used to generate cancer-associated inversions with high frequency and fidelity.
Discussion
Here, we describe PETI, a simple and accurate method for inducing targeted translocations and inversions in programmable manner using PE2 nuclease and paired pegRNAs. First, we showed that PETI could induce programmed DNA recombination in episomal plasmid of reporter constructs and reporter integrated stable cell lines. Subsequently, we used PETI to induce accurate translocations containing the desired sequences in the genomes of human cells. Finally, we generated precise cancer-associated translocations (NPM1-ALK and KIF5B-ALK fusions) and inversions (EML4-ALK fusion variants) using PETI.
Although PETI enables programmable genomic rearrangements in living cells, challenges remain to improve the performance of PETI. As the prime editing efficiency depends on the activity of pegRNAs, the efficiency and accuracy of PETI may be closely related to the composition pegRNAs in a site-specific manner. Indeed, the length of RTT affected the accuracy of PETI with different trends at the two target sites (Figure S13). Therefore, optimization of pegRNAs for each target locus is required to improve the performance of PETI.
Although previous papers have reported that Cas9 with a pair of gRNAs could induce inversions and translocations in living system, we believe that PETI has several advantages over Cas9. Cas9 frequently generate indels at the rearranged genomic breakpoint junctions.8 The majority of disease-associated genomic breakpoints occur in intron region indels at these sites do not affect protein expression, but genomic rearrangements between exon regions are found in tumors such as NAB2-STAT6 fusions,28 and precise editing by PETI might be required to disease modeling and therapeutic approach. PETI can also reduce undesired genomic rearrangement between genomic breakpoint junctions, thereby decreasing unwanted mutations in the genome compared Cas9 nuclease. Furthermore, PETI can install or remove specific sequences at the rearranged genomic breakpoint junctions. As well as small size of 3–6 bp insertion, PETI can introduce an attP sequence of 40 bp at the breakpoint junction (Figure S14). This programmable characteristic of PETI offers opportunities for further applications such as gene tagging and insertion of epitope sequences.
Newly developed prime editing technologies such as Prime-Del, PEDAR, and twinPE can produce large chromosomal deletions or inversions.22,23,29 Prime-Del use PE2 with a paired pegRNAs and PEDAR use PE2 nuclease (PE-Cas9) with a paired pegRNAs for targeted genomic deletion up to ∼10 kb. Although PETI is similar to PEDAR in that it uses PE2 nuclease, PETI enables large-scale genome rearrangement in living cells. Another method, called twinPE, uses PE2 with a paired pegRNAs and Bxb1 recombinase to generate chromosomal deletions and inversions.29 To induce inversions using this method, Bxb1 recognition sites attB and attP (39 and 50 bp, respectively) are first inserted into the genome and then, Bxb1 recombinase induces DNA recombination at attB and attP sites, resulting in deletions or inversions. Although twinPE provides precise deletions and inversions without unwanted DNA cleavage, it requires four pegRNAs (a paired pegRNAs for attB insertion and a paired pegRNAs for attP insertion) and additional Bxb1 recombinase. In addition, inversions generated using twinPE contain attB and attP sequences at the inversion junction. In contrast, the PETI method does not leave a scar at such junctions and can even insert or remove specific bases. On the basis of these results, we anticipate that PETI-mediated chromosomal rearrangements will be a powerful approach for not only generating but also correcting disease-associated inversions and translocations.
Materials and methods
Plasmid construction
The pCMV-PE2 (Addgene plasmid #132775) and pU6-pegRNA-GG-acceptor (Addgene plasmid #132777) plasmids were a gift from David Liu. pegRNA-encoding plasmids were generated by Golden Gate assembly using a custom acceptor plasmid according to methods described in a previous paper (Table S1).30 pCMV-PE2 was modified to encode pCMV-PE2 nuclease by reverting the H840A mutation in the Cas9 sequence to H840 using a Q5 Site-Directed Mutagenesis Kit (New England Biolabs). The coding sequences of eGFP and mCherry were cloned into pUC19 and p3s backbones, respectively, to generate pUC19-TL-eGFP and p3s-TL-EFS-mCherry plasmids for the episomal reporter assay. pLX-sgRNA (Addgene plasmid #50662) was used to make viral vector of these reporter constructs, pLX-TL-mcherry and pLX-TL-eGFP. Briefly, pLX-sgRNA vector was digested BamHI and NheI restriction enzyme and reporter constructs were cloned into them by Gibson Assembly. pLX-TL-mcherry vector was obtained by further changing blasticidin markers to puromycin markers.
Human cell culture and transfection
HEK293T cells (American Type Culture Collection [ATCC] CRL-11268) were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and verified using an STR profile. For the episomal reporter assay, HEK293T cells were seeded onto 48-well plates and transfected with 500 ng of plasmid (50 ng each of the pUC19-TL-eGFP and p3s-TL-EFS-mCherry plasmids, 200 ng of PE2-, PE2 nuclease-, or Cas9-encoding plasmid, and 200 ng each of two pegRNA- or sgRNA-encoding plasmids) using 1.5 μL Lipofectamine 2000 (Life Technologies). The GFP-positive cells were analyzed by flow cytometry and fluorescence microscopy 72 h after transfection. For endogenous mutagenesis, HEK293T cells were seeded onto 24-well plates and transfected with appropriate plasmids (for the PE2 experiments, the PE2-or PE2 nuclease-encoding plasmid [1,500 ng] and the two pegRNA-encoding plasmids [each 250 ng]; for the Cas9 experiments, the Cas9-encoding plasmid [1,500 ng] and the two sgRNA-encoding plasmids [each 250 ng]) using 3 μL Lipofectamine 2000. After 96 h, genomic DNA was isolated with a DNeasy Tissue Kit (Qiagen) according to the manufacturer’s instructions. To generate reporter stale cell lines, 2 × 105 HEK293T cells were seeded onto 24-well plates and 500 ng viral vector (pLX-TL-mcherry and pLX-TL-eEGFP), 300 ng plasmids of psPAX2, and 200 ng plasmids of pMD2.G were transfected one day after cell seeding. Medium was changed 24 h after transfection, and supernatant containing viral particle was harvested 72 h after transfection. The virus particle was filtered using a 0.45 μm filter and stored at −70°C until used. Serially diluted virus particle from pLX-TL-mcherry vector was infected in HEK293T cells, and puromycin was treated 24 h after infection for 3 days. Then, virus particle from pLX-TL-eEGFP vectors was transduced in these cells and blasticidin was treated 24 h after transduction for 3 days. Cells with one copy of pLX-TL-mcherry and pLX-TL-eGFP cassette were selected to stable cell reporter assay.
Multiplex PCR and sequencing
Targeted genomic sites were amplified using four primers, which could amplify not only sequences from wild-type chromosomes but also the junctions of rearranged genomic regions, and Phusion polymerase (New England BioLabs). To produce sequencing libraries, the resulting PCR amplicons were ligated with NEBNext Adaptors (New England BioLabs) and amplified using NEBNext Multiplex Oligos for Illumina (New England BioLabs) according to the manufacturer’s instructions. Sequencing libraries were then subjected to paired-end sequencing using an Illumina sequencing platform (iSeq or MiSeq).
RNA extraction and RT-PCR
RT-PCR was performed to amplify oncogenic fusion transcripts. Total RNA was extracted from transfected cells with a RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Total RNA (1 μg) was reverse transcribed using the TOPscript cDNA Synthesis Kit (Enzynomics) according to the manufacturer’s instructions. PCR amplification was performed using Taq DNA Polymerase (Life Technologies), and PCR products were analyzed by gel electrophoresis.
Intracellular staining of ALK proteins and flow cytometry
Cells were washed in phosphate-buffered saline twice, resuspended in 4% formaldehyde, and incubated for 15 min at room temperature for fixation. Fixed cells were washed in PBS twice and permeabilized in ice-cold 90% methanol for 30 min in ice. Then, cells were washed in 0.5% BSA and stained with 1:500 anti-ALK (#3633T; Cell Signaling Technology) for 1 h at room temperature, washed in 0.5% BSA, and stained with 1:500 anti-rabbit Alexa Fluor 488 secondary antibody (ab150077; Abcam) for 30 min at room temperature. Cells were washed in 0.5% BSA twice, resuspended in PBS, and analyzed using BD FACSAria II flow cytometry.
Statistical analyses
All results from experiments with two or three independent replicates are expressed as mean ± SEM. p values were calculated using the two-tailed Student’s t test.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (2020R1A2C2101714 to D.K.; 2017M3A9B4062419, 2021R1C1C1007162, and 2018R1A5A2020732 to Y.K.; and 2021R1C1C2013270 to J.K.), the Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korean government (Ministry of Science and ICT, Ministry of Health & Welfare [21A0202L1-12 to D.K.]), and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI21C1314 to D.K.).
Author contributions
Y.K. and D.K. supervised the research. J.K., H.-Y.H., and H.R. performed the experiments. J.K., Y.K., and D.K. carried out bioinformatics analyses. D.K. and Y.K. wrote the manuscript. All authors approved the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.09.008.
Contributor Information
Daesik Kim, Email: dskim89@skku.edu.
Yongsub Kim, Email: yongsub1.kim@gmail.com.
Supplemental information
Data and code availability
Sequencing data were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with BioProject accession code PRJNA804661.
References
- 1.Weckselblatt B., Rudd M.K. Human structural variation: mechanisms of chromosome rearrangements. Trends Genet. 2015;31:587–599. doi: 10.1016/j.tig.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stankiewicz P., Lupski J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 3.Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunet E., Simsek D., Tomishima M., DeKelver R., Choi V.M., Gregory P., Urnov F., Weinstock D.M., Jasin M. Chromosomal translocations induced at specified loci in human stem cells. Proc. Natl. Acad. Sci. USA. 2009;106:10620–10625. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee H.J., Kweon J., Kim E., Kim S., Kim J.S. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012;22:539–548. doi: 10.1101/gr.129635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piganeau M., Ghezraoui H., De Cian A., Guittat L., Tomishima M., Perrouault L., René O., Katibah G.E., Zhang L., Holmes M.C., et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013;23:1182–1193. doi: 10.1101/gr.147314.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y., Kweon J., Kim A., Chon J.K., Yoo J.Y., Kim H.J., Kim S., Lee C., Jeong E., Chung E., et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 2013;31:251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- 8.Maddalo D., Manchado E., Concepcion C.P., Bonetti C., Vidigal J.A., Han Y.C., Ogrodowski P., Crippa A., Rekhtman N., de Stanchina E., et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D.U., Jung I., Wu H., Zhai Y., Tang Y., et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162:900–910. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi P.S., Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat. Commun. 2014;5:3728. doi: 10.1038/ncomms4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres R., Martin M.C., Garcia A., Cigudosa J.C., Ramirez J.C., Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- 12.Ghezraoui H., Piganeau M., Renouf B., Renaud J.B., Sallmyr A., Ruis B., Oh S., Tomkinson A.E., Hendrickson E.A., Giovannangeli C., et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell. 2014;55:829–842. doi: 10.1016/j.molcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Q., Zong Y., Xue C., Wang S., Jin S., Zhu Z., Wang Y., Anzalone A.V., Raguram A., Doman J.L., et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020;38:582–585. doi: 10.1038/s41587-020-0455-x. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y., Li X., He S., Huang S., Li C., Chen Y., Liu Z., Huang X., Wang X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020;6:27. doi: 10.1038/s41421-020-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosch J.A., Birchak G., Perrimon N. Precise genome engineering in Drosophila using prime editing. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2021996118. e2021996118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petri K., Zhang W., Ma J., Schmidts A., Lee H., Horng J.E., Kim D.Y., Kurt I.C., Clement K., Hsu J.Y., et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat. Biotechnol. 2022;40:189–193. doi: 10.1038/s41587-021-00901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu W., Yang Y., Yang B., Krueger C.J., Xiao Q., Zhao S., Zhang L., Kang G., Wang F., Yi H., et al. A design optimized prime editor with expanded scope and capability in plants. Nat. Plants. 2022;8:45–52. doi: 10.1038/s41477-021-01043-4. [DOI] [PubMed] [Google Scholar]
- 19.Jang H., Jo D.H., Cho C.S., Shin J.H., Seo J.H., Yu G., Gopalappa R., Kim D., Cho S.R., Kim J.H., Kim H.H. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat. Biomed. Eng. 2022;6:181–194. doi: 10.1038/s41551-021-00788-9. [DOI] [PubMed] [Google Scholar]
- 20.Zhi S., Chen Y., Wu G., Wen J., Wu J., Liu Q., Li Y., Kang R., Hu S., Wang J., et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2022;30:283–294. doi: 10.1016/j.ymthe.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu P., Liang S.Q., Zheng C., Mintzer E., Zhao Y.G., Ponnienselvan K., Mir A., Sontheimer E.J., Gao G., Flotte T.R., et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021;12:2121. doi: 10.1038/s41467-021-22295-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J., Chen W., Suiter C.C., Lee C., Chardon F.M., Yang W., Leith A., Daza R.M., Martin B., Shendure J. Precise genomic deletions using paired prime editing. Nat. Biotechnol. 2022;40:218–226. doi: 10.1038/s41587-021-01025-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang T., Zhang X.O., Weng Z., Xue W. Deletion and replacement of long genomic sequences using prime editing. Nat. Biotechnol. 2022;40:227–234. doi: 10.1038/s41587-021-01026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H.J., Kim E., Kim J.S. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker J.D. Chromosome translocations and assessing human exposure to adverse environmental agents. Environ. Mol. Mutagen. 2010;51:815–824. doi: 10.1002/em.20561. [DOI] [PubMed] [Google Scholar]
- 26.Hallberg B., Palmer R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016;27:iii4–iii15. doi: 10.1093/annonc/mdw301. [DOI] [PubMed] [Google Scholar]
- 27.Zhang S.S., Nagasaka M., Zhu V.W., Ou S.H.I. Going beneath the tip of the iceberg. Identifying and understanding EML4-ALK variants and TP53 mutations to optimize treatment of ALK fusion positive (ALK+) NSCLC. Lung Cancer. 2021;158:126–136. doi: 10.1016/j.lungcan.2021.06.012. [DOI] [PubMed] [Google Scholar]
- 28.Chmielecki J., Crago A.M., Rosenberg M., O'Connor R., Walker S.R., Ambrogio L., Auclair D., McKenna A., Heinrich M.C., Frank D.A., Meyerson M. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat. Genet. 2013;45:131–132. doi: 10.1038/ng.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anzalone A.V., Gao X.D., Podracky C.J., Nelson A.T., Koblan L.W., Raguram A., Levy J.M., Mercer J.A.M., Liu D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022;40:731–740. doi: 10.1038/s41587-021-01133-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kweon J., Yoon J.K., Jang A.H., Shin H.R., See J.E., Jang G., Kim J.I., Kim Y. Engineered prime editors with PAM flexibility. Mol. Ther. 2021;29:2001–2007. doi: 10.1016/j.ymthe.2021.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with BioProject accession code PRJNA804661.