

Abstract
Impairment of innate immune cell function and metabolism underlies immunosuppression in sepsis; however, a promising therapy to orchestrate this impairment is currently lacking. In this study, high levels of NOD-like receptor family CARD domain containing-3 (NLRC3) correlated with the glycolytic defects of monocytes/macrophages from septic patients and mice that developed immunosuppression. Myeloid-specific NLRC3 deletion improved macrophage glycolysis and sepsis-induced immunosuppression. Mechanistically, NLRC3 inhibits nuclear factor (NF)-κB p65 binding to nuclear factor of activated T cells 5 (NFAT5), which further controls the expression of glycolytic genes and proinflammatory cytokines of immunosuppressive macrophages. This is achieved by decreasing NF-κB activation—co-induced by TNF-receptor-associated factor 6 (TRAF6) or mammalian target of rapamycin (mTOR)—and decreasing transcriptional co-activator p300 activity by inducing NLRC3 sequestration of mTOR and p300. Genetic inhibition of NLRC3 disrupted the NLRC3-mTOR-p300 complex and enhanced NF-κB binding to the NFAT5 promoter in concert with p300. Furthermore, intrapulmonary delivery of recombinant adeno-associated virus harboring a macrophage-specific NLRC3 deletion vector significantly improved the defense of septic mice that developed immunosuppression upon secondary intratracheal bacterial challenge. Collectively, these findings indicate that NLRC3 mediates critical aspects of innate immunity that contribute to an immunocompromised state during sepsis and identify potential therapeutic targets.
Keywords: sepsis-induced immunosuppression, host immune defense, macrophages, NLRC3, immunometabolism, gene therapy
Graphical abstract
This study connects NLRC3 expression in myeloid cells to the mechanism of sepsis-induced immunosuppression, where NLRC3 inhibits the binding of NF-kB to NFAT5 in concert with p300, thereby regulating metabolic reprogramming of macrophages, and shows that NLRC3 gene therapy targeting intrapulmonary macrophage improves lung defenses against secondary infections.
Introduction
Sepsis is a dysregulated host immune response to disseminated infection characterized by an acute inflammatory response and a prolonged hyporesponsiveness or immunosuppression.1,2 These infections are frequently encountered in the intensive care unit (ICU), accounting for 4%–17% of ICU admissions,3,4 and are responsible for more than 8 million deaths worldwide each year,1 with this form of immunosuppression accounting for 85% of sepsis-related deaths.1 In the setting of immunosuppression, deactivation of modulatory pathways has been considered an important cause of abnormal host response, which is characterized by reduced responsiveness of immune cells to pathogens5 or monocyte human leukocyte antigen (HLA)-DR expression,5,6 increased secondary bacterial infections, and an elevated risk of mortality after sepsis.5 Thus, the current view in the field is that targeting sepsis-induced immunosuppression could be a cure for sepsis, but the challenge remains to identify appropriate targeted therapy.
Sepsis-induced monocyte/macrophage dysfunction is one of the manifestations of sepsis-induced immunosuppression.5,7,8 During infection, the macrophage immune response is initiated when the pattern recognition receptors (PRRs) in the cell membrane of macrophages recognize microbial molecular patterns of external pathogens (pathogen-associated molecular patterns [PAMPs]). PAMPs recognition activates immediate innate host defense mechanisms and primes adaptive immune responses to protect the host from microbial infection.9 However, prolonged exposure to PRR ligands such as lipopolysaccharide (LPS) affects the signal transduction inside the cell and induces an immunosuppressive state known as endotoxin tolerance.10 The endotoxin tolerance of monocytes/macrophages contributes to the induction of whole-body immunosuppression because it recapitulates several key features of sepsis-induced immunosuppression.5,11 Among these features, metabolic reprogramming plays an essential role in inducing and maintaining immune tolerance during sepsis.12 Genome-wide transcriptome analysis of blood from patients with sepsis revealed that the metabolic shift to glycolysis was inhibited upon microbial reinvasion, thereby underlying the immunosuppressive phase.12 In an in vitro model that imitates sepsis-induced immunosuppression, metabolic defects in human monocytes were partially restored with interferon-γ through enhancing the glycolysis.12 Increased glycolysis has been postulated as a common feature of microbial ligand-activated macrophages, as it delivers energy to support antimicrobial responses.12,13,14,15 These lines of evidence regarding the role of aerobic glycolysis in monocytes/macrophages offer new insight into biological therapies for sepsis-induced immunosuppression if targeting PRRs, but further studies are still needed.16,17,18
Most of the best-studied PRRs are receptors or sensors that drive innate immune responses to microbial ligands and subsequently initiate inflammatory signaling cascades; however, these PRRs can easily elicit disproportionate inflammation and severe tissue injury.19 By contrast, a few PRRs in the myeloid lineage have a negative role in regulating innate immune responses, and deficiency of these molecules can strengthen the defensive capability of the host, but a promising therapeutic result has not yet been forthcoming.19,20,21 NOD-like receptor (NLR) family CARD domain containing-3 (NLRC3) is a less studied novel negative cytoplasmic PRR that is expressed in various immune tissues and cells. As a sensor of microbial ligands, NLRC3 acts as a fine-tuning rheostat to T cell responses during viral infection, and deficiency of this receptor enhances protection against lymphocytic choriomeningitis virus and Mycobacterium tuberculosis infection.21,22 NLRC3 is also a negative regulator of signaling pathways activated by Toll-like receptors (TLRs), and negatively regulates TLR4-dependent nuclear factor κB (NF-κB) activation by binding tumor necrosis factor receptor-associated factor 6 (TRAF6) in activated immune cells.20,23 During cancer development, TRAF6 sequesters NLRC3 and mammalian target of rapamycin (mTOR) to negatively regulate mTOR pathways.24,25 mTOR pathway serves as a master regulator of cell metabolism, and its activation is highly dependent on the phosphorylation of mTOR. Activation of mTOR pathway can elicit the subsequent activation of numerous downstream targets, thereby regulating several transcription factors involved in the regulation of metabolic pathway genes, including glycolytic genes.12,21 Additionally, mTOR can also interact with transcriptional co-activators such as p300 to control transcription and cell metabolism.26 However, there is limited understanding of the mechanism underlying the regulation of NLRC3 on aerobic glycolysis, and whether NLRC3 inhibits the innate immune signaling to drive sepsis-induced immunosuppression has yet to be elucidated.
Here, we found that high NLRC3 expression correlates with the glycolytic defects of the monocyte/macrophage from septic patients and mice that developed immunosuppression. Genetic inhibition of myeloid-specific NLRC3 disrupts the NLRC3-mTOR-p300 complex and enhances NF-κB binding to the NFAT5 promoter in concert with the p300 to further improve the glycolysis of cells and sepsis-induced immunosuppression. Intrapulmonary delivery of recombinant adeno-associated virus (rAAV) harboring macrophage-specific NLRC3 deletion vector (rAAV-SP-NLRC3) significantly improved the defense of septic mice that developed immunosuppression upon secondary intratracheal bacterial challenge, suggesting that NLRC3 may be a therapeutic target for sepsis-induced immunosuppression.
Results
NLRC3 is upregulated in clinical immunotolerant monocytes and in macrophages that underwent sepsis-induced immunosuppression
NLRC3 is a negative innate sensor that guards against excessive acute inflammation in mouse models of sterile sepsis,20 but its function in the host’s defense response after undergoing sepsis-induced immunosuppression has not been fully characterized. We firstly measured NLRC3 expression in monocytes from 35 patients with bacterial sepsis (Figures 1A–1E). Monocytes from septic patients had significantly higher NLRC3 levels than those from control subjects (Figure 1E). The demographic and clinical characteristics of these subjects are listed in Table S1. Low HLA-DR expression and decreased levels of proinflammatory cytokines (e.g., tumor necrosis factor α [TNF-α]) in monocytes after sepsis are considered a hallmark of sepsis-induced immunosuppression.5,27 These hallmarks were analyzed (Figures 1B–1D) and revealed an overall decrease in HLA-DR expression in monocytes from septic patients (Figures 1B and 1C), and the HLA-DR expression inversely correlated with induction of NLRC3 expression (r2 = −0.5451, p = 0.0007; Figure 1F). Additionally, we observed significantly impaired production of TNF-α in monocytes challenged with LPS in vitro (Figure 1D), and the decreased TNF-α was inversely correlated with induction of NLRC3 expression (r2 = −0.7132, p < 0.0004; Figure 1H). These findings suggest that NLRC3 elevation in monocytes is specifically correlated with induction of immunosuppression during sepsis.
Figure 1.

NLRC3 is upregulated in clinical immunotolerant monocytes and macrophages that underwent post-CLP immunosuppression
(A) Schematic overview of experimental design for (B) to (I). (B and C) Flow cytometry assessment of HLA-DR expression on circulating monocytes from septic patients (n = 35) and non-septic donors (n = 29). (D) ELISA for TNF-α in the culture supernatants from septic patients (n = 35) and non-septic donor monocytes (n = 29) stimulated with or without LPS (10 ng/mL) in vitro for 12 h (data are presented in a violin plot and were compared with a t test). (E) NLRC3 mRNA levels in monocytes of septic patients (n = 35) and non-septic donors (n = 29). (F) Correlation assay between HLA-DR levels and change in TNF-α production (ΔTNF-α) in monocytes of septic patients (n = 35). (G–I) Correlation assay between NLRC3 levels and HLA-DR levels (G), and changes in TNF-α production (ΔTNF-α) in monocytes (H) and SOFA scores (I) of septic patients (n = 35). (J) Treatment schematic for (K) and (L). (K and L) ELISA for TNF-α in the supernatants of NLRC3WT (LysM-Cre-NLRC3fl/fl) and NLRC3ΔMac (LysM-Cre+ NLRC3fl/fl) mouse AMs (K) and PMs (L) stimulated with or without LPS in vitro (n = 5). ΔTNF-α: RPMI-stimulated samples versus LPS-stimulated samples. Circles represent individual participants. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant (two-way ANOVA or Student’s t test). See also Table S3 and Figures S1–S6.
To further explore the regulatory role of NLRC3 in sepsis-induced immunosuppression, as previously described,7,28,29,30 we established a cecal ligation and puncture (CLP) model with impaired host immunity that most accurately resembles conditions in patients (Figures S1 and S2A). In this model, alveolar macrophages (AMs) and peritoneal macrophages (PMs) from mice after undergoing CLP had reduced expression of TNF-α in response to a subsequent LPS challenge in vitro (Figure S2C), which is analogous to what we observed with clinical monocyte tolerization, while the NLRC3 expression in AMs and PMs of sepsis-surviving mice increased 12 h after CLP and peaked at 48–72 h (Figure S3A). NLRC3-specific immunoblotting further confirmed upregulated NLRC3 protein expression in PMs 48 h after CLP (Figure S3B). These experiments suggest that NLRC3 expression in macrophages may have an important regulatory role in establishing immunosuppression in septic mice.
Sepsis-induced immunosuppression alters the host’s immune response, leading to increased susceptibility of septic host to nosocomial pneumonia.7,28,30,31 In the clinic cohort, we identified septic patients with culture-proven secondary infection (n = 19). To confirm whether induction of NLRC3 expression in macrophages correlated with impairment of septic host defenses, the antimicrobial role at the infection site was evaluated. There was a decrease in the host’s ability to clear the bacteria of the lungs and blood of CLP mice after Pseudomonas aeruginosa infection (Figures S1C and S1D), and the septic mice challenged with a secondary infection of P. aeruginosa intratracheally (i.t.) (3 × 109 colony-forming units [CFU] mL−1) at 48 h displayed a significant increase in mortality compared with sham mice subjected to infection or CLP without undergoing infection (Figure S1B).
NLRC3 depletion in myeloid cells alleviates sepsis-induced immunosuppression
To address the role of macrophage NLRC3 expression in orchestrating sepsis-induced immunosuppression, myeloid-specific NLRC3-deficient mice were generated by crossing NLRC3 floxed mice with LysM-Cre mice (Figures S4A and S4B). We confirmed that the Nlrc3 gene was specifically deleted in bone marrow-derived macrophages (BMDMs), AMs, and PMs isolated from NLRC3-deficient LysM-Cre+ NLRC3fl/fl (NLRC3ΔMac) mice but was unaffected in control LysM-Cre-NLRC3fl/fl mice (NLRC3WT) (Figures S4C and S4D). Myeloid cell population counts in the peritoneal cavities and spleens of NLRC3WTand NLRC3ΔMac mice were similar (Figures S5A and S5B), suggesting that NLRC3 deletion does not affect myeloid cell development or maturation. The mortality of NLRC3ΔMac mice was not significantly different from that of their wild-type (WT) counterparts 48 h after CLP (Figure 2B), but surviving septic NLRC3ΔMac mice had a significantly improved survival rate in response to secondary P. aeruginosa challenge at 48 h post CLP (Figure 2B). The bacterial burdens in the lungs and blood of NLRC3ΔMac mice undergoing CLP challenged with a secondary infection of P. aeruginosa were significantly lower than those in NLRC3WT mice Figure 2C). The TNF-α and interleukin-6 (IL-6) levels in lung homogenates from septic NLRC3ΔMac mice were significantly higher than those of NLRC3WT mice, but IL-1β levels showed no difference (Figure 2D). Lung section staining showed that lung inflammation was exacerbated in NLRC3ΔMac mice at 24 h after the secondary P. aeruginosa challenge (Figures 2E and 2F).
Figure 2.

NLRC3 depletion in myeloid cells alleviated sepsis-induced immunosuppression
(A) Schematic overview of experimental design for (B) to (I). (B) Kaplan-Meier survival curves. All mice were monitored for 10 days after secondary P. aeruginosa challenge (n = 16 mice/group). (C–F) Bacterial loads in the lung and blood (C), ELISA for TNF-α, IL-1β, and IL-6 in lung homogenate (D), histopathological images of lung tissues (E), and histological injury scores (F); samples were collected at 24, 48, or 96 h after the secondary infection. Scale bar represents 100 μm (n = 6 mice/group). (G–I) Percentages of lung-infiltrating cells that were neutrophils (CD11b+ Gr-1+) or monocyte-macrophages (CD11b+ Gr-1−) (G and H). Expression levels of CD86, MHC II, and CD206 were detected on monocyte-macrophages (CD11b+ Gr-1−), with representative histogram with the corresponding median fluorescence intensity (MFI) (I). All samples were collected at 24 h after the infection challenge; assays were conducted by flow cytometry (n = 6 mice/group). Circles represent individual mice. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant (two-way ANOVA or Student’s t test and log-rank test for survival). See also Figures S4–S6.
To further evaluate the role of macrophage NLRC3 deficiency in mice subjected to CLP during secondary infection, we detected the expression of cytokines and cell-surface molecules in macrophages from septic mice with LPS stimulation in vitro. AMs and PMs from septic NLRC3ΔMac mice showed higher levels of TNF-α (Figures 1K and 1L) and IL-6 (Figures S6A–S6B) in response to LPS challenge in vitro than did macrophages from NLRC3WT septic mice. Moreover, monocyte-macrophages in the lungs of septic mice also showed upregulation of CD86 and major histocompatibility complex class II (MHC II) expression relative to those from NLRC3WT mice (Figure 2I), although there were no significant differences in the overall numbers (Figure 2H). This evidence suggests that NLRC3 depletion in myeloid cells leads to stronger immune responses, alleviates sepsis-induced immunosuppression, and further confers protection against secondary pneumonia with P. aeruginosa in septic mice.
NLRC3 depletion improves metabolic pathways in immunotolerant macrophages during sepsis
The increased immune response by macrophages to eliminate pathogens during the acute phase of infection is characterized by robust alterations in the expression of genes encoding inflammation and immunological effector mechanisms. To characterize the transcriptome alterations by which NLRC3 loss regulates pathogen infection, we performed RNA sequencing (RNA-seq) to identify the genes involved in NLRC3 regulation of microbial ligand stimulation in macrophages. We noted robust alterations in the BMDM transcriptome 12 h after LPS treatment, such as differential expression of genes encoding TNF-α, cytokine interaction receptors, TLRs, metabolic pathway signaling molecules (Figure 3A), and some genes encoding products involved in glycolytic enzyme (Figure 3B). We further validated changes in genes encoding glycolytic enzymes using quantitative PCR (qPCR) and found that several mRNAs including Glut1, Hk2, and Ldha were significantly upregulated in AMs and PMs of sham-surgery NLRC3ΔMac mice subjected to LPS challenge than did macrophages from NLRC3WT mice (Figures 3C, S10, S11A, and S12).
Figure 3.

NLRC3 functions within macrophage to drive glycolysis defects during sepsis-induced immunosuppression
(A and B) Total RNA of each mouse was isolated from 1.0 × 106 BMDMs (n = 3 mice/each group) and analyzed by RNA-seq. (A) Left: scatterplot of the KEGG pathway enrichment analysis of the differentially expressed genes (DEGs) in immunometabolism-associated pathways. Right: the Rich factor is the ratio of DEG numbers annotated in this pathway term to all genes; the size of the symbols represents the number count of DEGs, and higher values indicate greater intensiveness; coloring of the p values indicates the significance of the Rich factor, ranging from −1 to 1. (B) Heatmap showing the relative amounts of RNA of the three pairwise comparisons. (C and E) qPCR detection of the mRNA expression levels of representative NLRC3-regulated genes involved in glycolysis in macrophages: (C) AMs with LPS in vitro challenge after CLP or sham operation (n = 5 mice/each group), and (E) tolerant or naive RAW 264.7 with null vector, stable NLRC3 deficiency (shNLRC3), or overexpression (ovNLRC3) following LPS stimulation. The results of mRNA expression were normalized to β-actin, and log2 values were used to calculate correlations. Each column in the heatmap represents the ratio of normalized expression (n = 5). (D and F) Lactate production in AMs and PMs following LPS in vitro challenge in the indicated genotypes after CLP or sham operation (D) in tolerant or naive BMDMs transduced with shNLRC3 or ovNLRC3 following LPS stimulation (F). (G–I) Analysis of the extracellular acidification rate (ECAR) of tolerant or naive RAW 264.7 macrophages transduced with null or shNLRC3 or ovNLRC3 vector after LPS stimulation. (n = 5). (J–L) Kaplan-Meier survival curves (n = 16) (J); schematic overview of experimental design (K); bacterial loads in the lung (left) and blood (right) from surviving NLRC3ΔMac (LysM-Cre+ NLRC3fl/fl) mice with either 2-DG or PBS after P. aeruginosa intratracheal administration (L). Circles represent individual mice. Graphs show the mean ± SE of experimental replicates and are representative of at least three independent experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant (two-way ANOVA or Student’s t test and log-rank test for survival). See also Figures S7–S13.
As a result of the functional reprogramming of macrophages during microbial ligand-induced immunosuppression, most inducing genes are transcriptionally silenced and not expressed upon restimulation;5,32,33 broad cellular glycolytic metabolism defects in the monocytes of septic patients underlie the immunosuppressive phase. In determining whether the above changes could be identified in macrophages from sepsis-induced immunosuppression hosts subjected to secondary LPS stimulation, we found that the mRNA levels of Glut1, Pfk, Hk2, and Ldha were significantly upregulated in AMs (Figures 3C and S10) and PMs (Figures S11A and S12) from septic NLRC3ΔMac mice following in vitro LPS challenge compared with those from septic NLRC3WT mice. The enhanced transcription of glycolytic genes increases the capacity of cells to execute glycolysis. The gene transcription data were thus confirmed by lactate release, which is an inevitable product of glycolysis. Substantial increases in lactate production were detected in culture supernatants of macrophages from NLRC3ΔMac mice subjected to CLP (Figure 3D) and monocytes from septic patients with immunosuppression after LPS stimulation in vitro compared with those of control host (Figures S11D and S11E). These results demonstrate a shift toward aerobic glycolysis (known as the Warburg effect) and thus illustrate that induction of the Warburg effect in macrophages from septic hosts occurred in immunosuppression after NLRC3 depletion.
To broadly assess the ability of NLRC3 to inhibit glycolysis in macrophages in the setting of immunosuppression during sepsis, we generated an in vitro tolerance model using LPS. RAW 264.7 cells were transduced with recombinant lentivirus expressing a stable short hairpin RNA (shRNA) that targets the mRNA encoding NLRC3 (shNLRC3) or overexpressing a vector encoding NLRC3 (ovNLRC3) before being treated with LPS. We found substantially lower expression of NLRC3 in RAW 264.7 cells (Figure S14A), while NLRC3-overexpressing macrophages (Figure S14A) that encountered tolerance showed lower mRNA levels of Glut1, Hk2, Pfk, and Ldha, while shNLRC3 resulted in higher expression levels (Figure 3E) after secondary LPS challenge (Figure S13) and a substantial increase in lactate production in culture supernatants when tolerant BMDMs (Figure 3F) or RAW 264.7 cells (Figure S11B) with NLRC3 deficiency were restimulated with LPS. Extracellular acidification rate (ECAR) analysis can directly and continuously detect changes in aerobic glycolysis in living cells and reflect the glycolysis rate of the cells.34,35,36 Consistent with endpoint assays such as lactate levels in supernatants, the basal ECARs, glycolytic reserve, and capacity were significantly elevated in immunotolerant macrophages with stable NLRC3 deficiency after 12 h of LPS stimulation (Figures 4G and 4H) compared with cells overexpressing NLRC3. These data suggest that NLRC3 depletion could alleviate the decrease in glycolytic activation in immunosuppressive macrophages.
Figure 4.

mTOR or TRAF6 activation is only part of the mechanism by which NLRC3 regulates metabolic pathways in tolerant macrophages
(A and B) (A) Schematic of treatments (upper), and immunoblot to detect phosphorylated mTOR, S6K, and 4EBP1 in tolerant or naive RAW 264.7 macrophages transduced with shNLRC3, ovNLRC3, or null vector (lower), and (B) in PMs with or without LPS in vitro challenge after CLP operation (n = 4). (C and I) Seahorse XFe 24 monitored ECAR in tolerant RAW 264.7 with stable NLRC3 deficiency in the presence of mTOR inhibitor INK-128 (C) or NF-κB inhibitor BAY11-7082 (I) or vehicle after LPS restimulation; and in naive macrophages with stable NLRC3 deficiency in the presence of INK-128 (C) or NF-κB inhibitor BAY11-7082 (I) or vehicle with no LPS stimulation (n = 5). (D, J, and K) ELISA for lactate production, TNF-α, IL-6, and IL-1β in tolerant BMDMs of NLRC3ΔMac (LysM-Cre+ NLRC3fl/fl) mice in the presence of INK-128 (D) or NF-κB inhibitor BAY11-7082 (J and K) or vehicle after LPS restimulation; and in naive macrophages of NLRC3ΔMac in the presence of INK-128 (D) or NF-κB inhibitor BAY11-7082 (J and K) or vehicle with no LPS stimulation (n = 5). (E) Immunoprecipitation of IκB and p65 and of TRAF6, IκB, and p65 from the tolerant or naive RAW 264.7 macrophages transduced with null or shNLRC3 or ovNLRC3 vector after 90-min LPS restimulation, followed by immunoblot to detect K63-linked ubiquitination (K63-Ub) of TRAF6. (F) Immunoblot of IκB, and p65 in PMs with or without LPS in vitro challenge after CLP operation (n = 4). (G) RAW 264.7 macrophages with stable NLRC3 deficiency were transfected with shTRAF6 or shNS (control) vector; Seahorse XFe 24 monitored ECAR in these tolerant cells following LPS stimulation or naive cells with no LPS stimulation (n = 5). (H) ELISA for lactate production in tolerant BMDMs of NLRC3ΔMac mice with shTRAF6 or shNS (control) vector after restimulation (n = 5), and in naive BMDMs of NLRC3ΔMac mice with shTRAF6 or shNS vector in the absence of LPS stimulation. Circles represent individual mice. Graphs show the mean ± SE of experimental replicates and are representative of at least three independent experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant (two-way ANOVA or Student’s t test). See also Figures S14 and S15.
To further confirm the role of glycolysis in host immune defense during sepsis-induced immunosuppression, we inoculated the glycolytic inhibitor 2-deoxyglucose (2-DG) into septic NLRC3ΔMac mice before P. aeruginosa infection. Treatment of septic NLRC3ΔMac mice with 2-DG significantly increased mortality (Figure 3J) and bacterial loads in the lung and blood (Figure 3L) compared with the control group, indicating a direct role of macrophage glycolysis in NLRC3-driven sepsis-induced immunosuppression.
Immunometabolism by NLRC3 deficiency is not completely dependent on mTOR or TRAF6 signaling
We next explored the specific molecular pathways used by NLRC3 to orchestrate the glycolytic metabolism of macrophages in the setting of immunotolerance. Because mTOR is found to increase transcription of glycolytic enzymes, thereby enhancing the capacity of cells to execute glycolysis,12,21 and given the finding that NLRC3 inhibits mTOR pathways in colon epithelial cells24,25 and the gene transcription data (Figure S8), we assessed whether mTOR signaling highly depends on the phosphorylation of mTOR and the downstream effectors S6K and 4EBP1. We observed that shNLRC3 enhanced S6K and 4EBP1 phosphorylation of tolerant macrophages upon secondary LPS challenge, whereas macrophages with NLRC3 overexpression showed lower phosphorylation (Figure 4A). Moreover, freshly isolated PMs from NLRC3WT mice that had undergone CLP 48 h were stimulated with LPS in vitro and showed decreased phosphorylation of S6K and 4EBP1. Conversely, macrophages from septic NLRC3ΔMac mice showed enhanced phosphorylation of S6K and 4EBP1 (Figure 4B). Notably, the small-molecule active-site mTOR kinase inhibitor INK128 37 did not completely inhibit the ECARs or lactate and proinflammatory cytokine production of tolerant macrophages with NLRC3 loss upon secondary LPS challenge (Figures 4C and 4D). These lines of evidence suggest that mTOR activation may be involved in sensing the cellular energy status of tolerant macrophages with NLRC3 deficiency but is not the only molecule that activates the downstream immunometabolism.
TLR-NF-κB systems are involved in most key metabolic pathways involved in the Warburg effect in tumor cells.38,39 Association of NLRC3 with TRAF6 leads to auto-ubiquitylation of the latter protein; this is a critical step in inhibiting TLR-dependent activation of NF-κB signaling.20,21 We observed more rapid K63-linked ubiquitination of TRAF6 in tolerant macrophages with stable NLRC3 deficiency than in cells stably overexpressing NLRC3, and expression levels of TRAF6 and the downstream effector NF-κB were more abundant in the former cell population (Figure 4E). NF-κB activation was also enhanced in macrophages from septic NLRC3ΔMac mice compared with WT mice (Figure 4F), but the ECARs and lactate production were not completely inhibited by TRAF6 knockdown (Figure S14B) in tolerant macrophages with NLRC3 deficiency upon secondary LPS challenge (Figures 4G and 4H). These findings suggest that immunometabolic changes due to NLRC3 deficiency do not completely depend on the association between TRAF6 and NLRC3, but rather that co-activation of both TRAF6 and mTOR pathways is needed. Thus, inhibition of either TRAF6 or mTOR is insufficient to adequately inhibit the immunometabolic effects; inhibition of both is required.
NLRC3 deletion affects NF-κB-mediated immunometabolism without directly interacting with NF-κB subunits
As a point of cross-talk of the downstream effectors between TRAF6 and mTOR signaling pathways, NF-κB functional inhibition results in impaired expression of the gene encoding the glycolytic enzyme and subsequent reduction of glycolysis in tumor cells.40 We found that the NF-κB inhibitor BAY11-7082, which antagonizes I-κB kinase-β preventing nuclear translocation of NF-κB, abolished the induction of a glycolytic response and proinflammatory cytokine production by NLRC3 knockdown (Figures 4I–4K). These results suggest that the effect of NLRC3 on glycolysis may require NF-κB signaling and subsequent translocation.
The key subunits of NF-κB form a homo- or heterodimer with other NF-κB subunits to activate the target gene transcription involved as the endpoint in an array of signal transduction events.41 To establish whether the p65 subunit transmitted these glycolytic metabolic effects, we used shRNA-mediated knockdown to inhibit them in NLRC3-deficient macrophages (Figure S14C). Loss of p65 or c-Rel could not completely reduce ECAR levels and lactate production of tolerant macrophages with NLRC3 deficiency upon secondary LPS challenge (Figure S15). These results suggest that the effect of NLRC3 on glycolysis may be independent of the forms of NF-κB dimer with its subunit and subsequent transduction in immunotolerant macrophages, and that another mechanism is required in the context of NF-κB-dependent glycolysis.
p300-Mediated NFAT5 signaling is required to activate NLRC3−/−-tolerant macrophage glycolysis
The effects of NLRC3 on signal transduction pathways in activated immune cells were orchestrated, whereby in addition to the NF-κB signaling the nuclear factor of activated T cell (NFAT) was also a major factor.23 The NFAT family consists of five members, NFAT1 to NFAT5, which share a conserved DNA-binding domain that is structurally related to the NF-κB family members42,43 and play a key role in the control of cytokine gene expression in T cells. We thus examined the expression levels of them in NLRC3−/− LPS-tolerant RAW264.7 macrophages (Figure S18B) and macrophages from septic NLRC3ΔMac mice (Figure S18C) upon LPS secondary challenge. Given that NFAT5 was most highly expressed (Figures S18A and S18B) and could regulate specific genes but also others that are inducible by NF-κB and NFAT1 to NFAT4,44 and that the RNA-seq data from the NCBI Gene Expression Omnibus (GEO)45 shows an enrichment of genes involved in glycolysis relevant to NFAT5 expression in BMDM macrophages with LPS stimulation (Figures 5A, S16, and S17), we thus focused on the NFAT5 isoform. PMs from NLRC3ΔMac mice that had undergone CLP 48 h and were stimulated with LPS in vitro showed a much stronger NFAT5 expression, and LPS-tolerant macrophages with NLRC3 deficiency showed more enhanced NFAT5-dependent reporter activity than that of NLRC3-overexpression cells (Figures 5B and 5C). Furthermore, monocytes from septic patients with immunosuppression also had lower NFAT5 expression after LPS stimulation in vitro compared with those of control hosts (Figure S18D). These data suggest that NFAT5 activation plays a critical role in activating NLRC3−/−-tolerant macrophage glycolysis.
Figure 5.

p300-mediated NFAT5 signaling is required for activation of NLRC3−/−-tolerant macrophage glycolysis
(A) The heatmap closely related to glycolysis is based on RNA-seq data comparing NFAT5 shRNA (NFAT5KD) and transduced empty vector (NFAT5Ctrl) RAW 264.7 (left), and NFAT5 KO BMDMs (NFAT5 KO) and NFAT5 WT BMDMs (NFAT5WT) treated with LPS (right) from the NCBI GEO database.45 Each column in the heatmap represents the ratio of normalized expression (n = 3). (B) Immunoblot to detect NFAT5 levels in PMs with or without LPS in vitro challenge after CLP or sham operation (n = 4). (C) Tolerant RAW 264.7 with stable NLRC3 deficiency (shNLRC3) or overexpression (ovNLRC3) were transfected with NFAT5 consensus sequences fused to a GFP reporter construct. Flow cytometry was performed to analyze NFAT5-dependent GFP expression (n = 3). (D, E, and H) (D) Tolerant macrophages were stably transfected with ovNLRC3 or shNLRC3 vector and restimulated with LPS, where macrophages (naive) transfected with ovNLRC3 or shNLRC3 vector without LPS served as the control groups; ChIP assays were conducted in these cells after 90-min secondary LPS stimulation, with exon14 used as a negative control (n = 3); (E) Flow cytometry monitored the GFP expression level in these macrophages following transfection with GFP reporter system containing two NF-κB binding sites upstream of NFAT5 gene (n = 3). (H) Immunoprecipitated p300 and lysate histone H3 were analyzed with anti-acetyl-lysine and anti-acetyl-histone H3 in these cells, respectively (n = 3). (F) Seahorse XFe 24 monitored ECAR in tolerant RAW 264.7 with stable NLRC3 deficiency in the presence of KRN2 or vehicle after LPS restimulation, and in naive macrophages with stable NLRC3 deficiency in the presence of KRN2 or vehicle without LPS stimulation (n = 5). (G and L) ELISA for lactate production and TNF-α, IL-6, and IL-1β in tolerant BMDMs of NLRC3ΔMac (LysM-Cre+ NLRC3fl/fl) mice in the presence of KRN2 (G) or cerulenin (L) or vehicle after undergoing LPS restimulation, and in naive macrophages of NLRC3ΔMac in the presence of KRN2 (G) or cerulenin (L) or vehicle in the absence of LPS stimulation (n = 5). (I) Human embryonic kidney (HEK) 293T cells were transfected with the p300, HA-NLRC3, and mTOR plasmids. p300 immunoprecipitates were analyzed for p300, HA, and mTOR expression by immunoblot (n = 3). (J) Tolerant macrophages with stable NLRC3 deficiency treated with vehicle or INK-128 before LPS rechallenge (n = 3). Naive macrophages with stable NLRC3 deficiency without LPS treatment served as the control group. Immunoprecipitated p300 and lysate histone H3 were analyzed with anti-acetyl-lysine and anti-acetyl-histone H3, respectively. (K) Tolerant macrophages with stable NLRC3 deficiency were treated with vehicle or cerulenin before LPS rechallenge, with naive macrophages transfected with shNLRC3 without undergoing LPS treatment as the control group. Cell lysates were immunoprecipitated (IP) using anti-p65 antibody. The immunoprecipitates and cell lysates were immunoblotted for p300, NFAT5, and p65 (n = 3). Circles represent individual mice. Graphs show the mean ± SE of experiment replicates and are representative of at least three independent experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant (two-way ANOVA or Student’s t test). See also Figures S16–S18.
NF-κB binding to the NFAT5 promoter is one of the main mechanisms modulating NFAT5 function.46 We found that NLRC3 deficiency markedly increased the affinity of NF-κB p65 binding to the NFAT5 promoter in LPS-tolerant macrophages upon LPS secondary challenge and validated the interaction between NF-κB and NF-κB consensus binding sites in the NFAT5 promoter region with chromatin immunoprecipitation (ChIP) assays (Figure 5D). We observed a similar increase in the activity of a reporter vector that includes the upstream site (base pairs −3,000 to +1) of NFAT5 that contains the NF-κB consensus sequences (Figure 5E). To better understand the role of NFAT5 in the effect of NLRC3 on glycolysis pathways, we inhibited NFAT5 with the specific inhibitor KRN2 that blocks the interaction between NF-κB and its binding site in the regulatory region of the NFAT5 gene in response to secondary LPS stimulation. This treatment abolished the induction of a glycolytic response by TLR signaling activation and resulted in a significant decrease in ECARs and lactate production in activated immunotolerant NLRC3−/− macrophages in response to LPS secondary challenge (Figures 5F and 5G), with a similar decrease in TNF-α, IL-1β, and IL-6 (Figure 5G). These findings suggest that NF-κB binding to NFAT5 might be essential for NLRC3-deficient-mediated immunometabolism in tolerant macrophages in response to secondary LPS stimulation, but the mechanism by which NLRC3 deficiency enhances NF-κB binding to the NFAT5 promoter region is not completely understood.
NFAT5 connects DNA-bound NF-κB to the histone acetyltransferase p300 when p300 is activated and is critical for the target gene transcription after NF-κB nuclear translocation.47 We thus examined intracellular p300 activity by measuring the acetylation of histone H3 and p300. The results showed that NLRC3 loss significantly increased the p300 activity of LPS-tolerant macrophages in response to secondary LPS stimulation (Figure 5H). p300 is predominantly nuclear but can shuttle between the cytoplasm and nucleus,26,48 and it can be activated to control cell metabolism through direct interaction with mTOR and subsequent mTOR-dependent phosphorylation.26 During colorectal tumorigenesis, NLRC3 regulates mTOR signaling by directly associating with the protein and modulating mTOR phosphorylation.24,25 One possibility is that NLRC3 loss disrupts the association with mTOR and p300, after which mTOR-mediated phosphorylation activates p300, thereby leading to subsequent NFAT5 activation. We overexpressed NLRC3, p300, and mTOR in 293T cells and performed co-immunoprecipitation experiments with p300, and observed that NLRC3, p300, and mTOR were in the same complex (Figure 5I). Consistent with this, INK128 reduced the p300 activity of tolerant macrophages with NLRC3 loss in response to secondary LPS challenge (Figure 5J). Similarly, after using the cerulenin to specifically disrupt the p65-NFAT5-p300 interaction, the NLRC3−/− LPS-tolerized macrophages following scondary LPS treatment exhibited a decrease in the co-precipitation of NFAT5 and p300 in p65 immunoprecipitation assays- (Figure 5K), in the glycolytic activity, and in the level of TNF-α, IL-6, and IL-1β (Figure 5L). These results suggest that NLRC3 loss enhances p300 activity by directly disrupting the association between mTOR and p300, which increases NF-κB binding to NFAT5 after NF-κB translocation, and that the NF-κB-NFAT5 complex controls macrophage metabolism and then modulates sepsis progression.
NLRC3 gene therapy targeting intrapulmonary macrophages improves sepsis-induced suppression of lung immune defenses
Although the benefit of NLRC3 knockdown in enhancing sepsis host immunity has been appreciated, NLRC3 has not yet been developed into a therapeutic target. rAAVs have emerged as attractive, highly versatile, gene delivery agents owing to their safe, stable, and transgene expression, and rAAVs have become the gene therapy vector of choice for human clinical trials.49 To further assess whether NLRC3 knockdown could be used as a potential therapeutic intervention against secondary infection in a septic host experiencing immunosuppression, we used an AAV system (Figure 6A) harboring macrophage-specific synthetic promoter 146 with shRNA targeting NLRC3 (AAV-SP146-miR30-shNLRC3-eGFP, AAV-SP-shNLRC3) to deliver the gene delivery agents according to the experimental setup in Figure 6B. NLRC3 expression in AAV-infected mice was confirmed on day 28 after infection. As shown in Figures 6C and 6D, AAV-SP-shNLRC3 was successfully delivered and expressed in AMs and lung macrophages (LMs), and significantly decreased the expression of NLRC3 in macrophages from mice treated with AAV-SP-shNLRC3 compared with macrophages from the control mice, as well as in their lung epithelial cells (Figure 6E). As sepsis-induced immunosuppression is evidenced by the suppression of host antibacterial defense in the lung, we investigated the role of AAV-SP-shNLRC3 in host defense against secondary intrapulmonary P. aeruginosa challenge in septic mice. As previously observed in the NLRC3 knockout mice, treatment with AAV-SP-shNLRC3 in mice undergoing CLP had significantly increased survival rate following secondary pulmonary P. aeruginosa challenge compared with the control group (Figure 6F). Sham-operated mice infected with Pseudomonas had 100% survival in both the AAV-SP-shNLRC3 and AAV-Ctrl shRNA groups (Figure 6F). Similar to previous observations, lung and blood from AAV-SP-shNLRC3-infected mice undergoing CLP displayed a reduced bacterial load after secondary P. aeruginosa infection (Figure 6G) compared with AAV-Ctrl shRNA groups, and lung homogenates from AAV-SP-shNLRC3-infected mice had higher levels of TNF-α and IL-6 than those of control groups (Figure 6H). Collectively, these results suggest that AAV-SP-shNLRC3 confers protection against secondary challenge with P. aeruginosa in septic mice.
Figure 6.

NLRC3 gene therapy targeting intrapulmonary macrophages improves sepsis-induced suppression of lung immune defense
(A) Full sequence map for AAV-SP146-miR30-shNLRC3-eGFP (AAV-SP-shNLRC3). (B) Schematic overview of experimental design for (C) to (H). Mice were administrated intratracheally with AAV-SP-shNLRC3 or AAV-Ctrl shRNA (nonsense control shRNA), and 28 days later were subjected to CLP or sham operation and secondary intrapulmonary P. aeruginosa infection. (C and D) Flow cytometric analysis of GFP expression in AMs from bronchoalveolar lavage fluid (C) and in lung macrophages (LMs) from homogenates (D) of uninfected and AAV-SP-shNLRC3-infected mice on day 28. (E) RT-PCR detected NLRC3 expression in AMs, LMs, and lung epithelial cells of AAV-SP-shNLRC3 or AAV-Ctrl shRNA-infected mice. (F–H) Surviving mice with AAV-SP-shNLRC3 or AAV-Ctrl shRNA were subjected to secondary intrapulmonary P. aeruginosa infection after CLP or sham operation: Kaplan-Meier survival curves (n = 19 mice/group) (F), bacterial loads in the lung (left) and blood (right) (G), ELISA for TNF-α, IL-1β, and IL-6 in lung homogenate (H); survival curves were monitored for 10 days, and samples were collected after the secondary infection challenge. i.t., intratracheal. Circles represent individual mice. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant (two-way ANOVA or Student’s t test and log-rank test for survival).
Discussion
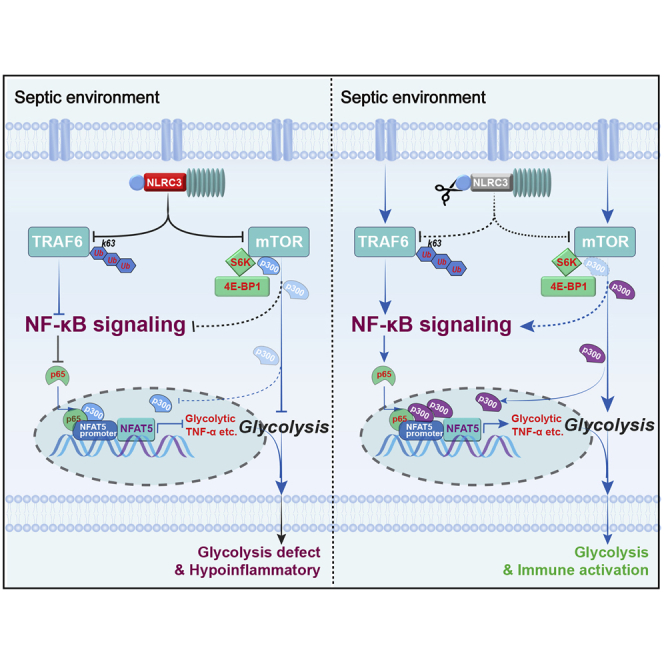
Immunometabolic modulation is critical for orchestrating the host immune defense during sepsis-induced immunosuppression.12,15,17 NLRC3 moderately mitigates CD4+ T cell activation and exerts an immunosuppressive effect in response to viruses.21 However, little is known about the regulatory role of NLRC3 in immunometabolism and innate immune defenses in the setting of sepsis. Our results demonstrate that NLRC3 expression in monocytes correlates with the immunosuppressive states of septic patients, and NLRC3 expression drives glycolytic defects of immunotolerant macrophages by inhibiting NF-κB p65 binding to NFAT5 after NF-κB translocation co-induced by both TRAF6 and mTOR in concert with mTOR-dependent p300 deactivation (Figure 7). Macrophage-specific deletion of NLRC3 could retune this pathway and protect a mouse model of sepsis by developing immunosuppression against secondary bacterial infection.
Figure 7.

Schematic model depicting how NLRC3 drives glycolytic defects and septic immunosuppression
(Left) NLRC3 expression of immunosuppressive macrophages increases in the context of sepsis-induced immunosuppression. On one hand, increased NLRC3 inhibits NF-κB translocation co-induced by both TRAF6- and mTOR-dependent signaling. On the other hand, NLRC3 association with mTOR and p300 inhibits mTOR-mediated phosphorylation dependent on p300 activity. Finally, the p300 activity and the NF-κB binding to the NFAT5 promoter region was decreased, thereby preventing the NF-κB-NFAT5 complex from controlling the expression of genes encoding glycolytic enzymes and proinflammatory cytokines. (Right) Macrophage-specific NLRC3 deletion increases the p300 activity and further enhances NF-κB binding to the NFAT5 promoter region following NF-κB translocation, and thereby the enhanced NF-κB-NFAT5 complex elicits fine-tuning of proinflammatory cytokine production and glycolysis while the septic host achieves an enhanced protective immune response against secondary bacterial challenge.
Septic patients who have undergone immunosuppression are more susceptible to secondary infections as a result of functional reprogramming of myeloid cells,5 which is characterized by hyporesponsiveness and metabolic defects. Our findings reveal NLRC3 as an inducer of monocyte and macrophage hyporesponsiveness during sepsis, whereby monocytes and macrophages exhibit low TNF-α production and glycolysis; myeloid NLRC3-deletion mice were more resistant to secondary bacterial challenge, with strikingly lower lung and blood bacterial loads and an increased survival rate compared with WT mice. However, a previous study demonstrated that NLRC3−/− mice have enhanced responses, including proinflammatory cytokine release and greater body temperature, during LPS-induced sterile shock than WT mice.20 Thus, it is unclear whether NLRC3 deficiency in macrophages will lead to uncontrolled tissue damage and a poor prognosis. In the present study, the mortality rate of septic NLRC3ΔMac mice 48 h after CLP was not strikingly different from that of their WT counterparts, and even splenic histopathology did not reveal significant differences. Although NLRC3ΔMac mice showed enhanced pulmonary inflammation as indicated by a higher histopathology score and a striking increase in cytokine levels (TNF-α, IL-1β, and IL-6), the pulmonary histopathology and inflammation gradually recovered after 2 days of secondary infection. Importantly, to avoid systemic adverse events as much as possible, we developed NLRC3 gene therapy targeting intrapulmonary macrophages, for which an AAV delivery strategy harboring a macrophage-specific promoter that restricts NLRC3 expression to the monocyte/macrophage lineage was designed to confer protection against secondary challenge with P. aeruginosa in septic mice.
Mechanistically, NLRC3 limits immune responses and the cellular glycolytic metabolism of immunotolerant macrophages during LPS rechallenge by suppressing NF-κB signaling. Previous studies revealed that NLRC3 reduced NF-κB activation in macrophages and T cells by attenuating K63-linked ubiquitination of TRAF6 in response to viruses,20,21 but it is unclear whether NLRC3 drives immunotolerant macrophage hyporesponsiveness via the same mechanism. After tolerization, NLRC3−/− macrophages showed heightened polyubiquitination of TRAF6 and increased NF-κB activity upon LPS rechallenge. Preventing activation of TRAF6, an upstream mediator of NF-κB, did not significantly reduce NF-κB activity or glycolysis in NLRC3-deficient tolerant macrophages following LPS rechallenge, suggesting that TRAF6 activation may not be the only mechanism that senses the immunometabolic status. mTOR is a central immunometabolic element in proinflammatory macrophages,50,51 and the associations of NLRC3 with TRAF6 and mTOR can influence NLRC3 function in the regulation of mTOR activation in colon epithelial cells.24,25 However, enhanced glycolytic activity and cytokine production by NLRC3 deficiency in immunotolerant macrophages were not restored after pharmacological inhibition of mTOR. These findings suggest that inhibition of mTOR or TRAF6 activation alone could not restore the enhanced immunometabolic response of immunotolerant macrophages induced by NLRC3 loss. Intriguingly, inhibiting NF-κB nuclear translocation, as a point of cross-talk of the downstream effectors linking TRAF6 and mTOR signaling, could reduce immunometabolic activity, suggesting that NLRC3 might impair the immune and metabolic processes in immunotolerant macrophages by inhibiting NF-κB translocation co-induced by mTOR and TRAF6 activation in the setting of immunosuppression during sepsis.
The mechanisms by which NF-κB regulates immune inflammatory responses in activated immune cells are well described,40 but their role in the immunometabolism of NLRC3-deficient immune cells is less studied. NF-κB is a master transcription factor; when active, it exerts immunologic defense functions by binding to specific DNA sequences in target genes to modulate their transcription. However, knocking down key subunits of the NF-kB pathway did not markedly enhance glycolytic or inflammatory activity elicited by LPS rechallenge in tolerant NLRC3-deficient macrophages. Intriguingly, we identified an additional transcription factor, NFAT5, whose DNA-binding domain shares structural homology with NF-κB52 and has two NF-κB consensus binding sites, as a key regulator of glycolytic activity. We also demonstrated that inducing NF-κB binding to the NFAT5 promoter is required for cell immune function in immunotolerant macrophages with NLRC3 deficiency following LPS rechallenge. Indeed, NFAT5 was also required to induce TNF-α and IL-6 gene expression in response to different stimulation thresholds, and it also controls glycolysis and MHC II expression of macrophages.45,53,54 Our findings show that promoting NF-κB binding to NFAT5 in macrophages can modulate immune responses and the glycolysis of immunotolerant macrophages with deficient NLRC3 expression. Levels of TNF-α, IL-6, and MHC II in lung tissue from NLRC3-deficient septic mice with immunosuppression were enhanced in response to secondary infection. The proglycolytic effect of NFAT5 in NLRC3-deficient immunotolerant macrophages was attenuated with NFAT5 inhibitors. These findings reveal a molecular mechanism for orchestrating immunometabolic activity associated with NFAT5-p65 function in immunotolerant macrophages with NLRC3 deficiency.
Nuclear NFAT5-p65-p300 complexes that connect DNA-bound NF-κB to the histone acetyltransferase p300 after p300 activation are critical for orchestrating NFAT5-p65 function.47 Our results demonstrated that p300 activity is increased in LPS-NLRC3−/−-tolerant macrophages compared with WT macrophages in response to secondary LPS stimulation. When the p65-NFAT5-p300 interaction was disrupted with a specific inhibitor, the glycolytic activity of LPS-NLRC3−/− tolerant macrophage was decreased. However, as a transcriptional co-activator with histone acetyltransferase activity, p300 is predominately localized in the nucleus;48 how the cytoplasmic sensor NLRC3 modulates NFAT5-p65 remains poorly understood. NLRC3 deletion induces mTOR co-localization with lysosomes,25 leading to auto-activation of mTOR, and lysosomal distribution of p300 with mTOR was also observed in HeLa cells.26 Our co-immunoprecipitation analysis revealed that NLRC3, p300, and mTOR were in the same complex. After blocking mTOR activity with inhibitors, p300 activity of immunotolerant macrophages with NLRC3 loss was reduced upon secondary LPS challenge. When p300 was inhibited, the amount of p300 immunoprecipitated with p65 was dramatically lowered, suggesting that hypoacetylation of H3 by NLRC3 occurs when p300 binding is inhibited. These data show that the effects of NLRC3 occur when histone acetylation is prevented by inhibition of p300, thus leading to the inhibition of NF-κB-independent NFAT5. Activated p300 can acetylate cytoplasmic substrates or act as scaffold protein to shuttle proteins from the cytoplasm to the nucleus for gene transcription. This has been demonstrated in numerous biological processes26,48 such as mTOR-mediated autophagy. We thus postulate that nucleocytoplasmic shuttling of p300 may contribute to NFAT5-dependent recruitment of p300 in NLRC3-deficient immunotolerant macrophages. However, the exact mechanism by which NLRC3 modulates p300-NF-κB-NFAT5 signaling needs further investigation.
Although this study showed that NLRC3 deficiency improved lung host defense responses in an experimental model of sepsis-induced immunosuppression in response to acute secondary bacterial infection by regulating macrophage function, and was associated with improved survival in the septic host, it is premature to conclude that myeloid-specific NLRC3 loss is protective for all septic patients because this model primarily serves as a model of immunosuppression rather than a clinically relevant model of infection in critically ill septic patients. However, very few sepsis patients succumb to early death; the majority of critically ill patients survive sepsis and rapidly develop immunosuppression,55,56 thus becoming susceptible to opportunistic secondary infections,2,31 which are the primary cause of death among critically ill patients. The experimental model in the current study in which a secondary intratracheal bacterial challenge was conducted, at one level, simulates the pathophysiological process. More importantly, as high NLRC3 expression correlated with secondary infections in patients with sepsis, NLRC3 expression in monocytes or macrophages may be a reliable parameter to monitor susceptibility to secondary infections or perform sepsis stratification, although this merits future investigation. Also, NLRC3 gene therapy targeting intrapulmonary macrophages improved lung antibacterial host defense and prognosis in septic mice; for septic hosts with high NLRC3 expression in monocytes, targeting NLRC3 in specific cells is likely to be a promising candidate therapy that orchestrates sepsis-induced immunosuppression.
In conclusion, we identified NLRC3 as a driver of monocyte and macrophage tolerance because its inhibition could improve immunometabolism in both cell types in the immunosuppressive period of sepsis. Loss of NLRC3 in macrophages intrinsically enhanced the protective immune response against secondary bacterial infection, in that NLRC3 deficiency activated cellular glycolytic pathways and cytokines by regulating NF-κB-independent NFAT5 signaling in an mTOR-dependent-on-p300 activation manner. Collectively, these results indicate that NLRC3 is a potential target for therapeutic interventions against secondary infection in polymicrobial sepsis.
Materials and methods
Mice
Six- to eight-week-old male C57BL/6 WT mice were obtained from Beijing HFK Bioscience (Beijing, China) and weighed between 20 and 25 g.
The Nlrc3 conditional knockout mouse model (named as Nlrc3fl/+ C57BL/6) was created by Beijing Biocytogen (Beijing, China). In brief, we employed a circular donor vector, and the loxP sites were introduced into introns 1 and 3. Cas9 mRNA, sgRNAs, and donor vector were mixed and co-injected into the cytoplasm of one-cell stage fertilized eggs (C57BL/6) to generate Nlrc3fl/+ C57BL/6 chimeric mice that were mated with C57BL/6 mice to generate Nlrc3fl/+ C57BL/6 mice. Southern blotting with two independent probes (3′ probe [NcoI, WT 4.1 kb, targeted allele 2.8 kb] and LR probe [5′] [NdeI, WT 5.7 kb, targeted allele 2.8 kb]) were used to identify correctly targeted Nlrc3fl/+ C57BL/6 mice. F0 offspring were genotyped using two pairs of primers: L-GT-F1, L-GT-R1 and R-GT-F1, R-GT-R1 (Table S2).
Myeloid-cell-specific NLRC3 knockout mice were generated by crossing NLRC3 floxed mice with LysM-Cre mice (#004781; The Jackson Laboratory, Bar Harbor, ME, USA). Male mice were used for experiments at 8–9 weeks of age. In all experiments, including BMDM studies, NLRC3WT littermates served as controls for NLRC3ΔMac mice. Mice were maintained under specific pathogen-free conditions and a 12:12-h light/dark cycle. They received food and water ad libitum.
Patients and samples
Patients who were admitted to Union Hospital and fulfilled the clinical criteria for sepsis-357 were screened for eligibility. Sepsis-3 defines sepsis as life-threatening organ dysfunction caused by a dysregulated host response to infection. Organ dysfunction can be identified as an acute change in total Sequential Organ Failure Assessment (SOFA) score of 2 or more points due to the infection. Blood samples of septic patients were collected on days 1–2 of hospitalization. Exclusion criteria included pregnancy or breast feeding, age >80 or <18 years, malignancy, organ transplantation, human immunodeficiency virus/acquired immunodeficiency syndrome, autoimmune diseases, and immunosuppressive medication use during the last month. Age- and sex-matched volunteers without sepsis for elective surgery served as controls. Demographic information and clinical variables of patients, including age, sex, infection site, types of infection, SOFA score, length of hospital and ICU stay, and 28-day mortalities were retrieved from available medical records (Table S3). SOFA grades the function of six organ systems on a scale of 0–4 depending on the degree of dysfunction using objective measurements. The maximum SOFA scores were the highest (worst) scores within 24 h of sepsis diagnosis. Hospital-acquired infection was diagnosed if the patient had a positive culture of a new pathogen obtained from lower respiratory tract specimens (bacterial pneumonia), blood samples (bacteremia), or urine (urinary tract infection) collected ≥48 h after ICU admission.58
PBMCs were isolated by dilution of the blood in pyrogen-free PBS and density gradient centrifugation over Ficoll/Paque (catalog [cat.] #17-1440-03; GE Healthcare, Chicago, IL, USA). Cells were washed twice in PBS and resuspended in RPMI 1640 medium (cat. #12-167F; Invitrogen, Carlsbad, CA, USA) containing 10% (v/v) fetal bovine serum (FBS) (cat. #TMS-013-B; Millipore, Billerica, MA, USA), 10 mM L-glutamine, and 50 U/mL penicillin/streptomycin (cat. #15070-063; HyClone, Logan, UT, USA). Cells were counted in a Coulter counter (Coulter Electronics, Hialeah, FL, USA) and adjusted to 5 × 106 cells/mL.
Monocytes were separated from other cells using the plastic adherence method.29 Monocyte HLA-DR (cat. #307619; BioLegend, San Diego, CA, USA) expression was measured with flow cytometry. Moreover, monocytes were also stimulated with RPMI culture medium containing Escherichia coli LPS (055: B5; 10 ng/mL; cat. # L6529; Sigma, St. Louis, MO, USA) for 12 h in the presence of 10% serum, and supernatants were stored at −80°C until cytokine and lactate measurements were performed.
We identified septic patients in an immunosuppression phase as indicated by low HLA-DR expression on monocytes (<30%) and low TNF-α levels in supernatants. A total of 35 septic patients met the above conditions. Detailed clinical information is presented in Table S3.
Cell preparation
Murine AMs and PMs were harvested by lavage as previously described.59 Cells were resuspended in RPMI 1640 containing 2 mM L-glutamine, 1% penicillin/streptomycin, and 10% FBS, and cultured for 2 h. Cells were washed with RPMI 1640 to remove nonadherent cells; only adherent monolayer cells were used.
BMDMs from the indicated mice femurs and tibias were obtained using 30 ng/mL recombinant macrophage colony stimulating factor (cat. # 315-02; PeproTech, Rocky Hill, NJ, USA) as described previously.60 Adherent BMDMs were detached on day 6 and replated in multiwell plates for analysis.
RAW 264.7 (TIB-71) cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). Before use, mycoplasma contamination was negative for all cells. NLRC3 stably overexpressing (ovNLRC3) and knockdown RAW 264.7 cells (shNLRC3) were selected using 8 μg/mL polybrene (cat. # H9268; Sigma) as described previously.61 The lentiviral vectors harboring shNLRC3, shTRAF6, p65, c-Rel, or overexpressing a vector encoding NLRC3 were constructed by Genechem (Shanghai, China), and were used to infect the RAW 264.7 cells or BMDMs as described by the manufacturer. These cells were grown in RPMI 1640 or Dulbecco’s minimum essential medium (DMEM) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin at 37°C, 95% humidity, and 5% CO2. HEK293 cells (ATCC #3216) were also cultured in DMEM with 10% FBS at 37°C under 5% CO2.
For primary AM or PM stimulation in vitro, 100 ng/mL LPS was added for the indicated time. To induce immune tolerance, RAW 264.7 cells or BMDMs were preincubated with 100 ng/mL LPS. After 24 h, cells were washed with PBS, cultured in medium (2 h), and rechallenged with 100 ng/mL LPS for the desired amount of time. In such experiments, unstimulated (naive) BMDMs or RAW 264.7 were incubated in the medium. After 24 h, cells were washed and challenged with 100 ng/mL LPS to produce responsive cell controls. Pharmacological inhibition of mTOR (300 nmol/L; INK128, cat. #S281104; Selleck Chemicals, Houston, TX, USA), inhibition of NF-κB (300 μM; BAY 11-7082, cat. #S2913; Selleck Chemicals), disruption of NFAT5-p65-p300 interaction (10 μM; cerulenin, cat. # C2389; Sigma), and inhibition of NFAT5 (0.8 μM, KRN2, cat. #248260-75-5; Sigma) were achieved by pretreating cells with inhibitors for 1 h before LPS activation unless otherwise indicated.10,21
Bacterial strains and culturing conditions
P. aeruginosa (ATCC #27853) was obtained as previously described.7,8,28,62 In brief, P. aeruginosa was incubated overnight in Luria-Bertani medium at 37°C with constant shaking (100 cycles min−1). Bacteria were washed and diluted in PBS. The concentration of bacteria was determined by reading the optical density (OD) at 600 nm and then plotting the OD on a reference curve verified by quantitative culture of the inoculum. The bacteria were then diluted to the desired concentration.
Polymicrobial sepsis model and secondary infection
For polymicrobial sepsis induction, the CLP model was performed as previously described.29,30,60 In brief, male C57BL/6 mice ranging from 22 to 28 g (8–10 weeks old) or littermates of the same sex NLRC3fl/flLysM-cre− (NLRC3WT) and NLRC3fl/flLysM-cre+ (NLRC3ΔMac) mice matched by age and body weight underwent CLP. Approximately 50% of the cecum was ligated and punctured twice with a 20-gauge needle to induce severe sepsis. Saline (1 mL) was injected subcutaneously for resuscitation immediately after closing the abdomen. A broad-spectrum carbapenem antibiotic (imipenem-cilastatin, 25 mg/kg; Merck, Kenilworth, NJ, USA) was given beginning 6 h after CLP and then every 12 h for the first 4 days.63 Sham-treated mice received the same operation without puncture.
For the secondary infection model, sepsis-surviving mice were anesthetized and infected with 20 μL (3 × 109 CFU mL−1) of P. aeruginosa intratracheally (i.t.) on day 2 after CLP as described previously.29,30 Control mice were administered with 20 μL of PBS i.t. Mice were monitored for 10 days or sacrificed 6–120 h after CLP and 12–96 h after i.t. challenge to collect serum or tissue.
AM and PM stimulation
Adherent AMs and PMs from CLP and sham mice were stimulated ex vivo with or without 100 ng/mL LPS (cat. # L6529; Sigma) at 37°C and 5% CO2 for indicated times. Cell lysates and supernatants were harvested for mRNA expression or ELISAs and measurement of lactate levels.
qPCR
Total RNA was extracted from macrophages using TRIzol reagent as previously described.64 cDNA synthesis was performed using PrimeScript RT reagent kit with gDNA eraser (RR047B; TaKaRa, Kusatsu, Japan). qPCR was run using SYBR Premix Ex Taq II Assay (RR820A; TaKaRa) on a C1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA). The primers are presented in Table S2. Target gene expression was normalized to β-actin, and fold changes were calculated via the 2–ΔΔCt method.
Histological assays
Mice were sacrificed at the indicated time points. After cardiac perfusion with PBS, the left upper of lung and spleen tissues were obtained, fixed in 4% paraformaldehyde overnight, and embedded in paraffin, and 4-μm sections were cut and stained with hematoxylin and eosin. The slides were imaged using a microscope. Lung injury was scored by a blinded investigator from 0 (absent) to 4 (severe) with the following parameters: necrosis or formation of abscess, interstitial inflammation, endothelialitis, bronchitis, edema, thrombi, pleuritis, and percentage of the lung surface demonstrating confluent (diffuse) inflammatory infiltrate, as previously described.65
Total lung leukocyte preparation
Whole-lung samples were harvested from euthanized mice and collagenase digested as previously described.7 The digestion buffer was prepared with RPMI, 10% fetal calf serum, 1 mg/mL collagenase (Roche, Basel, Switzerland), and 30 mg/mL DNase (Sigma-Aldrich), and the lung slurries were enzymatically digested for 30 min at 37°C. Samples were then centrifuged through a 40% Percoll gradient to enrich for leukocytes. Viable cells were counted on a hemocytometer by trypan blue exclusion and then cultured in RPMI, followed by flow cytometry (as described below).
Bacterial counts
Bacterial loads in lung and blood were determined as previously described.7,66 In brief, lungs were harvested after pulmonary vasculature perfusion with PBS containing 5 mM EDTA and then homogenized for 1 min with a homogenizer (Ultra-Turrax T25; Ika, Staufen, Germany) in 1 mL of sterile saline. Homogenates were then serially diluted 1:5 in PBS. Blood was collected from the right ventricle with a heparinized syringe at 24 h after P. aeruginosa challenge, then serially diluted 1:2 with PBS, after which 10 μL of each dilution was plated on blood agar to determine lung and blood CFU.
ELISA
The concentrations of TNF-α (cat. #500850; Cayman Chemical, Ann Arbor, MI, USA), IL-6 (cat. # 583371; Cayman Chemical), and IL-1β (cat. # EMC001b; Neobioscience, Beijing, China) in mouse serum, cell culture supernatants, and lung homogenates were determined by ELISA according to the manufacturer’s instructions. The lung tissues (right lobes) were obtained and homogenized in 2 mL of PBS with 0.05% Tween 80, and homogenized supernatants were filtered (0.22-μm pore size).
Metabolism assays
Lactate levels in the supernatants were measured using a Lactate Assay Kit (cat. #700510; Cayman Chemicals) according to the manufacturer’s instructions. In brief, the supernatants were collected and deproteinated, and neutralized acid was used to remove any precipitated salts. The reaction mix was added to the samples, and the average fluorescence of each sample was measured on a microplate reader (OD 570 nM) to calculate the lactate concentration.
ECARs were measured with a Seahorse XFe24 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA) as previously described.34 In brief, macrophages were seeded into XF-24 cell culture microplates (2 × 105 cells/well in 200 μL, cat. #102342-100) and then stimulated with LPS with or without the inhibitors (BAY 11-7082 or INK128 or KRN2) for the indicated time. The cells were washed with XF Running Buffer and then placed at 37°C for 1 h in the absence of CO2. Glycolysis was measured with the XF Glycolysis Stress Test Kit (cat. #103020-100; Agilent Technologies) according to the manufacturer’s instructions, and the macrophages were measured over time and exposed to glucose, oligomycin, and 2-DG for ECAR measurement at the indicated time points. ECARs were recorded three times for each condition. The basal ECAR, glycolysis (ECAR after glucose addition), glycolytic capacity (maximal ECAR after subtracting the ECAR following 2-DG exposure), and glycolytic reserve (difference between oligomycin-induced maximal ECAR and glucose-induced glycolytic flux) were calculated.
Flow cytometry
Single-cell suspensions of lung tissue, spleen tissue, and peritoneal cavity were harvested as previously described8,29 and washed in ice-cold PBS containing 1% FBS. Cells were preincubated with anti-FcgRII/FcgRIII antibodies (cat. #101320; BioLegend) and fixable viability dye eFluor 780 (cat. #565388; BD Biosciences, Franklin Lakes, NJ, USA) in DMEM with 2% FBS for 15 min at 4°C, then stained with fluorescently labeled antibodies for 30 min at 4°C. The cells were fixed and permeabilized with the IC Staining Buffer Kit (cat. #00-5523-00; eBioscience, Hatfield, UK) for intracellular transcription factor before antibody labeling. Stained cells were washed twice with 2 mL of fluorescence-activated cell sorting buffer (Dulbecco’s PBS containing 1% FBS and 2 mM EDTA). Flow cytometry antibodies and other reagents are listed in Table S4. Flow cytometry data were acquired on a FACSDiva flow cytometer (BD Biosciences) and further analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
Ubiquitination assay
Ubiquitination experiments were performed as described previously.21,35,67 In brief, cells were lysed with 1% SDS isotonic buffer, and lysed cell samples were boiled at 95°C for 15 min to remove any noncovalent interactions. Lysates were cleared by centrifugation (15,000 × g, 4°C, 15 min) and diluted with 1% Triton X-100 buffer. Lysates were immunoprecipitated with anti-TRAF6 antibody (cat. #sc-8409; Santa Cruz Biotechnology, Dallas, TX, USA), incubated overnight at 4°C with gentle shaking, after which protein G agarose beads (cat. #9863; Cell Signaling Technology, Danvers, MA, USA) were added, and incubation was continued for another 4 h at 4°C. The beads were washed and resuspended in sample buffer. Immunoprecipitates were separated on mini-gels (Invitrogen), and TRAF6 and K63 ubiquitination of TRAF6 were detected by immunoblotting with an antibody for either K63 Ub (cat. #5621; Cell Signaling Technology) or TRAF6 (cat. #sc-8409; Santa Cruz Biotechnology).
Immunoprecipitation and immunoblotting
Co-immunoprecipitation was performed as described previously.25 In brief, cells were collected with ice-cold PBS and lysed by three freeze-thaw cycles in PBS containing 0.1% Triton X-100 and protease and phosphatase inhibitors, then centrifuged at 13,000 rpm for 15 min at 4°C. The supernatants were incubated with 10 μL of Protein A/G agarose beads (cat. #20423; Pierce, Rockford, IL, USA) for 2 h to remove endogenous immunoglobulin G and centrifuged at 5,000 rpm for 5 min to remove the beads. For immunoprecipitation, protein samples were incubated with 3 μg of primary antibodies at 4°C for 12 h on a rocking platform. Protein A/G agarose beads (20 μL) were then added to enrich the primary antibody/protein complex for another 4 h on a rocking platform. The beads were washed five times with PBS containing 0.1% Triton X-100. Immunocomplexes were eluted using 2× SDS sample buffer and boiled at 100°C for 5 min.
Immunoblotting was performed as described previously.65 Cells were lysed in lysis buffer including proteinase/phosphatase inhibitors (cat. #MSSAFE; Sigma), and nuclear and cytoplasmic proteins were extracted. Cleared lysates were separated using 8%–15% SDS-PAGE. Proteins were transferred onto polyvinylidene fluoride membranes (cat. #IPVH00010; Millipore). The membranes were blocked with 5% non-fat dry milk (cat. #9999; Cell Signaling Technology) in TBST buffer (cat. #9997; Cell Signaling Technology) at room temperature and then incubated with the indicated primary antibodies at 4°C overnight. The antibody to mouse NLRC3 (cat. #ab77817; Abcam, Cambridge, UK) was used at 1:500 dilution in 5% non-fat milk. The other antibodies are listed in Table S4 and were used at 1:1,000 dilution; all secondary antibodies were used at a 1:40,000 dilution. Band densities were normalized to β-actin expression.
NFAT5 transcriptional activity assay
A green fluorescent protein (GFP)-NFAT5 promoter reporter system with two NF-κB binding sites and a NFAT5 transcriptional reporter with consensus sequence (TGG AAA ATT ACC G) were constructed as previously described46 and then transfected into NLRC3 stable knockdown RAW 264.7 with Lipofectamine 3000 (cat. #L3000015; Invitrogen). The cells were then made tolerant and rechallenged by LPS for the indicated time. Reporter gene activity was assessed by measuring GFP expression using a FACSDiva flow cytometer (BD Biosciences).
Chromatin immunoprecipitation
ChIP assays were performed according to the manufacturer’s protocol (Pierce Agarose ChIP kit, cat. #26156; Pierce).35 In brief, macrophages were harvested and fixed with 1% formaldehyde for 5 min at 37°C. The chromatin was purified by micrococcal nuclease digestion, after which soluble chromatin-containing lysates were immunoprecipitated with an anti-p65 antibody (cat. #8242; Cell Signaling Technology). The DNA sequences were amplified by PCR with specific primers to analyze p65/NF-κB binding capacity. Primers used to amplify NFAT5-bound regions were 5′-TTT GGA GGA TCC CTC TTC AC-3′ and 5′-ACA AGT CAA GAA GGG CCA AG-3′. Enrichment of DNA fragments was normalized against an NFAT5 exon region using the following control primers: 5′-GCG AGA TGA TGT CAC TTC AG-3′ and 5′-GTG GAA GTT TGA CTG TGG AC-3′.46
RNA-seq analysis
Total RNA was isolated from 1.0 × 106 BMDMs as previously described.1,64 RNA samples were obtained from BMDMs with or without LPS treatment in three independent WT and NLRC3−/− mice. RNA concentration and integrity were verified using a Nanodrop analyzer (Agilent). High-quality RNA was used for cDNA library construction. RNA-seq libraries were generated with the KAPA Stranded RNA-Seq Kit for Illumina with multiplexing primers, according to the supplier’s protocols, after which RNA-seq was conducted with the Illumina Nova sequencer (Illumina, San Diego, CA, USA). Differential gene expression analysis was performed using DESeq R package (ver. 1.10.1) with raw gene counts output from Rsubread. Counts for NLRC3 were based on number of RNA sequences in the deleted region of NLRC3 in NLRC3−/− mouse: from the final 103 bp of exon 2 to the first 35 bp of exon 4.20 The RNA-seq data used to identify targeting gene expression patterns of NFAT5 were obtained from the NCBI’s GEO database (GEO: GSE76554 and GSE49604).45,53 Raw sequence data were downloaded and analyzed with HISAT2 (ver. 2.0.5.2). Heatmaps of gene expression were generated using the pheatmap R package, and differential expression was analyzed using DESeq, GOseq, and R packages. Subsequent gene ontology/Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed based on differentially expressed genes identified in the previous step (adjusted p < 0.05) using the limma package (ver. 3.36.5), clusterProfiler package (ver. 3.8.1), and org.Hs.eg.db package (ver. 3.6.0) of R software.
Recombinant adeno-associated virus and intrapulmonary targeting interference
An rAAV vector carrying macrophage-specific NLRC3 deletion plasmid was generated by Hanbio Biotechnology (Shanghai, China). The AAV9 system harbors a macrophage-specific synthetic promoter 146,68 an miR30-based shRNA targeting NLRC3,69 a cytomegalovirus promoter, and an enhanced GFP reporter. The nucleotide shRNA of NLRC3 was cloned using the following sequence: 5′-GAG AAC CAC GGT CTG CAC CAT ATT A-3′. Four-week-old male mice were administered with AAV-SP-shNLRC3 or AAV-Ctrl shRNA virus at a dose of 1 × 1011 viral particles in 20 μL of PBS per mouse via intratracheal injection. GFP expression levels in infected macrophages were detected with flow cytometry and NLRC3 expression by real-time PCR on day 28 after AAV injection. The survival rate, bacterial load, and cytokine levels were assessed as described above.
Quantification and statistical analysis
Data are shown as mean ± standard error (SE). When multiple experiments using different numbers of animals were pooled for statistical analysis, the exact number (n) is indicated in the figure legend. Two-sided Student’s t tests were used to compare two groups. For multiple comparisons, one- or two-way ANOVA was performed followed by post hoc Bonferroni tests for two comparisons between groups. The correlation analysis was performed by Pearson rank correlation tests. Kaplan-Meier survival curves were compared using log-rank tests. All statistical tests were two-tailed with significance set at p < 0.05. All data were analyzed using GraphPad Prism software (ver. 8.3.0) (GraphPad Software, San Diego, CA, USA).
Ethics statement
This protocol was approved by the Institutional Review Board of Tongji Medical College (No. 2019 (S1001)). Informed consent for the studies was obtained according to the Declaration of Helsinki. All animal protocols were performed in accordance with the institutional guidelines approved by the Tongji Medical College, Huazhong University of Science and Technology Animal Care and Use Committee.
Acknowledgments
This study was supported by National Natural Science Foundation of China (nos. 81772047, 81971818, 82002026, and 82002099). We thank Bioacme Biotechnology (Wuhan, China) for RNA- sequencing.
Author contributions
Conceptualization: Y.S., S.Ya., and J.X. methodology: J.X., C.G., Y.Q., X.Zh., S.Yu., S.Ya, and Y.S. investigation: J.X., Y.H., Z.P., X.F., H.X., M.S., D.S., and C.G. visualization: J.X, Y.S., S.Ya., Y.H., Y.Q., and X.Zh. supervision: S.Yu, S.Ya., and Y.S. writing—original draft: J.X., S.Ya., and Y.S. writing—review & editing: J.X., Y.Q., X.Zh., S.Ya., and Y.S.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.08.023.
Supplemental information
Data and materials availability
The authors declare that all data are available in the article and supplemental information from the corresponding author upon reasonable request. The RNA-seq data have been deposited in the NCBI GEO repository with the accession numbers GEO: GSE211989, GSE49604, and GSE76554.
References
- 1.Nedeva C., Menassa J., Duan M., Liu C., Doerflinger M., Kueh A.J., Herold M.J., Fonseka P., Phan T.K., Faou P., et al. TREML4 receptor regulates inflammation and innate immune cell death during polymicrobial sepsis. Nat. Immunol. 2020;21:1585–1596. doi: 10.1038/s41590-020-0789-z. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss R.S., Monneret G., Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuk S.A., Kim H., Abutaleb N.S., Dieterly A.M., Taha M.S., Tsifansky M.D., Lyle L.T., Seleem M.N., Yeo Y. Nanocapsules modify membrane interaction of polymyxin B to enable safe systemic therapy of Gram-negative sepsis. Sci. Adv. 2021;7:eabj1577. doi: 10.1126/sciadv.abj1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteban A., Frutos-Vivar F., Ferguson N.D., Peñuelas O., Lorente J.A., Gordo F., Honrubia T., Algora A., Bustos A., García G., et al. Sepsis incidence and outcome: contrasting the intensive care unit with the hospital ward. Crit. Care Med. 2007;35:1284–1289. doi: 10.1097/01.CCM.0000260960.94300.DE. [DOI] [PubMed] [Google Scholar]
- 5.Seeley J.J., Baker R.G., Mohamed G., Bruns T., Hayden M.S., Deshmukh S.D., Freedberg D.E., Ghosh S. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature. 2018;559:114–119. doi: 10.1038/s41586-018-0253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otto G.P., Sossdorf M., Claus R.A., Rödel J., Menge K., Reinhart K., Bauer M., Riedemann N.C. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care. 2011;15:R183. doi: 10.1186/cc10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng J.C., Cheng G., Newstead M.W., Zeng X., Kobayashi K., Flavell R.A., Standiford T.J. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J. Clin. Invest. 2006;116:2532–2542. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roquilly A., McWilliam H.E.G., Jacqueline C., Tian Z., Cinotti R., Rimbert M., Wakim L., Caminschi I., Lahoud M.H., Belz G.T., et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity. 2017;47:135–147.e5. doi: 10.1016/j.immuni.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Iwasaki A., Medzhitov R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langston P.K., Nambu A., Jung J., Shibata M., Aksoylar H.I., Lei J., Xu P., Doan M.T., Jiang H., MacArthur M.R., et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 2019;20:1186–1195. doi: 10.1038/s41590-019-0453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavaillon J.M., Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng S.C., Scicluna B.P., Arts R.J.W., Gresnigt M.S., Lachmandas E., Giamarellos-Bourboulis E.J., Kox M., Manjeri G.R., Wagenaars J.A.L., Cremer O.L., et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016;17:406–413. doi: 10.1038/ni.3398. [DOI] [PubMed] [Google Scholar]
- 13.Krejčová G., Danielová A., Nedbalová P., Kazek M., Strych L., Chawla G., Tennessen J.M., Lieskovská J., Jindra M., Doležal T., Bajgar A. Drosophila macrophages switch to aerobic glycolysis to mount effective antibacterial defense. Elife. 2019;8:e50414. doi: 10.7554/eLife.50414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucey T.M., Verma J., Harrison P.F., Snelgrove S.L., Lo T.L., Scherer A.K., Barugahare A.A., Powell D.R., Wheeler R.T., Hickey M.J., et al. Glucose homeostasis is important for immune cell viability during Candida challenge and host survival of systemic fungal infection. Cell Metab. 2018;27:988–1006.e7. doi: 10.1016/j.cmet.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van den Bossche J., O'Neill L.A., Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. 2017;38:395–406. doi: 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 16.He Y.J., Xu J.Q., Sun M.M., Fang X.Z., Peng Z.K., Pan S.W., Zhou T., Wang Y.X., Shang Y. Glucocorticoid-induced leucine zipper: a promising marker for monitoring and treating sepsis. Front. Immunol. 2020;11:606649. doi: 10.3389/fimmu.2020.606649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies R., O'Dea K., Gordon A. Immune therapy in sepsis: are we ready to try again? J. Intensive Care Soc. 2018;19:326–344. doi: 10.1177/1751143718765407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delano M.J., Ward P.A. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J. Clin. Invest. 2016;126:23–31. doi: 10.1172/JCI82224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiersinga W.J., Leopold S.J., Cranendonk D.R., van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5:36–44. doi: 10.4161/viru.25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider M., Zimmermann A.G., Roberts R.A., Zhang L., Swanson K.V., Wen H., Davis B.K., Allen I.C., Holl E.K., Ye Z., et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat. Immunol. 2012;13:823–831. doi: 10.1038/ni.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchimura T., Oyama Y., Deng M., Guo H., Wilson J.E., Rampanelli E., Cook K.D., Misumi I., Tan X., Chen L., et al. The innate immune sensor NLRC3 acts as a rheostat that fine-tunes T cell responses in infection and autoimmunity. Immunity. 2018;49:1049–1061.e6. doi: 10.1016/j.immuni.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu S., Du X., Huang Y., Fu Y., Yang Y., Zhan X., He W., Wen Q., Zhou X., Zhou C., et al. NLRC3 negatively regulates CD4+ T cells and impacts protective immunity during Mycobacterium tuberculosis infection. Plos Pathog. 2018;14:e1007266. doi: 10.1371/journal.ppat.1007266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conti B.J., Davis B.K., Zhang J., O'Connor W., Jr., Williams K.L., Ting J.P.Y. CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J. Biol. Chem. 2005;280:18375–18385. doi: 10.1074/jbc.M413169200. [DOI] [PubMed] [Google Scholar]
- 24.Karki R., Malireddi R.K.S., Zhu Q., Kanneganti T.D. NLRC3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle. 2017;16:1243–1251. doi: 10.1080/15384101.2017.1317414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karki R., Man S.M., Malireddi R.K.S., Kesavardhana S., Zhu Q., Burton A.R., Sharma B.R., Qi X., Pelletier S., Vogel P., et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature. 2016;540:583–587. doi: 10.1038/nature20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan W., You Z., Xu Y., Zhou L., Guan Z., Peng C., Wong C.C.L., Su H., Zhou T., Xia H., Liu W. mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol. Cell. 2017;68:323–335.e6. doi: 10.1016/j.molcel.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 27.Mithal L.B., Arshad M., Swigart L.R., Khanolkar A., Ahmed A., Coates B.M. Mechanisms and modulation of sepsis-induced immune dysfunction in children. Pediatr. Res. 2022;91:447–453. doi: 10.1038/s41390-021-01879-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao J., Xu F., Lin S., Song Z., Zhang L., Luo P., Xu H., Li D., Zheng K., Ren G., Yin Y. IL-27 controls sepsis-induced impairment of lung antibacterial host defence. Thorax. 2014;69:926–937. doi: 10.1136/thoraxjnl-2014-205777. [DOI] [PubMed] [Google Scholar]
- 29.Dan C., Jinjun B., Zi-Chun H., Lin M., Wei C., Xu Z., Ri Z., Shun C., Wen-Zhu S., Qing-Cai J., Wu Y. Modulation of TNF-α mRNA stability by human antigen R and miR181s in sepsis-induced immunoparalysis. EMBO Mol. Med. 2015;7:140–157. doi: 10.15252/emmm.201404797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoetzenecker W., Echtenacher B., Guenova E., Hoetzenecker K., Woelbing F., Brück J., Teske A., Valtcheva N., Fuchs K., Kneilling M., et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 2011;18:128–134. doi: 10.1038/nm.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim E.Y., Ner-Gaon H., Varon J., Cullen A.M., Guo J., Choi J., Barragan-Bradford D., Higuera A., Pinilla-Vera M., Short S.A., et al. Post-sepsis immunosuppression depends on NKT cell regulation of mTOR/IFN-γ in NK cells. J. Clin. Invest. 2020;130:3238–3252. doi: 10.1172/JCI128075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mages J., Dietrich H., Lang R. A genome-wide analysis of LPS tolerance in macrophages. Immunobiology. 2007;212:723–737. doi: 10.1016/j.imbio.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Foster S.L., Hargreaves D.C., Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 34.Vaeth M., Maus M., Klein-Hessling S., Freinkman E., Yang J., Eckstein M., Cameron S., Turvey S.E., Serfling E., Berberich-Siebelt F., et al. Store-operated Ca(2+) entry controls clonal expansion of T cells through metabolic reprogramming. Immunity. 2017;47:664–679.e6. doi: 10.1016/j.immuni.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuk J.M., Kim T.S., Kim S.Y., Lee H.M., Han J., Dufour C.R., Kim J.K., Jin H.S., Yang C.S., Park K.S., et al. Orphan nuclear receptor ERRα controls macrophage metabolic signaling and A20 expression to negatively regulate TLR-induced inflammation. Immunity. 2015;43:80–91. doi: 10.1016/j.immuni.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Voss K., Hong H.S., Bader J.E., Sugiura A., Lyssiotis C.A., Rathmell J.C. A guide to interrogating immunometabolism. Nat. Rev. Immunol. 2021;21:637–652. doi: 10.1038/s41577-021-00529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F., Ye X.J., Chen M.Y., Li H.C., Wang Y.F., Zhong M.Y., Zhong C.S., Zeng B., Xu L.H., He X.H., Ouyang D.Y. Inhibition of NLRP3 inflammasome activation and pyroptosis in macrophages by taraxasterol is associated with its regulation on mTOR signaling. Front. Immunol. 2021;12:632606. doi: 10.3389/fimmu.2021.632606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mauro C., Leow S.C., Anso E., Rocha S., Thotakura A.K., Tornatore L., Moretti M., De Smaele E., Beg A.A., Tergaonkar V., et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011;13:1272–1279. doi: 10.1038/ncb2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Londhe P., Yu P.Y., Ijiri Y., Ladner K.J., Fenger J.M., London C., Houghton P.J., Guttridge D.C. Classical NF-κB metabolically reprograms sarcoma cells through regulation of hexokinase 2. Front. Oncol. 2018;8:104. doi: 10.3389/fonc.2018.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kracht M., Müller-Ladner U., Schmitz M.L. Mutual regulation of metabolic processes and proinflammatory NF-κB signaling. J. Allergy Clin. Immunol. 2020;146:694–705. doi: 10.1016/j.jaci.2020.07.027. [DOI] [PubMed] [Google Scholar]
- 41.Meylan E., Dooley A.L., Feldser D.M., Shen L., Turk E., Ouyang C., Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 43.Müller M.R., Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 2010;10:645–656. doi: 10.1038/nri2818. [DOI] [PubMed] [Google Scholar]
- 44.Aramburu J., López-Rodríguez C. Regulation of inflammatory functions of macrophages and T lymphocytes by NFAT5. Front. Immunol. 2019;10:535. doi: 10.3389/fimmu.2019.00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi S., You S., Kim D., Choi S.Y., Kwon H.M., Kim H.S., Hwang D., Park Y.J., Cho C.S., Kim W.U. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J. Clin. Invest. 2017;127:954–969. doi: 10.1172/JCI87880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han E.J., Kim H.Y., Lee N., Kim N.H., Yoo S.A., Kwon H.M., Jue D.M., Park Y.J., Cho C.S., De T.Q., et al. Suppression of NFAT5-mediated inflammation and chronic arthritis by novel κB-binding inhibitors. EBioMedicine. 2017;18:261–273. doi: 10.1016/j.ebiom.2017.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee H.H., Sanada S., An S.M., Ye B.J., Lee J.H., Seo Y.K., Lee C., Lee-Kwon W., Küper C., Neuhofer W., et al. LPS-induced NFκB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci. Rep. 2016;6:24921. doi: 10.1038/srep24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Tu K., Liu D., Guo L., Chen Y., Li Q., Maiers J.L., Liu Z., Shah V.H., Dou C., et al. p300 acetyltransferase is a cytoplasm-to-nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming Growth factor β-stimulated hepatic stellate cells. Hepatology. 2019;70:1409–1423. doi: 10.1002/hep.30668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye L., Park J.J., Dong M.B., Yang Q., Chow R.D., Peng L., Du Y., Guo J., Dai X., Wang G., et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat. Biotechnol. 2019;37:1302–1313. doi: 10.1038/s41587-019-0246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jing C., Castro-Dopico T., Richoz N., Tuong Z.K., Ferdinand J.R., Lok L.S.C., Loudon K.W., Banham G.D., Mathews R.J., Cader Z., et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc. Natl. Acad. Sci. USA. 2020;117:15160–15171. doi: 10.1073/pnas.2000943117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang S., Nakanishi Y., Kioi Y., Okuzaki D., Kimura T., Takamatsu H., Koyama S., Nojima S., Nishide M., Hayama Y., et al. Semaphorin 6D reverse signaling controls macrophage lipid metabolism and anti-inflammatory polarization. Nat. Immunol. 2018;19:561–570. doi: 10.1038/s41590-018-0108-0. [DOI] [PubMed] [Google Scholar]
- 52.Lopez-Rodríguez C., Aramburu J., Rakeman A.S., Rao A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc. Natl. Acad. Sci. USA. 1999;96:7214–7219. doi: 10.1073/pnas.96.13.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buxadé M., Lunazzi G., Minguillón J., Iborra S., Berga-Bolaños R., Del Val M., Aramburu J., López-Rodríguez C. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J. Exp. Med. 2012;209:379–393. doi: 10.1084/jem.20111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buxadé M., Huerga Encabo H., Riera-Borrull M., Quintana-Gallardo L., López-Cotarelo P., Tellechea M., Martínez-Martínez S., Redondo J.M., Martín-Caballero J., Flores J.M., et al. Macrophage-specific MHCII expression is regulated by a remote Ciita enhancer controlled by NFAT5. J. Exp. Med. 2018;215:2901–2918. doi: 10.1084/jem.20180314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mira J.C., Gentile L.F., Mathias B.J., Efron P.A., Brakenridge S.C., Mohr A.M., Moore F.A., Moldawer L.L. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit. Care Med. 2017;45:253–262. doi: 10.1097/CCM.0000000000002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brakenridge S.C., Efron P.A., Cox M.C., Stortz J.A., Hawkins R.B., Ghita G., Gardner A., Mohr A.M., Anton S.D., Moldawer L.L., Moore F.A. Current epidemiology of surgical sepsis: discordance between inpatient mortality and 1-year outcomes. Ann. Surg. 2019;270:502–510. doi: 10.1097/SLA.0000000000003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G.R., Chiche J.D., Coopersmith C.M., et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3) Jama. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Vught L.A., Klein Klouwenberg P.M.C., Spitoni C., Scicluna B.P., Wiewel M.A., Horn J., Schultz M.J., Nürnberg P., Bonten M.J.M., Cremer O.L., et al. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. Jama. 2016;315:1469–1479. doi: 10.1001/jama.2016.2691. [DOI] [PubMed] [Google Scholar]
- 59.Xu Y., Meng C., Liu G., Yang D., Fu L., Zhang M., Zhang Z., Xia H., Yao S., Zhang S. Classically activated macrophages protect against lipopolysaccharide-induced acute lung injury by expressing amphiregulin in mice. Anesthesiology. 2016;124:1086–1099. doi: 10.1097/ALN.0000000000001026. [DOI] [PubMed] [Google Scholar]
- 60.Kim E., Beon J., Lee S., Park S.J., Ahn H., Kim M.G., Park J.E., Kim W., Yuk J.M., Kang S.J., et al. Inositol polyphosphate multikinase promotes Toll-like receptor-induced inflammation by stabilizing TRAF6. Sci. Adv. 2017;3:e1602296. doi: 10.1126/sciadv.1602296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuk J.M., Shin D.M., Lee H.M., Kim J.J., Kim S.W., Jin H.S., Yang C.S., Park K.A., Chanda D., Kim D.K., et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011;12:742–751. doi: 10.1038/ni.2064. [DOI] [PubMed] [Google Scholar]
- 62.Hoogerwerf J.J., Leendertse M., Wieland C.W., de Vos A.F., de Boer J.D., Florquin S., van der Poll T. Loss of suppression of tumorigenicity 2 (ST2) gene reverses sepsis-induced inhibition of lung host defense in mice. Am. J. Respir. Crit. Care Med. 2011;183:932–940. doi: 10.1164/rccm.201006-0934OC. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H., Zeng L., Xie M., Liu J., Zhou B., Wu R., Cao L., Kroemer G., Wang H., Billiar T.R., et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020;27:556–570.e6. doi: 10.1016/j.chom.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu J., Hu H., Chen B., Yue R., Zhou Z., Liu Y., Zhang S., Xu L., Wang H., Yu Z. Lycopene protects against hypoxia/reoxygenation injury by alleviating ER stress induced apoptosis in neonatal mouse cardiomyocytes. PLoS One. 2015;10:e0136443. doi: 10.1371/journal.pone.0136443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu J., Li H.B., Chen L., Wang Y.X., Lu S., Li S.N., Cui S.N., Xiao H.R., Qin L., Hu H., et al. BML-111 accelerates the resolution of inflammation by modulating the Nrf2/HO-1 and NF-κB pathways in rats with ventilator-induced lung injury. Int. Immunopharmacol. 2019;69:289–298. doi: 10.1016/j.intimp.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 66.de Tymowski C., Heming N., Correia M.D.T., Abbad L., Chavarot N., Le Stang M.B., Flament H., Bex J., Boedec E., Bounaix C., et al. CD89 is a potent innate receptor for bacteria and mediates host protection from sepsis. Cell Rep. 2019;27:762–775.e5. doi: 10.1016/j.celrep.2019.03.062. [DOI] [PubMed] [Google Scholar]
- 67.Xiong Y., Qiu F., Piao W., Song C., Wahl L.M., Medvedev A.E. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IkappaB kinase gamma and increases A20 expression. J. Biol. Chem. 2011;286:7905–7916. doi: 10.1074/jbc.M110.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang W.S., Kwon J.S., Kim H.B., Jeong H.Y., Kang H.J., Jeong M.H., Cho J.G., Park J.C., Kim Y.S., Ahn Y. A macrophage-specific synthetic promoter for therapeutic application of adiponectin. Gene Ther. 2014;21:353–362. doi: 10.1038/gt.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fellmann C., Hoffmann T., Sridhar V., Hopfgartner B., Muhar M., Roth M., Lai D.Y., Barbosa I.A.M., Kwon J.S., Guan Y., et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data are available in the article and supplemental information from the corresponding author upon reasonable request. The RNA-seq data have been deposited in the NCBI GEO repository with the accession numbers GEO: GSE211989, GSE49604, and GSE76554.