

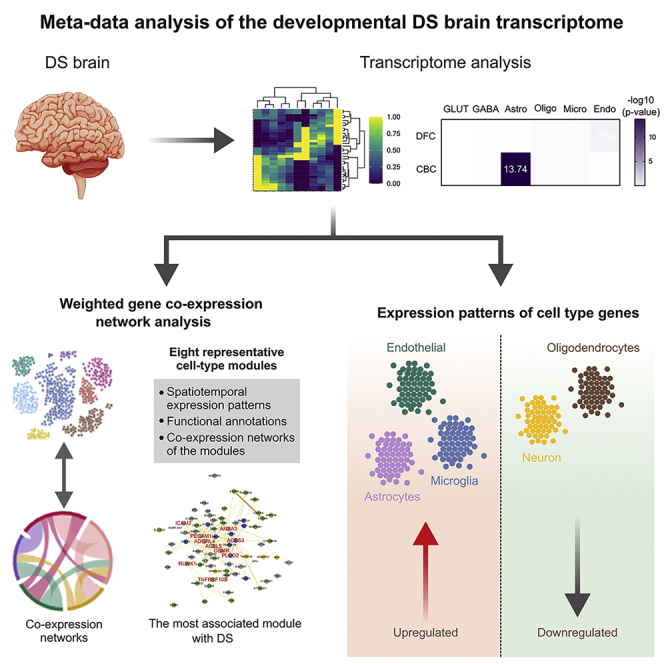
Summary
Down syndrome (DS) is the most common genetic cause of intellectual disability and increases the risk of other brain-related dysfunctions, like seizures, early-onset Alzheimer’s disease, and autism. To reveal the molecular profiles of DS-associated brain phenotypes, we performed a meta-data analysis of the developmental DS brain transcriptome at cell type and co-expression module levels. In the DS brain, astrocyte-, microglia-, and endothelial cell-associated genes show upregulated patterns, whereas neuron- and oligodendrocyte-associated genes show downregulated patterns. Weighted gene co-expression network analysis identified cell type-enriched co-expressed gene modules. We present eight representative cell-type modules for neurons, astrocytes, oligodendrocytes, and microglia. We classified the neuron modules into glutamatergic and GABAergic neurons and associated them with detailed subtypes. Cell type modules were interpreted by analyzing spatiotemporal expression patterns, functional annotations, and co-expression networks of the modules. This study provides insight into the mechanisms underlying brain abnormalities in DS and related disorders.
Subject areas: Developmental neuroscience, Transcriptomics
Graphical abstract
Highlights
-
•
Cell type-specific spatiotemporal co-expression modules are interpreted
-
•
Astrocyte, microglia, and endothelial cell associated genes are upregulated in the DS
-
•
Neuron and oligodendrocyte-associated genes are downregulated in the DS brain
-
•
The most related module with DS was enriched in endothelial cell-associated genes
Developmental neuroscience; Transcriptomics.
Introduction
Down syndrome (DS) is the most common genetic disorder causing intellectual disability; it occurs in approximately 1 per 800−1,200 live births.1,2 DS results from trisomy of human chromosome 21 (HSA21) and affects a wide range of phenotypes in many organ systems. Representative characteristics of DS include physical appearance, neurological symptoms, heart disease, cancer, and gastrointestinal problems.3,4 Among them, intellectual disability is the most common phenotype in individuals with DS, implying that almost all individuals with DS have abnormal brain phenotypes.
Because of the sustained influence of the extra copy of HSA21, the prenatal brain with DS develops abnormally.5 Its structural characteristics, such as the size of several brain regions, their connectivity, and the number or morphology of specific cell populations, differ from those of the normal brain.6 These developmental and structural differences and associated changes in gene expression patterns caused by a third copy of HSA21 affect various functions of the brain, leading to many brain-related symptoms. Reports have shown that individuals with DS have a higher incidence of seizures than euploid individuals and are more likely to develop early-onset Alzheimer’s disease in their 40s.7 In addition, associations between DS and psychiatric disorders, particularly autism spectrum disorders, have been reported.8,9,10
Although trisomy of HSA21 is the definitive underlying cause of DS and its associated symptoms, the mechanisms by which supernumerary HSA21 triggers these symptoms remain unclear. Substantial efforts have been made to identify and elucidate the roles of candidate genes on HSA21, but further work is needed to clarify the genotype−phenotype relationships. With the development of functional genomics, questions about the roles of the additional HSA21-coding and-non-coding sequences on the whole transcriptome have arisen.6 In a previous study that characterized gene expression in varied regions of postmortem human brains of DS and euploid controls ranging in age from 14 weeks post-conception to 42 years old, approximately 5% of differentially expressed genes (DEGs) were on HSA21, and the rest were on the other chromosomes.11 Therefore, to decipher the exact mechanisms of DS phenotypes, an investigation of the integrated features of gene expression in all chromosomes is required.
Tissue transcriptome data consist of aggregate gene expression data from heterogeneous cell types; nevertheless, the gene expression signatures are quite different across various cell types.12,13 Most biological phenomena, including brain development, function, and disease onset, are defined by the complex interactions between different cell types.14,15 Therefore, when intact tissue transcriptome data is used to investigate the biological mechanisms, there is a need to establish a method for clarifying the relevant cell types of differentially expressed transcripts.
Previous co-expression network analysis of DS, which is based on the human brain transcriptome, showed the important contributions of oligodendrocyte-lineage cells.11 Our aim in this study was to explore specific cell types associated with the abnormal brain development of DS. We first performed a co-expression network analysis to investigate the network of genes on all chromosomes across brain development without bias, and genes with spatiotemporally similar expression patterns were grouped together into gene modules. Cell-type enrichment analysis on the gene co-expression modules made it possible to identify gene modules related to specific brain cell types. The spatiotemporal expression patterns, associated cell types, and functional annotations of co-expression modules provided insights into the role of specific cell types in biological processes or mechanisms related to DS.
Results
Cell-type enrichment analysis on DEGs
In this study, we used DS human brain transcriptome data, GSE59630,11 from the Gene Expression Omnibus (GEO) repository. These data include DS and matched control samples from various brain regions, spanning many different developmental time points (Table S1). DEGs of these data were identified in the dorsolateral prefrontal cortex (DFC) and cerebellar cortex (CBC) in a previous study using the paired t-test (false discovery rate (FDR)-adjusted p-value <0.1).11 We focused on these two regions because they have the widest range of developmental periods and are associated with cognition and motor coordination in DS phenotypes.16,17 Cell-type enrichment analysis on DEGs was conducted using published cell type-enriched gene lists.18 We divided neuron-enriched genes into glutamatergic neuron- and GABAergic neuron-enriched genes and their subtypes (Table S2). This was based on the overlaps between cell type-enriched genes18 and genes that were co-expressed with marker genes of glutamatergic or GABAergic neurons and their subtypes.19 DEGs in the CBC were significantly enriched with astrocyte-enriched genes (Benjamini−Hochberg-adjusted (BHA) p = 1.8 × 10−14). Although the DEGs in the DFC were not significantly enriched with any cell type-enriched genes, the most enriched cell type was endothelial cell (BHA p = 3.0 × 10−1) (Figure S1).
Cell type-enriched genes were differentially expressed in DS
To determine the differential expression patterns of cell type-enriched genes (Table S2), we calculated the expression differences of these genes in each region at different developmental stages using a sliding window approach. Generally, genes associated with astrocyte, microglia, and endothelial cells were upregulated in DS, whereas oligodendrocyte- and neuron-enriched genes were downregulated (Figure 1). Expression differences usually increased with age, except for the microglia- and endothelial cell-enriched genes in the CBC data. Microglia- and endothelial cell-enriched genes were gradually more expressed in the DFC of DS than in control samples; however, this pattern was reversed and the expression differences gradually reduced with age in the CBC. Furthermore, microglia-enriched genes were less expressed in the CBC of DS in the last sliding window. Overall, glutamatergic and GABAergic neuron-enriched genes showed similar patterns. However, in the fetal and infancy stages (periods 5–9; human brain developmental periods were previously described in Kang et al.19), the expression patterns of these neuron types were dissimilar. Glutamatergic neuron-enriched genes were similarly expressed in DS and control DFC, but GABAergic neuron-enriched genes were less expressed in the DS DFC than in the control DFC in this period. Genes enriched in subtypes of glutamatergic and GABAergic neurons showed similar differential expression patterns to whole glutamatergic and GABAergic neuronal genes (Figures S2 and S3).
Figure 1.

Cell type-enriched genes were differentially expressed in DS
Differential expression patterns of each cell type-related gene between DS and control are displayed on a log2 scale across brain regions and developmental stages. A sliding window approach was used to investigate the temporal expression pattern. Human brain developmental periods were based on the criteria presented in Kang et al.19 Genes associated with astrocytes, microglia, and endothelial cells showed upregulated patterns in DS, whereas oligodendrocyte- and neuron-enriched genes showed downregulated patterns in DS. Expression differences were mostly increased in older brains. Differences in glutamatergic neuron- and GABAergic neuron-related genes appeared in fetal and infancy stages. There were no significant expression differences in glutamatergic neuron-enriched genes; however, GABAergic neuron-enriched genes showed significant expression differences in fetal and infancy stages. ∗, p< 0.05 (paired t-test).
Cell type-enriched weighted gene co-expression network analysis (WGCNA) modules were identified
We performed WGCNA20 to compare and analyze the brain transcriptomes of DS individuals and matched controls at the system level from an unbiased perspective. Module detection by gene clustering resulted in 57 co-expression gene modules. To establish cleaner modules, we allocated genes with a module membership (kME) value >0.7 in each module. This gene list in each module was used in subsequent analyses (Table S3). We calculated module−trait relationships using the module eigengene, which is the first principal component of each module,21 and filtered out some modules that were more correlated with other confounding factors (RIN, PMI, race, and sex) than main factors (brain region, developmental stage, and disease status) (Table S4). Finally, we obtained 43 modules significantly related to specific traits, like brain region, developmental stage, and disease status (Table S4).
Cell-type enrichment analysis was conducted on all WGCNA modules (Figure 2A) using the cell type-enriched genes (Table S2). Cell type-enriched modules (BHA p< 1 × 10−2) consisted of a large proportion of genes that were mainly expressed in those cell types and they did not usually have overlap between different cell types. Although glutamatergic and GABAergic neurons are neuron subclasses, enriched modules for these subtypes did not overlap. Oligodendrocyte- and microglia-enriched genes were exclusively enriched in one module for each cell type (module(M)14 and M41, respectively). However, there were many enriched modules for astrocytes and neurons. By contrast, endothelial cell-enriched genes were less enriched in modules than other cell type-enriched genes.
Figure 2.

Cell type-enriched WGCNA modules were identified
(A and B) Cell-type enrichment analysis with cell type-enriched genes of brain cell types (A) and neuronal subtypes (B) was conducted on 43 co-expressed gene modules constructed by WGCNA. The-log10 (p-value) value is shown (BHA p< 0.05, Fisher’s exact test). Cell type-enriched modules (BHA p< 1 × 10−2) for each cell type hardly overlapped, even in glutamatergic and GABAergic neurons. Oligodendrocyte- and microglia-enriched genes were exclusively enriched in one module for each cell type, but many modules were enriched for astrocytes and neurons.
We performed cell-type enrichment analysis using the genes of glutamatergic and GABAergic neuron subtypes (Figure 2B). M21 was enriched for layers (L) 2–4 and L6 glutamatergic neuronal genes. M24 and M26 were enriched for L2−4 and L4 glutamatergic neuronal genes. M24 was more enriched for L2−4 glutamatergic neuronal genes than for L4 glutamatergic neuronal genes; however, the opposite was the case in M26. L1 neuronal genes were less enriched in modules than other glutamatergic neuron subtypes but were significantly enriched in M17 and M18. M23 was mostly enriched for genes correlated with CALB1 and NOS1. Genes correlated with the pan-GABAergic markers, GAD1 and GAD2, and PVALB were mainly enriched in M32. Furthermore, M32 was enriched for genes correlated with CALB1 and NOS1. No module was significantly enriched for genes correlated with CALB2. Genes correlated with CCK and VIP were specifically enriched in M42. Next, we chose cell type-enriched modules for each cell type (astrocytes: M5 and M29; oligodendrocytes: M14; microglia: M41; glutamatergic neurons: M24 and M26; GABAergic neurons: M32 and M42) and characterized these modules.
Characterization of cell type-enriched modules
Cell type-enriched modules showed varied gene expression patterns
First, we identified the expression patterns of genes in cell type-enriched modules across developmental stages, brain regions, and disease statuses (Figures 3, 4, 5, and 6). Genes in the astrocyte modules, M5 and M29, were expressed at low levels during the fetal stages and were upregulated in DS brains, especially at later stages (Figure 3). M5 genes were expressed at similar levels for the DFC and CBC (Figures 3A and 3C), whereas M29 genes were more expressed in the DFC than in the CBC (Figures 3B and 3D). The expression of genes in the oligodendrocyte module, M14, gradually increased with age and was generally downregulated in DS (Figures 4A and 4C). These expression differences were more remarkable in the DFC than in the CBC. The microglia module (M41) genes were more expressed in postnatal DFC than fetal DFC and were specifically upregulated in the later stages of DS DFC (Figures 4B and 4D).
Figure 3.

Gene expression patterns of astrocyte modules
(A–D) Gene expression patterns of astrocyte modules (M5 and M29) are visualized in a line graph of module eigengenes (A and B) and a gene expression heatmap (C and D). Genes in astrocyte modules were relatively less expressed during prenatal stages and were upregulated in DS. Gene expressions of M5 genes were similar between the DFC and CBC, whereas M29 genes were less expressed in the CBC than in the DFC.
Figure 4.

Gene expression patterns of oligodendrocyte and microglia modules
(A–D) Gene expression patterns of oligodendrocyte and microglia modules (M14 and M41, respectively) are visualized in a line graph of module eigengenes (A and B) and a gene-expression heatmap (C and D). M14 genes became gradually more expressed with age and were downregulated in DS, especially in the DFC. M41 genes were highly expressed in the older DFC of DS.
Figure 5.

Gene expression patterns of glutamatergic neuron modules
(A–D) Gene expression patterns of glutamatergic neuron modules (M24 and M26) are visualized in a line graph of module eigengenes (A and B) and a gene expression heatmap (C and D). M24 and M26 genes showed high expression levels during fetal stages, unlike other modules. In addition, they were more expressed in the DFC than in the CBC and were downregulated in DS. M24 was more constrained by developmental stages and disease status than M26, which was more constrained by brain region.
Figure 6.

Gene expression patterns of GABAergic neuron modules
(A–D) Gene expression patterns of GABAergic neuron modules (M32 and M42) are visualized by a line graph of module eigengenes (A and B) and a gene expression heatmap (C and D). M32 and M42 genes were repressed during fetal stages and were downregulated in DS. Genes in M32 showed similar expression patterns between the DFC and CBC, but genes in M42 were substantially repressed in the CBC.
Genes of glutamatergic neuron modules (M24 and M26) were more expressed in the DFC than in the CBC and, unexpectedly, were highly expressed during fetal stages. This implies that they are related to development (Figure 5). These genes were downregulated in the brains of DS individuals. M24 genes were more affected by disease status and developmental stage than M26 (Figure 5). Genes in M32, one of the GABAergic modules, were repressed during fetal stages and more expressed during later stages (Figures 6A and 6C). They were downregulated in DS individuals and showed few regional differences. Genes in the other GABAergic module (M42) also showed low expression during the fetal stages (Figures 6B and 6D). With the exception of the prenatal stages, they were consistently expressed across all stages. The expression of M42 genes in the CBC was lower than in the DFC during the fetal stages. These genes showed few expression differences between control and DS brains. All selected cell-type modules showed unique gene expression patterns and, therefore, could affect different phenotypes of DS.
Biological roles of cell type-enriched modules were disclosed
To interpret the biological function of these cell-type modules, we conducted gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Figures 7 and 8, and Tables S5, S6, S7, S8, S9, S10, S11, and S12). M5 genes were enriched for growth-related terms like “regulation of cell proliferation” (BHA p = 1.1 × 10−3), “heart growth” (BHA p = 7.1 × 10−4), “growth factor binding” (p = 8.5 × 10−4), “fibroblast growth factor binding” (p = 3.0 × 10−4), and “signaling pathways regulating pluripotency of stem cells” (p = 1.3 × 10−2) (Figure 7A and Table S5). They were expected to be expressed outside of cells and be involved in cell–cell interactions, as they were enriched for the terms “extracellular vesicle” (BHA p = 1.6 × 10−3) and “cell junction” (BHA p = 4.7 × 10−2). The other astrocyte module, M29, was enriched for categories related to biomolecule transport and metabolism, such as “nitrogen compound transport” (p = 5.6 × 10−4), “organonitrogen compound catabolic process” (p = 2.4 × 10−4), “carboxylic acid transport” (p = 7.2 × 10−4), “carboxylic acid biosynthetic process” (p = 5.3 × 10−4), and “dicarboxylic acid metabolic process” (p = 3.5 × 10−4) (Figure 7B and Table S6). The GO and the KEGG pathway enrichment analysis results of M14 and M41 were consistent with the cell-type enrichment analysis results (Figures 7C and 7D, and Tables S7 and S8). The oligodendrocyte module, M14, was enriched for “axon ensheathment” (BHA p = 3.1 × 10−8), “myelin sheath” (p = 3.9 × 10−3), and “compact myelin” (p = 4.0 × 10−3) (Figure 7C and Table S7). The microglia module, M41, was enriched for many immune response-related terms, like “immune response” (BHA p = 9.3 × 10−30), “leukocyte activation” (BHA p = 1.5 × 10−17), “inflammatory response” (BHA p = 1.8 × 10−13), and “myeloid leukocyte activation” (BHA p = 1.8 × 10−12) (Figure 7D and Table S8).
Figure 7.

Functional annotation of glial cell type-related modules
(A–D) Functional enrichment analysis with genes related to GO terms and KEGG pathways on the astrocyte (A and B), oligodendrocyte (C), and microglia (D) modules. The five most significant terms (p< 0.05) are shown in each category. GO: gene ontology, BP: biological process, CC: cellular component, MF: molecular function, KEGG: Kyoto Encyclopedia of Genes and Genomes pathway. M5 genes were enriched for growth-related terms (“regulation of cell proliferation”, “heart growth”, “growth factor binding”, “fibroblast growth factor binding”, and “signaling pathways regulating pluripotency of stem cells”) and cell–cell interaction-related terms (“extracellular vesicle” and “cell junction”). M29 was enriched for biomolecule transport and metabolism-related terms (“nitrogen compound transport”, “organonitrogen compound catabolic process”, “carboxylic acid transport”, “carboxylic acid biosynthetic process”, and “dicarboxylic acid metabolic process”). M14 was enriched for oligodendrocyte-related terms (“axon ensheathment”, “myelin sheath”, and “compact myelin”), and M41 was enriched for immune-related terms (“immune response”, “leukocyte activation”, “inflammatory response”, and “myeloid leukocyte activation”).
Figure 8.

Functional annotation of neuronal cell type-related modules
(A–D) Functional enrichment analysis with genes related to GO terms and KEGG pathways on the glutamatergic (A and B) and GABAergic (C and D) neuron modules. The five most significant terms (p< 0.05) are shown in each category. GO: gene ontology, BP: biological process, CC: cellular component, MF: molecular function, KEGG: Kyoto Encyclopedia of Genes and Genomes pathway. M24 and M26 were enriched for neurodevelopment-related terms (“cell projection morphogenesis”, “muscle organ development”, and “growth cone” in M24, and “neuron development”, “neuron projection development”, “central nervous system neuron differentiation”, and “filopodium” in M26). M24 was enriched for a dendrite-related term (“dendritic shaft”), whereas M26 was enriched for axon-related terms (“axon”, “delayed rectifier potassium channel activity”, “ephrin receptor activity”, and “axon guidance”). M32 was enriched for synapse-related terms (“secretion by cell”, “regulation of neurotransmitter levels”, “presynapse”, “syntaxin-1 binding”, and “synaptic vesicle cycle”). M42 was enriched for calcium–calmodulin signaling-related terms (“cAMP metabolic process”, “calmodulin binding”, and “calcium signaling pathway”).
All neuron modules were enriched for neuron-related terms, consistent with the cell-type enrichment analysis results (Figure 8 and Tables S9, S10, S11, and S12). M24 and M26 were enriched for development-related terms, including “cell projection morphogenesis” (p = 1.3 × 10−4), “muscle organ development” (p = 4.4 × 10−3), and “growth cone” (p = 1.0 × 10−2) in M24 and “neuron development” (BHA p = 6.0 × 10−13), “neuron projection development” (BHA p = 2.0 × 10−12), “central nervous system neuron differentiation” (BHA p = 2.6 × 10−7), and “filopodium” (p = 6.1 × 10−3) in M26 (Figures 8A and 8B, and Tables S9 and S10). These results are consistent with the expression patterns of these modules—the modules’ expressions were characteristically upregulated during the fetal stages (Figure 5). The difference between the M24 and M26 results is that M24 was enriched for “dendritic shaft” (p = 1.6 × 10−2) and M26 was enriched for axon-related terms, like “axon” (BHA p = 3.9 × 10−2), “delayed rectifier potassium channel activity” (BHA p = 4.6 × 10−2), “ephrin receptor activity” (BHA p = 1.4 × 10−2), and “axon guidance” (BHA p = 7.8 × 10−7). As such, we inferred that M24 and M26 are related to neurite development and axon guidance, respectively. M26 was also enriched for “cognition” (BHA p = 5.0 × 10−7), and for glutamate-related categories like “glutamate receptor activity” (p = 2.2 × 10−3) and “glutamatergic synapse” (p = 2.8 × 10−3). GO analysis results are consistent with cell-type enrichment analysis. One of the GABAergic modules, M32, appeared to be related to synaptic vesicle and release of neurotransmitters, because it was enriched for “secretion by cell” (BHA p = 3.5 × 10−4), “regulation of neurotransmitter levels” (BHA p = 3.4 × 10−5), “presynapse” (BHA p = 1.5 × 10−5), “syntaxin-1 binding” (BHA p = 2.9 × 10−2), and “synaptic vesicle cycle” (BHA p = 5.9 × 10−4) (Figure 8C and Table S11). M32 was also enriched for “learning or memory” (BHA p = 3.5 × 10−2) and “GABAergic synapse” (BHA p = 1.7 × 10−3). M42 was enriched for diverse terms related to calcium-calmodulin signaling, like “cAMP metabolic process” (BHA p = 3.2 × 10−2), “calmodulin binding” (p = 2.4 × 10−3), and “calcium signaling pathway” (BHA p = 2.6 × 10−2) (Figure 8D and Table S12). Thus, we inferred the biological roles of co-expression gene modules through GO and pathway enrichment analysis.
Co-expression networks were visualized with hub genes of modules
We visualized the intramodular co-expression networks in each module to exhibit the relationship among genes in the module and the position of hub genes in each network (Figures 9 and 10). In every co-expression module network, the top 10 hub genes with the highest kME value were in the center. Their expression patterns were correlated with many other genes in the module. A large proportion of genes in the networks of cell type-enriched modules was most expressed in the associated cell type (M5: 74.8%, M14: 71.4%, M24: 64.4%, M26: 54.6%, M29: 54.2%, M32: 62.3%, M41: 90.3% and M42: 44.8%) in the Brain RNA-seq database.22 This tendency was strongest within the hub genes (Figures 9 and 10).
Figure 9.

Co-expression networks and hub genes of glial cell type-related modules
(A–D) Intramodular co-expression networks of the astrocyte (A and B), oligodendrocyte (C), and microglia (D) modules. Nodes indicate genes. The width and blueness of edges are proportional to the co-expression level between two genes. The 10 genes with the highest kME values were selected as hub genes and marked with large red labels. The nodes of genes that showed higher expression in the cell type of each module when compared with other cell types are colored yellow.
Figure 10.

Co-expression networks and hub genes of neuronal cell type-related modules
(A–D) Intramodular co-expression networks of glutamatergic neuron (A and B) and GABAergic neuron (C and D) modules. Nodes indicate genes, and the width and blueness of edges are proportional to the co-expression level between two genes. The 10 genes with the highest kME values were selected as hub genes and marked with large red labels. The nodes of genes that showed higher expression in the cell type of each module when compared with other cell types are colored yellow.
We investigated the cell type in which the hub genes were most expressed, as well as the GO and KEGG pathway terms that included the most hub genes. In the case of M5, seven hub genes were most expressed in astrocytes. The hub genes were mainly enriched for the “extracellular vesicle” pathway (ALDH6A1, PLPP3, ATP1A2, PLSCR4, NDRG2, and IL6ST) (Figures 7A and 9A, and Table S5). Nine hub genes in M29 were mainly expressed in astrocytes. The hub genes were most enriched for the “organonitrogen compound catabolic process” (GPC5, GLUD1, and NT5E), “nitrogen compound transport” (SLC1A2, SLC15A2, and GLUD1), and the “intrinsic component of plasma membrane” (SLC39A12, GPC5, and SLC15A2) (Figures 7B and 9B, and Table S6). Nine of the M14 hub genes were most expressed in oligodendrocytes. The most relevant term was “extracellular region part” (TMEM63A, HSPA2, and ASPA) (Figures 7C and 9C, and Table S7). Furthermore, nine of the M41 hub genes were mostly expressed in microglia; they were most enriched for “immune response” (NCKAP1L, C3AR1, ITGB2, CYBB, PTPRC, C3, and FYB) and the “cell surface receptor signaling pathway” (NCKAP1L, C3AR1, ITGB2, CYBB, PTPRC, ADORA3, and FYB) (Figures 7D and 9D, and Table S8).
The network of M24 looked dispersed because it had few gene pairs whose co-expression correlation was above the threshold, even though the threshold was identical to that of other neuron modules (Figure 10A). Eight hub genes were chiefly expressed in neurons. The most associated terms with the hub genes were “synapse” and “synapse part”, which were enriched for the same hub genes (SYT13, PRKAR2B, GABRB3, GABRA3, and DCLK1) (Figures 8A and 10A, and Table S9). All hub genes in M26 were primarily expressed in neurons. The terms “neuron development” and “neuron projection development” were equally enriched for most genes (PLK2, PAK3, MEF2C, KALRN, SLIT2, and LRRC4C) (Figures 8B and 10B, and Table S10). M32 had nine hub genes principally expressed in neurons. The term enriched for the most hub genes was “synapse part” (GABRA1, NSF, FAIM2, and SV2B) (Figures 8C and 10C, and Table S11). Six hub genes of M42 were highly expressed in neurons. The hub genes were enriched for diverse terms as the result of GO and KEGG pathway enrichment (Figures 8D and 10D, and Table S12). The neuronal hub genes of M42 were most enriched for “nervous system development” (GDA and NGEF) and “calmodulin binding” (RASGRF2 and RGS4). These results agree with the GO and KEGG pathway enrichment analysis and provide a basis for future studies.
The most DS-associated module was related to endothelial cells, microglia, and astrocytes
We identified the most disease-related module, M40, using multiple linear regression (Table S4). M40 was significantly enriched with microglia- and endothelial cell-related genes in cell-type enrichment analysis (Figure 2). The most DS-associated M40 module was highly overexpressed in DS brain, especially in DFC, and more expressed in postnatal periods than in fetal periods (Figures 11A and 11B). M40 was enriched with immune-response- and vasculature-associated terms like “immune response” (BHA p = 1.9 × 10−4), “leukocyte migration” (BHA p = 9.0 × 10−6), “positive regulation of vasculature development” (BHA p = 8.9 × 10−8), “cytokine–cytokine receptor interaction” (p = 1.1 × 10−2), “HIF-1 signaling pathway” (p = 6.2 × 10−3), and “adipocytokine signaling pathway” (p = 1.6 × 10−2) (Figure 11C). Most M40 genes (89.7%) were mainly expressed in three cell types: endothelial cell, microglia, and astrocyte (Figure 11D). Six of the ten hub genes were mainly expressed in endothelial cell, and two of them were mostly expressed in astrocyte; the other two were mostly expressed in microglia.
Figure 11.

Characterization of the most DS-associated module
(A–D) Gene expression patterns of the most DS-associated module (M40) are visualized in a line graph of module eigengenes (A) and a gene expression heatmap (B). Functional enrichment analysis with genes related to GO terms and KEGG pathways on M40 (C). The five most significant terms (p< 0.05) are shown in each category. GO: gene ontology, BP: biological process, CC: cellular component, MF: molecular function, KEGG: Kyoto Encyclopedia of Genes and Genomes pathway. Intramodular co-expression networks of M40 (D). Nodes indicate genes, and the width and darkness of edges are proportional to the co-expression level between two genes. The 10 genes with the highest kME values were selected as hub genes and are marked with large red labels. The nodes of genes that showed higher expression in a brain cell type are colored (astrocyte: blue, microglia: green, endothelial cell: grey, and oligodendrocyte: yellow), and the nodes of genes that are not related to any cell type are colored white. Genes of M40 were upregulated in DS and related to cell signaling, immune response, vasculature, and extracellular space.
Discussion
In this study, we present a comprehensive interpretation of the human DS brain transcriptome by applying co-expression network analysis and associating it with cell-type enrichment analysis. Co-expression gene modules related to major brain cell types showed unique expression patterns across age, brain region, and disease status. Through these analyses, we describe systemic changes in the DS brain at the cell type or the biological process level that other DEG analyses have not presented.
Cell-type enrichment analysis for DEGs showed differences by brain regions (Figure S1). DEGs from DFC and CBC tend to be rich in endothelial cell-associated genes and astrocyte-associated genes, respectively. These regional differences do not seem to be due to differences in the composition of each cell type between brain regions.23 Instead, they may be due to differences in the molecular context of function between brain regions. For example, cerebellar astrocytes are involved in the progression and onset of ataxia;24 cerebellar ataxia is one of the causes of prevalent hypotonia in DS. On the other hand, DEG, which is enriched in endothelial cell genes in DFC, is predicted to have an impact on the vascular pathology of early-onset Alzheimer’s disease seen in DS; microhemorrhage, cerebral amyloid angiopathy, and inflammation are observed in the DS brain in molecular and imaging studies.25,26,27 Although other cell types are also associated with DS symptoms, they are not enriched in DEGs. This is likely because of the complexity of datasets, which comprise samples of various developmental stages. Therefore, we investigated differential expression patterns of cell type-related genes across the developmental stages in each brain region (Figure 1). The differential expression patterns of cell type-related genes roughly demonstrated the state of each major brain cell type. In the DFC regions, genes related to astrocytes, microglia, and endothelial cells were upregulated as development progressed, whereas neuron- and oligodendrocyte-related genes were downregulated. This may imply an increase in the immune reaction through microglial activation28 or alteration of the blood−brain barrier29 and a decrease in neural transmission by regulating synaptic functions30 or by reducing myelination.31 Consistent with the results, neuroinflammatory changes associated with Alzheimer’s disease have been detected in the DS brain,32,33 and neural transmission is slowed in the DS brain.11
Neuroinflammation in the DS brain has been actively studied in recent years.34,35 Thus, we further investigated expression patterns of immune cell-related genes (Figure S4). We used an immune cell-related gene set from a previously published study that presented immune cell gene signatures.36 Immune cell-related genes, including myeloid and lymphoid genes, were expressed similarly to microglia and endothelial cell genes, which suggests the possibility of their cooperation in neuroinflammation (Figures 1 and S4). Peripheral immune infiltration and blood–brain barrier leakage in the DS brain have not been well studied. However, Alzheimer’s disease, which frequently occurs in individuals with DS, has been verified to result in peripheral immune cell infiltration in the brain.37 We also visualized expression patterns of more subdivided immune cell type-related genes (Figures S5–S8). The features were roughly similar to the overall expression patterns of myeloid and lymphoid genes. Nonetheless, some immune cell-type genes showed a unique expression trajectory. For example, Type 1 helper T cell genes were mostly downregulated in the DS brain, unlike other immune cell-type genes (Figures S7 and S8). This contradicts the peripheral immune system of DS.38 Further studies using single-cell RNA sequencing may be needed to elucidate the precise role of peripheral immune cells in the DS brain.
The WGCNA enabled a systematic analysis of the transcriptome by grouping co-expressed genes that were likely involved in the same biological process. Significant enrichment of cell type-related genes in some of the WGCNA modules indicated that cell type-related genes were highly co-expressed, especially in microglia and oligodendrocytes. Neuron- and astrocyte-associated genes were also enriched but were comparatively more distributed than other cell-type genes across many modules. This implies that these cell types have more diverse roles in DS brain development. Eight cell-type modules introduced in this paper were identified to be related to DS through multiple linear regression (Table S4).
The expression patterns of the two astrocyte modules, M5 and M29, were similar. Genes in these modules were repressed in fetal DFC tissues, were relatively highly expressed in postnatal samples, and showed decreased expression only in the late-adult brain of the control compare to DS (Figure 3), consistent with previous findings.39,40,41,42,43 However, M29 was only expressed well in the DFC and was suppressed in the CBC, whereas M5 was expressed well in both regions. Six of the 10 hub genes in M5 (ALDH6A1, PLPP3, ATP1A2, PLSCR4, NDRG2, and IL6ST) were included in the gene list related to the GO term “extracellular vesicle”. Studies have shown that exosome secretion is increased in the DS brain and alleviates endosomal pathology,44 and these exosomes include biomarkers for Alzheimer’s disease.45 The hub genes of M29 were mostly included in nitrogen-related terms, like “organonitrogen compound catabolic process” (GPC5, GLUD1, and NT5E) and “nitrogen compound transport” (SLC1A2, SLC15A2, and GLUD1). S100B in HSA21 and amyloid-beta can induce the generation of nitrogen species.46,47 Moreover, astrocytes detoxify reactive nitrogen species, which is related to the pathology of Parkinson's disease.48
The expression of M14, the oligodendrocyte module, increased gradually from the prenatal stages to middle age (Figures 4A and 4C), corresponding to the known patterns of myelination.49 In addition, the degree of temporal variance in expression was higher in the DFC than in the CBC. Unlike astrocyte-enriched genes, oligodendrocyte-enriched genes were downregulated in DS. A detailed investigation of the oligodendrocyte-enriched genes has been conducted in a previous report, and nine of the 10 hub genes in M14 were identical to those found in previous research.11 Among the hub genes of M14, MYRF, which is downregulated in the DS brain, is important in oligodendrocyte progenitor cell maturation and myelination.50 Another downregulated hub gene, ASPA, is known to cause Canavan disease, which shows some similar symptoms to DS, like intellectual disability, low muscle tone, damage to neurons, and loss of white matter in the brain.51
The microglia module M41 showed increased expression in DFCs at the postnatal stage, when microglia participate in synaptic pruning during brain development.52 Genes belonging to this module were expressed higher in the DFC of adult DS brains compared to the euploid controls (Figures 4B and 4D). Seven hub genes of M41 were related to immune response; this was concordant with previous research that investigated the relevance between DS and pathological hallmarks of Alzheimer’s disease, particularly inflammation.32,33 Neuroinflammation in the DS brain has been discussed in the context of the early onset and high incidence of Alzheimer’s disease in DS patients.32,33,53,54 Recently, a study has shown that resolving inflammation in the brain of the Ts65Dn DS model mouse reverses memory loss. Therefore, inflammation has been suggested as a novel target for DS and Alzheimer’s disease.55
Two glutamatergic neuron modules, M24 and M26, showed similar expression patterns across brain regions and developmental stages. The genes of those modules showed a higher level in the DFC than in the CBC. Of interest, they were highly expressed in the fetal brain, suggesting a role for glutamatergic neurons in the early neurodevelopment of the cerebral cortex.56 In addition, the glutamatergic neuron module genes were repressed in the DS brain, which may be related to neuronal loss and relevance to Alzheimer’s disease.57,58,59 Downregulation of these modules agrees with previous studies describing neuronal development disabilities in DS.5,57,60 M24 showed a greater difference with age and trisomy of HSA21 compared with M26. Hub genes of M24 were mostly enriched for synapse-related terms, and dendritic shaft-related genes were enriched for M24. M26 hub genes were mostly enriched for neuron development, and axon-related terms were enriched in the functional annotation results in this module. Previous studies have discovered abnormalities in DS dendrites and axons; more defects in dendrites than axons.61,62,63,64,65,66,67 This is consistent with the more affected expression patterns of genes in M24 than those in M26 by disease status. A GO term, “cognition”, was related to M26. This is concordant with the cognitive defects of mice overexpressing Dyrk1a, which has been implicated in the pathology of DS and Alzheimer’s disease, in glutamatergic neurons.68 The neuronal subtype enrichment analysis results are also supported by previous research that has demonstrated dendritic defects in L3 pyramidal cells in the prefrontal cortex of Ts65Dn mice.69 The data for these two modules presented in this paper will provide a molecular genetic basis for subsequent studies of neurodevelopmental and synaptic abnormalities in DS.
Like other neuron modules, GABAergic neuron modules (M32 and M42) were downregulated in DS but were more expressed during the postnatal stages, unlike glutamatergic neuron modules. This is concordant with the neurogenesis stage in brain development.56 M32 showed few regional differences when compared with M42. The gap between gene expression in the control and DS brain widens from the early postnatal stage, which insinuates a deficiency in GABA system development.70,71 This deficiency is implicated in many DS symptoms and related diseases.65,72,73,74,75,76,77,78,79,80,81 According to the enriched terms of M32, these genes are related to the release of neurotransmitters, especially GABA. Excitation–inhibition imbalance and over-inhibition have been implicated in the cognitive impairment of DS. Moreover, inhibitory synapses and their release of neurotransmitters, like GABA, are considered a cause of this imbalance.82 GABA release is increased in Ts65Dn mouse brain;63 attenuating this can alleviate cognitive dysfunctions.80 A recent study using the Ts2Cje DS model mouse has found that GABA release is increased in distal and decreased in proximal dendrites,67 respectively. M32 was enriched for parvalbumin-positive GABAergic neuron-related genes. Parvalbumin-positive cells are altered in DS and related neurodevelopmental disorders.65,78,80,83 In addition, M42 was enriched for calcium-calmodulin-related genes, which have been known to relate to the premature differentiation of neurons in the DS mouse model.84
We further analyzed the DS brain transcriptome using cell type-related genes from the developing brain (Figure S9) and cerebellum (Figure S10). For this analysis, we utilized the enriched genes in replicating and quiescent fetal neurons,12 and differentially expressed genes of cerebellum-specific cell types.13 Genes enriched in replicating fetal neurons were slightly less expressed in DS DFC than in euploid control at early stages, whereas they were more expressed in DS DFC than in euploid control at later stages (Figure S9A). Quiescent fetal neuron genes showed the opposite patterns (i.e., more expressed in early-stage brains of DS than euploid control and less expressed in later-stage brains of DS than euploid control). Reduced proliferation of neural stem cells and hypocellularity in the brain of DS fetuses has been reported in previous studies.5,85,86 Therefore, increased expression of replicating fetal neuron genes in later developmental stages of DFC was rather unexpected. This may be a counteraction of excessive neurodegeneration or a novel feature of the aged DS cerebral cortex. Further studies are needed to reveal the role of replicating neuron genes upregulated in DS DFC of later developmental stages. Cell-type enrichment analysis with fetal neuron genes identified M4 as a replicating fetal neuron module, and M13 and M20 as quiescent fetal neuron modules (Figure S9B). M4 module was downregulated in fetal DS brains and upregulated in old DS brains, whereas the expression patterns of M13 and M20 were not changed in DS brains (Figure S9C). Unlike in DFC, M4 genes were highly expressed after birth in CBC, which is concordant with the known timing of neurogenesis in different brain regions.87 The M4 module is enriched with cell-cycle-related terms in functional annotation (Figure S11A).
The differential expression pattern of cerebellum-specific cell-type genes in DS brain development was downregulated in both DFC and CBC (Figure S10A). This expression pattern of cerebellum-specific cell-type genes was similar to the expression pattern of the cortical neuron-enriched genes in the DFC of DS. Of interest, astrocyte and OPC-enriched genes showed a tendency to increase in the DS brain, especially in early development, and the increased expression of OPC-related genes in DS was consistent with the defects of oligodendrocyte development in the DS brain.11 Cell-type enrichment analyses on 43 co-expression modules using cerebellum-specific cell-type genes identified three modules that showed significant correlation; M8 was enriched in cerebellum-specific OPC, M17 was enriched in cerebellum-specific astrocyte and granule cells, and M23 was enriched mainly in Purkinje cell genes (Figure S10B). All these modules show an association with linear regression to DS and are highly expressed in CBC (Figure S10C and Table S4). GO analysis showed that M8, elevated in expression at the fetal stage of DS, is associated with chromosome organization (Figure S11B). M17 and M23, downregulated in the DS brain, are associated with synaptic transmission and cerebellar ataxia (Figures S11C and S11D), which is one of the causes of hypotonia.9
The most DS-associated co-expression module, M40, was upregulated in the DS brain and related to endothelial cells, microglia, and astrocyte and also associated with immune response and vasculature development in functional annotation (Figures 2 and 11). This implies that interactions between those cell types were implicated in DS brain phenotype and possible dysregulation of the blood-brain barrier, which includes blood vessels and astrocytes, and the immune system, which includes microglia, immune cells, and astrocytes. This was concordant with the recent review which described immune and cerebrovascular contributions to Alzheimer’s disease in DS.88 The enriched term including the largest number of hub genes in M40 was “extracellular region part”, which implies that M40 may have a role in extracellular signaling like cell-cell interactions. These results provide a data-based rationale for focusing on the relevance of this function in future research about DS pathophysiology. Considering the association between Alzheimer’s and Down’s syndrome, the hippocampus is one of the regions most implicated in Alzheimer’s disease. Although the number of samples was too small (three for each of the control and DS) to analyze, we analyzed hippocampus data to investigate whether the Alzheimer’s disease-related findings are repeated in the hippocampus (Figure S12). DEGs in the hippocampus (p< 0.01) were also significantly enriched with endothelial cell genes, and the expression patterns of cell-type genes and cell-type modules were similar to those of DFC. In-depth studies about the hippocampus with more biological replicates and other regions related to Alzheimer’s disease are needed to understand the mechanisms of early-onset Alzheimer’s disease in DS.
This research provides a resource for investigating the roles of brain cell types in DS brain development, especially in two regions of DFC and CBC. Associated gene expression patterns were described by systemic network analysis using WGCNA. Each co-expressed gene module showed unique expression features that were cell type- and biological role-specific. This study will assist researchers in elucidating the underlying mechanisms of DS and related neurological or psychiatric disorders by providing a systemic view into the DS brain condition. Combining this resource with other single-cell level analyses or brain organoid studies is expected to contribute to investigating the mechanisms of DS brain pathology.89,90,91
Limitations of the study
In this study, we used the bulk transcriptome of the DS brain and inferred the dysregulated biological roles of cell types based on DS-associated cell type-enriched gene co-expression modules. We did not directly analyze the DS brain transcriptome at the single-cell level in this study; however, the comparisons with the single-nucleus transcriptome in the DS brain will confirm and complement the dysfunction of cell types in the DS brain. Although there are multiple brain regions in the published DS brain transcriptome data we used, we focused on two brain regions because of the limitations of sample numbers in some regions. Further studies on other brain regions will clarify the cell type contributions and molecular mechanisms of DS brain development.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| DS brain transcriptome | (Olmos-Serrano et al., 2016)11 | GEO: GSE59630https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59630 |
| Software and algorithms | ||
| R (version 4.0.4) | The R Project | https://www.r-project.org/ |
| WGCNA package (version 1.70.3) | (Zhang and Horvath, 2005)20 | https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/ |
| DAVID 6.8 | (Huang da et al., 2009)92 | |
| DAVID 2021 | (Sherman et al., 2022)93 | https://david.ncifcrf.gov/ |
| Cytoscape (version 3.8.2) | (Shannon et al., 2003)94 | https://cytoscape.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hyo Jung Kang (hyokang@cau.ac.kr).
Materials availability
This study did not generate new unique reagents.
Method details
Data preprocessing
The series matrix file of GSE59630 from NCBI GEO was downloaded and used in the following analyses. Exon IDs were converted to the official gene symbol using the gene ID conversion of Database for Annotation, Visualization and Integrated Discovery (DAVID 6.8) to label the genes in figures and tables.92 Descriptions of developmental periods are summarized in Table S1.19
Cell-type enrichment analysis
The cell type-enriched gene list from Allen et al.18 and the gene list co-expressed with brain cell type marker genes from Kang et al.19 were used to compose our own cell type-enriched gene list (Table S2). Cell type-enriched gene lists for glial cell types were identical to the gene lists from Allen et al.18 Neuronal cell type-enriched gene lists were composed of genes overlapping with the gene list of Kang et al.19 Analyses on neuronal cell types were implemented using parts of separate gene lists (glutamatergic and GABAergic). Further analyses for subtypes were also performed, except for L5 glutamatergic neurons, because there were no overlapped genes between neuron-enriched genes in Allen et al.,18 and genes co-expressed with FEZF2, BCL11B, OTX1, and ETV1, which are markers of L5 glutamatergic neurons. The top 20 enriched genes for replicating and quiescent fetal neurons12 were used for cell-type analysis of fetal neuron genes and DEGs of cerebellum-specific cell types13 were used for cell-type analysis of cerebellum-specific cell-type genes.
Cell-type enrichment analyses on DEGs and WGCNA modules were performed using our own cell type-enriched gene list and Fisher’s exact test. DEGs of DFC and CBC were previously defined18 using the paired t-test (FDR-adjusted p-value <0.1). The-log10 (BHA p-value) values of Fisher’s exact test were calculated for each regional DEG and module. Enrichment analysis with neuronal subtype-enriched genes was only conducted on WGCNA modules because no significant enrichment for neuronal genes was found in the cell-type enrichment analysis on DEGs. DEGs of hippocampus were defined using the paired t-test with lower threshold (p-value <0.01) because no DEGs were defined with the same threshold of the DEGs in DFC and CBC.
Differential expression patterns of cell type-enriched genes
To determine the expression patterns of cell type-enriched genes in control and DS brains, we visualized expression differences of cell type-enriched genes across brain regions and developmental stages. Differences in expression values between DS and matched control sample data of cell type-enriched genes were calculated on a log2 scale. Data were separated by brain region. We used a sliding window approach for the developmental stages to diminish the uniqueness of each sample and emphasize the temporal pattern. Three developmental stages were grouped and are displayed together. A paired t-test was used to estimate the significance of expression differences between DS and control samples at each sliding window.
WGCNA
We used the WGCNA package (version 1.70.3) in R (version 4.0.4) to construct a signed gene co-expression network transcriptome from all samples, including not only DFC and CBC but also other brain regions of GSE59630 data, and to form 57 co-expression modules. When building the signed type network and forming co-expression modules, we set the soft-thresholding power to 21, according to the criteria presented in the WGCNA paper.20 The minimum module size was set to 20 genes, and the minimum cut height of the gene cluster dendrogram for the merging modules was set to 0.15. In accordance with the WGCNA protocol, the expression of each gene belonging to each module was summarized as the eigengene, the first principal component of each module. The kME of each gene belonging to each module was defined by the correlation between the module eigengene and the gene’s expression value. Genes were reassigned based on kME. Only those with a kME >0.7 for each module were used for further analysis (Table S3). The 10 genes with the highest kME values were defined as the hub genes of each module. Among the 57 modules, we filtered out those affected more by RIN, PMI, race, and sex than brain region, disease status, and developmental period. Module−trait relationships were calculated by multiple linear regression, and 43 modules were selected based on the R-squared value and used for further analyses (Table S4).
Gene and eigengene expression visualization
Gene expression patterns were visualized as line graphs and heatmaps according to the developmental stage, brain region, and disease status of the genes belonging to the selected module. The first principal components of the modules, the eigengenes, were represented by points according to age. A line graph was obtained by subsequent local regression in R (version 4.0.4). The gene expression heatmap was obtained by the expression value of each gene in a module, and a gene dendrogram was drawn.
GO and KEGG pathway enrichment analysis
The genes of each module were submitted to DAVID 6.8. Enrichment analysis was performed on three GO categories and the KEGG pathway. Similar terms were excluded by clustering enriched terms with medium stringency. The-log (p-value) of each term was calculated. The five most significantly enriched terms for each category were plotted. In the analysis of fetal neuron and cerebellum-specific cell-type genes, DAVID 202193 was used because the DAVID 6.8 has been retired since June 2, 2022.
Cytoscape
We exported the intramodular co-expression data for each module obtained from WGCNA (version 1.70.3) as an input file for Cytoscape (version 3.8.2) from R (version 4.0.4) and imported it into Cytoscape to create co-expression networks.94 Gene symbols of the hub genes are enlarged and are colored red. The nodes of genes corresponding to the cell type of each module were colored in yellow, while the others are colored in grey using Brain RNA-seq database.22 The width and darkness of edges are proportional to the co-expression level between two genes. In the analysis of M40, node colors are matched with all brain cell types, and the nodes which are not related to any cell type are colored white.
Quantification and statistical analyses
Fisher’s exact test and Benjamini–Hochberg adjustments were performed using R (version 4.0.4). All paired t-tests were performed in the one-tailed mode. Correlations were calculated using the cor function in R (version 4.0.4). Transcriptomes from 116 samples (58 pairs, all regions) were used in constructing co-expression networks using WGCNA (version 1.70.3). Data from 24 DFC samples (12 pairs) and 20 CBC samples (10 pairs) were used in detailed analyses.
Acknowledgments
This research was supported by the Chung-Ang University Graduate Research Scholarship in 2016 and the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (NRF-2017R1A2B4012237, 2018M3C7A1024150).
Author contributions
Conceptualization, H.J.K.; Methodology, H.J.K., S.S., and J.K.; Software, S.S. and J.K.; Formal Analysis, S.S. and J.K.; Investigation, S.S.; Resources, H.J.K.; Writing – Original Draft, S.S.; Writing – Review and Editing, H.J.K.; Visualization, S.S. and J.K.; Supervision, H.J.K.; Funding Acquisition, H.J.K.
Declaration of interests
The authors declare no competing interests.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105884.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Presson A.P., Partyka G., Jensen K.M., Devine O.J., Rasmussen S.A., McCabe L.L., McCabe E.R.B. Current estimate of Down Syndrome population prevalence in the United States. J. Pediatr. 2013;163:1163–1168. doi: 10.1016/j.jpeds.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Graaf G., Buckley F., Skotko B.G. Estimates of the live births, natural losses, and elective terminations with Down syndrome in the United States. Am. J. Med. Genet. 2015;167A:756–767. doi: 10.1002/ajmg.a.37001. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal Gupta N., Kabra M. Diagnosis and management of Down syndrome. Indian J. Pediatr. 2014;81:560–567. doi: 10.1007/s12098-013-1249-7. [DOI] [PubMed] [Google Scholar]
- 4.Asim A., Kumar A., Muthuswamy S., Jain S., Agarwal S. Down syndrome: an insight of the disease. J. Biomed. Sci. 2015;22:41. doi: 10.1186/s12929-015-0138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haydar T.F., Reeves R.H. Trisomy 21 and early brain development. Trends Neurosci. 2012;35:81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dierssen M. Down syndrome: the brain in trisomic mode. Nat. Rev. Neurosci. 2012;13:844–858. doi: 10.1038/nrn3314. [DOI] [PubMed] [Google Scholar]
- 7.Malt E.A., Dahl R.C., Haugsand T.M., Ulvestad I.H., Emilsen N.M., Hansen B., Cardenas Y.E.G., Skøld R.O., Thorsen A.T.B., Davidsen E.M.M. Health and disease in adults with Down syndrome. Tidsskr. Nor. Laegeforen. 2013;133:290–294. doi: 10.4045/tidsskr.12.0390. [DOI] [PubMed] [Google Scholar]
- 8.DiGuiseppi C., Hepburn S., Davis J.M., Fidler D.J., Hartway S., Lee N.R., Miller L., Ruttenber M., Robinson C. Screening for autism spectrum disorders in children with Down syndrome: population prevalence and screening test characteristics. J. Dev. Behav. Pediatr. 2010;31:181–191. doi: 10.1097/DBP.0b013e3181d5aa6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santoro J.D., Pagarkar D., Chu D.T., Rosso M., Paulsen K.C., Levitt P., Rafii M.S. Neurologic complications of Down syndrome: a systematic review. J. Neurol. 2021;268:4495–4509. doi: 10.1007/s00415-020-10179-w. [DOI] [PubMed] [Google Scholar]
- 10.Richards C., Jones C., Groves L., Moss J., Oliver C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatr. 2015;2:909–916. doi: 10.1016/S2215-0366(15)00376-4. [DOI] [PubMed] [Google Scholar]
- 11.Olmos-Serrano J.L., Kang H.J., Tyler W.A., Silbereis J.C., Cheng F., Zhu Y., Pletikos M., Jankovic-Rapan L., Cramer N.P., Galdzicki Z., et al. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron. 2016;89:1208–1222. doi: 10.1016/j.neuron.2016.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darmanis S., Sloan S.A., Zhang Y., Enge M., Caneda C., Shuer L.M., Hayden Gephart M.G., Barres B.A., Quake S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lake B.B., Chen S., Sos B.C., Fan J., Kaeser G.E., Yung Y.C., Duong T.E., Gao D., Chun J., Kharchenko P.V., Zhang K. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thion M.S., Ginhoux F., Garel S. Microglia and early brain development: an intimate journey. Science. 2018;362:185–189. doi: 10.1126/science.aat0474. [DOI] [PubMed] [Google Scholar]
- 15.Allen N.J., Lyons D.A. Glia as architects of central nervous system formation and function. Science. 2018;362:181–185. doi: 10.1126/science.aat0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lott I.T. Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 2012;197:101–121. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krinsky-McHale S.J., Silverman W., Gordon J., Devenny D.A., Oley N., Abramov I. Vision deficits in adults with Down syndrome. J. Appl. Res. Intellect. Disabil. 2014;27:247–263. doi: 10.1111/jar.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen M., Wang X., Burgess J.D., Watzlawik J., Serie D.J., Younkin C.S., Nguyen T., Malphrus K.G., Lincoln S., Carrasquillo M.M., et al. Conserved brain myelination networks are altered in Alzheimer's and other neurodegenerative diseases. Alzheimers Dement. 2018;14:352–366. doi: 10.1016/j.jalz.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 21.Langfelder P., Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Sloan S.A., Clarke L.E., Caneda C., Plaza C.A., Blumenthal P.D., Vogel H., Steinberg G.K., Edwards M.S.B., Li G., et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Bartheld C.S., Bahney J., Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J. Comp. Neurol. 2016;524:3865–3895. doi: 10.1002/cne.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerrato V. Cerebellar astrocytes: much more than passive bystanders in ataxia pathophysiology. J. Clin. Med. 2020;9:757. doi: 10.3390/jcm9030757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Head E., Phelan M.J., Doran E., Kim R.C., Poon W.W., Schmitt F.A., Lott I.T. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol. Commun. 2017;5:93. doi: 10.1186/s40478-017-0499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Head E., Powell D.K., Schmitt F.A. Metabolic and vascular imaging biomarkers in down syndrome provide unique insights into brain aging and alzheimer disease pathogenesis. Front. Aging Neurosci. 2018;10:191. doi: 10.3389/fnagi.2018.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helman A.M., Siever M., McCarty K.L., Lott I.T., Doran E., Abner E.L., Schmitt F.A., Head E. Microbleeds and cerebral amyloid angiopathy in the brains of people with down syndrome with Alzheimer's disease. J. Alzheimers Dis. 2019;67:103–112. doi: 10.3233/Jad-180589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xing C., Li W., Deng W., Ning M., Lo E.H. A potential gliovascular mechanism for microglial activation: differential phenotypic switching of microglia by endothelium versus astrocytes. J. Neuroinflammation. 2018;15:143. doi: 10.1186/s12974-018-1189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lécuyer M.A., Kebir H., Prat A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochim. Biophys. Acta. 2016;1862:472–482. doi: 10.1016/j.bbadis.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Flavell S.W., Greenberg M.E. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emery B., Lu Q.R. Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harbor Perspect. Biol. 2015;7:a020461. doi: 10.1101/cshperspect.a020461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilcock D.M. Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer's disease. Curr. Gerontol. Geriatr. Res. 2012;2012:170276. doi: 10.1155/2012/170276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilcock D.M., Griffin W.S.T. Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flores-Aguilar L., Iulita M.F., Kovecses O., Torres M.D., Levi S.M., Zhang Y., Askenazi M., Wisniewski T., Busciglio J., Cuello A.C. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain. 2020;143:3653–3671. doi: 10.1093/brain/awaa326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed M.M., Johnson N.R., Boyd T.D., Coughlan C., Chial H.J., Potter H. Innate immune system activation and neuroinflammation in down syndrome and neurodegeneration: therapeutic targets or partners? Front. Aging Neurosci. 2021;13:718426. doi: 10.3389/fnagi.2021.718426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ní Chasaide C., Lynch M.A. The role of the immune system in driving neuroinflammation. Brain Neurosci. Adv. 2020;4 doi: 10.1177/2398212819901082. 2398212819901082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franciotta D., Verri A., Zardini E., Andreoni L., De Amici M., Moratti R., Nespoli L. Interferon-gamma- and interleukin-4-producing T cells in Down's syndrome. Neurosci. Lett. 2006;395:67–70. doi: 10.1016/j.neulet.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 39.Palmer A.L., Ousman S.S. Astrocytes and aging. Front. Aging Neurosci. 2018;10:337. doi: 10.3389/fnagi.2018.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen J., Torres C. Astrocyte senescence: evidence and significance. Aging Cell. 2019;18:e12937. doi: 10.1111/acel.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boisvert M.M., Erikson G.A., Shokhirev M.N., Allen N.J. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018;22:269–285. doi: 10.1016/j.celrep.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen C., Jiang P., Xue H., Peterson S.E., Tran H.T., McCann A.E., Parast M.M., Li S., Pleasure D.E., Laurent L.C., et al. Role of astroglia in Down's syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014;5:4430. doi: 10.1038/ncomms5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dossi E., Vasile F., Rouach N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018;136:139–156. doi: 10.1016/j.brainresbull.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gauthier S.A., Pérez-González R., Sharma A., Huang F.K., Alldred M.J., Pawlik M., Kaur G., Ginsberg S.D., Neubert T.A., Levy E. Enhanced exosome secretion in Down syndrome brain - a protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol. Commun. 2017;5:65. doi: 10.1186/s40478-017-0466-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamlett E.D., Goetzl E.J., Ledreux A., Vasilevko V., Boger H.A., LaRosa A., Clark D., Carroll S.L., Carmona-Iragui M., Fortea J., et al. Neuronal exosomes reveal Alzheimer's disease biomarkers in Down syndrome. Alzheimers Dement. 2017;13:541–549. doi: 10.1016/j.jalz.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J., Castets F., Guevara J.L., Van Eldik L.J. S100 beta stimulates inducible nitric oxide synthase activity and mRNA levels in rat cortical astrocytes. J. Biol. Chem. 1996;271:2543–2547. doi: 10.1074/jbc.271.5.2543. [DOI] [PubMed] [Google Scholar]
- 47.White J.A., Manelli A.M., Holmberg K.H., Van Eldik L.J., Ladu M.J. Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005;18:459–465. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 48.Rizor A., Pajarillo E., Johnson J., Aschner M., Lee E. Astrocytic oxidative/nitrosative stress contributes to Parkinson's disease pathogenesis: the dual role of reactive astrocytes. Antioxidants. 2019;8:265. doi: 10.3390/antiox8080265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williamson J.M., Lyons D.A. Myelin dynamics throughout life: an ever-changing landscape? Front. Cell. Neurosci. 2018;12:424. doi: 10.3389/fncel.2018.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forbes T.A., Goldstein E.Z., Dupree J.L., Jablonska B., Scafidi J., Adams K.L., Imamura Y., Hashimoto-Torii K., Gallo V. Environmental enrichment ameliorates perinatal brain injury and promotes functional white matter recovery. Nat. Commun. 2020;11:964. doi: 10.1038/s41467-020-14762-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matalon R., Delgado L., Michals-Matalon K. In: GeneReviews((R)) Adam M.P., Mirzaa G.M., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Amemiya A., editors. 1993. Canavan disease. [Google Scholar]
- 52.Menassa D.A., Gomez-Nicola D. Microglial dynamics during human brain development. Front. Immunol. 2018;9:1014. doi: 10.3389/fimmu.2018.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castro P., Zaman S., Holland A. Alzheimer's disease in people with Down's syndrome: the prospects for and the challenges of developing preventative treatments. J. Neurol. 2017;264:804–813. doi: 10.1007/s00415-016-8308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lott I.T., Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019;15:135–147. doi: 10.1038/s41582-018-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hamlett E.D., Hjorth E., Ledreux A., Gilmore A., Schultzberg M., Granholm A.C. RvE1 treatment prevents memory loss and neuroinflammation in the Ts65Dn mouse model of Down syndrome. Glia. 2020;68:1347–1360. doi: 10.1002/glia.23779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim J.Y., Paredes M.F. Implications of extended inhibitory neuron development. Int. J. Mol. Sci. 2021;22:5113. doi: 10.3390/ijms22105113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Becker L., Mito T., Takashima S., Onodera K. Growth and development of the brain in Down syndrome. Prog. Clin. Biol. Res. 1991;373:133–152. [PubMed] [Google Scholar]
- 58.Sawa A. Neuronal cell death in Down's syndrome. J. Neural. Transm. Suppl. 1999;57:87–97. doi: 10.1007/978-3-7091-6380-1_6. [DOI] [PubMed] [Google Scholar]
- 59.Watson-Scales S., Kalmar B., Lana-Elola E., Gibbins D., La Russa F., Wiseman F., Williamson M., Saccon R., Slender A., Olerinyova A., et al. Analysis of motor dysfunction in Down Syndrome reveals motor neuron degeneration. PLoS Genet. 2018;14:e1007383. doi: 10.1371/journal.pgen.1007383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baburamani A.A., Patkee P.A., Arichi T., Rutherford M.A. New approaches to studying early brain development in Down syndrome. Dev. Med. Child Neurol. 2019;61:867–879. doi: 10.1111/dmcn.14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaufmann W.E., Moser H.W. Dendritic anomalies in disorders associated with mental retardation. Cerebr. Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- 62.Martinez de Lagran M., Benavides-Piccione R., Ballesteros-Yañez I., Calvo M., Morales M., Fillat C., Defelipe J., Ramakers G.J.A., Dierssen M. Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cerebr. Cortex. 2012;22:2867–2877. doi: 10.1093/cercor/bhr362. [DOI] [PubMed] [Google Scholar]
- 63.Cramer N., Galdzicki Z. From abnormal hippocampal synaptic plasticity in down syndrome mouse models to cognitive disability in down syndrome. Neural Plast. 2012;2012:101542. doi: 10.1155/2012/101542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simmons A.B., Bloomsburg S.J., Sukeena J.M., Miller C.J., Ortega-Burgos Y., Borghuis B.G., Fuerst P.G. DSCAM-mediated control of dendritic and axonal arbor outgrowth enforces tiling and inhibits synaptic plasticity. Proc. Natl. Acad. Sci. USA. 2017;114:E10224–E10233. doi: 10.1073/pnas.1713548114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulz J.M., Knoflach F., Hernandez M.C., Bischofberger J. Enhanced dendritic inhibition and impaired NMDAR activation in a mouse model of down syndrome. J. Neurosci. 2019;39:5210–5221. doi: 10.1523/JNEUROSCI.2723-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiotto A.M.A., Migliorero M., Pallavicini G., Bianchi F.T., Gai M., Di Cunto F., Berto G.E. Neuronal cell-intrinsic defects in mouse models of down syndrome. Front. Neurosci. 2019;13:1081. doi: 10.3389/fnins.2019.01081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Valbuena S., García Á., Mazier W., Paternain A.V., Lerma J. Unbalanced dendritic inhibition of CA1 neurons drives spatial-memory deficits in the Ts2Cje Down syndrome model. Nat. Commun. 2019;10:4991. doi: 10.1038/s41467-019-13004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brault V., Nguyen T.L., Flores-Gutiérrez J., Iacono G., Birling M.C., Lalanne V., Meziane H., Manousopoulou A., Pavlovic G., Lindner L., et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 2021;17:e1009777. doi: 10.1371/journal.pgen.1009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dierssen M., Benavides-Piccione R., Martínez-Cué C., Estivill X., Flórez J., Elston G.N., DeFelipe J. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cerebr. Cortex. 2003;13:758–764. doi: 10.1093/cercor/13.7.758. [DOI] [PubMed] [Google Scholar]
- 70.Schmidt M.J., Mirnics K. Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology. 2015;40:190–206. doi: 10.1038/npp.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kilb W. Development of the GABAergic system from birth to adolescence. Neuroscientist. 2012;18:613–630. doi: 10.1177/1073858411422114. [DOI] [PubMed] [Google Scholar]
- 72.Wong C.G.T., Bottiglieri T., Snead O.C., 3rd GABA, gamma-hydroxybutyric acid, and neurological disease. Ann. Neurol. 2003;54(Suppl 6):S3–S12. doi: 10.1002/ana.10696. [DOI] [PubMed] [Google Scholar]
- 73.Kleppner S.R., Tobin A.J. GABA signalling: therapeutic targets for epilepsy, Parkinson's disease and Huntington's disease. Expert Opin. Ther. Targets. 2001;5:219–239. doi: 10.1517/14728222.5.2.219. [DOI] [PubMed] [Google Scholar]
- 74.Pearl P.L., Hartka T.R., Cabalza J.L., Taylor J., Gibson M.K. Inherited disorders of GABA metabolism. Future Neurol. 2006;1:631–636. doi: 10.2217/14796708.1.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim Y.S., Yoon B.E. Altered GABAergic signaling in brain disease at various stages of life. Exp. Neurobiol. 2017;26:122–131. doi: 10.5607/en.2017.26.3.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kwakowsky A., Calvo-Flores Guzmán B., Govindpani K., Waldvogel H.J., Faull R.L. Gamma-aminobutyric acid A receptors in Alzheimer's disease: highly localized remodeling of a complex and diverse signaling pathway. Neural Regen. Res. 2018;13:1362–1363. doi: 10.4103/1673-5374.235240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rissman R.A., Mobley W.C. Implications for treatment: GABAA receptors in aging, Down syndrome and Alzheimer's disease. J. Neurochem. 2011;117:613–622. doi: 10.1111/j.1471-4159.2011.07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramamoorthi K., Lin Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol. Med. 2011;17:452–462. doi: 10.1016/j.molmed.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Potier M.C., Braudeau J., Dauphinot L., Delatour B. Reducing GABAergic inhibition restores cognitive functions in a mouse model of Down syndrome. CNS Neurol. Disord.: Drug Targets. 2014;13:8–15. doi: 10.2174/18715273113126660185. [DOI] [PubMed] [Google Scholar]
- 80.Contestabile A., Magara S., Cancedda L. The GABAergic hypothesis for cognitive disabilities in down syndrome. Front. Cell. Neurosci. 2017;11:54. doi: 10.3389/fncel.2017.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zorrilla de San Martin J., Delabar J.M., Bacci A., Potier M.C. GABAergic over-inhibition, a promising hypothesis for cognitive deficits in Down syndrome. Free Radic. Biol. Med. 2018;114:33–39. doi: 10.1016/j.freeradbiomed.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 82.Créau N. Molecular and cellular alterations in Down syndrome: toward the identification of targets for therapeutics. Neural Plast. 2012;2012:171639. doi: 10.1155/2012/171639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Filice F., Vörckel K.J., Sungur A.Ö., Wöhr M., Schwaller B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain. 2016;9:10. doi: 10.1186/s13041-016-0192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mouton-Liger F., Thomas S., Rattenbach R., Magnol L., Larigaldie V., Ledru A., Herault Y., Verney C., Créau N. PCP4 (PEP19) overexpression induces premature neuronal differentiation associated with Ca(2+)/calmodulin-dependent kinase II-delta activation in mouse models of Down syndrome. J. Comp. Neurol. 2011;519:2779–2802. doi: 10.1002/cne.22651. [DOI] [PubMed] [Google Scholar]
- 85.Guidi S., Bonasoni P., Ceccarelli C., Santini D., Gualtieri F., Ciani E., Bartesaghi R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008;18:180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guidi S., Ciani E., Bonasoni P., Santini D., Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol. 2011;21:361–373. doi: 10.1111/j.1750-3639.2010.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stagni F., Giacomini A., Guidi S., Ciani E., Bartesaghi R. Timing of therapies for Down syndrome: the sooner, the better. Front. Behav. Neurosci. 2015;9:265. doi: 10.3389/fnbeh.2015.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martini A.C., Gross T.J., Head E., Mapstone M. Beyond amyloid: immune, cerebrovascular, and metabolic contributions to Alzheimer disease in people with Down syndrome. Neuron. 2022;110:2063–2079. doi: 10.1016/j.neuron.2022.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palmer C.R., Liu C.S., Romanow W.J., Lee M.H., Chun J. Altered cell and RNA isoform diversity in aging Down syndrome brains. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2114326118. e2114326118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang X.Y., Xu L., Wang J., Hong Y., Wang Y., Zhu Q., Wang D., Zhang X.Y., Liu C.Y., Fang K.H., et al. DSCAM/PAK1 pathway suppression reverses neurogenesis deficits in iPSC-derived cerebral organoids from patients with Down syndrome. J. Clin. Invest. 2021;131:e135763. doi: 10.1172/JCI135763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stamoulis G., Garieri M., Makrythanasis P., Letourneau A., Guipponi M., Panousis N., Sloan-Béna F., Falconnet E., Ribaux P., Borel C., et al. Single cell transcriptome in aneuploidies reveals mechanisms of gene dosage imbalance. Nat. Commun. 2019;10:4495. doi: 10.1038/s41467-019-12273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 93.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., Imamichi T., Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.