

Abstract
EFEMP1 R345W is a dominant mutation causing Doyne honeycomb retinal dystrophy/malattia leventinese (DHRD/ML), a rare blinding disease with clinical pathology similar to age-related macular degeneration (AMD). Aged Efemp1 R345W/R345W knock-in mice (Efemp1ki/ki) develop microscopic deposits on the basal side of retinal pigment epithelial cells (RPE), an early feature in DHRD/ML and AMD. Here, we assessed the role of alternative complement pathway component factor B (FB) in the formation of these deposits. RNA-seq analysis of the posterior eyecups revealed increased unfolded protein response, decreased mitochondrial function in the neural retina (by 3 months of age) and increased inflammatory pathways in both neural retina and posterior eyecups (at 17 months of age) of Efemp1ki/ki mice compared with wild-type littermate controls. Proteomics analysis of eye lysates confirmed similar dysregulated pathways as detected by RNA-seq. Complement activation was increased in aged Efemp1ki/ki eyes with an approximately 2-fold elevation of complement breakdown products iC3b and Ba (P < 0.05). Deletion of the Cfb gene in female Efemp1ki/ki mice partially normalized the above dysregulated biological pathway changes and oral dosing of a small molecule FB inhibitor from 10 to 12 months of age reduced sub-RPE deposits by 65% (P = 0.029). In contrast, male Efemp1ki/ki mice had fewer sub-RPE deposits than age-matched females, no elevation of ocular complement activation and no effect of FB inhibition on sub-RPE deposits. The effects of FB deletion or inhibition on Efemp1ki/ki mice supports systemic inhibition of the alternative complement pathway as a potential treatment of dry AMD and DHRD/ML.
Introduction
Abnormal complement activation contributes to the pathogenesis of age-related macular degeneration (AMD) (1–4), a blinding disease affecting millions of older individuals worldwide. An early clinical hallmark of AMD is drusen (protein and lipid aggregates between the RPE and Bruch’s membrane). Complement proteins are present in drusen (5,6). However, the molecular/cellular changes and the role of complement in the formation of drusen remain unclear. Currently, there are no approved treatments for dry AMD and AMD-like macular dystrophies. A mechanistic understanding of sub-RPE deposit/drusen precursor formation may shed light on AMD etiology and aid in the development of AMD therapies.
The missense mutation R345W in the EFEMP1 gene encoding the Fibulin-3 protein is responsible for the development of Doyne honeycomb retinal dystrophy, also known as malattia leventinese (DHRD/ML) (7). DHRD/ML can be misdiagnosed as AMD as both share a number of pathological features including macular drusen/sub-RPE deposits, RPE degeneration and progression to choroidal neovascularization and geographic atrophy. The main difference is that DHRD/ML has an earlier age of onset (30–40 years versus 60–70 years in AMD) (7–10). DHRD/ML patients can have a unique ‘honeycomb’ pattern of drusen extending radially from the central macula into the periphery. AMD drusen are heterogeneous with a variety of shapes and patterns. Based on its ophthalmoscopic, electrophysiologic and genetic findings, DHRD/ML has been considered as one of the macular dystrophic diseases most similar to AMD (8). At the molecular level, many drusen components are shared between DHRD/ML and AMD such as TIMP3, vitronectin, amyloid P and complement proteins although DHRD/ML drusen reported to have a distinct laminated appearance with collagen type IV (11,12). Aberrant accumulation of EFEMP1 protein overlaying soft drusen or in drusen has been reported in both DHRD/ML and AMD (12–14). The EFEMP1 R345W substitution, an autosomal dominant mutation, is thought to increase protein misfolding leading to impairment of the proteostasis network, protein aggregation and drusen formation (15,16). Deposits between RPE cells and Bruch’s membrane in aged Efemp1 R345W knock-in (Efemp1ki/ki) mice, a mouse model of DHRD/ML, supports the causal role of the R345W mutation in DHRD/ML (17,18). Deposits in AMD patients have been defined primarily as basal laminar deposits containing long-spaced collagen (19,20) or basal linear deposits. The sub-RPE deposits in Efemp1ki/ki mice are similar to basal laminar deposits (17). Analysis of Efemp1 R345W mutant mice may reveal pathways underlying pathogenic mechanisms for early stages of AMD, DHRD/ML as well as other macular drusen dystrophies such as Sorsby fundus dystrophy (SFD) and Late onset retinal degeneration (LORD). Interestingly, deletion of the complement C3 gene, an AMD-associated gene (1) and a central node in all 3 complement pathways (alternative, classical and lectin) substantially reduces sub-RPE deposits in Efemp1ki/ki mice (11).
Most complement genes implicated in AMD from GWAS studies are components of the alternative complement pathway (AP) (1,3). Complement factor B (FB) is a key AP component essential for the proteolytic activity of the C3/C5 convertase. Loss-of-function alleles in the CFB gene confer protection from AMD (2,21). We hypothesized that AP complement activation may drive basal deposit/drusen accumulation and that genetic or pharmaceutical disruption of FB could reduce sub-RPE deposit formation in Efemp1ki/ki mice.
In the study reported here, we first characterized Efemp1 R345W-induced ocular changes in biological pathways via RNA-seq, revealing an upregulation of the unfolded protein response (UPR) and an innate immune response combined with dysregulation of mitochondrial metabolism. We found elevated ocular complement activation in Efemp1ki/ki eyes via western blot analysis. Proteomic profiling studies showed that many of the pathological pathway changes in Efemp1 R345W mutant mice were absent if the mice also had a deletion in Cfb gene. Finally, pharmacological inhibition of the AP complement pathway using an oral, low molecular weight FB inhibitor reduced sub-RPE deposits in female Efemp1ki/ki mice. Our results implicate AP complement activation as a critical driver of sub-RPE deposit accumulation in Efemp1ki/ki mice.
Results
The Efemp1 R345W mutation accelerates sub-RPE deposit accumulation and induces molecular changes in aged mice
Aged mice carrying the Efemp1 R345W mutation reportedly develop sub-RPE deposits, mimicking drusen precursor in human patients of DHRD/ML and AMD (17,18). To confirm the phenotype, we first performed transmission electron microscope (TEM) analysis of eyes from 16-month-old Efemp1R345W/R345W knock-in mice (Efemp1ki/ki) and their littermate wild-type mice (Efemp1+/+) as controls. Aged Efemp1ki/ki mice displayed extensive amorphous deposits between the RPE cells and Bruch’s membrane (Fig. 1A). As reported, small sub-RPE deposits could also be observed in some regions of aged wild-type mice, where basal infoldings are disrupted or missing (22,23). These would be categorized as basal laminar deposits which could be considered as ‘normal aging’ deposits in humans. Deposits found in aged Efemp1+/+ mice are morphologically similar to those found in Efemp1ki/ki, but they are generally smaller in size and less extensive than in age-matched Efemp1ki/ki mice (covering Bruch’s membrane length in TEM images ~10% versus >~50%), suggesting that the R345W mutation-induced protein misfolding likely accelerates age-related proteostasis impairment in RPE cells.
Figure 1.

Sub-RPE deposits and dysregulated biological pathways in eyes of Efemp1ki/ki mice. (A) Representative TEM images of RPE, Bruch’s membrane and choroid area in 16-month-old female Efemp1+/+ and Efemp1ki/ki mice, and 16-month-old Efemp1ki/ki mice develop more sub-RPE deposits than Efemp1+/+ wild-type mice, and 9 eyes of Efemp1+/+ mice and 10 eyes from Efemp1ki/ki mice were processed and analyzed by TEM. Extensive deposits were observed in 9 out of the 10 eyes from Efemp1ki/ki mice between RPE and Bruch’s membrane (BrM). Patchy/isolated minor deposits were observed in 4 out of the 10 eyes from aged Efemp1+/+ mice. GSEA of RNA-seq data from posterior eyecups (B) and neural retina (C) tissues of 17-month-old female Efemp1+/+ and Efemp1ki/ki mice. NES stands for normalized enrichment score which is the differential expression of pathway gene set between Efemp1+/+ and Efemp1ki/ki values. Upregulated pathways have positive NES value and down-regulated pathways have negative NES value. FDR = false discovery rate. Top 10 Efemp1 R345W-dysregulated Hallmark pathways with FDR < 0.1 at 17-month-old were included in the bar graphs and NES values of 3- and 17-month-old were stacked in the bar graphs. *FDR ≤ 0.05, **FDR ≤ 0.01, ***FDR ≤ 0.001. (D) Heatmap of differential expressed genes in Reactome UPR gene set between Efemp1+/+ and Efemp1ki/ki mice in posterior eye cup at 3, 9 and 17 months of age. The differential expression value between Efemp1+/+ and Efemp1ki/ki was expressed as t-statistic (t). A positive t value in red means an increase of gene expression in Efemp1ki/ki posterior eyecups compared with Efemp1+/+. Negative t values in blue indicate decreased expression. Only genes in the UPR signaling pathway that have P-value less than 0.05 were included in the heatmap when compared between 17-month-old Efemp1+/+ and Efemp1ki/ki mice. Most of these UPR genes were elevated in Efemp1ki/ki starting at 3 months of age. (E) TaqMan analysis of gene expression of RPE-specific Rpe65 and Lrat in posterior eyecups of both Efemp1+/+ and Efemp1ki/ki mice at 18 months of age. Relative gene expression levels were normalized by Gapdh gene expression in each sample. *P < 0.05, **P < 0.01, ***P < 0.001.
To identify the molecular pathway changes associated with sub-RPE deposit accumulation, we performed RNA-seq analysis of the neural retina and posterior eyecups (whole eye without lens and retina) from 3- to 17-month-old Efemp1+/+ and Efemp1ki/ki mice. The top 10 differentially expressed pathways between Efemp1+/+ and Efemp1ki/ki were determined using hallmark gene sets by gene set enrichment analysis (GSEA) (Fig. 1B for posterior eyecups, Fig. 1C for neural retina). These 10 pathways were selected based on statistical significance obtained from the analysis of 17-month-old mice (Supplementary Material, Table S1). Hallmark gene sets represent generalized and well-defined biological processes condensing thousands of original overlapping gene sets (24). Follow-up analysis with additional gene sets (KEGG, WIKI, GO-MF) is presented in Supplementary Material, Tables S2–S4. As expected from cell culture studies with EFEMP1 R345W (15,16), the UPR, the acute ER stress response to protein misfolding, is one of the top upregulated pathways in posterior eyecups at both 3 and 17 months of age (Fig. 1B and Supplementary Material, Fig. S1A). The majority of differentially expressed UPR genes, particularly the key ER chaperone gene Hspa5, are up-regulated as early as 3 months of age, with a greater difference in 17-month-old posterior eyecups (Fig. 1D). Single-cell RNA-seq data of human RPE/choroid identified 3 main cell types expressing EFEMP1 mRNA including fibroblasts, endothelial cells and the RPE (Supplementary Material, Fig. S1B) (25). Correspondingly, misfolding-stressed RPE cells in Efemp1ki/ki mice consistently expressed RPE-signature genes at a lower level, including the visual cycle genes Rpe65 and Lrat compared with Efemp1+/+ littermate controls (Supplementary Material, Fig. S1C) and confirmed by TaqMan analysis (Fig. 1E). In keeping with the subclinical nature of the deposits in early AMD and DHRD/ML, aged Efemp1ki/ki mice retain relatively normal RPE morphology with proper intercellular junction integrity on the tight-junction protein ZO-1 staining of RPE/choroid flat mounts. Color fundus photographs of aged Efemp1ki/ki mice also are generally normal (data not shown).
Research studies identify and categorize the types of molecular and cellular damage into 9 hallmarks of aging (26). Loss of proteostasis due to protein misfolding is one of the four primary types of damage in aging, triggering downstream processes leading to mitochondrial dysfunction. In Efemp1ki/ki mice, mitochondrial oxidative phosphorylation emerges as the most significantly down-regulated pathway in the neural retina, but not in the posterior eyecups (Fig. 1C, Supplementary Material, Fig. S1E). Mitochondrial defects in neural retina may reflect photoreceptor dysfunction secondary to impairment of proteostasis in the RPE. Indeed mitochondria are enriched in the photoreceptor inner segments, supporting the high energy requirement of phototransduction (27). Inflammaging, a chronic and low grade inflammation in old age, is a prominent cause of altered intercellular communication, one of the integrative hallmark responses following accumulation of primary and secondary aging types of damage (26). Several innate immune signaling pathways are upregulated in posterior eyecups and/or the neural retina of Efemp1ki/ki mice, especially at 17 months of age (Fig. 1B,C). Inflammatory cytokines and microglia/macrophage surface markers such as Tnf and Cd14 are moderately increased in the posterior eyecups at 17 months but not 3–9 months of age (Supplementary Material, Fig. S1F and G), suggesting that impaired proteostasis promotes the inflammatory response.
The early response to the Efemp1R345W misfolding mutation in young 3-month-old mice is the activation of UPR, down-regulation of neural retina mitochondrial pathways and a repression of the RPE-signature gene expression prior to sub-RPE deposit formation. Moderate upregulation of cytokine and microglia/macrophage gene expression at 17 months of age coincides with extensive sub-RPE deposit accumulation, a possible consequence of an increase in protein aggregates.
Complement is activated in eye tissues of Efemp1ki/ki mice
Complement and coagulation cascades were among the top upregulated pathways in neural retina of Efemp1ki/ki mice using GSEA KEGG and WikiPathways gene sets, although not with hallmark gene sets (Supplementary Material, Tables S1, S2B and S3B). Since different complement gene sets cover many pathways with a broad range of biological function, we further explored key complement cascade components in the RNA-seq dataset. In the neural retina, some of alternative and classical complement pathway genes were moderately upregulated in 17-month-old Efemp1ki/ki mice compared with Efemp1+/+ mice (Fig. 2A–C), with around 200% induction for C3 and C4b and 15% for Cfb. In posterior eyecups, Itgam was the only complement gene significantly induced in aged Efemp1ki/ki mice (Fig. 2B and C). Itgam (Cd11b) binds to Itgb2 (Cd18) forming complement receptor 3 (CR3), the myeloid cell receptor for complement activation product iC3b. Microglia CR3 activation plays a critical role in complement-dependent synapse pruning during brain development and in Alzheimer’s disease (28). Cd46 was the sole negative complement regulator with increased expression (Supplementary Material, Fig. S2A).
Figure 2.

Complement is activated in ocular tissues of female Efemp1ki/ki mice. Heatmaps of differential complement gene expression in neural retina (A) and posterior eyecups (B) in female Efemp1ki/ki mice versus in Efemp1+/+ mice at 3, 9 and 17 months of age. The differential expression value of each comparison was expressed as t-statistic (t). Most complement components from the RNA-seq study were increased (positive t-value in red) in both tissues when comparing Efemp1ki/ki to Efemp1+/+. (C) Examples of complement C3, Cfb, C4b and Itgam gene expression in neural retina and posterior eyecups from 17-month-old female Efemp1ki/ki mice and Efemp1+/+ mice. Relative gene expression levels are graphed as TPM reads. *P < 0.05, **P < 0.01, ***P < 0.001. (D) Ocular and systemic complement activation levels in 18-month-old female and male Efemp1ki/ki mice and Efemp1+/+ mice. Neural retina and posterior eye cup extracts and plasma from 18-month-old male and female Efemp1+/+ (n = 5–8) and Efemp1ki/ki (n = 5–8) mice were evaluated for complement activation products iC3b or C3d, and Ba by western blot analysis. Values are graphed relative to Efemp1+/+ levels per tissue and gender. Complement C3 and FB breakdown products iC3b and Ba were elevated about 2-fold (P < 0.05) in the eyes but unchanged in plasma of naive female Efemp1ki/ki mice as compared with Efemp1+/+ controls. However, ocular iC3b and Ba levels were not increased in the male Efemp1ki/ki mice, suggesting a gender-dependent response to the Efemp1 R345W mutation.
To assess any correlation between complement gene expression and complement protein activation, whole eye lysates (lens removed) from Efemp1ki/ki mice and littermate controls were evaluated by western blot. Complement factor D and C3b/Bb proteases cleave full-length FB and C3 proteins, respectively, to generate the breakdown products Ba, C3d and iC3b, levels of which are direct readouts for AP complement activity. C3d is the predominant C3 breakdown product in mouse plasma, while iC3b is the main C3 activation fragment in mouse eye tissue (29). Ocular Ba and iC3b levels were elevated 2-fold (P < 0.05), but serum levels remained unchanged in 18-month-old female Efemp1ki/ki mice compared with Efemp1+/+ mice (Fig. 2D), suggesting that the R345W mutation has an ocular-specific effect. Plasma and ocular levels of full-length C3alpha and FB were similar in female Efemp1ki/ki versus Efemp1+/+ mice (Supplementary Material, Fig. S2B). Surprisingly, ocular Ba and iC3b levels were not increased in male mice carrying the R345W mutation, indicating a gender difference (Fig. 2D).
Cfb deletion reduced Efemp1 R345W mutation-associated eye pathology
Garland et al. (11) previously reported that deletion of C3 prevents sub-RPE deposit formation in Efemp1ki/ki mice. C3 is the central node for all three complement pathways (alternative, classical and lectin pathway). We investigated the direct contribution of the alternative complement pathway in promoting sub-RPE deposit accumulation in Efemp1ki/ki mice by creating an Efemp1ki/ki Cfb−/− double mutant mouse line. Complement factor B is the essential catalytic subunit of C3 and C5 convertases with Cfb deletion abolishing alternative complement pathway activation in mice (29). Lack of FB protein in Efemp1ki/ki Cfb−/−mice was confirmed by western blot analysis (Supplementary Material, Fig. S3). Eyes of 18-month-old female mice with the 4 genotypes (Efemp1+/+:Cfb+/+control, Efemp1ki/ki:Cfb+/+, Efemp1+/+:Cfb−/− and Efemp1ki/ki:Cfb−/−double mutant) were examined by TEM. There were extensive sub-RPE deposits in Efemp1ki/ki:Cfb+/+ mice (Fig. 3A). In this cohort of Efemp1+/+:Cfb+/+ mice, the sub-RPE deposits were very small that could be seen only at higher magnification (not shown). No sub-RPE deposits were observed in Efemp1ki/ki:Cfb−/− and Efemp1+/+:Cfb−/− mice. Similar to C3, Cfb deletion appears sufficient to prevent the formation of sub-RPE deposits in Efemp1ki/ki mice, implicating the alternative complement pathway activation as a key driver of sub-RPE deposit accumulation.
Figure 3.

Cfb deletion reduces sub-RPE deposits and ameliorates pathway dysregulated in the eyes of Efemp1ki/ki mice. (A) Representative TEM images of RPE-choroid area in eyes collected from 18-month-old Efemp1+/+:Cfb+/+, Efemp1ki/ki:Cfb+/+, Efemp1+/+:Cfb−/− and Efemp1ki/ki:Cfb−/− mice. Extensive sub-RPE deposits were observed in Efemp1ki/ki:Cfb+/+ but not in Efemp1+/+:Cfb−/− or in Efemp1ki/ki:Cfb−/− mice. Only very few small deposits were detected in Efemp1+/+:Cfb+/+ (not shown). One eye per genotype was evaluated. White arrows indicate the location of sub-RPE deposits. All four images have the same magnification. Arrows indicate the sub-RPE deposit location. BrM = Bruch’s membrane, Scale bar = 0.5 μm. (B) Heatmap of pathway changes in eye tissues from Efemp1+/+:Cfb+/+ to Efemp1ki/ki:Cfb+/+, Efemp1ki/ki:Cfb−/− or Efemp1+/+:Cfb−/−. Whole eye lysates (lens removed) of 18-month-old female Efemp1+/+:Cfb+/+, Efemp1ki/ki:Cfb+/+, Efemp1+/+:Cfb−/− and Efemp1ki/ki:Cfb−/− mice were characterized by TMT proteomics profiling (IQ Proteomics). Pathway changes in each comparison were expressed as NES. Differential protein expression of GSEA hallmark pathways between Efemp1ki/ki:Cfb+/+ versus Efemp1+/+:Cfb+/+ with P < 0.1 was included. Dysregulated pathways include increased interferon response, complement, IL6 and UPR and decreased oxidative phosphorylation in Efemp1ki/ki:Cfb+/+mice compared with Efemp1+/+:Cfb+/+ controls. All dysregulated pathways by Efemp1 R345W mutation were either lessened or even reversed when Cfb was deleted. *P < 0.1, **P < 0.05, ***P < 0.01. (C) Examples of representative ocular protein levels in key pathways in the four genotypes: A (blue) = Efemp1+/+:Cfb+/+, B (red) = Efemp1ki/ki:Cfb+/+, C (green) = Efemp1ki/ki:Cfb−/− and D (purple) = Efemp1+/+:Cfb−/− mice. Protein levels measured as ‘abundance’ of total peptide signals for each protein. CR3 (Itgam and Itgb2) in complement/inflammatory pathways as well as Hspa5 and Hsp90b1 in UPR signaling were increased in Efemp1ki/ki:Cfb+/+ compared with in Efemp1+/+:Cfb+/+, then returned to wild-type control levels in Efemp1ki/ki:Cfb−/− mice. Mitochondrial oxidative phosphorylation proteins Ndufb7 (NADH:Ubiquinone Oxidoreductase Subunit B7) and Uqcrfs1 (Ubiquinol-Cytochrome C Reductase, Rieske Iron–Sulfur Polypeptide 1) were decreased in Efemp1ki/ki:Cfb+/+ compared with in Efemp1+/+:Cfb+/+, then partially returned to wild-type levels in Efemp1ki/ki:Cfb−/−.
To further investigate the effect of complement FB deficiency on specific biological pathways, proteomics profiling was performed using protein lysates of the whole eye minus lens from the four genotypes. Dysregulated protein pathways in hallmark gene sets are listed in Fig. 3B. Consistent with our RNA-seq data, some of the key Efemp1R345 mutation-dysregulated pathways in gene expression (Fig. 1B and C) were also altered at the protein level compared with littermate controls (Fig. 3B). Up-regulated pathways in Efemp1ki/ki eyes included IL-6 signaling, interferon response and UPR. Oxidative phosphorylation was again the top down-regulated pathway (Fig. 3B).
Importantly, deletion of Cfb in Efemp1ki/ki mice partially rescued most Efemp1R345W mutation dysregulated pathways (Fig. 3B). For example, Cfb deletion reduced Efemp1ki/ki-mediated upregulation of proteins of the immune response/complement CR3 complex, and even the upstream-UPR proteins (Itgam/Cd11b, Itgb2/Cd18, Hspa5, Hsp90b1) (Fig. 3C). Conversely, Cfb deletion partially reversed the repression of mitochondrial/energy metabolism proteins (Ndufb7, Uqcrfs1) (Fig. 3B and C). Deletion of Cfb in the Efemp1+/+ control strain had no significant effects on any of the pathways altered in Efemp1ki/ki mice (Fig. 3B). These results indicate that AP blockade via Cfb deletion exerts a broad protective effect on Efemp1 R345W mutation-associated pathway dysregulation affecting ocular homeostasis.
An oral FB inhibitor reduced sub-RPE deposit accumulation in Efemp1ki/ki mice
We subsequently assessed whether the accumulation of sub-RPE deposits could be ameliorated by pharmacologic inhibition of FB. Previously, we have shown that systemic administration of the low molecular weight complement factor B inhibitor, Iptacopan/LNP023, potently blocks LPS-induced complement activation in mice in a dose- and time-dependent manner and in both plasma and eye tissues (30,31). Here, we investigated the effects of a new factor B inhibitor, DB-82-CB77, described and synthesized in the patent application WO2013/192345. DB-82-CB77 exhibits dose-dependent and sustained inhibition of LPS-induced systemic and ocular complement activity in young wild-type mice (Fig. 4A and B). DB-82-CB77 fully inhibits ocular complement activation as soon as 4 h and up to 12–16 h post-administration as measured by quantification of the complement products C3d and iC3b levels in young and older C57BL/6 mice after a single dose or repeated daily dosing of 60 mg/kg for 8 weeks (Fig. 4B, Supplementary Material, Fig. S4). Hence, this dosing regimen provides complete AP complement inhibition for at least 16 h every day.
Figure 4.

Oral complement FB inhibitor DB-82-CB77 potently inhibited complement activation and reduced sub-RPE deposit accumulation in female Efemp1ki/ki mice. (A) Dose response relationship of oral small molecule FB inhibitor/FB compound, DB-82-CB77, in the mouse LPS-induced complement activation model, and 7-week-old female C57BL/6 mice were challenged with LPS or PBS control i.p. Three and half hours later, mice were orally administered with either vehicle or DB-82-CB77 at doses ranging from 1 to 30 mg/kg, plasma and eye tissues were collected 4 h after compound dosing and analyzed for complement activities by western blot. Inhibition of LPS-induced C3d + iC3b generation by DB-82-CB77 is expressed as % inhibition of C3d + iC3b SEM which is estimated as 100 × [1 − (compound group − average PBS group)/(average LPS group − average PBS group)]. DB-82-CB77 inhibited LPS-induced ocular and systemic C3d + iC3b generation in a dose-dependent manner. **P < 0.01, ***P < 0.001, ****P < 0.0001. (B) A time course of a single 60 mg/kg oral dose DB-82-CB77 in mouse LPS-induced complement activation model. Complement C3d + iC3b levels were expressed relative to baseline PBS-treated group. LPS treatment induced over 2-fold complement levels above the PBS level. Near complete AP inhibition lasted out to 16 h after single dose of DB-82-CB77 in ocular tissues and partial inhibition in plasma at 16 h. **P < 0.01, ***P < 0.001. (C) DB-82-CB77 inhibits plasma basal complement levels in Efemp1ki/ki mice. Male and female 10-month-old Efemp1ki/ki mice were dosed orally with either vehicle (veh) or DB-82-CB77 (FB compound) daily for 8 weeks. Plasma samples were collected 4 h post-dosing with DB-82-CB77 and analyzed for complement breakdown products by western blot. Plasma C3d and Ba levels were expressed relative to male vehicle group values. Female Efemp1ki/ki mice have higher basal plasma C3d and Ba levels than male Efemp1ki/ki mice. Plasma C3d and factor Ba were reduced (P < 0.05) in both male and female Efemp1ki/ki mice. *P < 0.05, **P < 0.01. (D) TEM mages are representative of both vehicle (left) and DB-82-CB77 (right)-treated mice. Images are from comparable quadrants of retinal sections. Deposits are outlined in yellow. Arrows indicate Bruch’s membrane. The white dots in Bruch’s membrane are unstained collagen fibrils. Magnification = 18 500.(E) Average sub-RPE deposit area in female and male Efemp1ki/ki mice treated with either vehicle or FB inhibitor DB-82-CB77. Electron microscopy was performed on 14–16 eyes per group. Images of basal laminar deposits were collected at a direct magnification of 18 500. Sub-RPE deposit area in each image was determined using ImageJ. All deposit area from the entire temporal/ventral and dorsal/nasal retinal section were summed as the total deposit area per eye. Eyes from female Efemp1ki/ki mice dosed with DB-82-CB77 for 8 weeks showed a 65% reduction in sub-RPE basal deposit area (P = 0.029) comparing with female vehicle controls, using one-way AVOVA Dunnett’s multiple comparison test. Female Efemp1ki/ki mice have 64% more sub-RPE deposits than male Efemp1ki/ki mice (P = 0.038).
SEM which is estimated as 100 × [1 − (compound group − average PBS group)/(average LPS group − average PBS group)]. DB-82-CB77 inhibited LPS-induced ocular and systemic C3d + iC3b generation in a dose-dependent manner. **P < 0.01, ***P < 0.001, ****P < 0.0001. (B) A time course of a single 60 mg/kg oral dose DB-82-CB77 in mouse LPS-induced complement activation model. Complement C3d + iC3b levels were expressed relative to baseline PBS-treated group. LPS treatment induced over 2-fold complement levels above the PBS level. Near complete AP inhibition lasted out to 16 h after single dose of DB-82-CB77 in ocular tissues and partial inhibition in plasma at 16 h. **P < 0.01, ***P < 0.001. (C) DB-82-CB77 inhibits plasma basal complement levels in Efemp1ki/ki mice. Male and female 10-month-old Efemp1ki/ki mice were dosed orally with either vehicle (veh) or DB-82-CB77 (FB compound) daily for 8 weeks. Plasma samples were collected 4 h post-dosing with DB-82-CB77 and analyzed for complement breakdown products by western blot. Plasma C3d and Ba levels were expressed relative to male vehicle group values. Female Efemp1ki/ki mice have higher basal plasma C3d and Ba levels than male Efemp1ki/ki mice. Plasma C3d and factor Ba were reduced (P < 0.05) in both male and female Efemp1ki/ki mice. *P < 0.05, **P < 0.01. (D) TEM mages are representative of both vehicle (left) and DB-82-CB77 (right)-treated mice. Images are from comparable quadrants of retinal sections. Deposits are outlined in yellow. Arrows indicate Bruch’s membrane. The white dots in Bruch’s membrane are unstained collagen fibrils. Magnification = 18 500.(E) Average sub-RPE deposit area in female and male Efemp1ki/ki mice treated with either vehicle or FB inhibitor DB-82-CB77. Electron microscopy was performed on 14–16 eyes per group. Images of basal laminar deposits were collected at a direct magnification of 18 500. Sub-RPE deposit area in each image was determined using ImageJ. All deposit area from the entire temporal/ventral and dorsal/nasal retinal section were summed as the total deposit area per eye. Eyes from female Efemp1ki/ki mice dosed with DB-82-CB77 for 8 weeks showed a 65% reduction in sub-RPE basal deposit area (P = 0.029) comparing with female vehicle controls, using one-way AVOVA Dunnett’s multiple comparison test. Female Efemp1ki/ki mice have 64% more sub-RPE deposits than male Efemp1ki/ki mice (P = 0.038).
The earliest appearance of sub-RPE deposits in Efemp1ki/ki mice was reported at 12 months of age (18); therefore, we elected to initiate daily treatment in 10-month-old animals to ensure sufficient detection of sub-RPE deposits in 12-month-old mice at the end of the 8-week study. Ten-month-old male and female Efemp1ki/ki mice were treated by daily oral gavage with either vehicle or 60 mg/kg of DB-82-CB77 for 8 weeks. Eyes and plasma were collected 6–9 h after the final dose. Plasma levels of basal complement breakdown products, C3d and Ba, were significantly reduced in DB-82-CB77-treated mice (Fig. 4C), confirming systemic inhibition of the spontaneous/tick-over complement activity. Basal systemic complement C3d and Ba levels in male Efemp1ki/ki mice were 30–50% lower than in females. Maximum inhibition of the spontaneous/tick-over complement activities by FB inhibitor DB-82-CB77, which is distinct from the exaggerated LPS or other danger signal-induced complement activation, was in the 30–40% range (data not shown).
Sub-RPE deposits were measured as the total area of deposits across the entire thin section from the temporal-ventral and nasal-dorsal regions at the level of the optic nerve for each eye as outlined in representative TEM images (Fig. 4D). Female mice (n = 16) showed on average a 65% reduction in deposit area compared with the vehicle group of female mice (n = 15; P = 0.029), while no differences were identified in males (vehicle n = 14, treated n = 15) (Fig. 4E). As sub-RPE deposits could have already accumulated by 10 months of age, a greater therapeutic potential may be achieved with an earlier and longer dosing regimen. In summary, systemic AP complement inhibition with an oral FB inhibitor for 8 weeks reduced sub-RPE deposit formation in female Efemp1ki/ki mice.
Sub-RPE deposits were 64% smaller in vehicle-treated Efemp1ki/ki males compared with females (Fig. 4E). The reduced basal complement activity in males versus females in plasma (Fig. 4C) is consistent with a lower AP activity leading to slower sub-RPE deposit accumulation and supports a critical role of AP complement activation in the formation of sub-RPE deposits. Lack of an apparent treatment effect with FB inhibitor in male mice may be related the slower deposit growth rate in males and reduced ocular complement activation compared with females (Fig. 2D).
Discussion
Age-related diseases are commonly associated with protein misfolding and aggregation. Monogenic inherited dystrophies with macular drusen similar to AMD can be caused by misfolding mutations of secreted proteins such as EFEMP1 R345W in DHRD/ML (8). In this study, we found that the Efemp1 R345W mutation in the eyes of mice exacerbates the dysregulation of aging-associated pathways including the alternative complement pathway (AP) as illustrated in Figure 5. AP complement activation together with other protein misfolding-induced cellular processes (such as UPR, innate immune response and disrupted mitochondrial metabolism) contribute to the formation and accumulation of pathological sub-RPE deposits. Our experiments suggest that Cfb KO or FB inhibition reduces sub-RPE deposit accumulation, although the effect was apparent only in female Efemp1ki/ki mice. Cfb KO also normalizes multiple pathological pathways in the eyes of Efemp1ki/ki mice including upstream degenerative events such as UPR signaling, demonstrating the importance of the AP in this pathology. Given the similarities between human AMD and Efemp1ki/ki mice in sub-RPE deposit morphology, this mouse model may provide further insights in the role of complement in drusen accumulation (formation, clearance or both) for both DHRD/ML and AMD. Our data demonstrate efficacy of alternative complement pathway inhibition in the Efemp1ki/ki mouse model, suggesting therapeutic potential in human macular dystrophies and AMD, particularly at the very early stage of the disease.
Figure 5.

Potential roles of AP complement and FB in sub-RPE deposit accumulation in Efemp1ki/ki mice. Aging-associated diseases and EFEMP1 R345W mutation are commonly associated with protein misfolding/proteostasis impairment, leading to ER stress/UPR activation. The Efemp1 R345W mutation prone to protein misfolding accelerates aging by exacerbating the dysregulation of aging associated pathways including complement activation of the alternative pathway in the back of the eye. Other Efemp1 R345W dysregulated pathways include innate immune/inflammatory responses and mitochondrial metabolism. AP complement activation together with other protein misfolding-induced pathways contribute to the accumulation of sub-RPE deposits by promoting deposit formation and/or interfering its clearance. Complement FB likely plays an important role in this process since Cfb KO or FB inhibitor was able to reduce sub-RPE deposit accumulation. AP complement activation may also influence upstream pathogenic pathways as Cfb KO reversed multiple pathological pathway changes in the eyes of Efemp1 ki/ki mice including UPR. This illustration was created with BioRender.com.
Complement activation is one of main, but not the sole, pathogenic mechanisms necessary for AMD. Complement, together with impaired aging-mediated proteostasis, dysregulated processing of extracellular matrix and deficient reverse cholesterol transport, likely drives AMD disease progression. Similarly, in DHRD/ML, complement activation could be one of the main responders following protein misfolding by the EFEMP1 R345W mutation, resulting in sub-RPE deposit accumulation. Complement activation is also increased with advancing age (29) and therefore may contribute to normal aging associated sub-RPE deposit accumulation.
Our observations are consistent with human RPE culture studies reporting reduced secretion of EFEMP1 R345W protein and accumulation of protein aggregates within endoplasmic reticulum (14,15,32,33). The EFEMP1 R345W protein co-localizes with the master regulator of ER homeostasis, HSPA5, in ER lumen causing UPR activation (15). The EFEMP1 R345W protein relies on the HSP90B1, a chaperone protein, to form proper structure for secretion (34). Our study provides in vivo validation of DHRD/ML Efemp1 R345W misfolding-prone mutation inducing both UPR mRNA and protein expression such as Hsp5a and Hsp90b1. The R345W mutation is associated with decreased RPE signature gene expression such as Rpe65 and Lrat, consistent with the ER stress response observed in human RPE cell culture (32). Other than the reduced expression of RPE visual cycle genes, we found no changes of dedifferentiation gene expression in bulk tissues from 18-month-old Efemp1ki/ki posterior eyecups. However, proteomics data (Fig. 3B) suggest a moderate increase of proteins involved in epithelial-mesenchymal transition at the pathway level. It is likely that at the ages examined, Efemp1ki/ki mice have very early signs of RPE de-differentiation but they have not yet progressed to the hypertrophic stage as described in an epithelial-mesenchymal transition model of RPE mitochondrial ablation (35).
Interestingly, R345W induced local/ocular complement activation in eye tissues but not in plasma. The 2-fold increase of complement activation in whole eye lysate in female Efemp1ki/ki mice may result from both systemic and local complement components activated in the sub-RPE space. We previously reported a gradual increase of AP complement activation with age in plasma and eye tissue of C57BL/6 wild-type mice (29). The Efemp1 R345W mutation accelerated age-dependent complement activation in the eyes of female mice, likely via impairing proteostasis.
Garland et al. demonstrated that genetic knockout of C3, but not of C5, substantially reduced the accumulation of sub-RPE deposits in the same strain of Efemp1ki/ki mice (11,22). The complement FB breakdown product Bb is the catalytic subunit of C3 and C5 convertases in complex with C3b which functions upstream of C5 in the AP complement pathway. Our finding of reduced sub-RPE deposits and Efemp1 R345W dysregulated pathways by Cfb deletion further confirms the important role of AP complement in macular diseases such as AMD. In addition to genetic deletion, we have demonstrated that the accumulation of sub-RPE deposits can be reduced by pharmacological inhibition of FB protease in female Efemp1ki/ki mice. Both Garland and our findings are consistent with clinical trial results showing a therapeutic benefit of C3 inhibition with drugs such as APL-2 in geographic atrophy (GA). In a phase 2 trial of APL-2, the growth of GA was reduced by 20–29% (36). Interestingly, the clinical benefit of C5 inhibition in GA is less clear with one phase 3 trial of Zimura (an inhibitor of the cleavage of C5) demonstrating reduced GA growth while two earlier trials of C5 inhibitors did not (NCT01255462, NCT01527500, NCT02686658, NCT04435366) (37,38). A second Zimura phase 3 trial projected to complete in 2022 will yield more information on the role of the terminal complement pathway on atrophy lesion growth. While C3 and C5 may have a role in the late stage of AMD, our data suggest that AP activation also contributes to drusen formation in earlier stages of the disease.
The reason for sex differences in complement activation between male and female Efemp1ki/ki mice is unknown. Female Efemp1ki/ki mice have higher complement activity and accumulate more sub-RPE deposits than males (Fig. 2D, Fig. 4C and E). Systemic complement activity varies by sex, mouse strain, detection method and even according to which specific complement component is used to measure the complement activity (39). Sex differences in the activity of the complement system in mice may not be rodent-specific. Sex differences in blood complement activity have been reported in a healthy Caucasian population with lower C3 levels in females compared with males (40). A recent study found higher levels of FB and FI in females than males with iAMD, although the ratios of activation products to full-length FB were similar between iAMD and control groups (41). The different complement responses to the Efemp1 R345W mutation between male versus female mice highlight the importance of assessing sex effects on the biology of preclinical models.
The gender difference in developing sub-RPE deposits in Efemp1ki/ki mice is intriguing. There is published evidence that sex hormones influence the transcriptome in advanced age (42). Also, expression of the EFEMP1 gene may be directly regulated by estrogen (43). In addition to our observation of higher complement activity in female mice, differentially expressed genes in inflammation-related pathways were higher in the retinal and sensory neurons of female mice than males (42,44). Expression of genes linked to oxidative phosphorylation or mitochondrial metabolism is higher in male mice (44), a phenomenon also observed in human tissues (45). Higher inflammatory response and weaker mitochondrial metabolism in aged females are two hallmarks of aging. Accelerated aging with impaired proteostasis may lead to more deposit accumulation in female mice. We note that AMD tends to be more common in women than men (65% cases are in women, https://www.nei.nih.gov/learn-about-eye-health/outreach-campaigns-and-resources/eye-health-data-and-statistics/age-related-macular-degeneration-amd-data-and-statistics) (46,47). Future studies examining gene and protein expression differences between male and female Efemp1ki/ki mice might provide further insights about the gender differences in susceptibility to sub-RPE deposits.
The sub-RPE deposits in aged Efemp1ki/ki mice mimic drusen precursors in DHRA/ML and AMD patients, an early change not visible on fundoscopy. The formation of the sub-RPE deposit in Efemp1ki/ki was a slow process, taking almost a year for detection by TEM, but without accompanying changes in RPE morphology or overt changes in gene expression (18). Despite the mild pathology, we were able to determine R3435W-induced transcript and protein pathway changes as well as complement FB inhibition of sub-RPE deposit formation.
Our data suggest that AP complement activation could play a causal role in age-related and impaired proteostasis-driven eye diseases including AMD and rare macular dystrophies with autosomal dominant drusen such as DHRD/ML, SFD and LORD. Although multiple pathogenic pathways may be involved in a complex disease such as AMD, human genetics and GA clinical trial results suggest that complement activation is critical for the initiation and the progression of AMD (36,37). Our findings from the evaluation of Cfb deletion in Efemp1ki/ki mice support FB inhibition as a therapeutic candidate for AMD patients. Characterization of AMD postmortem eyes identified the interface between neural retina and the choroid, i.e. RPE/Bruch’s membrane as the site of complement activation in AMD (48–51), with complement activity in that region derived from both systemic circulating and local production of an overlapping set of complement components. The ocular complement activation in Efemp1ki/ki mice implicates the involvement of local protein misfolding. Two clinical studies may shed new light on the role of local and systemic inhibition of complement factor B in AMD. A phase 2 trial in GA patients (NCT03815825) is evaluating a subcutaneously administered antisense oligonucleotide reducing liver but not ocular FB production, therefore reducing amount of systemic factor B available to activate the complement system. A different approach, currently in an intermediate AMD phase 2 trial, is evaluating an oral FB inhibitor with ocular bioavailability (Iptacopan/NCT05230537), thereby inhibiting both local and systemic complement activity.
Materials and methods
Mice
All animal experiments at the Novartis Institutes for BioMedical Research adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Novartis Institutes for BioMedical Research Animal Care and Use Committee. All animal experiments at the Massachusetts Eye and Ear Infirmary (MEEI) were approved by the MEEI, the Harvard Medical School Animal Care and Use Committee and the Novartis Institutes for BioMedical Research Animal Care and Use Committee. The point mutation knock-in mice (Efemp1R345W/R345W mice) were made in Eric Pierce’s lab and have been previously described (12). The Efemp1+/+ and Efemp1ki/ki mice in the C57BL/6 J strain background without the Crbrd8 mutation were generated from heterozygous × heterozygous cross breeding. The Cfb−/− mice in the C57BL/6 J strain background also did not carry the Crbrd8 mutation. Efemp1ki/ki mice were crossed with Cfb−/− to generate heterozygotes for both genes: Efemp1+/ki:Cfb+/−. Efemp1+/+:Cfb+/+, Efemp1ki/ki: Cfb+/+, Efemp1+/+:Cfb−/− and Efemp1ki/ki:Cfb−/− mice were generated from cross breeding with male and female Efemp1+/ki:Cfb+/− mice. Genotypes of each mouse strain were confirmed by PCR for the Efemp1 mutation and Cfb deletion. Mice were housed in a pathogen-free facility on a 12 h light and dark cycle and were fed standard laboratory chow and sterile water ad libitum. At indicated ages, eyes and plasma were collected for transmission electron microscopy (TEM), RNA-seq, proteomics and western blots.
Transmission electron microscopy
Eye globes were collected from 16-month-old female Efemp1+/+ and Efemp1ki/ki mice, fixed in Karnovsky’s fixative (Electron Microscopy Sciences, Hatfield, PA), embedded and sectioned for TEM at either Schepens/MEEI Morphology Core or Novartis Fort Worth electron microscopy facilities. Eyes were collected from 18-month-old female Efemp1+/+:Cfb+/+, Efemp1ki/k: Cfb+/+, Efemp1+/+:Cfb−/− and Efemp1ki/ki:Cfb−/− mice, and processed at the electron microscopy facility of the UMass Medical School. TEM analysis for eyes of 12-month-old male and female EFEMP1ki/ki mice from FB compound study was similar as described previously (11,22). Eyes from vehicle and compound-treated female and male groups (N = 14–16 per group) were processed at the Schepens/MEEI Morphology Core. Ultrathin sections (70 nm) were collected onto single slot formvar–carbon coated grids. The grids were imaged at a direct magnification of 18 500 using an FEI Tecnai G2 Spirit TEM (FEI, Hillsboro, Oregon) at 80 kV. The microscope was interfaced with an AMT XR41 digital CCD camera (Advanced Microscopy Techniques, Woburn, Massachusetts) for digital TIFF file image acquisition.
RNA-seq
Total RNA was isolated from homogenized neural retina and posterior eye cup tissue of 3-, 9- and 17-month-old Efemp1+/+ and Efemp1ki/ki mice (1 eye per mouse, 6–8 mice per group) using RNeasy micro columns (Qiagen, Germantown, MD) including a DNase I treatment step. There were 6–8 samples per genotype in each age group. RNA quality was assessed by the 18S/28S rRNA bands in capillary electrophoresis (Bioanalyzer, Agilent Technologies, Santa Clara, CA). The libraries were built with Illumina TruSeq® Stranded mRNA Sample Preparation kit on Hamilton STAR including a polyA selection step, using 200 ng total RNA input for each sample, and 2 μl of 1:200 ERCC RNA Spike-In Mix 1 was added to each sample (Ambion, 4 456 740), and 200 ng Universal Mouse Reference RNA (Agilent, 740 100) was used as positive control and water was used negative control. Libraries were checked for quality using Agilent Tapestation D1000 Tape, and 76-bp paired-end sequencing was performed on Illumina HiSeq 4000 at Analytical Sciences and Imaging facility in Novartis Institute of BioMedical Research (NIBR), capturing an average of 25 million reads per sample. Read alignment and quantification were run using an internal wrapper pipeline for salmon (52) and the mouse reference genome downloaded from GENCODE (GRCm38.p4). Principal component analysis was performed to assess intra-group and inter-group sample variations. Differential expression analysis was analyzed using edgeR-limma-voom workflow (53). All data process and analyses were performed in custom python and R scripts. The raw fastq files, processed data and metadata can be found on Gene Expression Omnibus (GEO) (accession number GSE210492) (54). Functional GSEA was carried out using differential gene list ranked by t-score for each comparison (3,24). Hallmark gene sets were utilized first for pathway enrichment analysis. Additional GSEA analysis included gene sets from KEGG, Wiki pathway and GO-molecular functions. The GEO ID for published single cell RNA-seq of RPE/choroid is GSE135922.
TaqMan
Eighteen-month-old female Efemp1+/+ and Efemp1ki/ki mice, neural retina and posterior eye cups were collected (n = 12 per genotype). Messenger RNAs were prepared using TurboCapture 96 mRNA kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol and was used directly in cDNA synthesis by High-capacity cDNA reverse Transcription kit (Thermo Fisher, Bedford, MA) according to the manufacturer’s protocol. Real-Time PCR reactions for Rpe65 (Mm01320362_m1, cat# 4331182) and Lrat (Mm00469972_m1, Cat# 4331182) were carried out on a ViiA 7 machine (Thermo Fisher) with TaqMan Gene Expression Assays using TaqMan Fast Advanced Master mix from Thermo Fisher. All data normalized to mouse Gapdh (Cat# 4352339E) expression. The delta–delta Ct method was used as described by Thermo Fisher to determine the relative levels of mRNA expression between experimental samples and controls.
Western blotting
Gel electrophoresis and western blot methods were described in Crowley et al., 2018 (29). Primary antibodies were anti-mouse C3d (R & D Systems, Minneapolis, MN) and anti-human FB antibody (Quidel, San Diego, CA). Anti-mouse tubulin and albumin antibodies (Abcam, Cambridge, UK) were used as loading controls to normalize protein levels for eye tissue and plasma, respectively.
Tandem mass tag-based proteomic profiling
Whole eyes minus lens collected from 18-month-old female Efemp1+/+:Cfb+/+. Efemp1ki/ki:Cfb+/+, Efemp1+/+:Cfb−/− and Efemp1ki/ki:Cfb−/− mice were pooled (3 eyes/sample, 3 samples per genotype). One of the 3 Efemp1+/+:Cfb−/− samples was not analyzed due to TMT11 plex limitation of 11 samples total. Sample preparation and LC–MS analysis were done at IQ Proteomics (Cambridge, MA). The analysis was performed similarly as previously published (55). Briefly, protein tissue extract was reduced, alkylated and ethanol-chloroform precipitation was performed to remove contaminants. Recovered protein pellet was digested with trypsin and labeled with Tandem Mass Tags (TMT) reagents. Pooled labeled peptides were separated into 12 fractions using high pH reversed-phase chromatography and desalted prior to mass spec analysis. LC–MS was performed in the SPS mode on Thermo Lumos instrument. Resulting raw mass spec data files were processed by ProteomeDiscoverer 2.1 (Thermo Fisher, Waltham, MA) searching against Uniprot mouse canonical database (downloaded in February 2018) and internally developed scripts. GSEA was carried out using differentially abundant protein list ranked by t scores (mean fold change value across replicates divided by its standard deviation) of each protein for each comparison (3,24). Hallmark gene sets were utilized for initial analysis. The raw mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (56) partner repository with the dataset identifier PXD035772.
Complement factor B low molecular weight inhibitor and compound study in mice
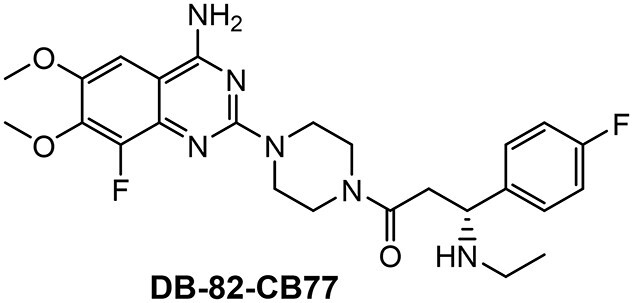
The molecular structure of the complement factor B inhibitor DB-82-CB77 is shown below. The synthesis of the compound is publicly available in patentWO2013/192345 (Example 84). A detailed synthesis protocol will be provided upon request. The IUPAC (International Union of Pure and Applied Chemistry) name for DB-82-CB77 is (R)-1-[4-(4-Amino-8-fluoro-6,7-dimethoxy-quinazolin-2-yl)-piperazin-1-yl]-3-ethylamino-3-(4-fluoro-phenyl)-propan-1-one.
Efficacy studies of FB inhibitors in the mouse model of LPS-induced complement activation were performed as described previously (29,30). For the dose response study, 7-week-old female C57BL/6Ncrl mice were challenged with LPS, 3.5 h later, FB compound DB-82-CB77 was administered orally at 1, 3, 6, 10, 20 or 30 mg/kg. Plasma and eye tissues were collected 4 h after compound dosing for assessment of complement activation. Percentage inhibition of C3d + iC3b by DB-82-CB77 = 100 × [((average LPS − average PBS) − (X − average PBS))/(average LPS − average PBS)]. X = individual value in compound treated group. For the time course study, 7-week-old female C57BL/6 Ncrl mice were dosed once with DB-82-CB77 at 60 mg/kg and samples collected at 4, 8, 10, 12, 16 and 24 h post-dosing. LPS challenge was consistently fixed at 7.5 h before sample collection so that all groups had equal LPS induction time regardless of compound dosing time.
Ten-month-old male and female Efemp1ki/ki mice were orally administered daily with 60 mg/kg DB-82-CB77 or vehicle (0.5% methylcellulose 0.5% Tween 80) for 8 weeks. There were four groups and on average 15 mice per group: female Efemp1ki/ki mice, vehicle; females, DB-82-CB77; males, vehicle and males, DB-82-CB77. Six to nine hours after last compound dose, plasma samples were collected after euthanasia for systemic complement by western blots and eyes were collected for TEM analysis of sub-RPE deposit.
TEM imaging analysis of sub-RPE deposit
Basal laminar deposit formation between the Bruch’s membrane and RPE was systematically assessed by analyzing total deposits within a 70-nm-thin retinal section that extends from the ora serrate in the temporal/ventral quadrant through the optic nerve to the ora serrate of the dorsal/nasal quadrant. TEM images were collected for all deposits within the retinal sections. The areas of the deposits were analyzed using ImageJ software on images that were collected at a direct magnification of 18 500 or were normalized to that magnification. The deposits were outlined manually in each image, areas were measured by ImageJ and the areas of all deposits summed per mouse.
Statistical and data analysis
Data are presented as mean ± standard error of mean (SEM). In heatmaps and bar graphs for differentially expressed pathways, values were expressed as normalized enrichment scores (NESs) using GSEA method for RNA-seq data (false discovery rate < 0.05 significance) and tandem mass tag-based proteomic (TMT proteomics) (P < 0.1 significance) (24). Differential RNA expression values were expressed in heatmaps as ‘t’ for t-score = (sample mean − population mean)/(sample standard deviation/square root (sample size)). RNA (transcript per million—TPM) and protein (abundance) levels in bar graphs (P < 0.05 significance). RNA-seq differential expression analysis using a moderated t-test was run using the limma-voom P package (adjusted P-value < 0.05, |log2FC| > 1 significance). To decrease the false discovery rate, P-values were adjusted using the Benjamini–Hochberg procedure. Relative protein abundance in TMT proteomics dataset was calculated using limma method (57). Specifically, difference in protein abundance expressed as log2 fold change (L2FC) between individual treatments was calculated as difference between means of replicates and corresponding P-values were estimate using limma’s moderated t-test. For comparisons of means between Efemp1+/+ and Efemp1ki/ki mice for complement levels and for comparisons of means between Efemp1+/+ and Efemp1ki/ki mice for sub-RPE deposit area, statistical analysis used was one-way ANOVA Dunnett’s multiple comparison (P < 0.05 significance).
Supplementary Material
Acknowledgements
We thank Patricia Pearson, Philip Seifert, David Cantu-Crouch, Gregory Hendricks, Chia-Ling Huang, Tyler Burks, Ryan Anderson, John Halupowski and Vanessa Davis for technical supports. We thank Magali Saint-Geniez and Chris Wilson for helpful suggestions on the manuscript.
Conflict of Interest statement: The following authors were Novartis employees (E) at the time of this research study: Maura A. Crowley (E), Holger Sellner (E), Angela Banks (E), Lin Fan (E), Tomas Rejtar (E), Natasha Buchanan (E), Omar Delgado (E), YongYao Xu (E), Sandra Jose (E), Christopher M. Adams (E), Muneto Mogi (E), Karen Wang (E), Chad Bigelow (E), Stephen Poor (E), Karen Anderson (E), Bruce Jaffee (E), Ganesh Prasanna (E), Cynthia Grosskreutz (E), Thaddeus P. Dryja (E), Sha-Mei Liao(E). Donita L. Garland, Rosario Fernandez-Godino, Eric A. Pierce performed the FB inhibitor study at the Ocular Genomics Institute at Massachusetts Eye and Ear, Harvard Medical School in collaborations with Novartis Institutes for BioMedical Research.
Conflict of Interest statement. The authors declare no conflict of interest.
Contributor Information
Maura A Crowley, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Donita L Garland, Ocular Genomics Institute at Massachusetts Eye and Ear, Harvard Medical School, Boston, MA 02114, USA.
Holger Sellner, Global Discovery Chemistry, Novartis Institutes for BioMedical Research, Basel, Switzerland.
Angela Banks, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Lin Fan, Analytical Sciences and Imaging, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Tomas Rejtar, Analytical Sciences and Imaging, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Natasha Buchanan, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Omar Delgado, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Yong Yao Xu, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Sandra Jose, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Christopher M Adams, Global Discovery Chemistry, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Muneto Mogi, Global Discovery Chemistry, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Karen Wang, Analytical Sciences and Imaging, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Chad E Bigelow, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Stephen Poor, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Karen Anderson, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Bruce D Jaffee, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Ganesh Prasanna, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Cynthia Grosskreutz, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Rosario Fernandez-Godino, Ocular Genomics Institute at Massachusetts Eye and Ear, Harvard Medical School, Boston, MA 02114, USA.
Eric A Pierce, Ocular Genomics Institute at Massachusetts Eye and Ear, Harvard Medical School, Boston, MA 02114, USA.
Thaddeus P Dryja, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Sha-Mei Liao, Ophthalmology, Novartis Institutes for BioMedical Research, Cambridge, MA 02319, USA.
Funding
Novartis Institutes for BioMedical Research.
Reference
- 1. Fritsche, L.G., Igl, W., Bailey, J.N., Grassmann, F., Sengupta, S., Bragg-Gresham, J.L., Burdon, K.P., Hebbring, S.J., Wen, C., Gorski, M. et al. (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet., 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tan, P.L., Bowes Rickman, C. and Katsanis, N. (2016) AMD and the alternative complement pathway: genetics and functional implications. Hum. Genomics, 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armento, A., Ueffing, M. and Clark, S.J. (2021) The complement system in age-related macular degeneration. Cell. Mol. Life Sci., 78, 4487–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fleckenstein, M., Keenan, T.D.L., Guymer, R.H., Chakravarthy, U., Schmitz-Valckenberg, S., Klaver, C.C., Wong, W.T. and Chew, E.Y. (2021) Age-related macular degeneration. Nat. Rev. Dis. Primers., 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson, D.H., Radeke, M.J., Gallo, N.B., Chapin, E.A., Johnson, P.T., Curletti, C.R., Hancox, L.S., Hu, J., Ebright, J.N., Malek, G. et al. (2010) The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog. Retin. Eye Res., 29, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hollyfield, J.G., Salomon, R.G. and Crabb, J.W. (2003) Proteomic approaches to understanding age-related macular degeneration. Adv. Exp. Med. Biol., 533, 83–89. [DOI] [PubMed] [Google Scholar]
- 7. Stone, E.M., Lotery, A.J., Munier, F.L., Heon, E., Piguet, B., Guymer, R.H., Vandenburgh, K., Cousin, P., Nishimura, D., Swiderski, R.E. et al. (1999) A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet., 22, 199–202. [DOI] [PubMed] [Google Scholar]
- 8. Saksens, N.T., Fleckenstein, M., Schmitz-Valckenberg, S., Holz, F.G., den Hollander, A.I., Keunen, J.E., Boon, C.J. and Hoyng, C.B. (2014) Macular dystrophies mimicking age-related macular degeneration. Prog. Retin. Eye Res., 39, 23–57. [DOI] [PubMed] [Google Scholar]
- 9. Hulleman, J.D. (2016) Malattia Leventinese/Doyne honeycomb retinal dystrophy: similarities to age-related macular degeneration and potential therapies. Adv. Exp. Med. Biol., 854, 153–158. [DOI] [PubMed] [Google Scholar]
- 10. Marmorstein, L. (2004) Association of EFEMP1 with malattia leventinese and age-related macular degeneration: a mini-review. Ophthalmic Genet., 25, 219–226. [DOI] [PubMed] [Google Scholar]
- 11. Garland, D.L., Fernandez-Godino, R., Kaur, I., Speicher, K.D., Harnly, J.M., Lambris, J.D., Speicher, D.W. and Pierce, E.A. (2014) Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Hum. Mol. Genet., 23, 52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sohn, E.H., Wang, K., Thompson, S., Riker, M.J., Hoffmann, J.M., Stone, E.M. and Mullins, R.F. (2015) Comparison of drusen and modifying genes in autosomal dominant radial drusen and age-related macular degeneration. Retina, 35, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grillo, S.L., Etzel, J.D., Weber, S.R., Ondeck, C., Wang, W., Zhao, Y., Barber, A.J. and Sundstrom, J.M. (2021) Descriptive analysis of Fibulin-3 and the extracellular vesicle marker, Alix, in drusen from a small cohort of postmortem human eyes. Exp. Eye Res., 203, 108422. [DOI] [PubMed] [Google Scholar]
- 14. Marmorstein, L.Y., Munier, F.L., Arsenijevic, Y., Schorderet, D.F., McLaughlin, P.J., Chung, D., Traboulsi, E. and Marmorstein, A.D. (2002) Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A., 99, 13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roybal, C.N., Marmorstein, L.Y., Vander Jagt, D.L. and Abcouwer, S.F. (2005) Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest. Ophthalmol. Vis. Sci., 46, 3973–3979. [DOI] [PubMed] [Google Scholar]
- 16. Hulleman, J.D., Kaushal, S., Balch, W.E. and Kelly, J.W. (2011) Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol. Biol. Cell, 22, 4765–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fu, L., Garland, D., Yang, Z., Shukla, D., Rajendran, A., Pearson, E., Stone, E.M., Zhang, K. and Pierce, E.A. (2007) The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum. Mol. Genet., 16, 2411–2422. [DOI] [PubMed] [Google Scholar]
- 18. Marmorstein, L.Y., McLaughlin, P.J., Peachey, N.S., Sasaki, T. and Marmorstein, A.D. (2007) Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum. Mol. Genet., 16, 2423–2432. [DOI] [PubMed] [Google Scholar]
- 19. Kliffen, M., Mooy, C.M., Luider, T.M. and de Jong, P.T. (1994) Analysis of carbohydrate structures in basal laminar deposit in aging human maculae. Invest. Ophthalmol. Vis. Sci., 35, 2901–2905. [PubMed] [Google Scholar]
- 20. Knupp, C., Amin, S.Z., Munro, P.M., Luthert, P.J. and Squire, J.M. (2002) Collagen VI assemblies in age-related macular degeneration. J. Struct. Biol., 139, 181–189. [DOI] [PubMed] [Google Scholar]
- 21. Thakkinstian, A., McEvoy, M., Chakravarthy, U., Chakrabarti, S., McKay, G.J., Ryu, E., Silvestri, G., Kaur, I., Francis, P., Iwata, T. et al. (2012) The association between complement component 2/complement factor B polymorphisms and age-related macular degeneration: a HuGE review and meta-analysis. Am. J. Epidemiol., 176, 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garland, D.L., Pierce, E.A. and Fernandez-Godino, R. (2021) Complement C5 is not critical for the formation of sub-RPE deposits in Efemp1 mutant mice. Sci. Rep., 11, 10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stanton, J.B., Marmorstein, A.D., Zhang, Y. and Marmorstein, L.Y. (2017) Deletion of Efemp1 Is Protective Against the Development of Sub-RPE Deposits in Mouse Eyes. Invest. Ophthalmol. Vis. Sci., 58, 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liberzon, A., Birger, C., Thorvaldsdottir, H., Ghandi, M., Mesirov, J.P. and Tamayo, P. (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Voigt, A.P., Mulfaul, K., Mullin, N.K., Flamme-Wiese, M.J., Giacalone, J.C., Stone, E.M., Tucker, B.A., Scheetz, T.E. and Mullins, R.F. (2019) Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. U. S. A., 116, 24100–24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopez-Otin, C., Blasco, M.A., Partridge, L., Serrano, M. and Kroemer, G. (2013) The hallmarks of aging. Cell, 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cooper, L.L., Hansen, R.M., Darras, B.T., Korson, M., Dougherty, F.E., Shoffner, J.M. and Fulton, A.B. (2002) Rod photoreceptor function in children with mitochondrial disorders. Arch. Ophthalmol., 120, 1055–1062. [DOI] [PubMed] [Google Scholar]
- 28. Hong, S., Beja-Glasser, V.F., Nfonoyim, B.M., Frouin, A., Li, S., Ramakrishnan, S., Merry, K.M., Shi, Q., Rosenthal, A., Barres, B.A. et al. (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crowley, M.A., Delgado, O., Will-Orrego, A., Buchanan, N.M., Anderson, K., Jaffee, B.D., Dryja, T.P. and Liao, S.M. (2018) Induction of ocular complement activation by inflammatory stimuli and intraocular inhibition of complement factor D in animal models. Invest. Ophthalmol. Vis. Sci., 59, 940–951. [DOI] [PubMed] [Google Scholar]
- 30. Mainolfi, N., Ehara, T., Karki, R.G., Anderson, K., Mac Sweeney, A., Liao, S.M., Argikar, U.A., Jendza, K., Zhang, C., Powers, J. et al. (2020) Discovery of 4-((2S,4S)-4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)be nzoic Acid (LNP023), a factor B inhibitor specifically designed to be applicable to treating a diverse array of complement mediated diseases. J. Med. Chem., 63, 5697–5722. [DOI] [PubMed] [Google Scholar]
- 31. Schubart, A., Anderson, K., Mainolfi, N., Sellner, H., Ehara, T., Adams, C.M., Mac Sweeney, A., Liao, S.M., Crowley, M., Littlewood-Evans, A. et al. (2019) Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. U. S. A., 116, 7926–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou, M., Weber, S.R., Zhao, Y., Chen, H., Barber, A.J., Grillo, S.L., Wills, C.A., Wang, H.G., Hulleman, J.D. and Sundstrom, J.M. (2020) Expression of R345W-Fibulin-3 Induces Epithelial-Mesenchymal Transition in Retinal Pigment Epithelial Cells. Front. Cell Dev. Biol., 8, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woodard, D.R., Nakahara, E. and Hulleman, J.D. (2021) Clinically-identified C-terminal mutations in fibulin-3 are prone to misfolding and destabilization. Sci. Rep., 11, 2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hulleman, J.D. and Kelly, J.W. (2015) Genetic ablation of N-linked glycosylation reveals two key folding pathways for R345W fibulin-3, a secreted protein associated with retinal degeneration. FASEB J., 29, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao, C., Yasumura, D., Li, X., Matthes, M., Lloyd, M., Nielsen, G., Ahern, K., Snyder, M., Bok, D., Dunaief, J.L. et al. (2011) mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Invest., 121, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liao, D.S., Grossi, F.V., El Mehdi, D., Gerber, M.R., Brown, D.M., Heier, J.S., Wykoff, C.C., Singerman, L.J., Abraham, P., Grassmann, F. et al. (2020) Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: a randomized phase 2 trial. Ophthalmology, 127, 186–195. [DOI] [PubMed] [Google Scholar]
- 37. Jaffe, G.J., Westby, K., Csaky, K.G., Mones, J., Pearlman, J.A., Patel, S.S., Joondeph, B.C., Randolph, J., Masonson, H. and Rezaei, K.A. (2021) C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: a randomized pivotal phase 2/3 trial. Ophthalmology, 128, 576–586. [DOI] [PubMed] [Google Scholar]
- 38. Yehoshua, Z., de Amorim Garcia Filho, C.A., Nunes, R.P., Gregori, G., Penha, F.M., Moshfeghi, A.A., Zhang, K., Sadda, S., Feuer, W. and Rosenfeld, P.J. (2014) Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology, 121, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kotimaa, J., Klar-Mohammad, N., Gueler, F., Schilders, G., Jansen, A., Rutjes, H., Daha, M.R. and van Kooten, C. (2016) Sex matters: Systemic complement activity of female C57BL/6J and BALB/cJ mice is limited by serum terminal pathway components. Mol. Immunol., 76, 13–21. [DOI] [PubMed] [Google Scholar]
- 40. Gaya da Costa, M., Poppelaars, F., van Kooten, C., Mollnes, T.E., Tedesco, F., Wurzner, R., Trouw, L.A., Truedsson, L., Daha, M.R., Roos, A. et al. (2018) age and sex-associated changes of complement activity and complement levels in a healthy Caucasian population. Front. Immunol., 9, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marin, A.I., Poppelaars, F., Wagner, B.D., Palestine, A.G., Patnaik, J.L., Holers, V.M., Frazer-Abel, A.A., Mathias, M.T., Manoharan, N., Fonteh, C.N. et al. (2022) Sex and age-related differences in complement factors among patients with intermediate age-related macular degeneration. Transl. Vis. Sci. Technol., 11, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du, M., Mangold, C.A., Bixler, G.V., Brucklacher, R.M., Masser, D.R., Stout, M.B., Elliott, M.H. and Freeman, W.M. (2017) Retinal gene expression responses to aging are sexually divergent. Mol. Vis., 23, 707–717. [PMC free article] [PubMed] [Google Scholar]
- 43. Blackburn, J., Tarttelin, E.E., Gregory-Evans, C.Y., Moosajee, M. and Gregory-Evans, K. (2003) Transcriptional regulation and expression of the dominant drusen gene FBLN3 (EFEMP1) in mammalian retina. Invest. Ophthalmol. Vis. Sci., 44, 4613–4621. [DOI] [PubMed] [Google Scholar]
- 44. Mecklenburg, J., Zou, Y., Wangzhou, A., Garcia, D., Lai, Z., Tumanov, A.V., Dussor, G., Price, T.J. and Akopian, A.N. (2020) Transcriptomic sex differences in sensory neuronal populations of mice. Sci. Rep., 10, 15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lopes-Ramos, C.M., Chen, C.Y., Kuijjer, M.L., Paulson, J.N., Sonawane, A.R., Fagny, M., Platig, J., Glass, K., Quackenbush, J. and DeMeo, D.L. (2020) Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep., 31, 107795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith, W., Mitchell, P. and Wang, J.J. (1997) Gender, oestrogen, hormone replacement and age-related macular degeneration: results from the Blue Mountains Eye Study. Aust. N. Z. J. Ophthalmol., 25, S13–S15. [DOI] [PubMed] [Google Scholar]
- 47. Nuzzi, R., Scalabrin, S., Becco, A. and Panzica, G. (2018) Gonadal hormones and retinal disorders: a review. Front Endocrinol (Lausanne), 9, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Demirs, J.T., Yang, J., Crowley, M.A., Twarog, M., Delgado, O., Qiu, Y., Poor, S., Rice, D.S., Dryja, T.P., Anderson, K. et al. (2021) Differential and altered spatial distribution of complement expression in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci., 62, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chirco, K.R., Flamme-Wiese, M.J., Wiley, J.S., Potempa, L.A., Stone, E.M., Tucker, B.A. and Mullins, R.F. (2018) Evaluation of serum and ocular levels of membrane attack complex and C-reactive protein in CFH-genotyped human donors. Eye (Lond.), 32, 1740–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Johnson, L.V., Ozaki, S., Staples, M.K., Erickson, P.A. and Anderson, D.H. (2000) A potential role for immune complex pathogenesis in drusen formation. Exp. Eye Res., 70, 441–449. [DOI] [PubMed] [Google Scholar]
- 51. Mullins, R.F., Russell, S.R., Anderson, D.H. and Hageman, G.S. (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J., 14, 835–846. [PubMed] [Google Scholar]
- 52. Patro, R., Duggal, G., Love, M.I., Irizarry, R.A., and Kingsford, C. (2020) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods., 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Law, C.W., Alhamdoosh, M., Su, S., Dong, X., Tian, L., Smyth, G.K. and Ritchie, M.E. (2016) RNA-seq analysis is easy as 1-2-3 with limma. Glimma and edgeR. F1000Research, 5, ISCB Comm J-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frankish, A., Diekhans, M., Ferreira, A.M., Johnson, R., Jungreis, I., Loveland, J., Mudge, J.M., Sisu, C., Wright, J., Armstrong, J. et al. (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res., 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Geri, J.B., Oakley, J.V., Reyes-Robles, T., Wang, T., McCarver, S.J., White, C.H., Rodriguez-Rivera, F.P., Parker, D.L., Jr., Hett, E.C., Fadeyi, O.O. et al. (2020) Microenvironment mapping via Dexter energy transfer on immune cells. Science, 367, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Perez-Riverol, Y., Bai, J., Bandla, C., Garcia-Seisdedos, D., Hewapathirana, S., Kamatchinathan, S., Kundu, D.J., Prakash, A., Frericks-Zipper, A., Eisenacher, M. et al. (2022) The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res., 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ritchie, M.E., Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W. and Smyth, G.K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.