

Abstract
Background:
Subtypes of pulmonary arterial hypertension (PAH) differ in both fundamental disease features and clinical outcomes. Angiogenesis and inflammation represent disease features that may differ across subtypes and are of special interest in connective tissue disease-associated PAH (CTD-PAH). We compared inflammatory and angiogenic biomarker profiles across different etiologies of PAH and related them to clinical outcomes.
Methods:
Participants with idiopathic PAH, CTD-PAH, toxin-associated PAH (tox-PAH), or congenital heart disease-associated PAH (CHD-PAH) were enrolled into a prospective observational cohort. Baseline serum concentrations of 33 biomarkers were related to three-year mortality, echocardiogram, REVEAL score, and six-minute walk distance (6MWD). Findings were validated using plasma proteomic data from the UK PAH Cohort Study.
Results:
112 patients were enrolled: 45 idiopathic, 27 CTD-PAH, 20 tox-PAH, 20 CHD-PAH. Angiogenic and inflammatory biomarkers were distinctly elevated within the CTD-PAH cohort. Six biomarkers were associated with mortality within the entire PAH cohort: interleukin-6 (IL-6, HR:1.6, 95% CI:1.18–2.18), soluble fms-like tyrosine kinase 1 (sFlt-1, HR:1.35, 95% CI:1.02–1.80), placental growth factor (PlGF, HR:1.55, 95% CI:1.07–2.25), interferon gamma-induced protein 10 (IP-10, HR:1.44, 95% CI:1.04–1.99), tumor necrosis factor-beta (TNF-β, HR:1.81, 95% CI:1.11–2.95), and NT-proBNP (HR:2.19, 95% CI:1.52–3.14). Only IL-6 and NT-proBNP remained significant after controlling for multiple comparisons. IL-6, IP-10, and sFlt-1 significantly associated with mortality in CTD-PAH, but not non-CTD-PAH subgroups. In the UK cohort, IP-10, PlGF, TNF-β, and NT-proBNP significantly associated with five-year survival.
Conclusions:
Levels of angiogenic and inflammatory biomarkers are elevated in CTD-PAH, compared with other etiologies of PAH, and may correlate with clinical outcomes including mortality.
Introduction:
Pulmonary arterial hypertension (PAH) is characterized by pulmonary vascular remodeling and leads to progressive right heart failure and ultimately death within a median of approximately six years from diagnosis.1 PAH subtypes include, but are not limited to, idiopathic (IPAH), connective tissue disease-associated (CTD-PAH), toxin-associated (tox-PAH), and congenital heart disease-associated (CHD-PAH).2 Patients across PAH subtypes differ in survival, symptom progression, and hemodynamics.3–7 Variation between subtypes in fundamental features like vasodilator responsivity,3,8,9 rates of bone morphogenic protein receptor type 2 (BMPR2) mutation,10–12 and response to medical therapy8,9,13,14 suggests that differences in outcomes may be driven by underlying pathophysiological divergence. Increasingly, the field has recognized the need for precision-medicine approaches to PAH.15,16 Despite this recognition, personalized or subtype-specific medication strategies have yet to be developed, and guideline-based treatment algorithms do not distinguish by PAH subtype.17,18 Treatment personalization is limited by our incomplete understanding of disease pathophysiology, and further obscured by disease heterogeneity both across and within subtypes.15,16,19
Peripheral serum biomarkers have emerged as a promising tool that offers the possibility of investigating disease heterogeneity at multiple levels and informing diagnosis and prognosis of PAH. Biomarkers of inflammation and angiogenesis are especially intriguing as they reflect likely mechanisms of disease which may be therapeutically targetable.20 Specific inflammatory and angiogenic markers are elevated in PAH and variably predict patient outcomes21–26; however, few studies have compared biomarker concentrations across PAH subtypes and related them to clinical outcomes. Examining PAH patients in aggregate may obscure important signals within subtypes and delay recognition of separately targetable disease endotypes in PAH. Conversely, robust findings within one subtype may drive statistical significance in the aggregate group, leading to inappropriate application of therapy to all patients, including subgroups that may not benefit. These issues are critical for understanding PAH pathobiology and moving towards personalized medicine for individuals with PAH.
As such, the aim of this study was to evaluate the angiogenic and inflammatory biomarker profiles of patients across four PAH subtypes within a well-characterized prospective observational cohort of patients with PAH. We hypothesized that these markers would systematically vary by PAH etiology and would have distinct relationships with mortality, echocardiographic findings, disease severity, and six-minute walk distance (6MWD).
Methods:
Data Collection and Study Procedures
Participants from the University of Washington pulmonary vascular disease clinic with an established diagnosis of PAH were enrolled in the Seattle Right Ventricle Translational Science (Servetus) cohort from April 2014 through May 2016. PAH diagnostic criteria were based on guidelines from the 5th World Symposium on Pulmonary Hypertension including a mean pulmonary artery pressure (mPAP) ≥ 25 mmHg, pulmonary capillary wedge pressure (PCWP) ≤ 15 mmHg, and pulmonary vascular resistance (PVR) > 3 Wood Units measured up to one year before study entry.27 Participants with IPAH, tox-PAH, CTD-PAH, or CHD-PAH were included in this analysis. At enrollment, demographics were recorded, New York Heart Association (NYHA) functional class was assessed using a standardized decision aid, 6MWD was completed, and REVEAL 2.0 score was calculated.28 Echocardiograms obtained up to six months before the index visit were read by one of two cardiologists using a standardized research protocol including measurement of RV basal diameter in diastole. Blood samples were collected on enrollment, processed, and frozen at −80°C using a standardized protocol.29 Samples were thawed only once for the current analysis and run in a single batch. An array of biomarkers was assayed using a Meso Scale Discovery multiplex immunoassay. N-terminal pro-brain natriuretic peptide (NT-proBNP) was measured for all participants and served as a “control” marker that was expected to correlate with clinical outcomes. Survival was monitored for 36 months from enrollment and included research questionnaires that were collected every four months. Vital status was available for all participants at study completion.
Statistics
To compare biomarker levels across subtypes, biomarker concentrations were standardized by z-score by subtracting the sample mean and then dividing by SD. Biomarker concentrations across subtypes were compared using Kruskal–Wallis one-way analysis of variance with Dunn’s comparison testing. Data was organized into a heatmap using GraphPad Prism (version 9.3.0 for Windows, GraphPad Software).
For relationships with mortality, biomarker levels were standardized within their own distribution by dividing each biomarker by the standard deviation (SD) for that marker. For each individual biomarker, separate Cox proportional hazards models were run to estimate relationships of a one-SD difference with the hazard of mortality over three years. Age, gender, and PAH etiology were considered in a fully adjusted model that was determined a priori. An additional exploratory model including NT-proBNP was performed to assess whether associations were independent of an established marker of severity. For biomarkers associated with mortality in the cohort at-large, analyses were repeated separately for each of the four subtypes of PAH. Linear regression estimated associations between a one-SD difference in biomarkers (exposure) and 6MWD (meters), right ventricular basal diameter (RVD, mm), tricuspid annular plane systolic excursion (TAPSE, mm), or REVEAL score. Covariates included age, gender, height, weight, and PAH etiology. For mortality-associated biomarkers, Spearman’s rank correlation coefficients were calculated.
A p-value ≤ 0.05 was defined as significant in the primary interpretation to avoid Type II error and a ‘false negative’ in this hypothesis-generating cohort. Primary analyses were complete case analyses of biomarkers with >90% capture. Given the possibility that “missing” biomarkers might represent biomarkers below the lower limit of detection, sensitivity analyses replaced “missing” biomarkers with a value equal to the lower limit of detection in the cohort. Statistical analysis and data illustration were performed with STATA 15.1 (StataCorp) and GraphPad Prism software.
Confidence in the results and external validation
Given that multiple biomarkers were tested, results in the Servetus cohort were also evaluated against a more restrictive false discovery rate (FDR) of 5% using a Benjamini-Hochberg procedure. In addition, for biomarkers associated with mortality in Servetus, a focused validation using the UK PAH Cohort Study was performed.30,31 Proteomic data for 357 patients with idiopathic or heritable PAH was obtained on plasma samples, using an aptamer-based assay (SomaScan 4). For each significant biomarker in the Servetus cohort, Cox regression analyses corrected for age and sex, were used to validate or refute associations with all-cause five-year mortality or lung transplant in the UK Cohort.30
Results:
PAH Patient Characteristics
A total of 112 patients with pulmonary arterial hypertension (PAH) were included in this study (Table 1). The largest group was patients with IPAH. Nearly all tox-PAH participants were a result of methamphetamine use and most CTD-PAH patients had systemic sclerosis (70.4%). Age varied by subtype, with CTD-PAH participants being the oldest and CHD-PAH participants the youngest. Most patients were female (82% overall). While the majority of CTD-PAH, IPAH, and CHD-PAH patients were NYHA Functional Class I or II, a higher percentage of tox-PAH patients were functional class III. Mortality at 3-years was highest in CTD-PAH (33%) and lowest in IPAH (9%). Most patients were on PAH-directed therapy at the time of enrollment (Table 1).
Table 1.
Baseline patient characteristics
| Patient Characteristics | All PAH (n=112) |
CTD-PAH (n=27) |
IPAH (n=45) |
Tox-PAH (n=20) |
CHD-PAH (n=20) |
|---|---|---|---|---|---|
| Age (year) | 51.6 (14.4) | 58.6 (11.5) | 53.4 (14.4) | 47.8 (8.8) | 41.8 (16.9) |
| Female sex | 82% (92) | 89% (24) | 76% (34) | 90% (18) | 84% (16) |
| BMI (kg/m2) | 29.1 (7.3) | 27.6 (6.5) | 30.8 (7.7) | 32.1 (6.8) | 24.1 (4.8) |
| NYHA Functional Class | |||||
| I/II | 66% (57) | 65% (13) | 71% (25) | 44% (7) | 75% (12) |
| III | 22% (25) | 30% (6) | 20% (7) | 50% (8) | 25% (4) |
| IV | 6% (5) | 5% (1) | 9% (3) | 6% (1) | 0% (0) |
| 6MWD (meters) | 366 (107) | 334 (105) | 387 (119) | 358 (84) | 372 (102) |
| Deceased | 17% (19) | 33% (9) | 9% (4) | 15% (3) | 15% (3) |
| PAH Therapy | |||||
| Monotherapy | 32% (36) | 41% (11) | 27% (12) | 35% (7) | 30% (6) |
| Dual Therapy | 35% (39) | 33% (9) | 38% (18) | 40% (8) | 25% (5) |
| Triple Therapy | 19% (21) | 19% (5) | 24% (11) | 10% (2) | 15% (3) |
| No Therapy | 14% (16) | 7% (2) | 11% (5) | 15% (3) | 30% (6) |
| Right Heart Catheterization | All PAH (n=95) |
CTD-PAH (n=25) |
IPAH (n=36) |
Tox-PAH (n=18) |
CHD-PAH (n=16) |
| RAP (mmHg) | 8.9 (5.8) | 7.9 (5.4) | 9.9 (5.7) | 10.1 (5.9) | 6.7 (6.2) |
| mPAP (mmHg) | 47.1 (12.7) | 42.8 (10.8) | 49.5 (11.4) | 51.0 (11.2) | 44.1 (17.3) |
| PCWP (mmHg) | 10.4 (3.4) | 9.6 (3.5) | 11.2 (3.4) | 11.2 (2.7) | 9.1 (3.8) |
| CI (L/min/m2) | 2.6 (0.8) | 2.3 (0.6) | 2.6 (0.7) | 2.3 (0.7) | 3.3 (0.6) |
| PVR (Wood units) | 8.8 (4.8) | 9.5 (5.3) | 8.6 (4.4) | 10.0 (5.0) | 6.6 (4.3) |
| Echocardiography | All PAH (n=88) |
CTD-PAH (n=16) |
IPAH (n=39) |
Tox-PAH (n=18) |
CHD-PAH (n=13) |
| RV Diameter (mm) | 46 (9) | 42 (8) | 47 (10) | 47 (7) | 45 (1) |
| TAPSE (mm) | 21 (6) | 20 (4) | 22 (6) | 19 (6) | 20 (6) |
Data are presented as mean (standard deviation) or % (n). Functional class was available for 88 patients (79%) and 6MWD data for 98 (87.5%). For the 95 patients undergoing right heart catheterization, right atrial pressure data was available for 86 patients (91%), wedge pressure for 84 (88%), cardiac index for 73 (77%) and pulmonary vascular resistance for 70 (74%). Of the 88 patients with an echocardiogram, RV diameter and TAPSE were not measurable in 2 patients. PAH: pulmonary arterial hypertension; IPAH: idiopathic PAH; CTD-PAH: connective tissue disease-associated PAH; Tox-PAH: toxin-associated PAH; CHD-PAH: congenital heart disease-associated PAH; BMI: body mass index; NYHA: New York Heart Association; 6MWD: 6-minute walk distance; RAP: right atrial pressure; mPAP: mean pulmonary artery pressure; PCWP: pulmonary capillary wedge pressure; CI: cardiac index; PVR: pulmonary vascular resistance; RV: right ventricle; TAPSE: tricuspid annular plane systolic excursion.
Biomarker Concentrations Across PAH Subtypes and Individual Patients
Each biomarker was available for over 90% of patients; patient-level data for missing measurements is shown in the online supplement. Comparison of standardized concentrations for the 33 biomarkers across subtypes revealed a distinct CTD-PAH biomarker profile (Figure 1). Mean concentrations were highest in the CTD-PAH group in 25 out of 33 surveyed biomarkers. Overall, 13 biomarkers were significantly different across groups, and in all cases CTD-PAH was higher than one or more groups. These 13 biomarkers were: interleukin (IL)-7, IL-10, IL-12p40, tumor necrosis factor-alpha (TNF-α), TNF-β, interferon gamma-induced protein 10 (IP-10), monocyte chemoattractant protein-1 (MCP-1), and placental growth factor (PlGF) at p<0.01, and IL-17, serum amyloid A (SAA), soluble intracellular adhesion molecule-1 (sICAM-1), soluble vascular cell adhesion marker (sVCAM-1), and vascular endothelial growth factor (VEGF) at p<0.05 (Figure 1). While CTD-PAH had the highest average age, biomarker levels did not increase with age within this subtype, and age did not appear to explain observed biomarker differences. Participant-level data for each biomarker was tabulated as a heatmap and showed higher levels of angiogenic and inflammatory biomarkers throughout the CTD-PAH cohort (Figure S1).
Figure 1: Heatmap of biomarker concentrations by PAH subtype.
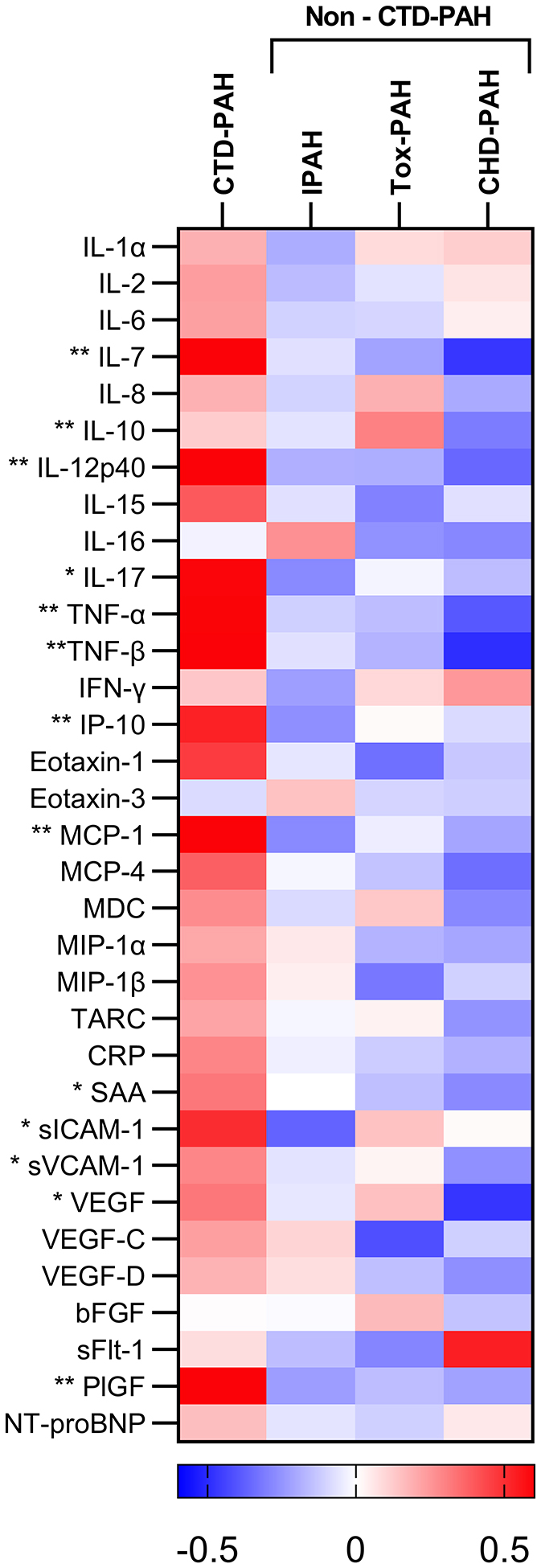
Heatmap displays row Z scores for serum markers of angiogenesis and inflammation across subtypes of pulmonary arterial hypertension. Asterisks denote biomarkers that significantly differ across subtype: * p≤0.05, ** p≤0.01. PAH: pulmonary arterial hypertension; IPAH: idiopathic PAH; CTD-PAH: connective tissue disease-associated PAH; Tox-PAH: toxin-associated PAH; CHD-PAH: congenital heart disease-associated PAH.
Biomarkers Associated with Mortality in All PAH
Biomarkers were investigated for their association with mortality in the full cohort of 112 enrolled PAH patients. Six of 33 biomarkers were associated with mortality: IL-6 (adjusted HR:1.6, 95%CI:1.18–2.18), IP-10 (adjusted HR:1.44, 95%CI:1.04–1.99), TNF-β (adjusted HR:1.81, 95%CI:1.11–2.95), soluble fms-like tyrosine kinase 1 (sFLT-1, adjusted HR:1.55, 95%CI:1.07–2.25), PlGF (adjusted HR:1.55, 95%CI:1.07–2.25), and NT-proBNP (adjusted HR:2.19, 95%CI:1.05–1.99, (Figure 2). Exploratory models evaluated the association with mortality in individuals with otherwise similar levels of NT-proBNP at baseline. After accounting for differences in NT-proBNP, IP-10 was not independently associated with mortality. The relationships of the other biomarkers with mortality were qualitatively similar after adjustment by NT-proBNP, but were attenuated, and only TNF-β remained statistically significant (HR:1.71, 95%CI:1.07–2.72; Figure S2). Sensitivity analysis performed by setting all missing biomarkers to the lowest detectable level did not influence the association with mortality. Accounting for multiple hypothesis testing using a threshold FDR < 5%, only IL-6 and NT-proBNP remained significant. Spearman testing did not suggest strong correlation among mortality-associated biomarkers (Figure S3).
Figure 2: Biomarker associations with mortality in the pooled PAH cohort.
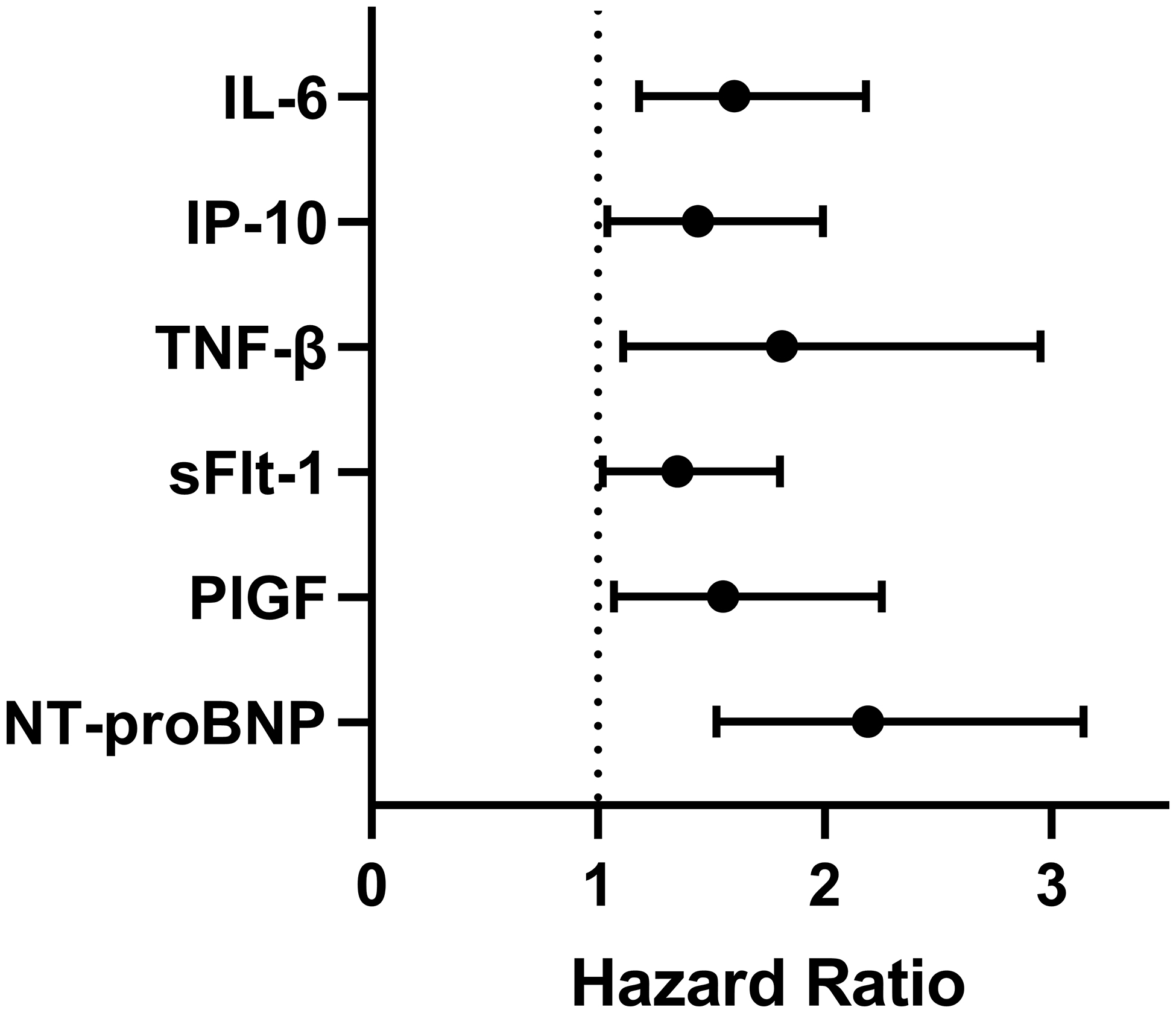
Cox proportional hazards models adjusted for age, sex, and pulmonary arterial hypertension etiology were run evaluating biomarker associations with mortality. The hazard ratio for a one-standard-deviation change in biomarker level against mortality is shown. Definition of abbreviations: IL-6: interleukin-6; IP-10: interferon gamma-induced protein 10; TNF-β: tumor necrosis factor-beta; sFlt-1: soluble fms-like tyrosine kinase 1; PlGF: placental growth factor; NT-proBNP: N-terminal pro-brain natriuretic peptide.
Validation of Biomarker Associations with Mortality
Using data from the UK PAH Cohort Study (n=357), four of six proteins from the Servetus cohort were associated with worse adjusted five-year transplant-free survival: NT-proBNP (p<0.001), TNF-β (p=0.009), IP-10 (p<0.001), and PlGF (p=0.05). IL-6 and sFLT-1 did not correlate with survival (Table S1).
Biomarker Association with Mortality by PAH Subtype
Having identified associations with mortality in the overall cohort and significantly different biomarker distribution in CTD-PAH compared to other subtypes, we performed subgroup analyses to determine if biomarker levels were differentially associated with mortality in CTD-PAH compared with non-CTD-PAH (pooled IPAH, tox-PAH, and CHD-PAH patients). There were similar numbers of deaths in the two groups with nine deaths among CTD-PAH participants and ten deaths among non-CTD PAH participants. Elevation of IL-6 (adjusted HR:1.71, 95%CI:1.12–2.62), IP-10 (adjusted HR:1.53, 95%CI:1.06–2.19), and sFlt-1 (adjusted HR:1.92, 95%CI:1.13–3.29) were associated with mortality in CTD-PAH, while associations did not reach significance in non-CTD-PAH but in many cases were qualitatively similar (Figure 3). NT-proBNP elevation was associated with mortality in both CTD-PAH (HR:2.97, 95%CI:1.45–6.06) and non-CTD-PAH (HR:1.88, 95%CI:1.20–2.96). Serum concentrations for three of the six biomarkers (IP-10, TNF-β, and PIGF) were significantly different in CTD-PAH compared to the other subtypes (Figure S4).
Figure 3: Biomarker association with mortality across different subgroups.
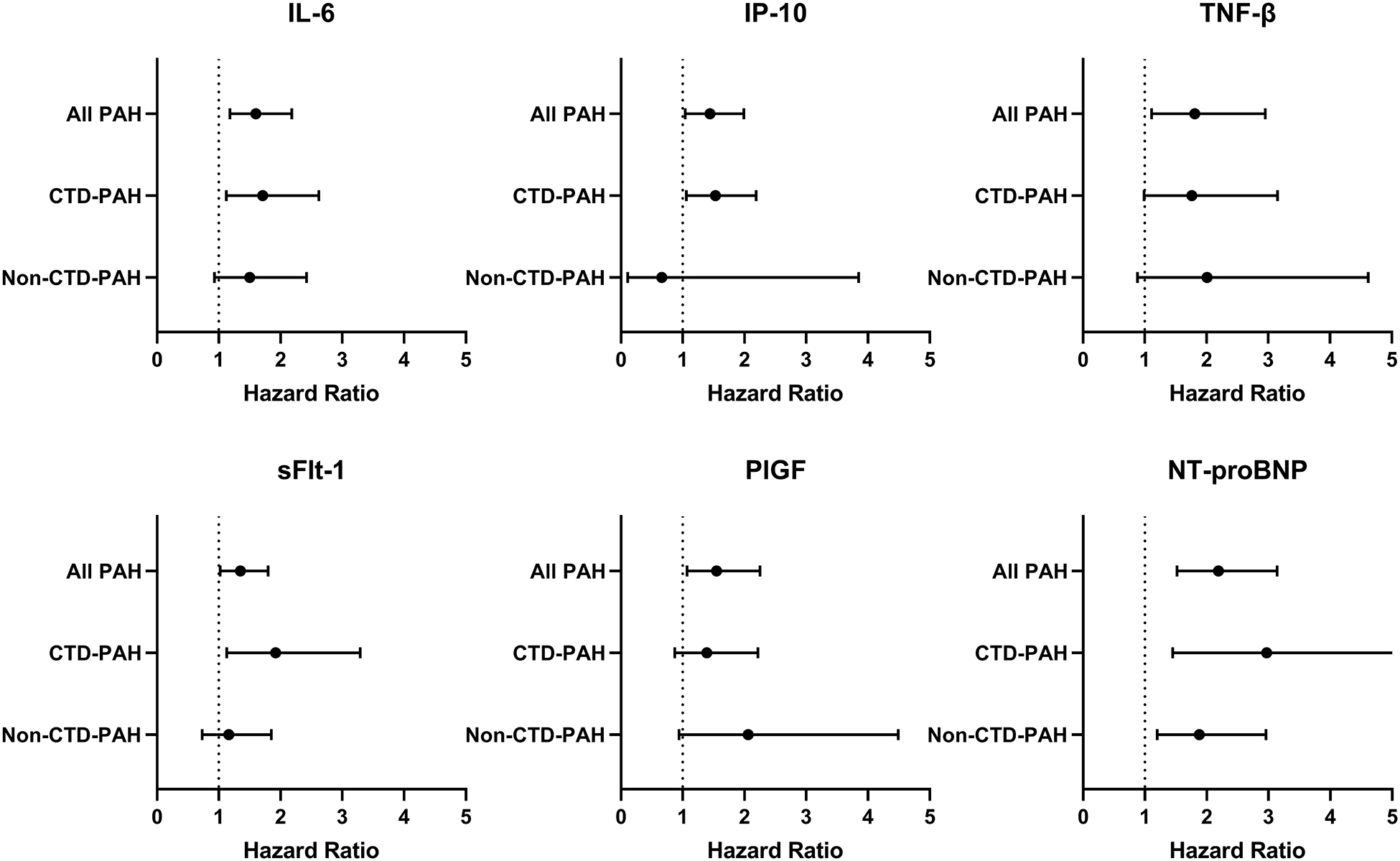
Cox proportional hazards models adjusted for age, sex, and pulmonary arterial hypertension etiology were performed separately for the CTD-PAH and non-CTD-PAH subgroups. The non-CTD-PAH subgroup includes patients with idiopathic, congenital heart disease-associated, and toxin-associated PAH. Definition of abbreviations: PAH: pulmonary arterial hypertension; CTD-PAH: connective tissue disease-associated PAH; IL-6: interleukin-6; IP-10: interferon gamma-induced protein 10; TNF-β: tumor necrosis factor-beta; sFlt-1: soluble fms-like tyrosine kinase 1; PlGF: placental growth factor; NT-proBNP: N-terminal pro-brain natriuretic peptide.
Biomarker Association with Markers of Disease Severity
For the six markers associated with mortality, linear regression was performed to evaluate associations with REVEAL risk score, right heart structure, right heart function, and exercise capacity. Increased IL-6, Flt-1, PlGF, IP-10, and NT-proBNP were associated with significant increases in REVEAL 2.0 score in the overall cohort of PAH patients (Figure 4). Elevated NT-proBNP was also associated with increased RVD (p<0.01), decreased TAPSE (p<0.01), and 6MWD (p<0.01). IL-6 was associated with increased RVD (p=0.04) and decreased 6MWD (p<0.01), and PlGF with decreased 6MWD (p=0.02). When stratified into CTD-PAH and non-CTD-PAH, NT-proBNP continued to be associated with outcomes in both groups. Among those with CTD-PAH (but not those without), sFLT-1 was associated with REVEAL score and IP-10 and IL-6 with 6MWD. Among those with non-CTD-PAH, PlGF and IL-6 were associated with a worse REVEAL score (Table S2).
Figure 4: Biomarker associations with echocardiographic and clinical outcomes.
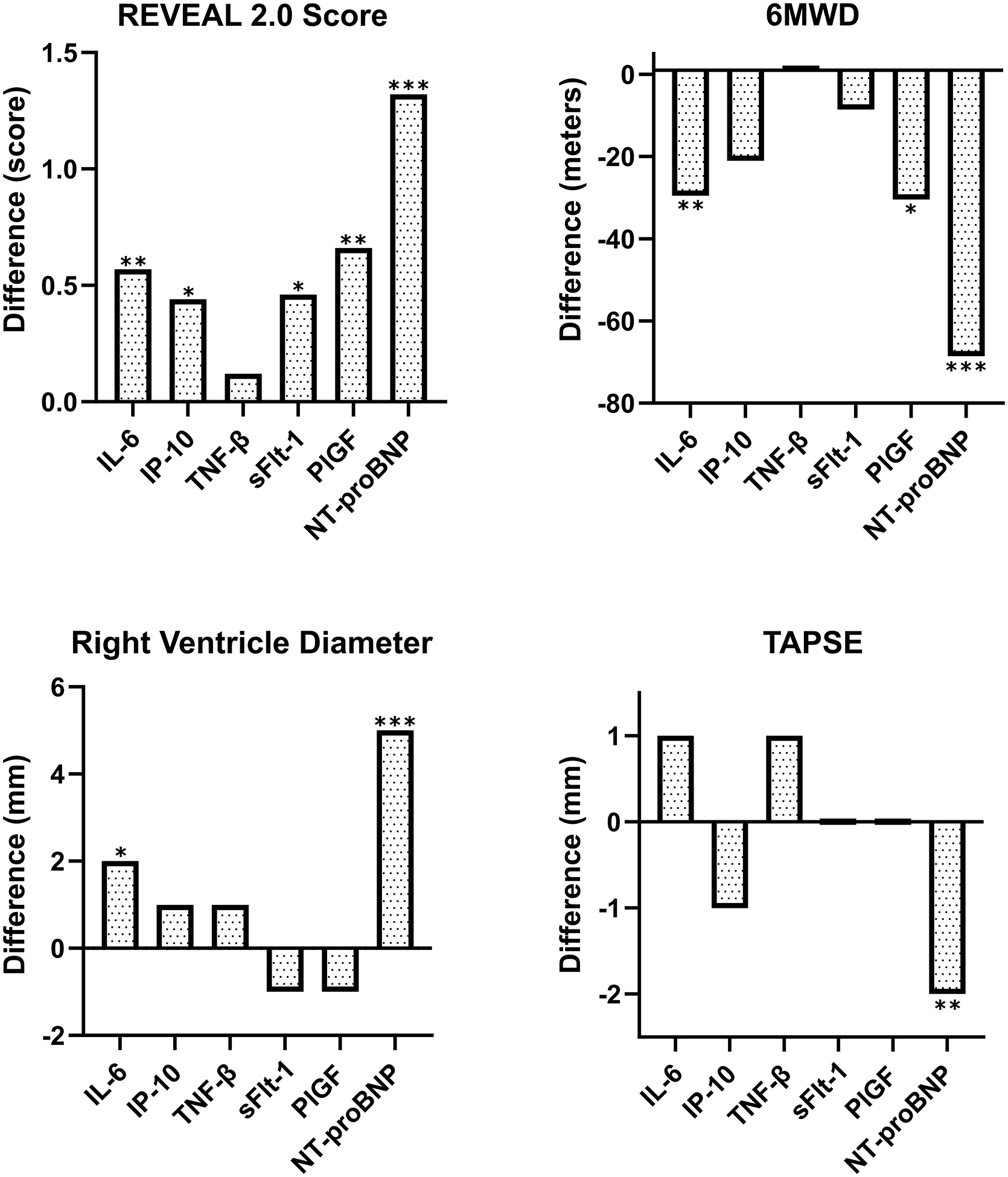
Linear regression models were run on biomarker associations with REVEAL 2.0 score, right ventricle (RV) diameter, tricuspid annular plane systolic excursion (TAPSE), and 6-minute walk distance (6MWD). Models were adjusted for age, sex, etiology, and height. Graphs display the difference in REVEAL score, RV diameter, TAPSE, or 6MWD associated with a standard deviation change in biomarker concentration (* p≤0.05; ** p≤0.01; *** p≤0.001).
Discussion:
In this single-center prospective cohort, we found that circulating levels of angiogenic and inflammatory cytokines were elevated in CTD-PAH, compared with three other subtypes of PAH. Significant heterogeneity in cytokine expression was observed at the level of both patient and subtype. Several biomarkers were associated with clinical outcomes, including mortality, and were validated using a second cohort. Interestingly, we identified distinct biomarker associations by subtype, such as those seen with sFLT-1 in CTD-PAH. Although large-scale proteomic analyses have made important recent contributions to understanding PAH, granular distinctions about protein levels and protein-outcome associations by PAH subtype are not yet well reported. This awareness has important implications for understanding heterogeneity in PAH.
Inflammation and angiogenesis are both involved in PAH pathogenesis, and prior studies have identified elevated circulating markers of inflammation and angiogenesis in PAH compared with control participants.25,26,32,33 Our research extends these studies and identifies differential expression of inflammatory and angiogenic cytokines among individual subtypes of PAH. While there was significant heterogeneity among patients, even within PAH subtypes, we identified relatively consistent elevation of cytokines in the CTD-PAH group compared to the other groups. Indeed, all 13 biomarkers that were statistically different across subtypes were elevated in CTD-PAH. Prior studies have reported on increased concentrations of individual cytokines in CTD-PAH, including IL-6,23,34 but our work is among the first to provide in-depth comparison of multiple biomarker levels across several subtypes of PAH. Mildly elevated markers of endothelial activation or vascular inflammation may be seen in patients with CTD at baseline and be associated with risk for PAH.35,36 Although speculative, elevation of these markers before overt disease develops may reinforce a mechanistic explanation for our observations.
Relating biomarker levels to clinical outcomes, we identified six markers that associated with higher three-year mortality: NT-proBNP, IL-6, IP-10, TNF-β, sFlt-1, and PlGF. In complementary and reassuring analyses, five of these six markers were also strongly associated with REVEAL score. While IL-625,32,34 and sFlt-123 have previously been associated with mortality in PAH, to our knowledge associations of TNF-β, IP-10, and PlGF with mortality have not been described before. In our single-institution cohort with limited power, only IL-6 and NT-proBNP were significant after accounting for multiple comparisons with a more stringent FDR threshold; nevertheless, focused validation in idiopathic/heritable PAH using the UK PAH Cohort Study reinforced the initial observation that IP-10, TNF-β, PlGF, and NT-proBNP had significant associations with survival. Although speculative, a mechanistic explanation for these associations is plausible. IL-6 can cause vascular inflammation and remodeling, and lead to pulmonary vascular lesions in murine models.37 Abnormalities in vascular endothelial growth factor (VEGF) signaling are implicated in angiogenesis in PAH, and VEGF pathway members PlGF and sFLT-1 are both implicated in deranged angiogenesis in preeclampsia and atherosclerosis.38–40 In addition, IP-10 provokes both vascular inflammation and impaired angiogenesis,24 and endothelial-derived TNF-β promotes vascular inflammation.41
Highlighting the potential for subtype-specific relationships, we found that sFLT-1 was associated with mortality in the full Servetus cohort; however, this relationship was predominantly seen in individuals with CTD-PAH, and no association was observed in those with non-CTD-PAH (Figure 3). While sFLT-1 was not “validated” in the UK cohort, this may be enitrely consistent with our findings given the lack of individuals with CTD-PAH in the UK Cohort.
IL-6 was also not validated in the UK cohort, which is curious given that association of IL-6 with PAH outcomes is widely reported.25,32,34,37 Importantly, a prior publication found poor correlation between SomaScan and two other methods of IL-6 detection.42 As aptamer-based assays are more widely embraced in protein research, this discordance with IL-6 may reinforce the importance of complementary studies using both immunoassay-based approaches (such as Servetus) and aptamer-based approaches (such as the UK cohort). High-dimensional proteomic analysis is a cutting-edge tool with substantial promise to elucidate complex mechanisms in PAH.30,31,43 Using aptamer-based approaches, such studies examine thousands of proteins using an unbiased approach that can suggest unexpected associations and optimize risk prediction.30 On the other hand, the use of immunoassays targeting a focused set of biomarkers, such as in our primary analyses, has unique strengths and weaknesses relative to a hypothesis-neutral proteomic approach. In addition to the noted differences across assays, a focused approach reduces the power needed to suggest a significant result, which can be important in a rare disease like PAH where relatively small sample sizes, even in large collaborations, limit power.
In addition to the iterative identification of additional biomarkers of interest, the key finding of this study is the heterogeneity across patients and subtypes of PAH which remains underreported. Understanding this heterogeneity is important to identify more precise approaches to PAH prognostication and treatment.15–18 Despite a recognition that more precision is needed, studies have generally focused on prognostication or treatment for PAH patients in aggregate. Our results support the intuitive paradigm that there may be sufficient heterogeneity within PAH to warrant more focused investigation of subtype-specific biological pathways. Approaches targeting angiogenesis or inflammation may have disproportionate benefit in CTD-PAH – a hypothesis that finds support in reports on small numbers of patients with PAH associated with systemic lupus erythematosus or mixed connective tissue disease who improved with immunosuppression.14,44 Finally, our study identified significant heterogeneity on a patient level as well as between subtypes. Recent exciting work using machine-learning identified disease endotypes within PAH that were not explained by etiology.45
Our study has several important limitations. Most notably, we were limited by low mortality and sample size which precluded firm conclusions about relationships with mortality in tox-PAH or CHD-PAH. Multiple comparisons increased the probability of identifying a relationship by chance alone and while we are reassured by the UK validation, within Servetus alone only IL-6 and NT-proBNP were associated with mortality after correction for multiple comparisons (FDR 5%). In addition, it should be noted that in the UK cohort, PlGF’s association barely met our significance threshold with a p-value of 0.0503 that rounded to 0.05 using three significant figures to determine significance.46 It is noteworthy that an alternative explanation for our findings would be differences in severity of illness by subtype, rather than PAH subtype itself. Reassuringly, we obtained similar results following adjustment by NT-proBNP; however, the possibility of residual confounding by severity persists. Finally, we did not record information about whether patients were on immunosuppressing medications, which may bias interpretation of biomarker levels especially within the CTD-PAH cohort.
In summary, in a single-center cohort, we observed elevated inflammatory and angiogenic biomarker levels in CTD-PAH when compared to three other PAH subtypes, along with distinct associations with survival within CTD-PAH versus non-CTD-PAH subtypes. We corroborate prior work suggesting IL-6 is associated with PAH outcomes and newly identify IP-10, TNF-β, and PlGF as associated with mortality in in both a discovery and validation cohort. We cautiously suggest sFlt-1 may be uniquely important in CTD-PAH and deserve further evaluation in this context. These results should encourage further research into subtype heterogeneity in PAH, particularly as mechanism-specific therapies are tested.
Supplementary Material
Acknowledgments:
The authors would like to acknowledge the patients who contributed to this research. We also appreciate the assistance of the UK Pulmonary Arterial Hypertension Cohort Study Consortium: Aman J1, Knight J2, Hanscombe KB3, Gall H4, Ulrich A1, Bogaard HJ5, Church C6, Coghlan JG7, Condliffe R8, Corris PA9, Danesino C10, Elliott CG11, Franke A12, Howard LS1, Graf S13, Ghio S14, Gibbs JSR1, Houweling AC5, Kiely DG8, Kovacs G15, LaudesM12, Lawrie A8, MacKenzie Ross RV16, Moledina S17, Newnham M13, Olschewski A15, Olschewski H15, Peacock AJ18, Pepke-Zaba J19, Scelsi L14, Seeger W4, Shaffer CM20, Sitbon O21, Suntharalingam J16, Swietlik EM13, Toshner M13, Treacy C13, Vonk Noordegraaf A5, WaisfiszQ5, Wort SJ1, Trembath RC3. (1) National Heart and Lung Institute, Imperial College London, London, UK. (2) Data Science Institute, Lancaster University, Lancaster, UK. (3) Genetics and Molecular Medicine, King’s College London, London, UK. (4) University of Giessen and Marburg Lung Center, Giessen, Germany. (5) VU University Medical Center, Amsterdam, Netherlands. (6) Golden Jubilee National Hospital, Glasgow, UK. (7) Royal Free Hospital, London, UK. (8) Royal Hallamshire Hospital, Sheffield, UK. (9) University of Newcastle, Newcastle, UK. (10) University of Pavia, Pavia, Italy. (11) Intermountain Medical Center, Murray, UT, USA. (12) University of Kiel, Kiel, Germany. (13) Department of Medicine, University of Cambridge, Cambridge, UK. (14) Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. (15) Ludwig Boltzmann Institute for Lung Vascular Research, Graz, Austria. (16) Royal United Hospitals Bath NHS Foundation Trust, Bath, UK. (17) Great Ormond Street Hospital, London, UK. (18) Golden Jubilee 1National Hospital, Glasgow, UK. (19) Royal Papworth Hospital, Cambridge, UK. (20) Vanderbilt University School of Medicine, Nashville, TN, USA. (21) University Paris-Sud, Université Paris-Saclay, Le Kremlin-Bicêtre, Paris, France.
This publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under Award Number KL2TR002317 and KL2TR000421. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors also acknowledge support from the American Heart Association (AHA) Mentored Clinical & Population Research Program (15MCPRP25550044) to PJL, the AHA Career Development Award to SGR (20CDA35310387), the British Heart Foundation Intermediate Basic Science Research fellowship (FS/15/59/31839) and Academy of Medical Sciences Springboard fellowship (SBF004\1095) to CJR, and the British Heart Foundation (RE/18/4/34215) to MJW.
Financial Disclosure Statement:
No authors have a direct conflict of interest related to this manuscript. Dr. Rayner receives research support from the NIH, AHA, and Bayer pharmaceuticals, and previously received research support from United Therapeutics and consulting fees from Verathon. Dr. Leary receives research support from the NIH, AHA, and Bayer; salary support from the Cystic Fibrosis Therapeutic Development network; and has received consulting fees from Bayer. Dr. Zheng and Dr. Altemeier receive funding from the NIH. Dr. Morrell received funding support from the British Heart Foundation, Medical Research Council and NIH. Dr Wilkins receives payment from Acceleron, Novartis, Janssen and MorphogenIX for consultancy and data adjudication, and research funding from the British Heart Foundation, The UK National Institute of Health Research, and the NIH. CJR has received consulting fees from United Therapeutics and Janssen (Johnson & Johnson). All other authors report no disclosures or conflicts of interest.
List of non-standard abbreviations:
- 6MWD
Six-minute walk distance
- BMPR2
Bone morphogenic protein receptor type 2
- CHD-PAH
Congenital heart disease-associated PAH
- CTD-PAH
Connective tissue disease-associated PAH
- IL
Interleukin
- IP-10
Interferon gamma-induced protein 10
- IPAH
Idiopathic PAH
- mPAP
Mean pulmonary artery pressure
- NT-proBNP
N-terminal prohormone of brain natriuretic peptide
- PAH
Pulmonary arterial hypertension
- PCWP
Pulmonary capillary wedge pressure
- PlGF
Placental growth factor
- PVR
Pulmonary vascular resistance
- RVD
Right ventricular basal diameter
- sFlt-1
Soluble fms-like tyrosine kinase 1
- TAPSE
Tricuspid annular plane systolic excursion
- TNF
Tumor necrosis factor
- Tox-PAH
Toxin-associated PAH
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35(5):1079–1087. doi: 10.1183/09031936.00072709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1). doi: 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamanian RT, Hedlin H, Greuenwald P, et al. Features and Outcomes of Methamphetamine-associated Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2018;197(6):788–800. doi: 10.1164/rccm.201705-0943OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher MR, Mathai SC, Champion HC, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54(9):3043–3050. doi: 10.1002/art.22069 [DOI] [PubMed] [Google Scholar]
- 5.Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448–456. doi: 10.1378/chest.11-1460 [DOI] [PubMed] [Google Scholar]
- 6.Dimopoulos K, Wort SJ, Gatzoulis MA. Pulmonary hypertension related to congenital heart disease: a call for action. Eur Heart J. 2014;35(11):691–700. doi: 10.1093/eurheartj/eht437 [DOI] [PubMed] [Google Scholar]
- 7.Gall H, Felix JF, Schneck FK, et al. The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36(9):957–967. doi: 10.1016/j.healun.2017.02.016 [DOI] [PubMed] [Google Scholar]
- 8.Montani D, Savale L, Natali D, et al. Long-term response to calcium-channel blockers in nonidiopathic pulmonary arterial hypertension. Eur Heart J. 2010;31(15):1898–1907. doi: 10.1093/eurheartj/ehq170 [DOI] [PubMed] [Google Scholar]
- 9.Halliday SJ, Hemnes AR, Robbins IM, et al. Prognostic value of acute vasodilator response in pulmonary arterial hypertension: beyond the “classic” responders. J Heart Lung Transplant. 2015;34(3):312–318. doi: 10.1016/j.healun.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limsuwan A, Choubtum L, Wattanasirichaigoon D. 5’UTR repeat polymorphisms of the BMPR2 gene in children with pulmonary hypertension associated with congenital heart disease. Heart Lung Circ. 2013;22(3):204–210. doi: 10.1016/j.hlc.2012.09.004 [DOI] [PubMed] [Google Scholar]
- 11.Roberts KE, McElroy JJ, Wong WPK, et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J. 2004;24(3):371–374. doi: 10.1183/09031936.04.00018604 [DOI] [PubMed] [Google Scholar]
- 12.Morse J, Barst R, Horn E, Cuervo N, Deng Z, Knowles J. Pulmonary hypertension in scleroderma spectrum of disease: lack of bone morphogenetic protein receptor 2 mutations. J Rheumatol. 2002;29(11):2379–2381. http://www.ncbi.nlm.nih.gov/pubmed/12415595 [PubMed] [Google Scholar]
- 13.Rhee RL, Gabler NB, Sangani S, Praestgaard A, Merkel PA, Kawut SM. Comparison of Treatment Response in Idiopathic and Connective Tissue Disease-associated Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2015;192(9):1111–1117. doi: 10.1164/rccm.201507-1456OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez O, Sitbon O, Jaï’S X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. Chest. 2006;130(1):182–189. doi: 10.1378/chest.130.1.182 [DOI] [PubMed] [Google Scholar]
- 15.Newman JH, Rich S, Abman SH, et al. Enhancing Insights into Pulmonary Vascular Disease through a Precision Medicine Approach. A Joint NHLBI-Cardiovascular Medical Research and Education Fund Workshop Report. Am J Respir Crit Care Med. 2017;195(12):1661–1670. doi: 10.1164/rccm.201701-0150WS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dweik RA, Rounds S, Erzurum SC, et al. An official American Thoracic Society Statement: pulmonary hypertension phenotypes. Am J Respir Crit Care Med. 2014;189(3):345–355. doi: 10.1164/rccm.201311-1954ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galiè N, Humbert M, Vachiery J-L, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endor. Eur Heart J. 2016;37(1):67–119. doi: 10.1093/eurheartj/ehv317 [DOI] [PubMed] [Google Scholar]
- 18.Humbert M, Farber HW, Ghofrani HA, et al. Risk assessment in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019;53(6). doi: 10.1183/13993003.02004-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halliday SJ, Hemnes AR. Identifying “super responders” in pulmonary arterial hypertension. Pulm Circ. 2017;7(2):300–311. doi: 10.1177/2045893217697708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1). doi: 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Säleby J, Bouzina H, Lundgren J, Rådegran G. Angiogenic and inflammatory biomarkers in the differentiation of pulmonary hypertension. Scand Cardiovasc J. 2017;51(5):261–270. doi: 10.1080/14017431.2017.1359419 [DOI] [PubMed] [Google Scholar]
- 22.Al-Naamani N, Palevsky HI, Lederer DJ, et al. Prognostic Significance of Biomarkers in Pulmonary Arterial Hypertension. Ann Am Thorac Soc. 2016;13(1):25–30. doi: 10.1513/AnnalsATS.201508-543OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malhotra R, Paskin-Flerlage S, Zamanian RT, et al. Circulating angiogenic modulatory factors predict survival and functional class in pulmonary arterial hypertension. Pulm Circ. 2013;3(2):369–380. doi: 10.4103/2045-8932.110445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heresi GA, Aytekin M, Newman J, Dweik RA. CXC-chemokine ligand 10 in idiopathic pulmonary arterial hypertension: marker of improved survival. Lung. 2010;188(3):191–197. doi: 10.1007/s00408-010-9232-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122(9):920–927. doi: 10.1161/CIRCULATIONAHA.109.933762 [DOI] [PubMed] [Google Scholar]
- 26.Tiede SL, Gall H, Dörr O, et al. New potential diagnostic biomarkers for pulmonary hypertension. Eur Respir J. 2015;46(5):1390–1396. doi: 10.1183/13993003.00187-2015 [DOI] [PubMed] [Google Scholar]
- 27.Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42–50. doi: 10.1016/j.jacc.2013.10.032 [DOI] [PubMed] [Google Scholar]
- 28.Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest. 2019;156(2):323–337. doi: 10.1016/j.chest.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 29.Bild DE, Bluemke DA, Burke GL, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156(9):871–881. doi: 10.1093/aje/kwf113 [DOI] [PubMed] [Google Scholar]
- 30.Rhodes CJ, Wharton J, Swietlik EM, et al. Using the Plasma Proteome for Risk Stratifying Patients with Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2022;205(9):1102–1111. doi: 10.1164/rccm.202105-1118OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harbaum L, Rhodes CJ, Wharton J, et al. Mining the Plasma Proteome for Insights into the Molecular Pathology of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2022;205(12):1449–1460. doi: 10.1164/rccm.202109-2106OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Humbert M, Monti G, Brenot F, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151(5):1628–1631. doi: 10.1164/ajrccm.151.5.7735624 [DOI] [PubMed] [Google Scholar]
- 33.Hemnes A, Rothman AMK, Swift AJ, Zisman LS. Role of biomarkers in evaluation, treatment and clinical studies of pulmonary arterial hypertension. Pulm Circ. 2020;10(4):2045894020957234. doi: 10.1177/2045894020957234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simpson CE, Chen JY, Damico RL, et al. Cellular sources of interleukin-6 and associations with clinical phenotypes and outcomes in pulmonary arterial hypertension. Eur Respir J. 2020;55(4). doi: 10.1183/13993003.01761-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wienke J, Mertens JS, Garcia S, et al. Biomarker profiles of endothelial activation and dysfunction in rare systemic autoimmune diseases: implications for cardiovascular risk. Rheumatology (Oxford). 2021;60(2):785–801. doi: 10.1093/rheumatology/keaa270 [DOI] [PubMed] [Google Scholar]
- 36.Kolstad KD, Khatri A, Donato M, et al. Cytokine signatures differentiate systemic sclerosis patients at high versus low risk for pulmonary arterial hypertension. Arthritis Res Ther. 2022;24(1):39. doi: 10.1186/s13075-022-02734-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toshner M, Rothman AMK. IL-6 in pulmonary hypertension: Why novel is not always best. Eur Respir J. 2020;55(4). doi: 10.1183/13993003.00314-2020 [DOI] [PubMed] [Google Scholar]
- 38.Khurana R, Moons L, Shafi S, et al. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation. 2005;111(21):2828–2836. doi: 10.1161/CIRCULATIONAHA.104.495887 [DOI] [PubMed] [Google Scholar]
- 39.Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350(7):672–683. doi: 10.1056/NEJMoa031884 [DOI] [PubMed] [Google Scholar]
- 40.Luttun A, Tjwa M, Moons L, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8(8):831–840. doi: 10.1038/nm731 [DOI] [PubMed] [Google Scholar]
- 41.Chen P-Y, Qin L, Li G, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab. 2019;1(9):912–926. doi: 10.1038/s42255-019-0102-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim SY, Lee JH, Welsh SJ, et al. Evaluation of two high-throughput proteomic technologies for plasma biomarker discovery in immunotherapy-treated melanoma patients. Biomark Res. 2017;5(1):32. doi: 10.1186/s40364-017-0112-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhodes CJ, Wharton J, Ghataorhe P, et al. Plasma proteome analysis in patients with pulmonary arterial hypertension: an observational cohort study. Lancet Respir Med. 2017;5(9):717–726. doi: 10.1016/S2213-2600(17)30161-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jais X, Launay D, Yaici A, et al. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum. 2008;58(2):521–531. doi: 10.1002/art.23303 [DOI] [PubMed] [Google Scholar]
- 45.Sweatt AJ, Hedlin HK, Balasubramanian V, et al. Discovery of Distinct Immune Phenotypes Using Machine Learning in Pulmonary Arterial Hypertension. Circ Res. 2019;124(6):904–919. doi: 10.1161/CIRCRESAHA.118.313911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aguinis H, Vassar M, Wayant C. On reporting and interpreting statistical significance and p values in medical research. BMJ evidence-based Med. 2021;26(2):39–42. doi: 10.1136/bmjebm-2019-111264 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.