

Abstract
TRAF7 somatic mutations are rare and have been reported in meningiomas, intraneural perineuriomas, and mesotheliomas. Triggered by an index case of an unclassified low-grade mesenchymal tumor with TRAF7 mutation as the only genetic alteration, we searched our files and identified two additional cases with similar features. The tumors arose in two females and one male, aged 63–75 years old (median 67 years). They were infiltrative deep soft tissue masses involving the shoulder, chest wall and thigh, measuring 7.0–9.1 cm in greatest dimensions. One tumor was locally aggressive, and two were associated with lung and bone metastases. The tumors displayed alternating fibrous and myxoid stroma with mild to moderate cellularity and consisted of uniform spindle cells with open chromatin, inconspicuous nucleoli and scant cytoplasm. Significant mitotic activity or necrosis were not present. However, the metastatic tumor of one case showed an epithelioid morphology and brisk mitotic activity. Immunohistochemically, the tumors showed nonspecific and focal SMA or CD34 expression. By DNA sequencing, all three cases harbored TRAF7 missense mutations involving the C-terminal WD40 domains as the only somatic mutations, showed non-recurrent focal copy number alterations, and were negative for gene fusions by targeted RNA sequencing. On methylation profiling, the tumors clustered with the undifferentiated sarcoma and myxofibrosarcoma methylation classes and were distinct from morphologic mimics. On follow-up (5–36 months), two patients died of disease following aggressive chemotherapeutic regimens. We describe a novel TRAF7-mutated mesenchymal tumor characterized by aggressive clinical behavior despite the histologic appearance of a low-grade fibromyxoid spindle cell tumor.
Keywords: TRAF7, mesenchymal neoplasms, fibromyxoid, low grade, methylation, sarcomas
Introduction
TRAF7 is a member of the tumor necrosis factor TNF receptor-associated factor (TRAF) family of intracellular proteins family. It contains an N-terminal RING finger domain, a zinc finger domain, a coiled-coiled domain, and seven WD40 repeats at its C terminus.1,2 TRAF7 is a signaling adaptor of various inflammatory pathways.3 By interacting with MEKK3/MAP3K3 via its WD40 domains, TRAF7 activates the mitogen activated protein kinase (MAPK)-AP1 pathway.2 TRAF7 has also been shown to promote polyubiquitination of p65/RelA of the nuclear factor-κB (NF-κB) pathway, and to act as a proapoptotic E3 ligase by mediating p53 ubiquitination, thus acting as an inhibitor for these pathways.4–6
Somatic mutations in TRAF7 are rarely reported in tumors and have been identified in non-NF2 meningiomas,7,8 intraneural perineuriomas,9 adenomatoid tumors,10 and a small percentage of malignant pleural mesothelioma.11 Germline mutations of TRAF7 have been detected in patients with developmental delay and cardiofacial defects.12,13
Herein, we report three cases of mesenchymal tumors with TRAF7 mutations being the sole oncogenic driver, detailing the clinicopathologic characteristics and examining the genomic and epigenetic features of these tumors.
Materials and Methods
Study Cohort
Triggered by an index case (case 2) of a low-grade appearing fibromyxoid tumor with TRAF7 mutation being the only genetic alteration, we searched the archival files of the Department of Pathology Memorial Sloan Kettering Cancer Center for mesenchymal tumors with TRAF7 mutation being the sole somatic mutation. Clinical data, including age, sex, anatomic site and treatment information were retrieved from clinical chart review. Hematoxylin and eosin–stained slides from resection specimens were rereviewed. Histopathologic parameters, including architectural patterns (solid, fascicular, whorling, infiltrative), cytomorphology, background stroma and vasculature, degree of cytologic atypia, mitotic activity, and tumor necrosis were evaluated. The study was approved by the Institutional Review Board.
Immunohistochemistry
The relevant antibodies and the dilutions used in this study are as follows: ALK D5F3 (Cell Signaling Technology clone D5F3, 1:400), CD163 (Ventana clone SP67, undiluted), CD34 (Ventana clone QBEnd10, undiluted), CDK4 (Invitrogen clone DCS-31, 1:300), MDM2 (Millipore IF2, 1:50), SMA (Cell Marque clone 1A4, undiluted), desmin (Ventana clone DE-R-11, undiluted), S100 (Cell Marque clone 4C4.9, 1:600), GLUT1 (Ventana clone pAb, undiluted), EMA (Ventana DE-R-11, undiluted), INI1 (BD Bioscience clone 25/BAF47, 1:200), BRG1 (Santa Cruz Technology clone B-7, 1:250), MUC4 (Cell Marque clone 8G7, 1:100), and STAT6 (Cell Marque clone EP325, 1:250).
Targeted DNA Sequencing
Detailed descriptions of MSK-IMPACT, a hybridization capture-based targeted DNA NGS assay, were described previously.14 Unstained recut slides from archival formalin-fixed, paraffin-embedded (FFPE) blocks were used for sequencing following extraction of DNA.
Methylation Profiling and Unsupervised Clustering Analysis
Methylation profiling was performed on the three cases with TRAF7 mutations. Details on methylation profiling were published previously.15 Briefly, genomic DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue sections for each of the samples. Next, 250 ng of genomic DNA was subjected to bisulfite conversion and processed on the Illumina (San Diego, CA) methylation EPIC/850k platform according to manufacturer’s instructions.
We used several external, publicly available data sets to enhance the analysis. We obtained raw IDAT files for 233 samples from the Heidelberg sarcoma methylation classifier reference cohort [Gene Expression Omnibus (GEO) study accession number GSE140668],16 including the following methylation classes: angiomatoid fibrous histiocytoma (AFH), control (reactive tissue), dermatofibrosarcoma protuberans (DFSP), desmoid type fibromatosis, inflammatory myofibroblastic tumor (IMT), low-grade fibromyxoid sarcoma (LGFMS), malignant peripheral nerve sheath tumor (MPNST), myopericytoma, myxofibrosarcoma, neurofibroma, nodular fasciitis, ossifying fibromyxoid tumor (OMFT), schwannoma (SCHW), solitary fibrous tumor (SFT), and undifferentiated sarcoma, 20 meningioma (MNG) samples from the Heidelberg central nervous system (CNS) methylation classifier reference and validation cohort (GSE 109381),17 and 79 mesothelioma (MESO) samples from the Heidelberg mesothelioma study (GSE164269).18
IDAT processing and data analysis samples was performed using R version 4.1.0 and the “minfi” package version 1.38.0.19 Normalization was performed using the preprocess Illumina function and probes with a detection P value > 0.01 were filtered as were single nucleotide polymorphisms (SNP)-related probes, and probes on sex chromosomes. Methylation levels were measured using beta values for all cases.
For unsupervised clustering, dimensionality reduction was performed by the T-distributed stochastic neighborhood embedding (t-SNE) method.20 After normalizing the input data matrix (centering the mean of each column to zero), the top 10,000 most variable CpGs by variance were analyzed using the “Rtsne” package version 0.15 with the following non-default parameters: perplexity = 10, max_iter = 5000, and theta = 0.
Results
Clinical summary
The three cases included two females and one male, aged 63 to 75 years old (median 67 years). The tumors were infiltrative masses involving deep soft tissue of the shoulder, chest wall, and thigh, ranging in greatest dimension between 7.0 and 9.1 cm (median 7.8 cm). Case 1 was a tumor at the right shoulder that was locally aggressive and infiltrative, involving deep soft tissue, bone and underlying lung parenchyma. Following re-resection of locally recurrent tumor, the patient received multiple chemotherapy regimens over the period of 36 months. However, he developed disease progression (multiple lung, spine and pelvic ramus lesions increasing in size highly suggestive of metastases on imaging) and ultimately succumbed to disease. Case 2 was a superoposterior chest wall mass that extends to the trapezius muscle (Figure 1A). She underwent radical surgical resection and remained free of tumor at 9-month follow-up. Case 3 was a left posterior thigh mass (Figure 1B) that has already metastasized to bilateral lungs, pleura and mediastinum at presentation. She received radiation therapy to the dominant lobulated mass on the right lung followed by chemotherapy but died of disease 5 months following surgery. The detailed clinical course is presented in Table 1.
Figure 1. Radiologic and low power features.
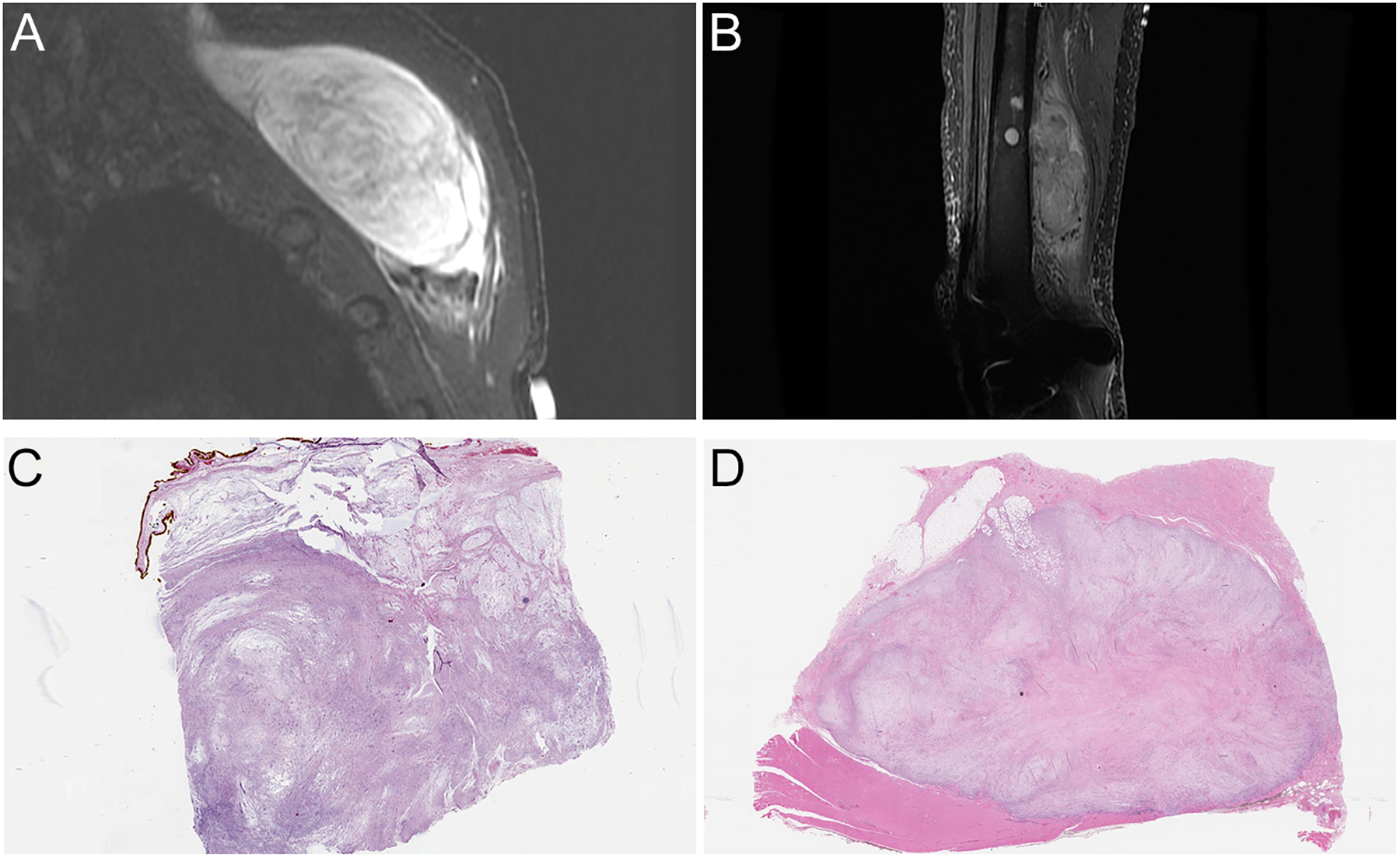
A, Magnetic resonance imaging (MRI) showing sagittal view of a left posterior chest wall mass (STIR sequence) (case 2). B, MRI showing a sagittal view of a large thigh mass involving skeletal muscle and femur (STIR sequence) (case 3). C,D, Corresponding whole slide imaging showing relative circumscribed, multinodular neoplasm with alternating fibrous and myxoid stroma (C: case 2; D: case 3).
Table 1.
Clinical Summary
| Case | Age (years) | Sex | Primary Site | Size (cm) | Presentation | Treatment | Status at last follow-up | Follow-up period (months) |
|---|---|---|---|---|---|---|---|---|
| 1 | 67 | Male | Right shoulder infiltrative mass involving soft tissue, bone and lung | 7.8 | Infiltrative shoulder mass | Marginal resection, subsequent wide resection (close margin) of local recurrence, gemcitabine/docetaxel × 3 months-->pazopanib × 2 months-->doxorubicin/olaratumab × 7 cycles-->pebrolizumab--->bevacizumab | DOD (distant lung, spine, pelvic metastases) | 36 |
| 2 | 63 | Female | Left superoposterior chest wall deep to trapezius muscle | 7.0 | Left shoulder mass with gradual increase in size over 1 year | Radical resection | ANED | 9 |
| 3 | 75 | Female | Left posterior thigh | 9.1 | Cough and fatigue for 3 months. CT scan reveals lung and mediastinal masses | Radiation to lung, doxil × 3 months, pazopanib | DOD (distant lung, pleural/mediastinal metastases) | 5 |
ANED: alive with no evidence of disease; DOD: dead of disease.
Histopathologic and immunohistochemical features
The tumors were relatively circumscribed but unencapsulated at low power, and focally infiltrating surrounding fat and skeletal muscles. All three cases were low-grade appearing spindle cell neoplasms, displaying alternating fibrous and myxoid stroma, reminiscent of LGFMS (Figures 1C–D). On higher magnification, the tumors were moderately cellular and consisted of uniform spindled cells arranged in loosely formed fascicles. Scattered thin-walled, slightly dilated vasculature and occasional admixed lymphocytes or neutrophils were present (Figure 2A–E). Case 1 also showed scattered ropy collagen strands and focal entrapment and infiltration of adipose tissue by spindled cells, similar to honeycombing of fat seen in DFSP (Figure 2C). The neoplastic cells consisted of fusiform nuclei with open chromatin, inconspicuous nucleoli, scant cytoplasm and indistinct cell borders. Mitotic activity and tumor necrosis were not evident. Interestingly, the pleural metastatic lesion of case 3 showed a more epithelioid morphology consisting of cells with slightly irregular and eccentric nuclei, inconspicuous nucleoli and moderate amount of eosinophilic cytoplasm. Frequent mitotic activity (greater than 10 per 10 high power fields) but not tumor necrosis was identified (Figure 2F).
Figure 2. Histologic morphology.
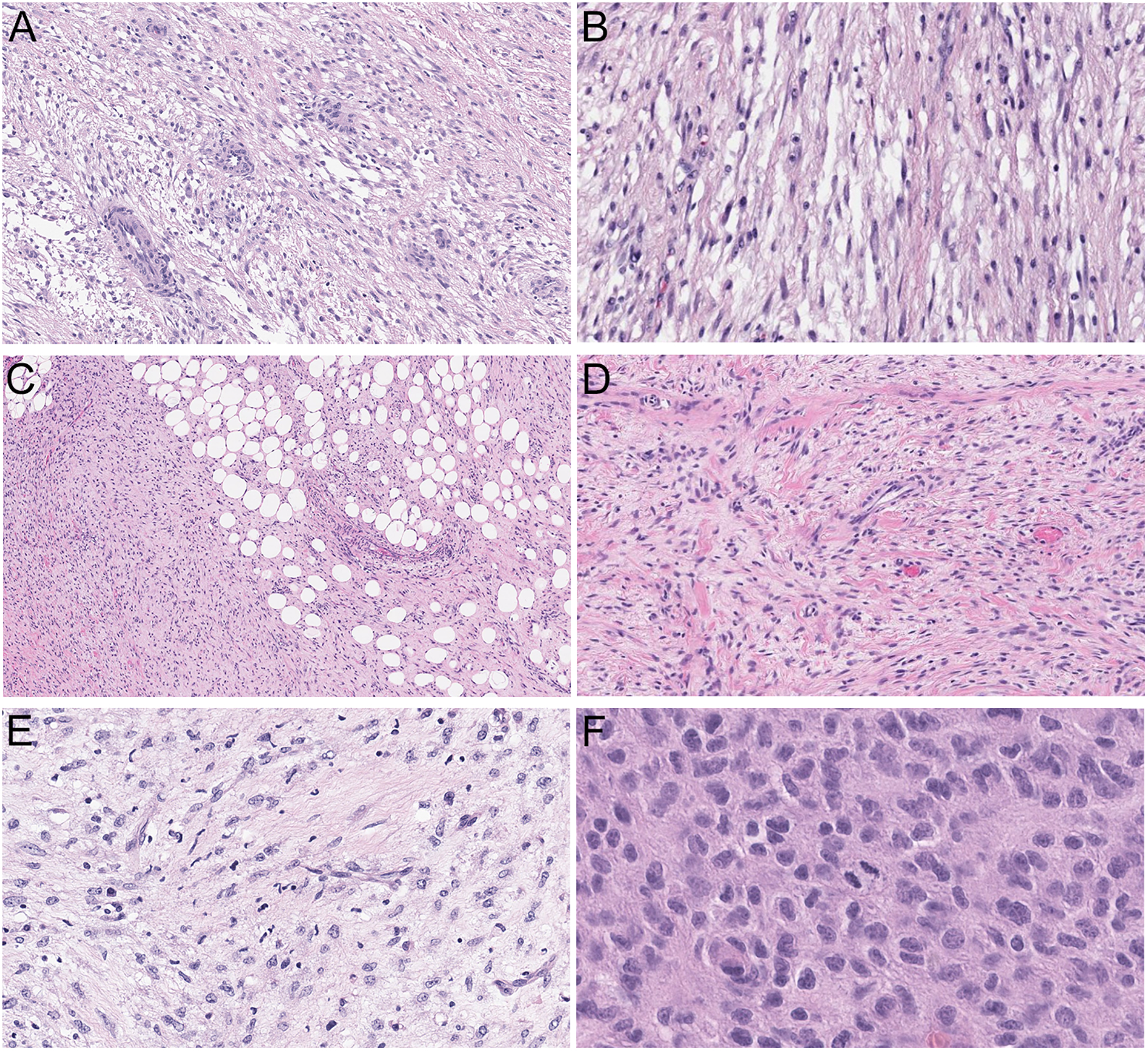
A-E, Low-grade fibromyxoid tumors consisting of fusiform cells arranged in loosely formed fascicles set in a collagenous to myxoid stroma (A-B: case 2, 100X and 200X; C-D: case 1, 100X and 200X; E: case 3). A, D, Admixed thin-walled, round to linear vasculature. B, E, Occasional lymphocytes and neutrophils admixed with tumor cells. C, Focal honeycombing of fat, reminiscent of dermatofibrosarcoma protuberans. F, The metastatic tumor of case 3 displayed a more prominent epithelioid morphology with irregular, eccentric nuclei and moderate amount of eosinophilic cytoplasm with increased mitotic activity (400X).
By immunohistochemistry, cases 1 and 2 had nonspecific focal SMA expression, while case 2 also showed CD34 and GLUT1 staining. The tumors were negative for cytokeratins, S100, desmin, MUC4, STAT6, and beta-catenin. Case 3 showed nonspecific staining for CD34, weak/focal MDM2, WT1 and PR, and was negative for cytokeratins, histiocytic markers, SMA, desmin, ALK D5F3, CDK4, and retained SMARCB1 (INI1) and SMARCA4 (BRG1) staining in the metastatic lesion (Table 2).
Table 2.
Immunohistochemical (IHC) and Molecular Findings
| Case | IHC (+) | IHC (−) | TRAF7 (NM_032271) mutation | TRAF7 mutation variant allele frequency | Copy number alterations | Sarcoma methylation classifier | Calibration score |
|---|---|---|---|---|---|---|---|
| 1 | SMA | Desmin, S100, melan-A, beta-catenin, MUC4, CD34, keratins, STAT6, beta catenin | c.1192C>T (p.P398S), c.1730A>G (p.Y577S) | 6.1%, 8.0% | CDKN2A/B loss | Undifferentiated sarcoma (USARC) | 0.79771 |
| 2 | CD34, SMA (focal), GLUT1 | SOX10, S100, EMA, pan-NTRK, ALK-1, MUC4, STAT6, desmin, beta-catenin and ER. | c.1606G>A (p.G536S) | 21.5% | 1p, 12q12–13, 16q arm level loss | Undifferentiated sarcoma (USARC) | 0.98635 |
| 3 | WT1, CD34, PR, scattered MDM2 | CK, histiocytic CD1a CD163 CD68, calretinin, melanocytic markers, PAX8, SMA, desmin, ALK D5F3, SMARCA4 retained, SMARCB1 retained, CDK4 | c.1896C>G (p.N632K) | 14.2% | 12q arm level gain, 20q13 gain, 16q12 gain | Undifferentiated sarcoma (USARC) | 0.33081 |
Molecular Findings
By DNA sequencing using MSK-IMPACT, a hybridization capture-based targeted NGS panel (Cheng 2015), out of a denominator of ~4000 mesenchymal tumors with DNA sequencing data, TRAF7 mutations were detected as the sole mutation in three cases of mesenchymal tumors, all performed in the primary tumor. All three were missense mutations as follows: p.P398S and p.Y577S in case 1, p. G536S in case 2, and p.N632K in case 3 (Table 2, Figure 3A). In all three cases, the TRAF7 mutations are located at the WD40 domains of the encoded TRAF7 protein, and were the sole mutations detected. Other genomic alterations identified included focal copy number alterations that were non-recurrent across the 3 cases: loss of CDKN2A/CDKN2B in case 1, focal arm level losses (1p, 12q12-13, 16q) in case 2, and arm level gains (12q, 16q12, 20q13) in case 3 (Table 2). All three cases were negative for gene fusions by targeted RNA sequencing using Archer technology.21
Figure 3. Mutational and methylation profiling.
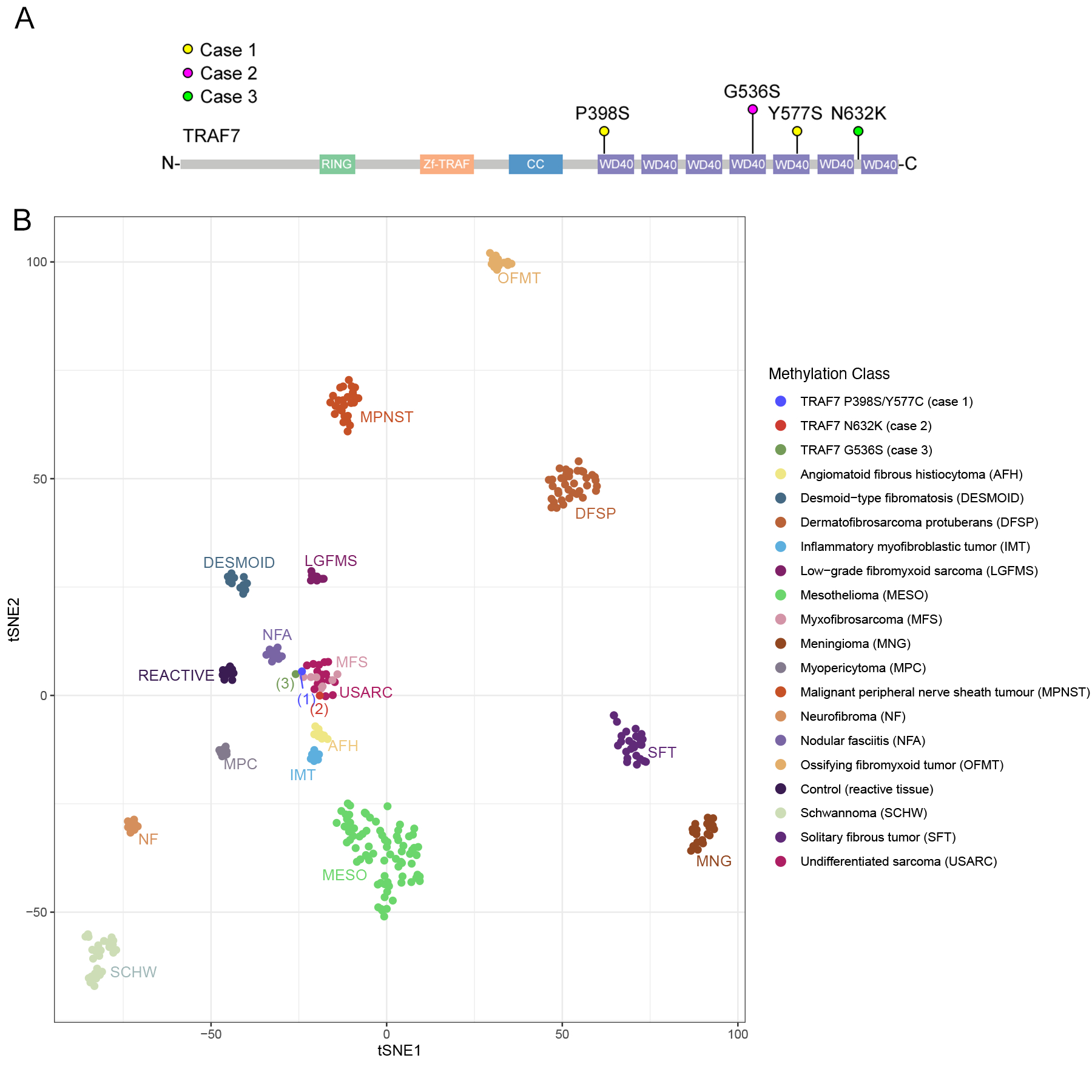
A, TRAF7 mutations schematic diagrams. Lollipop plot depicting TRAF7 mutations from the 3 cases of TRAF7-mutated sarcomas. All 4 mutations were missense mutations located at the C-terminal WD40 domains. Abbreviation: RING: RING finger domain (E3 ubiquitin ligase enzymatic domain); Zf-TRAF (TRAF-type ZINC finger domain); CC: coiled-coiled domain; WD40: WD40 repeats. B, T-distributed stochastic neighborhood embedding (t-SNE) plots showing unsupervised clustering of the 3 TRAF7-mutated tumors along with methylation classes of various mesenchymal tumors, meningiomas and mesotheliomas.16−18
Next, to shed light on the possible histogenesis of TRAF7-mutated tumors, we performed DNA methylation profiling using the Illumina EPIC 850k array. By unsupervised clustering using t-SNE, we then compared the methylation profiles of the TRAF7-mutated tumors with those of various mesenchymal tumors, meningiomas, and mesotheliomas.16–18 The three TRAF7-mutated cases clustered together with the undifferentiated sarcoma and myxofibrosarcoma methylation classes, but not other tumors including DFSP, desmoid-type fibromatosis, IMT, LGFMS, MPNST, neurofibroma, OFMT, SFT, mesothelioma, or meningioma (Figure 3B). This was corroborated by the findings applying the sarcoma methylation classifier to the TRAF7-mutated cases, with undifferentiated sarcoma being the methylation class with the highest calibration scores (0.33–0.99) in each case (Table 2). None of the TRAF7-mutated cases showed a calibration score > 0.15 to any methylation classes using the CNS tumor methylation classifier.
Discussion
Triggered by an index case of a low-grade mesenchymal tumor where a TRAF7 mutation was identified as the only genetic alteration, we searched our archival files and identified a total of three cases with similar morphologies and where TRAF7 mutations were likely the sole oncogenic driver.
The TRAF family of intracellular proteins were identified originally as signaling adaptors that are coupled to the cytoplasmic domains of TNF receptors. By acting as scaffolds and/or enzymatic proteins, TRAFs mediate TNFR signaling by regulating MAPKs and activating downstream transcription factors such as NF-κB, JNK and p38.2 The TRAF proteins have since been discovered to mediate signaling of diverse pathways, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), T cell receptors (TCR), interleukin 1 receptors, interferon receptors, and transforming growth factor-β (TGFβ) receptors.1 Aside from their physiologic roles in regulating immune responses and embryonic development, the role of the TRAF proteins in oncogenesis as a tumor suppressor protein has been increasingly recognized.2
Like other TRAF proteins, TRAF7 possesses the conserved RING finger domain followed by a zinc finger domain at the N terminal. The RING finger domain constitutes the core of the ubiquitin ligase catalytic domain. However, unlike the other TRAF members (TRAF1-6), instead of an extensive TRAF domain at the C terminal (which mediates interactions with partner proteins), the C terminal of TRAF7 is replaced by seven repeats of the WD40 domain.2 TRAF7 was shown to potentiate MEKK3-mediated AP1 and CHOP activation and induce apoptosis.22 By acting as a E3 ligase and promoting ubiquitin-proteasome mediated degradation of p53, TRAF7 has also been shown to promote tumorigenesis in breast cancer and hepatocellular carcinoma.5,6
Somatic mutations in TRAF7 are rarely reported in tumors and have been identified in meningiomas, intraneural perineuriomas, adenomatoid tumors of the male and female genital tracts, and a very small percentage of malignant pleural mesothelioma.7–11 In a whole exome sequencing study of 300 non-NF2 meningiomas, TRAF7 mutations were detected in nearly 25% of all cases, co-occurring with KLF4 or AKT1 mutations.7 In another study, 30 secretory meningiomas were subjected to whole exome sequencing or Sanger sequencing, and co-occurring TRAF7 and KLF4 mutations were detected in 29/30 cases.8 The authors also detected TRAF7 mutations in 7/89 non-secretory meningiomas. The TRAF7 and KLF4 mutations were mutually exclusive with NF2 mutations. In all cases, the TRAF7 mutations were restricted to the WD40 domains. In a study of intraneural perineurioma, 10 of 16 cases shared 3 TRAF7 mutations: p.L519P and p.H521R within exon 17, and p.S561R within exon 18, all within its WD40 domain,9 similar to the three cases in our current study. On the other hand, a study of 24 soft tissue perineuriomas by whole exome sequencing and OncoScan did not identify any TRAF7 mutations,23 suggesting that TRAF7 mutations may be exclusive to intraneural perineuriomas but not soft tissue perineuriomas. Further, in a comprehensive genomic study of 216 malignant pleural mesothelioma, TRAF7 mutations, primarily in the WD40 domain, were detected in 5/216 (2%) of cases and mutually exclusive with NF2 alterations.11 Similarly, in a cohort of 31 adenomatoid tumors of the male and female genital tracts, all tumors harbored somatic TRAF7 missense mutations within the recurrent WD40 domain hotspots.10 On the other hand, de novo germline missense mutations of TRAF7 in the WD40 repeats and coiled-coiled domains have been detected in patients with developmental delay, congenital anomalies and dysmorphic features.12,13 The phenotypic consequences of these TRAF7 missense mutations are not clear cut. None of these studies looked at whether the presence of mutation affected the expression of TRAF7 itself. Some authors have speculated that missense variants are more consistent with a gain-of-function or dominant negative effect.13 In vitro experiments in cells overexpressing TRAF7 missense variants led to reduced phosphorylation of ERK1/2,12 which is downstream of MEKK3, and activation of the NF-κB pathway.10 These findings are consistent with the knowledge that TRAF7 activates the JNK-AP1 pathway by binding to MEKK3/MAP3K3 via its WD40 domains,2 but negatively regulates the NF-κB pathway by promoting Lys-29-linked polyubiquitination of RelA/p65.4
In terms of morphology, the differential diagnosis of the TRAF7-mutated fibromyxoid tumors is broad and includes a wide range of benign and low-grade spindle cell mesenchymal tumors. LGFMS characteristically displays alternative fibrous and myxoid stroma at low magnification, and contains uniform spindled cells arranged in loosely formed fascicles admixed with delicate arcade-like thin-walled vasculature. Most LGFMS, however, express MUC4 by immunohistochemistry and harbor FUS::CREB3L1/2 (or EWSR1::CREB3L1/2 in the hybrid LGFMS-sclerosing epithelioid fibrosarcoma variant) fusions.24–26 A subset of MUC4-negative LGFMS harbors YAP1::KMT2A.27,28 Desmoid-type fibromatosis is a moderately cellular spindled tumor consisting of uniform myofibroblastic type cells arranged in long sweeping fascicles and admixed with linear, thin-walled blood vessels. The neoplastic cells express nuclear beta-catenin by immunohistochemistry.29 On DNA sequencing, desmoid-type fibromatosis harbors mutations in beta-catenin and the adenomatous polyposis coli gene APC.30 Conventional DFSP consists of uniform elongated spindle cells arranged in storiform pattern and classically shows honeycombing of fat, which was observed in one of the cases in this study. However, DFSP (apart from the fibrosarcomatous variant) is usually diffusely and strongly positive for CD34 and harbors PDGFB or PDGFD rearrangements.31–33 Myxoid SFT is a rare morphological variant of SFT and consists of bland spindle cells arranged haphazardly in a myxoid, richly vascularized stroma.34 Like all other morphologic variants of SFTs, myxoid SFT consistently expresses NAB2::STAT6 fusions.35 Perineurioma also contains long spindle cells arranged in concentric whorls, and GLUT1 and EMA is positive in the elongated cytoplasmic processes of the cells.36 Consistent, recurrent genetic alterations have thus far not been demonstrated in soft tissue perineuriomas. However, VGLL3 rearrangements have been shown in hybrid schwannoma-perineuriomas.37 In a recent report of RREB1::MRTFB fusion positive extra-glossal mesenchymal neoplasms, at least one case showed a perineurioma-like morphology with spindled cells arranged in whorls and storiform pattern.38 Other benign peripheral nerve sheath tumors, such as neurofibromas may enter the differential diagnosis, but would be express neuroectodermal markers such as S100 and SOX10. Finally, the emerging family of kinase-fusion positive mesenchymal neoplasms involving NTRK, ROS1, RET, BRAF, and RAF1 may show features seen in the cases in this study. These tumors display a wide histopathologic spectrum. At the low- to immediate-grade end of the spectrum, these soft tissue tumors may share a lipofibromatosis-like neural tumor pattern, an infantile fibrosarcoma-like phenotype, or being reminiscent of low-grade MPNST.39 Many of these tumors coexpress CD34 and S100 and are negative for SOX10. However, the detection of gene fusions involving one of these kinases establishes the diagnosis.
Despite a low-grade fibromyxoid spindle cell morphology, the three TRAF7-mutated tumors showed an unusually aggressive clinical behavior, with two out of three patients succumbing to disease within 5 to 36 months. To better understand the biology of TRAF7-mutated tumors, we also performed unsupervised clustering of the methylation profiles of a wide range of mesenchymal tumors from the sarcoma methylation classifier based on morphologic mimics, as well as meningioma from the CNS tumor methylation classifier and mesotheliomas from a separate methylation profiling study.16–18 We selected tumors that could potentially enter the differential diagnosis by histomorphology: DFSP, desmoid-type fibromatosis, IMT, LGFMS, MPNST, neurofibroma, OFMT, SFT, etc. We also included meningiomas since TRAF7 mutations are common in this tumor type, and mesotheliomas since TRAF7 mutations have been reported in a small percentage of pleural mesotheliomas and two of our cases involved the chest wall/lung. The three cases of TRAF7-mutated fibromyxoid tumors clustered with the undifferentiated sarcoma and myxofibrosarcoma methylation classes but not with any of the other aforementioned methylation classes, suggesting an epigenetic signature most related to undifferentiated sarcoma. One limitation of this study is the lack of inclusion of perineurioma as one of the methylation classes in unsupervised clustering, as this tumor type has not yet been included in published methylation profiling studies.
Given the clinically aggressive behavior of the few cases reported thus far, awareness of this entity may prompt additional genomic analysis beyond fusion panels to assess for mutations. The clinical behavior of TRAF7-mutated fibromyxoid tumors appears to be out of proportion with their relatively bland morphology and lack of mitotic activity or necrosis. Because of this, we advise for caution before drawing conclusions on the benignity or clinical behavior of mesenchymal tumors that lack a specific diagnosis. Additionally, we suggest keeping this entity on one’s radar and consider sending out materials for mutational analysis when an otherwise monotonous mesenchymal tumor with a low-grade fibromyxoid morphology lack specific immunophenotype or harbor no recurrent gene rearrangements. Nonetheless, we acknowledge that a specific diagnosis of this rare entity may not be readily attainable in most practice settings without mutational analysis using a panel that includes TRAF7, given the considerable overlap in morphology with other mesenchymal tumors. Although presumably rare, the true incidence of these tumors is currently unknown. We suspect this entity may be underdiagnosed in fusion-negative, monotonous mesenchymal tumors with a low-grade fibromyxoid morphology and an undifferentiated phenotype. Additional studies would be warranted to truly assess the incidence and clinicopathologic spectrum of this entity.
In conclusion, we describe for the first time, a novel TRAF7-mutated mesenchymal tumor characterized by aggressive clinical behavior despite having a histologic appearance of a low-grade fibromyxoid spindle cell tumor.
Supported by:
P50 CA217694 (CRA), P30 CA008748 (CRA), Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA)
Reference
- 1.Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. 2013;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zotti T, Scudiero I, Vito P, Stilo R. The Emerging Role of TRAF7 in Tumor Development. J Cell Physiol. 2017;232:1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. [DOI] [PubMed] [Google Scholar]
- 4.Zotti T, Uva A, Ferravante A, et al. TRAF7 protein promotes Lys-29-linked polyubiquitination of IkappaB kinase (IKKgamma)/NF-kappaB essential modulator (NEMO) and p65/RelA protein and represses NF-kappaB activation. J Biol Chem. 2011;286:22924–22933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L, Wang L, Zhang S, et al. Downregulation of ubiquitin E3 ligase TNF receptor-associated factor 7 leads to stabilization of p53 in breast cancer. Oncol Rep. 2013;29:283–287. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, Zhang X, Dong W. TRAF7 contributes to tumor progression by promoting ubiquitin-proteasome mediated degradation of P53 in hepatocellular carcinoma. Cell Death Discov. 2021;7:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reuss DE, Piro RM, Jones DT, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013;125:351–358. [DOI] [PubMed] [Google Scholar]
- 9.Klein CJ, Wu Y, Jentoft ME, et al. Genomic analysis reveals frequent TRAF7 mutations in intraneural perineuriomas. Ann Neurol. 2017; 81:316–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goode B, Joseph NM, Stevers M, et al. Adenomatoid tumors of the male and female genital tract are defined by TRAF7 mutations that drive aberrant NF-kB pathway activation. Mod Pathol. 2018;31:660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bueno R, Stawiski EW, Goldstein LD, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48:407–416. [DOI] [PubMed] [Google Scholar]
- 12.Tokita MJ, Chen CA, Chitayat D, et al. De Novo Missense Variants in TRAF7 Cause Developmental Delay, Congenital Anomalies, and Dysmorphic Features. Am J Hum Genet. 2018;103:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castilla-Vallmanya L, Selmer KK, Dimartino C, et al. Phenotypic spectrum and transcriptomic profile associated with germline variants in TRAF7. Genet Med. 2020;22:1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benhamida JK, Hechtman JF, Nafa K, et al. Reliable Clinical MLH1 Promoter Hypermethylation Assessment Using a High-Throughput Genome-Wide Methylation Array Platform. J Mol Diagn. 2020;22:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertero L, Righi L, Collemi G, et al. DNA methylation profiling discriminates between malignant pleural mesothelioma and neoplastic or reactive histologic mimics. J Mol Diagn. 2021;23:834–846. [DOI] [PubMed] [Google Scholar]
- 19.Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Maaten L, Hinton G. Visualizing data using t-SNE. JMLR 2008; 9(86):2569–2605. [Google Scholar]
- 21.Benayed R, Offin M, Mullaney K, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019; 25:4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu LG, Li LY, Shu HB. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem. 2004;279:17278–17282. [DOI] [PubMed] [Google Scholar]
- 23.Carter JM, Wu Y, Blessing MM, et al. Recurrent Genomic Alterations in Soft Tissue Perineuriomas. Am J Surg Pathol. 2018;42:1708–1714. [DOI] [PubMed] [Google Scholar]
- 24.Panagopoulos I, Storlazzi CT, Fletcher CD, et al. The chimeric FUS/CREB3l2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chromosomes Cancer. 2004;40:218–28. [DOI] [PubMed] [Google Scholar]
- 25.Mertens F, Fletcher CD, Antonescu CR, et al. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab Invest. 2005;85:408–415. [DOI] [PubMed] [Google Scholar]
- 26.Prieto-Granada C, Zhang L, Chen HW, et al. A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer. 2015;54:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao YC, Lee JC, Zhang L, et al. Recurrent YAP1 and KMT2A Gene Rearrangements in a Subset of MUC4-negative Sclerosing Epithelioid Fibrosarcoma. Am J Surg Pathol. 2020;44:368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puls F, Agaimy A, Flucke U, et al. Recurrent Fusions Between YAP1 and KMT2A in Morphologically Distinct Neoplasms Within the Spectrum of Low-grade Fibromyxoid Sarcoma and Sclerosing Epithelioid Fibrosarcoma. Am J Surg Pathol. 2020;44:594–606. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya B, Dilworth HP, Iacobuzio-Donahue C, et al. Nuclear beta-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol. 2005;29:653–659. [DOI] [PubMed] [Google Scholar]
- 30.Alman BA, Li C, Pajerski ME, Diaz-Cano S, Wolfe HJ. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors). Am J Pathol. 1997;151:329–334. [PMC free article] [PubMed] [Google Scholar]
- 31.Simon MP, Pedeutour F, Sirvent N, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997;15:95–98. [DOI] [PubMed] [Google Scholar]
- 32.Dadone-Montaudié B, Alberti L, Duc A, , et al. Alternative PDGFD rearrangements in dermatofibrosarcomas protuberans without PDGFB fusions. Mod Pathol. 2018;31:1683–1693. [DOI] [PubMed] [Google Scholar]
- 33.Dickson BC, Hornick JL, Fletcher CDM, Demicco EG, Howarth DJ, Swanson D, Zhang L, Sung YS, Antonescu CR. Dermatofibrosarcoma protuberans with a novel COL6A3-PDGFD fusion gene and apparent predilection for breast. Genes Chromosomes Cancer. 2018;57:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Saint Aubain Somerhausen N, Rubin BP, Fletcher CD. Myxoid solitary fibrous tumor: a study of seven cases with emphasis on differential diagnosis. Mod Pathol. 1999;12:463–471. [PubMed] [Google Scholar]
- 35.Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirose T, Tani T, Shimada T, Ishizawa K, Shimada S, Sano T. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293–298. [DOI] [PubMed] [Google Scholar]
- 37.Dickson BC, Antonescu CR, Demicco EG, et al. Hybrid schwannoma-perineurioma frequently harbors VGLL3 rearrangement. Mod Pathol. 2021;34:1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agaimy A, Din NU, Dermawan JK, et al. RREB1::MRTFB Fusion Positive Extra-glossal Mesenchymal Neoplasms: A Series of Five Cases Expanding Their Anatomic Distribution And Highlighting Significant Morphological and Phenotypic Diversity. Genes Chromosomes Cancer. 2022. Jun 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antonescu CR. Emerging soft tissue tumors with kinase fusions: An overview of the recent literature with an emphasis on diagnostic criteria. Genes Chromosomes Cancer. 2020;59:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]