

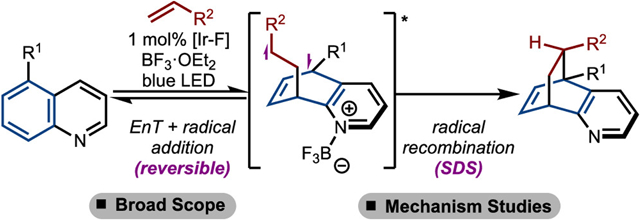
Abstract
Photochemical dearomative cycloaddition has emerged as a useful strategy to rapidly generate molecular complexity. Within this context, stereo- and regiocontrolled intermolecular para-cycloadditions are rare. Herein, a method to achieve photochemical cycloaddition of quinolines and alkenes is shown. Emphasis is placed on generating sterically congested products and reaction of highly substituted alkenes and allenes. In addition, the mechanistic details of the process are studied, which revealed a reversible radical addition and a selectivity-determining radical recombination. The regio- and stereochemical outcome of the reaction is also rationalized.
Graphical Abstract
INTRODUCTION
Cycloaddition reactions are among the most efficient and atom-economical approaches to quickly build up molecular complexity.1 Arene–alkene cycloaddition (AAC) reactions represent an ideal means of converting planar arenes into three-dimensional architectures.2 However, most thermally induced dearomative cycloadditions are difficult to realize experimentally due to high kinetic barriers and competing reverse processes. Photochemically driven processes have allowed for the development of AAC as a photon allows for the reaction to proceed via excited-state intermediates.3
Photochemical cycloadditions between arenes and unsaturated systems (typically alkenes and acetylenes) can lead to the formation of ortho, meta, and para regioisomeric products (Scheme 1A).4 Under direct irradiation, the singlet S1 excited state of the arene is generated, and the formation of meta-cycloadducts dominates via a concerted cycloaddition.5 However, if the triplet T1 excited state is generated, arenes can behave more like diradicals, allowing access to ortho and para adducts via stepwise radical cycloaddition. Computational studies by Houk established this general paradigm,6 which was further confirmed by Cornelisse and co-workers.7 As the S1 excited state of the arene can be easily accessed with simple UV-light irradiation, meta cycloadditions have been extensively studied and applied in the synthesis of various natural products.3b,8,9
Scheme 1.
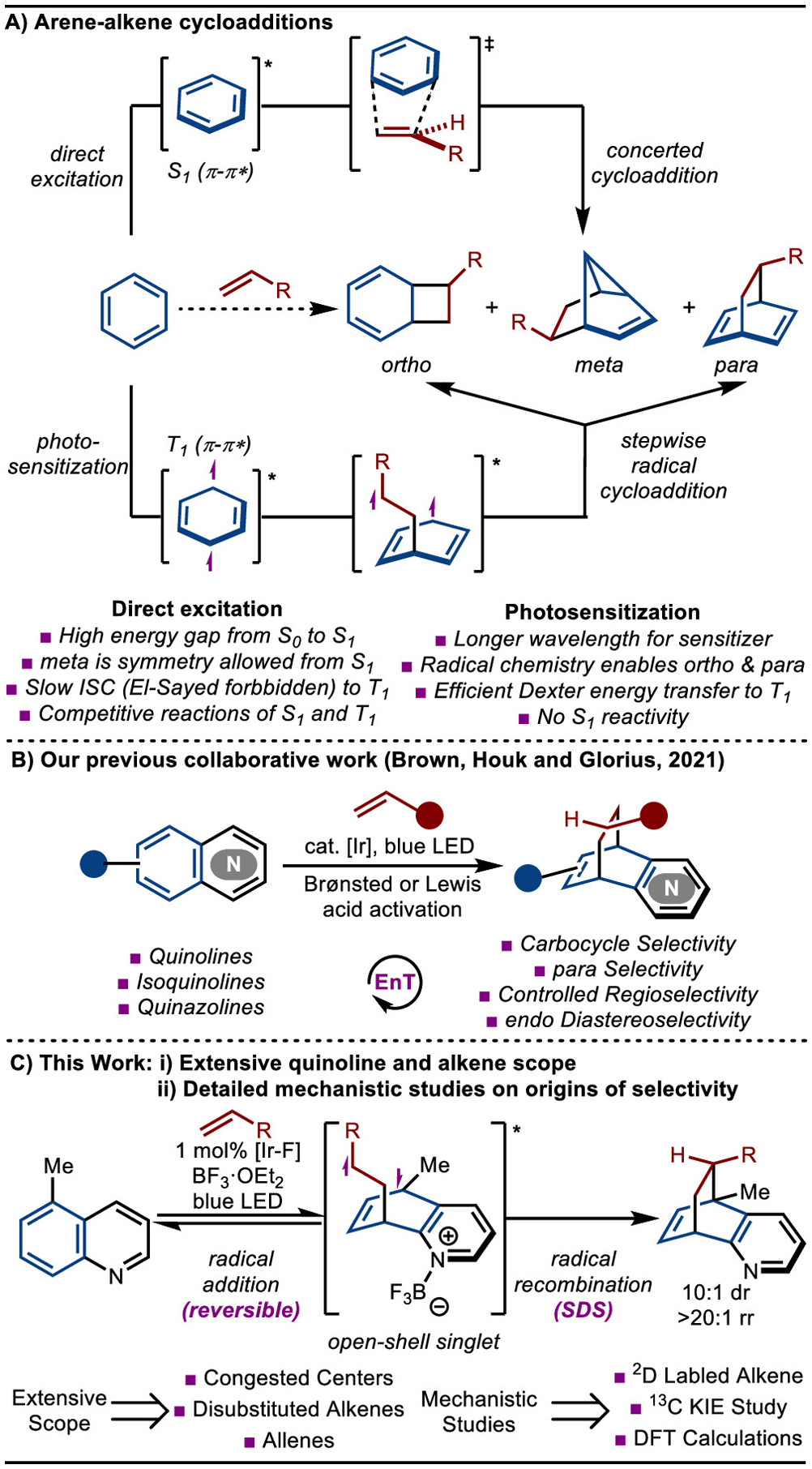
Arene–Alkene Cycloaddition
Efficient access to the T1 state of the arene through direct excitation is challenging for several reasons. First, the gap between S0 and S1 can be large, especially for S1 (π−π*).10 As a result, short-wavelength irradiation with Hg lamps is usually required. Second, the transition from S1 to T1 may be difficult because of the relatively fast relaxation of S1 to the ground state. Third, in accordance with El-Sayed’s rule, a forbidden ISC would be necessary for the conversion of S1 (π−π*) to T1 (π−π*).11 Finally, since meta cycloaddition can easily proceed via the S1 state, there will likely be a competition among multiple cycloaddition pathways even if the T1 state can be accessed. Despite these challenges, several ortho- and para-cycloadditions have been developed. For example, ortho-cycloadditions have been reported for systems in which the ISC to the T1 (π−π*) is facile. However, these systems are limited to specialized substrates with tethered alkenes.12 Recently, developments have been made with phenanthrene derivatives in an intramolecular cycloaddition.13 Only a few examples showed that electron-poor naphthalene derivatives could react with electron-deficient alkenes to yield the para cycloadducts.14
An alternative method to access the T1 state of the arene is by visible-light mediated triplet–triplet energy transfer (EnT).15 Because the T1 state is accessed directly without having to generate S1, only stepwise ortho-and para-cycloadditions are observed. Examples of EnT-induced dearomative ortho-cycloadditions include reactions of indoles16 as reported by You,17 Oderinde,18 Fu,19 and Zhang.20 In addition, early work from Meggers demonstrated the dearomatization of benzofuran and benzothiophene.21 Oderinde and co-workers later applied this strategy to electron-deficient indoles, benzofurans, benzothiophenes, and azaindoles.22 Glorius also realized the ortho cycloaddition of benzothiophene via an endergonic EnT.23 para-Cycloaddition is still rare under EnT conditions and mainly focuses on the sensitization of alkenes instead of arenes.24
Recently, our groups developed a selective intermolecular para-cycloaddition of bicyclic azaarenes with various activated and unactivated alkenes initiated by EnT (Scheme 1B).25 This reaction displays good selectivity toward the formation of one isomer and affords valuable functionalized aromatic heterocycles that have been established as privileged motifs in pharmaceutically active compounds.26 Additional studies revealed a novel photochemical cascade with certain substitution patterns.27 In this study, we greatly expand upon the substrate scope to more challenging examples such as disubstituted alkenes and allenes under Lewis acid-mediated conditions developed in the Brown lab (Scheme 1C). In addition, we demonstrate that targets containing tetrasubstituted carbons can be accessed readily using this protocol. Finally, through a combination of mechanistic experiments and density functional theory (DFT) calculations, we present a detailed analysis of the reaction mechanism and reveal the energy landscape of the transformation.
RESULTS AND DISCUSSION
Initially, we employed 2-iPr-thioxanthone (ITX, ET = 65.4 kcal/mol)28 as the photosensitizer for the reaction between quinoline (ET = 61.9 kcal/mol) and 1-hexene, which resulted in <2% conversion. A similar result was obtained with [Ir(dFCF3ppy)2dtbbpy]PF6 (Ir–F, ET = 60.1 kcal/mol). Addition of 25 mol % HNTf2 resulted in the formation of the para cycloadducts 2 and 3, albeit in low yields (Table 1, entry 3). Further studies showed that Lewis acids were more effective (Table 1, entries 4–5). We found that 125 mol % BF3·OEt2 was required for full conversion (Table 1, entry 6), likely due to product inhibition. Finally, while the higher-energy sensitizer ITX gave a similar result as [Ir(dFCF3ppy)2dtbbpy]PF6 (Table 1, entry 7), the use of lower-energy sensitizers led to diminished yields.
Table 1.
Reaction Optimization

|
Reactions were run on a 0.1 mmol scale.
Combined yields (rr and dr) were determined by crude NMR for using CH2Br2 as the internal standard.
5 mol % cat., 395 nm LED, 36 h.
While evaluation of various Lewis acids, photosensitizers, and temperatures did not alter the selectivities, regioselectivity was found to be controlled by the polarity of the solvent and the position of the quinoline substituent (Scheme 2A). For these reactions, we define 2 as the 8-to-5 product and 3 as the 5-to-8 product based on the order of bond formation (vide infra). For quinoline (1), the 8-to-5 product 2 was the major regioisomer formed in all solvents evaluated, with the highest regioselectivity observed in toluene (12:1 rr). When 6-Me-quinoline was evaluated, a switch in regioselectivity occurred, yielding the 5-to-8 product as the major isomer. In addition, a similar correlation between regioselectivity and solvent polarity was observed, with MeCN yielding the highest regioselectivity for the 5-to-8 product (>20:1 rr).29
Scheme 2.

Regioselectivitya
aReactions were run on a 0.25 mmol scale with standard conditions; yield of the isolated product is reported as combined yields of both diastereomers, average of two runs; dr and rr were determined by NMR analysis of the unpurified mixture. *NMR yield.
To understand the impact of substituent position on regioselectivity, we tested quinolines with methyl groups at different positions (Scheme 2B).30,31 For quinolines with substitution on the pyridine ring (products 2, 4–7) or substitution on the 5- and 7-positions of the phenyl ring (products 8–9), the 8-to-5 product was favored. The 5-to-8 product was preferentially formed when the substituent was in the 6- or 8-position on the phenyl ring (products 10–11). A lower yield was found for formation of 5 and 11, as coordination of the Lewis acid was likely impeded (see 12). X-ray structures of 13 and 14 were obtained to confirm the identity of the major regioisomer.
We explored the reaction scope for 5-substituted quinolines due to both the high diastereo- and regioselectivities observed and the potential for constructing products containing sterically congested carbons (Scheme 3). Substituents with a wide range of steric and electronic properties were well-tolerated under the conditions. Various quaternary carbons were achieved with different sp3 (products 8, 15–16, 19) or sp2 (products 17–18) substituted quinolines. Heteroatom substituted centers with F (product 22), Cl (product 23), O (product 20), N (product 21), and Si (product 24) were also easily prepared in high yields and selectivities. It is notable that intermolecular cycloaddition is faster than an intramolecular process (products 25–26). This is likely due to the slow reaction between C8 and the terminal carbon of the alkene via an 8- or 9-membered transition state. The alkene scope was also broad with excellent functional group compatibility with alkenes (product 27), ethers (product 30), esters (product 29), and halides (product 28). All mono-substituted alkenes, regardless if activated (products 34–40) or unactivated (products 27–33), favored formation of the endo-diastereomer (dr ~ 10:1). Exclusive endo products (dr > 20:1) were found using styrenes regardless of the different substitution patterns. Even sterically demanding alkenes allowed for formation of the endo-products 31–33. With 1,2-disubstituted alkenes, stereo-convergence to the trans product was observed from either cis or trans alkene inputs (product 42–43), with the alkene substituent closer to the 5-position still being placed endo to the pyridine ring. Even for cis-cyclic alkenes like cis-cyclohexene, cis-cycloheptene, and cis-cyclooctene, the trans products 44–46 were isolated with high diastereoselectivity. The anti-structures 41·MeI, 44·MeI, and 46·MeI were confirmed by X-ray analysis. The product of cis-cyclooctadiene 47 was formed with lower dr, presumably because bond rotation was constrained by the internal alkene. For indene and dihydronaphthalene, bond rotation was even more constrained, and only the cis-exo-isomers were isolated (49 and 50). With respect to limitations, 1,1-di, tri-, and tetrasubstituted alkenes did not allow for product formation (products 51–52). Finally, the reaction could be easily scaled up to 1.2 mmol/h using a flow chemistry system (8).
Scheme 3.

Substrate Scopea
aReactions were run on a 0.25 mmol scale; yields of isolated products were reported as combined yields of both diastereomers; dr and rr were determined by NMR analysis of the unpurified mixture; average of two runs. Condition for methylation: MeI (5 equiv), dioxane, 50 °C, 12 h; *48 h; †yields of all for diastereo- and regioisomers; ‡>20:1 dr after purification.
In addition to alkenes, allenes also underwent cycloadditions smoothly with quinoline substrates under standard conditions (Scheme 4, products 53–58).32 Regardless of the allene substitution pattern, the 8-to-5 product was exclusively formed. In the case of nonsymmetric allenes, mixtures of alkene isomers were formed (54 and 56). With 1,3-disubstituted allenes or trisubstituted allenes, the diastereoselectivities varied from endo- to exo-selective (products 56–58).
Scheme 4.

Allene Scopea
aReactions were run on a 0.25 mmol scale; values of dr, rr, and E/Z were determined by NMR analysis of the unpurified reaction mixture. *Combined yield of both diastereomers. Average of two runs.
Based on our previous report,25 the mechanism of the reaction likely involves triplet–triplet energy transfer to a Lewis acid-activated quinoline followed by stepwise radical cycloaddition. However, there are several aspects that need to be considered in more detail (Scheme 5): (1) the role of the Lewis acid; (2) aromatic ring selectivity (benzene vs pyridine); (3) ortho, meta, and para selectivity; (4) 5-to-8 vs 8-to-5 regioselectivity; and (5) diastereoselectivity. We conducted further mechanistic studies to address all of these issues.
Scheme 5.

Selectivity Challenges
We first focused on determining the first nonreversible step of the reaction as it is important for understanding the origins of the observed selectivities.33,34 To probe this, we conducted the standard reaction with the E-2D-labeled alkene 59. If the intermolecular radical addition step was selectivity-determining, we would expect to isolate 60a and/or 60b with recovered unchanged 59 (Scheme 6A). However, while 60a and 60b were isolated in a 1:1 mixture, the excess unreacted alkene 59 was recovered as a 1:1 mixture of E- and Z-isomers. One interpretation of this result is if the first intermolecular radical addition step is reversible and the second intramolecular radical recombination step is selectivity-determining. However, the isomerization of 59 could arise by an alternative unknown pathway. Therefore, 13C kinetic isotope effects (KIEs) at natural abundance were determined using the methods developed by Singleton and co-workers (Scheme 6B).35 We initially carried out KIE measurements on 5-Me-quinoline, the standard substrate in all DFT calculations, and observed a strong KIE at C5 (1.017). However, overlapping signals in the 13C NMR spectra with C8 and C10 made it difficult to confirm the KIE value at C8. Thus, 5-OAc-quinoline was investigated and a similarly strong KIE at C5 was observed (1.019).
Scheme 6.
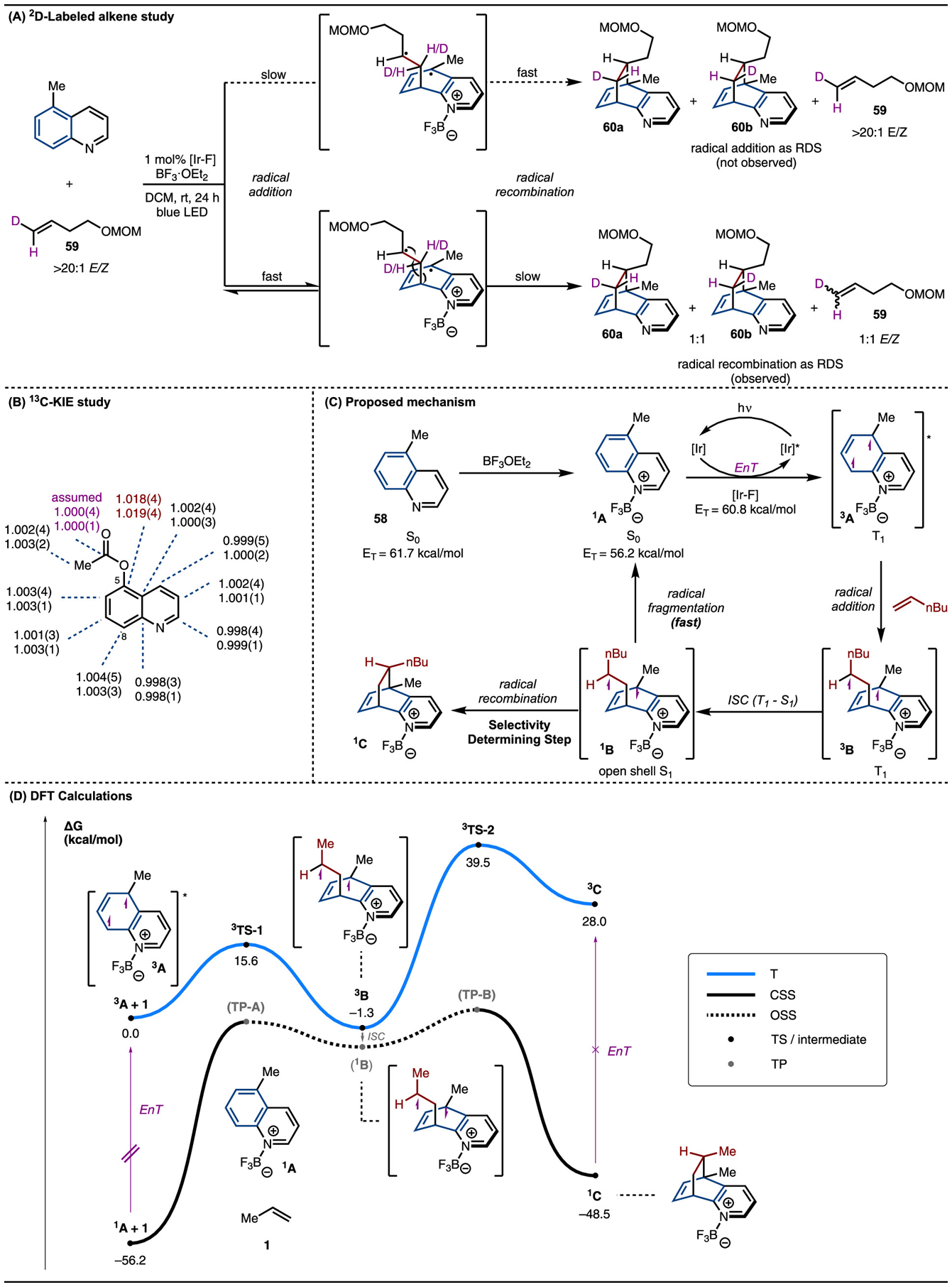
Selectivity-Determining Step
These mechanistic experiments strongly point to the first step being reversible and the intramolecular radical recombination being the selectivity-determining step. The observation that even cis-cyclic alkenes like cyclohexene preferentially form the trans product lends further support to the existence of a long-lived biradical intermediate that allows for bond rotation before recombination (see product 44, Scheme 3). A revised mechanism is therefore proposed in Scheme 6C. Photo-sensitization of the BF3 complex 1A by the excited triplet state of [Ir–F] generates 3A. Radical addition to 1-hexene then results in the formation of 3B, which upon intersystem crossing yields the singlet 1B. This singlet intermediate can then undergo either radical fragmentation to revert to starting materials 1A and 1-hexene or recombination to yield product 3C.
To further reveal the energetic landscape traversed by the reaction, which our experiments suggested was more complex than previously assumed,25,38 we performed DFT calculations (Scheme 6D). The three energy surfaces, triplet (T), open-shell singlet (OSS), and closed-shell singlet (CSS), were mapped out in detail along the reaction coordinates. At the beginning of the reaction, energy transfer from the excited photosensitizer to the ground-state quinoline–BF3 1A yields the triplet 3A. As the energy transfer process itself is not expected to impact the observed selectivities, we elected to focus our computational efforts on the subsequent transformations of 3A. Its reaction with propene (60) (used as a truncated computational model for 1-hexene) via 3TS-1 on the triplet surface forms the first C–C bond with a calculated activation free energy of 15.6 kcal/mol. While the formation of the triplet biradical intermediate 3B is slightly exergonic, our calculations showed that it would most likely undergo intersystem crossing (ISC) to the open-shell singlet 1B, which lies about 4–6 kcal/mol below 3B. The OSS and CSS surfaces intersect at two transition points (TPs) where the open-shell and closed-shell wave functions transition smoothly: TP-A, which leads back to separated reactants, and TP-B, which furnishes the cycloadduct. Instead of being determined by only a single triplet radical addition transition state (TS), the chemo- and diastereoselectivities of the reaction are therefore controlled by the rate of ISC from 3B to 1B, as well as the energies of the OSS to CSS TPs.36
Two possible roles were hypothesized for the requirement of Lewis acid complexation in this reaction: (1) facilitating energy transfer by lowering ET, and (2) accelerating the radical addition step by increasing the electrophilicity of the quinoline substrate. To probe the role of the Lewis acid in the energy transfer process,37 we performed Stern–Volmer quenching studies. It was found that quinoline and quinoline–BF3 complexes have similar quenching efficiencies (Scheme 7A). This result suggests that, while facilitating energy transfer, Lewis acid coordination is not strictly required for sensitization to happen. During the course of our study, Morofuji and Kano also showed that direct energy transfer to quinoline is feasible without the addition of an acid.38,39 In contrast, the idea that Lewis acid primarily assists in the radical addition step found experimental support. When the standard reaction was conducted with E-2D-labeled alkene 59 in the absence of BF3, we observed <2% yield of the product and no epimerization of the alkene starting material, which indicated that the radical addition barrier was significantly higher without the Lewis acid (Scheme 7B).
Scheme 7.
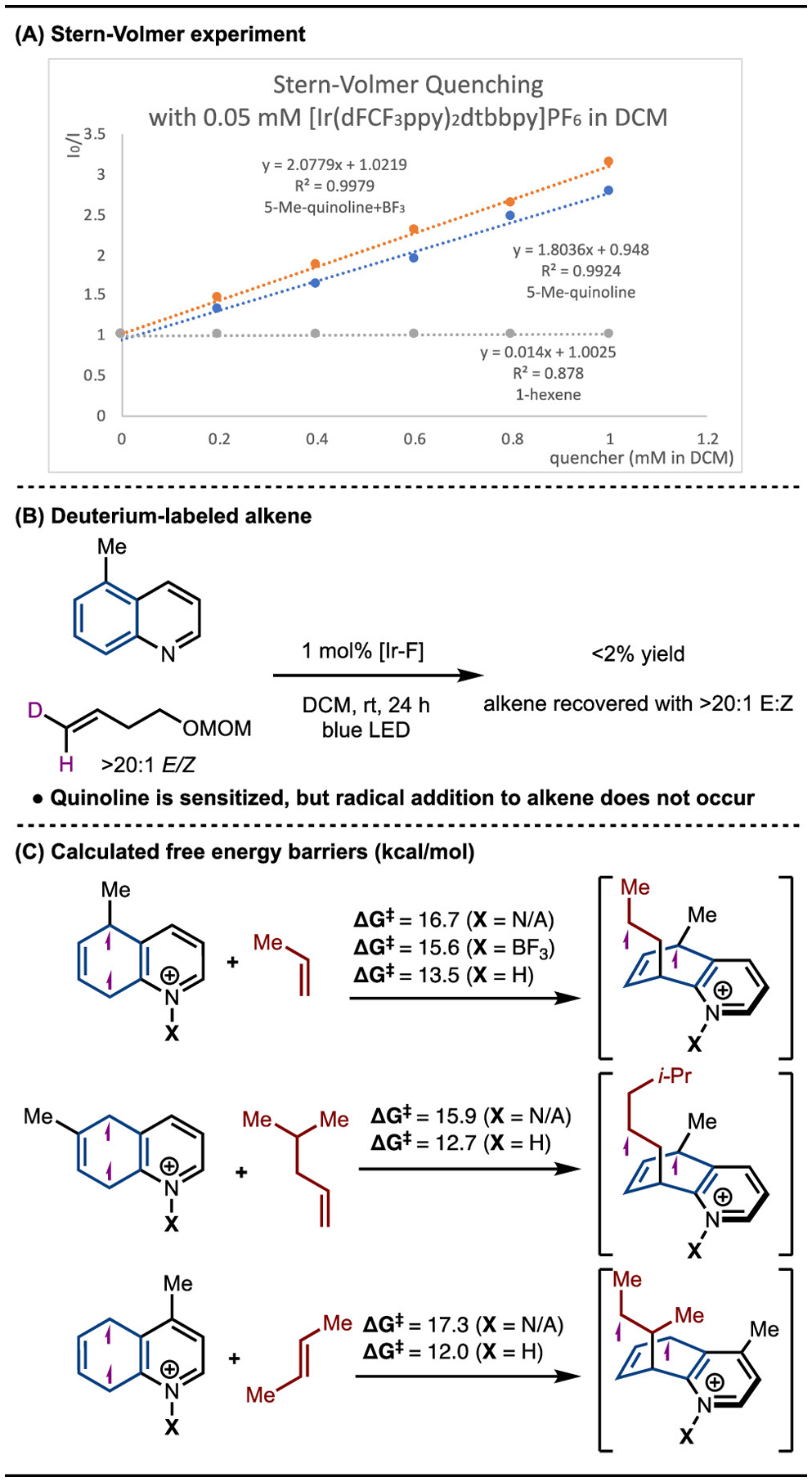
Role of Lewis Acid
To further verify that Lewis and/or Brønsted acid complexation has the effect of lowering the radical addition barriers, we calculated the radical addition TSs between various quinoline substrates and alkene partners. In all cases, the calculations confirmed that both Lewis and Brønsted acid complexation led to lower activation free energies (ΔG‡) for the radical addition step (Scheme 7C). The magnitude of this effect is dependent on the identity of the acid as well as the quinoline/alkene combination, ranging from a 1.1 kcal/mol decrease for 5-Me-quinoline/propene (with BF3 complexation) to 5.3 kcal/mol for the 3-Me-quinoline/2-butene (with H+ complexation) system investigated by Morofuji and Kano.38
Equipped with a deeper understanding of the reaction mechanism, we next sought to rationalize the observed selectivities. Our calculations of the triplet-state quinoline substrates indicate that there is a higher spin density on the benzene moiety than the pyridine moiety with or without Lewis acid complexation (Figure 1), which is likely linked to the experimentally observed regioselectivity for the benzene ring.
Figure 1.

Calculated spin densities.
We next turned our attention to rationalizing the observed para selectivity of the reaction over ortho (meta cycloadditions were not considered here as they are concerted and occur exclusively on the singlet energy surface). As our mechanistic studies revealed that the OSS to CSS TPs were likely the selectivity-determining structures in the reaction, we concentrated our efforts on locating and comparing the TPs for the para and ortho addition pathways. Because free energies cannot be calculated for these TPs as thermal contributions to energy from molecular vibrations cannot be obtained for nonstationary points on the potential energy surface,39 we present the electronic energies in Scheme 8A for the ortho- and para-additions originating from 3B. We found that for both the TP and the final cycloadduct, the calculated electronic energies of ortho were much higher (9 kcal/mol) than para. This result suggests that the formation of the para cycloadduct is likely kinetically favorable for this substrate.
Scheme 8.
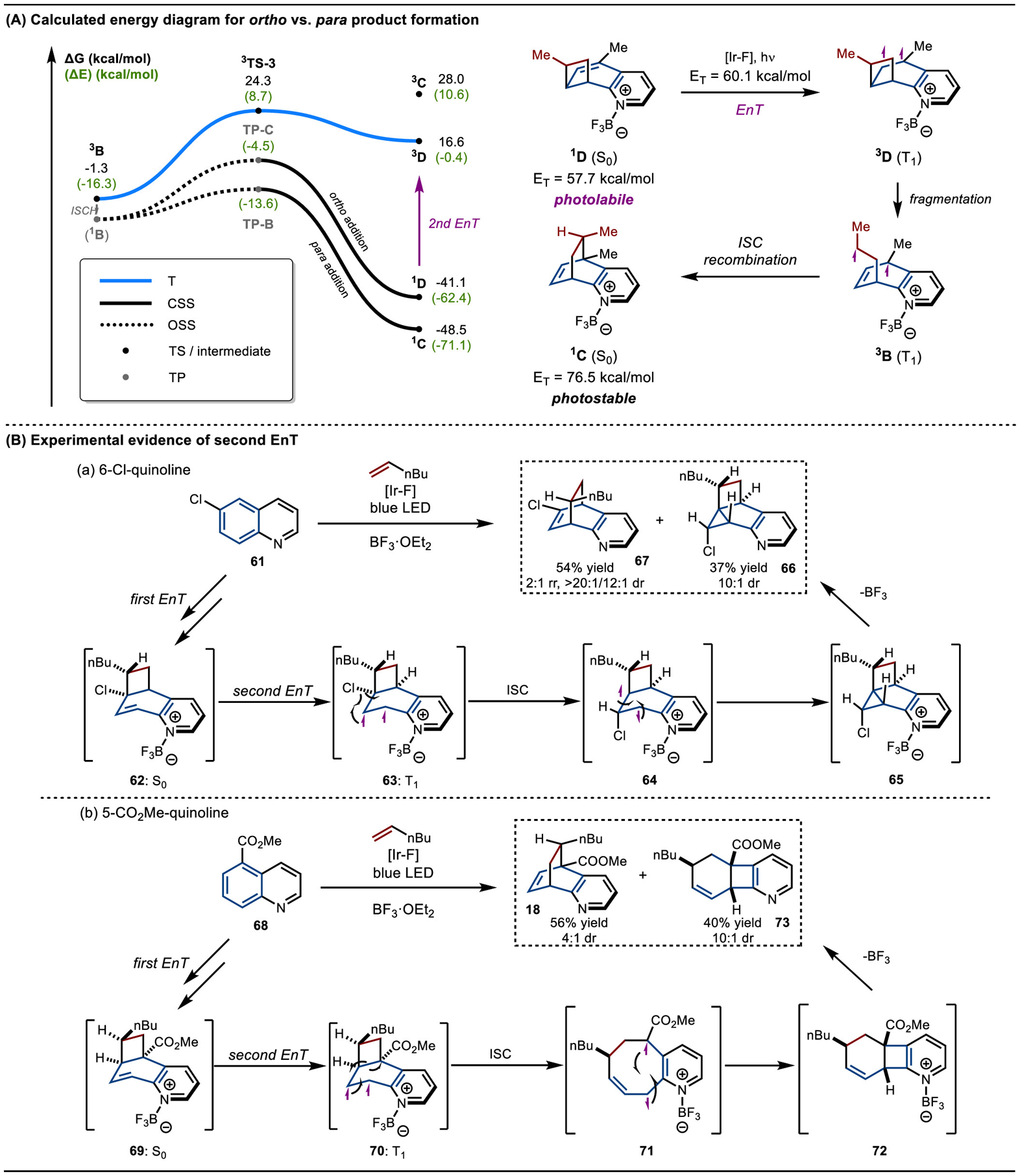
Ortho vs Para Selectivity
Furthermore, the ortho and para products also differ in their photostability under the reaction conditions. The ET of the ortho product 1D was calculated to be 57.7 kcal/mol, which is smaller than that of the photosensitizer [Ir–F] (60.1 kcal/mol). In contrast, the para product 1C had a calculated ET of 76.5 kcal/mol, meaning that it is likely photostable under the reaction conditions. We therefore reasoned that the ortho product can be sensitized and undergo radical fragmentation back to the starting materials, which funnels the reaction toward exclusive para product formation.
Although we were not able to isolate an ortho product and confirm the photosensitization-induced fragmentation, we observed two fused-ring products, which may suggest that an ortho cycloaddition does occur under the reaction conditions for certain substitution patterns (Scheme 8B).40 Cyclopropyl chloride 66 was isolated together with the para cycloadduct 67. The formation of 66 can be rationalized through a second energy transfer to the ortho cycloadduct 62, followed by C–Cl bond homolysis.41 The resulting diradical 64 undergoes recombination to deliver 65. In addition, in the case of 68, a second energy transfer can occur to fragment the central C–C bond. Subsequent recombination can then yield a new cyclobutane product 73. Overall, while formation of the para adducts is likely kinetically favored, for some substitution patterns, a photolabile ortho-adduct may be generated.
The 5-to-8 vs 8-to-5 regioselectivity of the reaction is also worthy of discussion. Having established through our mechanistic studies that the radical addition step is not selectivity-determining, we reasoned that the relative stability of the long-lived biradical intermediates could be key to explaining the regioselectivity. In the solvent-controlled regime (Scheme 2A), the biradical intermediate leading to the 5-to-8 product is expected to have a larger net dipole moment due to the alignment of partial dipoles (Scheme 9A). As a result, the 5-to-8 biradical intermediate would be better stabilized by more polar solvents. Our calculated dipole moment values (pcalc) for the biradical intermediates confirmed that the 5-to-8 biradical intermediate is more polar at 8.2 Debye, compared to 7.0 Debye for the 8-to-5 biradical intermediate. This is in good agreement with our experimental observations that higher-polarity solvents favor the 5-to-8 product.
Scheme 9.

5-to-8 vs 8-to-5 Regioselectivity
In the substitution-controlled regime (Scheme 2B), the stability of the biradical intermediates is primarily dictated by the degree of hyperconjugation. In these cases, the formation of the more stable biradical may render it less likely to undergo fragmentation back to starting materials. With an alkyl substituent in the 5- or 7-position, the biradical intermediate leading to the 8-to-5 product is better stabilized by hyper-conjugative donation than its 5-to-8 counterpart (Scheme 9B). This explains the preferential formation of the 8-to-5 product for the 5- and 7-substituted substrates. For quinoline substrates with substitution in the 6- or 8-position, the 5-to-8 biradical intermediate would be better stabilized by hyperconjugation, leading to the opposite regioselectivity.
Finally, we set out to rationalize the endo diastereoselectivity of the reaction, for which we proposed that London dispersion forces could be the key factor (Scheme 10).42 To verify that dispersive interactions favor the formation of the endo product diastereomer, we compared TP energies calculated at the B3LYP/def2-TZVPP, SMD(CH2Cl2) (not dispersion-corrected), and B3LYP-D3/def2-TZVPP, SMD(CH2Cl2) (dispersion-corrected) levels (Scheme 10B). Our results confirmed that the electronic energies of the TPs leading to the endo diastereomers are indeed lowered more compared to the exo diastereomers when a dispersion-corrected functional was applied. In the reaction between 5-Me-quinoline and propene, dispersion correction favors the endo-product-forming TP by 0.7 kcal/mol relative to its exo counterpart. When the alkene substituent is a larger tert-butyl group instead of the methyl group of propene, this effect is even more pronounced, with dispersion adding 1.8 kcal/mol favorability to the endo-product-forming TP. Noncovalent interaction (NCI) plots for the MECPs also showed that at these C–C-forming bond lengths, the alkene substituent is within the right distance range to have favorable dispersive interactions with the pyridine moiety of the quinoline when in the endo position. These computational results showed that dispersion indeed has the effect of steering diastereoselectivity toward the endo product isomer.
Scheme 10.
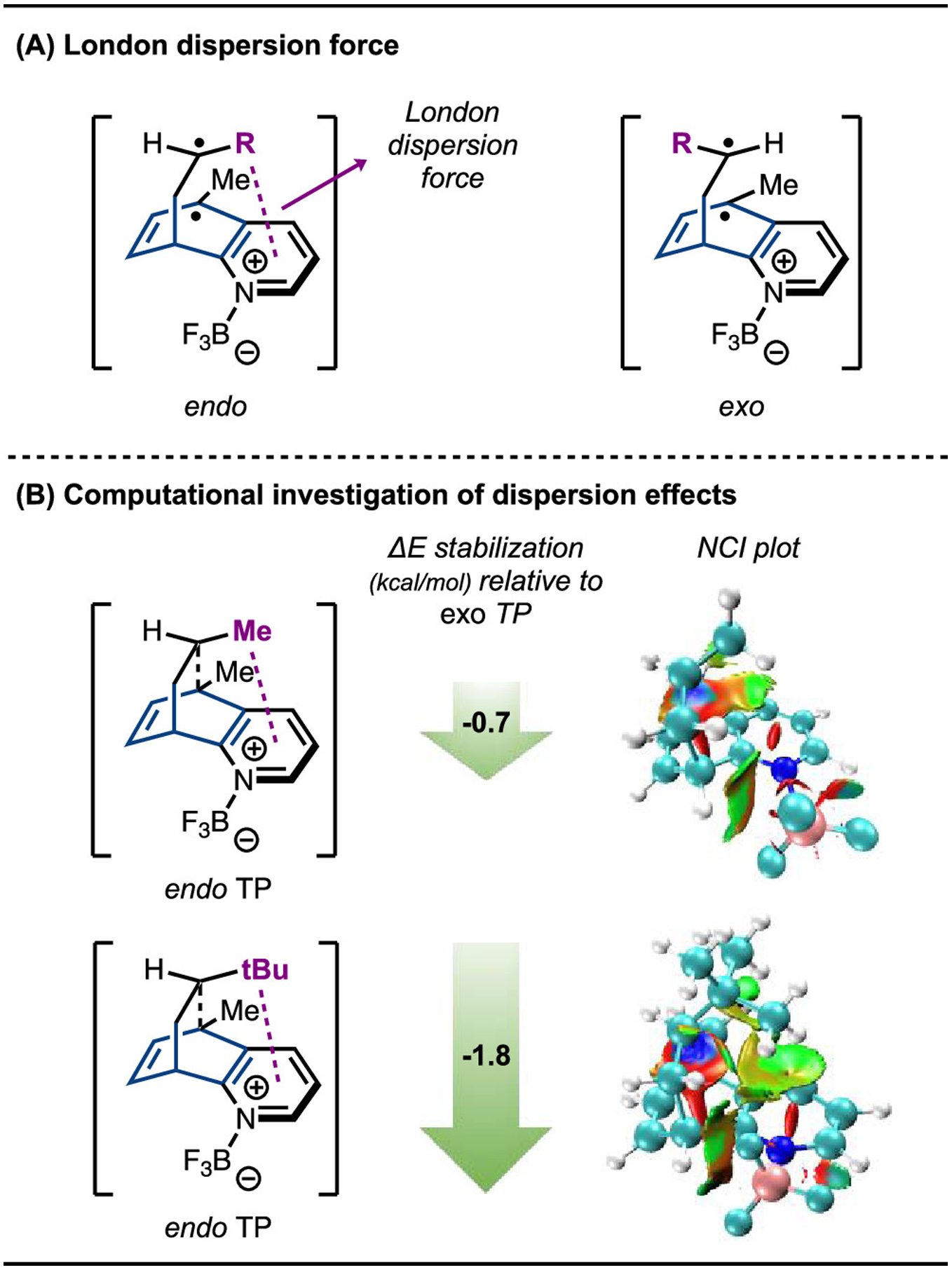
Endo vs Exo Diastereoselectivity
CONCLUSIONS
We described herein a dearomative para -cycloaddition of quinolines with a variety of activated and unactivated alkenes. The reaction was enabled by photosensitization of the Lewis acid-complexed substrates and displays high diastereo- and regioselectivities that are tunable through varying the solvent and substitution patterns. Our mechanistic studies revealed a complex multisurface energetic landscape traversed by the reaction. Using a combined experimental and computational approach, we also disclosed the origins of the observed regio- and diastereoselectivities. Given the emerging interest in bridged polycycles and heterocycles in medicinal chemistry, we anticipate that this method will find substantial use in facilitating the efficient synthesis of such scaffolds.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Indiana University and the NIH (R35GM131755) for financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund and the NSF MRI programs, CHE-1726633 and CHE-1920026. Support for the acquisition of the Bruker Venture D8 diffractometer through the Major Scientific Research Equipment Fund from the President of Indiana University and the Office of the Vice President for Research is gratefully acknowledged. S.C. is grateful to Oberlin College for financial support. DFT calculations were performed using the SCIURus, the Oberlin College HPC cluster (NSF MRI 1427949). Generous financial support by the University of Munster and the Deutsche Forschungsgemeinschaft (SFB 858) is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c07726.
Experimental procedures, analytical data for all new compounds (PDF)
Accession Codes
CCDC 2060640 and 2191096–2191099 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c07726
The authors declare no competing financial interest.
Contributor Information
Renyu Guo, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
Souvik Adak, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
Peter Bellotti, Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany.
Xinfeng Gao, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
W. Walker Smith, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
Sam Ngan Le, Department of Chemistry and Biochemistry, Oberlin College, Oberlin, Ohio 44074, United States.
Jiajia Ma, Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany.
K. N. Houk, Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California 90095, United States
Frank Glorius, Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany.
Shuming Chen, Department of Chemistry and Biochemistry, Oberlin College, Oberlin, Ohio 44074, United States.
M. Kevin Brown, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
REFERENCES
- (1).(a) Carruthers W; Baldwin JE Cycloaddition Reactions in Organic Synthesis, Elsevier Science, 1990. [Google Scholar]; (b) Kobayashi S; Jørgensen KA Cycloaddition Reactions in Organic Synthesis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001. [Google Scholar]; (c) Nishiwaki N Methods and Applications of Cycloaddition Reactions in Organic Syntheses; John Wiley & Sons: New York, 2014. [Google Scholar]
- (2).(a) Ritchie TJ; Macdonald SJF The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design? Drug Discovery 2009, 14, 1011–1020. [DOI] [PubMed] [Google Scholar]; (b) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (c) Lovering F Escape from Flatland 2: complexity and promiscuity. Med. Chem. Commun 2013, 4, 515–519. [Google Scholar]; (d) Mykhailiuk PK Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem 2019, 17, 2839–2849. [DOI] [PubMed] [Google Scholar]; (e) Subbaiah MAM; Meanwell NA Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design. J. Med. Chem 2021, 64, 14046–14128. [DOI] [PubMed] [Google Scholar]
- (3).For selected reviews see:; (a) Streit U; Bochet CG The Arene-Alkene Photocycloaddition. Beilstein J. Org. Chem 2011, 7, 525–542. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Remy R; Bochet CG Arene–Alkene Cycloaddition. Chem. Rev 2016, 116, 9816–9849. [DOI] [PubMed] [Google Scholar]; (c) Okumura M; Sarlah D Visible-Light-Induced Dearomatizations. Eur. J. Org. Chem 2020, 2020, 1259–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cheng Y-Z; Feng Z; Zhang X; You S-L Visible-Light Induced Dearomatization Reactions. Chem. Soc. Rev 2022, 51, 2145–2170. [DOI] [PubMed] [Google Scholar]
- (4).Gilbert A The Inter- and Intramolecular Photocycloaddition of Ethylenes to Aromatic Compounds. Pure Appl. Chem 1980, 52, 2669–2682. [Google Scholar]
- (5).Bryce-Smith D Orbital Symmetry Relationships for Thermal and Photochemical Concerted Cycloadditions to the Benzene Ring. J. Chem. Soc. D 1969, 806–808. [Google Scholar]
- (6).Houk KN Theory of Cycloadditions of Excited Aromatics to Alkenes. Pure Appl. Chem 1982, 54, 1633–1650. [Google Scholar]
- (7).van der Hart JA; Mulder JJC; Cornelisse J Funnels and barriers in the photocycloaddition of arenes to alkenes and dienes. J. Photochem. Photobiol. A 1995, 86, 141–148. [Google Scholar]
- (8).Cornelisse J The Meta Photocycloaddition of Arenes to Alkenes. Chem. Rev 1993, 93, 615–669. [Google Scholar]
- (9).(a) Wender PA; Ternansky R; deLong M; Singh S; Olivero A; Rice K Arene-Alkene Cycloadditions and Organic Synthesis. Pure Appl. Chem 1990, 62, 1597–1602. [Google Scholar]; (b) Kärkäs MD; Porco JA; Stephenson CRJJ Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev 2016, 116, 9683–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006; Chapter 16, pp 935–1000. [Google Scholar]
- (11).El-Sayed MA The Triplet State: Its Radiative and Nonradiative Properties. Acc. Chem. Res 1968, 1, 8–16. [Google Scholar]
- (12).Wagner PJ Photoinduced Ortho [2 + 2] Cycloaddition of Double Bonds to Triplet Benzenes. Acc. Chem. Res 2001, 34, 1–8. [DOI] [PubMed] [Google Scholar]
- (13).(a) Mattay J Selectivities in Photocycloadditions of Arenes to Olefins. J. Photochem 1987, 37, 167–183. [Google Scholar]; (b) Stegbauer S; Jandl C; Bach T Enantioselective Lewis Acid Catalyzed ortho Photocycloaddition of Olefins to Phenanthrene-9-carboxaldehydes. Angew. Chem., Int. Ed 2018, 57, 14593–14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Döpp D; Krüger C; Memarian HR; Tsay Y-H 1,4-Photocycloaddition of α-Morpholinoacrylonitrile to 1-Acylnaphthalenes. Angew. Chem., Int. Ed 1985, 24, 1048–1049. [Google Scholar]; (b) Ohkura K; Sugaoi T; Sakushima A; Nishijima K; Kuge Y; Seki K Thermodynamically Controlled Photocycloaddition of 5-Fluoro-1,3-dimethyluracil to Naphthalenes. Heterocycles 2002, 58, 595–600. [Google Scholar]
- (15).For selected reviews, see:; (a) Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]; (b) Zhou Q-Q; Zou Y-Q; Lu L-Q; Xiao W-J Visible-Light-Induced Organic Photochemical Reactions through Energy-Transfer Pathways. Angew. Chem., Int. Ed 2019, 58, 1586–1604. [DOI] [PubMed] [Google Scholar]; (c) Strieth-Kalthoff F; Glorius F Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem 2020, 6, 1888–1903. [Google Scholar]; (d) Großkopf J; Kratz T; Rigotti T; Bach T Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer. Chem. Rev 2022, 122, 1626–1653. [DOI] [PubMed] [Google Scholar]
- (16).Ma J; Schäfers F; Daniliuc C; Bergander K; Strassert CA; Glorius F Gadolinium Photocatalysis: Dearomative [2 + 2] Cycloaddition/Ring-Expansion Sequence with Indoles. Angew. Chem., Int. Ed 2020, 59, 9639–9645. [DOI] [PubMed] [Google Scholar]
- (17).(a) Zhu M; Zheng C; Zhang X; You S-L Synthesis of Cyclobutane-Fused Angular Tetracyclic Spiroindolines Via Visible-Light-Promoted Intramolecular Dearomatization of Indole Derivatives. J. Am. Chem. Soc 2019, 141, 2636–2644. [DOI] [PubMed] [Google Scholar]; (b) Zhu M; Huang X-L; Xu H; Zhang X; Zheng C; You S-L Visible-Light-Mediated Synthesis of Cyclobutene-Fused Indolizidines and Related Structural Analogs. CCS Chem. 2020, 2, 652–664. [Google Scholar]; (c) Zhu M; Zhang X; Zheng C; You S-L Visible-Light-Induced Dearomatization via [2+2] Cycloaddition or 1,5-Hydrogen Atom Transfer: Divergent Reaction Pathways of Transient Diradicals. ACS Catal. 2020, 10, 12618–12626. [Google Scholar]; (d) Zhu M; Huang X-L; Sun S; Zheng C; You S-L Visible-Light-Induced Dearomatization of Indoles/Pyrroles with Vinylcyclopropanes: Expedient Synthesis of Structurally Diverse Polycyclic Indo-lines/Pyrrolines. J. Am. Chem. Soc 2021, 143, 13441–13449. [DOI] [PubMed] [Google Scholar]
- (18).Oderinde MS; Mao E; Ramirez A; Pawluczyk J; Jorge C; Cornelius LAM; Kempson J; Vetrichelvan M; Pitchai M; Gupta A; Gupta AK; Meanwell NA; Mathur A; Dhar TGM Synthesis of Cyclobutane-Fused Tetracyclic Scaffolds via Visible-Light Photo-catalysis for Building Molecular Complexity. J. Am. Chem. Soc 2020, 142, 3094–3103. [DOI] [PubMed] [Google Scholar]
- (19).Zhang Z; Yi D; Zhang M; Wei J; Lu J; Yang L; Wang J; Hao N; Pan X; Zhang S; Wei S; Fu Q Photocatalytic Intramolecular [2 + 2] Cycloaddition of Indole Derivatives via Energy Transfer: A Method for Late-Stage Skeletal Transformation. ACS Catal. 2020, 10, 10149–10156. [Google Scholar]
- (20).Zhuang W; Cheng Y-Z; Huang X-L; Huang Q; Zhang X Visible-Light Induced Divergent Dearomatization of Indole Derivatives: Controlled Access to Cyclobutane-Fused Polycycles and 2-Substituted Indolines. Org. Chem. Front 2021, 8, 319–325. [Google Scholar]
- (21).Hu N; Jung H; Zheng Y; Lee J; Zhang L; Ullah Z; Xie X; Harms K; Baik M-H; Meggers E Catalytic Asymmetric Dearomatization by Visible-Light-Activated [2 + 2] Photocycloaddition. Angew. Chem., Int. Ed 2018, 57, 6242–6246. [DOI] [PubMed] [Google Scholar]
- (22).Oderinde MS; Ramirez A; Dhar TGM; Cornelius LAM; Jorge C; Aulakh D; Sandhu B; Pawluczyk J; Sarjeant AA; Meanwell NA; Mathur A; Kempson J Photocatalytic Dearomative Intermolecular [2 + 2] Cycloaddition of Heterocycles for Building Molecular Complexity. J. Org. Chem 2021, 86, 1730–1747. [DOI] [PubMed] [Google Scholar]
- (23).Strieth-Kalthoff F; Henkel C; Teders M; Kahnt A; Knolle W; Gómez-Suárez A; Dirian K; Alex W; Bergander K; Daniliuc CG; Abel B; Guldi DM; Glorius F Discovery of Unforeseen Energy-Transfer-Based Transformations Using a Combined Screening Approach. Chem 2019, 5, 2183–2194. [Google Scholar]
- (24).(a) Kishikawa K; Akimoto S; Kohmoto S; Yamamoto M; Yamada K Intramolecular photo[4+2]cycloaddition of an enone with a benzene ring. J. Chem. Soc., Perkin Trans 1 1997, 77–84. [Google Scholar]; (b) Ma J; Strieth-Kalthoff F; Dalton T; Freitag M; Schwarz JL; Bergander K; Daniliuc C; Glorius F Direct Dearomatization of Pyridines via an Energy-Transfer-Catalyzed Intramolecular [4 + 2] Cycloaddition. Chem 2019, 5, 2854–2864. [Google Scholar]
- (25).Ma J; Chen S; Bellotti P; Guo R; Schafer F; Heusler A; Zhang X; Daniliuc C; Brown MK; Houk KN; Glorius F Photochemical intermolecular dearomative cycloaddition of bicyclic azaarenes with alkenes. Science 2021, 371, 1338–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Taylor RD; MacCoss M; Lawson AD Rings in Drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (c) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (27).(a) Ma J; Chen S; Bellotti P; Wagener T; Daniliuc C; Houk KN; Glorius F Facile access to fused 2D/3D rings via intermolecular cascade dearomative [2 + 2] cycloaddition/rearrangement reactions of quinolines with alkenes. Nat. Catal 2022, 5, 405–413. [Google Scholar]; (b) Bellotti P; Rogge T; Paulus F; Laskar R; Rendel N; Ma J; Houk KN; Glorius F Visible-Light Photocatalyzed peri-(3 + 2) Cycloadditions of Quinolines. J. Am. Chem. Soc 2022, 144, 15662–15671. [DOI] [PubMed] [Google Scholar]
- (28).Elliott LD; Kayal S; George MW; Booker-Milburn K Rational Design of Triplet Sensitizers for the Transfer of Excited State Photochemistry from UV to Visible. J. Am. Chem. Soc 2020, 142, 14947–14956. [DOI] [PubMed] [Google Scholar]
- (29).Reichardt C; Welton T Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH Publishers, 2003; pp 470–475. [Google Scholar]
- (30).During the investigation we found it necessary to switch from the optimized solvent PhMe to CH2Cl2, and to dilute the reaction to [0.05 M] for improved solubility (especially for substituted quinolines).
- (31).Chénard E; Sutrisno A; Zhu L; Assary RS; Kowalski JA; Barton JL; Bertke JA; Gray DL; Brushett FR; Curtiss LA; Moore JS Synthesis of Pyridine– and Pyrazine–BF3 Complexes and Their Characterization in Solution and Solid State. J. Phys. Chem. C 2016, 120, 8461–8471. [Google Scholar]
- (32).For photochemical reaction with allenes, see:; (a) Haddaway K; Somekawa K; Fleming P; Tossell JA; Mariano PS Chemistry of Allene Cation Radicals Probed by the Use of Theoretical and Electron-transfer Photochemical Methods. J. Org. Chem 1987, 52, 4239–4253. [Google Scholar]; (b) Birbaum F; Neels A; Bochet CG Photochemistry of Allenyl Salicylaldehydes. Org. Lett 2008, 10, 3175–3178. [DOI] [PubMed] [Google Scholar]; (c) Streit U; Birbaum F; Quattropani A; Bochet CG Photocycloaddition of Arenes and Allenes. J. Org. Chem 2013, 78, 6890–6910. [DOI] [PubMed] [Google Scholar]
- (33). Several previous studies have proposed that the first step is the selectivity determining step. See refs 19, 22, 23.
- (34).For a recent study that highlights a selectivity determining radical recombination, see; Zhu M; Zheng C Post-Spin Crossing Dynamics Determine the Regioselectivity in Open-Shell Singlet Biradical Recombination. Org. Chem. Front 2022, 9, 995–1003. [Google Scholar]
- (35).(a) Singleton DA; Thomas AA High-Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc 1995, 117, 9357–9358. [Google Scholar]; (b) Beno BR; Houk KN; Singleton DA Synchronous or Asynchronous? An “Experimental” Transition State from a Direct Comparison of Experimental and Theoretical Kinetic Isotope Effects for a Diels–Alder Reaction. J. Am. Chem. Soc 1996, 118, 9984–9985. [Google Scholar]; (c) Hirschi JS; Takeya T; Hang C; Singleton DA Transition State Geometry Measurements from 13C Isotope Effects. The Experimental Transition State for the Epoxidation of Alkenes with Oxaziridines. J. Am. Chem. Soc 2009, 131, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]; For 13C KIE at natural abundance to probe a photochemical reaction, see:; Kuan K-Y; Singleton DA Isotope Effects and the Mechanism of Photoredox-Promoted [2 + 2] Cycloadditions of Enones. J. Org. Chem 2021, 86, 6305–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).It is also possible that the trajectory of approach of the alkene to the triplet quinoline diradical may influence the selectivity.
- (37).Daub ME; Jung H; Lee BJ; Won J; Baik M-H; Yoon TP Enantioselective [2+2] Cycloadditions of Cinnamate Esters: Generalizing Lewis Acid Catalysis of Triplet Energy Transfer. J. Am. Chem. Soc 2019, 141, 9543–9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Morofuji T; Nagai S; Chitose Y; Abe M; Kano N Protonation-Enhanced Reactivity of Triplet State in Dearomative Photocycloaddition of Quinolines to Olefins. Org. Lett 2021, 23, 6257–6261. [DOI] [PubMed] [Google Scholar]; The free energy barriers shown in Scheme 7C were re-calculated and differ from those calculated by the original authors.
- (39).Gaussian 16 Users Reference, Keyword: Freq, 2021. https://gaussian.com/man/.
- (40). see ref 27.; (b) Zhu M; Xu H; Zhang X; Zheng C; You S-L Visible-Light-Induced Intramolecular Double Dearomative Cycloaddition of Arenes. Angew. Chem., Int. Ed 2021, 60, 7036–7040. [DOI] [PubMed] [Google Scholar]
- (41).For triplet radical H atom abstraction, see:; ref 27.; (b) Xiong Y; Großkopf J; Jandl C; Bach T Visible Light-Mediated Dearomative Hydrogen Atom Abstraction/Cyclization Cascade of Indoles. Angew. Chem., Int. Ed 2022, 61, No. e202200555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).For selected reviews, see:; (a) Krenske EH; Houk KN Aromatic Interactions as Control Elements in Stereoselective Organic Reactions. Acc. Chem. Res 2013, 46, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wagner JP; Schreiner PR London Dispersion in Molecular Chemistry-Reconsidering Steric Effects. Angew. Chem., Int. Ed 2015, 54, 12274–12296. [DOI] [PubMed] [Google Scholar]; (c) Wheeler SE; Seguin TJ; Guan Y; Doney AC Noncovalent Interactions in Organocatalysis and the Prospect of Computational Catalyst Design. Acc. Chem. Res 2016, 49, 1061–1069. [DOI] [PubMed] [Google Scholar]; (d) Grimme S; Hansen A; Brandenburg JG; Bannwarth C Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev 2016, 116, 5105–5154. [DOI] [PubMed] [Google Scholar]; (e) Bursch M; Caldeweyher E; Hansen A; Neugebauer H; Ehlert S; Grimme S Understanding and Quantifying London Dispersion Effects in Organometallic Complexes. Acc. Chem. Res 2019, 52, 258–266. [DOI] [PubMed] [Google Scholar]; (f) Pollice R; Chen P A Universal Quantitative Descriptor of the Dispersion Interaction Potential. Angew. Chem., Int. Ed 2019, 58, 9758–9769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.