

Abstract
Eukaryotic cells decide in late G1 phase of the cell cycle whether to commit to another round of division. This point of cell cycle commitment is termed “Restriction Point” in mammals and “Start” in the budding yeast Saccharomyces cerevisiae. At Start, yeast cells integrate multiple signals such as pheromones and nutrients, and will not pass Start if nutrients are lacking. However, how cells respond to nutrient depletion after the Start decision remains poorly understood. Here, we analyze how post‐Start cells respond to nutrient depletion, by monitoring Whi5, the cell cycle inhibitor whose export from the nucleus determines Start. Surprisingly, we find that cells that have passed Start can re‐import Whi5 into the nucleus. In these cells, the positive feedback loop activating G1/S transcription is interrupted, and the Whi5 repressor re‐binds DNA. Cells which re‐import Whi5 become again sensitive to mating pheromone, like pre‐Start cells, and CDK activation can occur a second time upon replenishment of nutrients. These results demonstrate that upon starvation, the commitment decision at Start can be reversed. We therefore propose that cell cycle commitment in yeast is a multi‐step process, similar to what has been suggested for mammalian cells.
Keywords: cell division cycle, metabolism, nutrient signaling, quiescence, Saccharomyces cerevisiae
Subject Categories: Cell Cycle
Nutrient depletion after cell division commitment in late G1 phase causes nuclear re‐import and chromatin re‐association of the repressor Whi5, demonstrating that cell cycle commitment in yeast is actually a multi‐step process.
Introduction
Cells must coordinate growth and cell division with metabolism to proliferate and to maintain cellular homeostasis. This holds particularly true for single‐cell organisms like budding yeast, that are exposed to constantly changing nutrient supply (Smets et al, 2010). In yeast, nutrient supply directly affects metabolic rates and cell growth. Nutrient supply also impacts cell size, and entry and progression through the cell division cycle (Broach, 2012; Turner et al, 2012; Ewald, 2018; Qu et al, 2019). However, how the cell division cycle is responding to nutrients and which metabolic regulators interact with which components of the cell cycle machinery is still poorly understood (Ewald, 2018).
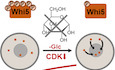
One of the most critical points of cell cycle signaling is the commitment point at the end of G1, termed “Restriction point” in mammals and “Start” in yeast (Morgan, 2007; Johnson & Skotheim, 2013; Pennycook & Barr, 2020). While the proteins involved in this decision have diverged, the architecture of cell cycle commitment is conserved from human to budding yeast (Cross et al, 2011; Johnson & Skotheim, 2013). In yeast, the cell cycle inhibitor Whi5 inhibits the transcription factor SBF (de Bruin et al, 2004; Fig 1A). This inhibition is partially relieved though Whi5 dilution by growth and activation of SBF‐dependent transcription by Cln3 (Wagner et al, 2009; Schmoller et al, 2015; Koivomagi et al, 2021). The partial activation of SBF leads to the transcription of the activators of CDK, the cyclins Cln1 and Cln2. The Cln1/2‐CDK complex hyperphosphorylates Whi5, which leads to its export from the nucleus and alleviation of SBF inhibition (Costanzo et al, 2004; Wagner et al, 2009). This positive feedback loop leads to full SBF activation, and the G1/S transition can proceed (Skotheim et al, 2008). When 50% of Whi5 has exited the nucleus, the positive feedback loop becomes self‐sustaining (Doncic et al, 2011). This is considered the point of irreversible cell cycle commitment, Start (Charvin et al, 2010; Doncic et al, 2011; Qu et al, 2019).
Figure 1. Post‐Start cells respond to starvation.
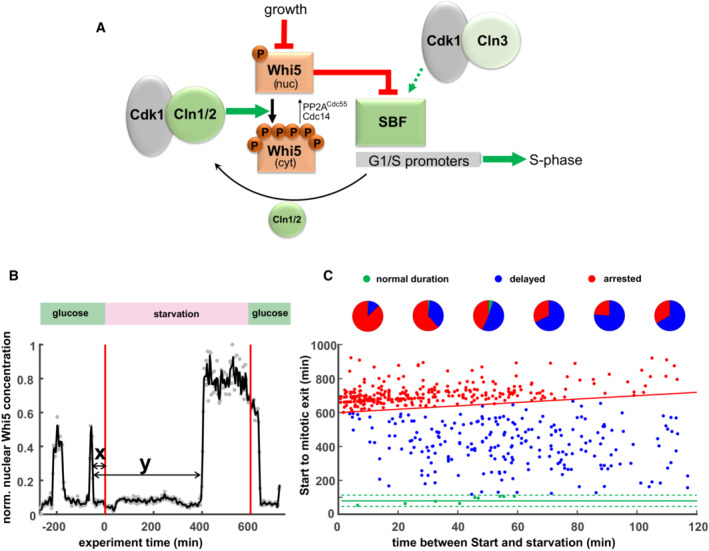
- Example trace of Whi5‐mCherry single yeast cell in a starvation experiment. Cells were grown for 8 h on glucose minimal medium, 10 h in starvation medium (1% sorbitol minimal medium), then 4 h on glucose minimal medium. Red lines indicate times of media switch. Gray dots represent original data; the black line represents data smoothed with a Savitzky–Golay filter. The arrows indicate the times used for the x‐ and y‐axis in graph (C).
- Cell cycle durations of single cells exposed to starvation. Cells were grown as described in (B). Each dot depicts a single cell (n = 579, from four independent experiments). The red line depicts the end of the 10‐h starvation period. Cells above this line continue their cycle only after glucose replenishment and are thus categorized as “permanently arrested.” The green lines depict the average duration in unperturbed cells ± 2 SD. Cells between the green and the red line are categorized as “delayed” cell cycle (such as the example cell in B). Pie charts depict the fractions of “permanently arrested” (red), “delayed” (blue), and “normal duration” (green) in 20‐min bins.
Leading up to Start, the cell integrates multiple internal and external signals—such as hormones and growth factors, stress, and cell size—to make the decision to commit to another round of DNA duplication and division (Morgan, 2007; Turner et al, 2012; Johnson & Skotheim, 2013). It is well established that also nutrient signaling can promote or prevent Start (Broach, 2012; Talarek et al, 2017; Ewald, 2018; Qu et al, 2019). Cells that do not have a sufficient supply of all essential nutrients will not pass Start and remain arrested in G1. However, what happens if essential nutrients are depleted after cells have passed Start, is largely unknown (Ewald, 2018).
Evidence from our work and others suggests that cells can respond to nutrients in all phases of the cell cycle. For example, S‐phase cells exposed to stress or a drop in glucose supply will transiently arrest the replication machinery by an inhibitory phosphorylation on Mrc1 (Duch et al, 2018). Wood et al (2020) found in single‐cell experiments that cells can enter a quiescence‐like state outside of G1 when responding to acute starvation, in agreement with earlier population‐based studies showing that even budded cells (Laporte et al, 2011) or cells in genetically induced cell cycle arrests (Wei et al, 1993) can enter quiescence. A recent report also demonstrated that cells can arrest their cell cycle in a “high CDK state” (Argüello‐Miranda et al, 2021) when facing nutrient perturbations. Despite these individual examples, a comprehensive picture of how cells proceeding through the cell cycle respond to nutrient signals is still lacking. Specifically, we do not know which cell cycle regulators receive nutrient‐dependent signals, or what phases of the cell cycle cells can delay or stably arrest in to wait for an improvement of nutrient supply.
Here, we investigated the response of post‐Start cells to acute carbon starvation. We analyzed thousands of single cells by monitoring Whi5 and other cell cycle regulators during live cell imaging. We found that starved cells can delay or stably arrest cell cycle progression. Surprisingly, cells that had already passed Start, could translocate Whi5 back into the nucleus when starved within the first 20 min post‐Start. This nuclear re‐entry of Whi5 corresponded to a reversal of Cdk1 activation and cells became sensitive to mating pheromone again. We conclude that Start remains reversible in response to nutrient perturbations for approximately 15–20 min. Thus, the current model of Start (Morgan, 2007; Charvin et al, 2010; Doncic et al, 2011; Bertoli et al, 2013; Johnson & Skotheim, 2013) is incomplete and Cdk1‐Cln1/2 activation is not the final point of cell cycle commitment.
Results
The impact of nutrient signaling on cell cycle progression downstream of Start is still largely unknown. Here, we investigated how acute carbon starvation impacts the yeast cell cycle using live cell imaging and microfluidics cultivation. We worked with prototrophic cells on glucose minimal medium without amino acids, to ensure cells do not have an alternative carbon source. To starve cells, we switched to medium containing 1% sorbitol, which is not metabolized by cells, but osmotically balances the medium. With this setup, we are confident to insulate the specific effect of carbon supply and the associated signaling. In our typical carbon starvation experiments, we first grew cells for several hours on glucose medium to adapt cells to the microfluidic environment and obtain data on the unperturbed cell cycle. Cells were then switched to starvation medium for 10 h (unless stated otherwise), after which glucose was replenished for at least 4 h.
Cells in the microfluidics cultivation platform grow asynchronously; therefore, we could observe cells in all phases of the cell cycle when the medium was switched to starvation conditions. Start was determined by monitoring endogenously fluorescently tagged Whi5. Specifically, Whi5's exit from the nucleus marks the Start transition (Doncic et al, 2011; Liu et al, 2015; Qu et al, 2019) which we confirmed for our strain and conditions (Fig EV1). We exposed cells harboring fluorescently labeled Whi5 to starvation, and analyzed those cells that had already passed Start before the switch. We determined the time that had passed between Start and when a cell was faced with carbon depletion (Fig 1B, x‐axis on Fig 1C), and the time that these cells spent from Start to the end of mitosis, as monitored by the re‐entry of Whi5 into mother and daughters (Fig 1C, y‐axis). We analyzed over 500 post‐Start cells (from four independent biological experiments) and found three different types of responses: A small fraction (< 2%) of cells completed their cell cycle (i.e. completed S/G2/M phases as evidenced by Whi5 re‐entering mother and daughter cell nucleus) with a similar duration as non‐starved cells (Fig 1C, green dots); 39% of cells completed their cell cycle, but with a much longer duration than non‐starved cells (Fig 1C, blue dots, we refer to these as “delayed cell cycle”); and 59% permanently arrested their cell cycle for the entire 10 h of the starvation period (Fig 1C, red dots). This arrest was typically stable even when the starvation period was extended to 16 h. Independent of their cell cycle or arrest state, almost all cells continued with normal cell cycles after glucose was replenished, indicating that cells do not lose viability or proliferative capacity during long‐term starvation‐induced arrest. The fraction of cells that consecutively stopped proliferation was below 1% in our assays, but this fraction gradually increased if the duration of starvation was extended.
Figure EV1. Commitment to mating pheromone occurs at Whi5 exit.
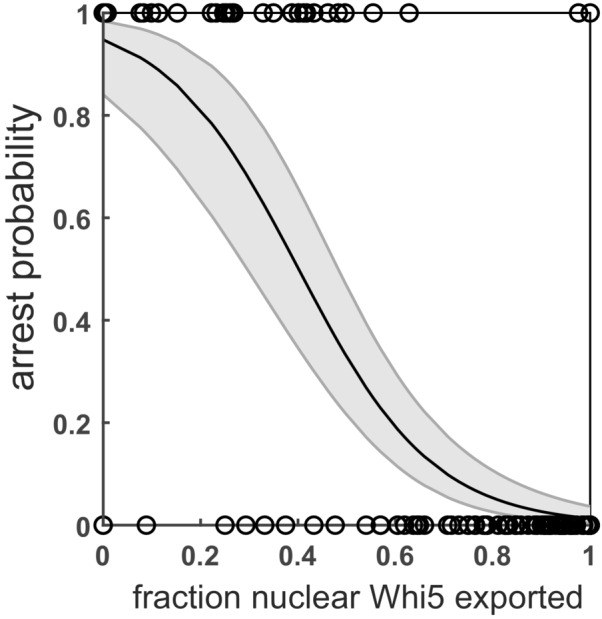
Cells were grown for 120 min on glucose minimal medium and then exposed to 1 μM α‐factor. x‐axis depicts the fraction of Whi5 that had been exported (relative to the peak value in the last G1) at the time point of the switch. Open circles represent 244 individual cells that either directly arrest in G1 (y = 1) or complete one more division before inducing a mating arrest (y = 0). A logistic regression was performed on these data to estimate the probability of arrest based on nuclear Whi5. Black line represents the result of the regression; the shaded areas represent the 95% confidence intervals.
Our data confirm recent studies reporting that cells respond to nutrient depletion in all phases of the cell cycle (Wood et al, 2020; Argüello‐Miranda et al, 2021); however, the mechanisms behind the cell cycle delay and arrest have not been determined. We decided to focus on those cells that enter a stable arrest to explore the mechanisms leading to and stabilizing such starvation‐induced arrests. When analyzing arrested cells, we noticed a surprising phenomenon: Cells that were exposed to starvation could translocate the Start inhibitor Whi5 back into the nucleus (Fig 2A and C, Movie EV1, Appendix Figs S1, S3 and S4). Given this surprising result, we wanted to confirm that Whi5 exit in these cells indeed corresponded to cells passing Start and activating the Cln1/2‐CDK‐positive feedback loop. We thus monitored Cln2 promoter activity using a fluorescent protein driven by the Cln2 promoter, but realized that the inherent delay caused by fluorescent maturation hindered sufficient time resolution. To increase time resolution for analyzing the promoter activity, we turned to the dPSTR system (developed by the Pelet lab; Aymoz et al, 2016) which turns a transcriptional signal into a localization change of a fluorescent reporter (Appendix Fig S2). With the Cln2‐dPSTR reporter we indeed observed that all analyzed cells activated Cln2 promoters at Whi5 exit (nuclear dPSTR peak after Whi5 exit was at least 50% of the last unperturbed cell cycle, 102 cells analyzed, example cell shown in Fig 2B). Thus, these cells had passed Start (as defined in the textbooks) before translocating Whi5 back into the nucleus when faced with starvation (Fig 2B and D). Based on > 800 observed cells, we calculated the probability of Whi5 re‐entry, given the time between Start and the exposure to starvation (Fig 2E). In the first 30 min, most of the cells re‐imported Whi5 (Figs 2E and 3D). As a control, we compared this starvation response to a response to mating pheromone. We exposed cells that had passed Start to high mating pheromone concentrations and monitored nuclear Whi5. We illustrate the difference with a heatmap showing the nuclear Whi5 profile of 50 cells each that were either starved or exposed to mating pheromone within 30 min after passing Start (Fig 2F). After passing Start, cells immediately become insensitive to mating pheromone and complete cell division; but cells remain sensitive to starvation and respond by re‐importing Whi5 and arresting cell cycle progression.
Figure 2. Whi5 re‐enters the nucleus upon starvation.
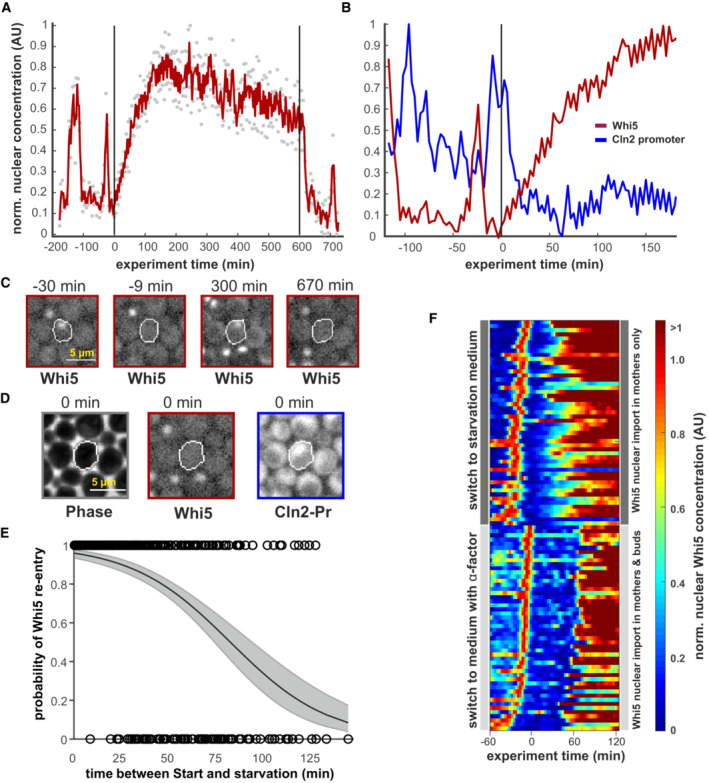
- Whi5‐mCherry trace of an example cell that re‐imports Whi5 upon starvation. Gray dots represent raw data, the red line results from a Savitzky–Golay smooth. Starvation was induced at t = 0 min and relieved after 10 h as indicated by the vertical black lines. Increase in nuclear fluorescence can be attributed mainly to nuclear import and not to overall increase in Whi5 (Appendix Figs S3 and S4).
- Images of the cell trace shown in (A) at indicated time points. Scale bar = 5 μm.
- Images from the cell shown in (C) at the time point immediately before starvation is induced. Absence of Whi5 and presence of the Cln2 reporter in the nucleus indicate that this cell had passed Start when it was exposed to starvation. See also Movie EV1. Scale bar = 5 μm.
- The probability of Whi5 nuclear re‐entry after starvation. Black circles indicate single cells (n = 849, from five independent experiments) that translocate Whi5 back into the nucleus. The x‐axis denotes the time that had passed since Whi5 exit until the medium was switched to starvation medium. The black line indicates a logistic regression of the single‐cell data, where the gray area denotes the 95% confidence intervals.
- Heatmap of nuclear Whi5 traces from 50 example cells (subset of E) that passed Start within 30 min before the switch to starvation. These are compared to 50 example cells that had the same initial conditions, but were then exposed to 10 μM α‐factor a t = 0 min. Traces were scaled to the maximum of the G1 peak before the switch.
Figure 3. Early and late Whi5 re‐entries.
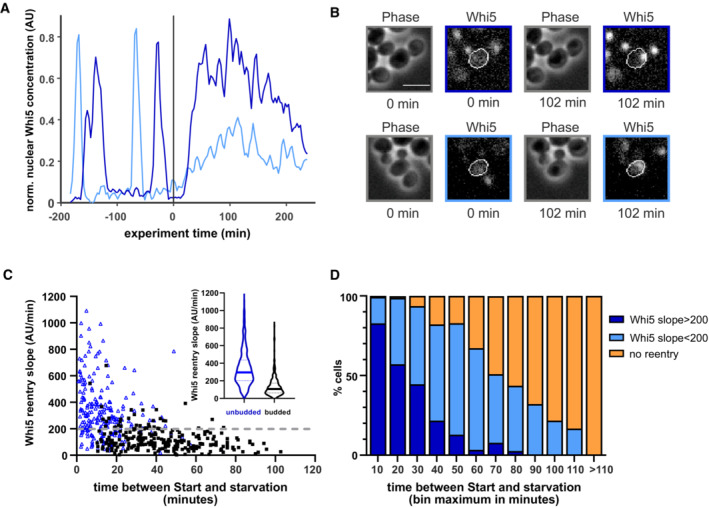
- Example traces of Whi5 of two cells exposed to starvation at t = 0 min. The dark blue line indicates a cell with a fast and steep re‐entry, and the light blue line indicates a cell with a slow re‐entry.
- Images of the two cells described in (A) at the indicated time points. Scale bar = 5 μm.
- We determined the slope of Whi5 nuclear re‐entry (regression of the first 30 min of nuclear Whi5 increase) in budded (black squares) and unbudded cells (blue triangle). We analyzed 680 cells from 5 replicate experiments. We picked 200 AU/min (dotted gray line) as the threshold between fast and slow re‐entries. Inset shows a violine plot of the slopes of all budded (black) and unbudded (blue) cells, where the solid lines depict the median of the distribution, and the dashed lines represent 25th and 75th percentiles.
- Distribution of fast, slow, and no re‐entries depending on how long after Start cells experience starvation (849 cells from five replicate experiments).
The nuclear re‐location of Whi5 after Start cannot be explained by the current model of Start, which states that Whi5 exit leads to irreversible commitment to cell cycle progression. However, Whi5 re‐entries have been noted, but not explained, in several previous studies (Liu et al, 2015; Litsios, 2017; Wood et al, 2020). We thus decided to examine when, how, and why Whi5 re‐entry occurs. While the probability for Whi5 re‐entry was highest in the first 10 min, the probability gradually dropped, but never reached zero. This seemed surprising, given that cells begin replicating their DNA at approximately 22 min after Start (Appendix Fig S6), so we did not understand the functional relevance of these late re‐entries. In this context, however, we noticed that there seemed to be two qualitatively distinct types of Whi5 re‐entries. Cells that were early in the cell cycle showed an immediate and steep Whi5 re‐entry. In contrast, cells that were later in the cell cycle and had a large bud showed a slow Whi5 re‐entry (example cells in Fig 3A and B). We hypothesized that these early and late nuclear re‐entries of Whi5 were mechanistically and functionally distinct.
We thus looked more closely at the dynamics of Whi5 re‐entry. We calculated the re‐entry slopes and determined the budding state of these cells (Fig 3C). Confirming our initial observation, the median slope of unbudded cells was approximately 2.5‐fold higher than in budded cells. However, there were some cells with small buds with very steep Whi5 re‐entries, and on the other hand cells with no obvious buds that showed slow re‐entries like budded cells. Thus, the budding event per se is not what distinguishes the two different types of Whi5 behavior.
Budding typically co‐occurs with the onset of replication (Appendix Fig S6), but the two are not mechanistically coupled (Pirincci Ercan et al, 2021; Appendix Fig S6B). We thus hypothesized that the difference between Whi5 behavior is due to the onset of replication, that is, the actual beginning of S‐phase. There is so far no canonical marker for observing S‐phase initiation in budding yeast; typically, the increase in histone concentration is used, although histone transcription and origin firing are not directly coupled. Unfortunately, upon starvation, the fluorescence from mTFP‐ or mCherry‐labeled histones increased in all phases of the cell cycle, likely due to sensitivity of the fluorophores to changes in the cellular environment (Appendix Fig S5). We therefore could not precisely determine the onset of histone production in our starved cells. However, the timing of histone production onset in unperturbed cells (Appendix Fig S6) corresponds well to the timing of the change in Whi5 behavior (Fig 3C). We therefore decided to group the cells according to their slope of Whi5 re‐entry, rather than by budding. We chose a cut‐off of 200 arbitrary intensity units/min (dotted gray line in Fig 3C) to separate “fast” re‐entries, which we interpret as pre‐replication cells, and “slow re‐entries,” which we interpret as cells that have started replication. Whether these cells arrest in S‐phase, or complete replication and arrest in G2, requires further investigation. None of these cells showed a Clb2 peak at the onset of or during starvation, indicating that all cells with Whi5 re‐entries arrest before the G2/M transition (Appendix Fig S8).
Neither the fast nor the slow re‐entries can be explained with our current understanding of Whi5 and Start regulation. We decided to focus on those cells with fast re‐entries, which mainly occur at the G1/S transition. Fast Whi5 re‐entries were very commonly observed: Within the first 20 min after Start, over 70% of cells translocated Whi5 back into the nucleus with a steep slope (Fig 3D); in the first 5 min after Start 99% of cells re‐imported Whi5 (Fig 2E). We hypothesized that this Whi5 re‐entry corresponds to a decrease in Cln1/2‐CDK activity. If Whi5 re‐entry indeed corresponds to a decrease in Cln1/2‐CDK activity, the cells would have to go through another round of the positive feedback loop of Whi5 exit and Cln1/2 activation after glucose replenishment. We therefore used the Cln2 promoter constructs to see what happens in these cells after glucose replenishment. Indeed, cells that had translocated Whi5 back at the onset of starvation, later showed another spike in Cln2 promoter activity once glucose was replenished, concurrent with Whi5 exit (Fig 4A–C). Those cells classified as “slow re‐entries” (Whi5 re‐entry slope < 200 AU/min) did not show a second round of Cln2 promoter activity before progressing with their cell cycle (Fig 4C, Appendix Fig S7). All of these cells did however show a peak of Clb2 expression, the cyclin required to enter mitosis, upon glucose re‐addition (Appendix Fig S8). This confirms that these cells had arrested in S or G2, and no longer required Cln1/2 activity to continue their cell cycle.
Figure 4. Early Whi5 re‐entries lead to functional Start reversal.
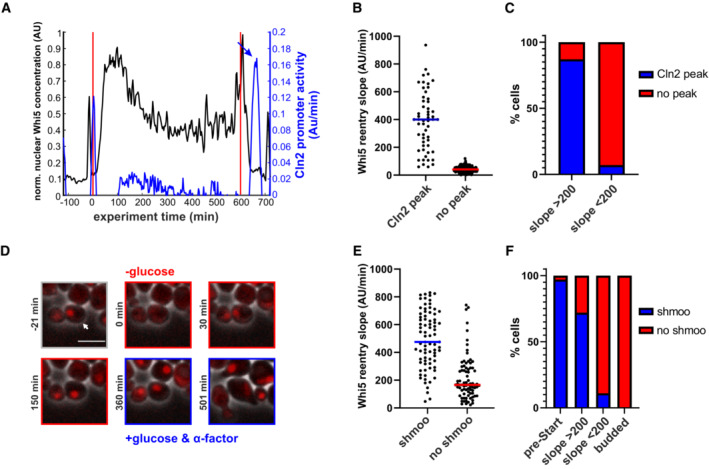
- Example cell showing that the Cln2 promoter (blue) fires upon glucose replenishment after Whi5 re‐entry (black). Vertical red lines indicate beginning and end of the starvation phase. The promoter activity was approximated by the change in fluorescence of a Cln2‐Promoter‐Neongreen construct (see also Appendix Fig S2). Arrow indicates Cln2 promoter activity peak after glucose replenishment.
- Whi5 re‐entry slopes of cells with and without Cln2 promoter activity after glucose replenishment (153 cells from three replicate experiments).
- Percentage of cells that show Cln2 promoter after glucose replenishment in cells that re‐imported Whi5 with slopes above or below our threshold of 200. Cells without Whi5 re‐entry never showed a Cln2 promoter activity peak after glucose replenishment. A Cln2 promoter peak after glucose replenishment was scored as such, if within 30 min after Whi5 exit, the slope of Cln2‐Promoter‐Neongreen fluorescence increase reached at least 50% of the pre‐starvation peak.
- Example cell that re‐imports Whi5 after starvation and responds to alpha factor after glucose replenishment. See also Movie EV2. Scale bar = 5 μm. Arrow indicates peak in Cln2 promoter activity after glucose replenishment.
- Whi5 re‐entry slopes of cells that shmoo or do not shmoo after glucose replenishment and alpha factor addition.
- Percentage of cells that respond to alpha factor addition by shmooing. The first bar describes cells that were arrested in a normal pre‐Start G1. The two middle bars include cells that exported and re‐imported Whi5 with no visible buds. The right bar includes all cells that were budded at the time of starvation and glucose replenishment.
These data on Whi5 localization and Cln2 promoter activity strongly suggested that activation of CDK at Start—supposedly the point of irreversible commitment—is in fact reversible for a certain duration if cells face starvation. To test whether cells with Whi5 re‐entries were functionally truly reversing cell cycle commitment, we tested their sensitivity to mating pheromone. We starved cells and then exposed them to the mating pheromone alpha‐factor at the same time when glucose was replenished. Most cells with Whi5 re‐entries during starvation later responded to alpha‐factor by shmooing, just like cells that were in a normal pre‐Start G1 state (Fig 4D–F, Movie EV2), albeit the fraction of shmooing cells was lower. This sensitivity to mating pheromone confirms that the positive feedback loop defining “Start” can be reversed within the first ~20 min. Thus, the current model of irreversible cell cycle commitment does not hold true vis‐à‐vis nutrient perturbations.
Having established the functional reversibility of the Whi5‐CDK‐positive feedback loop, we next wanted to understand the mechanism by which the G1/S transition is interrupted and Whi5 is translocated back to the nucleus. One plausible explanation is that the expression of the G1 cyclins is targeted by metabolic regulators to interrupt the G1/S transition. We thus investigated the possible role of known transcription factors involved in starvation responses and quiescence: The starvation‐responsive transcription factors Msn2 (Appendix Fig S9), Msn4, Xbp1 (Miles et al, 2013), have all been shown to bind the Cln1/2 promoters (data summarized on Yeastract; Monteiro et al, 2020); and the transcription factors Msa1 and Msa2 are important for arresting cells in G1 in preparation of quiescence (Miles et al, 2016). While these transcription factors are clearly relevant for long‐term starvation responses, they do not seem to be essential in driving Whi5 re‐entry during acute starvation (Fig 5). The xbp1, msa1msa2, and msn2msn4 deletion mutants all still translocated Whi5 back into the nucleus. Similarly, a deletion mutant of Rim15, a nutrient‐sensitive kinase that is upstream of these transcription factors, does not prevent Whi5 re‐entries upon starvation. We note, however, that the re‐entry dynamics of Whi5 appear different in the msn2msn4 mutant compared to wild‐type. The distinction between early and late Whi5 re‐entries based on the slope of Whi5 re‐import did not hold for all mutant cells. We observed both early re‐entries which were slow and late re‐entries with a very steep slope, for which we currently cannot offer a straightforward interpretation. Thus, while the initial decision to interrupt Start is probably not caused by these transcription factors, they likely contribute (directly or indirectly) to full Start reversal and the stabilization of the G1 arrest.
Figure 5. Deletion mutants still show Whi5 re‐entries.
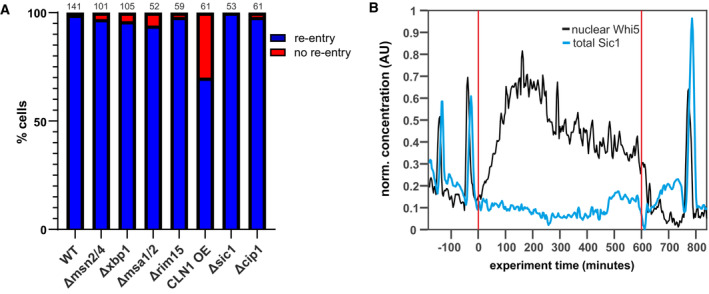
- We constructed the indicated mutants and monitored their Whi5 response to starvation. Depicted are the fractions of cells that re‐import Whi5 when starved within 15 min after Start. Numbers over the bars indicate total number of cells that were analyzed, cells were from at least three independent biological replicates.
- Example of a cell where Whi5 (black) re‐enters the nucleus upon starvation despite completed Sic1 (blue) degradation.
To test whether another transcription factor repressing cyclin expression may be essential for initiating Whi5 re‐entry, we expressed Cln1 constitutively from a strong synthetic promoter, which is active even under starvation (Δcln1Δcln2Δcln3, estradiol‐inducible‐Cln1, from Ottoz et al, 2014; Ewald et al, 2016; Zhang et al, 2019; see also Appendix Fig S10). Even in these cells overexpressing the activating cyclin, Whi5 still re‐entered the nucleus upon starvation in over 70% of the cells in the first 15 min (Fig 5A). The 30% reduction in cells re‐entering Whi5 is presumably caused by an overall acceleration of the G1/S transition due to high Cln1 levels. Thus, while repression of G1 cyclin expression is likely important for establishing and stabilizing quiescence, it is not essential for reversing Start under acute starvation.
Since Cln1/2 repression does not seem to be the initial target for interrupting Start, we wondered if the downstream inhibitor Sic1 could play a role. Sic1 is a well‐established inhibitor of CDK‐cyclin complexes and needs to be degraded to transition to S‐phase. Sic1 has been shown to be stabilized by Hog1 phosphorylation during the hyperosmolarity response (Escote et al, 2004; Adrover et al, 2011) and therefore seemed like an obvious target also for nutrient signaling. We thus fluorescently labeled Sic1 and determined the amount of Sic1 present in cells that translocated Whi5 versus cells that did not. However, Sic1 concentrations (total or nuclear) were not predictive of whether a cell reverses Start (Fig 5B and Appendix Fig S11). In agreement with this result, a deletion mutant of Sic1 did not lead to fewer Whi5 re‐entries (Fig 5A). In fact, due to a slightly slower cell cycle, the Sic1 deletion mutant translocated Whi5 back to the nucleus for an even longer time after Start. Similarly, a deletion mutant of the p21‐like inhibitor Cip1 (Chang et al, 2017) did not impact Whi5 translocations (Fig 5A).
We thus investigated whether Whi5 itself is the target of starvation signaling as it has been reported for high‐osmolarity signaling (Gonzalez‐Novo et al, 2015). Whi5 has 12 reported CDK phosphorylation sites, whose phosphorylation leads to Whi5 release from promoters and exit from the nucleus at Start (Taberner et al, 2009; Wagner et al, 2009). Additionally, there are at least seven non‐CDK sites, which have been confirmed by mutational analysis (Wagner et al, 2009 and Jan Skotheim, personal communication), but that have no clear function assigned yet. We thus wondered if one of these 7 phosphorylation sites could be targeted to cause Whi5 nuclear re‐import. We thus mutated all seven of these non‐CDK phosphorylation sites on Whi5, but this did not prevent Whi5 re‐entry upon starvation (Fig EV2A).
Figure EV2. Extended information for Fig 6 .
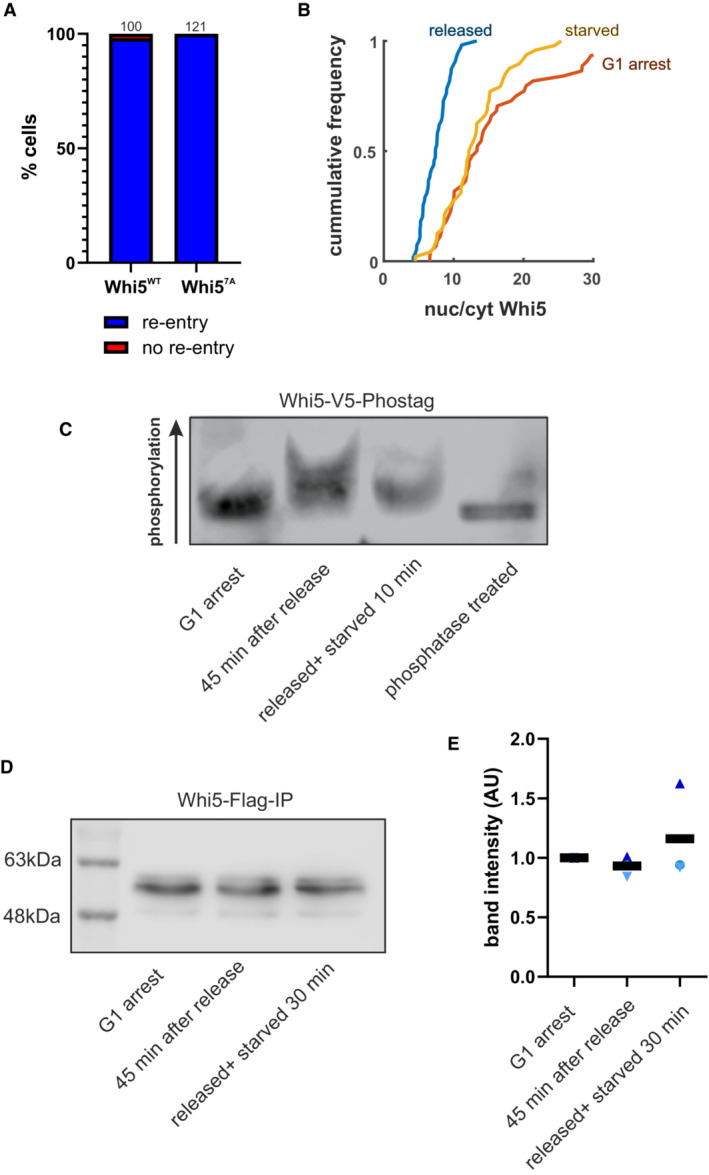
- We introduced wild‐type or the 7A phosphorylation site mutant (S78, S113, T114, S149, S276, T281, S288 mutated to alanine) Whi5 into a whi5‐deletion strain and determined its response to starvation. Bars indicate the fraction of cells that re‐import Whi5 when starved within 15 min after Start. Numbers over the bars indicate total number of cells that were analyzed.
- The histograms depicting Whi5 nuclear‐to‐cytoplasm ratios of Fig 6C are plotted as cumulated frequencies for direct comparison.
- Representative Western blot showing the amount of Whi5 protein pulled down in ChIP experiments (Fig 6E).
- Quantification of Western blots from three independent ChIP experiments as described in Fig 6E.
Since phosphorylation of the known non‐CDK sites on Whi5 did not seem to be responsible for Whi5 re‐entry, we decided to further investigate the phosphorylation of CDK sites on Whi5 in response to starvation. We thus turned to a bulk population‐based approach: We synchronized cells in G1 with our previously established system (Ewald et al, 2016), which is the same strain as used for constitutive Cln1 expression in Fig 5. As we reported previously, Whi5 leaves the nucleus approximately 35 min after inducing Cln1 expression by adding estradiol to the growth medium (Supplementary in Ewald et al, 2016). We released G1‐arrested cells growing on glucose minimal medium and after 45 min switched the cells to starvation medium. We harvested cells from the G1 arrest, 45 min after release immediately before starvation, and 10 and 30 min after the switch to starvation (Fig 6A–C). In these samples, we imaged Whi5‐GFP and confirmed that Whi5 had been exported from the nucleus and was re‐imported after starvation (Figs 6C and EV2B and C). In parallel samples, we then analyzed Whi5‐V5 by Phos‐tag (Kinoshita et al, 2006) Western blot. As reported previously (Wagner et al, 2009), Whi5 becomes hyperphosphorylated as CDK activity rises (Fig 6D, lane 2). Ten minutes after starvation however, these cells begin to lose their hyperphosphorylated form of Whi5 (Fig 6D, lane 3, Fig EV2C). This result suggests that Whi5 translocation is caused by dephosphorylation, most likely of CDK sites. Notably, we added the inducer for Cln1 expression in our starvation media, which confirms that Whi5 dephosphorylation is not dependent on cyclin repression. While our results do not exclude the possibility of additional phosphorylations on a previously uncharacterized site contributing to Whi5 re‐entry, they do strongly suggest that dephosphorylation of CDK sites on Whi5 is what drives Whi5 nuclear re‐entries.
Figure 6. Whi5 phosphorylation and DNA binding during starvation.
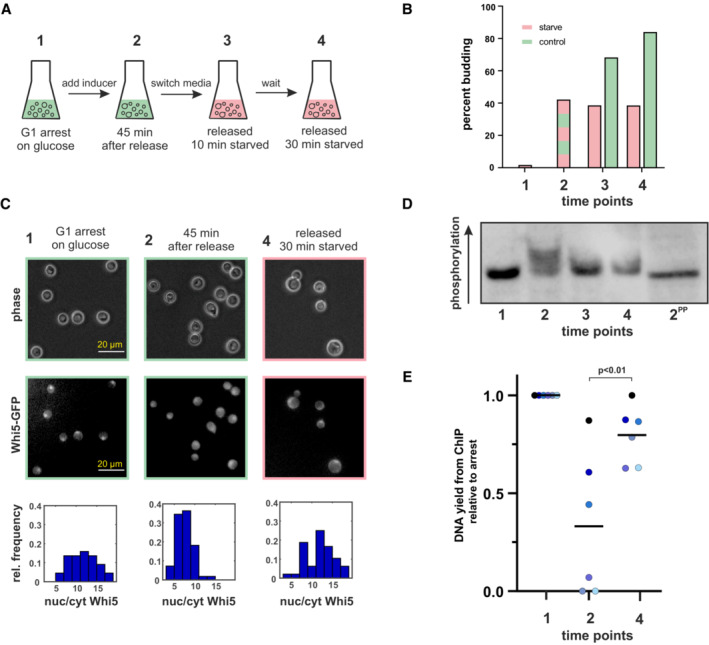
- Schematic of the experimental set‐up.
- Budding index of the cultures at the indicated time points. The green bars at t3 and t4 indicate control cultures that remained in glucose medium after 45 min.
- At the indicated time points samples from the cultures harboring Whi5‐GFP were imaged and the nuclear/cytoplasmic GFP concentration determined for 50 cells each. Bottom row depicts the resulting histogram (see also Fig EV2B for overlay), the upper rows show example images. Scale bar = 20 μm.
- Phos‐tag‐SDS–PAGE Western blot of Whi5‐V5. Numbers indicate the sampling time points shown in (A). 2PP indicates sample 2 (cells growing on glucose, 45 min after a G1 release) treated with phosphatase. See Fig EV2C for a blot from a replicate experiment.
- Cells from the experiment described in (A) were fixed at the indicated time points and chromatin‐immunoprecipitation with Whi5‐Flag as bait was performed. The graph reports the total DNA yield (normalized to t1) for six independent experiments. Black bars indicate the mean of the six replicates. P‐value based on a paired t‐test. The amount of Whi5 pulled down in the IP was similar across time points (Fig EV2E).
We next tested whether the dephosphorylated, re‐imported Whi5 also rebinds DNA. We used the same strain and set‐up as described for the phosphorylation assay: Cells were arrested in G1, released by inducing Cln1 expression, and starved 45 min after induction. Chromatin‐immunoprecipitation was then performed from these samples using the tagged Whi5. Consistently across six replicates, Whi5 pulled down less DNA after the release than in the G1 arrest. Once the released cells were starved, the amount of DNA bound to Whi5 increased again (P < 0.01, Fig 6E). We conclude that after starvation, Whi5 is dephosphorylated, re‐enters the nucleus, and rebinds promoters to re‐set the Whi5‐SBF‐CDK feedback loop, thus preventing the G1/S transition and re‐establishing a pre‐Start state.
However, these results did not tell us what the initial target of nutrient signaling is. It is possible that the specific activation of a phosphatase causes Whi5‐dephosphorylation and interruption of the feedback loop. However, we are not aware of a phosphatase that is strongly upregulated upon starvation and targets Whi5. So far, only two phosphatases have been reported to target Whi5, Cdc14, and Cdc55. Cdc14 dephosphorylates many Cdk1 sites including those on Whi5 (Taberner et al, 2009), but is active only during late mitosis. PP2A‐Cdc55 is the only previously reported phosphatase targeting Whi5 in G1 (Talarek et al, 2017; Baro et al, 2018). The deletion mutant of Cdc55 is very sick with irregular cell cycle progression and morphology. This phenotype is partially ameliorated by an additional Swe1 deletion (Harvey et al, 2011). We thus analyzed the cdc55 swe1 double deletion strain. These cells have normal morphology, but still show altered G1/S transition during growth on glucose. When starved, the cdc55 swe1 double deletion showed very pleiotropic phenotypes (Appendix Fig S12). However, those cells that exhibited normal G1 phases immediately before starvation, also showed Whi5 re‐entries (Appendix Fig S12A), indicating that Cdc55 is not essential for Whi5 dephosphorylation under starvation. Whether this re‐import in absence of Cdc55 is due to functional redundancy of phosphatases, or whether another phosphatase not yet associated with Whi5 is primarily responsible, remains to be shown. Since we could not find evidence for upregulation of Whi5 phosphorylation in published phosphatase deletion screens (e.g. Bodenmiller et al, 2010; Goldman et al, 2014), we currently consider it more likely that dephosphorylation of Whi5 is driven mainly by an inhibition of Cdk1 kinase activity, rather than by specific activation of a phosphatase.
Inhibition of Cdk1 could be mediated by the activity of Swe1. This kinase phosphorylates tyrosine 19, which keeps the CDK‐Clb2 complex inactive and thus prevents the G2/M transition. While this is the most prominent role of Swe1, other functions in the cell cycle have been described in yeast (Chauhan et al, 2015, 2016; Raspelli et al, 2018) and mammals (Moiseeva et al, 2019). We therefore investigated whether Swe1 could mediate Whi5 re‐entries. We found that swe1 deletion cells had difficulties responding to starvation in late cell cycle stages, which led to defects in mitosis (similar to the cdc55 swe1 double deletion shown in Appendix Fig S12B). However, cells that had just passed Start could re‐import Whi5 and arrest in G1 at the same frequency as wild‐type cells (Fig EV3A). In agreement with this, we detected only a very weak phosphorylation of CDK1 tyrosine 19 in cells starved early after Start (Fig EV3B and C). Thus, Swe1 likely plays a role in the cell's response to starvation, but not at the G1/S transition.
Figure EV3. The Swe1 kinase is not essential for Whi5 re‐entries.
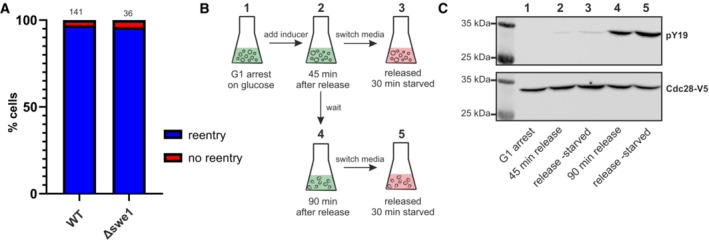
- We constructed a swe1 deletion mutant and monitored its Whi5 response to starvation (wild‐type control is the same data as in Fig 5A). Depicted is the fraction of cells that re‐imports Whi5 when starved within 15 min after Start. Numbers over the bars indicate total number of cells that were analyzed.
- Experimental setup for (C): Cells in liquid culture were arrested in G1 (time point 1) as described in Fig 6A. Cells were released by inducing Cln1 expression. After 45 min (2), half of the culture was transferred to starvation medium (3), and the other half continued to progress until 90 min (4). These cells were then also transferred to starvation medium (5).
- Cells from the experiment described in (B) were sampled at the indicated times and protein lysates were used for Western blotting. Upper blot shows the signal of a commercial antibody against phosphorylated tyrosine 19 on CDK. After the detection, the blot was stripped and re‐analyzed (lower blot) with an antibody detecting the V5‐tagged total CDK1 (Cdc28).
Discussion
In this work, we analyzed the starvation response of thousands of post‐Start cells. In agreement with recent studies, we show that cells can likely delay or arrest their cell cycle in any cell cycle stage (Wood et al, 2020; Argüello‐Miranda et al, 2021). We set out to understand how stable arrests are achieved and found that the cell cycle inhibitor Whi5 can re‐enter the nucleus in post‐Start cells. We showed that Whi5 re‐entries can be classified as fast and slow re‐entries, which likely corresponds to cells before and after entering S‐phase. The slow‐reentry cells are likely similar to the budded “high‐CDK quiescent cells” recently described by Argüello‐Miranda and colleagues (Argüello‐Miranda et al, 2021) although they induced starvation in a different way. Since it is not clear if Whi5 has any function in the later cell cycle, we focused on those cells re‐importing Whi5 with a steep slope, which are likely to be cells before replication. In the time window of 20 min (which is approximately 20% of the total time between Start and cytokinesis) over 70% of all cells respond to carbon starvation by re‐importing Whi5 with a steep slope (Fig 3).
We provide at least three lines of evidence that these cells are indeed reversing CDK activation and are “taking back” their commitment decision: Firstly, most cells that re‐import Whi5 upon starvation become sensitive to mating pheromone again (Fig 4), which is only expected from pre‐Start cells. Secondly, upon glucose replenishment these cells produce another peak of Cln2 expression, just like cells that have no history of passing Start (Fig 4). Thirdly, Whi5 re‐associates with DNA (Fig 6), thus likely inhibiting expression of G1/S genes again. Therefore, it seems highly likely that cells are indeed reversing Start (as defined in the textbooks). We note that Whi5 re‐entries have been previously mentioned in various contexts (Liu et al, 2015; Litsios, 2017; Wood et al, 2020), even though their cause or consequence have not been mechanistically studied. This shows that this is not an observation limited to our laboratory or to the strain investigated.
Are all cells that passed and reversed Start really identical to “normal” pre‐Start G1 cells after starvation? Several observations suggest that while cells reverse Cdk1 activation, they may retain some “memory” of passing Start. While many of these cells start shmooing in response to mating pheromone, the fraction of cells deciding to do so is 30% lower than the fraction in “normal” G1 cells at the tested concentration. Also, cells with tiny buds that have not yet started replicating can reverse (and later re‐activate) Cln1/2‐CDK activity, but they do not shmoo, and continue budding at the same site when re‐entering the cell cycle after glucose replenishment. We also note that cells with Whi5 re‐entries arrested with very different Sic1 concentrations. Those cells that had not started degrading Sic1 stabilized the protein and arrested with high Sic1 levels. Other cells that were starved after they had degraded Sic1 were arrested in a low Sic1 state and did not express any more Sic1 while in the G1 arrest. This may be relevant for the next cell cycle following nutrient upshift. Further molecular and biochemical studies are needed to fully understand the consequences of Whi5 re‐entry and Start reversal after starvation (and possible other perturbations).
Another interesting aspect of these findings is the relationship between cell cycle regulation, nutrient signaling, and pheromone signaling. The protein Far1, which is downstream of the pheromone sensing MAPK pathway and inhibits CDK, is normally expressed during G1, upregulated, and stabilized during pheromone response, but degraded if cells commit to the cell cycle, shortly after they pass Start. So how do cells that have passed Start and revert become sensitive to pheromone again (Fig 4)? Interestingly, during starvation, Far1 gets degraded in all cells, including those in “normal” G1 (Argüello‐Miranda et al, 2021 and our own preliminary results included in the response to the referees, see Review Process File). Once glucose returns, Far1 is re‐synthesized in unbudded cells, suggesting that both Far1 stability and expression are under control of glucose signaling. This is in good agreement with previous reports that showed that average Far1 concentrations are much lower on poor carbon sources than on glucose (Alberghina et al, 2004).
While we were not able to fully unveil the mechanism that translates nutrient sensing into interruption of the positive feedback loop and Start reversal, we can make several relevant conclusions: We first analyzed a series of transcriptional regulators that are known to be important for stationary phase and quiescence. However, none of these were essential for the acute starvation response of Whi5. In fact, a transcriptional mechanism seems unlikely, because overexpression of Cln1 did not prevent or delay re‐entries. We therefore turned away from transcription factors and searched for mechanisms directly acting on Whi5 and Cdk1. We showed that the mechanism of Whi5 re‐import is likely the dephosphorylation of the CDK1 sites, and not due to a regulatory phosphorylation on one of the previously reported (Wagner et al, 2009) non‐CDK sites. However, it is still possible that one of the many uncharacterized sites that have been picked up in mass spectrometry experiments (see e.g. biogrid.org; Oughtred et al, 2021 entries) could contribute to Whi5's response to starvation.
The observed dephosphorylation of Whi5 CDK sites could be caused by the upregulation of a starvation‐induced phosphatase. However, we did not find any evidence pointing in that direction. We therefore suggest that the cyclin‐CDK complex itself is the target of starvation signaling, and Whi5 re‐entry is the consequence rather than the primary cause of the positive feedback loop being interrupted. However, neither Sic1 and Cip1, the two inhibitors of CDK at the G1/S transition, nor the inhibitory kinase Swe1 appear to be essential for Start reversal. Other plausible mechanisms which we have not been able to systematically investigate within the scope of this study are, for example, an inhibitory post‐translational modification of CDK or the cyclins, the selective degradation of Start components (Morshed et al, 2020) or their sequestration in p‐bodies (van Leeuwen & Rabouille, 2019). Alternatively or additionally, the general biophysical/biochemical changes associated with starvation, such as drop in pH (Gutierrez et al, 2022), slowed diffusion rates (Joyner et al, 2016), and altered nuclear transport (Huang & Hopper, 2014; preprint: Heinrich et al, 2022), could affect the activity or stability of Start regulators.
If Whi5 export and CDK activation do not define the final commitment point, then what is the point‐of‐no‐return? Common sense dictates that cells should irreversibly commit to the cell cycle before attempting to replicate their DNA. Although we lack a good read‐out for replication initiation in budding yeast, our data on histone fluorescence intensity indeed suggest that cells with fast Whi5 re‐entries are pre‐replication. In mammalian cells, APC inactivation has been suggested as the point‐of‐no‐return (Cappell et al, 2016). In yeast cells, APC‐Cdh1 inactivation was recently placed 12 ± 3 min after Whi5 exit using an APC substrate fragment as a sensor (Ondracka et al, 2016). This time window is shorter than the ~20 min observed for Whi5 re‐entries in our experiments, but this difference could also be due to differences in strains and nutrient conditions. So, the role of the APC in commitment to S‐phase in yeast warrants further exploration.
The next step will be to find the missing pieces leading from Whi5 exit to irreversible commitment. As introduced above, the widely accepted textbook model states that the point‐of‐no‐return in the cell cycle is “Start” in yeast and the “Restriction Point” in mammalian cells (Morgan, 2007; Johnson & Skotheim, 2013; Pennycook & Barr, 2020). However, in mammalian cells, the notion that the restriction point is the universal cell cycle commitment point has been strongly challenged. Several studies suggested that there are at least two different commitment points: The restriction point for hormone and growth factor signaling, and a later point vis‐à‐vis nutrient and stress signals (Foster et al, 2010; Cappell et al, 2016; Patel et al, 2017; Pennycook & Barr, 2020; Yang et al, 2020). However, a complete and coherent model of all steps leading to cell cycle commitment in mammalian cells is still lacking. We now show that also in yeast commitment is a multi‐step process. Thus, our yeast experiments provide an excellent foundation to quantitatively and mechanistically study eukaryotic cell cycle commitment in a simple, tractable model.
Materials and Methods
Strain construction and cultivation
All strains were haploid W303 derivatives and most were prototrophic except for several mutants, which were partially auxotrophic as indicated in Appendix Table S1. Strains were constructed using standard PCR‐based homologous recombination. Whi5 phosphorylation mutants were generated by site‐directed mutagenesis on a plasmid, which was introduced into a Whi5 deletion mutant. See Appendix Table S1 for a detailed strain list with all genotypes. All strains constructed in this study are available from the corresponding author upon request.
For all reported experiments, yeast cells were grown in minimal medium without amino acids (1.7 g/l yeast nitrogen base without amino acids (US Biological), 5 g/l ammonium sulfate, 50 mM potassium phthalate, pH adjusted to 5 with KOH). Single amino acids were supplemented where necessary for strains and their corresponding controls (final concentrations histidine 5 mg/l, leucine 120 mg/l, uracil 20 mg/l). As a carbon source, 10 g/l glucose were added, which were then replaced by 10 g/l sorbitol in the starvation medium. Cells were incubated at 30°C on an orbital shaker at 200 rpm.
Cell cycle release and starvation experiments for Fig 6 were performed as follows: Strains derived from JE611c (Ewald et al, 2016; Zhang et al, 2019) were grown in a 15 ml pre‐culture on glucose minimal medium containing 40 nmol/l ß‐estradiol. A main culture was inoculated with OD 0.05 and grown overnight. Cells were arrested in G1 by filtering the culture and resuspending cells in estradiol‐free medium. After 5 h, arrest was verified by absence of budding, as observed under a light microscope. G1 cells were released into the cell cycle by addition of 200 nM ß‐estradiol. For starvation experiments, released cells were filtered 45 min after release, washed, and resuspended in sorbitol minimal medium (also supplemented with 200 nM estradiol).
Microfluidic cultivation
In preparation for live cell imaging experiments, cells were grown in 15 ml 1% glucose minimal medium overnight. The next morning, the cells were transferred to a fresh culture by applying 1:15 dilution and grown for at least another 5 h until reaching log phase (OD600 between 0.4 and 0.6).
Cells were sonicated at low power for 3 s and loaded onto a commercial microfluidics system (Y04C‐02 plates, CellASIC ONIX2 system, Merck). While loading the cells, the well which contained the starvation media was kept pressurized at 1.5 psi to avoid any backflow from the glucose‐containing wells into the glucose‐free media. During growth, the glucose media was supplied with a pressure of 3 psi. The well containing the starvation media was also pressurized with 0.5 psi to avoid backflow. Cells were grown inside the microfluidic chamber for the duration of at least two cell cycles before starting imaging. The temperature was kept constant at 30°C using an incubator chamber surrounding the imaging system (Okolab Cage Incubator, Okolab USA INC, San Bruno, CA).
The mating pheromone experiments were performed using the same setup and starting media as described above. For the commitment assay (Fig EV1), 1 μM α‐factor was added after 4 h of growth. For the control experiment in Fig 2, 10 μM α‐factor was added after 4 h of growth. In Fig 4, 500 nM α‐factor was added to the glucose media supplied after the starvation period. To prevent α‐factor from adhering to the microfluidic chamber, 20 μg/ml casein was added to all media with α‐factor.
For the growth of JE616, a final concentration of 1 μM β‐estradiol was added to both glucose and sorbitol minimal media during growth in the microfluidic chamber (ß‐estradiol adsorbs to the microfluidic chamber, which requires the use of higher concentrations than in shake flasks).
Microscopy
All live‐cell imaging experiments were performed on a Nikon Ti2 inverted epifluorescence microscope (Nikon Instruments, Japan) with a Lumencor SPECTRA X light engine (Lumencor, Beaverton, USA), a Photometrics Prime 95 (Teledyne Photometrics, USA) back‐illuminated sCMOS camera. The system was programmed and controlled by the Nikon software NIS Elements. Focus was maintained using the Nikon “Perfect Focus System.” The time‐lapse images were taken using a Nikon PlanApo oil‐immersion 60× objective (NA = 1.4) with a frequency of 3 min (2 min for experiments using the Cln2‐dPSTR construct). See Appendix Table S2 for optical filters and Appendix Table S3 for exposure settings. For all fluorophores and tagged proteins, we checked for absence of phototoxicity by comparing growth rates at varying exposures, frame rates, and in non‐exposed cells. Empty strains and single‐fluorophore expressing cells were used to control for autofluorescence and bleed‐through, respectively.
Image analysis and data processing
Cell segmentation, tracking, fluorescence quantification, and nuclear detection were performed using a custom‐built MATLAB script adapted from (Doncic et al, 2013; Wood & Doncic, 2019). The average background fluorescence (cell‐free area) for each image was determined and subtracted from the signal at each time point. Cell or nuclear volume was approximated using the 3/2 power of the segmented area (volume = pixels3/2). For protein concentrations during cell cycle progression, total fluorescence intensity divided by total approximated volume was used. For nuclear concentrations, the segmentation of the nucleus was achieved by applying a two‐dimensional Gaussian fit to the brightest pixels as described in (Doncic et al, 2011). The accuracy of this fitting method for Whi5‐based nuclear detection was verified using a fluorophore‐labeled histone as a nuclear marker.
Start was defined as the point where 50% of the Whi5 had exited the nucleus (Doncic et al, 2011). Whi5 re‐entries during starvation were defined as re‐importing Whi5 back into the nucleus after exporting at least 50% of nuclear Whi5. We note that none of our results qualitatively depend on the precise definition of Start. We verified for the wild‐type that results were nearly identical when Start was defined as complete Whi5 export. When calculating Whi5 re‐entry slopes, nuclear Whi5 concentration (fluorescent intensity/pixels3/2) was scaled between the minimal and maximal values of each cell. A linear regression was then fit to first 30 min of the increase.
For analysis of Cln2 promoter‐mNeongreen constructs, we called a peak, if the slope of the fluorescent increase (i.e. the activity of the promoter) post‐starvation was at least 50% of the last unperturbed cell cycle. For the Cln2 dPSTR constructs, we called a peak, if the nuclear fluorescence of the reporter reached at least 50% of the last unperturbed cell cycle.
Images were recorded at 12‐bit gray scale, then exported from NIS‐Elements software to 8‐bit tiffs. All data analysis was performed on these 8‐bit tiff images. For visualization purposes only, fluorescent images shown in figures and movies were denoised by setting the lowest 5% intensities of pixels to zero and then rescaling the resulting images to the full range of the gray scale.
Most MATLAB code used in this study has been published in (Doncic et al, 2013; Wood & Doncic, 2019); all code is available upon request.
SDS–PAGE Western blot
Ten milliliter of cell culture was harvested by centrifugation, frozen in liquid nitrogen and stored at −80°C. Cells were lysed by bead beating in urea buffer (20 mM Tris–HCl pH 7.5, 2 M Thiourea, 7 M Urea, 65 mM CHAPS, 65 mM DTT) supplemented with 1× EDTA‐free protease and phosphatase inhibitor cocktails (GoldBio). Two to three microgram of total protein were loaded onto 10% SDS‐polyacrylamide (29:1 Bio‐Rad) gels, run in SDS buffer for approximately 1.5 h at 15 mA and transferred overnight in a wet‐blot system (Bio‐Rad). The Cdk1 tyrosine 19 was detected by a commercial phospho‐specific antibody (Abcam, BLR101H, ab275958), and a secondary Anti‐Rabbit HRP Conjugate (Cell Signaling Technology, 7074S). After detection, the blot was stripped and Cdk1‐Flag was detected with a commercial Anti‐Flag antibody (Sigma‐Aldrich, product number: F1804), and an Anti‐Mouse HRP Conjugate (Promega, W4021). Luminescence was imaged on a Licor Odyssey FC.
Phos‐tag SDS–PAGE Western blot
Ten milliliter of cell culture was harvested by centrifugation, frozen in liquid nitrogen and stored at −80°C. Cells were lysed by bead beating in urea buffer (20 mM Tris–HCl pH 7.5, 2 M Thiourea, 7 M Urea, 65 mM CHAPS, 65 mM DTT) supplemented with 1× EDTA‐free protease and phosphatase inhibitor cocktails (GoldBio). Two to three microgram of total protein were loaded onto 10% SDS‐polyacrylamide (29:1 Bio‐Rad) gels with 10 μM Phos‐tag reagent (FUJIFILM Wako Chemicals) and 20 μM Mn2Cl. Gels were run in SDS buffer for approximately 2 h at 15 mA on ice. Gels were washed with 10 mM EDTA and then blotted on a dry blotting transfer system (iBlot2, Invitrogen). Whi5‐V5 was detected with a commercial Anti‐V5 mouse antibody (Bio‐Rad, MCA1360) and an Anti‐Mouse HRP Conjugate (Promega, W4021). Luminescence was imaged on a Licor Odyssey FC.
ChIP
The ChIP protocol was adapted from (Flick et al, 2003). Cell cycle release and starvation experiments were performed as described above. For starvation, released cells were filtered after 45 min, washed, and resuspended in a sorbitol minimal medium, supplemented with 200 nM estradiol. Cross‐links between DNA‐protein were introduced by incubating cells with 1% formaldehyde for 20 min at room temperature. 125 mM of glycine was added and incubated for 5 min to stop crosslinking. Cells were washed three times with ice‐cold TBS and cell pellets were frozen. Frozen cell pellet was resuspended in lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1% Triton, 0.1% Na Deoxycholate, 1 mM EDTA, and Protease Inhibitors). Cells were broken with zirconia beads 4 × 60 s at maximum speed in BeadBug homogenizer (Benchmark Scientific). After 15 min of centrifugation at maximum speed, the supernatant was discarded and the pellet (chromatin fraction) was resuspended in 400 μl of lysis buffer. The DNA was fragmented between 500 and 1,000 bp by sonication with Branson Sonifier 250. After clarification, the sonicated chromatin fraction was subjected to overnight immunoprecipitation with 80 μl anti‐FLAG beads (Sigma, A2220). The beads were then washed 1× with lysis buffer and 5× with wash buffer (10 mM Tris–HCl pH8.0, 250 mM LiCl, 0.75% NP‐40, 0.75% Na Deoxycholate, 1 mM EDTA). Protein‐DNA complex was eluted with 50 μl ChIP elution buffer (50 mM Tris–HCl pH8.0, 10 mM EDTA, 1% SDS), and crosslinking was reversed by incubating the eluate overnight at 65°C. DNA was purified using QIAGEN PCR purification kit (Cat. No. 28104). One microliter of the sample was loaded on an NP80 Spectrophotometer (Implen GmbH, Germany), and the concentration was determined by absorbance at 260 nm. To compare the ChIP‐DNA yield between replicates, the measured DNA concentrations were normalized to the concentrations from the arrested cells of the same experiment.
Author contributions
Deniz Irvali: Formal analysis; investigation; visualization; methodology; writing – original draft. Fabian P Schlottmann: Formal analysis; investigation; visualization; methodology; writing – review and editing. Prathibha Muralidhara: Investigation; writing – review and editing. Iliya Nadelson: Software; visualization; writing – review and editing. Katja Kleemann: Investigation. N Ezgi Wood: Software. Andreas Doncic: Resources; software. Jennifer C Ewald: Conceptualization; software; formal analysis; supervision; funding acquisition; investigation; visualization; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
PDF+
Acknowledgements
We thank the Linda Breeden, Kurt Schmoller, Jan Skotheim, Serge Pelet, Orlando Arguello‐Miranda, and Francesc Posas labs for strains and plasmids. We thank Lisa Dengler, Denise Dewald, Kurt Schmoller, Jan Skotheim, and Sege Pelet for helpful discussions. We gratefully acknowledge Tina Schneider and Stefanie Dommel for technical support. JCE gratefully acknowledges funding by the Institutional Strategy of the University of Tübingen (Deutsche Forschungsgemeinschaft ZUK 63) and from the Deutsche Forschungsgemeinschaft (Project 391105827). Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2023) 42: e110321
Data availability
Representative raw imaging data for each figure from this publication have been deposited to the Bioimage Archive database https://www.ebi.ac.uk/biostudies/studies/S‐BIAD449 and assigned the identifier S‐BIAD449. All raw data and all strains and constructs from this study are available from the corresponding author upon request.
References
- Adrover MA, Zi ZK, Duch A, Schaber J, Gonzalez‐Novo A, Jimenez J, Nadal‐Ribelles M, Clotet J, Klipp E, Posas F (2011) Time‐dependent quantitative multicomponent control of the G(1)‐S network by the stress‐activated protein kinase Hog1 upon osmostress. Sci Signal 4: ra63 [DOI] [PubMed] [Google Scholar]
- Alberghina L, Rossi RL, Querin L, Wanke V, Vanoni M (2004) A cell sizer network involving Cln3 and Far1 controls entrance into S phase in the mitotic cycle of budding yeast. J Cell Biol 167: 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argüello‐Miranda O, Marchand AJ, Kennedy T, Russo MAX, Noh J (2021) Cell cycle–independent integration of stress signals by Xbp1 promotes Non‐G1/G0 quiescence entry. J Cell Biol 221: e202103171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymoz D, Wosika V, Durandau E, Pelet S (2016) Real‐time quantification of protein expression at the single‐cell level via dynamic protein synthesis translocation reporters. Nat Commun 7: 11304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baro B, Jativa S, Calabria I, Vinaixa J, Bech‐Serra JJ, de LaTorre C, Rodrigues J, Hernaez ML, Gil C, Barcelo‐Batllori S et al (2018) SILAC‐based phosphoproteomics reveals new PP2A‐Cdc55‐regulated processes in budding yeast. Gigascience 7: giy047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RA (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O et al (2010) Phosphoproteomic analysis reveals interconnected system‐wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal 3: rs4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach JR (2012) Nutritional control of growth and development in yeast. Genetics 192: 73–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, Meyer T (2016) Irreversible APC(Cdh1) inactivation underlies the point of no return for cell‐cycle entry. Cell 166: 167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YL, Tseng SF, Huang YC, Shen ZJ, Hsu PH, Hsieh MH, Yang CW, Tognetti S, Canal B, Subirana L et al (2017) Yeast Cip1 is activated by environmental stress to inhibit Cdk1‐G1 cyclins via Mcm1 and Msn2/4. Nat Commun 8: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charvin G, Oikonomou C, Siggia ED, Cross FR (2010) Origin of irreversibility of cell cycle start in budding yeast. PLoS Biol 8: e1000284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan N, Visram M, Cristobal‐Sarramian A, Sarkleti F, Kohlwein SD (2015) Morphogenesis checkpoint kinase Swe1 is the executor of lipolysis‐dependent cell‐cycle progression. Proc Natl Acad Sci USA 112: E1077–E1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan N, Han G, Somashekarappa N, Gable K, Dunn T, Kohlwein SD (2016) Regulation of sphingolipid biosynthesis by the morphogenesis checkpoint kinase Swe1. J Biol Chem 291: 2524–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M (2004) CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117: 899–913 [DOI] [PubMed] [Google Scholar]
- Cross FR, Buchler NE, Skotheim JM (2011) Evolution of networks and sequences in eukaryotic cell cycle control. Philos Trans R Soc Lond B Biol Sci 366: 3532–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RA, McDonald WH, Kalashnikova TI, Yates J 3rd, Wittenberg C (2004) Cln3 activates G1‐specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117: 887–898 [DOI] [PubMed] [Google Scholar]
- Doncic A, Falleur‐Fettig M, Skotheim JM (2011) Distinct interactions select and maintain a specific cell fate. Mol Cell 43: 528–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Eser U, Atay O, Skotheim JM (2013) An algorithm to automate yeast segmentation and tracking. PLoS One 8: e57970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duch A, Canal B, Barroso SI, Garcia‐Rubio M, Seisenbacher G, Aguilera A, de Nadal E, Posas F (2018) Multiple signaling kinases target Mrc1 to prevent genomic instability triggered by transcription‐replication conflicts. Nat Commun 9: 379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escote X, Zapater M, Clotet J, Posas F (2004) Hog1 mediates cell‐cycle arrest in G1 phase by the dual targeting of Sic1. Nat Cell Biol 6: 997 [DOI] [PubMed] [Google Scholar]
- Ewald JC (2018) How yeast coordinates metabolism, growth and division. Curr Opin Microbiol 45: 1–7 [DOI] [PubMed] [Google Scholar]
- Ewald JC, Kuehne A, Zamboni N, Skotheim JM (2016) The yeast cyclin‐dependent kinase routes carbon fluxes to fuel cell cycle progression. Mol Cell 62: 532–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick KM, Spielewoy N, Kalashnikova TI, Guaderrama M, Zhu QZ, Chang HC, Wittenberg C (2003) Grr1‐dependent inactivation of Mth1 mediates glucose‐induced dissociation of Rgt1 from HXT gene promoters. Mol Biol Cell 14: 3230–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DA, Yellen P, Xu L, Saqcena M (2010) Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient‐sensing cell growth checkpoint(s). Genes Cancer 1: 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman A, Roy J, Bodenmiller B, Wanka S, Landry CR, Aebersold R, Cyert MS (2014) The calcineurin signaling network evolves via conserved kinase‐phosphatase modules that transcend substrate identity. Mol Cell 55: 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Novo A, Jimenez J, Clotet J, Nadal‐Ribelles M, Cavero S, de Nadal E, Posas F (2015) Hog1 targets Whi5 and Msa1 transcription factors to downregulate cyclin expression upon stress. Mol Cell Biol 35: 1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez JI, Brittingham GP, Karadeniz Y, Tran KD, Dutta A, Holehouse AS, Peterson CL, Holt LJ (2022) SWI/SNF senses carbon starvation with a pH‐sensitive low‐complexity sequence. Elife 11: e70344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SL, Enciso G, Dephoure N, Gygi SP, Gunawardena J, Kellogg DR (2011) A phosphatase threshold sets the level of Cdk1 activity in early mitosis in budding yeast. Mol Biol Cell 22: 3595–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich S, Hondele M, Marchand D, Derrer CP, Zedan M, Oswald A, Uliana F, Mancini R, Grunwald D, Weis K (2022) Condensation of a nuclear mRNA export factor regulates mRNA transport during stress. bioRxiv 10.1101/2022.01.30.478372 [PREPRINT] [DOI]
- Huang HY, Hopper AK (2014) Separate responses of karyopherins to glucose and amino acid availability regulate nucleocytoplasmic transport. Mol Biol Cell 25: 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A, Skotheim JM (2013) Start and the restriction point. Curr Opin Cell Biol 25: 717–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner RP, Tang JH, Helenius J, Dultz E, Brune C, Holt LJ, Huet S, Muller DJ, Weis K (2016) A glucose‐starvation response regulates the diffusion of macromolecules. Elife 5: e09376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita‐Kikuta E, Takiyama K, Koike T (2006) Phosphate‐binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5: 749–757 [DOI] [PubMed] [Google Scholar]
- Koivomagi M, Swaffer MP, Turner JJ, Marinov G, Skotheim JM (2021) G1 cyclin‐Cdk promotes cell cycle entry through localized phosphorylation of RNA polymerase II. Science 374: 347–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte D, Lebaudy A, Sahin A, Pinson B, Ceschin J, Daignan‐Fornier B, Sagot I (2011) Metabolic status rather than cell cycle signals control quiescence entry and exit. J Cell Biol 192: 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litsios A (2017) Metabolic‐rate dependent cell cycle entry and progression in Saccharomyces cerevisiae . University of Groningen
- Liu X, Wang X, Yang X, Liu S, Jiang L, Qu Y, Hu L, Ouyang Q, Tang C (2015) Reliable cell cycle commitment in budding yeast is ensured by signal integration. Elife 4: e03977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles S, Li L, Davison J, Breeden LL (2013) Xbp1 directs global repression of budding yeast transcription during the transition to quiescence and is important for the longevity and reversibility of the quiescent state. PLoS Genet 9: e1003854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles S, Croxford MW, Abeysinghe AP, Breeden LL (2016) Msa1 and Msa2 modulate G1‐specific transcription to promote G1 arrest and the transition to quiescence in budding yeast. PLoS Genet 12: e1006088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU, Bakkenist CJ (2019) WEE1 kinase inhibitor AZD1775 induces CDK1 kinase‐dependent origin firing in unperturbed G1‐ and S‐phase cells. Proc Natl Acad Sci USA 116: 23891–23893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro PT, Oliveira J, Pais P, Antunes M, Palma M, Cavalheiro M, Galocha M, Godinho CP, Martins LC, Bourbon N et al (2020) YEASTRACT plus: a portal for cross‐species comparative genomics of transcription regulation in yeasts. Nucleic Acids Res 48: D642–D649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO (2007) The cell cycle: principles of control. London: Primers in Biology New Science Press; [Google Scholar]
- Morshed S, Shibata T, Naito K, Miyasato K, Takeichi Y, Takuma T, Tasnin MN, Ushimaru T (2020) TORC1 regulates G1/S transition and cell proliferation via the E2F homologs MBF and SBF in yeast. Biochem Biophys Res Commun 529: 846–853 [DOI] [PubMed] [Google Scholar]
- Ondracka A, Robbins JA, Cross FR (2016) An APC/C‐Cdh1 biosensor reveals the dynamics of Cdh1 inactivation at the G1/S transition. PLoS One 11: e0159166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottoz DS, Rudolf F, Stelling J (2014) Inducible, tightly regulated and growth condition‐independent transcription factor in Saccharomyces cerevisiae . Nucleic Acids Res 42: e130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F et al (2021) The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 30: 187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Salloum D, Saqcena M, Chatterjee A, Mroz V, Ohh M, Foster DA (2017) A late G1 lipid checkpoint that is dysregulated in clear cell renal carcinoma cells. J Biol Chem 292: 936–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennycook BR, Barr AR (2020) Restriction point regulation at the crossroads between quiescence and cell proliferation. FEBS Lett 594: 2046–2060 [DOI] [PubMed] [Google Scholar]
- Pirincci Ercan D, Chretien F, Chakravarty P, Flynn HR, Snijders AP, Uhlmann F (2021) Budding yeast relies on G1 cyclin specificity to couple cell cycle progression with morphogenetic development. Sci Adv 7: eabg0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Jiang J, Liu X, Wei P, Yang X, Tang C (2019) Cell cycle inhibitor Whi5 records environmental information to coordinate growth and division in yeast. Cell Rep 29: 987–994 [DOI] [PubMed] [Google Scholar]
- Raspelli E, Facchinetti S, Fraschini R (2018) Swe1 and Mih1 regulate mitotic spindle dynamics in budding yeast via Bik1. J Cell Sci 131: jcs213520 [DOI] [PubMed] [Google Scholar]
- Schmoller KM, Turner JJ, Koivomagi M, Skotheim JM (2015) Dilution of the cell cycle inhibitor Whi5 controls budding‐yeast cell size. Nature 526: 268–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR (2008) Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454: 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smets B, Ghillebert R, De Snijder P, Binda M, Swinnen E, De Virgilio C, Winderickx J (2010) Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae . Curr Genet 56: 1–32 [DOI] [PubMed] [Google Scholar]
- Taberner FJ, Quilis I, Igual JC (2009) Spatial regulation of the start repressor Whi5. Cell Cycle 8: 3010–3018 [PubMed] [Google Scholar]
- Talarek N, Gueydon E, Schwob E (2017) Homeostatic control of START through negative feedback between Cln3‐Cdk1 and Rim15/Greatwall kinase in budding yeast. Elife 6: e26233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JJ, Ewald JC, Skotheim JM (2012) Cell size control in yeast. Curr Biol 22: R350–R359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen W, Rabouille C (2019) Cellular stress leads to the formation of membraneless stress assemblies in eukaryotic cells. Traffic 20: 623–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MV, Smolka MB, de Bruin RA, Zhou H, Wittenberg C, Dowdy SF (2009) Whi5 regulation by site specific CDK‐phosphorylation in Saccharomyces cerevisiae . PLoS One 4: e4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nurse P, Broek D (1993) Yeast cells can enter a quiescent state through G1, S, G2, or M phase of the cell cycle. Cancer Res 53: 1867–1870 [PubMed] [Google Scholar]
- Wood NE, Doncic A (2019) A fully‐automated, robust, and versatile algorithm for long‐term budding yeast segmentation and tracking. PLoS One 14: e0206395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood NE, Kositangool P, Hariri H, Marchand AJ, Henne WM (2020) Nutrient signaling, stress response, and inter‐organelle communication are non‐canonical determinants of cell fate. Cell Rep 33: 108446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Cappell SD, Jaimovich A, Liu C, Chung M, Daigh LH, Pack LR, Fan Y, Regot S, Covert M et al (2020) Stress‐mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. Elife 9: e44571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Winkler S, Schlottmann FP, Kohlbacher O, Elias JE, Skotheim JM, Ewald JC (2019) Multiple layers of phospho‐regulation coordinate metabolism and the cell cycle in budding yeast. Front Cell Dev Biol 7: 338 [DOI] [PMC free article] [PubMed] [Google Scholar]