

Abstract
The AKT‐mTOR pathway is a central regulator of cell growth and metabolism. Upon sustained mTOR activity, AKT activity is attenuated by a feedback loop that restrains upstream signaling. However, how cells control the signals that limit AKT activity is not fully understood. Here, we show that MASTL/Greatwall, a cell cycle kinase that supports mitosis by phosphorylating the PP2A/B55 inhibitors ENSA/ARPP19, inhibits PI3K‐AKT activity by sustaining mTORC1‐ and S6K1‐dependent phosphorylation of IRS1 and GRB10. Genetic depletion of MASTL results in an inefficient feedback loop and AKT hyperactivity. These defects are rescued by the expression of phosphomimetic ENSA/ARPP19 or inhibition of PP2A/B55 phosphatases. MASTL is directly phosphorylated by mTORC1, thereby limiting the PP2A/B55‐dependent dephosphorylation of IRS1 and GRB10 downstream of mTORC1. Downregulation of MASTL results in increased glucose uptake in vitro and increased glucose tolerance in adult mice, suggesting the relevance of the MASTL‐PP2A/B55 kinase‐phosphatase module in controlling AKT and maintaining metabolic homeostasis.
Keywords: AKT, cell cycle, glucose homeostasis, MASTL, mTOR
Subject Categories: Cell Cycle, Metabolism, Signal Transduction
The MASTL/Greatwall kinase is a new component of the feedback loop that restricts mTOR pathway activity in the presence of excessive signaling.
Introduction
Cellular responses to growth factors such as insulin are transduced into the cell through multiple signaling cascades including the PI3K‐AKT pathway (Efeyan et al, 2015). Upon stimulation, the PI3K‐AKT pathway activates the mechanistic target of rapamycin (mTOR) signaling through AKT‐dependent phosphorylation of the tuberous sclerosis complex 2 (TSC2, tuberin) protein. Upon activation, the mTORC1 complex phosphorylates critical regulators of ribosomes and translation such as the ribosomal S6 kinases (S6K1 and S6K2) and the eukaryotic translation factor 4E (eIF4E)‐binding protein 1 (4E‐BP1), thus driving the synthesis of proteins required for cell growth and proliferation (Laplante & Sabatini, 2012; Dibble & Cantley, 2015; Gonzalez & Hall, 2017; Saxton & Sabatini, 2017).
The mTORC1‐S6K1 axis also mediates potent feedback loops that restrain upstream signaling through insulin/IGF receptor. S6K1 inhibits insulin receptor substrate‐1 (IRS1) through priming phosphorylation that sensitizes IRS1 to further inhibitory phosphorylation by other kinases (Harrington et al, 2004). TSC1/2‐deficient cells are characterized by insulin/IGF‐1 resistance as a consequence of constitutive downregulation of IRS1 and IRS2 due to sustained mTOR activity (Harrington et al, 2004; Shah et al, 2004). Additional IRS1‐independent feedbacks have been proposed, including the mTORC1‐dependent phosphorylation of the growth factor receptor‐bound protein‐10 (GRB10), a negative regulator of insulin and IGF‐1 signaling (Hsu et al, 2011; Yu et al, 2011). Pharmacological inhibition of mTOR relieves the feedback inhibition of AKT (Harrington et al, 2004; Shah et al, 2004), limiting the efficacy of therapeutic strategies aimed to prevent mTOR signaling (O'Reilly et al, 2006; Tabernero et al, 2008).
Although the involvement of phosphatases in this feedback has been far less studied, evidence suggests that the action of mTOR in the phosphorylation and degradation of IRS1 results from the inhibition of PP2A (Hartley & Cooper, 2002). In fact, PP2A is able to dephosphorylate IRS1 (Clark et al, 2000) and inhibition of PP2A is sufficient to induce IRS1 degradation (Hartley & Cooper, 2002). The Serine/threonine kinase MASTL (also known as Greatwall) is a critical inhibitor of PP2A complexes during mitosis (Castilho et al, 2009; Vigneron et al, 2009; Glover, 2012). MASTL is activated by cyclin‐dependent kinase 1 (CDK1) complexes during mitotic entry leading to the phosphorylation of the endosulfines, endosulfine α (ENSA), and the 19 kD cAMP‐regulated phosphoprotein (ARPP19) (Gharbi‐Ayachi et al, 2010; Mochida et al, 2010). When phosphorylated, these small molecules function as specific inhibitors of phosphatase complexes in which PP2A activity is modulated by the B55 family of regulatory proteins (B55α, β, γ, and δ). Loss of MASTL results in mitotic defects such as defective chromosome condensation and segregation errors in Drosophila and mammalian cells (Burgess et al, 2010; Alvarez‐Fernandez et al, 2013; Wang et al, 2013; Diril et al, 2016).
In this manuscript, we report a mitotic‐independent function of the MASTL‐ENSA/ARPP19‐PP2A‐B55 axis in modulating the response to glucose and the feedback loops induced by sustained mTOR‐S6K1 activity. In conditions of nutrient excess and high mTOR signaling, MASTL inhibits PP2‐B55 activity thereby preventing the dephosphorylation of the feedback targets IRS‐1 and GRB10. These observations identify a new layer of control that interconnects a cell cycle module with the negative feedbacks regulating the AKT‐mTOR pathway, and suggest the possible use of MASTL inhibition to specifically limit the effects of these mTOR‐S6K1‐mediated feedback loops.
Results
MASTL regulates AKT inhibition by the mTORC1‐S6K1 axis
To study the effect of the MASTL‐ENSA/ARPP19‐PP2A‐B55 pathway in nutrient signaling, we first knocked down MASTL expression using specific short‐hairpin RNA (shMASTL) sequences in different human cell lines. Since MASTL inactivation may induce cell division defects, these experiments were performed shortly (72 h) after the induction of protein knockdown to avoid any interference with mitotic aberrations (Fig EV1A). Starvation of MDA‐MB‐231 cells for either growth factors or nutrients inhibited mTORC1 activity in control cells infected with a scrambled shRNA, as shown by decreased phosphorylation of its substrates S6K1 and 4E‐BP1 (Fig EV1B). However, MASTL knockdown resulted in increased phosphorylation of S6K1‐T389, and increased phosphorylation of S6 and 4E‐BP, especially in glucose‐starved cells. These signals were accompanied by a significant increase in the inhibitory phosphorylation of TSC2 on T1462 (Fig EV1B), suggesting that MASTL‐depleted cells are largely resistant to inhibit mTORC1 pathway in response to glucose deprivation.
Figure EV1. MASTL modulates AKT activity in response to glucose and insulin independently of its mitotic function.
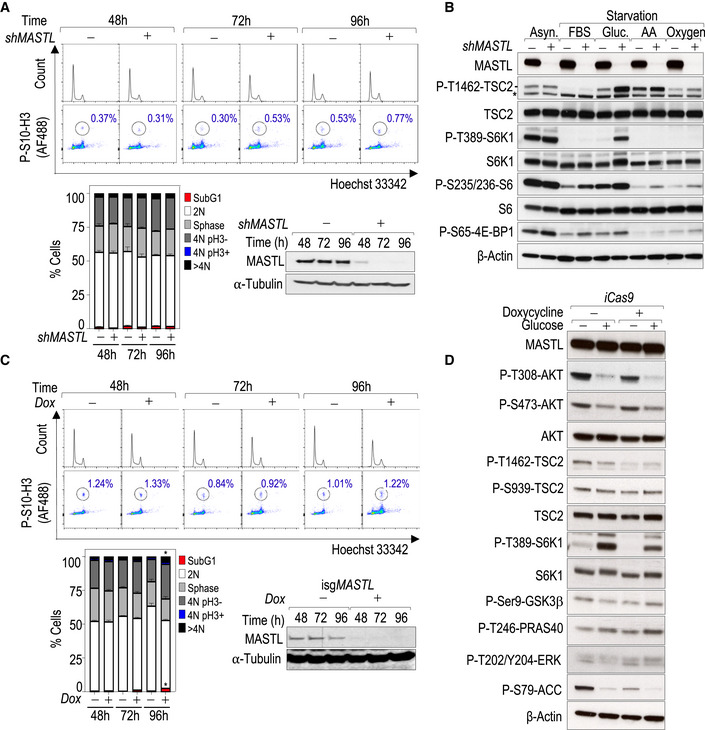
- Cell cycle analysis in RNA interference MASTL‐depleted cells. MDA‐MB‐231 cells were infected with shRNAs specific for MASTL (+) or scramble (−) as a control, and the cell cycle was analyzed 48, 72, and 96 h later. Mitotic cells were scored using immunodetection of P‐S10‐H3 (pH3) and total DNA was stained with Hoechst 33342 followed by flow cytometry analysis. The plot shows the quantification of the different phases of the cell cycle in control and MASTL‐depleted cells. Data are mean + SEM of three independent experiments. Significance was determined by two‐way ANOVA comparing MASTL‐depleted cells versus control cells (not significant at any time point). Representative immunoblotting to monitor MASTL depletion is also shown. α‐Tubulin was used as a loading control.
- Immunoblot analysis of asynchronous (Asyn.) MDA‐MB‐231 cells subjected to different starvations [elimination of fetal bovine serum (FBS), glucose (Gluc.), amino acids (AA), or oxygen] using the indicated antibodies in the absence (−) or presence (+) of shRNAs against MASTL. *Indicates unspecific band. β‐Actin was used as a loading control.
- Cell cycle analysis in inducible CRISPR‐Cas9 MASTL‐deleted cells. MASTL was ablated in MDA‐MB‐231 cells using the isgMASTL system, and cell cycle was analyzed 48, 72, and 96 h after doxycycline treatment. Mitotic cells were scored using immunodetection of P‐S10‐H3 (pH3) and total DNA was stained with Hoechst 33342 followed by flow cytometry analysis. The graph shows the quantification of the different phases of the cell cycle in control and MASTL‐depleted cells. Data are mean + SEM of three independent experiments. Significance was determined by two‐way ANOVA comparing MASTL‐depleted cells versus control cells (*P < 0.05 at 96 h for subG1 and > 4N). Representative immunoblotting to monitor MASTL depletion is also shown. α‐Tubulin was used as a loading control.
- Control of the Dox and Cas9 expression in the MDA‐MB‐231 isgMASTL system using a control clone that expresses Cas9 alone (iCas9). The same protocol as in Fig 1B was followed.
We next switched to an inducible CRISPR system recently established (isgMASTL) in which Cas9 was transcriptionally induced by doxycycline (Dox) in the presence of specific single‐guide RNAs (sgRNAs) against human MASTL sequences (Alvarez‐Fernandez et al, 2018). These assays were also performed at early time points after doxycycline addition (48–72 h) when no cell cycle defects are yet detected in MASTL‐deleted cells (Fig EV1C). In the presence of doxycycline, MASTL was efficiently ablated resulting in increased S6K1 T389 and TSC2 T1462 phosphorylation upon glucose deprivation (Fig 1A and B). In line with these observations, the TSC2‐inhibitory kinase AKT was strongly activated in MASTL‐null cells, as shown by increased phosphorylation on T308 and S473 residues. Other AKT substrates, like PRAS40, or GSK3, were also more phosphorylated in the absence of MASTL. No significant differences were observed in the phosphorylation of ERK1/2, another positive regulator of the mTORC1 pathway upstream of TSC2, or AMPK substrates such as ACC and Raptor, in these conditions (Fig 1B). The effect of MASTL ablation on the AKT‐TSC2‐S6K1 axis was specific as the induction of Cas9 alone in control cells did not modify the phosphorylation status of the components of the AKT‐S6K1 pathway (Fig EV1D).
Figure 1. MASTL modulates the AKT‐mTORC1 signaling pathway.
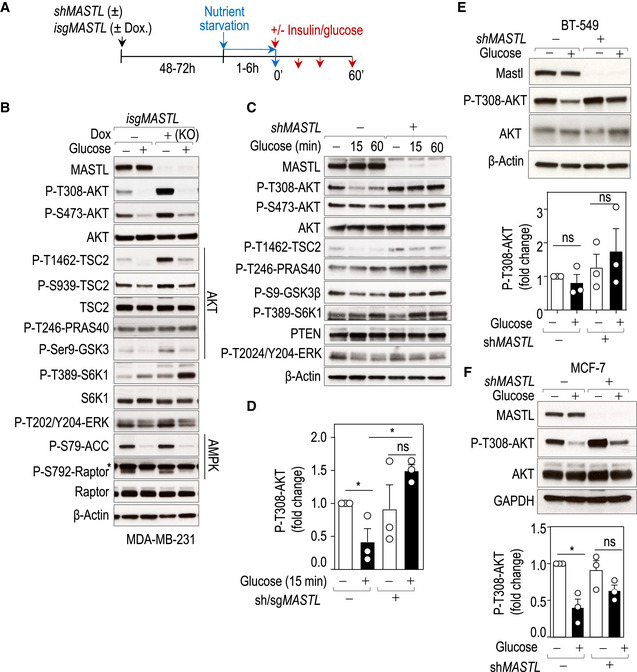
- Schematic representation of the protocol followed for the depletion of MASTL in human cell lines and different nutrient starvations. MASTL was knocked down (RNA interference; shMASTL) or knocked out (inducible CRISPR/Cas9; isgMASTL) and cells were nutrient starved for 1–6 h before analysis (time 0′). In case of re‐stimulation with glucose or insulin, cells were analyzed 10–60 min after re‐stimulation.
- MASTL was ablated in MDA‐MB‐231 cells using the isgMASTL system. Seventy‐two hours after Dox, cells were starved of glucose in the media in the presence of 10% dFBS for 1 h (−) and then re‐stimulated with 25 mM of glucose for 10 min before recovery (+). Whole‐cell lysates were blotted with the indicated antibodies. *Indicates unspecific band. β‐Actin was used as a loading control.
- MASTL was depleted in MDA‐MB‐231 cells using shRNAs against MASTL (+) or scrambled shRNAs (−) as control. Cells were starved from glucose for 2 h and re‐stimulated with 5 mM glucose for 15 min and 1 h. Total extracts were recovered and tested for the indicated antibodies. β‐Actin was used as a loading control.
- Quantification of phospho‐AKT T308 levels in glucose deprivation or glucose stimulation (15 min) conditions in MDA‐MB‐231 control cells and MASTL‐depleted cells either using shRNA or the inducible sgRNA. Bars displays mean data + SEM from three independent experiments. Significance determined by Student's t‐test comparing feedback activation in MASTL‐depleted cells versus control cells (ns, not significant; *P < 0.05).
- Immunoblot analysis with the indicated antibodies in BT‐549 cells upon glucose deprivation and 15 min glucose stimulation. MASTL knockdown was performed by infection with an shRNA for MASTL (+) or a scramble shRNA (−) as a control. β‐Actin was used as a loading control. The bar chart displays mean data + SEM from three experiments. Significance determined by Student's t‐test comparing feedback activation in MASTL‐depleted cells versus control cells (ns, not significant).
- Immunoblot analysis with the indicated antibodies in MCF‐7 cells upon glucose deprivation and glucose stimulation for 15 min. MASTL knockdown was performed by infection with an shRNA for MASTL (+) or a scramble shRNA (−) as a control. The bar chart displays mean data + SEM from three experiments. Significance determined by Student's t‐test comparing feedback activation in MASTL‐depleted cells versus control cells (ns, not significant; *P < 0.05).
How glucose regulates AKT signaling is not well understood. However, it has been shown that chronic activation of mTORC1 by glucose results in decreased AKT phosphorylation (Briaud et al, 2005). In a time‐course analysis of glucose stimulation, control cells responded to this glucose‐mediated feedback loop by inhibiting AKT activity. In contrast, MASTL‐null cells failed to reduce AKT T308 phosphorylation, despite having enhanced mTORC1 activity as shown by phospho‐S6K1 T389 (Fig 1C and D). Importantly, the effect of MASTL knockdown in the phosphorylation of AKT in response to glucose was also confirmed in two additional independent cell lines, BT‐549 and MCF‐7 (Fig 1E and F).
We next analyzed the role of MASTL in the regulation of AKT activity following mTORC1/S6K1 activation by insulin in a time‐course experiment (Fig 2A). PI3K/AKT‐mediated activation of mTORC1 is known to trigger a negative feedback loop ultimately leading to the attenuation of insulin signaling (Haruta et al, 2000; Greene et al, 2003; Carlson et al, 2004). Genetic ablation of MASTL did not modify the initial rise in AKT activity but prevented its downregulation after 30 min, despite having comparable levels of mTORC1 activity, resulting in sustained TSC2 and S6K1 phosphorylation (Fig 2A). A similar effect of MASTL knockdown was observed in a hepatocarcinoma‐derived cell line that also responds to insulin (HepG2; Fig EV2A), as well as in non‐transformed murine myoblasts (C2C12; Fig EV2B).
Figure 2. MASTL controls AKT phosphorylation in response to insulin signaling.
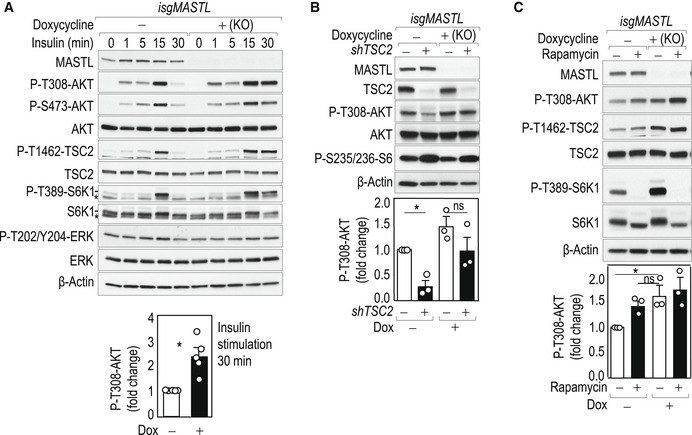
- Time‐course experiment at the indicated time points after 100 nM insulin stimulation in 6 h serum‐starved control and MASTL knockout MDA‐MB‐231 cells using the isgMASTL system. The plot shows the quantification of phospho‐AKT T308 after 30 min of insulin stimulation. Plots display mean data + SEM from five independent experiments. *P < 0.05, Student's t‐test.
- TSC2 was knocked down in control or MASTL‐null MDA‐MB‐231 cells using specific (+) or scrambled (−) shRNAs. Cells were starved for glucose for 1 h. The plot shows the quantification of phospho‐AKT T308. Data are mean + SEM from three independent experiments. ns, not significant; *P < 0.05, One‐way ANOVA.
- MDA‐MB‐231 control (−) or MASTL‐null (+) cells were treated with 100 nM rapamycin or vehicle as control. Cells were starved for glucose for 1 h and re‐stimulated with glucose for 15 min; rapamycin was added 15 min before glucose stimulation. The plot shows the quantification of phospho‐AKT T308. Data are mean + SEM from three independent experiments. ns, not significant; *P < 0.05, one‐way ANOVA.
Figure EV2. MASTL modulates AKT activity in response to insulin.
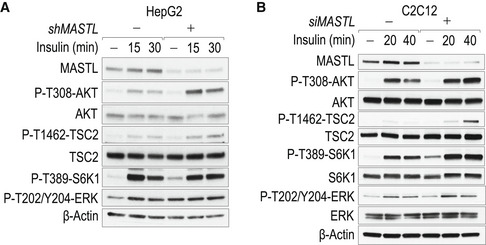
- HepG2 cells were depleted of MASTL, and serum was starved and stimulated with insulin for the indicated periods of time, following the protocol shown in Fig 1A. Whole‐cell lysates were probed with the indicated antibodies.
- Mouse C2C12 myoblasts were depleted of Mastl using specific siRNAs. Forty‐eight hours after transfection, cells were placed in differentiation medium for 24 h (2% horse serum), serum‐starved for 5 h, and stimulated with insulin for the indicated periods of time. Whole‐cell lysates were probed with the indicated antibodies.
Knockdown of TSC2 in MDA‐MB231 cells using specific shRNAs triggered activation of the feedback loop and AKT inhibition in control cells but not in MASTL‐depleted cells, in which AKT activity was maintained (Fig 2B). In line with these observations, acute mTORC1 inhibition by rapamycin treatment suppressed the effect of the negative feedback loop and boosted AKT phosphorylation (Fig 2C). Lack of MASTL mimicked the effect of rapamycin on AKT phosphorylation (Fig 2C), therefore suggesting a role for MASTL in the modulation of this feedback mechanism downstream of mTORC1.
MASTL depletion alters glucose metabolism in vitro and in vivo
The PI3K‐AKT pathway is a central regulator of insulin‐mediated glucose metabolism (Manning & Toker, 2017). Insulin stimulation following serum starvation increased GLUT4 translocation to the plasma membrane (PM), and MASTL knockdown further increased the percentage of cells positive for GLUT4 at the PM in this condition (Fig 3A). Similar results were obtained upon glucose starvation and re‐stimulation (Fig EV3), further supporting a role for MASTL in the regulation of AKT‐mediated GLUT4 translocation. In agreement with these data, depletion of MASTL led to a significant increase in glucose uptake compared with control cells (Fig 3B). Analysis of the extracellular medium indicated that both glucose consumption and lactate production were increased upon MASTL silencing but to the same extent so that the glycolytic index remained constant (Fig 3C and D). These results suggest that MASTL depletion does not induce a general metabolic switch, but rather a controlled and well‐defined upregulation of glycolysis under conditions of feedback‐mediated AKT regulation.
Figure 3. Metabolic alterations in MASTL‐depleted cells.
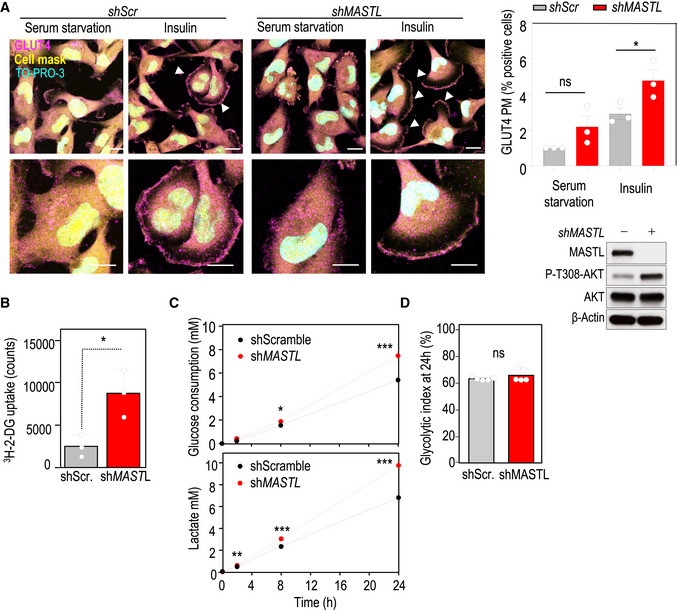
- Immunofluorescence for GLUT4 (magenta) in MDA‐MB‐231 cells in conditions of serum starvation and insulin stimulation. TO‐PRO‐3 (DNA) is in cyan and a cell mask is in yellow. Arrowheads indicate positive cells for GLUT4 enrichment at the plasma membrane (PM). Representative magnifications are also shown. Scale bars, 10 μm. The plot shows the quantification of the percentage of cells positive for GLUT4 translocation at the PM. Data are mean fold increase versus serum deprivation in scramble condition ± SEM from three independent experiments. ns, not significant; *P < 0.05, one‐way ANOVA. Control immunoblotting in conditions of insulin stimulation is also shown for the indicated antibodies.
- Cells were treated with specific shRNAs against MASTL (+) or scrambled shRNAs (−), and then serum starved before incubation with 2‐deoxy‐D‐[1‐3H]‐glucose. Bars show mean glucose uptake + SEM of three independent experiments. *P < 0.05, Student's t‐test.
- Control (shScr.) and MASTL‐depleted (shMASTL) HepG2 cells were incubated with 10 mM glucose and metabolites in the media were quantified by NMR at the indicated time points. The time course of glucose consumption (upper panel) and lactate production (lower panel) in cell media are indicated. Data are mean ± SD of five independent plates. *P < 0.05; **P < 0.01, ***P < 0.001, unpaired Student's t‐test.
- Glycolytic index at 24 h comparing control and MASTL‐depleted cells treated as in C calculated as −0.5 × ([Lac]/([Glc]0 − [Glc]). Data are mean ± SD of five independent plates. ns, not significant, unpaired Student's t‐test.
Figure EV3. Effect of MASTL depletion on cellular metabolism.
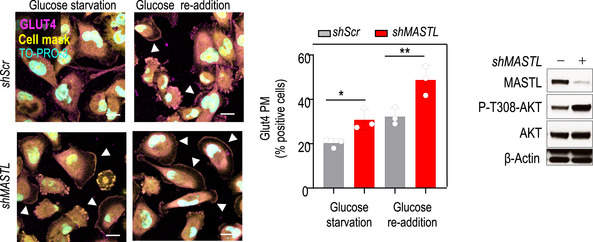
MASTL was knocked down using shRNAs for MASTL (+) and scramble shRNA (−) as a control in MDA‐MB‐231 cells, and immunofluorescence for GLUT4 (magenta) in conditions of glucose starvation and re‐addition was performed. TO‐PRO‐3 (DNA) is in cyan and a cell mask is in yellow. Arrowheads indicate positive cells for GLUT4 enrichment at the plasma membrane (PM). Scale bars, 10 μm. The graph shows the quantification of the percentage of cells positive for GLUT4 at the PM. The bar chart displays mean data ± SEM from thee independent experiments. Significance was determined by one‐way ANOVA (*P < 0.05; **P < 0.01). Control immunoblotting is shown in conditions of glucose re‐addition for the indicated antibodies.
Mastl is expressed in heart, skeletal muscle, white adipose tissue (WAT), and liver mouse tissues at comparable levels to the spleen, indicating that expression of this kinase is not restricted to proliferative tissues (Fig 4A). Interestingly, Mastl expression was modulated in vivo in response to food intake in muscle, epididymal WAT, and liver, in which Mastl mRNA levels increased in ad libitum‐fed mice compared to overnight fasted mice (Fig 4B), further suggesting a non‐mitotic role for Mastl in these tissues.
Figure 4. Mastl regulates glucose homeostasis in vivo .
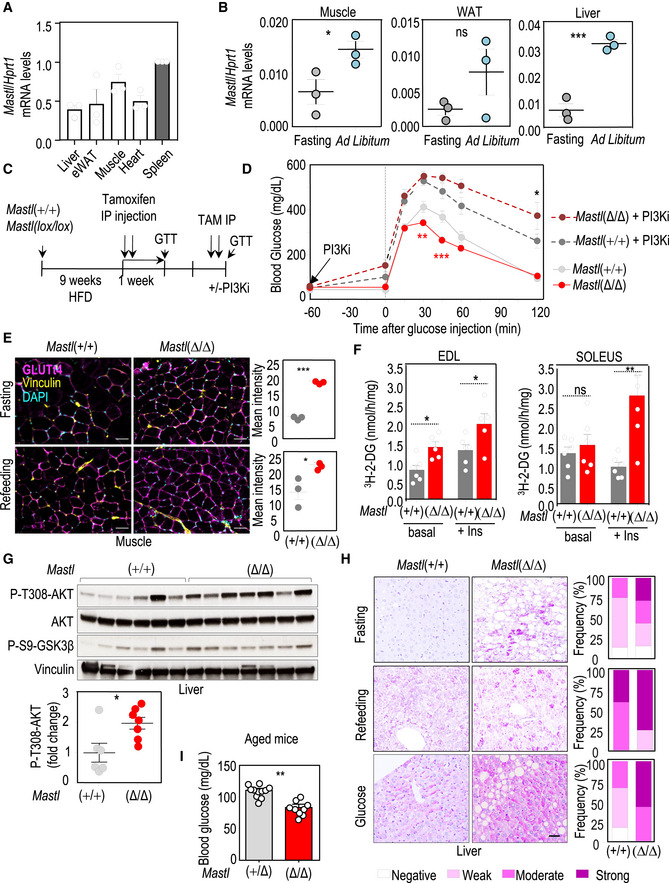
- RT–qPCR analysis of Mastl transcripts in different tissues from ad libitum mice. The graph represents the average + SEM of normalized Mastl mRNA level from three mice. Hprt1 was used as a housekeeping gene to normalize the Mastl expression level in each tissue.
- Relative levels of Mastl transcripts (normalized to Hprt mRNA levels) in muscle, epididymal white adipose tissue (WAT) and liver from wild‐type mice after fasting for 16 h or fed ad libitum. Data are mean ± SEM; ns, not significant; *P < 0.05; ***P < 0.001, Student's t‐test.
- Schematic representation of the protocol followed for the glucose tolerance test (GTT).
- Glucose tolerance test in Mastl(+/+) (n = 6) and Mastl(Δ/Δ) (n = 11) mice. Eight‐ to twelve‐week‐old male mice were fed high‐fat diet (HFD; 60% fat) for 9 weeks before being treated with tamoxifen to induce Mastl deletion. Treatment of Mastl(+/+) (n = 5) and Mastl(Δ/Δ) (n = 10) mice with the PI3K inhibitor ETP46992 (PI3Ki) was performed 1 h before glucose injection. Data are mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; two‐way ANOVA.
- GLUT4 staining (magenta) in skeletal muscle sections. Vinculin (membrane) and DAPI (DNA) staining are shown in yellow and cyan, respectively. Scale bars, 50 μm. Mice were fasted overnight for 16 h, and re‐fed for 2 h before sample collection. Quantification of the mean intensity [relative units ×103; n = 3 Mastl(+/+) and 3 Mastl(Δ/Δ)] is shown. Data points reflect the mean ± SEM; *P < 0.05; ***P < 0.001; unpaired Student's t‐test.
- Ex vivo glucose uptake in EDL and soleus muscles from Mastl(+/+) (n = 6) and Mastl(Δ/Δ) (n = 5) mice, in basal conditions and upon insulin stimulation (100 nM, 20 min). Mice were fasted overnight before muscle isolation, and data were normalized to muscle weight. Plots show the mean + SEM; ns, not significant; *P < 0.05; **P < 0.01; unpaired Student's t‐test.
- Immunoblot with the indicated antibodies in liver tissues from Mastl(+/+) (n = 6) and Mastl(Δ/Δ) (n = 7) mice. Mice were fasted overnight for 16 h, injected intraperitoneally with glucose (2 g/kg body), and sacrificed 30 min later for sample collection. Quantification of the relative fold change signal of phospho‐AKT T308 (normalized to total AKT level). n = 6 Mastl(+/+) and 7 Mastl(Δ/Δ) mice. Charts reflect the mean ± SEM; *P < 0.05; unpaired Student's t‐test.
- Representative images of periodic acid–Schiff (PAS) staining in liver sections from Mastl(+/+) and Mastl(Δ/Δ) mice. Scale bars, 20 μm. Mice were fasted overnight for 16 h, injected intraperitoneally with glucose (2 g/kg body), or re‐fed for 2 h, and sacrificed 30 min later for sample collection. Bars show the relative frequency of PAS staining intensity (n = 3 mice/genotype). Chi‐square test (***P < 0.001 in all conditions).
- Blood glucose levels in ad libitum Mastl(Δ/Δ) (n = 12) and Mastl(+/Δ) (n = 11) mice. Mice were treated with tamoxifen diet to induce Mastl depletion, and blood glucose concentration was determined 1 month later in the morning of ad libitum‐fed mice. Data are mean + SEM; **P < 0.01, unpaired Student's t‐test.
We directly tested the relevance of Mastl in glucose metabolism in vivo by using a Mastl conditional knockout model in which the murine Mastl gene can be excised by a tamoxifen‐induced Cre recombinase (Alvarez‐Fernandez et al, 2013). Although Mastl deletion results in embryonic lethality, adult Mastl(Δ/Δ) mice survived with no obvious phenotype until at least 5 months after continuous tamoxifen treatment, suggesting it is not an essential cell cycle kinase in adulthood (Fig EV4A). Young (8‐ to 12‐week‐old) control and Mastl(lox/lox) knockout mice were fed with high‐fat diet (HFD, 60% of calories derived from fat, ~4 kcal/kg) for 9 weeks before the induction of Mastl deletion by tamoxifen injection (Fig 4C). To avoid any interference with the potential proliferative defects induced by Mastl deletion, acute deletion was achieved in adult mice by intraperitoneal injection just before the metabolic assays. In these conditions, tamoxifen treatment and concomitant loss of Mastl did not affect the body weight of Mastl(Δ/Δ) compared to control Mastl(+/+) mice (Fig EV4B), and no significant defects were observed in highly proliferative tissues such as intestine (Fig EV4C). Although no statistical differences were observed in an insulin tolerance test (Fig EV4D), Mastl(Δ/Δ) mice displayed better glucose clearance (Fig 4D), Importantly, acute treatment with an orally bioavailable specific PI3K inhibitor known to inhibit AKT activity (Martínez González et al, 2012) completely rescued the observed differences in glucose clearance (Fig 4D). Of note, the effect of the inhibitor was even more pronounced in Mastl(Δ/Δ) mice that reached the highest and most sustained levels of hyperglycemia, compared to control mice (Fig 4D). These data also suggest that the enhanced glucose tolerance upon Mastl ablation might be a consequence of increased PI3K/AKT activity in Mastl(Δ/Δ) tissues responsible of glucose uptakes, such as muscle, WAT, or liver.
Figure EV4. Effect of MASTL ablation on glucose metabolism in vivo .
- Survival curve of adult Mastl(+/Δ) (n = 11) and Mastl(Δ/Δ) (n = 14) mice after continuous tamoxifen treatment. No significant differences were found in the log‐rank (Mantel–Cox) test.
- Plots representing the relative body weight of the mice included in the GTT assay from Fig 4. Body weight gain after 9 weeks of HFD (Left); body weight loss 1 week after tamoxifen injection (Middle); body weight loss after 16 h fasting (Right). n = 6 Mastl(+/+) and n = 11 Mastl(Δ/Δ). Error bars indicate SEM (unpaired student's t‐test).
- Representative images of the intestine from Mastl(+/+) and Mastl(Δ/Δ) mice showing the normal architecture of the epithelia by hematoxylin and eosin (H&E) staining (upper panel) and similar levels of proliferation (Ki67 staining, lower panels). Scale bars, 25 μm.
- Insulin tolerance test (ITT) in Mastl(+/+) (n = 6) and Mastl(Δ/Δ) (n = 11) mice. Data are mean ± SEM; ns, not significant; two‐way ANOVA.
- Immunoblot with the indicated antibodies in muscle tissues from Mastl(+/+) (n = 8) and Mastl(Δ/Δ) (n = 7) mice. Mice were fasted overnight for 16 h and sacrificed for sample collection. Quantification of the relative fold change signal of phospho‐AKT T308 and phospho‐AKT S473. Data are mean ± SEM; **P < 0.01; ***P < 0.001, unpaired Student's t‐test.
- RT–qPCR analysis of Mastl mRNA in liver tissues from Mastl(+/+) and Mastl(Δ/Δ) mice fasted overnight for 16 (n = 7 mice/genotype), and injected intraperitoneally with glucose (2 g/kg body) (n = 3 mice/genotype), or re‐fed for 2 h and sacrificed 30 min later for sample collection refeeding conditions (n = 4 mice/genotype). Hprt1 was used as a housekeeping gene to normalize Mastl expression level. Plots show the mean + SEM; ns, not significant; *P < 0.05; **P < 0.01, unpaired Student's t‐test.
- RT–qPCR analysis of Mastl mRNA in muscle tissues from Mastl(+/+) and Mastl(Δ/Δ) mice fasted overnight for 16 (n = 8 control mice and n = 5 Mastl KO mice), and injected intraperitoneally with glucose (2 g/kg body) (n = 4 control mice and n = 3 Mastl KO mice), or re‐fed for 2 h, and sacrificed 30 min later for sample collection refeeding conditions (n = 3 mice/genotype). Hprt1 was used as a housekeeping gene to normalize Mastl expression level. Plots show the mean + SEM; ns, not significant; *P < 0.05, unpaired Student's t‐test.
In agreement with these observations, expression of Glut4 at the plasma membrane (PM) increased in Mastl(Δ/Δ) muscles, most significantly in fasting conditions where the mTORC1 pathway is usually downregulated in control tissues, suggesting mTORC1 and Akt hyperactivity in the absence of Mastl (Fig 4E). Indeed, higher phosphorylation of Akt was detected in protein extracts from Mastl(Δ/Δ) muscles in fasting conditions (Fig EV4E). Since most insulin‐mediated glucose uptake occurs in skeletal muscle, isolated muscles derived from Mastl‐deficient mice were assessed for their capacity to uptake 3H‐2‐deoxy‐D‐glucose. As shown in Fig 4F, the rate of glucose uptake was significantly increased in Mastl(Δ/Δ) extensor digitorum longus (EDL) muscles in basal conditions, and even further upon insulin stimulation in both EDL and soleus muscles, representing fast‐ and slow‐twitch muscles, respectively (Fig 4F). These data are consistent with alleviation of mTORC1‐dependent feedback mechanisms affecting upstream insulin signaling in these muscles.
Phosphorylation of AKT was also increased in response to glucose stimulation in Mastl(Δ/Δ) livers (Fig 4G). AKT signaling in the liver promotes the conversion of glucose‐6‐phosphate into glycogen. Accordingly, liver sections from Mastl(Δ/Δ) mice showed a significant increase in periodic acid Schiff (PAS) staining, which detects glycogen and other polysaccharides, during fasting and also upon refeeding or glucose stimulation (Fig 4H).
Finally, 1‐year‐old Mastl(Δ/Δ) mice fed with chow diet ad libitum had a significantly reduced glycemia compared to control Mastl(+/Δ) littermates (Fig 4I), suggesting better glucose clearance in aged Mastl‐deficient mice. Altogether, these findings suggest that Mastl ablation improves glucose tolerance upon a stressful condition such as HFD, as well as basal glycemia in old mice.
MASTL controls AKT activity in an ENSA/ARPP19‐PP2A‐B55‐dependent manner
We next tested whether MASTL could regulate AKT‐mTORC1 signaling through the ENSA/ARPP19‐PP2A pathway. As shown in Fig 5A, the knockdown of ENSA and ARPP19 mimicked the effect of MASTL ablation and resulted in increased AKT phosphorylation thereby preventing the effect of glucose in the inactivation of AKT by the feedback loop. In addition, expression of ENSA and ARPP19 cDNAs with mutations that mimic MASTL‐dependent phosphorylation (ENSA‐S67D/ARPP19‐S62D) restored the downregulation of phospho‐AKT after MASTL ablation in conditions of glucose re‐addition (Fig 5B). The involvement of PP2A was then tested in similar assays using PP2A phosphatase inhibitors which we would expect to rescue the effect of MASTL inhibition. Although these inhibitors are not specific for PP2A/B55 complexes, treatment of cells with the general PP2A inhibitors fostriecin or okadaic acid led to decreased levels of AKT phosphorylation on T308 in conditions of insulin stimulation, partially restoring AKT inhibition in MASTL‐null cells (Fig 5C). Altogether these data indicate that the effect of MASTL on the regulation of AKT occurs through the ENSA/ARPP19‐mediated inhibition of PP2A.
Figure 5. MASTL mediates mTORC1/S6K1‐mediated AKT inhibition through the ENSA/ARPP19‐PP2A/B55 pathway.
- Co‐depletion of ENSA and ARPP19 proteins in the MDA‐MB‐231 cell line using specific siRNAs or control siRNAs against luciferase (Luc). Seventy‐two hours later transfection cells were starved for glucose for 1 h and re‐stimulated with glucose for 15 min. Total cell extracts were collected and analyzed for the indicated antibodies.
- isgMASTL cells were previously infected with lentiviral supernatants expressing FLAG‐ and HA‐tagged versions of ENSA and ARPP19 phosphomimetic mutants (S67D and S62D) or empty vector as a control. Cells were treated with Dox to induce MASTL CRISPR/Cas9‐dependent knockout, and 72 h later, cells were treated as specified in (A). The plot displays mean + SEM from three independent experiments. Significance determined by Student's t‐test comparing ENSA/ARPP19 overexpression versus control cells (ns, not significant; *P < 0.05).
- Control (Dox −) and MASTL knockout (Dox +) cells were treated with 5 μM fostriecin, 50 nM okadaic acid (OA), or vehicle (Veh) 10 min before insulin stimulation for 30 min. Total lysates were analyzed for the indicated antibodies and P‐T308‐AKT signal was quantified. β‐Actin was used as a loading control. The plot represents mean + SEM from three independent experiments, except for fostriecin treatment in which only data from two experiments were obtained, and no error bars are shown. *P < 0.05; Student's t‐test.
The MASTL‐PP2A/B55 axis modulates phosphorylation of the feedback targets IRS1 and GRB10
AKT signaling has been shown to be modulated by PP2A complexes (Fowle et al, 2019). However, the fact that MASTL inhibition and, as a consequence, PP2A/B55 activation lead to increased phosphorylation of AKT, which indicates that the effect we observe on the feedback loop is not mediated by direct dephosphorylation of AKT by PP2A/B55. Feedback inhibition of the PI3K/AKT pathway by mTORC1 involves mTORC1/S6K1‐dependent phosphorylation of the insulin receptor substrates, IRS1 and IRS2, as well as the adaptor protein GRB10. Whereas phosphorylation of IRS1 results in reduced protein levels as a consequence of ubiquitin‐dependent degradation (Sun et al, 1999; Rui et al, 2001), GRB10 phosphorylation prevents proteasomal‐mediated degradation thereby correlating with increased protein levels (Hsu et al, 2011; Yonghao et al, 2011). Interestingly, IRS1 stability is also regulated by PP2A complexes (Hartley & Cooper, 2002), although the precise mechanism remains unknown. As shown in Fig 6A, the knockdown of B55α and B55δ, the two ubiquitous B55 isoforms, led to increased phosphorylation of IRS1 on S616, a phosphoresidue that negatively regulates IRS1 function. Phosphorylation of GRB10 on S476 was also increased upon B55α,δ depletion, and, as expected, these phosphorylations correlated with decreased AKT activation in B55α,δ‐depleted cells (Fig 6A). On the contrary, ablation of MASTL prevented the phosphorylation of both IRS1 S616 and GRB10 S476, suggesting defective feedback signaling, and resulted in increased IRS1 and reduced GRB10 total protein levels (Fig 6B). Depletion of B55 subunits in MASTL‐null cells partially rescued the phosphorylation status of IRS1 and GRB10 and the levels of AKT activity, although these assays were limited by technical difficulties in the efficient elimination of these B55 proteins (Fig 6C).
Figure 6. The MASTL‐PP2A/B55 pathway controls the feedback loop at the level of adaptor proteins.
- MDA‐MB‐231 cells were co‐transduced with specific siRNAs against the PPP2R2A (B55α) and PPP2R2D (B55δ) transcripts (B55αδ) or against luciferase (Luc) as a control. Asynchronous cells were collected and blotted for the indicated antibodies. The histograms represent the quantifications of the specified phosphoresidues. Plots show mean + SEM from three independent experiments. *P < 0.05, Student's t‐test.
- Immunodetection of the indicated antigens in control (−) and MASTL knockout (+) cells starved from glucose and re‐stimulated with glucose for 15 min.
- siRNA‐mediated genetic depletion of B55 alpha and delta subunits (B55αδ) in control (Dox −) and MASTL‐null (Dox +) cells. The phosphorylation status of the different proteins was scored in conditions of glucose re‐addition. n = 3 independent experiments. The plot shows mean + SEM from three independent experiments. *P < 0.05, one‐way ANOVA.
- Immunoblot with the indicated antibodies in skeletal muscle (gastrocnemius) extracts. Mice were fasted overnight for 16 h, injected intraperitoneally with glucose (2 g/kg body), and sacrificed 30 min later for sample collection. Quantification of the relative fold change signal of total IRS1 and phospho‐S6K1 T389 [normalized to total S6K1 level; n = 6 Mastl(+/+) and 7 Mastl(Δ/Δ)]. β‐Actin or vinculin was used as a loading control. Data are mean ± SEM; *P < 0.05, unpaired Student's t‐test.
To further confirm these results in vivo, we analyzed the status of IRS in Mastl‐deficient muscle samples. Despite the variability in S6K1 phosphorylation signals among samples when taken from in vivo tissues, total IRS1 levels were overall higher in Mastl‐null tissues compared to control ones, in agreement with a deficient feedback loop due to Mastl deletion (Fig 6D).
Control of MASTL activity by the mTORC1‐S6K1 axis
As discussed above, glucose re‐addition following glucose starvation triggers mTORC1‐dependent AKT inhibition. In these conditions, phosphorylation of MASTL in one of the potential T‐loop activation sites (T194), used here as a surrogate of MASTL activation (Vigneron et al, 2011; Blake‐Hodek et al, 2012), was increased (Fig 7A). In agreement with these data, phosphorylation of MASTL on T194 was also increased in conditions of feedback activation in TSC2‐null cells, and was prevented by the mTOR inhibitors rapamycin and torin (Fig 7B). In vitro kinase assays using GFP‐MASTL immunocomplexes showed that MASTL activity over recombinant ARPP19 increased upon TSC2 knockout, and was partially prevented upon rapamycin and torin treatment (Fig EV5A). These data suggested that MASTL is under the control of the mTORC1‐S6K1 axis in a mitosis‐independent manner. In fact, both kinases were able to phosphorylate MASTL in assays with recombinant proteins (Fig 7C). Mass‐spectrometry analysis identified T710, S716/T718, T722 and S878 as possible sites phosphorylated by mTORC1 (Table EV1). Residues T710, T716/T718, and T722 were also phosphorylated by S6K1, although to a lesser extent. Among the four mTORC1/S6K potential sites, S878 contains a hydrophobic residue in position +1, fitting with the consensus motif for mTORC1 substrates (Kang et al, 2013; Robitaille et al, 2013). Interestingly, S878 was exclusively phosphorylated by mTORC1 and to a much higher extent compared to the other identified sites (150‐fold vs. less than 10‐fold for T710, T716/718, or T722; Fig 7D and Table EV1). These four sites are located in the C‐terminal part of MASTL and presented high conservation among species (Fig 7E).
Figure 7. The mTORC1/S6K1 pathway regulates MASTL kinase activity.
- MDA‐MB‐231 cells were starved for 1 h and re‐stimulated with glucose for 15 min and T194 phosphorylation was tested with a specific antibody. The plot shows the quantification of phospho‐MASTL T194. Data are mean + SEM from three independent experiments. ns, not significant; Student's t‐test.
- Cas9‐expressing control (−) cells or cells with CRIPSR/Cas9‐mediated TSC2 knockout (sgTSC2) were treated with rapamycin (Rap), Torin1 (Tor), or vehicle as control (V). MASTL‐knockdown cells (shMASTL) were used as an additional control of the specificity of the antibody. Inhibitors were added 15 min before glucose re‐stimulation for an additional 15 min and total cell lysates were harvested for immunoblot. The plot shows the quantification of phospho‐MASTL T194 as a mean from two independent experiments.
- In vitro kinase assay using purified GST‐human MASTL as a substrate, and recombinant mTORC1 and S6K1 kinases. Red arrowheads show the phosphorylation of MASTL in the presence of mTORC1 or S6K1. Note the appearance of other phosphorylation signals in mTORC1 and S6K1 reactions, independently of the presence of MASTL, which correspond to autophosphorylation of these kinases. Imperial staining shows the amount of GST‐MASTL protein used as a substrate in each reaction.
- Mass spectrometry analysis of samples treated as in panel (C). The plots represent the intensity of mTORC1‐S6K1‐regulated phosphosites on human MASTL. Data (mean and individual values from two technical replicates) were normalized to the total levels of MASTL and plotted as relative values to the condition of MASTL alone.
- Schematic representation of MASTL protein structure showing the potential mTORC1/S6K phosphorylation sites (red color) identified in the in vitro kinase assay shown in (C). The autophosphorylation residue previously reported to be required for MASTL activity is shown in blue. Orange boxes indicate the conserved kinase domains. Yellow boxes indicate additional sequences conserved in Mastl in different species. The C‐tail domain is shown in blue. The alignment of the amino acid sequence regions surrounding those phosphosites across different species is shown below. Mm, Mus musculus; Rr, Rattus rattus; Hs, Homo sapiens; Gg, Gallus gallus; Xl, Xenopus laevis; Dr, Danio rerio; Dm, Drosophila melanogaster.
- In vitro kinase activity of Mastl mutant isoforms. HEK‐293T cells were transiently transfected with the indicated GFP fusions of mouse Mastl mutants. Anti‐GFP immunocomplexes were subjected to a kinase reaction using recombinant ARPP19 (white circles) or ENSA (filled circles) as a substrate. The graph shows the relative catalytic activity of the indicated mutants. The kinase activity of GFP‐mMastl wild‐type was set as 1 (n = 3 independent experiments). Data are mean + SEM from three independent experiments. ***P < 0.001; one‐way ANOVA.
- isgMASTL MDA‐MB‐231 cells were transduced with the indicated Mastl mutants or GFP alone as a control and treated with doxycycline (Dox) to induce Mastl deletion. Cells were collected after glucose stimulation, and total lysates were blotted for the indicated antibodies.
- MDA‐MB‐231 cells were transduced with the indicated Mastl mutants or GFP alone as a control, and infected with scramble or shMASTL to deplete endogenous MASTL. Cells were fixed upon insulin stimulation for GLUT4 staining. Quantification of the percentage of cells positive for GLUT4 at the plasma membrane in each condition is shown. Data show the mean and individual values from two independent experiments.
Figure EV5. Regulation of MASTL activity by mTORC1.
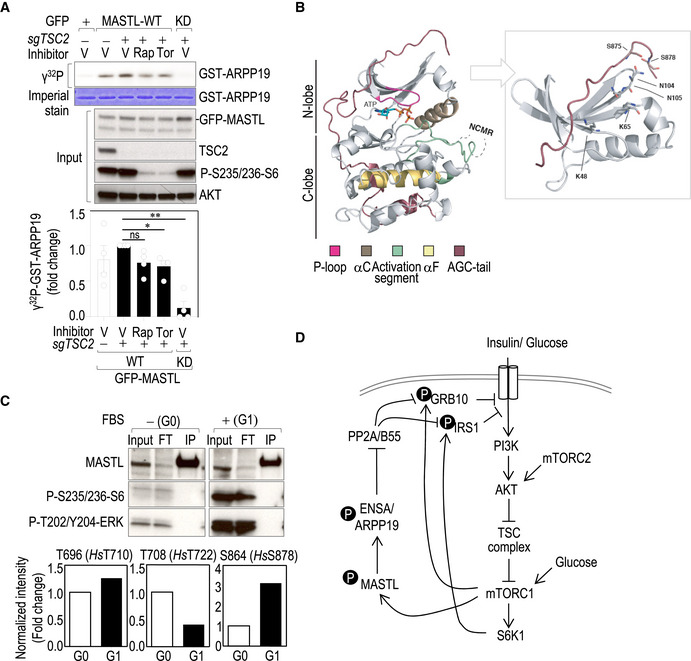
- In vitro kinase assay using GFP‐hMASTL immunocomplexes from asynchronous TSC2 knockout cells (sgTSC2) or control cells treated with the indicated inhibitors for 1 h. Immunoprecipitation of GFP alone was used as a control. GST‐ARPP19 was used as a substrate. The upper panel shows the radiograph of ARPP19 phosphorylation in the different conditions accompanied by the immunoblots of the whole‐cell extracts. Quantification of the relative kinase activity of MASTL is shown in the lower graph, in which MASTL activity in sgTSC2 cells was set as 1. Data are mean ± SEM from four independent experiments. Significance determined by paired Student's t‐test (ns, not significant; *P < 0.05; **P < 0.01).
- A homology structural model showing the position of S875 and S878 in the regulatory C‐tail domain of human MASTL. Ribbon diagram of the MASTL kinase domain (without the residues 181–727 coding for the NCMR; left panel) in complex with ATP. The protein fold displays the standard bilobal structure (N‐lobe and C‐lobe) with its respective AGC C‐tail (shown in brown). Please see the associated key for common structural kinase domain components. Molecular details for the phospho‐binding pocket of MASTL (right panel). Using the homology‐modeled AGC C‐tail of MASTL, it is possible to determine that amino acids Lys48, Lys65, Asn104, and Asn105 (stick representation) are positioned such that they could interact with the phosphorylated tail/liker reside (pSer875) and the mTOR‐phosphorylation site (pSer878). Note, in both panels, the MASTL protein has been homology‐modeled using MODELLER based on the structures of MASTL (PDB 5LOH2; https://www.rcsb.org/) for the kinase domain and of PKA and RSK2 (PDBs 1ATP and 4EL9, respectively) for the AGC C‐tail.
- Mouse embryonic fibroblasts (MEFs) were synchronized in G0 by serum starvation and then released for 6 h in the presence of serum (G1). Endogenous Mastl was immunoprecipitated with a specific antibody against Mastl (Abgent) and subjected to mass spectrometry to identify phosphorylated residues in Mastl in the indicated conditions. The upper panel shows the immunoblot of the immunoprecipitates and inputs probed with the indicated antibodies. The lower graphs show the relative quantification of the indicated phosphopeptides in both conditions.
- A model for the role of the MASTL‐ENSA/ARPP19‐PP2A/B55 pathway in the negative feedback loop that controls AKT activity in response to mTORC1‐S6K1 signaling. For simplicity, only the phosphoresidues in the negative feedback loop are shown.
MASTL belongs to the AGC kinase family but it differs from other family members in a 500‐aa‐long insertion of an unknown role in the activation loop that divides the kinase domain into two separate N‐terminal and C‐terminal fragments (Fig 7E). The C‐terminal tail (known as the turn motif in PKA or the tail/Z site in growth factor‐stimulated AGC kinases) is a critical region whose phosphorylation is required for conformational changes and kinase activity. In fact, autophosphorylation at S875 is considered a major requirement for MASTL activation, which requires some priming phosphorylations (Blake‐Hodek et al, 2012; Hermida et al, 2020), and S878, the main mTOR phosphorylation site identified in our study, locates next to it in the C‐terminal tail (Figs 7E and EV5B). Phosphorylation of S878 has been found in interphase with high occupancy and proposed as one of the potential priming phosphosites for MASTL activation (Hermida et al, 2020). We actually detected phosphorylation of MASTL S878 in non‐mitotic (G1) cells in response to growth factors in vivo (Fig EV5C and Table EV2). Mutation of the homologous residue in Xenopus (S886) results in a partial inactivation of the protein in mitotic extracts (about 60% kinase activity on its substrates; Blake‐Hodek et al, 2012). Importantly, we detected a similar reduction in phosphorylation of recombinant ARPP19 or ENSA (about 40% less activity compared to wild‐type Mastl) when the homolog mouse residue (S864) was mutated to alanine and immunopurified from interphasic cells (Fig 7F). Mutation of the autophosphorylation site (mouse S861A equivalent to human S875) almost completely abolished Mastl kinase activity, and we did not detect any further reduction in catalytic activity when both mutants were combined, suggesting that phosphorylation of S864 might work as a priming phosphorylation site for mTORC1‐dependent MASTL activation (Fig 7F).
To evaluate the relevance of the phosphorylation of MASTL by mTOR in the feedback in response to glucose stimulation, we performed reconstitution assays using MASTL knockout cells expressing GFP fusions with the mouse Mastl mutants resistant to the sgRNAs against human MASTL. Whereas the wild‐type version of Mastl was able to restore the phosphorylation of AKT in T308 to basal levels, mutation of Mastl in the mTOR site (S864A) was not able to reduce AKT activity to the level of control cells (Fig 7G). Similarly, this Mastl phosphomutant also failed to rescue the increment in GLUT4 at PM observed in Mastl‐deficient cells in response to insulin (Fig 7H). Collectively, these data suggest that mTORC1 might modulate the kinase activity of MASTL, through the phosphorylation of its C‐terminal region, thereby inhibiting PP2A‐B55 in order to restrain AKT activity in response to nutrients.
Discussion
The presence of negative feedback loops (Fig EV5D) that attenuate AKT activity upon sustained mTORC1‐S6K signaling reflects the need for fine‐tuning regulation of the AKT‐mTOR axis at different levels. Mechanistically, suppression of AKT depends on the mTORC1‐ and S6K1‐dependent phosphorylation of IRS1, IRS2, and GRB10 (Harrington et al, 2004; Shah et al, 2004; Hsu et al, 2011; Yu et al, 2011), as well as non‐cell‐autonomous effects such as the mTORC1‐dependent secretion of IGF‐binding protein 5 (IGFBP5) which blocks IGFR‐I activation in the extracellular space (Ding et al, 2016).
The participation of cellular phosphatases in these feedback loops has been far less studied. PP2A inhibition has been previously shown to increase IRS1 phosphorylation inducing its degradation (Clark et al, 2000; Hartley & Cooper, 2002). Because of the multiplicity of regulatory subunits, the involvement of specific PP2A complexes in different molecular pathways is not well established. Our data indicate that PP2A complexes containing the B55α and δ regulatory subunits modulate the phosphorylation status of IRS1 and GRB10, and are inhibited by MASTL‐ENSA/ARPP19 during feedback signaling. Intriguingly, it has been recently reported that B55β subunit regulates mTORC1 signaling by directly dephosphorylating 4EBP1 and p70S6K in HER2‐positive breast cancer cells suggesting some substrate specificity among B55 isoforms (Bao et al, 2020).
A few studies have previously linked MASTL with AKT signaling. MASTL overexpression in MCF10‐A cells is accompanied by increased phosphorylation of GRB10 on S104, and IRS1 on S312 (two residues also involved in the AKT‐mTORC1 negative feedback loop), and decreased phosphorylation of TSC2 on S939, an AKT site involved in the regulation of mTORC1 activity (Rogers et al, 2018). In contrast, exogenous expression of MASTL may also induce AKT hyperactivation in tumor cells (Vera et al, 2015). Intriguingly, this last effect is ENSA‐PP2A/B55 independent and, in contrast with our data, it affects the phosphorylation of AKT on S473, but not T308. This might suggest that the regulation of mTORC2 and/or PHLPP1, the main kinase/phosphatase responsible for AKT S473 phosphorylation may be PP2A independent. It is, therefore, possible that MASTL may influence AKT signaling differentially in cycling (Vera et al, 2015) or starved/G0 cells as suggested by our data. Interestingly, AKT may lead to MASTL phosphorylation and activation specifically during mitosis (Reshi et al, 2020), although whether this is mediated by direct phosphorylation and whether these data have any relevance in vivo in a physiological context have not been addressed so far.
The MASTL‐PP2A kinase‐phosphatase module is highly conserved across organisms, although its physiological functions have varied through evolution. In the budding yeast, the MASTL‐endosulfine (RIM15‐IGO1/2) axis is required for G1 arrest and entry into quiescence in response to nutritional deprivation by inducing the expression of a particular transcriptional response (Talarek et al, 2010; Bontron et al, 2013; Sarkar et al, 2014; Moreno‐Torres et al, 2015). In fission yeast, TORC1 activity leads to inhibition of the MASTL ortholog Ppk18 in the presence of nutrients. In that situation, cyclin B‐Cdk1 complexes are inhibited in a PP2A‐dependent manner delaying mitotic entry until cells reach the proper size (Chica et al, 2016). The particular control of quiescence and cell size in yeast makes it difficult to establish parallelism with the function of MASTL in mammals. However, our data suggest that this kinase has evolved to participate in nutrient sensing by regulating the Ensa‐PP2A/B55 pathway in the negative feedback loop that controls AKT‐mTOR activity (Fig EV5D). Moreover, our data also suggest that mTORC1 can directly phosphorylate MASTL to regulate its activity in response to nutrients, another parallelism with the function of the yeast MASTL ortholog. MASTL appears to limit AKT activation in response to glucose and insulin but not in response to other nutrients, such as amino acids, which are also sensed by the mTORC1 pathway. Unfortunately, how glucose controls activation of the AKT‐mTOR pathway is not completely understood (Liu & Sabatini, 2020; Orozco et al, 2020), and the observed selectivity of MASTL activity in glucose signaling deserves further studies. In Saccharomyces cerevisiae, RIM15 activity and cellular localization are regulated by PKA and mTORC1 (Pedruzzi et al, 2003; Wanke et al, 2005; Wei et al, 2008), and whether PKA control MASTL activity in mammals has not been explored yet.
In mammals, the insulin‐mediated activation of AKT is critical for proper glucose disposal and metabolic adaptations. One of the key roles of AKT is the regulation of glucose uptake into insulin‐regulated tissues (reviewed in Manning & Toker, 2017; Hoxhaj & Manning, 2019). This is partially mediated by the translocation of GLUT4 from vesicular intracellular compartments to the plasma membrane. By phosphorylating and inhibiting glycogen synthase kinase (GSK3), AKT promotes glucose storage in the form of glycogen. In agreement with these data, increased AKT activity as a consequence of MASTL deletion leads to enhanced GLUT4 translocation and glucose uptake in cells, resulting in enhanced glycolysis (Fig 3). In vivo, deletion of Mastl leads to increased AKT phosphorylation and glycogen accumulation in the liver, and enhanced GLUT4 translocation and glucose uptake in Mastl‐deficient muscle cells (Fig 4).
Insulin resistance is a pathological state of obesity and type 2 diabetes that occurs, at least in part, through chronic activation of mTORC1/S6K1 by nutrients and constitutive feedback activity that blocks AKT activation (Zoncu et al, 2011). Supporting the role of the mTORC1/S6K1 feedback loop in the pathogenesis of type 2 diabetes, S6K1‐deficient mice are protected from age‐ and obesity‐induced insulin resistance in the presence of reduced IRS1 phosphorylation (Um et al, 2004). In addition, GRB10‐deficient mice also exhibited enhanced glucose tolerance and insulin sensitivity (Wang et al, 2007), whereas IRS1‐deficient mice developed insulin resistance (Saltiel & Kahn, 2001). On the other hand, enhancing AKT signaling in skeletal muscle can improve systemic metabolism and overcome diet‐induced obesity (Izumiya et al, 2008). Ablation of Mastl is also able to improve glucose tolerance in a model of high‐fat diet (HFD)‐induced obesity and results in lower blood glucose levels in aged animals. The effect of Mastl ablation is likely mediated by increased PI3K/AKT signaling as it can be prevented upon treatment with a PI3K inhibitor. Of note, Ensa knockout mice have improved glucose tolerance and are more insulin sensitive (Wiles et al, 2002). In the same direction, haploinsufficiency of the B55α subunit of PP2A causes insulin resistance likely due to the defective insulin‐induced AKT stimulation in insulin‐responsive tissues (Goldsworthy et al, 2016). These observations are in line with a model in which MASTL phosphorylates ENSA to inhibit PP2A/B55 and AKT signaling (Fig EV5D). These data suggest the intriguing possibility that MASTL inhibition could be considered as a new potential target of intervention for metabolic diseases, such as obesity‐induced diabetes. Since MASTL does not participate in the central AKT‐mTOR axis, its inhibition could be used as a more specific strategy to downregulate the negative feedback loop without the need of inhibiting the central mTOR‐S6K kinases.
Materials and Methods
Cell lines
All human cancer cell lines were grown in DMEM supplemented with 10% FBS and gentamycin at 37°C with 5% CO2. MDA‐MB‐231, BT‐549, and MCF‐7 breast cancer cell lines were kindly provided by M.A. Quintela (CNIO, Spain). HepG2 and C2C12 cells were kindly provided by A. Efeyan (CNIO, Spain) and P. Muñoz‐Canoves (CNIC, Spain), respectively. The human embryonic kidney (HEK) 293‐T cell line was obtained from American Type Culture Collection (ATCC; #CRL‐3216). The MDA‐MB‐231 cell line has been DNA fingerprinted using the GenePrint 10 System (Promega). All cell lines were routinely tested for mycoplasma.
Genetically modified mouse models
Mastl conditional knockout mice were generated by crossing the Mastl(lox) allele with a mouse strain carrying a knockin of the CRE‐ERT2 recombinase inducible by tamoxifen (TAM) in the locus of the ubiquitous RNA polymerase promoter (RNAP2_CreERT2) (Alvarez‐Fernandez et al, 2013). Males (8‐ to 12‐week‐old) were fed for 9 weeks with a 60% high‐fat diet (HFD) (60% fat, 20% protein, and 20% carbohydrate; Research Diets; D12492). Mice were treated with TAM (Sigma; T5648) by intraperitoneal injection at a dose of 200 mg/kg twice 1 week before the assay.
All animals were maintained in a C57BL6/J‐129S mixed background. Mice were housed in the pathogen‐free animal facility of the CNIO (Madrid) and maintained under a standard 12‐h light–dark cycle, at 23°C with free access to food and water. All animal work and procedures were approved by the corresponding Ethics Committee of Animal Experimentation in our institution and local government (Instituto Carlos III and Comunidad de Madrid). The CNIO has earned accreditation through the AAALAC International Accreditation Program since 2016.
Plasmids, transfections, and lentiviral infections
Silencing of MASTL and TSC2 was performed using specific siRNA (ON‐TARGET SMARTpool, Dharmacon) or pLKO.1 lentiviral plasmids encoding shRNA sequences (Sigma), as previously described (Álvarez‐Fernández et al, 2017). An shRNA scramble (SHC002, Sigma) was used as a control. TSC2 was knocked out using the CRISPR‐Cas9 system. The sgRNA (GTGGCCTCAACAATCGCATC) against the human TSC2 gene (Bai et al, 2015) was subcloned into the lentiCRISPR v2 lentiviral vector (Addgene #52961). For lentiviral transduction, pLKO plasmids were co‐transfected into HEK293‐T cells along with packaging plasmids pMDLg/pRRE (Addgene #12251), pRSV‐Rev (Addgene #12253), and pMD2.g (Addgene #12259) expression plasmids using Lipofectamine 2000 (Invitrogen) following manufacturer's instructions. Virus‐containing supernatants were collected 48 and 72 h after transfection and target cells were infected in the presence of 4 μg/ml polybrene. Cells were analyzed 48–72 h later, and knockdown efficiencies were evaluated by immunoblotting using specific antibodies. For the conditional knockout MASTL gene in the MDA‐MB231 cell line, we used a doxycycline‐inducible CRISPR‐Cas9 system (Álvarez‐Fernández et al, 2017). isgMASTL clones constitutively express the sgRNA targeting the exon 1 of MASTL (TCCTTCTTGCTTCCCGCGGT), and Cas9 expression is induced by doxycycline (Dox) at a concentration of 2 μg/ml. Experiments were performed 48–72 h after Dox addition. To knock down PPP2R2A and PPP2R2D transcripts, specific siRNAs (SI02225825 for PPP2R2A and SI02759148 for PPP2R2D) were purchased from Qiagen. ON‐TARGET plus SMARTpool siRNAS against human ENSA (L‐011852‐00) and ARPP19 (L‐015338‐00) were purchased from Dharmacon. A siRNA against luciferase was used as a control (Dharmacon). Transfections were performed using Hiperfect (Qiagen), according to manufacturer's instructions.
For transient overexpression, the full‐length cDNA of mouse Mastl was amplified by PCR using mouse cDNA as a template and subcloned into the pDEST 3.1‐GFP vector (Gateway system, Invitrogen). Kinase‐dead mutation (G43S) and phosphorylation mutants (S861A and S864A) were generated by site‐directed mutagenesis. For rescue assays, full‐length human cDNAs for ENSA, and ARPP19 were amplified by PCR using cDNA clones (MGC:839; IMAGE:2820528 for ENSA and MGC:5468; IMAGE:3451558 for ARPP19) and subcloned as N‐terminal flag‐ and HA‐tag fusions, respectively, into the lentiviral vector pLVX.puro (Clontech). Phosphomimetic mutations (ENSA‐S67D and ARPP19‐S62D) were generated by site‐directed mutagenesis. Lentivirus production was performed as indicated above, and host cells were infected with viral supernatants and selected with puromycin (1 μg/ml).
mRNA analysis
Total RNA was extracted from mouse tissues using Trizol (Sigma), according to the manufacturer's instructions. Reverse transcription from 1 μg of total RNA was performed with the SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme (Thermofisher Scientific), followed by quantitative PCR (qPCR), using QuantStudio™ 6 Flex Real‐Time PCR System (Applied Biosystems). Relative quantification of mRNA expression for Mastl was performed by real‐time PCR using TaqMan® probes (Mastl Mm01329243_m1; Mastl Mm01329246_m1; and Hprt1 Mm00446968_m1, Applied Biosystems). Data were analyzed using the comparative delta–delta Ct method and are expressed as the ratio between the expression of Mastl and the housekeeping gene Hprt1.
Nutrient deprivation assays
For nutrient deprivation experiments, cells were seeded into six‐well plates at a density of 300,000 cells per well 24 h before starvation. Actively proliferating cells were rinsed twice with Hank's balanced salt solution (HBSS) and incubated in DMEM without glucose (Gibco) or DMEM/Ham's F‐12 without amino acids (US Biological) supplemented with 10% dialyzed FBS (dFBS, Gibco) for 1 h, unless otherwise indicated. Stimulation with 5–25 mM glucose (Sigma) was performed for 10, 15, or 60 min, as indicated. For serum withdrawal, cells were rinsed twice with HBSS and incubated in a medium containing 0.1% FBS for 6 h and, when specified, stimulated with 100 nM insulin (Sigma). For oxygen starvation, cells were treated with the hypoxia‐mimetic agent 150 μM CoCl2 (Sigma) for 22 h in complete media. Incubation with inhibitors was performed at the time of starvation or 10–20 min before re‐stimulation. For mTOR inhibition, cells were treated with rapamycin (Santa Cruz Biotechnologies) or torin1 (Selleckem) at 100 and 250 nM, respectively. For PP2A inhibition, okadaic acid (Calbiochem) and fostriecin (Santa Cruz Biotechnologies) were used at 50 nM and 5 μM, respectively.
Immunodetection
For immunoblot analysis, cells were extracted on ice in cold lysis buffer (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 1 mM EGTA, 0.1% Triton X‐100, and 0.1% 2‐mercaptoethanol) in the presence of protease inhibitors (Roche) and phosphatase inhibitors (1 mM sodium orthovanadate, 50 mM sodium fluoride, and 20 mM β‐glycerophosphate). Mouse tissues were extracted in RIPA buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP‐40, 0.5% Sodium Deoxycholate, and 0.1% SDS) with protease and phosphatase inhibitors using a Precellys tissue homogenizer. Lysates were cleared by centrifugation at 15,000 g for 10 min at 4°C, and protein was quantified using the Bradford method. Protein extracts were mixed with sample buffer (350 mM Tris–HCl pH 6.8, 30% glycerol, 10% SDS, 0.6 M DTT, and 0.1% bromophenol blue), boiled for 5 min, and separated on TGX Criterion Bis–Tris acrylamide gels (BioRad). Proteins were then transferred to a nitrocellulose membrane (BioRad), blocked in PBS 0.1% Tween‐20 buffer containing 3% non‐fat dried milk, and probed overnight with primary antibodies (1:1,000 dilution) at 4°C and for 1 h at room temperature with peroxidase‐conjugated secondary antibodies. Blots were developed using an enhanced chemiluminescence reagent (Western Lightning Plus‐ECL; Perkin Elmer), exposed to an autoradiograph film, and developed using standard methods. Films were scanned and quantifications were performed using ImageJ.
Antibodies for western blot (WB) were obtained from the following sources: anti‐phospho‐T308 AKT (9275), phospho‐S473 AKT (9271), phospho‐T1462 TSC2 (3611), phospho‐S939 TSC2 (3615), phospho‐T389 S6K1 (9234), phospho‐S235/236 S6 (2211), phospho‐S612 IRS (3203), phospho‐S476 GRB10 (11817), phospho‐T202/Y204 ERK (9101), phospho‐S79 ACC (11818), phospho‐S792 Raptor (2083), phospho‐T246 PRAS40 (2640), phospho‐S9 GSK‐3β (9336), phospho‐S67/S62 Ensa/Arpp19 (5240), total AKT (9272), ERK (9102) TSC2 (4308), S6K1 (9202), S6 (2217), IRS1 (3407), GRB10 (3702), ACC (3676), Raptor (2280), and PTEN (9188) were from Cell Signaling Technology. Anti‐human and mouse MASTL (MABT372) and phospho‐S10 H3 (06‐570) were from Millipore. Anti‐mouse Mastl (AP14289c) was from Abgent and anti‐human MASTL was generated at the CNIO (available from Abcam, ab166647). Anti‐pan‐B55 (sc‐81606) was from Santa Cruz. Anti‐phospho‐T194 MASTL was a gift from H. Hochegger (Univ. of Sussex). Anti‐ENSA (180513) and HA (1424) were from Abcam. Anti β‐Actin (5441), FLAG (F7425), and Vinculin (V9131) were from Sigma. Anti‐GFP was from Roche (11814460001). Mouse monoclonal anti‐GAPDH was generated at the Monoclonal Antibodies Unit at the CNIO.
For immunofluorescence, MDA‐MB231 cells cultured on glass coverslips were fixed in 4% buffered formaldehyde (FA) for 10 min at RT. For GLUT4 immunodetection, FA was first quenched with 100 mM glycine for 10 min followed by permeabilization using 0.2% Triton X‐100 for 10 min, and blocking with 10% goat serum plus 1% BSA for 1 h at RT. Incubation with a specific antibody against GLUT4 (ab654, 1:200) was performed for 1 h at 37°C in combination with 1 μM HCS cell mask (Invitrogen; H32720) that stains the entire cell, and TO‐PRO™‐3 Iodide (T3605, Thermofisher) to visualize the nuclei.
For histological examination, samples were fixed in a solution of 4% of paraformaldehyde, embedded in paraffin, and cut into 2.5 μm sections. Then, sections were deparaffinized in xylene and rehydrated through a series of graded alcohols, stained with hematoxylin and eosin (H&E) and Periodic‐Schiff staining (PAS staining), following standard protocols. An additional immunohistochemical examination was performed using a specific antibody against Ki67 (Master Diagnostica). For immunofluorescence, rehydrated paraffin sections were boiled for 5 min in 10 mM citrate buffer at pH 6.0 for antigen retrieval, and then they were incubated 1 h at room temperature with normal goat serum blocking solution (2% goat serum, 1% BSA, 0.1% cold fish skin gelatine, 0.1% Triton X‐100, 0.05% Tween‐20, and 0.05% sodium azide in PBS at pH 7.2). Sections were incubated overnight at 4°C with the indicated primary antibodies: Glut4 (ab654, Abcam, 1:100) and Vinculin (V9131, Sigma, 1:200). Slides were then washed three times for 3 min in 0.05% of Tween‐20 in PBS and incubated with Alexa‐conjugated secondary antibodies (Molecular Probes) for 1 h at room temperature in 3% BSA in PBS. After washing three times for 3 min in PBS, cell nuclei were counterstained with 100 ng/ml of DAPI for 10 min at room temperature (Roche Applied Science). Slides were mounted on coverslips using Fluoromount mounting medium (DAKO). Images were captured using a laser scanning confocal microscope TCS‐SP5 (AOBS) Leica. Image analysis was performed using ImageJ software.
Flow cytometry
Flow cytometry analysis of DNA content was performed by cell fixation with cold 70% ethanol followed by staining with phospho‐histone H3 (Millipore) to detect mitotic cells and with 10 μg/ml Hoechst 33342 (Molecular Probes, Thermofisher). Data acquisition was performed with an LSR Fortessa analyzer (BD Biosciences).
In vitro kinase assays
Cells transiently expressing GFP‐mMastl were lysed in ELB buffer (50 mM Hepes pH 7.4, 150 mM NaCl, 5 mM EDTA, and 1% NP‐40) in the presence of protease and phosphatase inhibitors. Cell lysates were cleared by centrifugation at 15,000 g for 10 min at 4°C. Mastl immunocomplexes were purified with an anti‐GFP antibody (Roche), crosslinked to Dynabeads Protein A (Invitrogen). Immunoprecipitates were washed two times in ELB buffer, twice in high salt (500 mM NaCl) ELB buffer, followed by two additional washes in kinase buffer (20 mM Hepes pH 7.5 and 10 mM MgCl2). Equal amounts of beads were re‐suspended in kinase buffer and mixed with 500 ng of recombinant ARPP19 purified from HEK293T cells (kindly provided by A. Castro, Montpellier, France) in a final volume of 20 μl, in the presence of 50 μM ATP and 2 μCi 32P‐labeled ATP, and incubated for 20 min at 30°C. Reactions were stopped by the addition of sample buffer and boiling for 5 min. Samples were analyzed by SDS–PAGE in a 10% acrylamide gel, transferred to a nitrocellulose membrane, and developed by exposure to a radiosensitive screen. Images and quantifications were performed using ImageJ. Total MASTL in the reactions was quantified by immunoblot against MASTL on the transferred membrane.
For mTORC1 and p70S6K1 kinase assays, active recombinant proteins were purchased from Sigma (SRP0364 and P0066, respectively). Recombinant purified human MASTL protein was used as a substrate and was from Abnova (H00084930). The reaction was performed in a final volume of 30 μl consisting of kinase buffer (supplemented with 50 mM KCl for the mTOR assay), 200 ng of the upstream kinase, and 500 ng of MASTL, in the presence of 100 μM ATP and 2 μCi 32P‐labeled ATP, and incubated for 30 min at 30°C. Reactions were stopped by the addition of sample buffer and boiling for 5 min, and were analyzed by SDS–PAGE in an 8% acrylamide gel. Proteins in the gel were stained using Imperial Stain (Invitrogen) following the manufacturer's instructions. The gel was vacuum dried and exposed to a radiosensitive screen.
MASTL phosphorylation site mapping
mTORC1 and S6K1 in vitro kinase assays were performed as detailed above but in the absence of radiolabeled ATP. Samples in 5 M urea were doubly digested using the FASP procedure with some modifications. The resulting peptides were analyzed by LC–MS/MS using an Impact mass spectrometer (Bruker Daltonics). Raw files were searched against a Uniprot Homo sapiens database (20,187 sequences) using Andromeda as the search engine through the MaxQuant software. Peptide identifications were filtered by Percolator at 1% FDR using the target‐decoy strategy. Label‐free quantification was performed and extracted ion chromatograms for MASTL phosphopeptides were manually validated.
To map phosphoresidues on Mastl, endogenous mouse Mastl was immunoprecipitated from mouse embryonic fibroblasts (MEFs) using a polyclonal antibody against Mastl (Abgent; AP14289c) cross‐linked to Dynabeads Protein A. Cells were extracted in RIPA buffer as indicated above and samples were subjected to mass spectrometry analysis. Protein eluates were doubly digested using the FASP procedure with some modifications. The resulting peptides were analyzed by LC–MS/MS using a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). In addition, samples were subjected to TiO2 enrichment. Raw files were searched against a UniprotKB/TrEMBL Mus musculus database (UniProtKB/Swiss‐Prot/TrEMBL 43,539 sequences) using Sequest‐HT as the search engine through the Proteome Discoverer 1.4 (Thermo Scientific) software. Peptide identifications were filtered by Percolator at 1% FDR using the target‐decoy strategy. Label‐free quantification was performed with MaxQuant and extracted ion chromatograms for MASTL phosphopeptides were manually validated in Xcalibur 2.2 (Thermo Scientific).
Metabolite analysis by NMR
Concentrations of cell media metabolites were determined by NMR spectroscopy as described in (Domenech et al, 2015). Briefly, HepG2 cells were transduced with scramble or shMASTL lentiviruses. Forty‐eight hours post‐infection, cells were plated in DMEM containing 10 mM U‐13C‐glucose and 10% FBS, and samples were taken at variable times after incubation in a fresh medium. Glucose and lactate concentrations were measured from the sum of all resolved signals in the first increment of a 2D 1H‐1H NOESY experiment, using the H4 signal of glucose and the methyl signal of lactate. Concentrations were normalized to the number of cells in each condition.
Glucose uptake assays
Cells were serum‐deprived for 16 h, washed twice in KRH buffer (50 mM Hepes pH 7.4, 137 mM NaCl, 4.7 mM KCl, 1.85 mM CaCl2, 1.3 mM MgSO4, and 0.1% (w/v) BSA), and incubated for 5 min at 37°C in KRH buffer with 6.5 mM 2‐deoxy‐D‐[1‐3H]‐glucose (0.5 μCi) and 7 mM D‐[14C]‐mannitol (0.3 μCi) per well. Then, cells were washed four times with ice‐cold KRH buffer and lysed with 0.05 M NaOH for 2 h at 37°C. Cell lysates were mixed with 3 ml of liquid scintillation cocktail and radioactivity was quantified using a Perkin Elmer Wallac 1414 Liquid Scintillation counter. From the 14C‐specific radioactivity, the extracellular water volume present in the culture was calculated. This enables the extracellular 3H volume to be determined and subtracted from the total 3H radioactivity to calculate the intracellular amount of 2‐deoxy‐D‐[1‐3H]‐glucose transported.
Ex vivo muscle [3H]‐2‐deoxy‐D‐glucose uptake was assessed using methods previously described, with minor modifications (Hinkley et al, 2014; Ruiz et al, 2018). Briefly, soleus and extensor digitorum longus (EDL) muscles from both legs were dissected from overnight fasted and anesthetized mice with ketamine (100 mg/kg) and xylazine (10 mg/kg). Mice were euthanized by cervical dislocation after muscles had been removed and muscles were rapidly incubated for 60 min at 37°C in pre‐gassed (95% O2 and 5% CO2) Krebs–Ringer bicarbonate (KRB) solution containing the following (in mmol/l): 117 NaCl, 4.7 KCl, 2.5 CaCl2·2H2O, 1.2 KH2PO4, 1.2 MgSO4·7H2O, and 24.6 NaHCO3 and supplemented with 2 mmol/l pyruvate and 0.1% of BSA (RIA Grade). For insulin stimulation, muscles were incubated in the presence or absence of insulin (100 nM; Sigma) for 20 min at 37°C. Contralateral non‐stimulated muscles were used as controls. Glucose transport was assessed during 10 min at 37°C using 1 mM 2‐deoxyglucose (2‐DG) containing 1.5 μCi/ml 2‐deoxy‐D‐[1, 2‐3H]‐glucose (Perkin Elmer, Waltham, MA) and 7 mM mannitol containing 0.45 μCi/ml D‐[14C]‐mannitol (Perkin Elmer, Waltham, MA) in KRB with or without 100 nM insulin. The gas phase was maintained at 95% O2 and 5% CO2. After radioactive incubations, muscles were dried by putting them briefly on Whatman paper, flash‐frozen in liquid nitrogen, weighted, and solubilized for 30 min at 65°C in 300 μl 1 M NaOH. Non‐soluble particulates were precipitated by centrifugation at 10,000 g for 1 min and aliquots were removed for quantification of [3H] and [14C] labels in a liquid scintillator counter. Presence of [14C]‐mannitol was used to correct for the amount of extracellular [3H]‐2‐DG that was non‐specifically uptaken by the tissue but not actively transported into muscle cells. Radioactive counts were normalized to muscle weight.
Metabolic assays in mouse models
For glucose tolerance test assay (GTT) mice were fasted for 16 h and then administered an intraperitoneal glucose injection (2 g/kg body weight). Glycemia was measured by tail vein blood sampling using a glucometer and glucose strips (Bio Medical) in fasting and 15, 30, 45, 60, 90, and 120 min after glucose injection. The PI3K inhibitor CNIO ETP‐44692 (Martínez González et al, 2012) was administered to the 16 h fasted mice by oral gavage in a unique dose of 20 mg/kg 1 h before starting the GTT. ETP‐44692 was dissolved in 90% PEG‐300 and 10% N‐methyl‐2‐pyrrolidone (NMP). Mice were randomly selected from their pre‐determined conditions for the treatment. Insulin tolerance test (ITT) was performed in mice fasted for 6 h and then intraperitoneally injected with rapid‐acting insulin (0.75 U/kg body weight) (Actrapid, Novo Nordisk) followed by tail vein blood glucose sampling to measure glycemia at 0, 15, 30, 45, and 60 min after insulin injection.
Statistics
The mouse sample size was selected based on previous studies in our laboratory following the 3R recommendations. No blinding was done in this study. Statistical analysis was carried out using Prism 8 (GraphPad). Statistical tests were performed using two‐sided, unpaired Student's t‐test, paired Student's t‐test, one‐ or two‐way ANOVA (Bonferroni's multiple tests), or Chi‐square test according to specifications in the figures. Data with P > 0.05 were considered not statistically significant (ns); *P < 0.05; **P < 0.01; ***P < 0.001.
Author contributions
Belen Sanz‐Castillo: Formal analysis; investigation; visualization; methodology. Begoña Hurtado: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Diana Vara‐Ciruelos: Investigation; visualization. Aicha El Bakkali: Formal analysis; investigation; visualization; methodology. Dario Hermida: Investigation; visualization. Beatriz Salvador‐Barbero: Investigation. Diego Martínez‐Alonso: Investigation. José González‐Martínez: Investigation. Clara Santiveri: Investigation. Ramón Campos‐Olivas: Investigation; visualization; methodology. Pilar Ximénez‐Embún: Formal analysis; investigation; methodology. Javier Muñoz: Formal analysis; supervision; investigation; visualization; methodology. Mónica Álvarez‐Fernández: Conceptualization; resources; data curation; formal analysis; supervision; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Marcos Malumbres: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; visualization; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
PDF+
Acknowledgements
We are fully indebted to B. Manning (Harvard TH Chan School of Public Health, Boston) and A. Efeyan (CNIO) for suggestions and helpful discussions. We thank the Proteomics, Nuclear Magnetic Resonance, Microscopy, Cytometry, Comparative Pathology and Mouse Facility core services of the CNIO for their support. We thank H. Hochegger (University of Sussex) for antibodies, A. Castro (University of Montepellier) for recombinant proteins, A. Efeyan (CNIO), M.A. Quintela (CNIO), and P. Muñoz‐Canoves (CNIC and Pompeu Fabra University) for cell lines. BS‐C was supported by Foundation la Caixa, and AEB was funded by Comunidad de Madrid. MÁ‐F was supported by a young investigator grant from the Spanish Ministry of Economy and Competitiveness (MINECO; SAF2014‐60442‐JIN; co‐financed by FEDER funds). The Cell Division and Cancer lab of the CNIO is supported by grants from the MICIU (RTI2018‐095582‐B‐I00), Red de Excelencia iDIFFER (RED2018‐102723‐T), Comunidad de Madrid (B2017/BMD‐3884), and Worldwide Cancer Research (WCR‐20‐0155).
The EMBO Journal (2023) 42: e110833
Contributor Information
Mónica Álvarez‐Fernández, Email: monica.alvarez@ispasturias.es.
Marcos Malumbres, Email: malumbres@cnio.es.
Data availability
The MS proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD032796. All the datasets generated during the current study are available from the corresponding authors upon request.
References
- Alvarez‐Fernandez M, Sanchez‐Martinez R, Sanz‐Castillo B, Gan PP, Sanz‐Flores M, Trakala M, Ruiz‐Torres M, Lorca T, Castro A, Malumbres M (2013) Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc Natl Acad Sci USA 110: 17374–17379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez‐Fernández M, Sanz‐Flores M, Sanz‐Castillo B, Salazar‐Roa M, Partida D, Ali HR, Manchado E, Lowe S, VanArsdale T, Shields D et al (2017) Therapeutic relevance of the PP2A‐B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ 25: 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Fernandez M, Sanz‐Flores M, Sanz‐Castillo B, Salazar‐Roa M, Partida D, Zapatero‐Solana E, Ali HR, Manchado E, Lowe S, VanArsdale T et al (2018) Therapeutic relevance of the PP2A‐B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ 25: 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Xuan B, Liu H, Zhong J, Yu D, Qian Z (2015) Tuberous sclerosis complex protein 2‐independent activation of mTORC1 by human cytomegalovirus pUL38. J Virol 89: 7625–7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Oguz G, Lee WC, Lee PL, Ghosh K, Li J, Wang P, Lobie PE, Ehmsen S, Ditzel HJ et al (2020) EZH2‐mediated PP2A inactivation confers resistance to HER2‐targeted breast cancer therapy. Nat Commun 11: 5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake‐Hodek KA, Williams BC, Zhao Y, Castilho PV, Chen W, Mao Y, Yamamoto TM, Goldberg ML (2012) Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol Cell Biol 32: 1337–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontron S, Jaquenoud M, Vaga S, Talarek N, Bodenmiller B, Aebersold R, De Virgilio C (2013) Yeast endosulfines control entry into quiescence and chronological life span by inhibiting protein phosphatase 2A. Cell Rep 3: 16–22 [DOI] [PubMed] [Google Scholar]
- Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ (2005) Insulin receptor substrate‐2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)‐induced negative feedback down‐regulates protein kinase B‐mediated signaling pathway in b‐cells. J Biol Chem 280: 2282–2293 [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B‐Cdc2/PP2A balance. Proc Natl Acad Sci USA 107: 12564–12569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CJ, White MF, Rondinone CM (2004) Mammalian target of rapamycin regulates IRS‐1 serine 307 phosphorylation. Biochem Biophys Res Commun 316: 533–539 [DOI] [PubMed] [Google Scholar]
- Castilho PV, Williams BC, Mochida S, Zhao Y, Goldberg ML (2009) The M phase kinase Greatwall (Gwl) promotes inactivation of PP2A/B55delta, a phosphatase directed against CDK phosphosites. Mol Biol Cell 20: 4777–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chica N, Rozalen AE, Perez‐Hidalgo L, Rubio A, Novak B, Moreno S (2016) Nutritional control of cell size by the Greatwall‐endosulfine‐PP2A.B55 pathway. Curr Biol 26: 319–330 [DOI] [PubMed] [Google Scholar]
- Clark SF, Molero JC, James DE (2000) Release of insulin receptor substrate proteins from an intracellular complex coincides with the development of insulin resistance. J Biol Chem 275: 3819–3826 [DOI] [PubMed] [Google Scholar]
- Dibble CC, Cantley LC (2015) Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25: 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Bruick RK, Yu Y (2016) Secreted IGFBP5 mediates mTORC1‐dependent feedback inhibition of IGF‐1 signalling. Nat Cell Biol 18: 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diril MK, Bisteau X, Kitagawa M, Caldez MJ, Wee S, Gunaratne J, Lee SH, Kaldis P (2016) Loss of the Greatwall kinase weakens the spindle assembly checkpoint. PLoS Genet 12: e1006310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech E, Maestre C, Esteban‐Martinez L, Partida D, Pascual R, Fernandez‐Miranda G, Seco E, Campos‐Olivas R, Perez M, Megias D et al (2015) AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol 17: 1304–1316 [DOI] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, Sabatini DM (2015) Nutrient‐sensing mechanisms and pathways. Nature 517: 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowle H, Zhao Z, Grana X (2019) PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv Cancer Res 144: 55–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharbi‐Ayachi A, Labbe JC, Burgess A, Vigneron S, Strub JM, Brioudes E, Van‐Dorsselaer A, Castro A, Lorca T (2010) The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330: 1673–1677 [DOI] [PubMed] [Google Scholar]
- Glover DM (2012) The overlooked Greatwall: a new perspective on mitotic control. Open Biol 2: 120023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsworthy M, Bai Y, Li CM, Ge H, Lamas E, Hilton H, Esapa CT, Baker D, Baron W, Juan T et al (2016) Haploinsufficiency of the insulin receptor in the presence of a splice‐site mutation in PPP2R2A results in a novel digenic mouse model of type 2 diabetes. Diabetes 65: 1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Hall MN (2017) Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36: 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene MW, Sakaue H, Wang L, Alessi DR, Roth RA (2003) Modulation of insulin‐stimulated degradation of human insulin receptor substrate‐1 by serine 312 phosphorylation. J Biol Chem 278: 8199–8211 [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR et al (2004) The TSC1‐2 tumor suppressor controls insulin‐PI3K signaling via regulation of IRS proteins. J Cell Biol 166: 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley D, Cooper GM (2002) Role of mTOR in the degradation of IRS‐1: regulation of PP2A activity. J Cell Biochem 85: 304–314 [DOI] [PubMed] [Google Scholar]
- Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M (2000) A rapamycin‐sensitive pathway down‐regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate‐1. Mol Endocrinol 14: 783–794 [DOI] [PubMed] [Google Scholar]
- Hermida D, Mortuza GB, Pedersen AK, Pozdnyakova I, Nguyen T, Maroto M, Williamson M, Ebersole T, Cazzamali G, Rand K et al (2020) Molecular basis of the mechanisms controlling MASTL. Mol Cell Proteomics 19: 326–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkley JM, Ferey JL, Brault JJ, Smith CA, Gilliam LA, Witczak CA (2014) Constitutively active CaMKKα stimulates skeletal muscle glucose uptake in insulin‐resistant mice in vivo . Diabetes 63: 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxhaj G, Manning BD (2019) The PI3K‐AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 20: 74–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB et al (2011) The mTOR‐regulated phosphoproteome reveals a mechanism of mTORC1‐mediated inhibition of growth factor signaling. Science 332: 1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K (2008) Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7: 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM et al (2013) mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341: 364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149: 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, Sabatini DM (2020) mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21: 183–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169: 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez González S, Hernández AI, Varela C, Lorenzo M, Ramos‐Lima F, Cendón E, Cebrián D, Aguirre E, Gomez‐Casero E, Albarrán MI et al (2012) Rapid identification of ETP‐46992, orally bioavailable PI3K inhibitor, selective versus mTOR. Bioorg Med Chem Lett 22: 5208–5214 [DOI] [PubMed] [Google Scholar]
- Mochida S, Maslen SL, Skehel M, Hunt T (2010) Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330: 1670–1673 [DOI] [PubMed] [Google Scholar]
- Moreno‐Torres M, Jaquenoud M, De Virgilio C (2015) TORC1 controls G1‐S cell cycle transition in yeast via Mpk1 and the Greatwall kinase pathway. Nat Commun 6: 8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL et al (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66: 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco JM, Krawczyk PA, Scaria SM, Cangelosi AL, Chan SH, Kunchok T, Lewis CA, Sabatini DM (2020) Dihydroxyacetone phosphate signals glucose availability to mTORC1. Nat Metab 2: 893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi I, Dubouloz F, Cameroni E, Wanke V, Roosen J, Winderickx J, De Virgilio C (2003) TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol Cell 12: 1607–1613 [DOI] [PubMed] [Google Scholar]
- Reshi I, Nisa MU, Farooq U, Gillani SQ, Bhat SA, Sarwar Z, Nabi N, Fazili KM, Andrabi S (2020) AKT regulates mitotic progression of mammalian cells by phosphorylating MASTL leading to PP2A inactivation. Mol Cell Biol 40: e00366‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto‐Baschong C, Sauer U, Jenoe P, Hall MN (2013) Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339: 1320–1323 [DOI] [PubMed] [Google Scholar]
- Rogers S, McCloy RA, Parker BL, Gallego‐Ortega D, Law AMK, Chin VT, Conway JRW, Fey D, Millar EKA, O'Toole S et al (2018) MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 37: 4518–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Fisher TL, Thomas J, White MF (2001) Regulation of insulin/insulin‐like growth factor‐1 signaling by proteasome‐mediated degradation of insulin receptor substrate‐2. J Biol Chem 276: 40362–40367 [DOI] [PubMed] [Google Scholar]
- Ruiz A, Dror E, Handschin C, Furrer R, Perez‐Schindler J, Bachmann C, Treves S, Zorzato F (2018) Over‐expression of a retinol dehydrogenase (SRP35/DHRS7C) in skeletal muscle activates mTORC2, enhances glucose metabolism and muscle performance. Sci Rep 8: 636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799–806 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Dalgaard JZ, Millar JB, Arumugam P (2014) The Rim15‐endosulfine‐PP2ACdc55 signalling module regulates entry into gametogenesis and quiescence via distinct mechanisms in budding yeast. PLoS Genet 10: e1004456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T (2004) Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 14: 1650–1656 [DOI] [PubMed] [Google Scholar]
- Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ (1999) Insulin‐induced insulin receptor substrate‐1 degradation is mediated by the proteasome degradation pathway. Diabetes 48: 1359–1364 [DOI] [PubMed] [Google Scholar]
- Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, Martinelli E, Ramon Y, Cajal S, Jones S et al (2008) Dose‐ and schedule‐dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 26: 1603–1610 [DOI] [PubMed] [Google Scholar]
- Talarek N, Cameroni E, Jaquenoud M, Luo X, Bontron S, Lippman S, Devgan G, Snyder M, Broach JR, De Virgilio C (2010) Initiation of the TORC1‐regulated G0 program requires Igo1/2, which license specific mRNAs to evade degradation via the 5′‐3′ mRNA decay pathway. Mol Cell 38: 345–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J et al (2004) Absence of S6K1 protects against age‐ and diet‐induced obesity while enhancing insulin sensitivity. Nature 431: 200–205 [DOI] [PubMed] [Google Scholar]
- Vera J, Lartigue L, Vigneron S, Gadea G, Gire V, Del Rio M, Soubeyran I, Chibon F, Lorca T, Castro A (2015) Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. Elife 4: e10115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Brioudes E, Burgess A, Labbe JC, Lorca T, Castro A (2009) Greatwall maintains mitosis through regulation of PP2A. EMBO J 28: 2786–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Gharbi‐Ayachi A, Raymond AA, Burgess A, Labbe JC, Labesse G, Monsarrat B, Lorca T, Castro A (2011) Characterization of the mechanisms controlling Greatwall activity. Mol Cell Biol 31: 2262–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Balas B, Christ‐Roberts CY, Kim RY, Ramos FJ, Kikani CK, Li C, Deng C, Reyna S, Musi N et al (2007) Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo . Mol Cell Biol 27: 6497–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Galan JA, Normandin K, Bonneil E, Hickson GR, Roux PP, Thibault P, Archambault V (2013) Cell cycle regulation of Greatwall kinase nuclear localization facilitates mitotic progression. J Cell Biol 202: 277–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanke V, Pedruzzi I, Cameroni E, Dubouloz F, De Virgilio C (2005) Regulation of G0 entry by the Pho80‐Pho85 cyclin‐CDK complex. EMBO J 24: 4271–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, Longo VD (2008) Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet 4: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles VM, Baribault H, Zhang Q (2002) Transgenic mice containing alpha‐endosulfine gene disruptions. Patent Application number: 10016993 (Application #20020137203), pp 1–8
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villén J, Kubica N, Hoffman GR, Cantley LC, Gygi SP et al (2011) Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332: 1322–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]