

Abstract
Epithelial‐mesenchymal transition (EMT) is pivotal in the initiation and development of cancer cell metastasis. We observed that the abundance of glycosphingolipids (GSLs), especially ganglioside subtypes, decreased significantly during TGF‐β‐induced EMT in NMuMG mouse mammary epithelial cells and A549 human lung adenocarcinoma cells. Transcriptional profiling showed that TGF‐β/SMAD response genes and EMT signatures were strongly enriched in NMuMG cells, along with depletion of UDP‐glucose ceramide glucosyltransferase (UGCG), the enzyme that catalyzes the initial step in GSL biosynthesis. Consistent with this finding, genetic or pharmacological inhibition of UGCG promoted TGF‐β signaling and TGF‐β‐induced EMT. UGCG inhibition promoted A549 cell migration, extravasation in the zebrafish xenograft model, and metastasis in mice. Mechanistically, GSLs inhibited TGF‐β signaling by promoting lipid raft localization of the TGF‐β type I receptor (TβRI) and by increasing TβRI ubiquitination and degradation. Importantly, we identified ST3GAL5‐synthesized a‐series gangliosides as the main GSL subtype involved in inhibition of TGF‐β signaling and TGF‐β‐induced EMT in A549 cells. Notably, ST3GAL5 is weakly expressed in lung cancer tissues compared to adjacent nonmalignant tissues, and its expression correlates with good prognosis.
Keywords: epithelial‐mesenchymal transition, glycosphingolipids, ST3GAL5, transforming growth factor‐β, UDP‐glucose ceramide glucosyltransferase
Subject Categories: Cancer, Signal Transduction
Plasma membrane glycosphingolipid composition and related biosynthesis enzymes suppress dynamic EMT and malignant transformation in human epithelia.
Introduction
Transforming growth factor beta (TGF‐β) is the prototypical member of a multifunctional family of secreted dimeric cytokines containing more than 30 structurally and functionally related proteins that fulfill essential roles in embryogenesis and controlling tissue homeostasis. TGF‐β regulates the proliferation, differentiation, death, migration, and invasion of numerous cells in a highly contextual manner (Massague, 2008; Colak & Ten Dijke, 2017). TGF‐β initiates signaling by binding to the transmembrane TGF‐β type II receptor (TβRII), which has serine/threonine kinase activity (Massague, 2000; Shi & Massague, 2003). Thereafter, the TGF‐β type I receptor (TβRI) is recruited to form a heteromeric complex with TβRII and becomes transphosphorylated by the TβRII kinase (Heldin et al, 1997). Upon TβRI activation, intracellular signaling continues with the phosphorylation of Sma‐ and Mad‐related (SMAD) proteins, i.e., SMAD2 and SMAD3, by the TβRI kinase at the two carboxy‐terminal serine residues. These activated R‐SMADs form heteromeric complexes with a common SMAD mediator, SMAD4 (ten Dijke & Hill, 2004). Subsequently, these activated SMAD complexes translocate into the nucleus, where they interact with high‐affinity DNA binding transcription factors and chromatin remodeling proteins to modulate the transcription of target genes such as SMAD7, SERPINE1, and CCN2, which encode the SMAD7, PAI1, and CTGF proteins, respectively (Levy & Hill, 2005; Tzavlaki & Moustakas, 2020).
Every step of the TGF‐β signaling pathway is precisely controlled and participates in crosstalk with other signaling pathways. One key step in which the intensity and duration of TGF‐β signaling are determined is the endocytosis and intracellular sorting of cell surface TGF‐β receptors (Chen, 2009). Two major endocytic pathways regulate the internalization of TGF‐β receptors: clathrin‐mediated endocytosis and lipid‐raft‐ or caveolae‐mediated endocytosis. Clathrin‐dependent endocytosis is based on directing TGF‐β receptors to early endosomes enriched with the early endosomal antigen 1 (EEA1) and the SMAD anchor for receptor activation (SARA) proteins (Panopoulou et al, 2002). After entering early endosomes, TGF‐β receptors associate with the FYVE domain‐containing protein SARA, resulting in the enhancement of TGF‐β‐induced SMAD2/3 complex activation and the subsequent propagation of SMAD‐dependent signaling (Di Guglielmo et al, 2003). The internalized receptors can be sorted to recycling endosomes to return to the membrane in a Rab11‐dependent manner (Mitchell et al, 2004). For lipid raft‐mediated internalization, the TGF‐β receptors enter caveolin‐positive vesicles, where TβRI preferentially associates with SMAD7 (Zhao & Chen, 2014). SMAD7, a negative regulator of TGF‐β signaling, competes with SMAD2/3 for interaction with TβRI or recruits E3 ubiquitin ligases such as SMAD‐specific E3 ubiquitin protein ligase 1/2 (SMURF1/2) for ubiquitin‐dependent lysosomal degradation of the TGF‐β receptor complex, among other negative regulatory mechanisms (Hayashi et al, 1997; Nakao et al, 1997). Therefore, clathrin‐mediated endocytosis of TGF‐β receptors promotes TGF‐β/SMAD signaling, whereas the internalization mediated by caveolin terminates signaling.
Epithelial‐mesenchymal transition (EMT) is a dynamic and reversible morphological process in which epithelial cells lose their cell–cell contacts and apical‐basal polarity, to acquire a mesenchymal phenotype with enhanced cell motility (Katsuno et al, 2013). EMT can be characterized by decreased expression of epithelial markers, that is, E‐cadherin and β‐catenin, and increased expression of mesenchymal markers, that is, N‐cadherin, vimentin, SNAIL1/2, and ZEB (Lamouille et al, 2014). The transition from the epithelial to the mesenchymal state is often incomplete, and cells in the intermediate states display mixed E/M characteristics. This ability is referred to as epithelial‐mesenchymal plasticity (EMP) (Yang et al, 2020). EMT is a transient and reversible critical process during embryonic development (Nakajima et al, 2000) and wound healing (Barriere et al, 2015). Moreover, it plays an important role in pathological processes, specifically in cancer progression and fibrosis (Zeisberg et al, 2007; Derynck & Weinberg, 2019). TGF‐β is a potent inducer of EMT (Derynck et al, 2014). However, how dynamic TGF‐β receptor signaling pathways control EMT and how this process is precisely regulated and executed and contributes to cancer progression are incompletely understood.
Glycosylation involves the addition of sugars to proteins and lipids and is catalyzed by various enzymes, including glycosyltransferases (Xu et al, 2018). Protein glycosylation mainly involves the attachment of N‐linked glycans (N‐glycans), O‐linked glycans (O‐glycans), glycosaminoglycans (GAGs), or glycosylphosphatidylinositol (GPI) anchors to peptide backbones (Reily et al, 2019). Glycosphingolipids (GSLs) are the major class of glycolipids found in cell membranes. The biosynthesis of GSLs is a stepwise process starting with ceramide galactosylation to form galactosylceramide (GalCer) or glucosylation by the specific enzyme UDP‐glucose ceramide glucosyltransferase (UGCG) to form glucosylceramide (GlcCer) (Schnaar & Kinoshita, 2015). Subsequently, GlcCer is elongated by the addition of Gal, which is catalyzed by the enzymes B4GALT5/6, to form lactosylceramide (LacCer), the precursor for the formation of three GSL classes: gangliosides, (iso‐)globosides, and (neo‐)lacto‐series GSLs (Schnaar & Kinoshita, 2015). GSLs, together with cholesterol and selected proteins, including GPI‐anchored proteins and some transmembrane signaling proteins, such as receptor tyrosine kinases, are enriched in cell membrane microdomains called lipid rafts (Regina Todeschini & Hakomori, 2008). GSL‐mediated formation of lipid rafts may affect the localization of signaling molecules and thereby modulate cellular signaling responses. Indeed, several growth factor receptors, including the epidermal growth factor (EGF) receptor (EGFR), the insulin receptor, and the nerve growth factor receptor, are localized in membrane microdomains, and their signaling functions have been reported to be modulated by glycolipids (Kabayama et al, 2007; Kawashima et al, 2009; Coskun et al, 2011; Pontier & Schweisguth, 2012). Previous studies have shown that the aberrant expression of certain glycosyltransferases participating in the synthesis of N‐ and O‐ glycans can affect TGF‐β signaling and be involved in TGF‐β‐induced EMT (Li et al, 2016; Zhang et al, 2021). Although changes in GSLs during EMT have been reported (Guan et al, 2009), whether there is a functional link between GSLs and TGF‐β signaling or TGF‐β‐induced EMT remains unclear. This uncertainty can be attributed partially to the insufficient sensitivity and analytical depth of methodologies for GSL analysis used in previous studies.
In this study, we used two in vitro cellular EMT model systems, that is, mouse normal mammary gland epithelial NMuMG cells and human lung epithelial adenocarcinoma A549 cells, to investigate the alterations in and role of GSLs in TGF‐β‐induced EMT with a sensitive analytical glycomics platform. Our study showed a strong decrease in GSLs in these two cell lines during TGF‐β‐induced EMT. Furthermore, we showed that inhibition of GSL expression by inactivation of the glycosyltransferase UGCG promoted TGF‐β signaling and TGF‐β‐induced EMT using in vitro and in vivo models. Importantly, ST3GAL5, which catalyzes the synthesis of a‐series gangliosides, inhibited TGF‐β signaling responses. These gangliosides were found to determine the localization of TβRI in lipid rafts and control its ubiquitination and turnover. Strikingly, we found that ST3GAL5 is expressed at much lower levels in lung cancer tissues than in adjacent normal tissues and that its expression correlates with good prognosis in lung cancer patients.
Results
TGF‐β inhibits the expression of gangliosides in NMuMG and A549‐VIM‐RFP cells
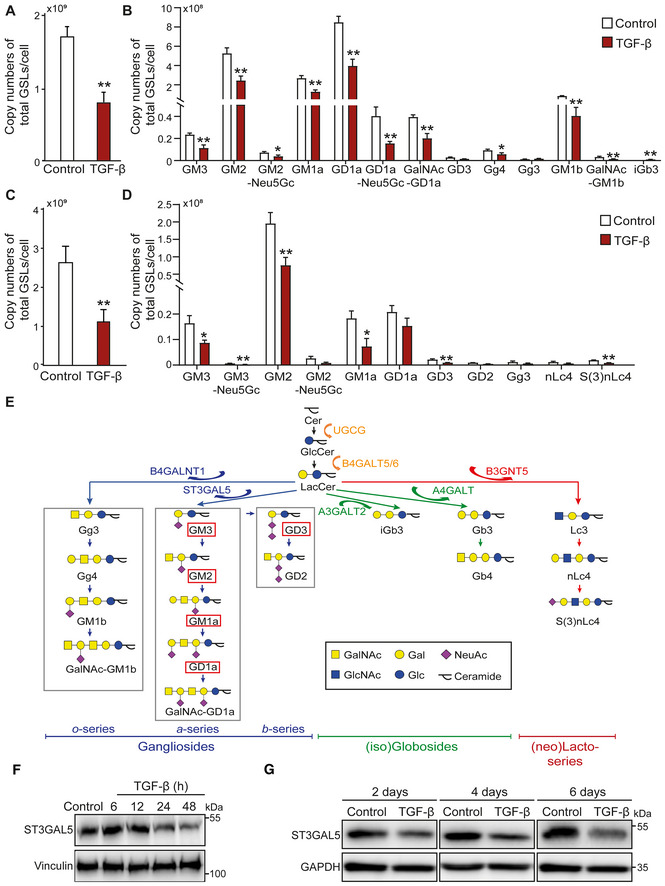
To investigate the TGF‐β‐induced changes in GSL‐glycans in cells undergoing EMT, we chose two established cell models with prominent TGF‐β‐induced EMT responses, i.e., NMuMG (Zhang et al, 2020a) and A549‐vimentin (VIM)‐red fluorescent protein (RFP) cell lines (Wang et al, 2020). We performed quantitative glycomic analysis of these cell lines using porous graphitized carbon (PGC) nano‐liquid chromatography‐electrospray ionization tandem mass spectrometry (nano‐LC‐ESI‐MS/MS) in negative ESI mode, allowing in‐depth determination of GSL profiles (Appendix Fig S1; Anugraham et al, 2015). The high separation power of PGC chromatography allows discrimination between glycan linkages and positional isomers (Anugraham et al, 2015; Zhang et al, 2020b). Identification of GSL‐glycans was performed based on the PGC retention time, the known MS/MS fragmentation patterns, the described biosynthetic pathways and manual inspection of the fragmentation spectra. All annotated structures with their relative abundances are listed in Appendix Table S1 and Fig S11. In addition to performing relative quantification, we spiked in GT1b as an internal standard to monitor the expression levels of GSL‐glycans. With this approach, we were able to provide a full profile of GSLs present on the cell membrane, to identify the most abundant glycans accounting for more than 98% of the relative intensity, and to quantify the expression of GSLs during TGF‐β‐induced EMT responses in two cell models. The most abundant GSLs in untreated NMuMG cells were gangliosides, including o‐, a‐, and b‐series gangliosides, along with a small amount of isogloboside 3 (iGb3) neo‐expressed after TGF‐β treatment (Appendix Fig S2A). Although the relative abundances of the individual gangliosides remained unaffected by TGF‐β treatment for 48 h (Appendix Fig S2B), the absolute quantities of these gangliosides, especially GM3, GM2, GM1a, GM1b, GD1a, Gg4, GalNAc‐GD1a, and GD1a‐Neu5Gc, were significantly decreased (Fig 1A and B). In parallel, the same glycosylation analysis was performed in A549‐VIM‐RFP cells (Appendix Fig S3A). We obtained a GSL‐glycan profile similar to that of NMuMG cells. In addition to the highly expressed a‐ and b‐series gangliosides, small amounts of (iso)globosides (iGb3) and (neo)lacto‐series GSLs (nLc4, and sialylated nLc4 [S(3)nLc4]), were detected in untreated A549‐VIM‐RFP cells (Appendix Fig S3A). Similar to the observations in NMuMG cells, large decreases in the absolute abundances of GSLs, including GM3, GM2, GM1, and GD3, were observed in TGF‐β‐stimulated A549‐VIM‐RFP cells compared to the corresponding control cells (Fig 1C and D). Most of the GSLs, such as GM3, GM1a, GD3, and GD2, showed no significant change in relative abundance, except for a slightly decrease in GM2 and increase in GD1a (Appendix Fig S3B). Figure 1E summarizes the GSL biosynthetic pathways and basal GSL expression patterns in the NMuMG and A549‐VIM‐RFP cell lines; the GSL types present in both cell lines were highly similar. Consistent with the glycomic profiling results, ST3GAL5 protein expression was significantly decreased by stimulating NMuMG and A549‐VIM‐RFP cells with TGF‐β (Fig 1F and G). Furthermore, TGF‐β reduced the gene expression levels of B4galnt1, B5galt6, and St3gal5, essential enzymes catalyzing the formation of gangliosides, in NMuMG cells (Appendix Fig S2C).
Figure 1. TGF‐β decreases the abundance of glycosphingolipid (GSL)‐glycans.
-
A, BAverage copy numbers of (A) total GSL‐glycans and (B) individual GSL‐glycans per cell in NMuMG cells stimulated with TGF‐β.
-
C, DAverage copy numbers of (C) total GSL‐glycans and (D) individual GSL‐glycans per cell in A549‐VIM‐RFP cells treated with TGF‐β.
-
EGSL biosynthesis scheme. GSLs present in both NMuMG cells, and A549‐VIM‐RFP cells are highlighted by red boxes.
-
F, GImmunoblot analysis of ST3GAL5 in (F) NMuMG and (G) A549‐VIM‐RFP cells treated with vehicle control or TGF‐β for the indicated time. Vinculin and GAPDH: loading controls.
Data information: Unless stated otherwise, TGF‐β (2.5 ng/ml) and vehicle control were applied for 48 h. The histograms show the mean ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01 based on unpaired Student's t‐test. GT1b: internal normalization control.
UGCG knockout leads to promotion of TGF‐β signaling and TGF‐β‐induced EMT in NMuMG cells
The synthesis of GSLs starts with the addition of glucose to ceramide and continues with the formation of more complex GSL structures catalyzed by specific glycosylation‐related enzymes. The glycosyltransferase UGCG is the first enzyme initiating this biosynthetic pathway (Allende & Proia, 2014). To investigate the role of GSLs in TGF‐β signaling, we generated Ugcg NMuMG knockout (KO) cells by clustered regularly interspaced short palindromic repeats (CRISPR)‐CRISPR‐associated protein 9 (Cas9) gene editing and picked two independent KO single‐cell clones. Depletion of Ugcg and its GSL products was confirmed by various functional experiments. We performed a UGCG enzyme activity assay by incubation of lysates of control (expressing Cas9) or KO cells with a BODIPY‐conjugated analog of the UGCG substrate ceramide followed by thin‐layer chromatography (TLC) of extracted GSLs (Fig EV1A). The data showed a near absence of BODIPY‐GlcCer in the lysates of Ugcg KO cells, demonstrating the high KO efficiency. By using cholera toxin subunit B (CTB) conjugated to the fluorophore Alexa Fluor 488 and fluorescence‐activated cell sorting (FACS) analysis, we detected a significant decrease in the abundance of GM1, a ganglioside subtype that interacts with CTB, in Ugcg KO NMuMG cells (Fig EV1B). Similarly, the glycomic profiling results and quantification of the absolute abundances of individual GSLs, indicated that all GSLs were depleted in Ugcg KO cells (Figs 2A and EV1C and D).
Figure EV1. related to Fig 2. KO of Ugcg promotes TGF‐β signaling, EMT and migration of NMuMG cells.
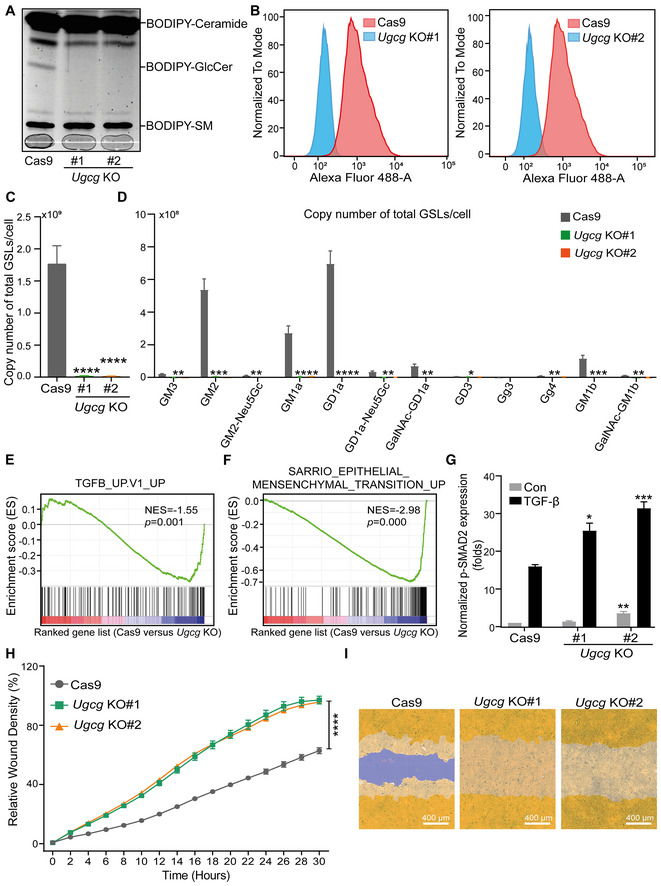
- Analysis of BODIPY‐conjugated ceramide, GlcCer, and sphingomyelin (SM) in two independent Ugcg KO NMuMG cell lines and the control NMuMG cell line using TLC. The light gray line at the bottom of the gel (that is marked with a pencil) was used to indicate the running lanes during the experiment (and where the samples were loaded).
- Detection of GM1 ganglioside by flow cytometry using Alexa Fluor 488‐conjugated CTB in two Ugcg KO (KO#1 and KO#2) NMuMG cell lines infected with one guide RNA and a Cas9 expression vector or with a Cas9 expression vector alone (control).
- Average absolute quantities of total GSL‐glycans per cell in NMuMG cells with Ugcg KO.
- Average absolute quantities of individual GSL‐glycans per cell in NMuMG cells with Ugcg KO.
- The human TGF‐β gene response signature was enriched in Cas9 control versus Ugcg KO NMuMG cells, as shown by GSEA. NES = −1.55, P = 0.001.
- GSEA of the human EMT gene signature enriched in Cas9 control versus Ugcg KO NMuMG cells. NES = −2.98, P = 0.000.
- Quantification of the p‐SMAD2 level in NMuMG cells, including the Cas9 group and the Ugcg KO groups with and without TGF‐β (2.5 ng/ml) treatment for 1 h. Tubulin: loading control.
- The time course of wound closure in NMuMG cells with or without Ugcg deficiency was analyzed by an IncuCyte system.
- Representative images of a scratch wound at the final time point (30 h) in the control group and two Ugcg KO groups of NMuMG cells. The region of the original scratch is indicated in purple, and the region of the cell is colored yellow. Scale bar = 400 μm.
Data information: All data are expressed as the mean ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. In (C, D, G), statistical analysis was based on unpaired Student's t‐test. In (H), statistical analysis was based on two‐way ANOVA.
Source data are available online for this figure.
Figure 2. KO of Ugcg, a key enzyme in the biosynthesis of GSLs, promotes TGF‐β signaling and TGF‐β‐induced EMT in NMuMG cells.
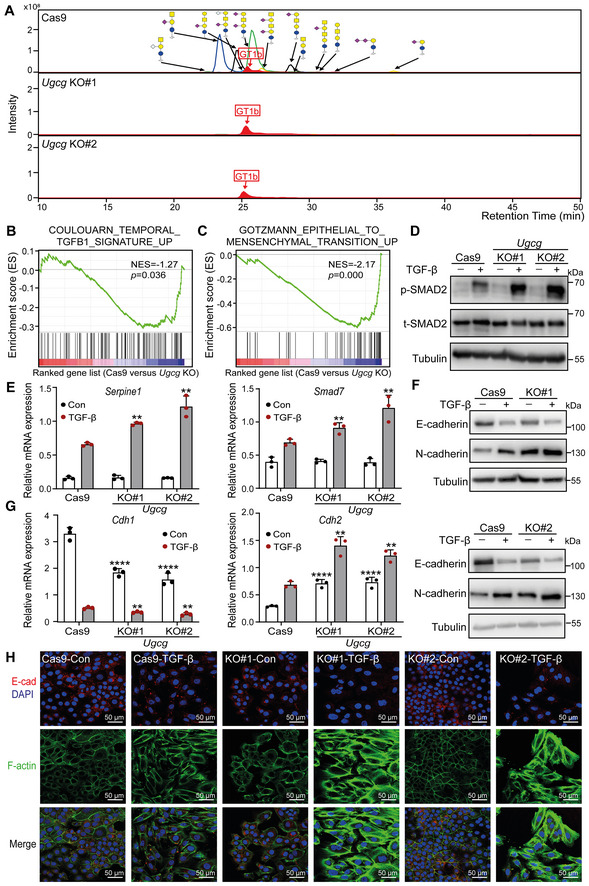
- Combined extracted ion chromatography of GSL glycans in Ugcg KO and control NMuMG cells. GT1b: internal normalization control.
- Mouse TGF‐β gene signatures were enriched in Cas9 control versus Ugcg KO NMuMG cells, as shown by GSEA. Normalized enrichment score (NES) = −1.27, P = 0.036.
- GSEA of mouse EMT gene signatures in Cas9 control versus Ugcg KO NMuMG cells. NES = −2.17, P = 0.000.
- Immunoblot analysis of p‐SMAD2 and total SMAD2 (t‐SMAD2) in NMuMG cells with or without Ugcg deficiency and treated with vehicle control or TGF‐β for 1 h. Tubulin: loading control.
- qRT–PCR analysis of TGF‐β target genes, including Serpine1 and Smad7, in Ugcg KO NMuMG or control NMuMG cells treated with vehicle control or TGF‐β for 6 h.
- Immunoblot analysis of E‐cadherin and N‐cadherin in Ugcg KO NMuMG or control NMuMG cells treated with vehicle control or TGF‐β for 48 h. Tubulin: loading control.
- Cdh1 and Cdh2 mRNA levels in Ugcg KO or control NMuMG cells after vehicle control or TGF‐β treatment for 48 h.
- Immunofluorescence analysis of the expression and localization of E‐cadherin (red) and the formation of F‐actin (green) after treatment with vehicle control or TGF‐β for 2 days. Nuclei were counterstained with DAPI (blue). Images were acquired with confocal microscopy. Scale bar = 50 μm.
Data information: TGF‐β was applied at a final concentration of 2.5 ng/ml. In (E, G), the data are expressed as the mean ± SD values from three biological replicates (n = 3). **P < 0.01; ****P < 0.0001 based on unpaired Student's t‐test.
Next, to obtain insight into signaling pathways and processes affected upon Ugcg depletion, we compared the transcriptional profiles in NMuMG cells with or without Ugcg KO by RNA sequencing (RNA‐seq) and performed gene set enrichment analysis (GSEA). Importantly, we observed that a mouse TGF‐β response signature (consisting of 109 genes) was significantly enriched in Ugcg‐depleted NMuMG cells, indicating a negative correlation between UGCG and TGF‐β signaling (Fig 2B). GSEA performed with a human TGF‐β response signature (containing 189 genes) showed a similar negative correlation, confirming the hypothesis that UGCG mediates the inhibition of TGF‐β signaling (Fig EV1E). In addition, we found that UGCG is also negatively correlated with the EMT process based on GSEA using the mouse and human EMT gene signatures (Figs 2C and EV1F). Thereafter, we examined the effect of Ugcg depletion on TGF‐β‐induced responses and EMT marker expression levels in NMuMG cells. Consistent with our RNA profiling results, we observed significant upregulation of basal and TGF‐β‐induced SMAD2 phosphorylation in Ugcg‐depleted NMuMG cells (Figs 2D and EV1G). Consistent with this, qRT–PCR analysis confirmed that upon loss of Ugcg, the expression levels of TGF‐β target genes, including Smad7 and Serpine1 (Fig 2E), were elevated. Regarding TGF‐β‐induced EMT, depletion of Ugcg inhibited the expression of the epithelial marker E‐cadherin and promoted the expression of the mesenchymal marker N‐cadherin at both the mRNA and protein levels (Fig 2F and G). The enhanced TGF‐β‐induced EMT upon Ugcg KO was further validated by the decreased E‐cadherin expression and increased filamentous (F)‐actin formation in Ugcg KO cells compared to control cells as measured by immunofluorescence staining with an anti‐E‐cadherin antibody and FITC‐conjugated phalloidin, respectively (Fig 2H).
Activation of EMT can endow cells with an enhanced ability to migrate (Zhang et al, 2020a). We thus further investigated the basal migration rate upon Ugcg KO in NMuMG cells. NMuMG cells with Ugcg depletion showed enhanced migration compared to control cells (Fig EV1H and I). Taken together, these results show that UGCG is a critical inhibitor of TGF‐β/SMAD signaling and EMT in NMuMG cells.
Eliglustat, an inhibitor of UGCG activity, promotes TGF‐β signaling, TGF‐β‐induced EMT, cell migration, extravasation, and early metastatic outgrowth
Having determined the inhibitory role of UGCG in TGF‐β signaling and EMT in mouse normal epithelial NMuMG cells, we expanded our study to human lung cancer A549 cells. To inactivate the glucosylceramide synthase activity of UGCG, we used the clinically approved GSL synthesis inhibitor eliglustat (Stirnemann et al, 2017). The effectiveness of eliglustat as a UGCG inhibitor was confirmed by measuring GM1 expression using FACS analysis (Appendix Fig S4A and B). Eliglustat treatment strongly promoted TGF‐β‐induced SMAD2 phosphorylation in A549‐VIM‐RFP cells (Fig 3A and B; Fig EV2A). Consistent with this finding, TGF‐β‐induced SMAD3‐dependent CAGA‐GFP transcriptional reporter activity was potently upregulated by eliglustat stimulation, and this effect was further blocked by the addition of the highly selective small molecule TβRI kinase inhibitor SB505124 (Fig EV2B). Furthermore, qRT–PCR analysis confirmed that eliglustat promotes TGF‐β signaling by increasing the expression levels of TGF‐β target genes including SMAD7, SERPINE1 and CCN2 (Fig EV2C). Moreover, the addition of eliglustat inhibited the expression of the epithelial marker E‐cadherin and enhanced the expression of mesenchymal markers, including N‐cadherin and vimentin, at both the protein and mRNA levels in either the absence or presence of exogenous TGF‐β (Figs 3C and EV2D). The eliglustat‐mediated promotion of TGF‐β‐induced EMT was further confirmed by the dynamic increase in RFP‐tagged vimentin expression (Fig 3D, Appendix Fig S4C) and enhanced F‐actin formation (Fig EV2E). We next investigated whether this UGCG inhibitor promotes the migration and invasion of A549‐VIM‐RFP cells. As expected, in the scratch assay, eliglustat treatment enhanced both basal and TGF‐β‐induced cell migration (Fig 3E; Appendix Fig S4D). Moreover, the eliglustat‐induced promotion of cell migration was completely blocked by the addition of SB505124, indicating that eliglustat‐induced cell migration requires TβRI signaling (Fig 3E; Appendix Fig S4D). Next, we investigated the effect of eliglustat on the extravasation of A549 cells using a zebrafish xenograft model system. We pretreated mCherry‐labeled A549 cells with eliglustat for 4 days and then injected them into the ducts of Cuvier of embryonic zebrafish. The number of extravascular cell clusters was determined 4 days after injection. Eliglustat‐treated cells/embryos exhibited greater invasive capability than untreated cells/embryos (Fig 3F and G). Furthermore, SB505124 significantly blocked the eliglustat‐induced extravasation of A549 cells (Fig 3F and G). In addition, the effect of eliglustat on the metastatic ability of A549‐Luc cells was investigated by injecting these cells into nude mice via the tail vein. Mice injected with A549‐Luc cells pretreated with eliglustat for 1 week prior to injection exhibited earlier metastasis than mice injected with vehicle control‐pretreated cells (Fig 3H). In addition, eliglustat significantly promoted early metastatic colonization of A549‐Luc cells in mice (Fig 3I and J; Appendix Fig S4E). We also examined the inhibitory effect of eliglustat on GSL biosynthesis in NMuMG cells by FACS analysis (Fig EV2F). Similar to the observations in A549 cells, we observed (slight) increases in TGF‐β/SMAD2 signaling and TGF‐β‐induced EMT after eliglustat addition (Fig EV2G and H).
Figure 3. Eliglustat, a UGCG inhibitor, enhances TGF‐β signaling and TGF‐β‐induced EMT, migration, extravasation and early metastatic outgrowth of A549 cells.
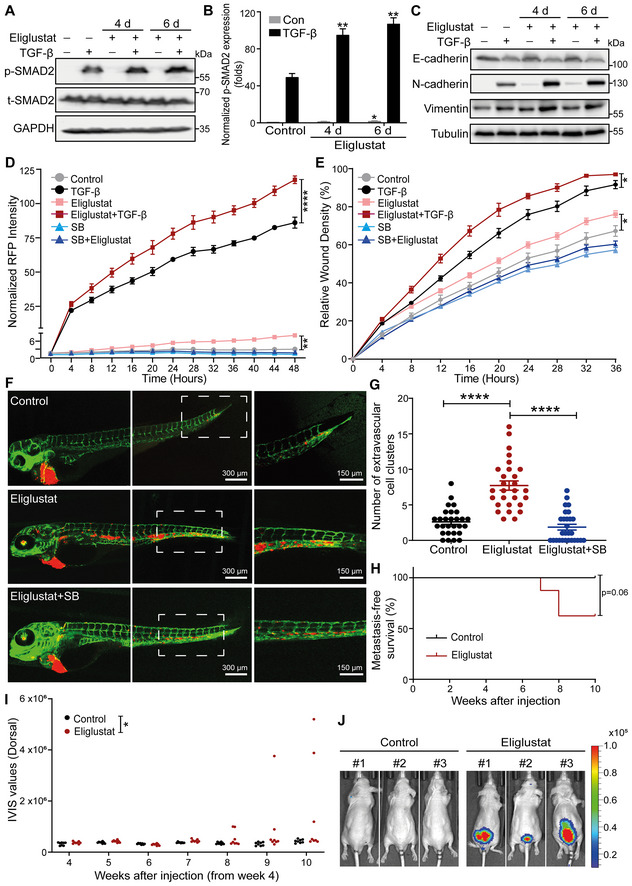
- Immunoblot analysis of p‐SMAD and t‐SMAD2 in A549‐VIM‐RFP cells pretreated with eliglustat for 4 or 6 days and then treated with vehicle control or TGF‐β for 1 h. GAPDH: loading control.
- Quantification of the p‐SMAD2 level in A549‐VIM‐RFP cells as shown in (A). The levels were normalized to that of GAPDH, and fold changes were then further normalized to the level of p‐SMAD2 in control cells without TGF‐β treatment.
- Immunoblot analysis of the epithelial marker E‐cadherin and mesenchymal markers N‐cadherin and vimentin in A549‐VIM‐RFP cells treated with eliglustat for 4 or 6 days and/or with vehicle control and/or TGF‐β for 2 days. Tubulin: loading control.
- Effect of eliglustat (pre)treatment for 4 days on vimentin expression in A549‐VIM‐RFP cells in response to TGF‐β and/or SB505124 (SB, 1 μM) treatment for the indicated times. The time course of RFP‐tagged vimentin expression was monitored with IncuCyte. The red object intensity was normalized to the red intensity at 0 h.
- A549‐VIM‐RFP cells were pretreated with eliglustat for 4 days and were then incubated with TGF‐β or SB505124 (SB, 1 μM) for the indicated times. The real‐time scratch assay results were analyzed with an IncuCyte system.
- mCherry‐labeled A549 cells were pretreated with eliglustat (2 μM) for 4 days and were then injected into ducts of Cuvier of zebrafish embryos. Representative images with magnified regions (outlined with dotted squares) of extravasated cells were acquired 4 days after injection by confocal microscopy. SB group zebrafish were treated with the inhibitor SB505124 (1 μM) in the egg water together with eliglustat for 4 days after injection with daily refreshment of the treatments. Scale bar = 300 or 150 μm.
- Quantification of the number of extravasated cell clusters from 28 embryos per group. ****P < 0.0001 based on unpaired Student's t‐test from two biological replicates (n = 2).
- Kaplan–Meier analysis of metastasis‐free survival in eight mice in the group injected with untreated A549‐Luc cells and eight mice injected with cells pretreated with eliglustat for 1 week. The log‐rank test was used for statistical analysis; P = 0.06.
- Analysis of the in vivo imaging system (IVIS) values from the fourth week post‐injection in eight mice in the control group and eight mice in the eliglustat pretreatment group.
- Whole‐body bioluminescence images (BLI) at 10 weeks of three mice injected with untreated A549‐Luc cells or cells pretreated with eliglustat for 1 week. BLI of all eight mice in each group are shown in Appendix Fig S4E.
Data information: TGF‐β and eliglustat were applied at final concentrations of 2.5 ng/ml and 2 μM, respectively. All data are expressed as the mean ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01; ****P < 0.0001. In (B, D, E), statistical analysis was based on two‐way ANOVA.
Figure EV2. related to Fig 3. Treatment with the UGCG inhibitor eliglustat promotes TGF‐β signaling and TGF‐β‐induced EMT.
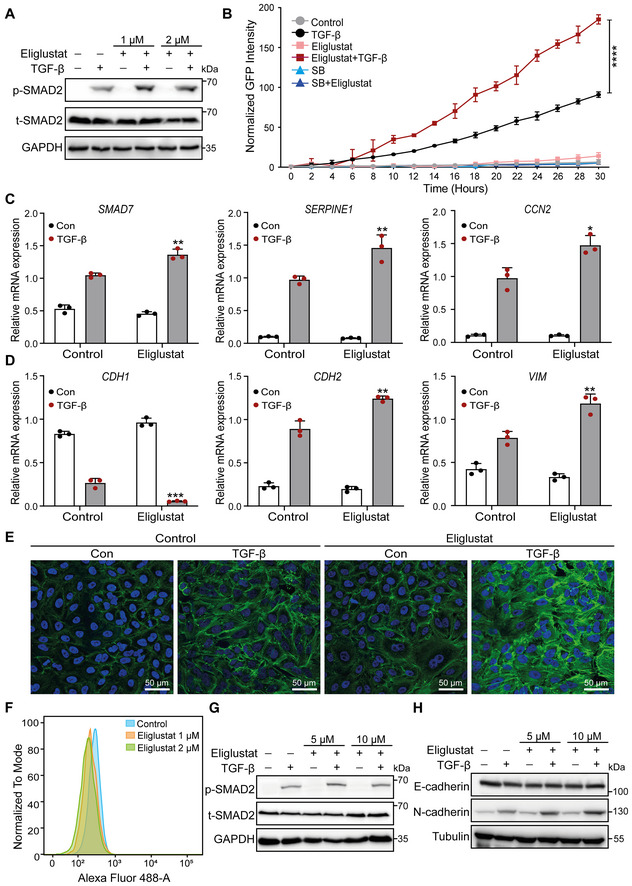
- Levels of p‐SMAD2 and t‐SMAD2 in A549‐VIM‐RFP cells pretreated with eliglustat (1 or 2 μM) for 4 days and then treated with TGF‐β or vehicle control for another 2 days, as shown by immunoblot analysis. GAPDH: loading control.
- Effect of eliglustat (2 μM) (pre)treatment for 4 days on SMAD3/SMAD4‐dependent (CAGA)12‐mediated transcriptional GFP reporter expression levels in A549‐VIM‐RFP cells transduced with the CAGA‐GFP lentiviral vector and treated with TGF‐β and/or SB505124 (SB, 1 μM) for the indicated times. The time course of the GFP signal was monitored with an IncuCyte system. The GFP object intensity was normalized to the green intensity at 0 h.
- qRT–PCR analysis of TGF‐β target genes, including SMAD7, SERPINE1 and CCN2, in A549‐VIM‐RFP cells pretreated with eliglustat (2 μM) for 6 days and then treated with vehicle control or TGF‐β for 6 h.
- CDH1, CDH2, and VIM mRNA levels in A549‐VIM‐RFP cells pretreated with eliglustat (2 μM) for 6 days and then treated with vehicle control or TGF‐β for 6 h.
- Alexa Fluor 488 phalloidin staining of F‐actin (green) in A549‐VIM‐RFP cells pretreated with eliglustat (2 μM) for 4 days and then treated with vehicle control or TGF‐β for 48 h. Nuclei were counterstained with DAPI (blue). Images were acquired with confocal microscopy. Scale bar = 50 μm.
- Detection of GM1 ganglioside expression in NMuMG cells upon treatment with eliglustat (1 or 2 μM) for 4 days, as analyzed by flow cytometry using Alexa Fluor 488‐conjugated CTB.
- Immunoblot analysis of p‐SMAD2 and t‐SMAD2 in NMuMG cells treated with eliglustat (5 or 10 μM) for 4 days and with vehicle control and/or TGF‐β for 1 h. GAPDH: loading control.
- Western blot analysis of the expression of the epithelial marker E‐cadherin and mesenchymal marker N‐cadherin in NMuMG cells (pre) treated with eliglustat (5 or 10 μM) for 4 days and with vehicle control and/or TGF‐β for 2 days. Tubulin: loading control.
Data information: TGF‐β was applied at a final concentration of 2.5 ng/ml. All data are expressed as the means ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. In (B), statistical analysis was based on two‐way ANOVA. In (C, D), statistical analysis was based on unpaired Student's t‐test.
Taken together, our data demonstrate that UGCG and UGCG‐defined GSLs inhibit TGF‐β signaling and EMT in mouse epithelial NMuMG cells and human lung cancer A549 cells, as well as the migration, extravasation, and metastasis of A549 cells.
Inhibition of GSL biosynthesis decreases the localization of TβRI in lipid rafts and protects TβRI from ubiquitination and increases its stability
Glycosphingolipids were found to impact signal transduction pathways by regulating the internalization of cell surface proteins. They can do so by facilitating lateral interactions between membrane‐anchored molecules, such as transmembrane protein receptors, in lipid rafts or caveolae membranes (Simons & Ikonen, 1997; Anderson & Jacobson, 2002; Mukherjee & Maxfield, 2004; von Zastrow & Sorkin, 2007). However, how GSLs control TGF‐β receptor signaling and TGF‐β‐receptor‐induced EMT is not well understood. Our immunoblot analysis of total TβRI expression in both NMuMG cells and A549‐VIM‐RFP cells showed that the genetic or pharmacological inactivation of GSLs had no effect on TβRI when the total level in cell lysates was examined (Appendix Fig S5A and B). However, when we specifically examined the cell surface TβRI pool, we observed a significant increase in cell surface TβRI levels in A549‐VIM‐RFP cells treated with eliglustat (Appendix Fig S5C). Therefore, we investigated whether inhibition of GSL synthesis can affect the partitioning of TβRI between lipid raft and non‐lipid raft microdomains in the plasma membrane. We performed sucrose density gradient ultracentrifugation to isolate the raft and nonraft fractions in Ugcg KO NMuMG cells and A549‐VIM‐RFP cells treated with the UGCG inhibitor eliglustat. The distinctive distribution of the specific lipid raft marker flotillin‐1 and that of nonraft markers, including β1‐integrin and EEA1, in nonraft fractions indicated the successful isolation of fractions containing lipid raft and non‐lipid raft microdomains (Fig 4A and C). UGCG depletion led to a decrease in lipid raft microdomains, which resulted in a decreased level of TβRI in raft‐containing fractions (fractions 3–5), in Ugcg KO cells compared to control NMuMG cells (Fig 4A and B). Consistent with this finding, inhibition of GSL biosynthesis in A549‐VIM‐RFP cells by the UGCG inhibitor eliglustat also decreased the TβRI levels in lipid raft fractions (fractions 3–6) (Fig 4C and D). Furthermore, we examined the effects of eliglustat on the ubiquitination of TβRI by overexpressing Myc‐tagged constitutively active TβRI (caTβRI) and HA‐tagged ubiquitin (HA‐Ub) in human embryonic kidney (HEK)293T cells. The UGCG inhibitor eliglustat mitigated SMAD7–SMURF2‐induced caTβRI ubiquitination in the presence of the proteasome inhibitor MG132 (Fig 4E, Appendix Fig S5D). The role of eliglustat in regulating the stability of TβRI was studied by examining TβRI expression after treatment with the protein synthesis inhibitor cycloheximide (CHX) in A549‐VIM‐RFP cells. The half‐life of the TβRI protein was prolonged by eliglustat treatment (Fig 4F and G). Taken together, these results suggest that mechanistically, the GSL‐induced inhibition of TGF‐β signaling is caused by favoring the localization of TβRI in lipid rafts, thereby triggering its ubiquitination and subsequent degradation.
Figure 4. Inhibition of GSL biosynthesis decreases the TβRI level in lipid rafts and inhibits the ubiquitination and degradation of TβRI.
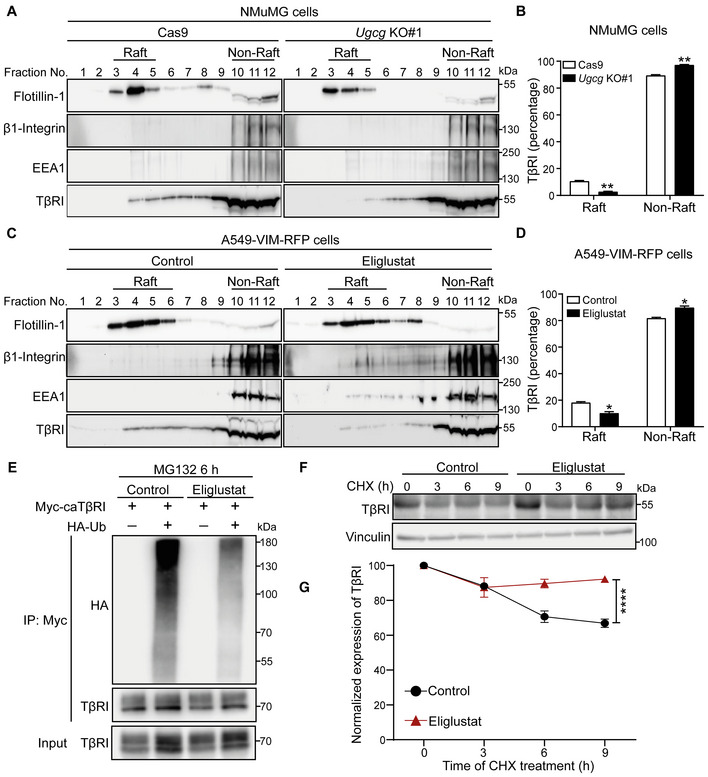
- Lysates from NMuMG control cells (Cas9‐expressing cells) and Ugcg KO cells were subjected to sucrose density gradient ultracentrifugation. The expression levels of flotillin‐1, β1‐integrin, EEA1, and TβRI in the sucrose gradient fractions were analyzed by immunoblotting. Fractions 3, 4, and 5 contained lipid rafts, whereas fractions 10–12 corresponded to the non‐lipid raft fractions.
- Quantification of TβRI percentages in lipid raft and non‐lipid raft fractions from Ugcg KO NMuMG and control cells.
- Isolation of lipid rafts from other cellular components in A549‐VIM‐RFP cells treated with the UGCG inhibitor eliglustat (2 μM) for 6 days using sucrose density gradient ultracentrifugation and measurement of flotillin‐1, β1‐integrin, EEA1, and TβRI levels in the sucrose gradient fractions using immunoblot analysis. Fractions 3–6 contained lipid rafts, whereas fractions 10–12 corresponded to the non‐lipid raft fractions.
- Quantification of TβRI percentages in lipid raft and non‐lipid raft fractions from A549‐VIM‐RFP cells treated with eliglustat (2 μM) for 6 days.
- Ubiquitination of TβRI was detected by immunoprecipitation (IP) of Myc‐tagged caTβRI from HA‐Ub‐transfected HEK293T cells with or without eliglustat (2 μM) treatment for 6 days. All groups were treated with MG132 (5 μM) for 6 h.
- Immunoblot analysis of TβRI expression levels in control and eliglustat (2 μM)‐treated A549‐VIM‐RFP cells treated with 50 μg/ml CHX for the indicated times. Vinculin: loading control.
- Quantification of TβRI expression levels with normalization to the t = 0 controls.
Data information: In (B, D), the data are expressed as the mean ± SD values from two biological replicates (n = 2). In (G), the data are expressed as the means ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01; ****P < 0.0001 based on unpaired Student's t‐test.
ST3GAL5‐mediated biosynthesis of a‐series gangliosides inhibits TGF‐β signaling and TGF‐β‐induced EMT
After showing the inhibitory effects of GSLs on TGF‐β signaling and TGF‐β‐induced EMT, we sought to elucidate the specific enzyme(s) involved in the biosynthesis of the GSLs found to be decreased in response to TGF‐β challenge. Since ~ 95% of the GSLs decreased by TGF‐β stimulation in A549‐VIM‐RFP cells were gangliosides, we focused our studies on the two critical enzymes B4GALNT1 and ST3GAL5, which catalyze the synthesis of o‐series and a‐ or b‐series gangliosides, respectively. To investigate the functional involvement of these enzymes, we generated B4GALNT1‐ or ST3GAL5‐depleted A549 cells using siRNA‐mediated transfection. The high efficiency of ST3GAL5 and B4GALNT1 knockdown was validated using qRT–PCR analysis (Appendix Fig S6A and B). Glycomic analysis of cells deficient in ST3GAL5 or B4GALNT1 using PGC nano‐LC‐ESI‐MS/MS confirmed the large decreases in specific ganglioside products produced by these two enzymes (Fig 5A; Appendix Fig S6C and D). ST3GAL5 knockdown cells showed significant decreases in GM3, GM2, and GM1a (Appendix Fig S6E), consistent with the findings after TGF‐β treatment. B4GALNT1 knockdown, however, induced a decrease in the total GSL level (Appendix Fig S6D) and led to an increase in the GM3 level (Appendix Fig S6E), an effect very different from that of TGF‐β on ganglioside levels.
Figure 5. ST3GAL5 inhibits TGF‐β signaling and TGF‐β‐induced EMT.
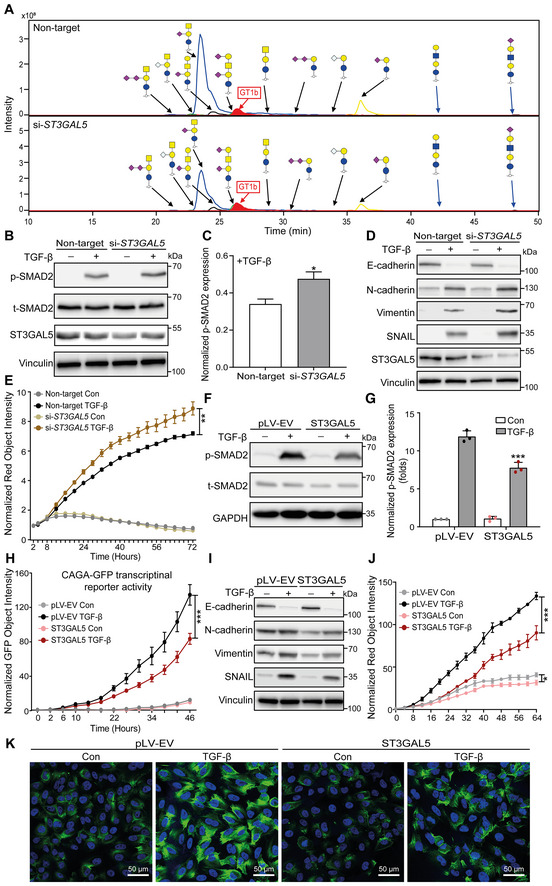
- Extracted ion chromatograms of GSL‐glycans released from A549‐VIM‐RFP cells transfected with nontargeting, ST3GAL5 or B4GALNT1 siRNA. GT1b: internal normalization control.
- Immunoblot analysis of p‐SMAD2, t‐SMAD2, and ST3GAL5 in A549‐VIM‐RFP cells with siRNA‐mediated ST3GAL5 knockdown or transfection of nontargeting siRNA and treated with vehicle control or TGF‐β for 1 h. Vinculin: loading control.
- Quantification of the p‐SMAD2 level in A549‐VIM‐RFP cells transfected with nontargeting or ST3GAL5 siRNA and treated with TGF‐β as shown in (B).
- Expression levels of ST3GAL5, the epithelial marker E‐cadherin, and mesenchymal markers, including N‐cadherin, vimentin, and SNAIL, in siRNA ST3GAL5‐depleted and nontargeting siRNA‐transfected A549‐VIM‐RFP cells treated with vehicle control or TGF‐β for 48 h. Vinculin: loading control.
- The time course of RFP‐tagged vimentin expression was monitored with an IncuCyte system in siRNA ST3GAL5 knockdown and nontargeting siRNA‐transfected A549‐VIM‐RFP cells treated with vehicle control or TGF‐β for the indicated times. The red object intensity was normalized to the red intensity at 0 h.
- Immunoblot analysis of p‐SMAD2 and t‐SMAD2 in A549‐VIM‐RFP cells transduced with empty vector (pLV‐EV) or the ST3GAL5 expression construct and stimulated with vehicle control or TGF‐β for 1 h. GAPDH: loading control.
- Quantification of the p‐SMAD2 level in A549‐VIM‐RFP cells transduced with pLV‐EV or the ST3GAL5 overexpression construct and treated with TGF‐β, as shown in (F).
- A549‐VIM‐RFP cells transduced with the CAGA‐GFP lentiviral vector and with the pLV‐EV control or ST3GAL5 expression construct were treated with vehicle control or TGF‐β for the indicated times. SMAD3/SMAD4‐dependent (CAGA)12‐mediated transcriptional GFP reporter expression levels were monitored with an IncuCyte system. The GFP object intensity was normalized to the green intensity at 0 h.
- Immunoblot analysis of the epithelial marker E‐cadherin and mesenchymal markers, including N‐cadherin, vimentin, and SNAIL, in A549‐VIM‐RFP cells transduced with the pLV‐EV control or ST3GAL5 expression construct and treated with vehicle control or TGF‐β for 48 h. GAPDH: loading control.
- Real‐time expression of RFP‐tagged vimentin was monitored with an IncuCyte system in A549‐VIM‐RFP cells transduced with pLV‐EV or the ST3GAL5 expression construct and treated with vehicle control or TGF‐β for the indicated times. The red object intensity was normalized to the red intensity at 0 h.
- Alexa Fluor 488 phalloidin staining of F‐actin (green) in pLV‐EV control‐ or ST3GAL5‐expressing A549‐VIM‐RFP cells after stimulation with vehicle control or TGF‐β for 48 h. Nuclei were counterstained with DAPI (blue). Images were acquired with confocal microscopy. Scale bar = 50 μm.
Data information: TGF‐β was applied at a final concentration of 2.5 ng/ml. In (C, E, G, H, J), the data are expressed as the mean ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01; ***P < 0.001. In (C), statistical analysis was based on unpaired Student's t‐test. In (E, G, H, J), statistical analysis was based on two‐way ANOVA.
Next, we investigated the effects of ST3GAL5 and B4GALNT1 knockdown on TGF‐β signaling and EMT in A549 cells. Depletion of ST3GAL5 enhanced TGF‐β‐induced SMAD2 phosphorylation (Fig 5B and C), while B4GALNT1 knockdown had no significant effect (Appendix Fig S7A). Knockdown of ST3GAL5 but not of B4GALNT1 enhanced the TGF‐β‐induced decrease in the level of the epithelial marker E‐cadherin and increases in the levels of mesenchymal markers, that is, N‐cadherin, vimentin, and SNAIL (Fig 5D; Appendix Fig S7B). This finding was further confirmed by the increase in TGF‐β‐induced RFP‐tagged vimentin expression upon ST3GAL5 depletion in A549‐VIM‐RFP cells (Fig 5E). Moreover, we overexpressed ST3GAL5 in A549‐VIM‐RFP cells using lentiviral transduction and confirmed the high mRNA and protein expression levels of ST3GAL5 in cells transduced with the ST3GAL5 construct (Fig EV3A and B). Ectopic expression of ST3GAL5 inhibited the TGF‐β signaling pathway by downregulating TGF‐β‐induced SMAD2 phosphorylation and SMAD3‐dependent CAGA‐GFP reporter activity, as well as decreasing the transcript levels of TGF‐β/SMAD target genes, including SMAD7, SERPINE1, and CCN2 (Fig 5F–H; Fig EV3C). TGF‐β‐induced EMT was also suppressed by ST3GAL5 overexpression, as demonstrated by the increased protein and mRNA expression levels of the epithelial marker E‐cadherin and decreased levels of mesenchymal markers, including N‐cadherin, vimentin, and SNAIL (Figs 5I and EV3D). Consistent with this finding, ectopic expression of ST3GAL5 significantly inhibited the dynamic changes in RFP‐tagged vimentin expression and F‐actin formation in A549‐VIM‐RFP cells (Fig 5J and K). All of these results indicate that ST3GAL5 but not B4GALNT is the key player in TGF‐β signaling and TGF‐β‐induced EMT.
Figure EV3. related to Fig 5. ST3GAL5 and its downstream products GM3, GM2, and GM1a inhibit TGF‐β signaling and TGF‐β‐induced EMT.
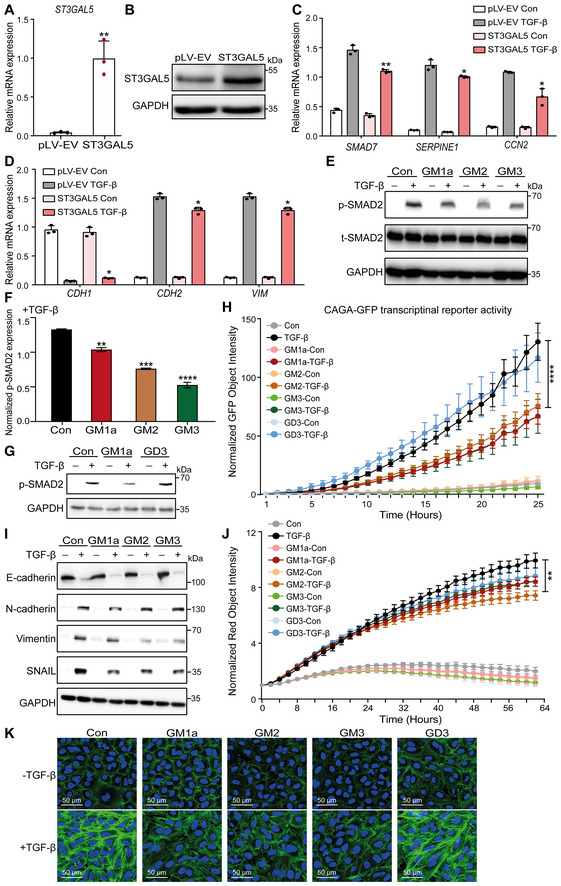
- qRT–PCR analysis of ST3GAL5 in A549‐VIM‐RFP cells lentivirally transduced with the pLV‐EV control or ST3GAL5 expression construct.
- Immunoblot analysis of ST3GAL5 expression in A549‐VIM‐RFP cells transduced with the pLV‐EV or ST3GAL5 expression construct. GAPDH, loading control.
- qRT–PCR analysis of TGF‐β target genes, including SMAD7, SERPINE1 and CCN2, in A549‐VIM‐RFP cells transduced with the pLV‐EV control or ST3GAL5 expression construct and treated with vehicle control or TGF‐β for 6 h.
- CDH1, CDH2 and VIM mRNA levels in A549‐VIM‐RFP cells transduced with or without the ST3GAL5 expression construct and treated with vehicle control or TGF‐β for 2 days.
- A549‐VIM‐RFP cells were preincubated with 50 μg/ml GM1a, GM2, or GM3 for 24 h and were then treated with vehicle control or TGF‐β for 1 h. Immunoblot analysis of p‐SMAD2 and t‐SMAD2 was then performed. GAPDH: loading control.
- Quantification of the p‐SMAD2 level in A549‐VIM‐RFP cells treated with 50 μg/ml GM1a, GM2, or GM3 in combination with TGF‐β (results shown in E).
- A549‐VIM‐RFP cells were pretreated with 50 μg/ml GM1a or GD3 for 24 h and were then treated with vehicle control or TGF‐β for 1 h. The levels of p‐SMAD2 were measured by western blotting. GAPDH: loading control.
- A549‐VIM‐RFP cells transduced with the CAGA‐GFP lentiviral vector were pretreated with 50 μg/ml GM1a, GM2, GM3 or GD3 for 24 h and were then treated with vehicle control or TGF‐β for the indicated times. SMAD3/SMAD4‐dependent (CAGA)12‐mediated transcriptional GFP reporter expression levels were monitored with an IncuCyte system. The GFP object intensity was normalized to the green intensity at 0 h.
- The expression levels of the epithelial marker E‐cadherin and mesenchymal markers, including N‐cadherin, vimentin, and SNAIL, in A549‐VIM‐RFP cells preincubated with 50 μg/ml GM1a, GM2, or GM3 for 24 h and then treated with vehicle control or TGF‐β for another 48 h were measured by immunoblotting. GAPDH: loading control.
- A549‐VIM‐RFP cells were pretreated with 50 μg/ml GM1a, GM2, GM3 or GD3 for 24 h and were then incubated with vehicle control or TGF‐β (2.5 ng/ml) for the indicated times. Real‐time expression of RFP‐tagged vimentin was monitored with an IncuCyte system, and the red object intensity was normalized to the red intensity at 0 h.
- Alexa Fluor 488 phalloidin staining of F‐actin (green) in A549‐VIM‐RFP cells preincubated with 50 μg/ml GM1a, GM2, GM3 or GD3 for 24 h and then treated with vehicle control or TGF‐β for another 48 h. Nuclei were counterstained with DAPI (blue). Images were acquired with confocal microscopy. Scale bar = 50 μm.
Data information: TGF‐β was applied at a final concentration of 2.5 ng/ml. The data are expressed as the mean ± SD values from three biological replicates (n = 3). *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. In (A, C, D, F), statistical analysis was based on unpaired Student's t‐test. In (H, J), statistical analysis was based on two‐way ANOVA.
ST3GAL5 catalyzes the conversion of LacCer into GM3, which is the precursor ganglioside for extension and further branching reactions to produce a‐series gangliosides, including GM2, GM1a, and GD1a and b‐series gangliosides such as GD3 and GD2 (Schnaar & Kinoshita, 2015). Both types of gangliosides were highly expressed in NMuMG and A549 cells (Fig 1E). Next, we examined the effects of exogenous addition of individual a‐ or b‐series gangliosides on TGF‐β signaling. As expected, addition of exogenous GM1a, GM2, or GM3 caused inhibition of TGF‐β/SMAD2 signaling in A549‐VIM‐RFP cells (Fig EV3E and F). However, exogenous addition of GD3 elicited no effect on this response (Fig EV3G). Consistent with this finding, A549 cells pretreated with GM1a, GM2, or GM3 exhibited significant inhibition of TGF‐β‐induced SMAD3‐dependent CAGA‐GFP reporter activity, while GD3‐pretreated cells showed no changes compared to control cells (Fig EV3H). Furthermore, exogenous addition of GM1a, GM2, or GM3 abrogated the TGF‐β‐induced expression of mesenchymal markers, including N‐cadherin, vimentin, and SNAIL (Fig EV3I). In addition, the TGF‐β‐induced promotion of RFP‐tagged vimentin expression was attenuated by addition of GM1a, GM2, or GM3 but not of GD3 (Fig EV3J). Moreover, upon stimulation with exogenous GM1a, GM2, or GM3, A549‐VIM‐RFP cells showed less F‐actin formation than control cells after TGF‐β treatment (Fig EV3K). In contrast, the exogenously added GD3 had no effect on F‐actin formation in A549‐VIM‐RFP cells (Fig EV3K). Taken together, these results indicate the pivotal role of ST3GAL5‐synthesized a‐series but not b‐series gangliosides in TGF‐β signaling and TGF‐β‐induced EMT.
ST3GAL5 promotes SMAD7‐SMURF2‐induced ubiquitination and degradation of TβRI
We next investigated the mechanism by which ST3GAL5 induces the inhibition of TGF‐β signaling. In HEK293T cells engineered to express Myc‐tagged caTβRI and HA‐Ub, knockdown of ST3GAL5 (but not of B4GALNT1) significantly decreased the ubiquitination of TβRI in the presence of the proteasome inhibitor MG132 (Fig 6A; Appendix Fig S7C). Conversely, ectopic expression of ST3GAL5 or addition of exogenous GM3 (but not of GM1a, GM2 or GD3) enhanced TβRI ubiquitination in HEK293T cells overexpressing Myc‐caTβRI and HA‐Ub (Figs 6B and EV4A; Appendix Fig S7D). As the endogenous TβRI level is much higher in MDA‐MB‐231 cells than in NMuMG cells and A549 cells, we used this former cell line to investigate the effect of gangliosides on TβRI ubiquitination. We observed that addition of exogenous GM1a, GM2, or GM3 to MDA‐MB‐231 cells infected with HA‐Ub lentivirus promoted the ubiquitination of TβRI (Fig EV4B). In addition, the GM3‐induced increase in TβRI ubiquitination was attenuated by knockdown of SMURF2, indicating that this E3 ligase is involved in the ubiquitination of TβRI (Fig EV4C). In addition, the half‐life of TβRI was shortened upon ST3GAL5 overexpression or GM3 treatment (Figs 6C and D, and EV4D and E). We also analyzed the cell surface expression of endogenous TβRI epitope tagged with a HiBiT sequence in MDA‐MB‐231 cells. Cell surface expression of TβRI was examined in these cells treated with GM3, the proteasome inhibitor MG132, or the lysosome inhibitor bafilomycin A1 (BafA1) alone or in combination. The quantitative results showed that GM3 treatment significantly decreased the expression of TβRI on the cell surface, and this effect was attenuated by addition of the proteasome inhibitor MG132 or the lysosome inhibitor BafA1, indicating that GM3‐induced degradation of TβRI is mediated through both the proteasomal and lysosomal pathways (Fig EV4F). These results demonstrate that ST3GAL5‐catalyzed a‐series gangliosides mitigate TGF‐β signaling responses and increase SMURF2‐induced TβRI ubiquitination by decreasing both the cell surface expression and stability of TβRI (Fig 6E).
Figure 6. ST3GAL5 promotes TβRI ubiquitination and degradation.
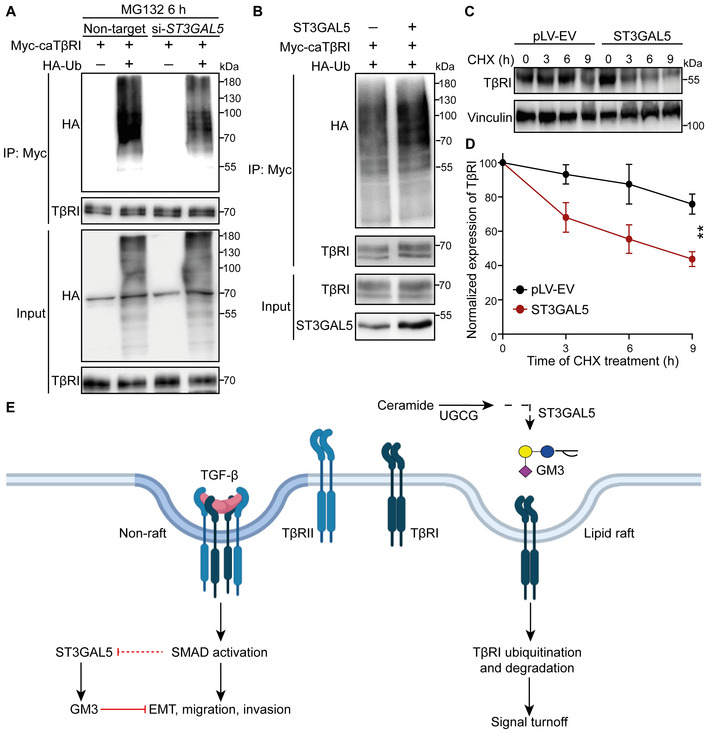
- HEK293T cells transfected with Myc‐tagged caTβRI, (HA‐Ub, and nontargeting siRNA or ST3GAL5 siRNA were collected for IP with an anti‐Myc antibody and immunoblot analysis. All groups were treated with MG132 (5 μM) for 6 h.
- Ubiquitination of TβRI was detected by IP of Myc‐tagged caTβRI from HA‐Ub‐transfected HEK293T cells with or without ST3GAL5 overexpression. All groups were treated with MG132 (5 μM) for 6 h.
- Western blot analysis of TβRI expression in A549‐VIM‐RFP cells transduced with the pLV‐EV control or ST3GAL5 expression construct and treated with CHX for the indicated times. Vinculin: loading control.
- Quantification of TβRI expression in pLV‐EV control‐ and ST3GAL5‐expressing A549‐VIM‐RFP cells. The results were normalized to the t = 0 controls. The data are shown as the mean ± SD of three independent experiments. **P < 0.01 based on two‐way ANOVA.
- Working model for UGCG–ST3GAL5–GM3‐mediated inhibition of TGF‐β signaling via induction of TβRI localization in lipid rafts. The dashed arrow indicates that the SMAD activation‐induced decrease in ST3GAL5 expression has not been formally shown.
Figure EV4. related to Fig 6. ST3GAL5 and its downstream products, including GM1a, GM2, and GM3, promote TβRI ubiquitination and degradation by increasing the TβRI level in lipid rafts.
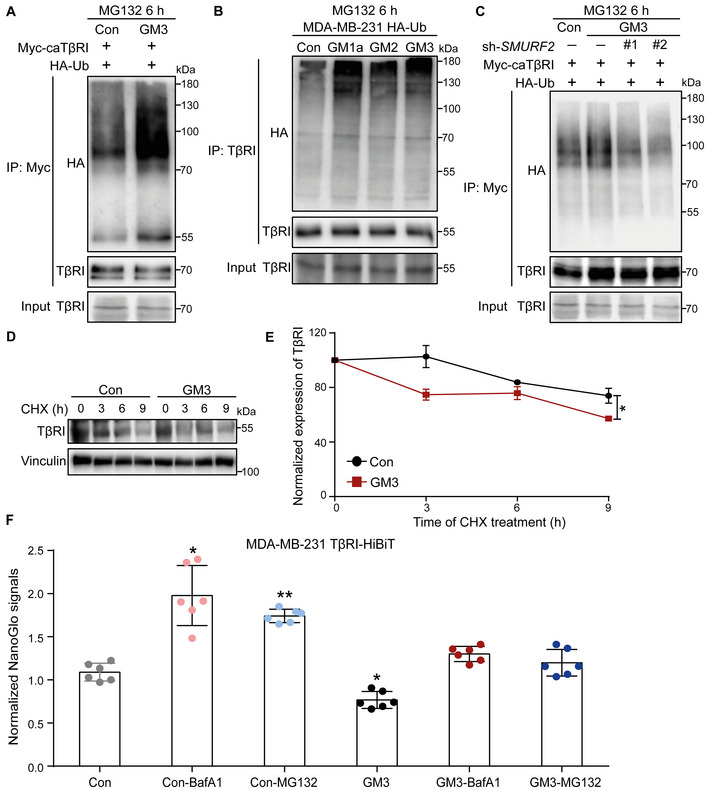
- Ubiquitination of TβRI was detected by IP of Myc‐tagged caTβRI from HA‐Ub‐transfected HEK293T cells with or without exogenous addition of GM3 (50 μg/ml) for 24 h. All groups were treated with MG132 (5 μM) for 6 h.
- Western blot analysis of whole‐cell lysates (Input) and immunoprecipitates from MDA‐MB‐231 cells stably expressing HA‐Ub with or without exogenous addition of 50 μg/ml GM1a, GM2 or GM3 for 24 h. Total ubiquitination of TβRI was evaluated. All groups were treated with MG132 (5 μM) for 6 h.
- HA‐Ub‐expressing HEK293T cells transfected with Myc‐tagged caTβRI and two SMURF2 shRNAs (sh#1 and sh#2) were treated with or without 50 μg/ml GM3 for 24 h. Cells were then harvested for IP with an anti‐Myc antibody followed by IB analysis of HA‐Ub and TβRI. All groups were treated with MG132 (5 μM) for 6 h.
- Western blot analysis of TβRI expression in A549‐VIM‐RFP cells with or without exogenous addition of GM3 (50 μg/ml) for 24 h followed by CHX treatment for the indicated times. Vinculin: loading control.
- Quantification of TβRI expression in the control and GM3‐treated groups of A549‐VIM‐RFP cells. Expression levels were normalized to the t = 0 controls. The data are shown as the mean ± SD of three independent experiments. *P ≤ 0.05 based on unpaired Student's t‐test.
- Measurement of TβRI‐HiBiT as evaluated by the Nano‐Glo signal in MDA‐MB‐231 cells with or without GM3 (50 μg/ml) challenge for 24 h followed by treatment with the proteasome inhibitor MG132 (5 μM) or lysosome inhibitor BafA1 (20 nM) for 6 h. The Nano‐Glo signal intensities were normalized to the GFP intensity in cells and are expressed as the mean ± SD of six biological replicates (n = 6). *P ≤ 0.05; **P < 0.01 based on unpaired Student's t‐test.
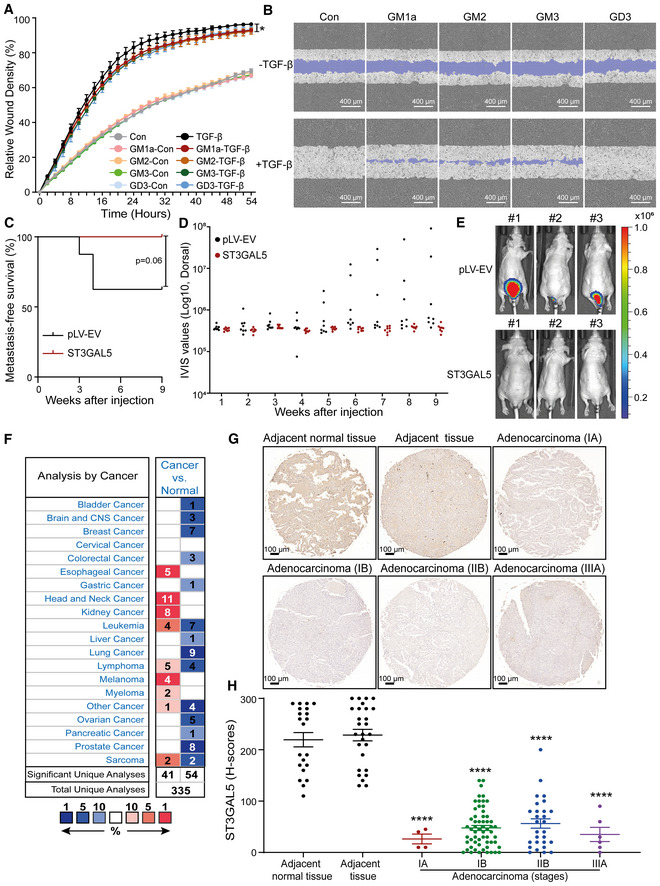
Ectopic expression of ST3GAL5 inhibits A549 cell migration, invasion, and metastatic outgrowth and is linked to a favorable prognosis in lung cancer
To investigate the function of ST3GAL5 and the gangliosides produced via its catalytic activity in A549 cell migration, we performed a scratch assay in A549 cells with siRNA‐mediated ST3GAL5 knockdown or transfection of control siRNA. While depletion of ST3GAL5 promoted the TGF‐β‐induced migration of A549‐VIM‐RFP cells (Fig 7A and B), knockdown of B4GALNT1 showed no effect on cell migration compared to that of nontargeting siRNA‐transfected control cells (Appendix Fig S8A and B). In contrast, overexpression of ST3GAL5 significantly inhibited TGF‐β‐induced migration (Fig 7C and D). Moreover, addition of GM1a, GM2, or GM3 but not GD3 decreased the migration ability of A549‐VIM‐RFP cells upon TGF‐β stimulation (Fig EV5A and B). Next, we analyzed the effect of misexpression of ST3GAL5 in lung cancer A549 cells on extravasation in a zebrafish xenograft model. mCherry‐expressing A549 cells with ST3GAL5 depletion or overexpression were injected into zebrafish embryos, and extravascular clusters were counted 4 days after injection. More cell clusters between blood vessels were observed in the group of zebrafish embryos injected with ST3GAL5 knockdown A549 cells than in the group of zebrafish embryos injected with nontarget siRNA‐transfected A549 cells (Fig 7E and F). This enhanced invasive ability of A549 cells was attenuated by addition of the TβRI inhibitor SB505124, implying that the increase in migration upon ST3GAL5 knockdown is at least partially caused by increased TβRI signaling (Fig 7E and F). Conversely, ectopic expression of ST3GAL5 strongly inhibited the extravasation of A549 cells in the zebrafish model (Fig 7G and H). Then, we used 5‐week‐old BALB/c nu/nu mice injected with A549‐Luc cells with or without ST3GAL5 overexpression to investigate the effects of ST3GAL5 on cell metastatic outgrowth ability. The overexpression efficiency of ST3GAL5 in A549‐Luc cells (derived from a single cell clone) was confirmed by qRT–PCR and immunoblot analysis, and the decreased migration ability of these cells was confirmed by a scratch assay (Appendix Fig S9A–C). Consistent with our findings in vitro and in the zebrafish xenograft model, we observed a trend in which the initial cell metastasis was detected later in mice injected with ST3GAL5‐overexpressing A549‐Luc cells than in mice injected with A549‐Luc control cells (Fig EV5C). Additionally, ectopic expression of ST3GAL5 inhibited (although not significantly, as determined by the significance cutoff of 0.05) the early metastasis of circulating A549 cells (Fig EV5D and E; Appendix Fig S9D).
Figure 7. ST3GAL5 inhibits A549 cell migration and invasion, and its expression is associated with good prognosis in lung cancer.
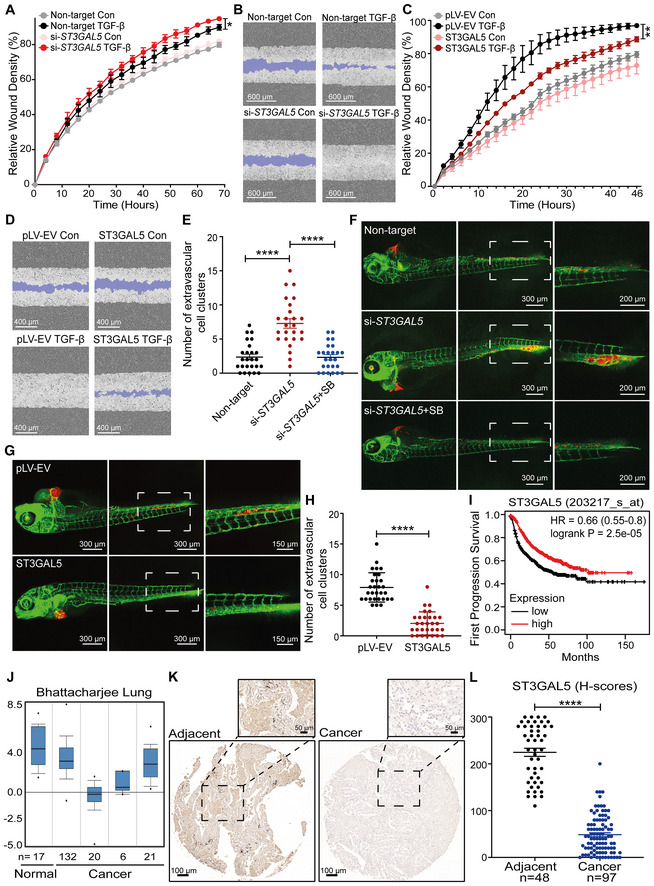
- Real‐time scratch assay results were analyzed by an IncuCyte system in ST3GAL5 siRNA‐depleted or nontargeting siRNA‐transfected A549‐VIM‐RFP cells treated with vehicle control or TGF‐β (2.5 ng/ml) for the indicated times. The relative wound density (closure) values are presented as the means ± SD values from three biological replicates (n = 3). *P ≤ 0.05 based on two‐way ANOVA.
- Representative images of a scratch wound at the final time point (68 h) in nontargeting siRNA‐transfected and ST3GAL5 siRNA knockdown A549‐VIM‐RFP cells treated with vehicle control or TGF‐β (2.5 ng/ml). The region of the original scratch is indicated in purple. Scale bar = 600 μm.
- The time course of wound closure was analyzed by an IncuCyte system in A549‐VIM‐RFP cells transduced with the pLV‐EV control or ST3GAL5 expression construct and treated with vehicle control or TGF‐β (2.5 ng/ml) for the indicated times. The relative wound density (closure) values are presented as the means ± SD values from three biological replicates (n = 3). **P < 0.01 based on two‐way ANOVA.
- Representative images of a scratch wound at the 46 h time point in A549‐VIM‐RFP cells transduced with the pLV‐EV control or ST3GAL5 ectopic expression construct. The original scratch region is indicated in purple. Scale bar = 400 μm.
- mCherry‐labeled A549 cells with ST3GAL5 siRNA depletion or transfection of nontargeting control siRNA were injected into ducts of Cuvier of zebrafish embryos. Zebrafish embryos in the SB group were treated with the inhibitor SB505124 (1 μM) in the egg water for 4 days after injection with daily refreshment of the treatment. The number of extravasated cell clusters was quantified in 25 embryos per group. ****P < 0.0001 based on unpaired Student's t‐test, n = 2.
- Representative images with magnified regions (outlined with dotted squares) of extravasated A549 cells in the indicated groups were acquired 4 days after injection by confocal microscopy. Scale bar = 300 or 200 μm.
- In vivo zebrafish extravasation experiments with mCherry‐labeled A549 cells with or without ectopic expression of ST3GAL5. Representative images with magnified regions (outlined with dotted squares) of extravasated cells were acquired 4 days after injection by confocal microscopy. Scale bar = 300 or 150 μm.
- The number of extravasated cell clusters was quantified in 30 embryos injected with A549 cells transduced with the pLV‐EV control or ST3GAL5 expression construct. ****P < 0.0001 based on unpaired Student's t‐test, n = 2.
- Kaplan–Meier survival curves showing the first progression survival of lung cancer patients in a publicly available lung cancer dataset according to ST3GAL5 expression (n = 982).
- Box plots of ST3GAL5 gene expression levels in lung cancer tissues and normal tissues in the Bhattacharjee Lung database. The central bands indicate the medium expression values of ST3GAL5, boxes indicate the expression level ranges of ST3GAL5. Data are presented as the means ± SDs from the indicated number of tissues.
- Representative images of ST3GAL5 immunohistochemistry in a human lung tissue microarray including normal and cancer tissues. Wide field and magnified images (outlined with dotted squares) are shown. Scale bar = 100 μm.
- Scatter plot showing the expression of ST3GAL5 in normal and cancerous lung tissues. Each point represents the H‐score of a single tissue sample with ST3GAL5 staining ranging from completely absent (H‐score 0) to very strong (H‐score 300). H‐scores are presented as the means ± SDs; normal tissues, n = 48; cancer tissues, n = 97; ****P < 0.0001 based on unpaired Student's t‐test.
Figure EV5. related to Fig 7. ST3GAL5 expression levels in lung cancer tissues, and ST3GAL5‐catalyzed a‐series gangliosides, but not B4GALNT1 or exogenous GD3, inhibit cell migration and metastasis.
- A549‐VIM‐RFP cells were pretreated with 50 μg/ml GM1a, GM2, GM3, or GD3 for 24 h and were then incubated with vehicle control or TGF‐β (2.5 ng/ml) for the indicated times. The time course of wound healing was analyzed by an IncuCyte system. The relative wound density (closure) values are presented as the means ± SD values from three biological replicates (n = 3). *P ≤ 0.05 based on two‐way ANOVA.
- Representative images of a scratch wound at the 50 h time point in A549‐VIM‐RFP cells with exogenous addition of 50 μg/ml GM1a, GM2, GM3, or GD3 and treatment with vehicle control or TGF‐β (2.5 ng/ml). The original scratch region is indicated in purple. Scale bar = 400 μm.
- Kaplan–Meier analysis of metastasis‐free survival of eight mice injected with A549‐Luc cells transduced with the pLV‐EV control or eight mice in the group with ST3GAL5 expression construct. The log‐rank test was used for statistical analysis; P = 0.06.
- Analysis of the IVIS values from eight mice in each indicated group.
- Whole‐body images (BLI) at 9 weeks of three mice injected with A549‐Luc cells transduced with the pLV‐EV control or ST3GAL5 expression construct. In Appendix Fig S9D, the BLI of eight mice in each group are shown.
- Oncomine database summary of ST3GAL5 gene expression in various cancers. The comparison indicates the number of datasets with statistically significant ST3GAL5 mRNA overexpression (red) and downregulation (blue) in cancer versus normal tissues. The colors indicate the best gene rank percentiles for the within‐data set analyses.
- A microarray containing tissues from lung cancer patients was stained with an anti‐ST3GAL5 antibody. Representative images of lung cancer biopsies with adenocarcinoma and adjacent normal tissues are shown. Scale bar = 100 μm.
- Scatter plot of ST3GAL5 expression in normal lung tissues and lung cancer tissues of different stages. Each point represents the H‐score from a single tissue sample with ST3GAL5 staining ranging from completely absent (H‐score: 0) to very strong (H‐score: 300). H‐scores are presented as the means ± SDs; adjacent normal tissue, n = 21; cancer‐adjacent lung tissue (adjacent tissue), n = 27; adenocarcinoma (stage IA), n = 4; adenocarcinoma (stage IB), n = 59; adenocarcinoma (stage IIB), n = 28; adenocarcinoma (stage IIIA), n = 6; ****P < 0.0001 based on unpaired Student's t‐test.
Next, given the critical role of ST3GAL5 in the regulation of cell migration and invasion, we investigated whether ST3GAL5 might be a relevant biomarker in early‐stage lung cancer. Using a publicly available cohort of 982 lung cancer patients, we found that low expression of ST3GAL5 was associated with a poor prognosis of survival until first progression (Fig 7I; Gyorffy et al, 2013). In addition, analysis of the mRNA expression level of ST3GAL5 using the Oncomine database showed that ST3GAL5 is frequently expressed at low levels in various human cancer tissues, including lung cancer tissues, compared to the corresponding adjacent normal tissues (Fig EV5F; Neapolitan et al, 2015; Cabarcas‐Petroski et al, 2020; Saha et al, 2020). This finding is consistent with the results obtained by analysis of the Bhattacharjee Lung database: low levels of ST3GAL5 were detected in lung cancer compared to adjacent normal tissues (Fig 7J; Bhattacharjee et al, 2001; Granville & Dennis, 2005). Moreover, we examined the ST3GAL5 protein level in a tissue microarray with 150 lung cancer tissues and adjacent phenotypically normal tissues derived from 50 patients by immunohistochemical (IHC) staining with an ST3GAL5‐specific antibody. ST3GAL5 was highly expressed in adjacent normal tissues compared to cancer tissues (Fig 7K). Moreover, ST3GAL5 was barely present in lung cancer tissues of different stages from stage IA to stage IIIA with increased growth and metastatic abilities (Fig EV5G). Quantification of the H scores for ST3GAL5 confirmed that ST3GAL5 levels are low in lung adenocarcinoma tissues compared to adjacent normal tissues (Figs 7L and EV5H). In conclusion, these results suggest that ST3GAL5 expression inhibits cell migration, invasion, and metastasis and is associated with good prognosis in lung cancer.
Discussion
TGF‐β‐induced EMT is mediated via a decrease in a‐series gangliosides
The NMuMG and A549 cell lines are frequently used models for TGF‐β‐induced EMT. Profiling GSLs in both cell lines in the absence of or upon several days of exposure to TGF‐β showed high similarities between GSLs in cells in the basal epithelial state and those in a TGF‐β‐induced mesenchymal state. The a‐series gangliosides, that is, GM3, GM2, GM1a, and GD1a, were the most abundant GSLs in both cell lines. Low levels of certain b‐series gangliosides, including GD3, were also detected in both NMuMG and A549 cells. Small amounts of (neo)‐lacto‐series GSLs, that is, nLc4 and S(3)nLc4, were detected in A549 cells but not in NMuMG cells. TGF‐β‐induced EMT in both cell lines coincided with striking decreases in all GSLs, especially a‐series gangliosides, consistent with the downregulation of ST3GAL5 (the key enzyme in a‐series ganglioside synthesis). A previous study reported on changes in the levels and composition of gangliosides in human normal bladder HCV29 cells and mouse NMuMG cells during TGF‐β‐induced EMT (Guan et al, 2009); significantly reduced levels of Gg4 and GM2, and slight but nonsignificant decreases in GM1 and GM3 levels were reported in response to TGF‐β stimulation. These findings are consistent with our results in NMuMG cells (Guan et al, 2009). Strikingly, we found that genetic Ugcg KO in NMuMG cells or pharmacological inhibition of GSL synthesis in both NMuMG and A549 cells promoted TGF‐β‐induced EMT. Therefore, it will be interesting to explore whether TGF‐β‐induced EMT in other pathophysiological contexts is accompanied by decreases in a‐series gangliosides.
UGCG and ST3GAL5 inhibit TGF‐β signaling and EMT
The production of a‐series gangliosides mediated by the glycotransferase UGCG and ST3GAL5 was identified as a critical negative modulator of TGF‐β signaling. In our study, analysis of the transcriptional profiles of wild‐type or Ugcg‐deficient NMuMG cells obtained by GSEA for TGF‐β gene response signatures and EMT gene signature sets revealed negative correlations between Ugcg expression and both TGF‐β signaling and EMT. Consistent with the results of genetic inactivation, pharmacologic inhibition of UGCG with eliglustat in A549 cells promoted TGF‐β signaling and TGF‐β‐induced EMT. Furthermore, we revealed the essential role of ST3GAL5‐synthesized a‐series gangliosides in inhibiting TGF‐β‐induced responses in A549 cells. ST3GAL5 knockdown but not depletion of B4GALNT1 mimicked the UGCG inactivation‐induced promoting effects on TGF‐β signaling, EMT and migration in A549 cells. Overexpression of ST3GAL5 or exogenous addition of ST3GAL5‐mediated a‐series gangliosides, including GM3, GM2, and GM1a, antagonized TGF‐β signaling and EMT in A549 cells. Consistent with this finding, UGCG inactivation or ST3GAL5 depletion promoted extravasation in zebrafish, while ectopic expression of ST3GAL5 showed the opposite effects, inhibiting the TGF‐β‐induced response. Moreover, our data showed that pretreatment of A549 cells with eliglustat prior to injection into mice and ectopic expression of ST3GAL5 in A549 cells promoted and inhibited metastatic outgrowth, respectively. Because the basal and TGF‐β‐induced migration/invasion mediated by UGCG‐ST3GAL5 inhibition were attenuated by the TGF‐β kinase inhibitor SB505124, it can be concluded that these responses are dependent on TβRI signaling. Thus, our findings narrow down the GSLs involved in TGF‐β signaling and the TGF‐β‐induced changes to cell phenotype to the specific a‐series subtype of gangliosides. In addition, these findings, taken together with the TGF‐β‐induced decrease in a‐series gangliosides, indicate that these gangliosides participate in a self‐enabling response of EMT promoted by TGF‐β.
Consistent with our results, chemoenzymatically synthesized GM3 effectively inhibited the migration of melanoma B16‐F10 cells (Li et al, 2021). In addition, GM3 synthase overexpressing A2780 ovarian carcinoma cells were characterized by increased levels of caveolin‐1 and reduced motility in vitro (Prinetti et al, 2011). Enhanced synthesis of GM3 also caused a decrease in the invasiveness of bladder cancer cells (Satoh et al, 2001). Furthermore, previous publications demonstrated that GM3 and GM1 overexpression was negatively correlated with tumorigenicity. In rat PC12 cells, GM1 significantly enhanced neurite outgrowth (Mutoh et al, 1995). Exogenous addition of GM3 to a human neuroblastoma cell line (NBL‐W) inhibited the invasion of glioma cells in rat brain slice cultures, an effect that manifested earlier than the inhibition of cell proliferation and induction of apoptosis (Fujimoto et al, 2005). Our findings, taken together with other reports, demonstrate the inhibitory role of UGCG‐ST3GAL5‐synthesized a‐series gangliosides in various cancer types. Notably, in human lens epithelial cells, expression of GM3 promoted TGF‐β signaling and TGF‐β‐induced EMT via potential interactions with TβRs (Kim et al, 2013). While we observed no significant differences in TGF‐β signaling and EMT upon treatment with exogenous GD3, others reported that ST8SIA1, an enzyme involved in GD3 and GD2 biosynthesis, plays an important role in the initiation and maintenance of EMT (Sarkar et al, 2015). Inhibition of this enzyme suppressed the invasion and motility of breast cancer cells as well as metastasis in mice (Sarkar et al, 2015). Therefore, the effects of specific gangliosides, such as GD3 and GM3, on EMT and metastatic capacity may vary across different cancer types. This variability might be explained by the previously reported evidence that EMT and metastatic ability can result from the expression of more complex gangliosides as a consequence of dynamic alterations in GSLs upon experimental modulation (Cumin et al, 2021).
Specific GSLs inhibit TGF‐β signaling by favoring TβRI localization into lipid rafts
From a mechanistic perspective, we observed that UGCG‐ST3GAL5‐synthesized a‐series gangliosides inhibited TGF‐β signaling by favoring the localization of TβRI into lipid rafts. Lipid raft localization of TβRI was linked to an increase in its ubiquitination and degradation. Ubiquitination of TβRI was previously shown to be regulated by the SMURF2‐SMAD7 complex and intricately linked to the lysosomal/proteasomal degradation of receptors (Kavsak et al, 2000; Di Guglielmo et al, 2003; Chen, 2009). Consistent with this observation, the SMAD7 and SMURF2 overexpression‐induced promotion of TβRI ubiquitination was abolished by eliglustat treatment. In addition, treating cells with the proteasome inhibitor MG132 or the lysosome inhibitor BafA1 reversed the decrease in cell surface TβRI resulting from exogenous GM3 addition, suggesting that GM3 enhances TβRI degradation via the proteasomal and lysosomal pathways.
Gangliosides have also been implicated in regulating the EGFR signaling pathway (Kim et al, 2021). We therefore explored the effect of eliglustat treatment and ST3GAL5 misexpression on EGFR signaling. Eliglustat treatment increased cell surface EGFR expression in A549 cells and promoted the activation of EGFR signaling (Appendix Fig S10B). Ectopic ST3GAL5 expression in A549 cells reduced the p‐EGFR level without changing the level of total EGFR (Appendix Fig S10A). This result is consistent with previous reports demonstrating that GM3 addition inhibited EGFR activity (Rebbaa et al, 1996; Kawashima et al, 2009). Notably, TGF‐β‐induced ERK1/2 phosphorylation was slightly promoted by ST3GAL5 overexpression (Appendix Fig S10C), consistent with a previous report in which lipid rafts were found to be necessary for ERK–MAPK activation (Zuo & Chen, 2009). Thus, GSL‐enriched lipid rafts may regulate TGF‐β‐induced SMAD and non‐SMAD signaling in opposite ways.
ST3GAL5 expression correlates with good prognosis in lung cancer
Notably, mining of RNA expression data in publicly accessible databases of lung cancer mRNA profiles revealed that ST3GAL5 expression correlates with favorable prognosis in lung cancer. Consistent with this finding, our immunohistochemical analysis of ST3GAL5 protein expression showed that ST3GAL5 is expressed at significantly lower levels in lung cancer tissues than in adjacent normal tissues. Our finding is consistent with a study in which the expression of ST3GAL5 was analyzed in bladder cancer patients by mining publicly available datasets; low expression of ST3GAL5 was reported to be associated with muscle invasion, high grade, and poor prognosis in patients with bladder cancer (Ouyang et al, 2020). Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis focused on ST3GAL5 showed its association with TGF‐β signaling (Ouyang et al, 2020). In breast cancer, high expression of ST8SIA1 but not ST3GAL5 was significantly correlated with poor overall survival (Kan et al, 2020). In addition, some gangliosides, that is, GM2, GD2, and GD3, are highly expressed in various human tumors (melanomas, gliomas, neuroblastomas, and breast cancers) but are not expressed or weakly expressed in normal tissues (Lloyd & Old, 1989; Hakomori, 1996; Ouyang et al, 2020). All these studies performed in different cancer subtypes suggest the pleiotropic nature of specific gangliosides and related enzymes in cancer progression.
In conclusion, we performed an in‐depth quantitative glycomic analysis of GSLs and demonstrated that TGF‐β induced a decrease in GSLs‐specifically, a‐series gangliosides‐in mouse normal epithelial NMuMG cells and human lung cancer A549 cells undergoing EMT. Since inhibition of ST3GAL5‐mediated a‐series ganglioside synthesis promotes TGF‐β signaling and TGF‐β‐induced EMT, cell migration, cell invasion, and early metastasis, and TGF‐β‐induced inhibition of these gangliosides enforces a TGF‐β‐induced EMT response. The a‐series gangliosides that are enriched in lipid rafts stimulate the ubiquitination and degradation of TβRI. Moreover, we revealed an association between high ST3GAL5 expression and good prognosis in lung cancer patients. It will be interesting to explore the potential of ST3GAL5 as a prognostic biomarker and as a molecular target for lung cancer therapy.
Materials and Methods
Cell culture
Mouse NMuMG epithelial cells, HEK 293T cells, human lung adenocarcinoma A549 cells, A549‐VIM‐RFP cell lines, and MDA‐MB‐231 breast cancer cells were originally from American Type Culture Collection (ATCC). A549‐Luc cells were purchased from PerkinElmer (Waltham, USA). All these cell lines were cultured in Dulbecco's modified Eagle medium (DMEM, 11965092, Thermo) with 10% fetal bovine serum (FBS, S1860‐500, BioWest) and 100 U/ml penicillin–streptomycin (15140148, Thermo). Cells were frequently tested for the absence of mycoplasma contamination, and all human cell lines were authenticated by short tandem repeat (STR) profiling.
Chemical and reagents
Ammonium bicarbonate (ABC), trifluoroacetic acid, cation exchange resin beads (AG50W‐X8), potassium hydroxide, sodium borohydride, and cycloheximide (CHX) were obtained from Sigma‐Aldrich (Steinheim, Germany). The UGCG inhibitor eliglustat was purchased from MedChemExpress (HY‐14885A). The gangliosides GM1a (1545), GM2 (1542), GM3 (1503), GD3 (1504), and GT1b (1548) were purchased from Matreya (State College, PA). SB505124 was purchased from Tocris (3263). HPLC SupraGradient acetonitrile (ACN) was obtained from Biosolve (Valkenswaard, The Netherlands), and other reagents and solvents such as chloroform, methanol, 2‐propanol, and glacial acetic acid were obtained from Merck (Darmstadt, Germany). The 50 mg tC18 reverse phase (RP) cartridges were from Waters (Breda, The Netherlands). Endoglycoceramidase I (EGCase I, recombinant clone derived from Rhodococcus triatomea and expressed in Escherichia coli) and 10× EGCase I buffer (500 mM HEPES, 1 M NaCl, 20 mM DTT, and 0.1% Brij 35, pH 5.2) were purchased from New England BioLabs Inc. (Ipswich, MA). Bafilomycin A1 (BafA1, B1793), cycloheximide (CHX, 01810) and MG132 (474787) were purchased from Sigma‐Aldrich. Biotin (21335) was obtained from Thermo. The concentration of TGF‐β was applied as 2.5 ng/ml, and the same volume of ligand buffer (4 mM HCl, 0.1% BSA) was used as a vehicle control.
Preparation of GSL‐glycan alditols
The extraction of GSLs and the preparation of GSL‐glycan alditols from cells were performed in triplicate as previously described (Zhang et al, 2020b) with slight modifications. Shortly, 2 × 106 cells were lysed by 200 μl of water. In prior to GSL extraction, 2.5 μl of 0.5 μM ganglioside GT1b in ethanol were added as a spiked internal standard to monitor sample preparation and normalize absolute quantification. Crude GSLs were extracted and dried under vacuum in an Eppendorf Concentrator 5301 (Eppendorf, Hamburg, Germany) at 30°C. The extracted GSLs were dissolved in 100 μl of methanol followed by an addition of 100 μl water in prior to solid phase extraction (SPE) purification on tC18 RP cartridges. Eluate was collected and dried in an Eppendorf Concentrator. Next, a mixture of EGCase I (12 mU, 2 μl), EGCase I buffer (4 μl), and water (34 μl) (pH 5.2) was added to each sample and incubated for 36 h at 37°C to release the glycan head group. The released glycans were further purified by tC18 RP cartridges. The flow‐through and wash fractions were collected and dried in an Eppendorf Concentrator.
The reduction and desalting of GSL‐glycans were carried out with slight modifications following the same procedure as described in previous studies (Jensen et al, 2012; Zhang et al, 2020b). In brief, GSL‐glycans were reduced to alditols in 30 μl of sodium borohydride (500 mM) in potassium hydroxide (50 mM) for 3 h at 50°C. After desalting, the glycan alditols were collected by combining flow‐through and eluate and dried in an Eppendorf Concentrator at 30°C. A PGC SPE clean‐up was performed to further purify the samples. The purified glycan alditols were re‐suspended in 200 μl of water prior to PGC nano‐LC‐ESI‐MS/MS analysis.
Analysis of GSL‐glycan alditols using PGC nano‐LC‐ESI‐MS/MS
The analysis of glycan alditols was performed using PGC nano‐LC‐ESI‐MS/MS following a method described previously (Jensen et al, 2012, Zhang et al, 2020b). Measurements were performed on an Ultimate 3000U high‐performance liquid chromatography (HPLC) system (Thermo) equipped with a home‐packed PGC trap column (5 μm Hypercarb, 320 μm × 30 mm) and a home‐packed PGC nano‐column (3 μm Hypercarb 100 μm × 150 mm) coupled to an amaZon ETD speed ion trap (Bruker, Bremen, Germany). To analyze glycans, 2 μl of samples were injected. Separation was achieved with a linear gradient from 1% to 50% mobile phase B over 73 min at a 0.6 μl/min flow rate. The column was held at a constant temperature of 35°C.
Ionization was achieved using the nanoBooster source (Bruker) with a capillary voltage of 1,000 V applied, a dry gas temperature of 280°C at 3 l/min, and isopropanol‐enriched nitrogen at 3 psi. MS spectra were acquired within an m/z range of 340–1,850 in enhanced mode using negative ion mode. The smart parameter setting was set to m/z 900. MS/MS spectra were recorded using the three highest intensity peaks.
Glycan structures were assigned on the basis of the known MS/MS fragmentation patterns in negative‐ion mode (Karlsson et al, 2004a,b; Anugraham et al, 2015), elution order, and general glycobiological knowledge, with help of Glycoworkbench (Ceroni et al, 2008) and Glycomod (Cooper et al, 2001) software. Relative quantification of individual glycans was performed by normalizing the total peak area of all glycans within one sample to 100%. To estimate the glycan amount per cell, glycan intensity was normalized to the intensity of the internal standard GT1b. Afterward, assuming the complete release of glycans and similar response factors between released glycan and GT1b standard, the number of glycans per cell was estimated. Structures are depicted according to the Consortium of Functional Glycomics (CFG). The blue square is N‐acetylglucosamine; the yellow square is N‐acetylgalactosamine; the blue circle is glucose; the yellow circle is galactose; the red triangle is fucose; the purple diamond is N‐acetylneuraminic acid; and the gray diamond is N‐glycolylneuraminic acid. In the figures containing glycan structures, black arrows indicate gangliosides, green arrows indicate globosides, and blue arrows indicate neolacto series GSL.
Quantitative real‐time‐polymerase chain reaction (qRT–PCR)
Total RNA extraction was prepared using the NucleoSpin RNA II kit (740955, BIOKÉ). The concentration of RNA was determined by NanoDrop™ 2000/2000c (Thermo), and a total of 1 μg of RNA was retro‐transcribed using the RevertAid First Strand cDNA Synthesis Kit (K1621, Thermo). Real‐time reverse transcription‐PCR was conducted with SYBR Green (GoTaq DNA Polymerase, PROMEGA) using the CFX Connect Detection System (1855201, Bio‐Rad). The DNA primer sequences that were used to detect the expression of specific genes are listed in Appendix Table S2. All target gene expression levels were normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). If not stated otherwise, results are shown as biological triplicates with statistical analysis.
Ugcg knockout by CRISPR/Cas9
The mouse sgRNAs targeting Ugcg (forward: 5′‐CACCTCGTGCTCTTCGTGGTGCTG‐3′; reverse: 5′‐AAACCAGCACCACGAAGAGCACGA‐3′) were provided by Dr. Robbert M Spaapen (Sanquin Research, Amsterdam) and were cloned into the lentiviral vectors lentiCRISPR_v2 (Addgene; Jongsma et al, 2021). The design and cloning of sgRNA were performed according to the general protocols obtained from the Feng Zhang lab (Sanjana et al, 2014; Shalem et al, 2014). The pLV‐Cas9‐blasticidin plasmid (CAS9BST, Sigma) was used as a negative control.
For lentivirus production, the cloned l lentiCRISPR_v2 with Ugcg sgRNA inserts or the pLV‐Cas9 plasmid (Hunter et al, 2019) was co‐transfected into HEK293T cells with three packaging plasmids: pVSVg, psPAX2, and pAdv. The lentivirus was harvested at 48 h post‐transfection and filtered through a 0.45 μm polyethersulfone (PES) filter. Viruses were either directly used for infection or quickly stored at −80°C to avoid loss of titer.
To generate the stable NMuMG cells with Ugcg knockout, we first prepared a 1:1 dilution of the pLV‐Cas9 lentivirus in completed DMEM with 5 ng/ml polybrene (Sigma). Afterward, NMuMG cells were infected with the lentiviral dilution at a low cell density (30%), and cells were selected with 4 μg/ml blasticidin (R21001, Invitrogen) for 1 week after 24 h of infection to generate NMuMG cells with Cas9 expression. Once the Cas9 expression was validated, the NMuMG cells were further infected with Ugcg sgRNA lentivirus and selected by DMEM medium with 1 μg/ml puromycin for 1 week. Thereafter, two single cell colonies were picked up under microscope, and cells were then cultured and expanded.
UGCG activity assay
Cas9 and Ugcg KO clones were pelleted and resuspended in Buffer Q (10 mM Tris 7.0, 10 mM NaCl and 0.5 mM EDTA). Cells were lysed by passing them 10 times through a 26G needle. Post‐nuclear supernatant was collected by centrifuging for 5 min at 900 g. A post‐nuclear supernatant equivalent to 100 × 103 cells in 50 μl was combined with Buffer Q supplemented with 3.2 mM UDP‐Glucose (Sigma cat# 94335) and 1 mM BSA‐coupled BODIPY‐C5‐Ceramide (ThermoFisher cat# B22650). Reactions were incubated for 1 h at 37°C with constant shaking. Next, samples were supplemented with 100 μl of 2% NaCl and subjected to standard Bligh‐Dyer lipid extractions (PMID: 13671378). Lipids were dried and resuspended in chloroform/methanol (2:1, v/v) and spotted on a thin‐layer chromatography plate. Plates were developed in chloroform/acetone/acetic acid/methanol/water (50:20:10:10:5, v/v/v/v/v). Dried plates were imaged using a GE Typhoon FLA 9500 imager using a 488 nm laser and 530/20 (BPB1) band‐pass filters.
Small interfering RNA (siRNA) and short hairpin (sh)RNA transfections
A549‐VIM‐RFP cells were transfected with nontargeting (4390843, Dharmacon, Lafayette, CO), ST3GAL5 (L‐011546‐00‐0005, Dharmacon) or B4GALNT1 (L‐011279‐00‐0005, Dharmacon) siRNAs mixed with transfection reagent DharmaFECT1 #1 (T‐2001, Dharmacon). The mixture was incubated in a serum‐free medium for 20 min at room temperature and then added to cells. After 3 days of transfection, the target gene knockdown efficiencies of the cells were analyzed using qRT–PCR.
SMURF2 shRNAs were obtained from Sigma (MISSION shRNA library). We tested six SMURF2 shRNAs; the two most effective shRNAs for HEK293T cells were used for further experiments. These were sh#1‐SMURF2 (TRCN0000003478, 5′‐CCACCCTATGAAAGCTATGAA‐3′) and sh#2‐SMURF2 (TRCN0000010792, 5′‐GCTGGATTTCTCGGTTGTGTT‐3′).
Lentiviral infection and generation of stable cell lines
The expression construct containing the human ST3GAL5 was a gift from Marco Trinchera (Indellicato et al, 2019) and the cDNA was inserted into the pLV‐CMV‐IRES‐PURO lentiviral vector.
Lentiviruses were produced by transfecting HEK293T cells with the ST3GAL5 plasmid, pLV empty vector or pLV‐mCherry, and three packaging plasmids that are pCMV‐G protein of the vesicular stomatitis virus (VSVG), pMDLg‐RRE (gag‐pol) and pRSV‐REV as described (Zhang et al, 2012). The viruses were harvested at 48 h (h) post‐transfection and filtered through a 0.45 μm polyethersulfone (PES) filter. Viruses were either directly used for infection or stored at −80°C as soon as possible to avoid loss of titer.
To generate A549 cells stably overexpressing ST3GAL5, we first prepared a 1:1 dilution of the lentivirus in DMEM complemented with 5 ng/ml of Polybrene (Sigma). Thereafter, A549 cells were infected with the lentiviral ST3GAL5 expression construct at a low cell density (30%). After infection for 48 h, cells were selected with 1 μg/ml of puromycin for 1 week in order to generate the A549 cells with ST3GAL5 overexpression.
Dynamic detection of RFP‐vimentin expression assay
A549‐VIM‐RFP cells (in which the red fluorescent protein coding region is closed with the VIMENTIN locus) were used as a model system to indicate EMT by analyzing the changes of RFP‐tagged vimentin expression. The near confluent cells with the indicated treatment were cultured in a 96‐well plate in the IncuCyte live cell imaging system. The RFP signals were captured every 2 or 4 h over a period of 48–72 h using a 10× objective. The RFP‐vimentin density was then analyzed by the IncuCyte software and normalized by the RFP signals at 0 h for each group. All the experiments were performed with biological triplicates, and representative results are shown.
CAGA‐GFP transcriptional response assay
A549‐VIM‐RFP cells were infected by pLV‐CAGA12‐green fluorescent protein (GFP) lentivirus and selected with 1 μg/ml puromycin for 1 week to obtain the cells stably expressing a SMAD3/4‐dependent CAGA12‐transcriptional GFP reporter. Thereafter, the A549‐VIM‐RFP cells with CAGA‐GFP were seeded in a 96‐well plate and incubated in the IncuCyte live cell imaging system (Essen BioScience). The CAGA12‐mediated transcriptional activity was measured every 1 h over a 25 h‐period using a 10× objective and normalized by the GFP signals at 1 h. All the experiments were performed with biological triplicates, and representative results are shown.
Ubiquitination, immunoprecipitation, immunoblotting, and biotinylation
HEK293T cells were transfected with Myc‐tagged constitutively active TβRI (Myc‐caTβRI) and HA‐ubiquitin (HA‐Ub) for 48 h and treated with 5 μM proteasome inhibitor MG132 for 6 h before harvesting. For the analysis of the endogenous ubiquitination of TβRI, MDA‐MB‐231 cells stably expressing HA‐Ub were used. Cells were then harvested and washed twice in cold phosphate buffered saline (PBS) with 10 mM N‐ethlmaleimide (NEM) and lysed in 1% sodium dodecyl sulfate (SDS)‐RIPA buffer (25 mM Tris–HCl, pH7.4, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, and 1% SDS) supplemented with protease inhibitors (11836153001, Roche) and 10 mM NEM for 10 min on ice. The lysates were centrifuged at 11 × 103 g for 10 min at 4°C, and the protein concentrations were then measured using the DC protein assay (Pierce). To prevent the detection of any ubiquitination of co‐immunoprecipitating proteins, the lysates were boiled for 5 min, diluted to 0.1% SDS in an RIPA buffer, and followed up with immunoprecipitation analysis. For the immunoprecipitation assay, equal amounts of protein were incubated with an anti‐Myc or anti‐ TβRI antibody overnight and the protein G‐Sepharose (GE Healthcare Bio‐Sciences AB) for 2 h at 4°C. Thereafter, beads were washed five times with a TNE buffer at 4°C and boiled with a sample buffer for 5 min. The immunoprecipitated proteins were then separated by SDS–PAGE. For immunoblotting, equal amounts of proteins were loaded on a gel, and proteins were separated by SDS polyacrylamide gel electrophoresis (PAGE). Afterward, proteins were transferred onto a 45 μm polyvinylidene difluoride (PVDF) membrane (IPVH00010, Merck Millipore) and analyzed using specific primary and secondary antibodies. Signals were visualized with chemiluminescence. The biotinylation analysis of cell surface receptors was performed as previously described (Budi et al, 2016). Cells were biotinylated for 40 min at 4°C, then the biotinylated cell surface receptors were precipitated with streptavidin beads and analyzed by immunoblotting. All experiments were performed with biological triplicates, and representative results are shown.
The antibodies used for immunoprecipitation (IP) and immunoblotting (IB) are as follows: phosphor‐SMAD2 1:1,000 (IB: 3108, Cell Signaling), total‐SMAD2 1:1,000 (IB: 3103S, Cell Signaling), GAPDH 1:1,000 (IB: MAB374, Millipore), Tubulin 1:1,000 (IB: 2148, Cell Signaling), E‐Cadherin 1:1,000 (IB: 610181, BD Biosciences), N‐cadherin 1:1,000 (IB: 610920, BD Biosciences), vimentin 1:1,000 (IB: 5741, cell signaling), SNAIL 1:1,000 (IB: 3879, cell signaling), vinculin (IB: V9131, Sigma), Flotillin‐1 1:1,000 (IB: sc‐25506, Santa Cruz), β1‐integrin 1:1,000 (IB: ab183666, Abcam), EEA1 1:1,000 (IB: 610457, BD Biosciences), ST3GAL5 1:1,000 (IB: NBP2‐20492, Novus Biologicals), c‐Myc 1:200 (IP: sc‐40, Santa Cruz), HA 1:1,000 (IB: 1583816, Roche), TβRI 1:1000 (IB: sc‐398, Santa Cruz), FLAG 1:1,000 (IB: 14793, Cell Signaling), EGFR 1:1,000 (IB: 06‐847, Merck), p‐EGFR 1:1,000 (IB: 2234, Cell Signaling), and p‐ERK1/2 1:1,000 (IB: 9101, Cell Signaling).
Separation of lipid raft and non‐lipid raft microdomains of plasma membranes
Sucrose density gradient analysis was performed as described previously (Gajate & Mollinedo, 2017). In brief, 1 × 108 cells were digested by trypsin and washed with ice‐cold PBS twice. The cells were then pelleted by centrifugation at 290 g for 4 min, and the supernatant was discarded. Thereafter, 1.4 ml of membrane raft isolation TNVE buffer (1% Triton X‐100, 10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM Na3VO4) containing a freshly added protease inhibitor cocktail was added to resuspend the cell pellet. The cell lysate was then transferred to a precooled tightfitting Dounce homogenizer and kept on ice for 60 min. Thereafter, cell homogenization was carried out with 10 strokes. The homogenized cells were transferred into a 15 ml plastic tube and centrifuged at 200 g for 8 min at 4°C. One milliliter of supernatant was mixed with 1 ml of 85% (w/v) sucrose in a TNEV buffer and transferred to the bottom of a 14 ml Beckman centrifuge tube (344060). Six milliliters of 35% (w/v) sucrose in the TNEV buffer were carefully added on top of the diluted cell lysate using a 1 ml pipette and followed up with 3.5 ml of 5% (w/v) sucrose in the TNEV buffer on its top. Once the centrifuge tubes are loaded, each tube was placed into the compatible ultracentrifuge rotor (SW 40 Ti, Beckman) and balanced with a TNEV buffer. The loaded tubes were centrifuged at 257,000 g for 18 h at 4°C with the brake off to ensure that deceleration did not disrupt the density gradient. After centrifugation, centrifuge tubes were carefully taken out from the rotor. One milliliter of each fraction was collected from the top of the gradient until the end of the tube. The individual fractions were subjected to SDS–PAGE and analyzed by immunoblotting analysis. All experiments were repeated three times in biologically independent experiments, and representative results are shown.
IncuCyte migration assay
Cells were seeded in the IncuCyte 96‐well Essen ImageLock plate (4379, Essen BioScience) and scratched using the IncuCyte WoundMaker (Essen BioScience). The scratched cells were washed with phosphate‐buffered saline (PBS) and then cultured with DMEM medium with 10% serum in the IncuCyte live cell imaging system. Images were acquired every 2 or 4 h over a 36–68 h period using a 10× objective. Relative wound density was analyzed by the IncuCyte cell migration software for each well. All experiments were performed with biological triplicates, and representative results are shown.
Nano‐Glo HiBiT lytic detection assay
MDA‐MB‐231 cells stably expressed green fluorescent protein (GFP) were generated as previously described (Deckers et al, 2006). The HiBiT tag, a small 11 amino acid peptide (Schwinn et al, 2018), was knocked in using CRISPR/Cas9 at the endogenous locus of TβRI, resulting a TβRI in which the HiBiT sequence is inserted within the extracellular domain at a location carboxy terminal of the signal peptide. This cell line can be used for the specific detection and quantification of the cell surface TβRI expression level by addition of large BiT (LgBiT) to the cells. The HiBiT‐TβRI cell line with the indicated treatment was incubated with the PBS/LgBiT/NanoGlo substrate mixture using the NanoGlo‐HiBiT Detection kit (N2420, Promega) for 15 min and the signals were measured using the VICTOR multilabel plate reader (2030‐0050, PerkinElmer). Thereafter, the plate was imaged in the IncuCyte live cell imaging system to determine the GFP intensity, which is a proxy for the number of live cells. NanoGlo signals were normalized to the GFP intensity.
Zebrafish extravasation assay
The A549 cells were infected with pLV‐mCherry lentivirus and subjected to FACS sorting to obtain a pool of cells that highly expresses mCherry. Thereafter, the mCherry‐labeled A549 cells were applied to the zebrafish embryo xenograft experiments. We used the transgenic green fluorescent zebrafish Tg fli:enhanced green fluorescent protein (EGFP) strain for our xenograft cancer model. The experiments were conducted in a licensed establishment for the breeding and use of experimental animals (Leiden University) and subject to internal regulations and guidelines, stating that advice is taken from the animal welfare body to minimize suffering for all experimental animals housed at the facility. The zebrafish assays described are not considered an animal experiment under the Experiments on Animals Act (Wod, effective 2014), the applicable legislation in the Netherlands in accordance with the European guidelines (EU directive no. 2010/63/EU) regarding the protection of animals used for scientific purposes, because nonself‐eating larvae where used. Therefor a license specific for these assays on zebrafish larvae (< 5 days) was not required. The zebrafish extravasation assay was performed as previous described (Ren et al, 2017). Briefly, A549‐mCherry cells were transfected with the indicated siRNAs for 3 days or pretreated with indicated inhibitors for 6 days. Thereafter, approximately 400 cells were injected into the ducts of Cuvier of zebrafish embryos at 48 h post‐fertilization. Four days after injection, the fish were fixed with 4% paraformaldehyde (PFA, 28908, Thermo Fisher Scientific) and imaged by inverted SP5 confocal microscopy (Leica Microsystems). The invasive cell clusters (more than five cells were defined as a cluster) between the vessels in the caudal hematopoietic tissue region were counted. The experiments were repeated twice in biologically independent experiments, and at least 25 injected embryos were included for quantification.
Immunofluorescence staining
Cells were seeded onto sterile 18 mm long square glass coverslips (631‐1331, Menzel Gläser) and cultured with complete DMEM. After the indicated treatment, the cells were fixed with 4% PFA for 30 min and permeabilized with 0.1% Triton X‐100 for 10 min at room temperature. Afterward, 5% bovine serum albumin (BSA) (A2058, Sigma‐Aldrich) in 0.1% PBS‐Tween was used for 1 h to block the cells. The 1:1,000 diluted primary antibody of E‐cadherin (610181, BD Biosciences) in PBS was incubated with cells for 1 h. Thereafter, the cells were washed with PBS three times and incubated with a 1:500 diluted Alexa Fluor 555 secondary antibody (A‐21422, Thermo) or 1:1,000 diluted Alexa Fluor 488 Phalloidin (A12379, Thermo) for 1 h. The cells were then washed with PBS three times and mounted with a VECTASHIELD antifade mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI, H‐1200; Vector Laboratories). SP8 confocal microscopy was used to capture images (Leica Microsystems). All experiments were performed in biological triplicates, and representative results are shown.
Flow cytometry
The cells with UGCG knockout or those treated with eliglustat were harvested and counted using a cell counter. Thereafter, 2 × 105 cells were incubated with 50 μl of the diluted FITC‐conjugated Cholera Toxin B subunit (CTB, C1655, Sigma) in PBS with the final concentration of 20 μg/ml for 30 min at 4°C. Afterward, 150 μl of PBS/bovine serum albumin (BSA) was added to the cells, and samples were further analyzed on BD flow cytometers (Canto, Fortessa, LSR II or ARIA II). FACS data were analyzed by FlowJo (Tree Star, Inc).
RNA sequencing
The RNA samples used for sequencing were prepared with three biological triplicates. Library preparation and sequencing were performed by the Beijing Genomics Institute (BGI) in China. Total RNA was extracted using the NucleoSpin RNA II kit (740955, BIOKÉ) following the manufacturer's instructions. Briefly, purified mRNA was poly‐A selected using oligo dT beads and fragmented with the fragmentation enzyme. The first and second strand synthesis was then performed using random N6‐primed reverse transcription. The synthesized cDNA was subjected to end‐repair and was then 3′ adenylated. After amplification by PCR and cyclization by splint oligo and DNA ligase, RNA‐seq libraries were sequenced on the DNBSEQ platform. RNA‐Seq files were processed using the open‐source BIOWDL RNA‐seq pipeline v5.0.0 (https://zenodo.org/record/5109461#.Ya2yLFPMJhE) developed at the LUMC. This pipeline performs FASTQ preprocessing (including quality control, quality trimming, and adapter clipping), RNA‐Seq alignment, read quantification, and optional transcript assembly. FastQC was used for checking raw read QC. Adapter clipping was performed using Cutadapt (v2.10) with default settings. RNA‐Seq reads' alignment was performed using STAR (v2.7.5a) on the GRCm39 mouse reference genome. The gene read quantification was performed using HTSeq‐count (v0.12.4) with setting “–stranded = no.” The gene annotation used for quantification was Ensembl version 104. Using the gene read count matrix, counts per million mapped (CPM) was calculated per sample on all annotated genes. In total, 11,218 genes with a higher CPM than 1 in at least 25% of all samples are kept for downstream analysis.
Dataset acquisition and processing
We used the Oncomine (www.oncomine.org) (Rhodes et al, 2004, 2007) public bioinformatics database to obtain a summary of ST3GAL5 expression in 20 types of cancer vs. healthy tissues. The threshold was designed with the following parameters: a fold change of 1.5, a P‐value of 0.05, and a gene ranking of 10%. Box plots of the ST3GAL5 gene expression levels in the lung cancer tissues and normal tissues in the Bhattacharjee Lung database were analyzed from the Oncomine bioinformatics database, and the P value for statistical significance was set up as 0.05 (Bhattacharjee et al, 2001). The Kaplan–Meier curve of the first progression survival of lung cancer patients linked with ST3GAL5 mRNA expression was analyzed from a public bioinformatics database Kaplan–Meier plotter (kmplot.com/analysis). The first progression (FP) survival of 982 patients was collected with a mean follow‐up of 180 months. When the ST3GAL5 gene was uploaded to the database, samples were divided into two cohorts according to the median expression of ST3GAL5 (high vs. low expression) to obtain the Kaplan–Meier survival plots. The log‐rank P‐value and the hazard ratio (HR) with 95% confidence intervals were calculated and displayed on the web page.
For GSEA, we used the GSEA tool. We uploaded all genes with normalized count information from RNA sequencing analysis and ran it under default conditions with the gene symbol annotation (Mootha et al, 2003; Subramanian et al, 2005). The mouse COULOUARN_TEMPORAL_TGFB1_SIGNATURE_ UP gene set (Coulouarn et al, 2008) and the human TGFB_UP.V1_UP gene set (Padua et al, 2008) were used as TGF‐β gene signatures. The mouse GOTZMANN_EPITHELIAL_TO_MESENCHYMAL_ TRANSITION_UP gene set (Gotzmann et al, 2006) and the human SARRIO_EPITHELIAL_MESENCHYMAL_TRANSITION_UP gene set (Sarrio et al, 2008) were used as EMT gene signatures.
Immunohistochemical (IHC) staining and evaluation
The formalin‐fixed paraffin‐embedded microarray of lung cancer tissues including matched lung cancer and adjacent normal tissues was purchased from Biomax (LC1504), and details of every tissue are listed in Appendix Fig S8. The tissue microarray was baked overnight at 37°C and then for 2 h at 60°C until the paraffin melted. The slide was then placed in a xylene bath for 5 min three times. Thereafter, the slide was rehydrated in fresh absolute ethanol for 5 min twice and rinsed in methanol with 0.5% hydrogen peroxide for 20 min to block the activity of endogenous peroxidases. Subsequently, the slide was transferred once through 96%, 70%, and 50% ethanol, respectively, for 3 min each and washed with PBS containing 0.1% Tween‐20. The washed slide was boiled in an antigen unmasking buffer (1.5 M Tris–HCl, pH 8.0, 0.5 M EDTA, 10% Tween‐20) for 30 min using a pressure cooker. The tissue microarray was then washed with PBS/Tween three times and incubated with a 1:100 diluted primary ST3GAL5 antibody in PBS containing 1% BSA overnight at 4°C. Thereafter, the 1:200 diluted biotinylated secondary antibody (E3053, DAKO) in PBS/BSA was added onto the tissue array for 30 min at room temperature after washing the slide with PBS/Tween three times. The slide was then incubated with Vectastain complex (PK‐6100, Vector Laboratories) for 30 min at room temperature and washed with PBS/Tween three times and with 0.5 M Tris–HCl, pH 7.4 once. A 0.05% 3,3′‐diaminobenzidine (DAB) substrate solution with freshly added 0.015% H2O2 in 0.5 M Tris–HCl was applied to the sections on the slide for < 5 min, until the desired color intensity was reached. Hematoxylin (517‐28‐2, KLINIPATH) was used to counterstain nuclei for 10 s. The slide was then rinsed in running tap water for 10 min. Subsequently, the tissue slide was dehydrated through 50%, 70%, 96%, and 100% ethanol, respectively, for 5 min each and washed with xylene twice, followed by further mounting with Entellan (107961, Merck Millipore). The color of the antibody staining in the tissue sections was imaged by a 3DHistech Pannoramic slide scanner.
The quantification of IHC staining was expressed as H scores, which were calculated as previous described (Zhou et al, 2014). H scores are between 0 and 300 and calculated by the following formula: 3 × the percentage of cells with strong staining + 2 × the percentage of cells with moderate staining + 1 × the percentage of cells with weak staining. The analysis of tissue sections was unbiased.
Mice xenograft model
Five‐week‐old male BALB/c nu/nu mice were ordered and acclimatized for 1 week in the Laboratory Animal Center (LAC) of the Netherlands Cancer Institute (Amsterdam, The Netherlands). The mice were anesthetized by the inhalation of isoflurane (0.8 l/min) and intravenous tail vein injected either with A549‐Luc cells pretreated with or without eliglustat for 1 week, or A549‐Luc cells infected with pLV‐EV control or ST3GAL5 expression vector. Eight mice were included in each group. Bioluminescence imaging was carried out once a week to monitor the growth of metastases. All the mice experiment procedures were approved by the Animal Welfare Committee of the Netherlands Cancer Institute (Amsterdam, The Netherlands).
Statistical analysis
Statistical analyzes were performed with a two‐way ANOVA, a Student's unpaired t‐test using Prism 8 software (GraphPad La Jolla, CA), or as indicated in the legends. P‐value is indicated by asterisks in the figures: *P ≤ 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. P ≤ 0.05 was considered statistically significant.
Author contributions
Peter ten Dijke: Conceptualization; supervision; funding acquisition; project administration; writing—review and editing. Jing Zhang: Conceptualization; data curation; formal analysis; validation; investigation; methodology; writing—original draft; writing—review and editing. Jin Ma: Validation. Hailiang Mei: Methodology. Cedrick C Agaser: Methodology. Birol Cabukusta: Methodology. Katarina Madunic: Methodology. Gerard van der Zon: Validation; methodology. Manfred Wuhrer: Supervision; project administration; writing—review and editing. Tao Zhang: Data curation; formal analysis; methodology; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Acknowledgments
We are grateful to Robbert M Spaapen for providing the UGCG KO plasmids and reagents and to Marlieke Jongsma for the anti‐EEA1 antibody and Frank Kruyt for gift of A549‐Luc cells. We thank Maarten A H van Dinther, and Midory Thorikay for technical assistance, Sijia Liu and Jiang Ren for help with injection of cancer cells into zebrafish embryos, and all members of our laboratories for valuable discussion. We thank Babak Mousavi Gourabi and Boudewijn PT Kruithof for help with IHC staining. We acknowledge the support of the Chinese Scholarship Council (CSC) to Jing Zhang and Jin Ma; the Cancer Genomics Centre in the Netherlands (CGC. NL), and the ZonMW grant (09120012010061) to Peter ten Dijke.
The EMBO Journal (2023) 42: e110553
Contributor Information
Tao Zhang, Email: t.zhang@lumc.nl.
Peter ten Dijke, Email: p.ten_dijke@lumc.nl.
Data availability
The raw mass spectrometric data files that support the findings of this study are available in GlycoPOST in mzXML format, with the identifier GPST000243, accessible via the following link: https://glycopost.glycosmos.org/entry/GPST000243. MS/MS spectra of glycan structures with fragmentation annotations are supplied in Appendix Fig S11.
Sequencing data has been deposited to a publicly available GEO data set with an accession number of GSE192691 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192691).
References
- Allende ML, Proia RL (2014) Simplifying complexity: genetically resculpting glycosphingolipid synthesis pathways in mice to reveal function. Glycoconj J 31: 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG, Jacobson K (2002) A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science 296: 1821–1825 [DOI] [PubMed] [Google Scholar]
- Anugraham M, Everest‐Dass AV, Jacob F, Packer NH (2015) A platform for the structural characterization of glycans enzymatically released from glycosphingolipids extracted from tissue and cells. Rapid Commun Mass Spectrom 29: 545–561 [DOI] [PubMed] [Google Scholar]
- Barriere G, Fici P, Gallerani G, Fabbri F, Rigaud M (2015) Epithelial mesenchymal transition: a double‐edged sword. Clin Transl Med 4: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M et al (2001) Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA 98: 13790–13795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budi EH, Xu J, Derynck R (2016) Regulation of TGF‐β receptors. Methods Mol Biol 1344: 1–33 [DOI] [PubMed] [Google Scholar]
- Cabarcas‐Petroski S, Meneses PI, Schramm L (2020) A meta‐analysis of BRF2 as a prognostic biomarker in invasive breast carcinoma. BMC Cancer 20: 1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM (2008) GlycoWorkbench: a tool for the computer‐assisted annotation of mass spectra of glycans. J Proteome Res 7: 1650–1659 [DOI] [PubMed] [Google Scholar]
- Chen YG (2009) Endocytic regulation of TGF‐β signaling. Cell Res 19: 58–70 [DOI] [PubMed] [Google Scholar]
- Colak S, Ten Dijke P (2017) Targeting TGF‐β signaling in cancer. Trends Cancer 3: 56–71 [DOI] [PubMed] [Google Scholar]
- Cooper CA, Gasteiger E, Packer NH (2001) GlycoMod‐‐a software tool for determining glycosylation compositions from mass spectrometric data. Proteomics 1: 340–349 [DOI] [PubMed] [Google Scholar]
- Coskun U, Grzybek M, Drechsel D, Simons K (2011) Regulation of human EGF receptor by lipids. Proc Natl Acad Sci USA 108: 9044–9048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulouarn C, Factor VM, Thorgeirsson SS (2008) Transforming growth factor‐β gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 47: 2059–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumin C, Huang YL, Everest‐Dass A, Jacob F (2021) Deciphering the importance of glycosphingolipids on cellular and molecular mechanisms associated with epithelial‐to‐mesenchymal transition in cancer. Biomolecules 11: 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G, ten Dijke P (2006) The tumor suppressor Smad4 is required for transforming growth factor β‐induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 66: 2202–2209 [DOI] [PubMed] [Google Scholar]
- Derynck R, Weinberg RA (2019) EMT and cancer: more than meets the eye. Dev Cell 49: 313–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Muthusamy BP, Saeteurn KY (2014) Signaling pathway cooperation in TGF‐β‐induced epithelial‐mesenchymal transition. Curr Opin Cell Biol 31: 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL (2003) Distinct endocytic pathways regulate TGF‐β receptor signalling and turnover. Nat Cell Biol 5: 410–421 [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS (2004) New insights into TGF‐β‐Smad signalling. Trends Biochem Sci 29: 265–273 [DOI] [PubMed] [Google Scholar]
- Fujimoto Y, Izumoto S, Suzuki T, Kinoshita M, Kagawa N, Wada K, Hashimoto N, Maruno M, Nakatsuji Y, Yoshimine T (2005) Ganglioside GM3 inhibits proliferation and invasion of glioma. J Neurooncol 71: 99–106 [DOI] [PubMed] [Google Scholar]
- Gajate C, Mollinedo F (2017) Isolation of lipid rafts through discontinuous sucrose gradient centrifugation and Fas/CD95 death receptor localization in raft fractions. Methods Mol Biol 1557: 125–138 [DOI] [PubMed] [Google Scholar]
- Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H et al (2006) A crucial function of PDGF in TGF‐β‐mediated cancer progression of hepatocytes. Oncogene 25: 3170–3185 [DOI] [PubMed] [Google Scholar]
- Granville CA, Dennis PA (2005) An overview of lung cancer genomics and proteomics. Am J Respir Cell Mol Biol 32: 169–176 [DOI] [PubMed] [Google Scholar]
- Guan F, Handa K, Hakomori SI (2009) Specific glycosphingolipids mediate epithelial‐to‐mesenchymal transition of human and mouse epithelial cell lines. Proc Natl Acad Sci USA 106: 7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, Lanczky A (2013) Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer. PLoS One 8: e82241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakomori S (1996) Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res 56: 5309–5318 [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL et al (1997) The MAD‐related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 89: 1165–1173 [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P (1997) TGF‐β signalling from cell membrane to nucleus through SMAD proteins. Nature 390: 465–471 [DOI] [PubMed] [Google Scholar]
- Hunter FW, Devaux JBL, Meng F, Hong CR, Khan A, Tsai P, Ketela TW, Sharma I, Kakadia PM, Marastoni S et al (2019) Functional CRISPR and shRNA screens identify involvement of mitochondrial electron transport in the activation of Evofosfamide. Mol Pharmacol 95: 638–651 [DOI] [PubMed] [Google Scholar]
- Indellicato R, Parini R, Domenighini R, Malagolini N, Iascone M, Gasperini S, Masera N, dall'Olio F, Trinchera M (2019) Total loss of GM3 synthase activity by a normally processed enzyme in a novel variant and in all ST3GAL5 variants reported to cause a distinct congenital disorder of glycosylation. Glycobiology 29: 229–241 [DOI] [PubMed] [Google Scholar]
- Jensen PH, Karlsson NG, Kolarich D, Packer NH (2012) Structural analysis of N‐ and O‐glycans released from glycoproteins. Nat Protoc 7: 1299–1310 [DOI] [PubMed] [Google Scholar]
- Jongsma MLM, de Waard AA, Raaben M, Zhang T, Cabukusta B, Platzer R, Blomen VA, Xagara A, Verkerk T, Bliss S et al (2021) The SPPL3‐defined glycosphingolipid repertoire orchestrates HLA class I‐mediated immune responses. Immunity 54: 132–150.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabayama K, Sato T, Saito K, Loberto N, Prinetti A, Sonnino S, Kinjo M, Igarashi Y, Inokuchi J (2007) Dissociation of the insulin receptor and caveolin‐1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci USA 104: 13678–13683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan JY, Moi SH, Hung WC, Hou MF, Chen FM, Shih SL, Shiau JP, Li CL, Chiang CP (2020) Comprehensive transcriptomic analysis identifies ST8SIA1 as a survival‐related Sialyltransferase gene in breast cancer. Genes (Basel) 11: 1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson NG, Schulz BL, Packer NH (2004a) Structural determination of neutral O‐linked oligosaccharide alditols by negative ion LC‐electrospray‐MSn. J Am Soc Mass Spectrom 15: 659–672 [DOI] [PubMed] [Google Scholar]
- Karlsson NG, Wilson NL, Wirth HJ, Dawes P, Joshi H, Packer NH (2004b) Negative ion graphitised carbon nano‐liquid chromatography/mass spectrometry increases sensitivity for glycoprotein oligosaccharide analysis. Rapid Commun Mass Spectrom 18: 2282–2292 [DOI] [PubMed] [Google Scholar]
- Katsuno Y, Lamouille S, Derynck R (2013) TGF‐β signaling and epithelial‐mesenchymal transition in cancer progression. Curr Opin Oncol 25: 76–84 [DOI] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol Cell 6: 1365–1375 [DOI] [PubMed] [Google Scholar]
- Kawashima N, Yoon SJ, Itoh K, Nakayama K (2009) Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J Biol Chem 284: 6147–6155 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Chung TW, Choi HJ, Kwak CH, Song KH, Suh SJ, Kwon KM, Chang YC, Park YG, Chang HW et al (2013) Ganglioside GM3 participates in the TGF‐β1‐induced epithelial‐mesenchymal transition of human lens epithelial cells. Biochem J 449: 241–251 [DOI] [PubMed] [Google Scholar]
- Kim DH, Triet HM, Ryu SH (2021) Regulation of EGFR activation and signaling by lipids on the plasma membrane. Prog Lipid Res 83: 101115 [DOI] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15: 178–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L, Hill CS (2005) Smad4 dependency defines two classes of transforming growth factor β (TGF‐ β) target genes and distinguishes TGF‐β‐induced epithelial‐mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol 25: 8108–8125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang X, Tan Z, Chen S, Guan F (2016) Role of glycans in cancer cells undergoing epithelial‐mesenchymal transition. Front Oncol 6: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Wang X, Dong P, Yu P, Zhang Y, Meng X (2021) Chemoenzymatic synthesis and biological evaluation of ganglioside GM3 and lyso‐GM3 as potential agents for cancer therapy. Carbohydr Res 509: 108431 [DOI] [PubMed] [Google Scholar]
- Lloyd KO, Old LJ (1989) Human monoclonal antibodies to glycolipids and other carbohydrate antigens: dissection of the humoral immune response in cancer patients. Cancer Res 49: 3445–3451 [PubMed] [Google Scholar]
- Massague J (2000) How cells read TGF‐β signals. Nat Rev Mol Cell Biol 1: 169–178 [DOI] [PubMed] [Google Scholar]
- Massague J (2008) TGFβ in cancer. Cell 134: 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell H, Choudhury A, Pagano RE, Leof EB (2004) Ligand‐dependent and ‐independent transforming growth factor‐β receptor recycling regulated by clathrin‐mediated endocytosis and Rab11. Mol Biol Cell 15: 4166–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E et al (2003) PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Maxfield FR (2004) Membrane domains. Annu Rev Cell Dev Biol 20: 839–866 [DOI] [PubMed] [Google Scholar]
- Mutoh T, Tokuda A, Miyadai T, Hamaguchi M, Fujiki N (1995) Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci USA 92: 5087–5091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Yamagishi T, Hokari S, Nakamura H (2000) Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: Roles of transforming growth factor (TGF)‐β and bone morphogenetic protein (BMP). Anat Rec 258: 119–127 [DOI] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH et al (1997) Identification of Smad7, a TGF‐β‐inducible antagonist of TGF‐β signalling. Nature 389: 631–635 [DOI] [PubMed] [Google Scholar]
- Neapolitan R, Horvath CM, Jiang X (2015) Pan‐cancer analysis of TCGA data reveals notable signaling pathways. BMC Cancer 15: 516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang S, Liu JH, Ni Z, Ding GF, Wang QZ (2020) Downregulation of ST3GAL5 is associated with muscle invasion, high grade and a poor prognosis in patients with bladder cancer. Oncol Lett 20: 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J (2008) TGFβ primes breast tumors for lung metastasis seeding through angiopoietin‐like 4. Cell 133: 66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulou E, Gillooly DJ, Wrana JL, Zerial M, Stenmark H, Murphy C, Fotsis T (2002) Early endosomal regulation of Smad‐dependent signaling in endothelial cells. J Biol Chem 277: 18046–18052 [DOI] [PubMed] [Google Scholar]
- Pontier SM, Schweisguth F (2012) Glycosphingolipids in signaling and development: from liposomes to model organisms. Dev Dyn 241: 92–106 [DOI] [PubMed] [Google Scholar]
- Prinetti A, Cao T, Illuzzi G, Prioni S, Aureli M, Gagliano N, Tredici G, Rodriguez‐Menendez V, Chigorno V, Sonnino S (2011) A glycosphingolipid/caveolin‐1 signaling complex inhibits motility of human ovarian carcinoma cells. J Biol Chem 286: 40900–40910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbaa A, Hurh J, Yamamoto H, Kersey DS, Bremer EG (1996) Ganglioside GM3 inhibition of EGF receptor mediated signal transduction. Glycobiology 6: 399–406 [DOI] [PubMed] [Google Scholar]
- Regina Todeschini A, Hakomori SI (2008) Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim Biophys Acta 1780: 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C, Stewart TJ, Renfrow MB, Novak J (2019) Glycosylation in health and disease. Nat Rev Nephrol 15: 346–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu S, Cui C, Ten Dijke P (2017) Invasive behavior of human breast cancer cells in embryonic zebrafish. J Vis Exp 55459 [DOI] [PMC free article] [PubMed]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM (2004) ONCOMINE: a cancer microarray database and integrated data‐mining platform. Neoplasia 6: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Kalyana‐Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead‐Beal C, Kulkarni P et al (2007) Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9: 166–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SK, Islam SMR, Kwak KS, Rahman MS, Cho SG (2020) PROM1 and PROM2 expression differentially modulates clinical prognosis of cancer: a multiomics analysis. Cancer Gene Ther 27: 147–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez‐Pena EQ, Taube JH, Chang JT, Miura N, Porter W, Sphyris N et al (2015) GD3 synthase regulates epithelial‐mesenchymal transition and metastasis in breast cancer. Oncogene 34: 2958–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D, Rodriguez‐Pinilla SM, Hardisson D, Cano A, Moreno‐Bueno G, Palacios J (2008) Epithelial‐mesenchymal transition in breast cancer relates to the basal‐like phenotype. Cancer Res 68: 989–997 [DOI] [PubMed] [Google Scholar]
- Satoh M, Ito A, Nojiri H, Handa K, Numahata K, Ohyama C, Saito S, Hoshi S, Hakomori SI (2001) Enhanced GM3 expression, associated with decreased invasiveness, is induced by brefeldin a in bladder cancer cells. Int J Oncol 19: 723–731 [PubMed] [Google Scholar]
- Schnaar RL, Kinoshita T (2015) Glycosphingolipids. In Essentials of glycobiology, 3rd edn, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH et al (eds), pp 125–135. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [PubMed] [Google Scholar]
- Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, Hall MP, Encell LP, Binkowski BF, Wood KV (2018) CRISPR‐mediated tagging of endogenous proteins with a luminescent peptide. ACS Chem Biol 13: 467–474 [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J (2003) Mechanisms of TGF‐β signaling from cell membrane to the nucleus. Cell 113: 685–700 [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387: 569–572 [DOI] [PubMed] [Google Scholar]
- Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A et al (2017) A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 18: 441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzavlaki K, Moustakas A (2020) TGF‐β signaling. Biomolecules 10: 487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Douglas D, Zhang J, Kumari S, Enuameh MS, Dai Y, Wallace CT, Watkins SC, Shu W, Xing J (2020) Live‐cell imaging and analysis reveal cell phenotypic transition dynamics inherently missing in snapshot data. Sci Adv 6: eaba9319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Uddin N, Wagner GK (2018) Covalent probes for carbohydrate‐active enzymes: from glycosidases to glycosyltransferases. Methods Enzymol 598: 237–265 [DOI] [PubMed] [Google Scholar]
- Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori G et al (2020) Guidelines and definitions for research on epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 21: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M, Sorkin A (2007) Signaling on the endocytic pathway. Curr Opin Cell Biol 19: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB et al (2007) Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nat Med 13: 952–961 [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou F, Drabsch Y, Gao R, Snaar‐Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX et al (2012) USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF‐β type I receptor. Nat Cell Biol 14: 717–726 [DOI] [PubMed] [Google Scholar]
- Zhang J, Thorikay M, van der Zon G, van Dinther M, Ten Dijke P (2020a) Studying TGF‐β signaling and TGF‐β‐induced epithelial‐to‐mesenchymal transition in breast cancer and Normal cells. J Vis Exp 10.3791/61830 [DOI] [PubMed] [Google Scholar]
- Zhang T, van Die I, Tefsen B, van Vliet SJ, Laan LC, Zhang J, Ten Dijke P, Wuhrer M, Belo AI (2020b) Differential O‐ and glycosphingolipid glycosylation in human pancreatic adenocarcinoma cells with opposite morphology and metastatic behavior. Front Oncol 10: 732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ten Dijke P, Wuhrer M, Zhang T (2021) Role of glycosylation in TGF‐β signaling and epithelial‐to‐mesenchymal transition in cancer. Protein Cell 12: 89–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Chen YG (2014) Regulation of TGF‐β signal transduction. Scientifica (Cairo) 2014: 874065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Drabsch Y, Dekker TJ, de Vinuesa AG, Li Y, Hawinkels LJ, Sheppard KA, Goumans MJ, Luwor RB, de Vries CJ et al (2014) Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF‐β signalling. Nat Commun 5: 3388 [DOI] [PubMed] [Google Scholar]
- Zuo W, Chen YG (2009) Specific activation of mitogen‐activated protein kinase by transforming growth factor‐β receptors in lipid rafts is required for epithelial cell plasticity. Mol Biol Cell 20: 1020–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]