

Abstract
Effective therapies are lacking for patients with advanced colorectal cancer (CRC). The CRC tumor microenvironment has elevated metabolic waste products due to altered metabolism and proximity to the microbiota. The role of metabolite waste in tumor development, progression, and treatment resistance is unclear. We generated an autochthonous metastatic mouse model of CRC and used unbiased multi-omic analyses to reveal a robust accumulation of tumoral ammonia. The high ammonia levels induce T cell metabolic reprogramming, increase exhaustion, and decrease proliferation. CRC patients have increased serum ammonia, and the ammonia-related gene signature correlates with altered T cell response, adverse patient outcomes, and lack of response to immune checkpoint blockade. We demonstrate that enhancing ammonia clearance reactivates T cells, decreases tumor growth, and extends survival. Moreover, decreasing tumor-associated ammonia enhances anti-PD-L1 efficacy. These findings indicate that enhancing ammonia detoxification can reactivate T cells, highlighting a new approach to enhance the efficacy of immunotherapies.
Keywords: tumor microenvironment, ammonia, immunotherapy, cancer metabolism
eTOC Blurb:
Bell et al identify that ammonia accumulates in colon tumors. The accumulated ammonia reduces tumor immune response by specifically inhibiting T cell function. Removing microenvironmental ammonia reactivates T cells, decreases tumor size, and re-sensitizes tumors to immunotherapy.
Graphical Abstract
INTRODUCTION
Colorectal cancer (CRC) is the second leading cause of cancer mortality worldwide1. Improvements in CRC patient survival stems in part from enhanced early detection due to increased rates of colonoscopy 2. However, 20% of patients with CRCs have metastatic disease at the time of diagnosis, and the 5-year overall survival rate in this patient subset is 10.5% 3,4. Moreover, immune checkpoint blockade (ICB) is not effective in the vast majority of patients with CRC 5. Recent work has revealed that the tumor microenvironment (TME) in many CRCs is often immunosuppressive. The TME is a hypoxic, nutrient-sparse environment with high concentrations of metabolic waste. Heterocellular metabolite exchange in the TME can lead to dysregulated immune cell effector function. Metabolic dysregulation in primary CRCs is also affected by intestinal microbes, as the gut microbiota generates unique metabolites and waste products.
Ammonia is a waste product that is generated at high levels from host and microbial cellular metabolism. Host-derived ammonia arises via glutamine and asparagine catabolism, cysteine and nucleotide biosynthesis6,7 Microbiota also produce large quantities of ammonia through breakdown of host proteins by microbial ureases 8. Due to its toxic effects on many cells, ammonia must be exported and processed by the liver through the urea cycle 9. Of note, some cancer cells can assimilate ammonia as a biosynthetic metabolite to drive amino acid metabolism 10. However, the role of extracellular ammonia in the TME is largely unstudied.
We employed an autochthonous metastatic model of CRC arising from concurrent mutations in tumor suppressor and oncogenes known to contribute to human CRC. Using multi-omic profiling to investigate the altered immune landscape and mechanisms of immune therapy resistance, we identified an important transcriptional network regulated by hepatic nuclear factor-4 (HNF4)α. HNF4α is a master regulator of genes encoding urea cycle factors, and disruption of HNF4α leads to increased ammonia accumulation 11. We identify a robust increase of ammonia in a metastatic colon cancer model and show increased TME ammonia levels reduce T cell activation and proliferation and lead to T cell exhaustion. Promoting enhanced TME ammonia detoxification inhibits tumor growth and activates the response to ICB in our mouse CRC model. Evidence implicates elevated ammonia levels in the TME of human CRC patients that do not respond to ICB and/or who have poorer outcomes. Collectively, our findings identify ammonia as a key immune-inhibitory waste product in CRC and highlight the potential of enhancing ammonia clearance as an approach to increase the effectiveness of ICB in CRC.
RESULTS
Mouse CRC model lacks response to multiple immune therapy approaches.
ICB therapy has recently led to clinical breakthroughs for some solid tumors, such as clear cell renal cell carcinoma, melanoma, non-small cell lung cancer, and mismatch repair defective (MMRD) CRCs and other MMRD cancers12,13. However, in the vast majority of CRCs, ICB is not effective 14. Unlike the situation in human CRC, some widely used syngeneic mouse CRC xenograft models as well as sporadic mouse CRC models are highly responsive to immunotherapies15. To understand how genetically engineered mouse models with variable defects in the murine homologues of tumor suppressor and oncogenes recurrently mutated in human CRC would respond to immune-based therapies, we pursued work with mice where tamoxifen treatment can induce target genes of interest in mouse colon epithelium to induce benign or malignant tumors 16. We utilized mice with a biallelic disruption of Apc (SingleMut), Apc and Tp53 (DoubleMut), or Apc, Tp53 and a Kras G12D knock-in allele (TripleMut) targeted to the colon epithelium specifically using CDX2-CreERT2. These mice were assessed for overall survival following one, two, three, or four daily doses of 50 mg/kg tamoxifen (Figure 1A and B, S1A). As expected, increasing tamoxifen doses led to progressively shorter survival times. Of note, for equivalent tamoxifen dosages, the average survival of the TripleMut is approximately 50% shorter than that of the DoubleMut. Moreover, TripleMut had increased numbers of tumors, and histologically more advanced tumors compared to SingleMut or DoubleMut (Figure 1C, 1D, 1E). The TripleMut, but not the SingleMut and DoubleMut mice developed lung and liver metastases (Figure 1F). We assessed the response of each mouse model to anti-Programmed-Dealth-Ligand-1 (PD-L1). We used a tamoxifen dosing strategy that led to mice from each model having an overall average survival of 30 days. SingleMut and DoubleMut demonstrated increased survival following anti-PD-L1 treatment (Figure 1G). However, TripleMut did not show improved survival following anti-PD-L1 treatment as compared to isotype control (Figure 1G). We next used an alternative form of immunotherapy, IL-17 bone marrow knockout, in the TripleMut mouse model. IL-17 promotes colorectal cancer by driving pro-tumor inflammation, generating an immunosuppressive microenvironment, and suppressing cytotoxic immune function17–19. Using bone marrow chimeras from IL-17 knockout mice, we found that the SingleMut mice had increased survival following disruption of IL-17 signaling as previously reported, but the TripleMut mouse model showed no response to transplantation with IL-17-disrupted immune cells (Figure S1B) 19 Together, these data reveal that the TripleMut model is resistant to immune-based therapeutic approaches.
Figure 1: Autochthonous metastatic mouse model of colon cancer is resistant to immunotherapy.

A) Schematic of SingleMut, DoubleMut, and TripleMut. B) Mice were induced with one injection of 50 mg/kg tamoxifen and then assessed for survival (n=8–10), C) tumor number (n=8–10), D) tumor progression (n=8–10), and E) histology (n=8–10). F) Mice were treated with low dose tamoxifen and lungs and livers were macroscopically assessed for metastasis (n=7). G) Mice were induced with a tamoxifen dose calculated to cause death around 30 days in untreated mice for each model, treated daily with intraperitoneal anti-PD-L1 (1 μg per mouse) and monitored for survival. (n=8–10). Statistics were calculated with one-way ANOVA or t-test. Survival significance is calculated using log-rank test. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM
Mice with advanced lesions develop an immunosuppressive microenvironment.
We sought to understand the differential responsiveness of the CRC models to immune-based therapies. Acquired and adaptive resistance to immunotherapy can result from dysfunction or deletion of CD8+ T cells. In these CRC models, when treated with three concurrent doses of tamoxifen, the whole colon becomes an adenomatous epithelium. RNA sequencing on tumor colon epithelium demonstrated differential global gene expression for each model (Table S1). Principal components analyses demonstrated that the TripleMut mice separated from the SingleMut and DoubleMut mice along PC1 (Figure 2A and B). Gene set enrichment analysis revealed a robust depletion in pathways essential for adaptive immune response or T cell function in TripleMut mice compared to the DoubleMut (Figure 2C). We performed multiplex immunohistochemical staining (OPAL) for CD3, arginase-1, CD8 and CK19 on paraffin sections from a different mouse cohort (Figure S2A). The TripleMut mouse model showed an increase in arginase positive cells (Figure S2B). We saw a decrease in the distance between CD3+ T cells and ARG-1+ myeloid cells, suggesting possible contacts between immunosuppressive macrophages and tumor-infiltrating T cells (Figure S2C). We next performed CyToF on a separate cohort of mice (Figure 2D). This experiment showed that TripleMut mice had increased CD44+/CD45− cells, indicating an increased proportion of cancer stem cells in this mouse model (Figure 2E). The TripleMut mouse model also displayed a significant decrease in CD45+ cells and CD3+ cells (Figure 2E). The number of CD4+ cells and CD8+ T cells in the TripleMut model was decreased, but not significantly (Figure S2D). We observed no significant changes in the CD11b+Ly6G+ or CD11b+ F4/80+ populations between the DoubleMut and TripleMut mice (Figure S2E). The TripleMut mouse model had a significant increase in CD4+ and CD8+ positive T cells expressing the inhibitory receptors CTLA-4 and PD-1 (Figure 2F and S2F). Overall, these findings suggest that TripleMut mice have fewer overall T cells and increased T cell dysfunction.
Figure 2: T cells are reduced in the TripleMut mice.

A) PCA analysis of RNA-sequencing of colon cancer mouse models induced with one dose of 100 mg/kg tamoxifen (n=3). B) Heat map and C) gene set enrichment of differentially expressed genes between mouse models (n=3). D) Representative TSNE plot of CyToF data of different mouse models induced with one dose of 100 mg/kg tamoxifen (n=3). E and F) Selected cell populations from CyToF data (n=3). *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM.
Ammonia accumulates in CRC due to dysregulation of the urea cycle.
To identify upstream regulators essential for the differential expression between the DoubleMut and TripleMut mice, a CHIP-SEQ transcription factor enrichment analysis in the genes differentially expressed between the DoubleMut and TripleMut from our previous RNA-SEQ experiment was performed 20. We screened the enriched transcription factors for differential expression between the DoubleMut and TripleMut, and for differential expression in CRC compared to normal colon in the publicly available TCGA datasets. We next screened the transcription factors for expression associated with differential survival in human patients, also in the TCGA dataset. Finally, we narrowed our list to transcription factors that had an association with KRAS using STRING 21. This analysis identified four potential candidates: GFI, HNF4α, RUNX1, and the IRF protein family (Figure 3A). Hepatocyte nuclear factor 4 α (HNF4α) is a master regulator of the urea cycle and is necessary to clear cellular ammonia 11. A previously described ammonia gene signature which consist of low expression of OTC, CPS1, HNF4a, ARG1, ASS1, and ASL and high expression of SLC4A11, GLS, and GLUD1 was altered in the TripleMut mice (Figure 3B and C)22. Metabolomics analysis, using PCA analysis on SingleMut, DoubleMut, and TripleMut mice, found the metabolic profiles clustered separately (Table S2, Figure S3A). Urea cycle intermediates were not targeted with this particular metabolomics analysis, but differentially abundant metabolites in the TripleMut identified a robust decrease in pyrimidine metabolism (Figure S3B and S3C). Dysregulated pyrimidine metabolism is a central feature in urea cycle dysfunction, as nitrogen is diverted towards carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase activation 22. We next mined The Cancer Genome Atlas (TCGA) to assess RNA expression of HNF4α and its direct target the ornithine transcarbamylase (OTC) in the cancer and normal colon. HNF4α and OTC were significantly downregulated in CRC compared to normal colon tissues (Figure 3D). These findings were confirmed in an institutional cohort of CRC patients (Figure 3E). Further, analysis of flow sorted epithelial and stromal compartments of dissected tumors from the SingleMut, DoubleMut, and TripleMut mice models demonstrated that Otc and Hnf4a levels were reduced specifically in the tumor epithelium (Figure 3F). Enteroids generated from the tumors of the induced TripleMut mice had the most significantly reduced Hnf4a and Otc expression compared to normal enteroids from wildtype mice or tumor enteroids from SingleMut and DoubleMut mice (Figure S3D). Similarly, the normal human colon cell line NCM460 highly expressed HNF4A and OTC compared to CRC-derived cell lines HCT116 and the SW480 (Figure S3E). Moreover, analysis of data from the Cancer Cell Line Encyclopedia and TCGA found that cell lines or patients with a KRAS mutation had lower levels of HNF4a (Figure S3F and S3G). To determine the role of mutant KRAS in the dysregulation of Hnf4aand Otc, a colon-epithelial doxycycline inducible Kras mutant mouse model was generated (Figure S3H). Mice expressing the mutant Kras allele had an increased prevalence of dysplasia, tumor formation, and increased GFP Kras reporter expression in their colons (Figure S3I and S3J). In the mutant Kras mice, Hnf4a and Otc expression were decreased, although this decrease was not significant (Figure S3K). This suggests that the downregulation of Hnf4a and Otc is due to a combinatorial effect of Apc and Tp53 loss as well as mutant Kras expression.
Figure 3: Ammonia metabolism is dysregulated in advanced colorectal cancer.

A) Schematic of transcription factor analysis. B) Schematic of urea cycle intermediates and enzymes. C) Heat map of expression of genes (GLS1 (Glutaminase-1), GLUD (Glutamate Dehydrogenase-1), OTC (Ornithine Transcarbamylase), CPS1 (Carbamoyl Phosphate Synthase-1), HNF4a (Hepatic Nuclear Factor 4 alpha), ARG (Arginase), ASS (Arginosuccinate Synthase), ASL (Arginosuccinate Lyase) and SLC4A11 (Solute Carrier Family 4 Member 11)) in the ammonia signature from RNA-Sequencing experiment. D) OTC and HNF4α expression in cancer and normal tissue from the TCGA dataset. E) Gene expression of HNF4α and OTC in normal and cancer patients. F) Gene expression of flow-sorted epithelial and stromal compartments. (n=6–8). G) Representative Nessler’s staining and H) quantification of mice induced with 100 mg/kg tamoxifen and sacrificed at endpoint (n=9). I) Mass spectrometry ammonia quantification of tumors from mice induced with 100 mg/kg tamoxifen. (n=3). J) Kaplan-Meier survival curves of cancer patients stratified by the ammonia gene signature. Data from TCGA. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM.
The downregulation of Hnf4a, Otc and other ammonia related genes in our cancer models, and the metabolic shift in these metabolites prompted us to assess ammonia accumulation in CRC. Nessler’s reagent is a stain that can detect free ammonia in tissues 23. Staining DoubleMut and TripleMut colons demonstrated that TripleMut mice had significantly increased epithelial ammonia staining compared to the DoubleMut (Figure 3G and H). To confirm these findings, we used mass spectrometry in an independent cohort of mice and found a robust increase in ammonia in TripleMut mice (Figure 3I). In three gastrointestinal carcinomas (colon, gastric, and hepatocellular) the ammonia gene signature was associated with decreased survival (Figure 3J) (Lánczky & Győrffy, 2021). However, the signature did not predict survival in most other cancer types, potentially suggesting a specificity for the gastrointestinal tract. Together, these data reveal: that 1) dysregulation of ammonia metabolism in murine and human CRC leads to intratumoral ammonia accumulation, and 2) that ammonia-related gene expression prognostically stratifies CRC patients.
Ammonia exposure increases exhausted T cells in vitro and in vivo.
We assessed how increased microenvironmental ammonia altered the growth of a panel of CRC cell lines and primary immune cells. Ammonia increased or did not change cell growth in several CRC cell lines, consistent with what was previously reported in breast cancer-derived cell lines 10(Figure 4A). While bone marrow-derived neutrophils and macrophages were unaffected by ammonia levels, in vitro activated splenic CD3+ T cells showed a strong reduction in proliferation at doses within the range of ammonia observed in colon tumors. We confirmed that primary hepatocytes can effectively clear ammonia following treatment over a period of 24 hours, while MC38 CRC cancer cell line did not accumulate ammonia following treatment. This is consistent with cancer cells rapidly using ammonia as a nitrogen source to support growth10. Ammonia accumulates within T cells and bone marrow derived macrophages and is not cleared after 48 hours (Figure S4A). Dose response studies demonstrated significantly diminished proliferation of CD4+ and CD8+ T cells (Figure 4B and S4B–C), reduced expression of the T cell activation marker CD25 (Figure 4C), and a significant increase in cell death after ammonia treatment (Figure 4D). Further, human T cells treated in vitro with ammonia had significantly reduced proliferation (Figure 4E). Similar to the TripleMut mice, ammonia treatment increased the exhausted T cell markers (PD1+TIM3+), and decreased production of interferon gamma (IFNγ) in T cells as assessed by flow cytometry (Figure 4F and S4D). Glutamate dehydrogenase, GLUD, was increased in T cells treated with ammonia, while expression of other components of glutamine synthesis and the urea cycle were not altered in T cells, macrophages, or a colon cancer cell line (Figure S4E–S4G). To confirm that ammonia drives an exhausted tumor microenvironment, we analyzed gene expression of PD-1 and CTLA-4 after ammonia treatment (Figure 4G). Consistent with the mouse data, ammonia significantly induced PD-1 expression, but not CTLA-4 in human T cells (Figure 4H and S4H). To determine whether an increase in systemic ammonia levels in mice alters T cell function, mice were placed on an ammonium acetate diet. After seven days, serum ammonia levels were increased (Figure 4I). T cells isolated from mice on ammonium acetate diet demonstrated decreased proliferation, as well as increased PD-1 and CTLA-4 expression, supporting the model that high levels of ammonia drive T cell exhaustion (Figure 4J and 4K). Interestingly, T cells treated with ammonia for 12 h, then replated with fresh media for 72 h, had a sustained decrease in T cell growth (Figure S4I). To confirm the long-term effects of ammonia on T cells, we treated mice with ammonium acetate or normal chow, isolated CD8+ T cells and adoptively transferred the cells into MC38 tumor bearing mice following ex vivo activation (Figure 4L). As expected, activated CD8+ T cells from control mice were able to reduce tumor growth, but T cells isolated from mice on the ammonium acetate chow did not suppress tumor growth (Figure 4M). Collectively, this data suggests that ammonia induces T cell exhaustion.
Figure 4: Ammonia increases exhausted T cells and reduces T cell viability.

A) Panel of cell types treated with indicated ammonia concentration for 72 h. T cells were stimulated with CD3 and CD28. Quantification of T cell B) proliferation (CFSE), C) activation (CD25), and D) cell death (7AAD and Annexin V). Ammonia levels range from 1–10 mM. E) Proliferation of human T cells treated with ammonia for 72 h. F) IFNγ and exhaustion markers in mouse T cells by flow (quantitation and representative image) after the indicated ammonia concentration for 72 h. G) RT-PCR for indicated exhaustion marker after the indicated ammonia concentration for 72 h. H) Gene expression of human T cells treated with ammonia for 72 h. I) Serum ammonia quantification in mice treated for 7 days with ammonium acetate diet (n=3). J) Proliferation and K) gene expression of CD3+ T cells isolated from the spleens of mice treated with ammonium acetate diet or control diet (n=3). L) Schematic illustrating the T cell transfer experiment in which mice were treated with control chow or ammonium acetate chow for 7 days. T cells were isolated, counted, and intravenously injected into MC38 tumor bearing mice. M) Tumors were monitored every 3 days (n=5–7). SingleMut mice were induced with 3 injections of 100 mg/kg tamoxifen and placed on ammonium acetate diet. At end point, N) mouse colon length O) intratumoral T cells were assessed (n=4). *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM. All experiments were performed in triplicates at least three times.
To understand if ammonia can drive tumorigenesis and alter tumor T cell response, an ammonium acetate diet was fed to the SingleMut mouse model. SingleMut mice on ammonium acetate compared to untreated SingleMut mice had decreased body weight, decreased survival, shorter colons, and decreased proportion of CD4+ and CD8+ T cells (Figure 4N and 4O). This data indicates that ammonia treatment durably reduces the proliferation and activation of T cells and contributes to tumor progression.
Ammonia inhibits the transsulfuration pathway to induce oxidative stress in T cells.
To determine the mechanisms by which ammonia alters T cell function, targeted metabolomics was performed. T cells isolated from wildtype mice and treated in vitro with ammonia, and T cells isolated from mice on an ammonium acetate diet clustered on PC1, indicating a similar metabolic response (Figure 5A, Table S3). Amino acids, urea cycle and nicotinamide metabolites were the most enriched and most in common between both treatment paradigms (Figure 5B–C). However, transcripts for key enzymes in glycolysis and the TCA cycle were not significantly altered in ammonia-treated T cells (Figure S5A).
Figure 5: Ammonia leads to oxidative stress and dysregulated methionine metabolism in T cells.

A) PCA analysis and B) heat maps annotated with significant pathways from pathway analysis of metabolomics in T cells isolated from wildtype mice, mice treated with ammonium acetate diet or wildtype T cells treated with 10 mM ammonia in vitro for 24 h (n=3). C) Schematic of the transsulfuration pathway with selected metabolite relative levels indicated. D) Absolute quantification of GSH in in vitro treated mouse T cells. E) 35S tracing of methionine flux into GSH in in vitro treated mouse T cells. F) T cells were treated for 24 hours with 10 mM ammonia, labeled for 1 hour with radiolabeled methionine, then processed for mass spectrometry and analyzed for label incorporation. T cells were treated with CBS inhibitor CH004 (doses from 5–10 μM and CSE inhibitor propargylglycine (doses from 3–6 mM) for 24 hours, then G) activation was assessed by flow cytometry for CD25 and H) cytokine production was assessed using ELISA. I) Effect of CBS inhibitor CH004 (doses from 0.015–20 μM) and CSE inhibitor propargylglycine (doses from .01–6 mM) on T cell proliferation over 72 hours. J) T cells were treated for 24 h with 10 mM NAC and 10 mM GEE then treated with 10 mM ammonia for 72 h before CFSE was acquired using flow cytometry. K) T cells were treated with ammonia or cotreated with ammonia and NAC, and ROS was assessed by flow cytometry. L) Mice were treated with ammonium acetate diet for 7 days, then splenic T cells were assessed for ROS by flow cytometry (n=3). M) CRC-derived cell lines were treated with 10 mM ammonia for 72 h, assessed for lipid ROS by flow cytometry. N) T cells were pretreated for 24 h with ferrostatin (2 μm) followed by cotreatment with 10 mM ammonia for 24 h. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM. All cell experiments were performed in triplicates at least three times.
Notably, there were robust changes in the transsulfuration pathway metabolites as indicated by arrows in this schematic (Figure 5C). To evaluate the dysregulation of the transsulfuration pathway, total protein methylation was assessed. Histone methylation was not affected in T cells treated with ammonia in vitro or in vivo (Figure S5B and S5C). CBS and MAT2A expression were also unchanged (Figure S5D). However, gene expression analysis identified betaine-homocysteine S-methyltransferase (BHMT) as an ammonia regulated gene in T cells (Figure S5E and S5F). Bhmt was unchanged in ammonia treated MC38 colorectal cancer cells (Figure S5G). Since glutathione was significantly reduced following ammonia treatment (Figure 5D), radiolabel flux analysis from [35S]-methionine to glutathione was assessed. [35S]-methionine incorporation into glutathione was significantly decreased following ammonia treatment (Figure 5E). Furthermore, ammonia treatment decreased the conversion of 13C5-methionine tracing into labeled S-adenosyl-methionine (Figure 5F). We cotreated T cells with the CSE inhibitor propargylglycine and the CBS inhibitor CH004 to examine whether the inhibition of the transsulfuration pathway was toxic to T cells. CSE/CBS inhibitor treatment decreased T cell activation, cytokine production, and cell proliferation (Figure 5G–5I). Interestingly, N-acetyl cysteine (NAC), and a cell permeable glutathione ethyl ester (GEE), reversed the effects of ammonia on T cell proliferation (Figure 5J), whereas S-adenosyl-methionine, amino acids, sodium sulfide, cystathionine, homocysteine, pyruvate, serine, or taurine did not rescue the ammonia-induced growth restriction in T cells (Figure S5H and S5I). We quantified metabolic changes, and in addition to dysregulation of the transsulfuration pathway, several TCA cycle intermediates were also altered (Figure S5J). Ammonia inhibits the electron transport chain and increases oxidative stress 24, and consistent with this data ammonia treatment reduced the oxygen consumption rate of T cells significantly (Figure S5K). A significant increase in ROS and lipid ROS was observed in CD4+ and CD8+ T cells after in vitro and in vivo treatment with ammonia (Figure 5K–L, S5L–S5M), but not in CRC cell lines (Figure 5M). Furthermore, the ferroptotic inhibitor ferrostatin rescued T cell growth inhibition by ammonia (Figure 5N). Together, these data indicate that high ammonia levels induce T cell oxidative stress via dysregulation of the transsulfuration pathway.
Cellular ammonia clearance reduces CRC growth.
Ornithine is used clinically in hyperammonemic patients to eliminate ammonia by stimulating both the urea cycle and glutamine synthesis 25. Specifically, ornithine is metabolized by the mitochondria of periportal hepatocytes. It activates carbamoyl-phosphate synthetase and drives the urea cycle26. During urea biosynthesis, the hepatocyte eliminates two ammonia molecules. Intraperitoneal injection of ornithine at clinically relevant doses to ammonium acetate chow-fed mice reduced serum ammonia and increased T cell proliferation (Figure 6A and B). Murine serum ammonia was found to be within 50–100 μM. Ornithine treatment in vitro has no effect on T cell proliferation or urea cycle gene expression or polyamine levels at physiologically relevant doses but can inhibit proliferation at supraphysiological doses (Figure S6A–S6C). Ornithine treatment led to a striking decrease in MC38 and CT26 syngeneic tumor growth (Figure 6C–D and Figure S6D–E). The reduction of tumor growth was T cell dependent, as ornithine did not alter growth of MC38 and CT26 tumors implanted in nude mice or following CD8 depletion (Figure 6E and Figure S6F–J). Decreased tumor growth was accompanied by significantly reduced Ki67 (Figure 6F–G and Figure S6K) and tumor ammonia levels (Figure 6H and S6L). We further found that ornithine decreased CD44+ tumor stem cells and increased CD4+ and CD8+ T cells in MC38 tumors (Figure S6M and S6N). To confirm these findings, an orthogonal approach to reducing systemic ammonia was utilized. Glycerol phenylbutyrate eliminates ammonia via conversion to phenylacetate that binds nitrogen, and conjugates with glutamine to form phenylactylglutamine which is excreted by the kidney 27. Glycerol phenylbutyrate treatment significantly reduced MC38 syngeneic tumor size (Figure S6O and S6P). To explore the source of the high levels of ammonia, MC38 tumor-bearing mice were treated with lactulose, a microbiota-dependent ammonia reducing agent 28. Lactulose treatment did not significantly reduce tumor size (Figure S6P). This suggests that the source of excess ammonia is likely the cancer epithelial cells rather than a dysbiotic microbiota (Figure S6Q). Ornithine supplementation to the ammonium acetate diet restored T cell function. Ammonia diet reduced T cell secretion of IFNγ, IL-2, and IL-6, but co-treatment with ornithine restored cytokine levels (Figure S6R).
Figure 6: Neutralizing ammonia reduces colorectal cancer tumor growth.

A) Serum ammonia or B) T cell proliferation was assessed in wildtype mice treated with ammonium acetate diet for 7 days or cotreated with ammonium acetate and daily intraperitoneal injection of 10 mM ornithine (n=3). C, D) Syngeneic tumor study in C57BL/6J or E) athymic nude mice with CRC-derived MC38 cells following ornithine treatment (10 mM ornithine intraperitoneally (n=8–10). Representative IHC F) image and G) quantitation of Ki67 in MC38 tumors treated with ornithine. H) Quantification of ammonia in MC38 tumors treated with ornithine. I) Serum ammonia levels in healthy, CRC, or metastatic CRC patients. J) Gene expression analysis for OTC and HNF4α in normal livers or livers with metastatic lesions K) Ammonia quantification from normal livers or metastatic tumors (n=3). L) Representative images and M) tumor weights of in the liver metastasis model treated with vehicle and intraperitoneal ornithine (n=8–10). N) Ammonia concentration in ornithine treated liver metastasis livers (n=3). *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM. All experiments were performed in triplicates at least three times.
To extend these findings to humans, serum ammonia levels were assessed in CRC patients and healthy controls. A significant increase in serum ammonia levels was found in CRC patients compared to healthy controls. Serum ammonia was further increased in metastatic CRC (Figure 6I). These results were reflected in the systemic ammonia levels in our murine colorectal cancer model (Figure S6S). Having seen a further increase in systemic ammonia levels in metastatic CRC patients, we utilized a previously established syngeneic liver metastasis mouse model 29. Expression of Otc and Hnf4a was significantly decreased in mice with liver metastasis and ammonia levels were increased (Figure 6J and K.). Mice treated with ornithine daily had reduced tumor size and maintained more normal liver (Figure 6L and 6M). Reduction in tumor size was accompanied by lower ammonia levels, confirming ammonia detoxification (Figure 6N).
High TME ammonia inhibits response to ICB.
Ornithine treatment rescues the ammonia driven T cell functional deficits seen in our CRC mouse models. We next evaluated whether ornithine could increase the efficacy of anti-PD-L1 immunotherapy in CRC. TripleMut mice were injected with 50 mg/kg tamoxifen, and immediately treated with ornithine or vehicle and co-treated with anti-PD-L1 or isotype control. Anti-PD-L1 or ornithine (p=0.07) alone did not improve mouse survival. However, ornithine and anti-PD-L1 co-treatment combined led to a significant increase in survival (Figure 7A). Analysis of ornithine only treated TripleMut mice demonstrated that these mice had a significant increase in CD3+ T cell infiltration into the tumors, with less of these T cells being exhausted than the controls (not significant) (Figure S7A).
Figure 7: Detoxifying ammonia reactivates immunotherapy in CRC.

A) Kaplan-Meier survival curves of TripleMut mice injected with 50 mg/kg tamoxifen, and then treated with daily intraperitoneal ornithine, vehicle, isotype control, or anti-PDL1. Significance is compared to vehicle isotype control (n=5–7). B and C) Flow cytometry analysis from TripleMut mice treated with ornithine, IgG, or anti-PD-L1 therapy for 7 days. (n=3). D) Human tumor samples were binned into OTC high, or OTC low and overall survival was assessed based on cytotoxic T cell scores. (n=62). E) Kaplan-Meier survival curves of PD-L1 treated cancer cohort stratified into high and low ammonia gene signature (n=426). F) Representative OTC staining and G) Kaplan-Meier patient survival based on OTC low and high staining following immunotherapy. Survival significance is calculated using log-rank test. H) Schematic summary of the role of ammonia accumulation in CRC progression. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 unless otherwise indicated. Data is presented as mean ± SEM. All experiments were performed in triplicates at least three times.
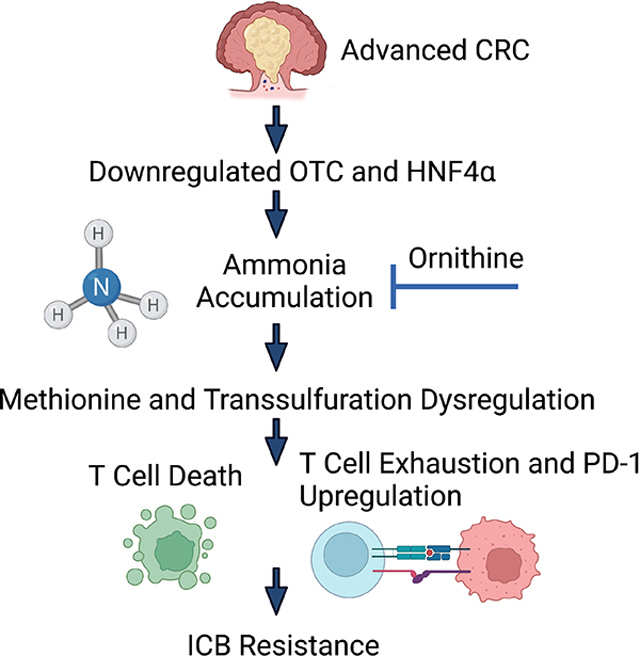
PD-L1 and ornithine co-treatment led to a significant reduction in T cell exhaustion and an increase in overall T cell infiltration (Figure 7B and 7C and S7B). We next used our ammonia gene signature to score the data from our RNA-SEQ experiment comparing the Single, Double, and TripleMut mouse models. A high ammonia score was associated with low CD4+ T cells in the TripleMut (Figure S7C). TIDE, an algorithm that correlates gene expression with T cell infiltration, demonstrated that tumors without a strong presence of cytotoxic T lymphocytes, typically have worse survival 30. However, we found that in tumors with low expression of OTC, the presence of cytotoxic T lymphocytes was no longer predictive of survival (Figure 7D). In addition, we found that a gene signature score associated with high ammonia levels predicted worse survival following ICB treatment in a large cohort of combined cancer types (Figure 7E). We assessed a small institutional cohort of MSI-high CRC patients treated with immunotherapy. Patients with low OTC staining (associated with high ammonia levels) by IHC had worse survival after PD-1 treatment (Figure 7F and 7G). OTC levels were not associated with differential response to traditional chemotherapy (Figure S7D). We hypothesize that in advanced CRC, OTC and HNF4α are downregulated which leads to ammonia accumulation, transsulfuration dysregulation in T cells, increased T cell exhaustion and ICB resistance (Figure 7H).
DISCUSSION
Altered cellular metabolism is a hallmark of cancers and impacts the tumor microenvironment, immune landscape, and therapy response. Moreover, metabolic reprogramming in tumors generates excess byproducts that must be counteracted by a heterocellular metabolic exchange in the tumor microenvironment. Here, using a highly metastatic model of CRC (TripleMut), we show that there is a robust downregulation of T cell fitness compared to more benign less progressive CRC mouse models (DoubleMut or SingleMut mice). These data are consistent with other models of CRC demonstrating a robust immunosuppressive tumor microenvironment 31. Through a multi-omic analyses integrating transcriptomic, CyTOF and unbiased metabolomics data, we identified a novel connection between ammonia metabolism and T cell function. The activity of HNF4α and its downstream target genes involved in ammonia clearance were robustly inhibited in CRC, leading to a buildup of extracellular ammonia. Consistent with the mouse model data, HNF4a signaling and the expression of ammonia clearance genes were downregulated and serum ammonia was increased in CRC patients. High extracellular ammonia decreased T cell anti-tumor function, while not altering viability or function of other immune cells or cancer cells. However, using several orthogonal approaches, neutralizing ammonia levels decreased tumor growth in a T cell dependent manner, whereas increasing tumor ammonia led to attenuation of T cell function and increased progression in the SingleMut mice.
A nutrient limited microenvironment alters T cell proliferation and function. Insufficient oxygen and waste removal occurs as tumors grow. Hypoxia inhibits CD4+ T cell function and promotes regulatory T cell activity 32 Accumulation of lactate suppresses cytotoxic T lymphocytes 33. Previous work has shown that ammonia accumulates in hepatocellular carcinoma 34 Urea cycle dysregulation is a common feature of tumors, and ammonia is a central nitrogen source to support cancer growth 10. However, little work has been performed on how altered ammonia metabolism in cancers affect stromal cells 22. Our data reveals that dysregulation of urea cycle enzymes is correlated with increased extracellular ammonia, which is a major mechanism that leads to an immunosuppressive tumor microenvironment in CRC. The tumor microenvironment is an important driver of immunosuppression and response to ICBs 35.
Recent work suggests a more complicated metabolic interaction where nutrients are selectively partitioned to immune and cancer cells based on uptake mechanisms 36. However, decreases in glutamine, lipids, and amino acids in the tumor microenvironment limit T cell growth, viability and/or function 37,38. Our metabolomics data from ammonia treated T cells implicated altered methionine metabolism. T cells proliferate rapidly and have a high metabolic requirement for methionine. Recent work has shown that methionine is necessary to drive T cell differentiation and proliferation39. Methionine can be converted to S-adenosylmethionine which is a common methyl donor. Tumors disrupt methionine metabolism and reduce S-adenosylmethionine in CD8+ T cells, which leads to impaired T cell immunity 37. Our work highlights the requirement of the transsulfuration pathway for the conversion of methionine to cysteine, which is the limiting substrate for glutathione synthesis. Radioactive and heavy isotope methionine tracing analysis in T cells revealed that ammonia decreased transsulfuration flux and lowered glutathione levels, which is associated with increased ROS. Currently, the specific enzymatic steps that are altered in the transsulfuration pathway are not clear. The isotope tracing experiment showed reduced ion abundance of m+5 labeled S-adenosyl-methionine, suggesting decreased MAT2a activity. However, several transsulfuration metabolic intermediates downstream of MAT2A did not rescue the ammonia phenotype. We observed decreased TCA and glycolytic intermediates and oxygen consumption, suggesting that ammonia may alter several pathways. Future work will assess key steps in the transsulfuration pathway and other metabolic programs to precisely define how ammonia impacts T cell fitness. Moreover, cancer cells and T cells have a less robust ability to clear ammonia than normal colon cells or hepatocytes, due to significant downregulation of the urea cycle. It is likely that the T cell vulnerability mediated by a decrease in the transsulfuration flux combined with the limited ability of T cells to clear exogenously produced ammonia leads to a specific metabolic vulnerability.
Although some work has shown that an immunoscore for T cell infiltration predicts survival, initial CRC approaches to ICB have been very disappointing 13. The majority of CRC is microsatellite stable, but 15% of cancer has high microsatellite instability (MSI) 40. However, in MSI high CRC, the objective response after PD-1 inhibitor is 40% compared to 0% in non MSI high tumors 5. Most CRCs are not MSI high and there is significant heterogeneity in immune infiltration 41. Our work demonstrates that clearance of ammonia decreases tumor growth and potentiates ICB response in CRC models that are refractory to immunotherapy. High OTC expression in patient samples is associated with ammonia clearance, and an improved response to immunotherapy. The data, including work done in T cell deficient models, points to an ammonia/T cell-axis that is central in CRC growth and the immunotherapy response. Reactivating ammonia clearance may be a viable therapeutic option to increase response to ICB in non MSI high CRC. However, more functional studies are needed to understand how ammonia in the tumor microenvironment may alter the numbers and/or function of other stromal cells.
Ammonia metabolism and accumulation is unique in colon cancer compared to other cancers due to two major sources of ammonia: cell autonomous metabolism and the microbiota. Many commensal microbes including Clostridia, Enterobacteria, and Bacillus spp produce large quantities of ammonia predominantly by amino acid deamination and urea hydrolysis by urease 42. Host cells generate ammonia via the conversion of L-glutamine to L-glutamate catalyzed by glutaminase 43. Ammonia is largely eliminated through the urea cycle. Ammonia accumulates rapidly in tumors due to poor vascularization and increased glutamine metabolism. Moreover, Kras mutations are known to drive glutamine metabolism, which is in line with TripleMut mice having the highest level of tumor ammonia and our most progressive and immunosuppressive model. Interestingly, activation of the Kras mutation by itself did not lead to increased ammonia levels or dysregulation of HNF4α signaling. We hypothesize that the metabolic changes leading to high ammonia result from the combinatorial effects of the Apc, Kras, and p53 mutationsTp53, which will be tested using the appropriate mouse models in the future.
Our work suggests the tumor protection derived from ammonia detoxification is T cell dependent. The temporal studies done in vivo suggest that initially, increased ammonia levels led to T cell exhaustion and increased expression of PD-1 and CTLA-4 on T cells. As the tumors progress, ammonia levels accumulate leading to a reduction in T cell numbers. The same phenomenon is seen in vitro, as markers of T cell exhaustion are upregulated early and prior to changes in cell viability. Higher levels of ammonia in vitro potently reduce T cell growth and lead to cell death. That said, more studies will be helpful in informing the time course of ammonia accumulation on T cell numbers and function in vivo. This is difficult to evaluate in late stage TripleMut mice due to the rapid progression of disease, and variability between mice. Detoxifying ammonia via ornithine treatment increased T cell numbers in a syngeneic CRC model. In the TripleMut model, which we consider to be more progressive and more immunosuppressive, we found that ornithine alone reduces T cell exhaustion markers and increases T cell numbers. In this advanced model, PD-L1 therapy alone is largely ineffective. Combined ornithine and PD-L1 therapy combined leads to a significant increase in total T cells and T cell activation, and a further decrease in markers of T cell exhaustion. The combination of ornithine and anti-PD-L1 immunotherapy also increased survival by 72%, indicating that the two-pronged approach to reactivate T cells is effective. Athymic nude mice and wildtype mice treated with a CD8 depleting antibody are less protected by ornithine, indicating that ornithine treatment is somewhat T cell dependent. Low levels of ammonia signature are associated with improved survival after anti-PD-L1 therapy in a large cohort of many cancer types. In addition, high OTC expression is associated with improved survival after ICB treatment in patients with MSI-high CRC.
Together, this suggests that ammonia detoxification could be an adjuvant to traditional cancer chemo and immunotherapy. Significantly more work is needed to identify the most effective treatment approach. Glycerol phenylbutyrate and ornithine are effective at reducing ammonia secondary to hepatic encephalopathy. However, dosing and route of ornithine requires optimization. Moreover, a recent paper engineered bacterium to hone to tumors and produce large quantities of arginine, and potentially a similar bacterial engineering approach could be used to target ornithine directly to tumors or other areas of high ammonia 44.
In sum, our results reveal dysregulation of the urea cycle in CRC, which leads to extracellular ammonia accumulation. Ammonia preferentially suppresses T cell growth and induces exhaustion and immunosuppressive markers. Ammonia detoxification is therefore a potential novel therapeutic option, particularly for metastatic and non-MSI high CRC.
STAR METHODS
Resource Availability:
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact is Dr. Yatrik Shah, who can be reached at shahy@umich.edu.
Material Availability:
All unique and stable reagents including mouse lines that were generated in the course of this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability:
Data:
Data generated in this study is available in the supplementary tables. Any raw data requested that was generated from this paper will be shared by the lead contact upon request.
Code:
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details:
Mice:
All mice used in this paper are in a predominant C57Bl/6J background. Males and females are equally represented, and littermates were randomly mixed in all experimental conditions. The mice were housed in a temperature controlled, specific pathogen free environment, with a 12 hour light/dark cycle. They were fed ad libitum with standard chow diet. Studied mice were between 4 to 6 weeks old. Mouse lines used were CDX2-ERT2Cre; Apcfl/fl mice; ROSA KRASG12D CDX2-ERT2Cre; Apcfl/fl mice, CDX2-ERT2Cre; Apcfl/fl, Tp53fl/fl, and Apcfl/fl; Tp53fl/fl; KrasLSLG12D mice. Nude mice from Jackson laboratories strain 007850 were used for the xenograft tumor studies. All animal studies were carried out in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International guidelines and approved by the University Committee on the Use and Care of Animals at the University of Michigan.
Human Subjects:
The study was approved by the institutional review boards at the University of Michigan (IRBMED, protocol number HUM00085066 and HUM00064405) and the Department of Health and Human Services, Food and Drug Administration (Research Involving Human Subjects Committee/RIHSC, protocol number14-029D). All protocol and applicable local regulatory requirements were meant. All adult male and female subjects provided written informed consent and review of their medical history.
Human Survival Analysis:
Human survival analysis was determined using the online tool KMplot45 This is a web based, registration-free survival analysis tool that can perform univariate and multivariate survival analysis. For both general survival and immunotherapy survival, we used our ammonia gene signature as an input. Significance is computed using the Cox-Mantel log rank test.
Cell Lines:
Human intestinal cell lines HCT116, RKO, SW480, and DLD1 were used for most experiments. Other cell lines used are MC38 and CT26. Cell lines have been STR-authenticated. All cells were maintained in complete DMEM medium (supplemented with 10% fetal bovine serum and 1% antibiotic/antimycotic agent) at 37°C in 5% CO2 and 21% O2.
Method Details:
Growth assays:
Adherent cell growth assays were performed using live cell imaging using the Cytation 5 Imaging Multi-Mode reader. Cells were plated down, treated 24 h later with indicated treatments, and immediately imaged and analyzed for cell number. Images were then taken every 24 h. Cytation software was used to quantify cell number.
Real time quantitative PCR:
One μg of total RNA, extracted using Trizol reagent from mouse tissues (intestinal epithelial scrapes), human intestine, primary T cells and cell lines, was reverse transcribed to cDNA using SuperScriptTM III First-Strand Synthesis System (Invitrogen). Real time PCR reactions were set up in three technical replicates for each sample. cDNA gene specific primers, SYBR green master mix was combined, and then run in QuantStudio 5 Real-Time PCR System (Applied BioSystems). The fold-change of the genes were calculated using the ΔΔCt method using Actb as the housekeeping gene mRNA. Primers are listed in Table S4.
Metabolomics:
For indicated mouse model, mouse was induced with Tamoxifen, and colon scrapes were collected day 10 after induction. Colon scrapes were snap-frozen in liquid nitrogen and then normalized by mass. Metabolites were extracted by adding dry-ice cold 80% methanol for 10 minutes before bead homogenization. Samples were clarified via high-speed centrifugation and supernatant extracted. Metabolite extracts were then lyophilized using a SpeedVac concentrator and resuspended in 50:50 methanol/water mixture for LC-MS analysis. For T-cell samples, cells were treated at 10 million cells per .5 ml of complete RPMI. Samples were then washed once with ice cold PBS, then incubated in dry-ice cold 80% methanol on dry ice for 10 min before homogenization. Samples were normalized by conducting BCA protein concentration on a technical replicate and volume normalizing across samples to achieve even concentrations. Metabolite extracts were then lyophilized using a SpeedVac concentrator and resuspended in 50:50 methanol/water mixture for LC-MS analysis. Data was collected in both negative and positive ion modes. For negative mode, previously published parameters were used46–48. For positive mode, an Agilent Technologies Triple Quad 6470 LC/MS system consisting of a 1290 Infinity II LC Flexible Pump (Quaternary Pump), 1290 Infinity II Multisampler, 1290 Infinity II Multicolumn Thermostat with 6 port valve and 6470 triple quad mass spectrometer was used. Agilent Masshunter Workstation Software LC/MS Data Acquisition for 6400 Series Triple Quadrupole MS with Version B.08.02 is used for calibration, compound optimization and sample data acquisition. A Waters Acquity UPLC HSS T3 1.8 μm VanGuard Pre-Column 2.1 × 5 mm column and a Waters UPLC BEH TSS C18 column (2.1 × 100mm, 1.7μm) was used with mobile phase A) consisting 0.1% formic acid in water; mobile phase (B) consisting of 0.1% formic acid in acetonitrile. The solvent gradient was: mobile phase (B) held at 0.1 % for 3 min, increased to 6 % at 12 min, 15 % at 15 min, 99% at 17 min and held for 2 min before reverting to the initial condition and held for 5 min. The column was held at 40 °C and 3 μl of sample was injected into the LC-MS with a flow rate of 0.2 ml/min. Calibration of 6470 QqQ MS was achieved through Agilent ESI-Low Concentration Tuning Mix. QQ data for both ion modes were preprocessed with Agilent MassHunter Workstation Quantitative Analysis Software (B0700). Metabolite chromatogram peaks were manually inspected. Each metabolite abundance level was median normalized across the sample population. Statistical significance was determined by a one-way ANOVA with a significance threshold of 0.05. Metaboanalyst was used for pathway enrichment analysis.
Polyamine Quantification:
Briefly, cells were washed with PBS, then lysed with methanol and homogenized. Samples were clarified by centrifugation and protein concentration was identified for normalization. Lyophilized pellets were resuspended in 50:50 water and methanol, then collected using previously published parameters 46 The peaks were confirmed with standards of spermidine, spermine, and putrescine (Sigma). The ion counts were normalized to protein concentration and to vehicle control. QQ data was analyzed with the Agilent MassHunter Quantitative Analysis platform.
[35S]-Methionine flux into GSH:
T cells (15 million cells in 0.5 mL of RPMI media) were cultured in a 12-well plate. L-[35S]-methionine (1175 Ci/mmol, NEG009A500UC, Perkin Elmer), was added (5 μCi/ml) and incubated for 24 h. Then, the T cell suspension was transferred to 1.5 ml tubes and centrifuged at 10,000 × g for 3 min. The supernatant was discarded, and the cell pellet was suspended in 150 μl of 1X PBS. One aliquot of the suspension (100 μl) was mixed with an equal volume of metaphosphoric acid solution (16.8 mg/ml) with EDTA and 150 mM NaCl while a second aliquot (30 μl) was mixed with an equal volume of RIPA lysis buffer (R0278, Sigma) with protease inhibitor cocktail for mammalian tissue extract (P8340, Sigma) for protein quantitation using the Bradford reagent. The RIPA lysis buffer and protease inhibitor was mixed in the ratio of 100:1. After vortexing for 5–10 sec, the samples were frozen on dry ice and stored at −20 °C. For HPLC analysis, samples were thawed on ice, centrifuged at 12,000 × g for 5 min and mixed with 15 μl of monoiodoacetic acid. The pH was adjusted to 7–8 (monitored using pH paper strips) and incubated for 1 h in the dark at room temperature. Then, the samples were mixed with an equal volume of Sangers reagent (15 μl/ml in absolute ethanol, 200 proof, Decon Laboratories), incubated at room temperature for 4 h and separated by HPLC using a Bondapak column (Agilent) as described previously49.
13C5 methionine labeling and mass spectrometry:
Cells were cultured in DMEM lacking glutamine, methionine, and cystine (Life Technologies, 21013–024). The media was supplemented with 10% FBS (R & D Systems, S1150H), penicillin and streptomycin (15140–122), 4 mM glutamine (Life Technologies, 25030–081), 0.2 mM cystine, and the heavy methionine (13C5 methionine, catalog- CLM-893-H-PK). T cells were plated 24 hours prior to labeling at 1.5 million cells per 6-well dish. Cells were treated with ammonia for 24 hours and labeled with heavy methionine for 1 hour. Cells were lysed with dry-ice cold 80% methanol and extracts were incubated at 80°C for 10 min. Samples were then centrifuged at 4000 rpm for 10 min at 4°C and the supernatant was stored at −80°C until further analyses. Protein concentrations were determined by processing a parallel 6-well dish and used to normalize metabolite fractions. Based on protein concentrations, normalized aliquots of the supernatants were transferred to a fresh microcentrifuge tube and lyophilized using an Equavap. Dried metabolite pellets from cells resuspended in 1:1 methanol: H2O before LC-MS/MS analysis. Samples were run in 5 replicates on an Orbitrap IDX with HILIC chromatography. All chromatography analysis was done with Skyline in the molecule interface.
Ammonium acetate diet:
Diet was made from pelleted chow mixed with 25% powdered ammonium acetate and water. It was mixed using a KitchenAid food mixer and dried in a dehydrator.
IL 17 bone marrow transplant:
Eight-week-old mice from the indicated genotype were lethally irradiated twice on the same day. Bone marrow was isolated from the indicated genotype from a separate donor mouse and 5 million cells were injected into the tail vein on the same day. Mice were monitored for diarrhea and weight loss daily. Engraftment was confirmed using RT-PCR.
T cell isolation:
CD4+ and CD8+ T cells were isolated from the spleen. Briefly, the spleen was removed from a sacrificed mouse, pushed through a sterile 70-micron filter using the plunger of a syringe, and then washed in PBS. Cells were incubated with the antibody and beads from the MojoSort CD3+, CD4+ and CD8+ T cell isolation kit, and then negative selection was achieved using a magnet. Cells were counted and plated in RPMI with FBS. T cell proliferation was performed under plate bound anti-CD3 and anti-CD28 stimulation. Cells were incubated for 45 minutes with MTT solution (5X concentrate stock: 5 mg/ml, in 1XPBS, pH 7.4) or 15 minutes with CFSE (5 μM). Absorbance was read at 570nm for MTT and CFSE was assessed by the Z2 flow cytometer.
CyToF:
The indicated mouse line was induced with tamoxifen and killed on day 10 after induction. The colons were incubated with 10 mM EDTA, and the epithelial cells were discarded. The colons were next incubated with 1 mg/ml of Type IV collagenase for 40 min, and then immune cells were isolated using a Percoll gradient. Cells were stained with indicated CyToF antibodies (Table 1), and a DNA intercalator after permeabilization with paraformaldehyde. Samples were acquired on the Helios (Fluidigm) and analyzed in CytoBank.
Ammonia Quantification:
Cells or tissues were deproteinated using a water-methanol-chloroform gradient. Lysates were then centrifuged rapidly to clear debris. Ammonia was then quantified using the L-Glutamate Dehydrogenase ammonia kit (Sigma). Ammonia was converted to indophenol using the Berthelot reaction, and then quantified using mass spectrometry as previously described50.
C11-BODIPY lipid ROS measurement:
Wildtype splenic T cells or CRC cells were incubated for 24 h with the indicated concentration of ammonia. Cells were harvested in HBSS, washed, and stained with 5 μM C11-BODIPY (ThermoFisher) at 37 °C for 30 minutes. Fluorescent intensity was measured on the FITC channel on the Z2 flow cytometer. A minimum of 20,000 cells were analyzed per condition, data was analyzed using FlowJo software (Tree Star). Values are MFI.
ROS Detection Assay:
ROS detection assay was performed as previously described16. Briefly, cell permeable free radical sensor carboxy-H2DCFDA (Invitrogen) was used. Cells were treated with indicated ammonia concentration for 24 h, then incubated with 10 μM carboxy-H2DCFDA in PBS at 37 °C for 45 min. Cells were then washed and resuspended in PBS. Mean fluorescent intensity was obtained from the Cytation 5 Imaging Multimode Reader.
Histology:
Tumor tissues were rolled and fixed in formalin for 24 h, then embedded in paraffin. Sections of 5 μm were stained for H&E and mounted with Permount Mounting Medium (Thermo Fisher Scientific). For immunohistochemistry, paraffin tissue sections underwent antigen retrieval, blocking in 5% goat serum in PBS, and probed with Ki67 antibody (Cell Signaling, 1:250), OTC antibody (ThermoFisher 1:200), or CD8 antibody (BD Bioscience 1:200). Sections were washed twice with PBST and incubated with HRP conjugated anti-rabbit IgG (1:500, catalog 7074S, Cell Signaling Technology) for 1 h. Sections were then washed with PBST and stained with DAB substrate. After the color change, the reaction was stopped with distilled water and dehydration was completed before mounting with Permount Mounting Medium. For immunofluorescence OPAL staining, samples were initially dehydrated, then stained with the indicated fluorescent OPAL antibody, then analyzed by multispectral imaging and analyzed for spatial and morphological context. Histological scoring of dysplasia was done by a blinded pathologist.
Nessler’s Reagent Staining:
Nessler’s reagent staining was performed as described in 51 Briefly, tissues were emebedded in paraffin was and sectioned into 5 microns. Samples were hydrated through 5 minute washes with alchohol. Samples were incubated 5 minutes with Nessler’s reagent, then washed for 10 seconds, then counterstained with hematoxylin, washed with running water for 60 seconds, then dehydrated briefly and mounted on slides for imaging. Nessler’s reagent is composed of 2 grams potassium iodide, 3 grams mercury iodide and 40 grams potassium hydroxide in 20 mL of water.
Immune Cell Identification:
Colons were removed, washed thoroughly in PBS, and then disassociated with 10 mM EDTA for 45 min. The remaining colon after this treatment was incubated with 1 mg/mL collagenase Type IV for 45 min. Immune compartment was obtained through a Percoll gradient, cells were stained using indicated antibody, and then acquired using flow cytometry.
RNA-SEQ analysis:
Indicated mouse line was induced with tamoxifen and tissues were collected on day 10 after injection. Colons were washed gently in PBS, scraped with a glass slide, and then the scraped tissue was flash frozen with liquid nitrogen. RNA was isolated from tissue as described above. Quality of fastq files was assessed using FastQC v0.11.8 and MultiQC v1.752. Reads were then aligned to GENCODE’s GRCm38.vM24 assembly using STAR v2.6.1a_08-2753,54. Aligned reads were counted using featureCounts v1.6.355. PCA-based clustering was used to identify outliers and then differential expression was performed using DESeq2 v1.30.156. Genes with an adjusted p-value < 0.05 were considered differentially expressed. All analysis was carried out on the University of Michigan Great Lakes HPC cluster.
Survival analysis
The TCGA data was DESeq normalized, and the mean expression of the genes included in the signature was used in the survival analysis employing overall survival data. The differential survival was assessed by a Cox proportional hazards regression and Kaplan-Meier plots were drawn to visualize the survival difference. The plots also include hazards rates and 95% confidence intervals.
Syngeneic and xenograft studies:
Wildtype or athymic nude mice of both sexes were inoculated with 2 million MC38 or CT26 cells. Cells were implanted into lower flanks, and treatment began at day 10 after palpable tumors. Tumor size was measured with digital calipers. At the endpoint roughly 30 days or until tumors were 2 cubic centimeters, mice were killed, and tumors excised. Tumor volume and weight were measured, and tissues were prepared for histology, IHC, and flow cytometry.
Liver metastasis model:
Liver metastasis model was performed as previously described29. Briefly mice were anesthetized using isoflurane. A small incision was made on the left side above the spleen. The spleen was located and gently pulled out. The top splenic artery was sutured off, and then a loose suture loop was placed around the other two vessels that run backwards into the liver (hepatic portal). MC38 cells were suspended in 100ul of PBS and injected into the spleen. After the injection, the suture loop around the vessels was tightened, and a splenectomy was performed.
Adoptive Transfer:
Mice were inoculated subcutaneously on both flanks with 2×106 MC38-OVA cells. When the tumors reached 50–75mm3 in size, 2×106 activated T cells from either ammonia fed OT1 mice, normal fed OT1 mice, or PBS alone was injected intravenously. Tumors size was monitored.
Quantification and Statistical Analysis:
In vitro experiments were validated in multiple cell lines. Each condition of each cell line experiment was performed in technical replicates and repeated three times to ensure reproducibility. Figures show a representative biological replicate unless otherwise indicated. Blinding was performed whenever appropriate. Sample description and identification was unavailable to the core personnel during data collection and analysis. Statistical details of all experiments can be found in the figure legends. The sample numbers are mentioned in each figure legend and denote biological replicates. Statistical details are reported in the figure legends. Results are expressed as the mean +/− standard error of the mean for all figures unless otherwise noted. Significance between 2 groups was tested using a 2 tailed unpaired t test. Significance among multiple groups was tested using a one-way ANOVA. For all ANOVAs the Tukey-Kramer test was used for posthoc multiple comparison, as this allows for the possibility of unequal sample sizes and takes into account the scatter of both groups. GraphPad Prism 7.0 was used for the statistical analysis. Statistical significance is described in the figure legends as: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Supplementary Material
Table S1: Mouse Model RNA Sequencing, Related to Figure 2. This table contains the fold change, significance, and base mean of the RNA sequencing of the colon epithelium of the different mouse models.
Table S2: Mouse Metabolomics, Related to Figure 3. This table contains the metabolomics from the isolated colon epithelium of the indicated mouse models.
Table S3, T Cell Metabolomics, Related to Figure 5. This table contains the metabolomics data for T cells treated with ammonia in vitro or in vivo.
Table S4: Primers and Antibodies, Related to STAR Methods. This table contains the list of primers used in this study as well as the CyToF antibodies used.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ki67 Antibody | Cell Signaling | Cat#12202T |
| InVivoMAb anti-mouse CD8 | BioXCell | Cat#BE0117 |
| Actin Antibody | Protein Tech | Cat# 60008-1-Ig |
| OTC Polyclonal Antibody | Invitrogen | Cat#PA5-28197 |
| CBS (D8F2P) Rabbit mAb | Cell Signaling | Cat#14782 |
| Gamma Cystathionase Rabbit Polyclonal Antibody | ProteinTech | Cat#12217 |
| Mat 1A/IIA Antibody (B-10) | Santa Cruz | Cat#166452 |
| Rat Anti-Mouse IFN-Gamma | BD Biosciences | Cat# 554411 |
| 7-AAD | BD Biosciences | Cat# 559925 |
| Rat Anti-Mouse TNF | BD Biosciences | Cat#561063 |
| Cytof Antibodies | Listed in Table 4. | |
| InVivoMAb anti-mouse PD-L1 | BioXCell | Cat#BE0101 |
| Di-methyl-histone H3 (Lys9) (D85B4) Rabbit mAb | Cell Signaling | Cat#4658 |
| Tri-methyl-histone H3 (Lys9) (D4W1U) Rabbit mAb | Cell Signaling | Cat#13969 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| N-acetyl-cysteine | Cayman Chemical | Cat#20261 |
| Sodium Sulfide | Cayman Chemical | Cat#10122555 |
| Cystathionine | Cayman Chemical | Cat# 16061 |
| Potassium Iodide | Sigma | Cat#7681-11-0 |
| Mercury Iodide | Sigma | Cat# 221090 |
| Ammonia Chloride | Sigma | Cat#213330 |
| Potassium Hydroxide | Sigma | Cat#221473 |
| Sodium Pyruvate | Sigma | Cat#P2256 |
| Lactulose | Sigma | Cat#61360 |
| Taurine | Sigma | Cat#T0625 |
| Serine | Sigma | Cat#S4500 |
| L-Ornithine Monohydrochloride | Sigma | Cat#O6503 |
| Ferrostatin-1 | Cayman Chemical | Cat#17729 |
| L-CC-Propargylglycine | Sigma | Cat#81838 |
| Glutathione Ethyl Ester | Cayman Chemical | Cat#14953 |
| Critical Commercial Assays | ||
| CellTrace CFSE Cell Proliferation Kit | ThermoFisher | Cat#C34554 |
| Ammonia Assay Kit | Sigma | Cat#MAK310 |
| Ammonia Assay Kit | Sigma | Cat#AA0100 |
| DCFDA/H2DCFDA Cellular ROS Assay Kit | AbCam | Cat#ab113851 |
| BODIPY (Lipid Peroxidation Sensor) | ThermoFisher | Cat#D3861 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | N/A |
| MC38 | ATCC | N/A |
| CT26 | ATCC | N/A |
| SW480 | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| Wildtype: C56Bl/6J | Jackson Labs | N/A |
| SingleMut: CDX2-ERT2Cre; Apcfl/fl | Jackson Labs | N/A |
| DoubleMut: CDX2-ERT2Cre; Apcfl/fl; Trp53fl/ | Jackson Labs | N/A |
| TripleMut: Apcfl/fl; Trp53fl/fl; KrasLSLG12D | Jackson Labs | N/A |
| Oligonucleotides | ||
| List of primers | Supplemental Table 4 | N/A |
Highlights:
Autochthonous metastatic mouse model of colon cancer is refractory to immunotherapy.
Ammonia accumulates in tumors and reduces T cell function in vitro and in vivo.
Ammonia decreases T cell transsulfuration leading to altered redox status.
Clearance of ammonia reactivates T cells and decreases colorectal cancer.
Acknowledgements
This work was funded by NIH grants: R01CA148828, R01CA245546, and R01DK095201 ( Y.M.S); R37CA237421, R01CA248160, and R01CA244931 (C.A.L); UMCCC Core Grant P30CA046592 (Y.M.S and C.A.L); and R35GM130183 (R.B). H.N.B was supported by T32 training grant (GM008322) and F30CA257292. R.K was supported by a postdoctoral fellowship from the American Heart Association (826245) and NIH grant F30CA257292. S.A.K was supported by an NIH F31 fellowship (F31CA247457) and NIH fellowship (F99CA264414). S.L.M was supported by CMB Graduate Program T32GM007315. R.S was supported by an American Physiological Society postdoctoral fellowship (032650). S.S was supported by the Crohn’s and Colitis Foundation Research fellowship award (623914) and the American Heart Association postdoctoral fellowship (19POST34380588). B.G. was supported by National Research, Development and Innovation Office (PharmaLab, RRF-2.3.1-21-2022-00015 and 2020-1.1.6-JÖVŐ-2021-00013).
Footnotes
Declaration of Interests: C.A.L. has received consulting fees from Astellas Pharmaceuticals, Odyssey Therapeutics, and T-Knife Therapeutics, and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1-pathway as a therapeutic approach (US Patent No: 2015126580-A1, 05/07/2015; US Patent No: 20190136238, 05/09/2019; International Patent No: WO2013177426-A2, 04/23/2015).
The authors declare no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, and Bray F (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca Cancer J Clin 71, 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Holt PR, Kozuch P, and Mewar S (2009). Colon cancer and the elderly: From screening to treatment in management of GI disease in the elderly. Best Pract Res Clin Gastroenterology 23, 889–907. 10.1016/j.bpg.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biller LH, and Schrag D (2021). Diagnosis and Treatment of Metastatic Colorectal Cancer. Jama 325, 669–685. 10.1001/jama.2021.0106. [DOI] [PubMed] [Google Scholar]
- 4.Araghi M, Arnold M, Rutherford MJ, Guren MG, Cabasag CJ, Bardot A, Ferlay J, Tervonen H, Shack L, Woods RR, et al. (2021). Colon and rectal cancer survival in seven high-income countries 2010–2014: variation by age and stage at diagnosis (the ICBP SURVMARK-2 project). Gut 70, 114–126. 10.1136/gutjnl-2020-320625. [DOI] [PubMed] [Google Scholar]
- 5.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. (2010). Phase I Study of Single-Agent Anti–Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol 28, 3167–3175. 10.1200/jco.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Pavlova NN, and Thompson CB (2017). Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. Embo J 36, 1302–1315. 10.15252/embj.201696151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Zhu H, Sun W, Yang X, Nie Q, and Fang X (2021). Role of glutamine and its metabolite ammonia in crosstalk of cancer-associated fibroblasts and cancer cells. Cancer Cell Int 21, 479. 10.1186/s12935-021-02121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Ruan J, Li D, Wang M, Han Z, Qiu W, and Wu G (2021). The Role of Intestinal Bacteria and Gut–Brain Axis in Hepatic Encephalopathy. Front Cell Infect Mi 10, 595759. 10.3389/fcimb.2020.595759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holeček M (2018). Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metabolism 15, 33. 10.1186/s12986-018-0271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, and Haigis MC (2017). Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946. 10.1126/science.aam9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoue Y, Hayhurst GP, Inoue J, Mori M, and Gonzalez FJ (2002). Defective Ureagenesis in Mice Carrying a Liver-specific Disruption of Hepatocyte Nuclear Factor 4α (HNF4α) HNF4α REGULATES ORNITHINE TRANSCARBAMYLASE IN VIVO *. J Biol Chem 277, 25257–25265. 10.1074/jbc.m203126200. [DOI] [PubMed] [Google Scholar]
- 12.Li B, Chan HL, and Chen P (2019). Immune Checkpoint Inhibitors: Basics and Challenges. Curr Med Chem 26, 3009–3025. 10.2174/0929867324666170804143706. [DOI] [PubMed] [Google Scholar]
- 13.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, and Diaz LA (2019). Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroentero 16, 361–375. 10.1038/s41575-019-0126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diao Z, Han Y, Chen Y, Zhang R, and Li J (2020). The Clinical Utility of Microsatellite Instability in Colorectal Cancer. Crit Rev Oncol Hemat 157, 103171. 10.1016/j.critrevonc.2020.103171. [DOI] [PubMed] [Google Scholar]
- 15.Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, Hermann-Kleiter N, Löwer M, Baier G, Krogsdam A, et al. (2018). Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun 9, 32. 10.1038/s41467-017-02424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell HN, Rebernick RJ, Goyert J, Singhal R, Kuljanin M, Kerk SA, Huang W, Das NK, Andren A, Solanki S, et al. (2021). Reuterin in the healthy gut microbiome suppresses colorectal cancer growth through altering redox balance. Cancer Cell 40, 185–200.e6. 10.1016/j.ccell.2021.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu D, Wu P, Huang Q, Liu Y, Ye J, and Huang J (2013). Interleukin-17: A Promoter in Colorectal Cancer Progression. Clin Dev Immunol 2013, 436307. 10.1155/2013/436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu G-Y, Österreicher CH, Hung KE, et al. (2012). Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258. 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chae W-J, Gibson TF, Zelterman D, Hao L, Henegariu O, and Bothwell ALM (2010). Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc National Acad Sci 107, 5540–5544. 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, Jagodnik KM, Kropiwnicki E, Wang Z, and Ma’ayan A (2019). ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res 47, W212–W224. 10.1093/nar/gkz446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2018). STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, gky1131. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, Auslander N, Keshet R, Stettner N, Silberman A, Agemy L, et al. (2018). Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 174, 1559–1570.e22. 10.1016/j.cell.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Shi R, Bian X, Zhou C, Zhao Y, Zhang S, Wu F, Waterhouse GIN, Wu L, Tung C, et al. (2019). Ammonia Detection Methods in Photocatalytic and Electrocatalytic Experiments: How to Improve the Reliability of NH3 Production Rates? Adv Sci 6, 1802109. 10.1002/advs.201802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar A, Welch N, Mishra S, Bellar A, Silva RN, Li L, Singh SS, Sharkoff M, Kerr A, Chelluboyina AK, et al. (2021). Metabolic reprogramming during hyperammonemia targets mitochondrial function and postmitotic senescence. Jci Insight 6, e154089. 10.1172/jci.insight.154089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies NA, Wright G, Ytrebø LM, Stadlbauer V, Fuskevåg O, Zwingmann C, Davies DC, Habtesion A, Hodges SJ, and Jalan R (2009). L-ornithine and phenylacetate synergistically produce sustained reduction in ammonia and brain water in cirrhotic rats. Hepatology 50, 155–164. 10.1002/hep.22897. [DOI] [PubMed] [Google Scholar]
- 26.Kircheis G, and Lüth S (2019). Pharmacokinetic and Pharmacodynamic Properties of l-Ornithine l-Aspartate (LOLA) in Hepatic Encephalopathy. Drugs 79, 23–29. 10.1007/s40265-018-1023-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berry SA, Lichter-Konecki U, Diaz GA, McCandless SE, Rhead W, Smith W, LeMons C, Nagamani SCS, Coakley DF, Mokhtarani M, et al. (2014). Glycerol phenylbutyrate treatment in children with urea cycle disorders: Pooled analysis of short and long-term ammonia control and outcomes. Mol Genet Metab 112, 17–24. 10.1016/j.ymgme.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrmann R, and Weber FL (1987). LACTULOSE AND AMMONIA. Lancet 330, 1333. 10.1016/s0140-6736(87)91224-4. [DOI] [PubMed] [Google Scholar]
- 29.Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, Rizvi SM, Qin A, Waninger JJ, Lang X, et al. (2021). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med 27, 152–164. 10.1038/s41591-020-1131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J, et al. (2019). KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 35, 559–572.e7. 10.1016/j.ccell.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, Pastille E, and Jendrossek V (2017). Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell Physiol Biochem 41, 1271–1284. 10.1159/000464429. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z-H, Peng W-B, Zhang P, Yang X-P, and Zhou Q (2021). Lactate in the tumour microenvironment: From immune modulation to therapy. Ebiomedicine 73, 103627. 10.1016/j.ebiom.2021.103627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bai C, Wang H, Dong D, Li T, Yu Z, Guo J, Zhou W, Li D, Yan R, Wang L, et al. (2021). Urea as a By-Product of Ammonia Metabolism Can Be a Potential Serum Biomarker of Hepatocellular Carcinoma. Frontiers Cell Dev Biology 9, 650748. 10.3389/fcell.2021.650748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bader JE, Voss K, and Rathmell JC (2020). Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell 78, 1019–1033. 10.1016/j.molcel.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT, et al. (2021). Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288. 10.1038/s41586-021-03442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawłowska A, et al. (2020). Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282. 10.1038/s41586-020-2682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang C-H, and Pearce EL (2016). Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol 17, 364–368. 10.1038/ni.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinclair LV, Howden AJ, Brenes A, Spinelli L, Hukelmann JL, Macintyre AN, Liu X, Thomson S, Taylor PM, Rathmell JC, et al. (2019). Antigen receptor control of methionine metabolism in T cells. Elife 8, e44210. 10.7554/elife.44210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gatalica Z, Vranic S, Xiu J, Swensen J, and Reddy S (2016). High microsatellite instability (MSI-H) colorectal carcinoma: a brief review of predictive biomarkers in the era of personalized medicine. Fam Cancer 15, 405–412. 10.1007/s10689-016-9884-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Picard E, Verschoor CP, Ma GW, and Pawelec G (2020). Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front Immunol 11, 369. 10.3389/fimmu.2020.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, An Y, Qin X, Wu X, Wang X, Hou H, Song X, Liu T, Wang B, Huang X, et al. (2021). Gut Microbiota-Derived Metabolites in Colorectal Cancer: The Bad and the Challenges. Frontiers Oncol 11, 739648. 10.3389/fonc.2021.739648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper AJL, and Jeitner TM (2016). Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomol 6, 16. 10.3390/biom6020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, Neumann J, James MJ, Geiger S, Jin W, et al. (2021). Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature 598, 662–666. 10.1038/s41586-021-04003-2. [DOI] [PubMed] [Google Scholar]
- 45.Lánczky A, and Győrffy B (2021). Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res 23, e27633. 10.2196/27633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennett BD, Yuan J, Kimball EH, and Rabinowitz JD (2008). Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc 3, 1299–1311. 10.1038/nprot.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chae TU, Kim WJ, Choi S, Park SJ, and Lee SY (2015). Metabolic engineering of Escherichia coli for the production of 1,3-diaminopropane, a three carbon diamine. Sci Rep-uk 5, 13040. 10.1038/srep13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singhal R, Mitta SR, Das NK, Kerk SA, Sajjakulnukit P, Solanki S, Andren A, Kumar R, Olive KP, Banerjee R, et al. (2021). HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest 131. 10.1172/jci143691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosharov E, Cranford MR, and Banerjee R (2000). The Quantitatively Important Relationship between Homocysteine Metabolism and Glutathione Synthesis by the Transsulfuration Pathway and Its Regulation by Redox Changes †. Biochemistry-us 39, 13005–13011. 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 50.Spinelli JB, Kelley LP, and Haigis MC (2017). An LC-MS Approach to Quantitative Measurement of Ammonia Isotopologues. Sci Rep-uk 7, 10304. 10.1038/s41598-017-09993-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gutiérrez-de-Juan V, Davalillo S.L. de, Fernández-Ramos D, Barbier-Torres L, Zubiete-Franco I, Fernández-Tussy P, Simon J, Lopitz-Otsoa F, Heras J. de las, Iruzubieta P, et al. (2017). A morphological method for ammonia detection in liver. Plos One 12, e0173914. 10.1371/journal.pone.0173914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2018). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, gky955–. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 56.Ren X, and Kuan P-F (2020). Negative binomial additive model for RNA-Seq data analysis. Bmc Bioinformatics 21, 171. 10.1186/s12859-020-3506-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mouse Model RNA Sequencing, Related to Figure 2. This table contains the fold change, significance, and base mean of the RNA sequencing of the colon epithelium of the different mouse models.
Table S2: Mouse Metabolomics, Related to Figure 3. This table contains the metabolomics from the isolated colon epithelium of the indicated mouse models.
Table S3, T Cell Metabolomics, Related to Figure 5. This table contains the metabolomics data for T cells treated with ammonia in vitro or in vivo.
Table S4: Primers and Antibodies, Related to STAR Methods. This table contains the list of primers used in this study as well as the CyToF antibodies used.
Data Availability Statement
Data:
Data generated in this study is available in the supplementary tables. Any raw data requested that was generated from this paper will be shared by the lead contact upon request.
Code:
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.