

Abstract
Type I toxin–antitoxin systems (T1TAs) are extremely potent bacterial killing systems difficult to characterize using classical approaches. To assess the killing capability of type I toxins and to identify mutations suppressing the toxin expression or activity, we previously developed the FASTBAC-Seq (Functional AnalysiS of Toxin–Antitoxin Systems in BACteria by Deep Sequencing) method in Helicobacter pylori. This method combines a life and death selection with deep sequencing. Here, we adapted and improved our method to investigate T1TAs in the model organism Escherichia coli. As a proof of concept, we revisited the regulation of the plasmidic hok/Sok T1TA system. We revealed the death-inducing phenotype of the Hok toxin when it is expressed from the chromosome in the absence of the antitoxin and recovered previously described intragenic toxicity determinants of this system. We identified nucleotides that are essential for the transcription, translation or activity of Hok. We also discovered single-nucleotide substitutions leading to structural changes affecting either the translation or the stability of the hok mRNA. Overall, we provide the community with an easy-to-use approach to widely characterize TA systems from diverse types and bacteria.
INTRODUCTION
Many bacteria encode lethal proteins in their genome alongside antidotes that are able to counteract their toxicity. These toxin–antitoxin (TA) systems are classified into different types according to the nature of the antitoxins and the mechanism of action of the toxin, extensively reviewed by Harms et al. (1). Type I TA systems (T1TAs) are RNA/RNA interacting systems. The antitoxin RNA, encoded on the opposite strand of the toxin mRNA, acts as a noncoding antisense RNA, mediating translation inhibition and/or mRNA degradation (2). The genomic location of predicted and validated T1TAs can be found in the T1TA database (db) that we recently developed (3).
The hok/Sok system has been the most studied T1TA. It was first discovered on Escherichia coli R1 plasmid where it acts by maintaining plasmid copies in a cell population through post-segregational killing of the plasmid-free cells (4,5). The Hok (host-killing) type I toxin is a small hydrophobic protein [52 amino acids (aa)] targeting the inner membrane and leading to cell death. The Sok (suppression of killing) antitoxin is an RNA that inhibits the production of Hok at the post-transcriptional level (6,7). The system is composed of a third gene, mok (modulation of killing), that overlaps with the hok coding sequence (CDS) and is required for hok translation (6). Indeed, the translation of mok, rather than the Mok product, was shown to be important for proper hok regulation and expression (6). For simplicity, the mok_hok bicistronic mRNA will be referred to as the hok mRNA throughout this paper.
Pioneering studies have demonstrated that hok mRNA translation requires a conformational switch between a full-length (FL) hok mRNA (translationally inactive) and a truncated (Tr) hok mRNA structure (translationally active) (8,9). First, a long-distance interaction between the 5′ and the 3′ untranslated regions (UTRs) strongly stabilizes the closed FL-hok mRNA. This interaction, described as the ‘translational activator element’/‘fold-back inhibitory element’ (tac/fbi) stem, prevents translation by sequestration of the Shine–Dalgarno sequence (SD) of mok by an ‘upstream complementary box’ (ucb) (9). Then, the hok mRNA undergoes a slow maturation at the 3′ end, triggering a structural rearrangement that renders the Tr-hok mRNA translatable by the formation of a tac/ucb stem and a mok SD/‘downstream complementary box’ (dcb) stem (10). Not only this rearrangement frees the mok SD for translation, it also allows the binding of the Sok RNA antitoxin (6,10). This binding entails rapid degradation of the hok mRNA/Sok RNA duplex by RNase III (10,11). In addition, to avoid co-transcriptional translation, a transient metastable structure is formed before the hok mRNA is fully transcribed, where the tac element pairs with a complementary region immediately downstream (12).
T1TAs have been predicted in pathogenic bacteria both in silico and from transcriptomic studies (e.g. Helicobacter pylori, Clostridioides difficile, Staphylococcus aureus) (13–16). Nonetheless, most of these systems still require functional characterization. The peculiarities of T1TAs make them difficult to study. Indeed, they are strongly regulated at the co- and post-transcriptional levels by key sequence and/or structure elements. Very often, these regulatory elements are difficult to predict and their study requires extensive genetic manipulation. Up to now, insights about TA systems from various types and bacteria were mostly gathered from studying the effects of toxin overexpression (17). Because many of these systems are chromosomally encoded, their expression should be studied at the chromosome, instead of using plasmid overexpression, to ensure studying native levels. To this end, we recently developed a strategy to study chromosomal TA expression named FASTBAC-Seq (Functional AnalysiS of Toxin–Antitoxin Systems in BACteria by Deep Sequencing) (18,19). Our approach enables us to (i) assess the death-inducing capabilities of a toxin and (ii) identify single-nucleotide substitutions that abrogate the toxin’s function or expression. We take advantage of the lethality induced by the expression of the toxin in the absence of the antitoxin to search for intragenic toxicity suppressor mutations. Using this method, we identified crucial elements that regulate the aapA3/IsoA3 T1TA in H. pylori. In brief, we introduced mutated PCR libraries of the aapA3/IsoA3 and of the aapA3/ΔIsoA3 loci in the H. pylori chromosome. In the aapA3/ΔIsoA3 transformants, the expression of the IsoA3 antitoxin was turned off by introducing point mutations in its promoter, leading to the constitutive synthesis of AapA3 toxin and cell death. We then performed a life and death selection and identified numerous nucleotide substitutions in the CDS, but also in the UTRs impacting AapA3 protein activity or the aapA3 toxin mRNA structure or translatability. The analysis of these mutations led to the discovery of novel regulatory elements of this system, notably the identification of functional metastable structures in the aapA3 mRNA (18). These transient structures inhibit co-transcriptional translation of the nascent aapA3 mRNA and prevent premature toxicity.
Here, we adapted and improved the FASTBAC-Seq method to be performed in the model organism E. coli, making it convenient to widely characterize TA systems from diverse types and bacteria. As a proof of concept, we investigated the well-studied plasmidic hok/Sok system, for which many intragenic regulatory elements have already been described and validated. Overall, we retrieved most of the already described toxicity determinants, identifying single-nucleotide mutations impeding the toxin mRNA synthesis and translation as well as the toxin activity. In particular, we detected many single-nucleotide substitutions that prevent the conformational change of the hok mRNA locking it in its inactive state, validating the previously published secondary structures of the FL- and Tr-hok mRNAs. Unexpectedly, several substitutions in the hok CDS were not interfering with protein activity but instead altered its expression. Overall, our method not only identified known and predicted regulatory elements, but also pointed to new elements in the CDS that act by an unknown mechanism. Delineating regulatory elements of TA is crucial to annotate these systems and develop better bioinformatic prediction tools.
MATERIALS AND METHODS
Escherichia coli strains, plasmids, oligonucleotides and culture conditions
All the E. coli strains used and constructed in the present study originate from the XTL632 strain ‘MG1655 galM<tetA-sacB>gpmA’ from the Court laboratory (20) and are listed in Supplementary Table S1. The plasmid and oligonucleotide sequences are listed in Supplementary Tables S2 and S3. Escherichia coli strains were grown on LB agar plates (Condalab) and in LB broth (Condalab) (with 30 μg/ml chloramphenicol when needed).
hok/Sok cassettes inserted in E. coli
The studied locus hok/Sok originates from the E. coli plasmid R1 minimal parB+ region (sequence corresponding to entry TA07123 in T1TAdb: https://d-lab.arna.cnrs.fr/t1tadb, from NCBI GenBank accession number X05813). The sequences of hok/Sok, hok/ΔpSok (containing point mutations inactivating Sok promoter) and Δphok/ΔpSok (containing point mutations inactivating both hok and Sok promoters) were synthesized (IDT-DNA, gBlocks, Supplementary Table S4) and inserted in E. coli by λ-red recombineering following the protocol from the Court laboratory (21) and summarized below. The strain XTL632 ‘MG1655 galM<tetA-sacB>gpmA’ was used as a recipient strain and its tetA-sacB cassette was replaced by the different wild-type (WT) and mutated hok/Sok cassettes. Note that the XTL632 strain contains five endogenous hok/Sok systems, namely hokA, hokB, hokC, hokD and hokE (3).
FASTBAC-Seq experiment
The synthesized DNA sequences hok/Sok, hok/ΔpSok and Δphok/ΔpSok were first amplified by three cycles of PCR with FA890 and FA891 using the Phusion High-Fidelity Hot Start DNA Polymerase (Thermo Fisher Scientific) to add the flanking regions necessary for recombination at the tetA-sacB site in E. coli, and then by 30 cycles of PCR with FA873 and FA874 using the Dream Taq DNA polymerase (Thermo Fisher Scientific) introducing mutations in the PCR fragments. After purification using the ‘gel extraction kit’ (Neobiotech), these PCR products were introduced in E. coli by electroporation. In brief, after an overnight culture with 50 μg/ml of hygromycin at 30°C, 180 rpm, the XTL632 strain was diluted 70-fold in fresh medium and grown during 2 h at 32°C (to reach 0.4 < OD600nm < 0.6) without antibiotics. Induction of the λ-red recombination functions was performed by heat shock at 42°C for 15 min. The cells were rendered competent using ice-cold water and 50 μl of competent cells were mixed with either 10 μl of 10 ng/μl PCR fragments (100 ng total) or H2O for the negative control, and electroporated at 1.8 kV in 0.1 cm cuvettes (Dutscher). The cells were recovered after 4 h at 32°C, 180 rpm, in 10 ml of LB medium, before being pelleted, washed with M9 medium and plated on ‘Tet/SacB counter-selection plates’ [prepared as described in (21): 4 g tryptone, 4 g yeast extract, 15 g Difco agar, 8 g NaCl, 8 g NaH2PO4·H2O, 100 ml of a 60% sucrose stock solution, 0.5 ml of a 48 mg/ml fusaric acid (Sigma) stock solution in ethanol, 32 ml of a 25 mM ZnCl2 stock solution) at 42°C. The total viable cells were determined by plating 50 μl of a 10–6 dilution in triplicate on LB agar plates. Ninety-six transformants from the ‘Tet/SacB plates’ were tested by colony PCR using FA873 and FA874, and the PCR products were sequenced. All the transformants tested had integrated the hok/Sok cassette. Escherichia coli strains with hok/Sok and Δphok/ΔpSok were checked for sequence integrity and frozen as EFD475 and EFD515, respectively. Insertion of the hok/ΔpSok was toxic and resulted only in transformants with mutations in the locus; individual mutants were frozen (Supplementary Table S1). This experiment (both PCR and transformations) was repeated five times independently and we retrieved from 3000 to 15 000 colonies (same number per experiment, according to the smallest number obtained). The genomic DNA of the mixture of colonies was extracted using the Quick Bacteria Genomic DNA Extraction Kit (Neobiotech) following the supplier’s recommendations. The hok/Sok locus was amplified by FA910 and FA911 (carrying Illumina adapters) using the Mix 2X KAPA HiFi HotStart (Roche), resulting in 486 nucleotide (nt) amplicons. The amplicons were sequenced at the Bordeaux Transcriptome Genome Platform (Cestas-Pierroton, France) by Illumina technology on a MiSeq V3 instrument in paired-end mode (2 × 300 nt with overlapping reads).
FASTBAC-Seq data analysis
Sequence data were processed and analyzed as in (19) with slight adjustments [software versions and parameters that differ from (19) are given below]. The numbers of total reads, merged reads and mapped reads are available in Supplementary Table S5. Briefly, the analysis pipeline included the following steps for each sample: (i) trimming low-quality 3′ ends of reads and discarding low-quality reads using cutadapt (https://cutadapt.readthedocs.io/en/stable/) and prinseq-lite (22); reverse reads (R2 reads) were also trimmed of their first seven bases (option ‘-trim_left 7’ in prinseq-lite) because there was a large proportion (>20%) of these reads with undefined bases in this region; (ii) recovering read pairs for which both mates passed the quality filtering steps using cmpfastq (https://github.com/iirisarri/scripts_main/blob/master/cmpfastq.pl) and assembling mates into a single sequence by means of PANDAseq 2.9 (23) run with options ‘-N -o 20 -O 0 -t 0.6 -A simple_bayesian -C empty’; (iii) aligning these assembled reads onto the 486 nt reference sequence (hok/Sok, hok/ΔpSok or Δphok/ΔpSok sequence, depending on the sample) by the BWA-MEM algorithm of BWA 0.7.17 (24) run with options ‘-A 1 -B 4 -O 6 -E 1 -L 10, 10’; (iv) extracting mapped sequences of length 486 nt and showing a single-nucleotide substitution compared to the reference using samtools 1.9 (25) and bamtools (https://github.com/pezmaster31/bamtools); and (v) performing statistical analyses of the differential distribution of single-nucleotide mutations between hok/Sok or Δphok/ΔpSok and hok/ΔpSok sequences. For substitutions, a ‘nucleotide-specific’ analysis to determine whether particular nucleotides were enriched at specific positions was carried out exactly as described in (19) using Trinity 2.10.0 (19,26), R 3.5.3 (https://www.R-project.org/) and DESeq2 1.22.2 (27). Mutations were considered as significantly over- or under-represented in the mutant versus the reference sample if the adjusted P-value (Padj, corresponding to the false discovery rate) was inferior or equal to 5% (Padj ≤ 0.05) and the log2 fold change (FC) was superior or equal to 1 or inferior or equal to − 1 (|log2 FC| ≥ 1) (Supplementary Table S6). We then combined the results of the differential distribution of mutations in hok/Sok versus hok/ΔpSok and Δphok/ΔpSok versus hok/ΔpSok (Supplementary Table S6). Note that the positions corresponding to the mutated nucleotides of hok or Sok promoters were not included in the analysis. Three positions had a negative log2 FC, meaning that they were more frequent in hok/Sok or Δphok/ΔpSok than in hok/ΔpSok and were not further studied.
Construction of plasmids expressing hok-FLAG variants
The synthesized DNA sequence hok/ΔpSok was amplified by FB64/FB66 and FB67/FB65 primer pairs to add the FLAG sequence and the two fragments were then assembled using FB64/FB65. The resulting fragment was cloned in pAZ3 digested with EcoRI and HindIII to generate the plasmid pEFD558, which was used as a template to construct the plasmids carrying mutated Hok*-FLAG variants. The mutations were introduced with a two-step PCR, using primers listed in Supplementary Table S3. The sequence integrity of all obtained plasmids was checked using FA858.
Total RNA extraction
Six hundred twenty-five microliters of Stop Solution (95% ethanol, 5% phenol, pH 4.5, 4°C) was added to 5 ml of exponentially growing cultures. The cells were immediately centrifuged and the pellets were stored at −80°C. After resuspension in 400 μl Lysis Solution (20 mM NaAc, pH 5.2, 0.1% SDS, 1 mM EDTA), the samples were transferred into 400 μl hot phenol (pH 5.2) and incubated for 10 min at 65°C. The samples were centrifuged for 10 min at 13 000 rpm at room temperature and the aqueous phase was transferred to a phase-locked gel tube (Eppendorf) containing an equal volume of chloroform/isoamyl alcohol. After 10 min of centrifugation at 13 000 rpm at room temperature, the aqueous phase was mixed with 2.5 volumes of 100% ethanol and 0.1 volume of 3 M NaAc (pH 5.2) and incubated overnight at −20°C. The precipitated RNAs were centrifuged for 30 min at 13 000 rpm at 4°C, washed with 70% ethanol, air-dried and resuspended in H2O. The RNA concentration was determined using a DeNovix and the RNA integrity was checked on 2% agarose gel stained with EtBr.
Northern blot
Ten micrograms of total RNA was separated on 8% polyacrylamide/7 M urea gels in 1× Tris–borate–EDTA (TBE). The RNAs were transferred to a Nylon membrane (Hybond-N, GE Healthcare Life Sciences) by electroblotting in 1× TBE at 8 V and 4°C overnight and cross-linked to the membrane by UV irradiation (302 nm) for 2 min in a UV cross-linker. The FB178, FA909, FB181 and FD211 (5S) oligonucleotides (Supplementary Table S3) were labeled at their 5′ end with 32P and incubated with the membranes in a modified Church Buffer (1 mM EDTA, 0.5 M NaPO4, pH 7.2, 7% SDS) overnight at 42°C. The membranes were washed two times for 5 min in 2× SSC and 0.1% SDS and revealed using a Pharos FX phosphorimager (Bio-Rad).
Western blot
Cell pellets from 1 ml culture reaching OD600nm between 0.5 and 0.8 were lysed in Lane Marker Reducing Sample Buffer 1× (Thermo Scientific) (100 μl for 1 OD600/ml) incubated at room temperature in an ultrasound water bath for 15 min. Samples were incubated with 25 units of Benzonase (Merck) at room temperature for 15 min. Ten microliters of each sample was separated on 16% SDS-PAGE gels (1 M Tris, pH 8.45, glycerol 10%, 0.1% SDS) in cathode buffer (0.1 M Tris, 0.1 M tricine, 0.1% SDS) and anode buffer (0.1 M Tris–HCl, pH 8.9) at 60 V for 30 min and at 160 V for 90 min. Proteins were transferred to a nitrocellulose membrane in transfer buffer (Tris–glycine 1×, ethanol 20%, SDS 0.05%) at 90 V for 90 min. The membrane with total proteins was stained with SYPRO™ Ruby Protein Gel Stain (Thermo Fisher Scientific) and detected using a Pharos FX phosphorimager (Bio-Rad). Membranes were blocked with 5% skimmed milk in TBS 1× and Tween 20 0.5%, then incubated with 1:800 monoclonal anti-FLAG M2 and 1:10 000 anti-rabbit IgG (whole molecule)-peroxidase (Sigma) and revealed using Western Clarity ECL (Bio-Rad) on a Chemidoc (Bio-Rad).
RESULTS AND DISCUSSION
Experimental strategy and application to the hok/Sok T1TA system
T1TA systems are small toxic genetic modules whose expression involves multiple levels of co- and post-transcriptional regulations (2). We recently developed the FASTBAC-Seq method, which is a powerful tool to quickly reveal key regulatory elements. The aim of the present study was to establish this method in the model organism E. coli. Indeed, the original method could only be carried out in the human gastric pathogen H. pylori (18,19), which hampered the analysis of multiple predicted TA systems.
Before going to the detailed study of the hok/Sok system, here is a brief summary of our revised experimental strategy summarized in three steps. (I) The first step is to generate mutated PCR libraries of TA and Δantitoxin (ΔA) loci from synthesized DNA fragments (Figure 1). In the ΔA fragment, the promoter of the antitoxin is lacking or inactivated by point mutations. It is essential to check that the mutations in the antitoxin promoter are effectively abrogating antitoxin expression, for instance, by using northern blot. (II) Then, these PCR libraries are transformed in E. coli and integrated by homologous recombination in a chromosomal region of interest containing a selection/counter-selection cassette. (III) Finally, the transformants are selected by the loss of the cassette and first analyzed by single colony PCR and Sanger sequencing. Because this method is designed for the study of death-inducing toxins, it is crucial to demonstrate the toxicity of the studied toxin when expressed from the chromosome. This is easily assessed by individually sequencing several ΔA transformants (Figure 1). If all surviving colonies contain inactivating mutations, it indicates that the ΔA construct is toxic. Once the toxicity is confirmed, loss-of-function mutants can be identified in a high-throughput manner. All transformants obtained from each biological replicate are pooled together and steps of DNA extraction, amplicon PCR and deep sequencing are performed (Figure 1). Finally, the mutants of interest can be reconstructed individually to validate their ability to grow and assess their effect on transcription and translation using routine molecular techniques.
Figure 1.
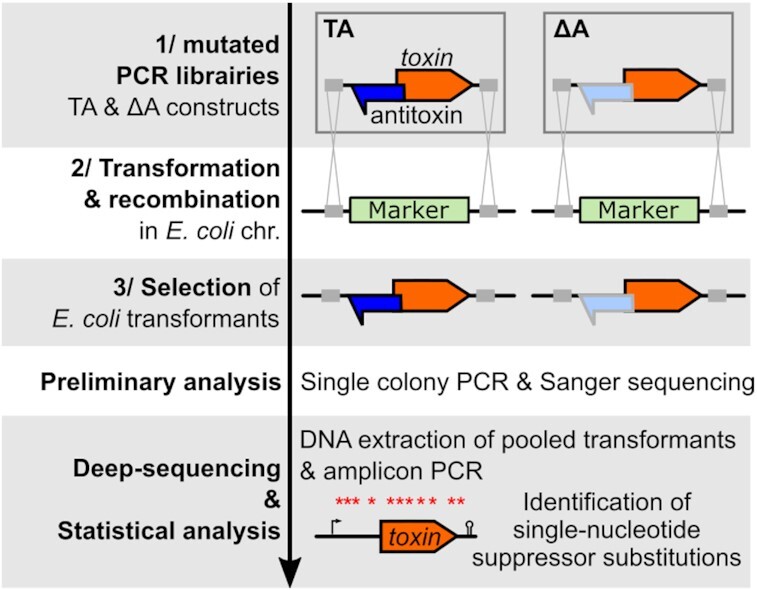
Strategy of FASTBAC-Seq in Escherichia coli. (1) The first step is to generate mutated PCR libraries from TA and ΔA sequences containing flanking regions (gray boxes) allowing recombination in the E. coli chromosome. To do this, chemically synthesized DNA fragments (TA and ΔA) are amplified using the Taq polymerase, generating mutations. The gray outline and lighter blue color of the antitoxin in the ΔA construct indicates the loss of expression due to point mutations in the promoter. (2) The PCR libraries are then introduced in an E. coli recipient strain containing a selection/counter-selection marker (in green) and (3) transformants are selected on appropriate media. Preliminary analysis: The TA and ΔA constructs of randomly chosen transformants are amplified and sequenced. If the TA loci of all ΔA transformants are mutated, it indicates that mutations are necessary for survival and therefore the expression of the toxin from the E. coli chromosome is toxic in the absence of the antitoxin. In contrast, only a proportion of TA should be mutated. Deep-sequencing & Statistical analysis: Several independent PCR reactions for each PCR library are transformed and integrated in the E. coli chromosome. All the colonies obtained from the same transformation are pooled together and the TA and ΔA loci are amplified by PCR after DNA extraction. The amplicon is sequenced using Illumina technology and mapped against the WT sequence, and the occurrence of single-nucleotide substitutions is compared between the TA and ΔA using statistical analysis. The substitutions significantly enriched in the ΔA relative to TA samples are annotated as loss-of-function substitutions, mapped over the TA locus and further studied.
To set up our new protocol, we chose the best characterized T1TA of E. coli, the hok/Sok system discovered on the R1 plasmid (5). We first designed three hok/Sok constructs to be integrated in the E. coli chromosome, namely the hok/Sok toxin–antitoxin (HS), hok/ΔpSok (HΔS), but also Δphok/ΔpSok (ΔHΔS) as an additional control (Figure 2A). In the HS locus, both the toxin and antitoxin RNAs are expressed and the antitoxin binds to the Tr-hok mRNA leading to its degradation. In HΔS, only the toxin RNA is expressed, as the transcription of the Sok antitoxin is turned off by mutations in its promoter region. The Hok protein should be produced in this construction. In ΔHΔS, the transcription of both the hok toxin mRNA and the Sok antitoxin RNA is inhibited by mutations in their respective promoters.
Figure 2.

Identification of single-nucleotide suppressor substitutions in the hok/Sok locus. (A) The hok/Sok locus consists of two overlapping CDSs, the leader peptide mok and the toxic protein hok, and of the Sok noncoding RNA. The mok_hok mRNA (or hok mRNA) is transcribed as an untranslatable FL mRNA that is processed at the 3′ end to form a translatable, truncated mRNA (Tr-hok). Here, the HS, HΔS and ΔHΔS constructs were amplified by PCR to induce mutations and individually introduced into the E. coli chromosome. The expected expression of each of the three constructs designed for FASTBAC-Seq is indicated. HS: The Sok antitoxin inhibits the Tr-hok mRNA translation by inducing degradation of the toxin/antitoxin RNA duplex by RNase III. HΔS: In the absence of the Sok antitoxin, Tr-hok mRNA can be translated and is toxic for the host cell. ΔHΔS: The locus with the promoters of both hok and Sok inactivated was used as a control; hok and Sok are not transcribed. (B) FASTBAC-Seq results. The differential distribution of single-nucleotide substitutions was statistically analyzed between the hok*/ΔpSok and the controls (ctrl) hok/Sok and Δhok/ΔpSok. We obtained loss-of-function substitutions (occurring statistically more in hok*/ΔpSok) well distributed over the hok/Sok locus. The arrow represents the promoter, and the ‘stem-loop’ represents the terminator. The CDSs of mok and hok are shown in orange colors. The zoomed gray box shows the location substitution leading to stop codons in mok or hok CDS.
We first amplified the three synthesized DNA fragments by PCR using a low-fidelity Taq DNA polymerase introducing mutations (Figure 1). The PCR libraries were then transformed in E. coli XTL632, a strain harboring a tetA-sacB selection/counter-selection cassette in its chromosome between the galM and gpmA genes (20) (Supplementary Figure S1). The tetA gene confers tetracycline resistance and fusaric acid sensitivity, while sacB confers sucrose sensitivity. We promoted E. coli recombination functions following a robust protocol (21). Briefly, we induced the expression of the λ-red recombination genes from the pSIM18 plasmid before preparing electrocompetent cells and transforming the constructs of interest by electroporation. We then selected the transformants in which the tetA-sacB selection cassette was replaced by the HS, HΔS or ΔHΔS constructs by plating on LB agar containing fusaric acid and sucrose (21) (Supplementary Figure S1). All the clones tested individually had integrated the constructs, confirming that this selection method was highly efficient.
Preliminary analysis: chromosomal expression of Hok is toxic
We sequenced two transformants for the HS and ΔHΔS PCR transformations, as well as 91 transformants for HΔS. As expected, we obtained unmutated sequences for HS and ΔHΔS; however, all HΔS colonies carried loss-of-function mutations in their locus (HΔS). Among them, we obtained 26 clones with a single-nucleotide substitution, 22 clones with a single deletion and the 43 other clones containing more complex mutations (one or several truncations, and several substitutions/deletions/insertions) (Supplementary Table S6). Two mutations were located in the hok promoter, one in the mok start codon and two led to a premature stop codon in hok. This demonstrated that the expression of a chromosomal copy of hok from the R1 plasmid using its native promoter is toxic for E. coli in the absence of the Sok antitoxin. Hok was previously shown to be toxic when expressed from a plasmid but never from the chromosome. Therefore, our work reveals that the level of chromosomal expression is not the limiting factor for these systems to be active. In addition, the five endogenous chromosomal hok systems were not deleted in the strain used in this study, showing that none of the chromosomal Sok RNAs could inhibit the expression of the studied hok.
As a first approach to understand the suppression mechanism of some of the substitutions, we inspected hok and Sok RNA expression by northern blot analysis (Figure 3A). As expected from previous studies, we observed the Sok RNA and two isoforms of the hok mRNA for the WT locus. Those two bands correspond to the FL inactive forms of the hok mRNA in which the SD of mok is sequestrated, impeding hok translation (10). The Tr-hok mRNA is absent in the WT as it is degraded together with the Sok antitoxin by RNase III (10). In ΔHΔS transformants, both the hok and Sok RNAs were absent validating that the mutations in the promoters inhibit transcription, as expected. In the various HΔS mutants, Sok was absent and the pattern of hok expression varied between the mutants. In addition to both FL-hok mRNAs, the translatable Tr-hok mRNA was present in most mutants indicating that these mutations impair Hok translation or activity. In the mutants located in the 3′ UTR (G345A and C358A), the Tr-hok mRNA was absent, preventing toxicity by destabilizing or inhibiting the production of this active form. To further understand the mechanism of loss of function, we constructed plasmids expressing selected Hok mutants tagged with a C-terminal FLAG (inhibiting toxicity) under arabinose induction. The mutated Hok (Hok*) protein production was then monitored by Western blotting analysis (Figure 3B). We observed extremely different patterns of Hok* expression although the mutated hok mRNAs were comparably expressed (Supplementary Figure S2). These results suggest that some of the mutations inhibit translation (e.g. A106G) while other interfere with Hok toxin activity (T204T and G311T).
Figure 3.
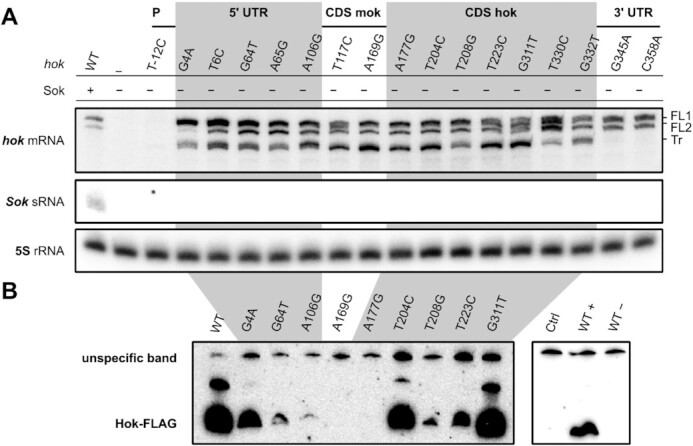
Characterization of hok suppressor substitutions. (A) Northern blot analysis of hok and Sok RNAs. The hok mRNA was revealed using a probe matching the hok mRNA 5′ UTR (FB178). The two FL forms of WT hok mRNA were detected in all transformants expressing hok. The translatable Tr-hok mRNA was present in most mutants, except for C345A and C358A. Sok was targeted with the probe FB213 and was absent in H*ΔS strains. 5S rRNA was revealed using probe FD211 and used as a loading control. P: promoter. (B) Western blotting of arabinose-induced Hok-FLAG protein mutants from pBAD plasmids (hok*-FLAG mRNA expression is stable; see Supplementary Figure S2). The upper signal corresponds to an unspecific band, as confirmed with pBAD vector control as well as induced (WT+) and uninduced (WT−) WT Hok-FLAG.
High-throughput analysis and overview of the results
The results described above showed that our transformation and selection method was efficient and suggested that it was suitable for discovering single-nucleotide suppressor mutants in a high-throughput manner. For this, we scaled up our analysis and prepared five biological replicates of each PCR sample (HS, HΔS, ΔHΔS). We pooled the transformants, extracted the DNA, amplified the locus by PCR and submitted our samples to deep sequencing. After assembling read pairs, we obtained 2.3 million reads of exactly 486 nt mapped to the amplicon for the HS, 1 million for HΔS and 1.8 million for ΔHΔS (Supplementary Table S5). About 30% of the reads were not mutated in the HS and ΔHΔS and around 15% were not mutated in HΔS. Considering the deadly phenotype of unmutated HΔS, the high proportion of unmutated sequences in these reads probably corresponds to amplification of dead cell DNA. One could consider replicating the transformants on a fresh plate before pooling the cells for DNA extraction to avoid this noise in the sequencing data.
To identify loss-of-function single-nucleotide substitutions, we retrieved all reads having only one substitution and performed differential distribution analysis between HS or ΔHΔS and HΔS sequences at each position of the amplicon for each possible nucleotide. The mutations were well distributed over the amplicon (Fig 2B), unlike in our previous study in H. pylori where a disproportionate peak of mutations was found at the PCR overlap region. In total, 162 single-nucleotide substitutions were statistically significantly enriched in the HΔS samples compared to the HS and ΔHΔS controls, located in 117 positions throughout the 486 nt amplicon (Supplementary Table S6). This included 128 single-nucleotide substitutions enriched in both comparisons and 34 additional substitutions identified only in one comparison, 17 in the HS versus HΔS and 17 in the ΔHΔS versus HΔS comparison. The fact that 79% of the substitutions were found in both conditions and that we retrieved all the substitutions identified during the preliminary analysis validated the strength of our study. To be fully exhaustive, we retained all significant substitutions, including the ones found in one comparison only.
Among the 162 total substitutions identified, 79 positions were substituted with only 1 nt, 31 with 2 nt and 7 with the 3 possible nt. In total, 2 substitutions mapped in the hok promoter, 31 in the hok mRNA 5′ UTR (upstream of the mok CDS), 20 in the mok CDS (upstream of the hok start codon), 87 in the hok CDS and 22 in the hok mRNA 3′ UTR (Figure 2B, Table 1, Supplementary Table S6).
Table 1.
Summary of the localizations of single-nucleotide loss-of-function substitutions identified
| Location of the substitutions | Number of substitutions (number of positions) | Predicted mechanism of action |
|---|---|---|
| Promoter | 2 (1) | Transcription inhibition |
| hok mRNA 5′ UTR | 31 (22) | |
| 10 (4) | Destabilization of the tac-stem | |
| 7 (7) | Stabilization of aSD pairing | |
| 2 (2) | Mutation of mok SD sequence | |
| 12 (9) | Unknown | |
| mok CDS | 20 (18) | |
| 4 (2) | Mok and Hok not translated | |
| 5 (5) | Mok truncated and Hok not translated | |
| 11 (11) | Unknown | |
| hok CDS | 87 (64) | |
| 81 (59) | Hok activity impaired | |
| 3 (2) | Hok not translated | |
| 3 (3) | Unknown | |
| hok mRNA 3′ UTR | 22 (12) | |
| 19 (9) | Degradation by RNase | |
| 3 (3) | Unknown |
The locations of all significant substitutions obtained by FASTBAC-Seq are indicated as well as their number, and the number of nucleotide positions mutated is given in parentheses. The predicted mechanism of action for those mutations is indicated. See Supplementary Table S6 for details. CDS: coding sequence.
Validation of the method: suppressor mutations in promoter and coding sequences
We validated many well-known determinants of hok/Sok regulation such as the promoter and the hok CDS. We could locate the toxin mRNA promoter sequence and identified the precise nucleotides required for its functionality. Indeed, we obtained four substitutions in the −10 box promoter sequence (TATGAT) of the hok gene, two from the deep sequencing analysis (A−11C/G) and two from the preliminary analysis (T−12C and T−7C). This is consistent with the conserved TANNNT (where N means any nucleotide) −10 box promoter sequence described for E. coli (28). We also confirmed that the hok_T−12C mutant was not transcribed in this strain (Figure 3A).
We next examined the 87 substitutions in the hok CDS. As expected, we obtained substitutions impairing Hok production and activity, located in the start (3) and stop codons (6) as well as mutations leading to a premature stop codon (18) and nonsynonymous amino acid changes (57) (Figure 2B, listed in Supplementary Table S6, Supplementary Figure S3). Unexpectedly, we also obtained three synonymous substitutions preserving the hok amino acid sequence.
The hok start codon was mutated on its second and third positions. As expected, substitution of the start codon from AUG to GUG/UUG/CUG was not observed as those codons all constitute valid alternative start options for translation (29). Six substitutions abrogated the hok stop codon, leading to the formation of a 17-aa longer protein. It is expected that these proteins are not toxic, as adding a C-terminal 8-aa FLAG-tag entails loss of toxicity. Indeed, in two of these mutants (T330C and G332T) the Tr-hok mRNA is present and may be translated (Figure 3A).
We monitored the abundance of the Hok*-FLAG mutants carrying nonsynonymous mutations, postulating that they should be expressed at a similar level to the WT Hok-FLAG protein. While the T204CC11R (T204C corresponds to amino acid substitution C11R) and G311TM46I mutants produced Hok protein levels comparable to the WT, T208GV12G and T223CL17P produced less Hok and Hok from A177GK2E was not detected (Figure 3B). Note that the hok mRNA was present at a comparable level in all those strains (Supplementary Figure S2). This unexpected result strongly suggested that not all CDS substitutions lead to the production of inactive Hok proteins, but some can also affect translation, Hok protein stability or Hok mRNA stability (although we did not have any example among the mutants tested by northern blot). Finally, the mechanism of action of the substitutions in the hok CDS leading to synonymous substitutions (A227UT18T, A320GE49E and U326CG52G) remains to be understood. These mutations may act at the structural level, as demonstrated for a synonymous substitution observed in the FASTBAC-Seq analysis of the aapA3 locus of H. pylori (18).
Translational regulation by the mok leader peptide
The start codon of the 70-aa mok leader peptide was affected by four substitutions T117C, T117G, G118T and G118A (Supplementary Table S6), validating that Mok translation is required for Hok translation. The presence of the Tr-hok mRNA in the T117C clone was consistent with the absence of translation (Figure 3A).
It was known that inserting a stop codon in mok after the hok start codon does not have any effect on hok translation. Therefore, it was proposed that only translation of mok up to hok translation initiation region is important to start hok translation (6). To test this, we tallied up the number of stop codons that could theoretically be generated by single-nucleotide substitutions in mok and hok and compared it with the actual number identified here. We obtained 18 out of the 20 codons from hok that could be theoretically changed to a stop codon by only one substitution and 7 out of 21 for mok, including 5 upstream of the hok start codon and only 2 downstream (Figure 2B, Supplementary Figure S3). Our data support that translation of the mok mRNA at least up to the hok SD is mandatory to allow Hok translation and that mok translation beyond the hok start codon is not necessary for hok to be translated. The two substitutions identified downstream of the hok start codon (G176T and T315A) inhibit toxicity likely by affecting the hok and not the mok CDS (mutation of the hok start codon and production of Hok_Y48N).
Thus, we analyzed the 20 substitutions identified in the 19 first amino acids of mok (located upstream the hok start codon). In addition to the four substitutions in mok start codon and the five premature stop codons, we identified six nonsynonymous and five synonymous changes in Mok. Amino acid substitutions in Mok should not affect Hok production (6); therefore, we hypothesize that these mutations affect the translatability of hok via other mechanisms such as interfering with sequences important for translation or affecting the toxin mRNA structure. Substitutions A163GG16G, A159GQ15R, A159GQ15Q, A166GE17E and A169GE18E are in the close vicinity of the hok SD sequence and could affect SD recognition. The pattern of expression of hok was tested in the Hok_A169GE18E synonymous mutant. The Tr-hok mRNA was observed; however, the flagged protein was not translated (Figure 3B). Future experiments will be required to understand the exact mechanism of action of these mutations.
Deciphering functional determinants of the hok mRNA structure
We obtained 31 substitutions in the hok mRNA 5′ UTR (upstream of the mok start codon) and 22 substitutions in the 3′ UTR. These substitutions could affect either specific sequences guiding translation, like the mok SD, or the structure of the translatable Tr-hok mRNA (18). Indeed, the hok 5′ UTR adopts a closed conformation in the FL-hok mRNA but also in the nascent hok mRNA, where a metastable structure is formed, and an open conformation in the Tr-hok (8). The hok mRNA is not translatable when it adopts the closed conformation since the mok SD is sequestrated in a stem-loop structure via pairing with the aSD (anti-SD or ucb). However, in the open conformation of the Tr-hok mRNA, the mok aSD is trapped by the tac-stem structure rendering the mRNA translatable (Figure 4A, Supplementary Figure S4).
Figure 4.
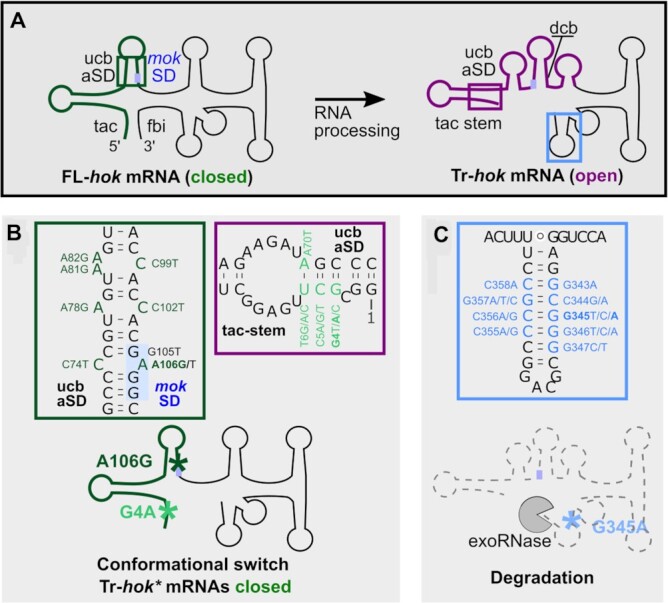
Single-nucleotide substitutions affect hok mRNA structural conformation and stability. (A) hok mRNA is transcribed and folded into a FL isoform with a closed conformation [mok SD (blue box) sequestrated] that is not translatable. Following maturation by RNase(s), a translatable Tr-hok mRNA form with an open conformation is produced. In the WT, this form is targeted by Sok, whereas in the absence of Sok, Hok is produced entailing bacterial cell death. tac: translational activator element; fbi: fold-back inhibitory element; ucb: upstream complementary box; dcb: downstream complementary box. The schematic structures are based on (8). (B) Several substitutions prevent the conformational change of Tr-hok mRNA from the closed to the open form, locking the hok* mRNA in the closed, untranslatable form with mok SD sequestrated even after processing. Those substitutions act by either stabilizing the closed structure (green box) or destabilizing the open structure (purple box). Green box: Seven single substitutions (dark green, including A106G) increased the stability of the mok SD/aSD stem-loop by creating extra base pairs locking Tr-hok mRNA in the closed conformation. Purple box: Ten single substitutions (four positions, light green including G4A) decreased the stability of the tac-stem-loop in the open conformation by disrupting base pairs. (C) Blue box: Nineteen substitutions (nine positions) affected negatively the stability of the last stem-loop at the 3′ end of Tr-hok mRNA (blue), therefore increasing its susceptibility to 3′-to-5′ exoRNases. See also Supplementary Figure S4.
We identified three mutations in the GAGG mok SD motif: G105T and A106G/T. While the G105T and A106T substitutions (Supplementary Figure S4, dark blue) likely impair the base pairing with the 16S rRNA SD complementary sequence, this should not be the case for the A106G substitution, which allows the formation of a GU wobble pair. In contrast, the A106G substitution strengthens the mok SD/aSD interaction by creating an extra base pair (Figure 4B, green box). Since the Tr-hok mRNA form was observed in this strain (Figure 3A), we hypothesize that the increased stability of the SD/aSD interaction prevents the conformational change and locks the truncated form in the closed conformation, thereby inhibiting the translation of mok, and consequently that of hok. Indeed, we monitored the abundance of Hok*_A106G-FLAG by Western blot and observed a very strong inhibition of Hok translation (Figure 3B). Strengthening this hypothesis, we found that the C74T substitution located in the mok aSD and opposite to A106G in the FL-hok mRNA conformation was also responsible for loss of function. Five additional substitutions could have the same mechanism of action (Figure 4B, green box).
We also found substitutions affecting the nucleotides G4, C5 and T6 (Figure 4B, purple box). For each of these nucleotides, a substitution into the three other possible nucleotides abolished hok toxicity, implying that those mutations act by breaking base pairs. These substitutions could be disturbing the formation of the open tac-stem structure as they base-paired with the ucb mok aSD sequence in the FL-hok mRNA. Both the FL- and the Tr-hok mRNA forms were present in the northern blot analyses of G4A and T6C, and the abundance of Hok*_G4A-FLAG was drastically reduced compared to the WT, validating the effect on Hok translation (Figure 3). We concluded that these mutations could prevent Tr-hok mRNA translation once again by preventing a conformational change between the FL- and the Tr-hok mRNA, this time by destabilizing the Tr-hok mRNA conformation. In addition, we identified a mutation opposite to T6 in the tac-stem, A70T (located in the mok aSD), which is also responsible for a loss of function. Substitutions of G71 and C72, which are also part of the mok aSD, were not retrieved in our analysis, suggesting their importance for the sequestration of the mok SD in the FL mRNA. Indeed, a mutation in those two positions would release the mok SD and allow Hok translation before even affecting the tac-stem formation.
Overall, the mutations in the hok mRNA 5′ UTR locking the toxin mRNA in an inactive conformation for translation initiation are reminiscent of loss-of-function mutations identified in aapA3 mRNA of H. pylori (18). These mutations acted at the mRNA folding level to trap transient structures in an untranslatable state.
Other mutations of the hok 5′ UTR led to loss of function but, based on our current knowledge of the mRNA conformation, we were not able to propose any possible mechanism of action. For example, in the G64T mutant, the Tr-hok mRNA was present and the abundance of the Hok protein was drastically reduced compared to the WT explaining its lack of toxicity (Figure 3, Supplementary Figure S4, red).
We obtained 22 substitutions in the hok mRNA 3′ UTR, among which 19 broke a CG pair in the terminal stem of the Tr-hok mRNA (including G345A and C358A). We observed that the Tr-hok mRNA form was absent in the G345A and C358A mutants (Figure 3A) showing that weakening the 3′ stem-loop affects the overall hok mRNA stability. This effect is probably due to the action of 3′-to-5′ exoRNases such as PNPase, as previously proposed (10) (Figure 4C, blue box).
Further considerations and possible limitations of the method
Although our method is a powerful tool to rapidly identify loss-of-function mutations and characterize T1TAs, a few considerations and limitations should be taken into account.
First, among the 20 codons from hok that could be theoretically changed to a stop codon by only one substitution, only 18 were mutated (Figure 2B, listed in Supplementary Table S6). In addition, 20% of the substitutions that we obtained were only found in comparing HΔS with HS or with ΔHΔS (10% in each). Altogether, these observations suggest that we might not have identified all loss-of-function mutants, probably because we did not collect enough transformants to avoid false negatives.
Second, mutations affecting the previously studied mechanism of hok regulation (conformational changes and stabilization of the untranslatable structure) have been smoothly annotated thanks to the fact that the hok mRNA structure was already well characterized. We recommend, when investigating an mRNA whose structure is unknown, to perform computational folding using alignment of homologous sequences in parallel to FASTBAC-Seq to facilitate the interpretation of the results (8,30–31). However, even with good knowledge of the mRNA structure, we also obtained several loss-of-function mutants for which we were unable to address the mechanism of action, such as synonymous mutations present in the hok CDS or in the 5′ UTR. It is therefore decisive to construct individual mutants, to prove their viability and investigate individually their mechanism of action.
Third, the mutation ΔpSok lies in the dcb sequence (Supplementary Figure S4) and may render the mok SD/dcb stem less prone to be formed (one less GC pair). So, during the design of the experiment, it is important to keep in mind that mutating the promoter of the antitoxin can also affect the WT toxin mRNA sequence encoded on the opposite DNA strand.
Lastly, we investigated here a plasmidic T1TA not expressed from the chromosome, and validated the lack of cross-talk with chromosomal hok. In other studies, note that if the T1TA of interest is already present on the host chromosome, it should be deleted from the receiver strain before starting the investigation, and if a homologue is present in the host a possible cross-talk should be investigated.
CONCLUSIONS
A plethora of T1TA systems have been bioinformatically predicted in 2010 by Fozo et al., but most of them have never been validated (32). Moreover, a precise characterization of the mechanism of action of the few systems studied in more detail is lacking. Here, we provide a genetic tool to study these mechanisms using high-throughput selection of single-nucleotide suppressor mutants of type I toxins in E. coli. T1TA toxicity has often been assessed under overexpression conditions. Our method is based on analyzing the chromosomal expression level of the toxin, thus securing against a potential toxic overexpression from a plasmid. This method, adapted from the FASTBAC-Seq of H. pylori, is based on uncovering loss-of-function mutants of the toxin by deep sequencing.
We validated our approach by revisiting the regulation of the well-characterized hok/Sok T1TA from E. coli. We obtained a good distribution of the substitutions over the locus and detected key regulatory elements embedded in the hok toxin mRNA at a nucleotide resolution. Then, we analyzed the mRNA and protein expression of several mutated transformants obtained during our preliminary analysis to validate our hypotheses concerning their mode of action. We found many cis-regulatory elements of the hok/Sok system that were already published, such as the promoter region, SD sequences and CDS boundaries, but also structural determinants found in the 5′ and 3′ UTRs. Indeed, comparably to what was published for aapA3 (18), we detected here important nucleotides involved either in stabilizing structures that are not compatible with translation or in destabilizing translatable structures. This conformation equilibrium is characteristic of these T1TA systems for which the protein expression is regulated by a series of transient and final structures sequestrating either the toxin SD sequence for aapA3 or the mok leader peptide SD for hok. Finally, in addition to these already described regulatory determinants, we uncovered other substitutions for which the mechanism of action was not obvious or is still to be determined, such as synonymous amino acid substitutions in the mok and hok CDSs, but also substitutions in the 5′ and 3′ UTRs (Supplementary Figure S4, in red).
Compared to other methods that have been developed to study 5′ UTRs, such as saturated mutagenesis followed by the monitoring of a reporter gene expression (33), our method is particularly adapted for the study of T1TAs. Although we are missing all mutations that have a positive effect on expression, we are not biased by the introduction of a reporter gene. Another major output provided by the use of FASTBAC-Seq is the capacity to reveal the conformational switch between the open and closed conformations of type I mRNAs. Therefore, we believe that our method could be suitable, with minor changes, for the study of other RNA structure-based systems, such as bacterial riboswitches and RNA thermometers. In these cases, a toxic CDS would be added downstream a library of mutated regulatory 5′ UTRs, allowing life/death selection.
While identifying intragenic suppressors, our method does not enable the identification of trans-acting determinants regulating the systems although T1TAs are known to be regulated by RNA editing protein (tadA) and RNases (RNase III, PNPase, RNase II) (10–11,34). Therefore, it could be of interest to combine our method with the recently described toxin activation–inhibition conjugation method to identify extragenic activators and inhibitors of TA systems (35).
Taken together, we add a valuable tool in the field of T1TA research. Indeed, although they are produced as highly stable and not translatable molecules, toxin mRNAs are in a fragile balance between structures governing their activities; one single-nucleotide change can freeze a structure in the active translatable (open) or inactive (closed) state. With this study, we hope to provide a framework for a better analysis of the TA system determinants involved in toxin function and regulation. It will be helpful to study type I from many bacteria, and also the study of other TA types, in this case focusing on protein activity domains.
DATA AVAILABILITY
DNA sequencing data and results of differential statistical analyses have been deposited to NCBI GEO under the accession number GSE207195. All datasets generated in this study are available within the supplementary data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Donald Court laboratory for providing strains and plasmids for the tetA-sacB selection/counter-selection experiments. We acknowledge in particular Corentin Pelletan for technical help and Erwan Guichoux from the Plateforme Génome Transcriptome de Bordeaux. We acknowledge Thibaud Renault and Andrés Escalera Maurer for critical reading of the manuscript. We acknowledge all present and past members of the ARNA laboratory for helpful discussions.
Contributor Information
Anaïs Le Rhun, Univ. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France.
Nicolas J Tourasse, Univ. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France.
Simon Bonabal, Univ. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France.
Isabelle Iost, Univ. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France.
Fanny Boissier, Univ. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France.
Fabien Darfeuille, Univ. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
INSERM (to ALR, NJT, SC and FD), Federation of European Biochemical Societies (to ALR), CNRS (to IO, ALR and FD), University of Bordeaux (to ALR, SB, FB and FD). Funding for open access charge: INSERM.
Conflict of interest statement. None declared.
REFERENCES
- 1. Harms A., Brodersen D.E., Mitarai N., Gerdes K.. Toxins, targets, and triggers: an overview of toxin–antitoxin biology. Mol. Cell. 2018; 70:768–784. [DOI] [PubMed] [Google Scholar]
- 2. Masachis S., Darfeuille F.. Type I toxin–antitoxin systems: regulating toxin expression via Shine–Dalgarno sequence sequestration and small RNA binding. Microbiol. Spectr. 2018; 6: 10.1128/microbiolspec.RWR-0030-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tourasse N.J., Darfeuille F.. T1TAdb: the database of type I toxin–antitoxin systems. RNA. 2021; 27:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gerdes K., Rasmussen P.B., Molin S.. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc. Natl Acad. Sci. U.S.A. 1986; 83:3116–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gerdes K., Larsen J.E., Molin S.. Stable inheritance of plasmid R1 requires two different loci. J. Bacteriol. 1985; 161:292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thisted T., Gerdes K.. Mechanism of post-segregational killing by the hok/sok system of plasmid R1. Sok antisense RNA regulates hok gene expression indirectly through the overlapping mok gene. J. Mol. Biol. 1992; 223:41–54. [DOI] [PubMed] [Google Scholar]
- 7. Gerdes K., Bech F.W., Jørgensen S.T., Løbner-Olesen A., Rasmussen P.B., Atlung T., Boe L., Karlstrom O., Molin S., von Meyenburg K.. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J. 1986; 5:2023–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steif A., Meyer I.M.. The hok mRNA family. RNA Biol. 2012; 9:1399–1404. [DOI] [PubMed] [Google Scholar]
- 9. Thisted T., Sørensen N.S., Gerdes K.. Mechanism of post-segregational killing: secondary structure analysis of the entire hok mRNA from plasmid R1 suggests a fold-back structure that prevents translation and antisense RNA binding. J. Mol. Biol. 1995; 247:859–873. [DOI] [PubMed] [Google Scholar]
- 10. Franch T., Gultyaev A.P., Gerdes K.. Programmed cell death by hok/sok of plasmid R1: processing at the hok mRNA 3′-end triggers structural rearrangements that allow translation and antisense RNA binding. J. Mol. Biol. 1997; 273:38–51. [DOI] [PubMed] [Google Scholar]
- 11. Gerdes K., Nielsen A., Thorsted P., Wagner E.G.. Mechanism of killer gene activation. Antisense RNA-dependent RNase III cleavage ensures rapid turn-over of the stable hok, srnB and pndA effector messenger RNAs. J. Mol. Biol. 1992; 226:637–649. [DOI] [PubMed] [Google Scholar]
- 12. Gultyaev A.P., Franch T., Gerdes K.. Programmed cell death by hok/sok of plasmid R1: coupled nucleotide covariations reveal a phylogenetically conserved folding pathway in the hok family of mRNAs. J. Mol. Biol. 1997; 273:26–37. [DOI] [PubMed] [Google Scholar]
- 13. Fozo E.M. New type I toxin–antitoxin families from “wild” and laboratory strains of E. coli: Ibs-Sib, ShoB-OhsC and Zor-Orz. RNA Biol. 2012; 9:1504–1512. [DOI] [PubMed] [Google Scholar]
- 14. Sharma C.M., Hoffmann S., Darfeuille F., Reignier J., Findeiß S., Sittka A., Chabas S., Reiche K., Hackermüller J., Reinhardt R.et al.. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010; 464:250–255. [DOI] [PubMed] [Google Scholar]
- 15. Maikova A., Peltier J., Boudry P., Hajnsdorf E., Kint N., Monot M., Poquet I., Martin-Verstraete I., Dupuy B., Soutourina O.. Discovery of new type I toxin–antitoxin systems adjacent to CRISPR arrays in Clostridium difficile. Nucleic Acids Res. 2018; 46:4733–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pichon C., Felden B.. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl Acad. Sci. U.S.A. 2005; 102:14249–14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fraikin N., Goormaghtigh F., Van Melderen L.. Type II toxin–antitoxin systems: evolution and revolutions. J. Bacteriol. 2020; 202:e00763-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masachis S., Tourasse N.J., Lays C., Faucher M., Chabas S., Iost I., Darfeuille F.. A genetic selection reveals functional metastable structures embedded in a toxin-encoding mRNA. eLife. 2019; 8:e47549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Masachis S., Tourasse N.J., Chabas S., Bouchez O., Darfeuille F.. FASTBAC-Seq: functional analysis of toxin–antitoxin systems in bacteria by deep sequencing. Methods Enzymol. 2018; 612:67–100. [DOI] [PubMed] [Google Scholar]
- 20. Li X., Thomason L.C., Sawitzke J.A., Costantino N., Court D.L.. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res. 2013; 41:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomason L.C., Sawitzke J.A., Li X., Costantino N., Court D.L.. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. 2014; 106:1.16.1–1.16.39. [DOI] [PubMed] [Google Scholar]
- 22. Schmieder R., Edwards R.. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011; 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Masella A.P., Bartram A.K., Truszkowski J.M., Brown D.G., Neufeld J.D.. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics. 2012; 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H. Aligning sequence reads, clone sequences and assemblycontigs with BWA-MEM. 2013; arXiv doi:16 March 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997. [Google Scholar]
- 25. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079.1000 Genome Project Data Processing Subgroup. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M.et al.. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013; 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mutalik V.K., Guimaraes J.C., Cambray G., Lam C., Christoffersen M.J., Mai Q.-A., Tran A.B., Paull M., Keasling J.D., Arkin A.P.et al.. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013; 10:354–360. [DOI] [PubMed] [Google Scholar]
- 29. Hecht A., Glasgow J., Jaschke P.R., Bawazer L.A., Munson M.S., Cochran J.R., Endy D., Salit M.. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017; 45:3615–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gultyaev A.P., Franch T., Gerdes K.. Coupled nucleotide covariations reveal dynamic RNA interaction patterns. RNA. 2000; 6:1483–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tourasse N.J., Darfeuille F.. Structural alignment and covariation analysis of RNA sequences. Bio Protoc. 2020; 10:e3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fozo E.M., Makarova K.S., Shabalina S.A., Yutin N., Koonin E.V., Storz G.. Abundance of type I toxin–antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010; 38:3743–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holmqvist E., Reimegård J., Wagner E.G.H.. Massive functional mapping of a 5′-UTR by saturation mutagenesis, phenotypic sorting and deep sequencing. Nucleic Acids Res. 2013; 41:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bar-Yaacov D., Mordret E., Towers R., Biniashvili T., Soyris C., Schwartz S., Dahan O., Pilpel Y.. RNA editing in bacteria recodes multiple proteins and regulates an evolutionarily conserved toxin–antitoxin system. Genome Res. 2017; 27:1696–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bobonis J., Mitosch K., Mateus A., Karcher N., Kritikos G., Selkrig J., Zietek M., Monzon V., Pfalz B., Garcia-Santamarina S.et al.. Bacterial retrons encode phage-defending tripartite toxin–antitoxin systems. Nature. 2022; 609:144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequencing data and results of differential statistical analyses have been deposited to NCBI GEO under the accession number GSE207195. All datasets generated in this study are available within the supplementary data.