


Abstract
Transcriptional reactivation of hTERT is the limiting step in tumorigenesis. While mutations in hTERT promoter present in 19% of cancers are recognized as key drivers of hTERT reactivation, mechanisms by which wildtype hTERT (WT-hTERT) promoter is reactivated, in majority of human cancers, remain unknown. Using primary colorectal cancers (CRC) we identified Tert INTeracting region 2 (T-INT2), the critical chromatin region essential for reactivating WT-hTERT promoter in CRCs. Elevated β-catenin and JunD level in CRC facilitates chromatin interaction between hTERT promoter and T-INT2 that is necessary to turn on hTERTexpression. Pharmacological screens uncovered salinomycin, which inhibits JunD mediated hTERT-T-INT2 interaction that is required for the formation of a stable transcription complex on the hTERT promoter. Our results showed for the first time how known CRC alterations, such as APC, lead to WT-hTERT promoter reactivation during stepwise-tumorigenesis and provide a new perspective for developing cancer-specific drugs.
Graphical Abstract
Graphical Abstract.
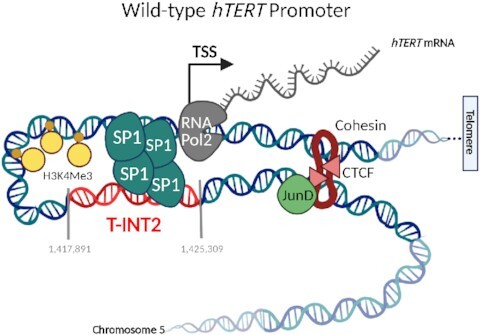
Reactivation of hTERTgene in cancer cells. Cancer-specific long-range chromatin interactions switch on wild-type hTERTpromoter.
INTRODUCTION
Expression of hTERT (human TElomerase Reverse Transcriptase), which encodes the catalytic subunit of the telomerase enzyme complex, is transcriptionally repressed in somatic cells (1). In up to 90% of human cancer cells, hTERT expression is reactivated, resulting in the reconstitution of telomerase activity that is required for replicative immortality (1). Persistent telomerase activity is also essential for the survival and maintenance of stem cells (2). Hence systemically inhibiting telomerase causes intolerable cytotoxicity (3). Targeting cancer cell-specific transcriptional reactivation of hTERT, which leaves its activity in stem cells intact, could provide an alternate way of making clinically usable telomerase inhibitors. It is now understood that 19% of human cancers harbour single-base mutations that create de novo binding sites for the ETS family of transcription factors in the proximal hTERT promoter, thereby driving its reactivation (4–6). A long-range chromatin interaction between these mutant (Mut)-hTERT promoters and a genomic region (Tert INTeracting region 1; T-INT1) located 260 kb upstream and consisting of multiple binding motifs for GA-binding protein (GABP)α/β (members of the ETS transcription-factor family) is essential for reactivation of the mutant hTERT promoters (7). Surprisingly, T-INT1 was found to have no role in regulating wild-type (WT)-hTERT promoters (7), which drive hTERT reactivation in the majority of cancers, raising the questions, if long-range chromatin interactions are only necessary to drive Mut-hTERT promoters, and how key genetic alterations or oncogenes known as drivers of the step-wise models of tumorigenesis contribute to telomerase reactivation in cancers with WT-hTERT promoters.
In this study, using colorectal cancer (CRC) as a model, we investigated the molecular mechanism of WT-hTERT reactivation. We find that the AP-1 family transcription factor JunD, is essential for the reactivation of WT-hTERT transcription in CRCs. Increased levels of JunD work by creating a conducive chromatin architecture to facilitate the formation of Sp1–Sp1 tetramers between the proximal WT-hTERT promoter and a distal regulatory chromatin region, which we name Tert INTeracting region 2 (T-INT2). Chemical screens uncovered salinomycin as a specific inhibitor of WT-hTERT promoter in CRCs. Salinomycin works by inhibiting the action of JunD on the formation of productive chromatin architecture required for the formation of Sp1-SP1 tetramers. Our results indicate, for the first time, how cancer-initiating oncogenes contribute to the reactivation of WT-hTERT promoters, the rate limiting step in transformation. Targeting such cancer-specific hTERT reactivation mechanisms could enable the development of novel cancer cell specific therapeutic strategies in CRC and in other malignancies.
MATERIALS AND METHODS
Cell lines and culture conditions
The A375 and HCT116 cell lines were a gift from Dr Shang Li (DUKE-NUS Medical School, Singapore). HCT116−146C and HCT116−146C>T isogenic cell lines were generated in our previous study (7). DLD1, Caco2, HT29 and Colo205 cell lines were a gift from Dr Ernesto Guccione (Institute of Molecular and Cell Biology, Singapore). All cells were maintained in DMEM supplemented with 10% fetal bovine serum, penicillin and streptomycin (Gibco). Patient-derived primary CRC cell lines were derived in the laboratory of Dr. Ramanuj DasGupta. Primary CRC cell lines were grown in six-well tissue-culture treated plates (Falcon) precoated using a Coating Matrix Kit (Gibco), and were maintained in DMEM/F12 medium (Gibco) supplemented with penicillin–streptomycin (Gibco), B27 (without vitamin A; Gibco), 20 ng/ml EGF (Gibco) and 10 ng/ml bFGF (Gibco).
ATAC sequencing
ATAC-seq library preparation was as previously described (8). Briefly, 50 000 cells were lysed for isolation of nuclei. Transposition reaction was performed using Nextera DNA library preparation kit (NEB). Samples were amplified by eight PCR cycles for library preparation. Primer dimers and long DNA fragments were cleaned by an AMPure XP beads purification step. DNA concentration was measured by Qubit fluorometric assay, and library quality was determined by Bioanalyzer. The library was sequenced in Nextseq High 76 bp paired-end configuration using an Illumina platform.
ATAC-seq data analysis
CRC PDC ATAC-Seq data was analysed by first filtering low-quality bases and adapter sequences using the Trimmomatic tool (9). Good-quality sequences were subsequently aligned to the human genome (GRCh38) using the HISAT2 aligner, and the alignment was sorted by read name using Samtools (10,11). With alignment files, peak calling was performed separately for each group (high-TERT and low-TERT) using the Genrich tool with options -j, -r, -e MT, which respectively adjust for ATAC-seq alignment, remove PCR duplicates and remove mitochondria reads. Significant peaks (false-discovery-rate-adjusted P-value <0.05) of both groups were then merged for downstream analysis to identify differential chromatic accessible regions (differential peaks). Homer getDifferentialPeaksReplicates.pl tool was used in differential-peak calling with parameter log2 fold change ≥0.5 (12). Visualization files in bigWig format were generated using deepTools bamCoverage (13).
4C and 3C assays and 4C-seq analysis
4C and 3C assays were performed as previously described (7). 3C qPCR primers are shown in Supplementary Table S1. 4C-seq analysis is described in the supplementary materials. FASTQ files of 4C-Seq were processed using the pipe4C analysis pipeline (14,15). In brief, the 5′ ends and 3′ ends of the sequencing reads were trimmed to the primary restriction enzyme (HindIII) location and/or secondary restriction enzyme (DpnII) location. Trimmed reads were then mapped against a human reference genome (GRCh38) using the Bowtie2 aligner. With reference to the fragment-end library created in silico, reads mapped per fragment end were then quantified and normalized. Lastly, peak calling was performed using the peakC R package (16), to identify significant interacting regions (window size = 31, P < 0.05).
Removal of hTERT interaction regions by CRISPR/Cas9 genome editing
The gRNAs targeting the 5′ and 3′ ends of the T-INT2 region were cloned into pX-458-GFP and pX458-dsRED (no-Cas9) vectors (17). Cells were transfected in 6-well plates using X-tremeGENE 9 transfection reagent (Roche). Double-positive cells expressing GFP and dsRED were sorted into 96-well plates (1 cell/well), and each clone was genotyped with T-INT2-specific primers. The T-INT2-SP1 cluster (560 bp) in the T-INT2 region, and the JunD-CTCF region (220 bp), were removed using the same strategy. All gRNAs are listed in Supplementary Table S1.
Targeting chromatin interactions by dCas9
T-INT2 specific gRNAs were designed to tile the entire interaction region. All gRNAs were cloned into the lentiGuide-Puro vector (Addgene #52963). Cells were transfected with a gRNA together with KRAB-dCas9 (Addgene #71237) or AM-dCas9 (Addgene #92220) vectors. Cells were harvested 48 h after transfection for ChIP and gene expression analysis. All gRNAs are listed in Supplementary Table S1.
Transfection
For RNA interference, cells were transfected with the dsi-RNAs dsi-control and dsi-JunD (Integrated DNA Technologies) using Lipofectamine reagent (Thermo). Transfection medium was replaced with DMEM supplemented with 10% FBS after 6 h, and cells were harvested after 36 h.
RNA isolation and expression analysis
RNA was extracted by the Trizol method, followed by column purification (Qiagen). cDNA synthesis was performed from 1 μg RNA using the Maxima First-strand cDNA synthesis kit (Thermo). qPCR was performed using 5 ng cDNA and SSO-Sybr Greener qPCR master mix (BioRAD). Data were normalized to actin and analysed by the 2−ΔΔCt delta-delta Ct method. All qPCR primers are listed in Supplementary Table S1.
Chromatin immunoprecipitation (ChIP) assays
ChIP was performed as described previously (18). Cells were cross-linked for 10 min using 1% formaldehyde. After sonication and pre-clearing, anti-Sp1 (Active-Motif; 39058), anti-H3K4Me3 (Millipore; 04-745), anti-CTCF (Active-Motif; 91285), anti-Pol2 (Millipore; 05-623) and IgG (Santa Cruz Biotechnology) antibodies were used for immunoprecipitation at a concentration of 1 μg per 1 million cells. ChIP eluate was used to perform ChIP-qPCR with specific primers. Primers are shown in Supplementary Table S1.
ChIP-seq data analysis
ChIP-seq data (bigWig file) for transcription factors JunD and CTCF in HCT116 lines were obtained from NCBI GEO GSE32465 and GSE30263, respectively.
Pharmaceutical drug screen analysis
Gene's expression (Cancer Cell Line Encyclopedia, genes TPM) and drug sensitivity datasets (PRISM repurposing screen, MFI log2 fold-change) were obtained from Broad Institute DepMap web portal (https://depmap.org/portal/download/), 21Q3 release (19). Pearson's correlation was used in the correlation analysis of genes expression with drugs sensitivity. Negative correlation means higher drug sensitivity with high expression of respective gene in the cells. Unpaired two-samples t-test was used in comparison of drug sensitivity in between CRC cells with high and low JunD expression.
Telomerase-activity assay (TRAP)
The TRAP assay was performed as previously described (20).
Cell proliferation and viability
Cell proliferation was measured using CCK8, according to the manufacturer's recommendations. Briefly, 2000 cells per well were seeded into 96-well plates. Cells were treated with 2 μM dimethyl sulphoxide (DMSO) and 5 μM salinomycin. Medium was replaced with CCK8 regent 2h before of each time point. Absorbance was measured at 450 nm.
Xenograft studies
HCT116 T-INT2-SP1-WT and T-INT2-SP1-KO cells (5 × 106) were subcutaneously injected into NOD/SCID mice. Tumour volume was monitored for 24 days.
Statistical analysis
The two-tailed Student's t-test was applied to compare two groups for the ChIP-qPCR, 3C-qPCR, qPCR and RT-TRAP experiments. The mean ± SD of each ChIP, 3C, gene expression, 4C and 3C assay were obtained from at least three independent experiments, as indicated in the figure legends.
RESULTS
Increased open chromatin and hTERT expression in CRCs correlate with JunD levels
Expression of AP-1 family of transcription factors is upregulated in CRCs (21). To evaluate the influence of members of the AP-1 family on hTERT expression, the rate limiting step in tumorigenesis, we analysed RNA-seq data from 292 CRC samples (22) and found a positive correlation between JunD and hTERT expression (Supplementary Figure S1A). It is widely perceived that detection of hTERT is challenging in RNA-seq experiments because of its low levels of mRNA, which often result in low correlation coefficients (23). The correlation between hTERT and JunD levels was also evident when validated by RT-qPCR using patient-derived CRC cells (PDCs) (n = 10) and fresh CRC patient samples (n = 20) (Figure 1A and B). Indeed increased JunD levels were seen in primary tumours as compared to their matched normal controls (Figure 1B, bottom panel). We hence evaluated whether JunD directly regulates hTERT transcription. Depletion of JunD using two different siRNAs reduced hTERT expression in HCT116 human CRC cells (Supplementary Figure S1B, C). Since most CRCs harbour no hTERT promoter mutations, we queried if JunD specifically regulates hTERT expression via the WT-hTERT promoters. Depletion of JunD in HCT116 isogenic cells HCT116−146C or HCT116−146C>T harbouring WT-hTERT or Mut-hTERT promoters respectively, showed that JunD specifically regulated the WT-hTERT promoter (Figure 1C).
Figure 1.

Colon cancer cells with high levels of expression of JunD and hTERT harbour a CTCF-occupied open chromatin region. (A, B) Gene expression correlation of JunD and hTERT is shown in patient derived CRC cells (n = 10) and (B) fresh CRC tumor samples (n = 20) by RT-qPCR analysis. Bottom panel in (B) shows JunD protein levels in normal-tumor matched samples by western-blot analysis. (C) Gene expression analysis of hTERT by RT-qPCR in isogenic HCT116 cells transfected with si-Control or si-JunD. (D, E) Gene expression analysis of hTERT and JunD genes by RT-qPCR in six PDCs. We selected the top 30% high JunD expressed samples based on panel (A) and grouped them as JunD-High (red). Similarly, lowest 30% of the PDC samples in panel (A) were selected as JunD-Low (green) group. Gene expression is calculated relative to the sample with lowest hTERT expression. (F) Differential accessible regions identified between cells with low and high JunD expression sequentially overlapped with normal samples, CRC-specific chromatin accessible regions, JunD and CTCF binding sites. Numbers of potential regulatory regions in chromosome 5 are listed in the Venn diagram. (G) Additional filter applied to eliminate overlapping peaks associated with embryonic stem cells from the consensus regions. The hits remained after filtering are indicated in a table. (H) Genome tracks show the ATAC-seq and ChIP-seq peaks at the candidate region (chr5:1 437 340–1 438 020) closest to the hTERT promoter (∼140 kb upstream) for the indicated samples.
In many cancer types, Jun family members form homo or hetero dimers and regulate proliferation by recruiting the CBP/p300 complex to specific promoters (24,25). Aside from other AP-1 members, JunD has also been shown to co-localize with CTCF on chromatin (26) and regulate chromatin compactness through alteration of CTCF occupancy and activity. To identify accessible chromatin regions that are influenced by JunD occupancy and which might have functional roles in regulating hTERT expression, we first analysed differential ATAC-seq regions in high-JunD and low-JunD patient-derived CRC cells (PDCs) (Figure 1D, E) all of whom had a WT-hTERT promoter (Supplementary Figure S1D) and compared them with ATAC-seq peaks from healthy colon cells. To identify JunD-dependent CRC-specific open chromatin regions, we overlapped our high-JunD-specific open chromatin regions with ATAC-seq peaks from public CRC samples. Subsequently, these regions were matched with JunD and CTCF chromatin immunoprecipitation sequencing (ChIP-seq) peaks leading to the identification of 25 candidate regions that might functionally regulate WT-hTERT activity (Figure 1F). Unlike in stem cells, where WT-hTERT promoter is constitutively active, hTERT reactivation in cancer cells is a consequence of oncogene mediated epigenetic alterations (27–31). To identify cancer-specific distal regulatory regions which drive WT-hTERT promoter in response to oncogenes, we compared accessible chromatin regions between CRC cells and embryonic stem cells. Among the 25 identified genomic regions, we observed 10 unique accessible regions co-enriched with JunD and CTCF that were not present in stem cells. We focused on the most significant, chr5:1 437 340–1 438 020 region located ∼140 kb upstream of the hTERT proximal promoter (Figure 1G). Genomic tracks of ATAC-seq and ChIP-seq peaks (for JunD and CTCF) are shown for chr5:1 437 340–1 438 020 (hereafter referred to as the JunD-CTCF region) (Figure 1H). These results suggested for the first time that distinct open chromatin regions and underlying mechanisms, not only between WT-hTERT and Mut-hTERT promoters but also between WT-hTERT promoters in cancer cells and stem cells, likely regulate hTERT expression.
The JunD-CTCF region functionally regulates WT-hTERT promoter in CRCs
To analyse the functional relevance of the JunD-CTCF region (Figure 2A) in regulating hTERT expression, we knocked out (KO) this region by CRISPR, creating HCT116JunD-CTCF-KO and DLD1JunD-CTCF-KO cells (Figure 2B). Interestingly, removal of this non-coding, distal DNA region located ∼140 kb upstream of the WT-hTERT promoter resulted in dramatic reduction of hTERT expression, suggesting that epigenetic regulation, possibly through alteration of chromatin architecture may regulate WT-hTERT promoter (Figure 2C, Supplementary Figure S1E). Five Sp1 binding sites in the proximal WT-hTERT promoter are the major determinants of its activity in cancer cells (32). We performed ChIP analysis to determine whether the reduction of hTERT expression observed in HCT116JunD-CTCF-KO cells was the result of the loss of Sp1 occupancy on the proximal hTERT promoter. Sp1, H3K4Me3 and Pol2 occupancy on the proximal hTERT promoter significantly reduced in HCT116JunD-CTCF-KO and DLD1JunD-CTCF-KO cells, which suggested that the distal JunD-CTCF region had a functional role in regulation of WT-hTERT promoter activity (Figure 2D, F and Supplementary Figure S1F). To assess whether the JunD binding site immediately upstream of JunD-CTCF region was essential for WT-hTERT regulation, we knocked down JunD in HCT116JunD-CTCF-WT and HCT116JunD-CTCF-KO cells and found that hTERT expression was specifically reduced in HCT116JunD-CTCF-WT cells with intact JunD-CTCF region (Figure 2G). To examine whether JunD regulated hTERT expression via opening of the chromatin, enabling CTCF to bind to the JunD–CTCF region, we created HCT116 and DLD cells (HCT116ΔJunD and DLD1ΔJunD) which had a 9-bp deletion in JunD binding motif within the JunD-CTCF region (Figure 2H, I). Similar to the observations in HCT116JunD-CTCF-KO and DLD1JunD-CTCF-KO cells where JunD-CTCF region was completely deleted, HCT116ΔJunD DLD1ΔJunD cells with targeted deletion of JunD sites showed marked reduction in hTERT expression (Figure 2J), when compared with HCT116WT-JunD cells where JunD site was intact. Compared to HCT116WT-JunD cells HCT116ΔJunD showed no JunD occupancy and reduced CTCF occupancy in the JunD-CTCF region (Figure 2K, L). Similar to deletion of the JunD-CTCF region, JunD-motif deletion also abrogated Sp1 binding in the proximal WT-hTERT promoter (Figure 2M). Sp1 molecules are known to form dimers or tetramers, either between adjacent or (via long-range chromatin interactions) distal Sp1 sites, leading to super-activation of promoters (33). Our results suggested that Sp1 occupancy on the proximal WT-hTERT promoter was regulated by a distal regulatory region enriched with CTCF binding sites. Because CTCF is known to regulate the formation of chromatin interactions, we postulated that Sp1 bound to the proximal WT-hTERT promoter might be tethering long-range Sp1–Sp1 dimers to stabilize and drive WT-hTERT expression.
Figure 2.

The JunD-CTCF region regulates hTERT promoter activity. (A) Schematic view of chromosome 5 with the hTERT promoter and JunD-CTCF region (chr5:1 437 340–1 438 020) identified by ATAC-seq and ChIP-seq analysis. The discontinuous line indicates the regions removed by genome editing with CRISPR/Cas9. (B) Genotyping results are shown for each of the HCT116JunD-CTCF-WT and HCT116JunD-CTCF-KO clones. (C) Comparison of hTERT expression levels in HCT116JunD-CTCF-WT and HCT116JunD-CTCF-KO clones by RT-qPCR analysis (D–F) ChIP was performed in HCT116JunD-CTCF-WT and HCT116JunD-CTCF-KO cells using antibodies against Sp1, H3K4Me3 and Pol2, followed by qPCR with primers specific to the proximal hTERT promoter region. (G) HCT116JunD-CTCF-WT and HCT116JunD-CTCF-KO cells were transfected with si-control, si-JunD#1 and si-JunD#2. The graph shows hTERT gene expression analysis by RT-qPCR after 36 h of transfection. (H) Schematic view of chromosome 5 indicating the hTERT promoter and JunD-CTCF region. The discontinuous line indicates the JunD binding site that was removed by genome editing with CRISPR/Cas9. (I) Genotyping results by Sanger sequencing are shown for HCT116WT-JunD and HCT116ΔJunD clones. (J) Comparison of hTERT expression levels in HCT116WT-JunD and HCT116ΔJunD cells by RT-qPCR analysis. (K–M) ChIP was performed in HCT116WT-JunD and HCT116ΔJunD cells using antibodies against JunD, CTCF and Sp1, followed by qPCR with primers specific to the JunD-CTCF region and the proximal hTERT promoter region. Enrichment was calculated by the % input method for ChIP-qPCR experiments. Ct values were normalized to actin in RT-qPCR analyses. Error bars indicate the mean ± SD of three independent CRISPR clones. P-values were calculated by Student's t-test (*P < 0.05; **P < 0.01; ***P < 0.001; n.s. not significant).
JunD is one of the factors that regulate CTCF occupancy seeds long-range chromatin interactions between the T-INT2 and WT-hTERT promoter in CRC
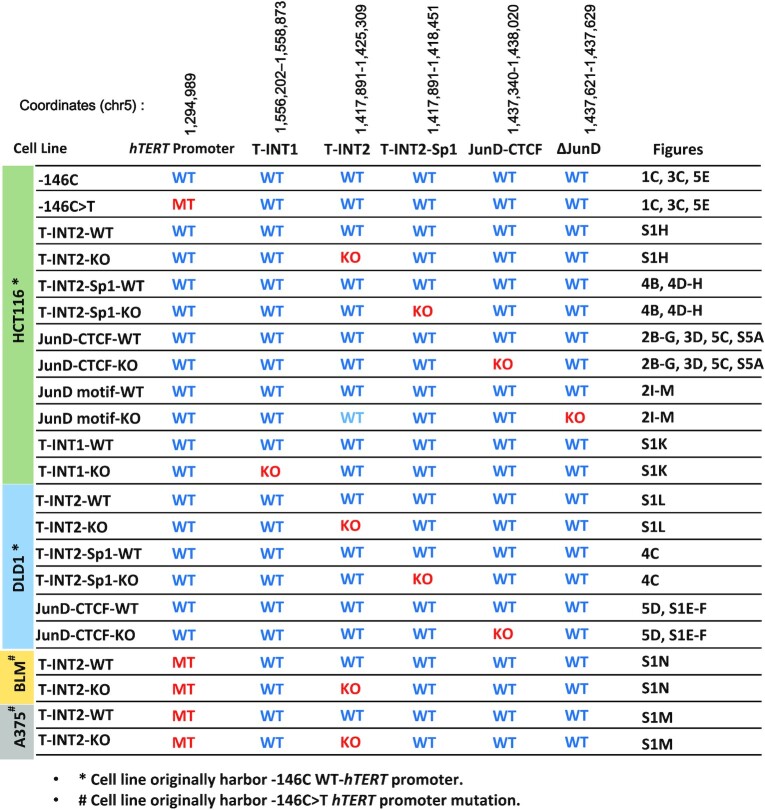
Given that increased occupancy of CTCF in the JunD-CTCF region, facilitated by JunD, enhanced hTERT expression, and because CTCF is known to mediate chromatin interactions, we performed circularized chromosome conformation capture sequencing (4C-seq) in CRC cells to identify chromatin interactions that functionally drive WT-hTERT promoters. We observed distinct chromatin interactions in CRC cells harbouring an active WT-hTERT promoter (Figure 3A). 4C-seq was used to distil the common interaction regions detected in all CRC cells and these were validated and quantified by chromosome conformation capture (3C) qPCR assays. A 7.5-kb T-INT2 region (hereafter referred to as Tert interacting region 2 [T-INT2]) with the genomic location chr5:1 417 891–1 425 309 was found to interact strongly with the proximal WT-hTERT promoter (Supplementary Figure S1G). T-INT2 was also found to form stronger interactions with the WT-hTERT promoter in PDCs with high hTERT expression than in samples with low hTERT expression (Figure 3B), thereby suggesting that it harbours functional elements key to drive hTERT. There was no significant difference in the interaction of T-INT1 with proximal WT-hTERT in CRCs, irrespective of their high or low hTERT levels (Figure 3B), further reiterating that distinct chromatin regions drive Mut-hTERT vs. WT-hTERT promoters; while T-INT1 drives Mut-hTERT, T-INT2 is needed to drive WT-hTERT promoters. To investigate whether any of the chromatin interactions detected in the 4C and 3C experiments functionally regulated the WT-hTERT promoter, we systematically knocked out T-INT2 (Supplementary Figure S1H) and all other chromatin regions, including T-INT1, in HCT116 cells, using two guide RNAs (gRNAs) targeting the 5′ and 3′ ends of each interacting chromatin region, and assayed for hTERT expression (Supplementary Figure S1I–K). T-INT2 region (chr5:1 417 891–1 425 309) is functionally key in driving WT-hTERT, since the removal of this region specifically led to the loss of hTERT expression in HCT116T-INT2-KO cells (Supplementary Figure S1H). Additionally, deletion of T-INT1 region, known to drive Mut-hTERT promoter (7), did not alter hTERT expression driven by the WT-hTERT promoter in HCT116T-INT1-KO cells (Supplementary Figure S1K). We also generated DLD1T-INT2-KO cells and observed a similar reduction of hTERT expression as in HCT116T-INT-KO cells (Supplementary Figure S1L). Notably, deletion of T-INT2 region in A375 and BLM cells (which harbour Mut-hTERT promoters) had no significant effect on hTERT expression (Supplementary Figure S1M, N). These results suggested that T-INT2 functionally regulates hTERT expression specifically via WT-hTERT promoters (Figure 3B). By contrast, T-INT1 did not have any role in the regulation of hTERT expression in these cells (Figure 3B and Supplementary Figure S1K). For easy reference, Table 1 summarizes the chromosome coordinates of all the edited regions, along with the hTERT promoter status in the edited cells and the figure numbers where these have been used in this study.
Figure 3.

JunD-mediated CTCF occupancy regulates WT-hTERT–T-INT2 long-range chromatin interaction. (A) 4C sequencing for long-range chromatin interactions with the hTERT promoter in four CRC cell lines. Significant interaction regions (P < 0.05) identified in each line are plotted and regions present in all lines are highlighted in red. Interaction regions are shown in dark-blue boxes, and were functionally validated by 3C-qPCR and CRISPR deletion experiments. (B) Relative chromatin interaction frequency between the hTERT promoter and the indicated chromatin regions identified by 4C were measured by 3C-qPCR in primary CRC samples. Mut-hTERT-specific chromatin interaction region T-INT1 (chr5:1,558,087) was used as a negative control. (C) Chromatin interaction frequency between the hTERT promoter and T-INT2 region was measured by 3C-qPCR in isogenic HCT116 cells transfected with si-Control or si-JunD. (D) Chromatin interaction frequency between the WT-hTERT promoter and T-INT2 was measured by 3C-qPCR in HCT116JunD-CTCF-WT and HCT116JunD-CTCF-KO cells. (E) Isogenic HCT116−146C and HCT116−146C>T cells were co-transfected with KRAB–dCas9 expression vector and different gRNAs targeting the T-INT2 region, or non-targeting control gRNA. RNA was isolated 2 days after transfection and gene expression levels of hTERT were measured by qPCR. The horizontal dotted line shows the mean hTERT expression change in HCT116−146C>T cells and is set as a threshold. The T-INT2 region (chr5:1 417 891–1 425 309) is shown in grey. Putative Sp1 motifs are shown as blue dots within the Sp1 cluster. (F) HCT116−146C and HCT116−146C>T cells were co-transfected with KRAB–dCas9 expression vector and non-targeting control gRNA (cont) or one of three different gRNAs targeting the region targeted by gRNA_a. Gene expression levels of hTERT were measured by qPCR. (G) HCT116–146C cells were co-transfected with dCas9, non-targeting control gRNA (cont) or three different gRNAs targeting the region nearby to gRNA_a. RNA was isolated two days after transfection and gene expression levels of hTERT were measured by qPCR. Ct values were normalized to actin. Error bars indicate mean ± SD of three independent experiments. P-values were calculated by Student's t-test (*P < 0.05; **P < 0.01).
Table 1.
Edited cell lines used in the study along with respective figure numbers, edited coordinates and hTERT promoter mutation status are shown
|
But how does T-INT2 region regulate WT-hTERT activity? Given the correlation between JunD and hTERT expression in cells with WT-hTERT promoters, we next analysed if JunD plays a key initiating role in the formation of long-range chromatin interactions between T-INT2 and proximal WT-hTERT promoter. Depletion of JunD in isogenic HCT116−146C or HCT116−146C>T cells with WT-hTERT and Mut-hTERT promoters, respectively, showed that T-INT2 interaction with proximal WT-hTERT promoter is specifically reduced in HCT116−146C cells (Figure 3C). Similarly, deletion of JunD-CTCF region inhibited the interaction between T-INT2 and proximal WT-hTERT promoter in HCT116JunD-CTCF-KO cells (Figure 3D). We analysed T-INT2 and JunD-CTCF regions for H3K4Me1 and H3K27Ac histone mark enrichment and showed that neither of these regions have enhancer features (Supplementary Figure S2). These results suggest that JunD enrichment in the JunD-CTCF region relaxes the chromatin structure and enables CTCF to bind and bend the chromatin to enhance the interaction between T-INT2 and proximal WT-hTERT promoter. This function of JunD was specifically relevant to WT-hTERT promoters, as JunD loss had no consequences on hTERT expression and chromatin interactions via Mut-hTERT promoters (Figures 1C and 3C).
Catalytically dead Cas9-induced steric hindrance identifies an Sp1 cluster 5′ of the T-INT2 region critical for activation of WT-hTERT promoter in CRCs
To define the core functional region within the 7.5-kb T-INT2 region, that drives WT-hTERT promoter, we designed eight dCas9 gRNAs spaced throughout the entire T-INT2 region (Figure 3E) and evaluated them in HCT116−146C and HCT116−146C>T cells. Each gRNA was co-transfected with a vector expressing the Krüppel-associated box repressor fused to dCas9 (KRAB–dCas9). In HCT116−146C cells, but not in HCT116−146C>T cells, we observed a significant reduction of hTERT expression in cells transfected with gRNA_a, which targets the 5′ region of T-INT2 (Figure 3E). Next, we designed three closely inter-spaced gRNAs that bind within the region targeted by gRNA_a. We observed a dramatic and consistent reduction in hTERT expression in cells transfected with gRNAs a1, a2 and a3 in HCT116−146C cells but not in HCT116−146C>T cells (Figure 3F). As the experiments were performed using dCas9 fused to KRAB, which is a transcriptional inhibitor itself and could recruit other epigenetic modifiers to the site, we repeated the experiment with a version of dCas9 without the KRAB and noted a similar reduction in hTERT expression in HCT116–146C cells co-transfected with gRNAs (control, a1, a2 and a3) and dCas9 (Figure 3G). A motif-prediction algorithm (34) identified five Sp1 and c-MYC binding motifs at the 5′ end of the T-INT2 region (Supplementary Figure S3A-B). Notably, the hTERT proximal promoter also harbours five Sp1 and c-MYC motifs (Supplementary Figure S3C-D). Regulatory factors that bind to the hTERT promoter, including GABPα/β, Sp1 and c-MYC, have all been shown to mediate chromatin interactions by juxtaposing enhancers to promoters (35–38). Notably, the gRNAs a1 and a2 targeted the regions immediately upstream and downstream of the Sp1 cluster in the T-INT2 region, suggesting that Sp1 subunits bound at the 5′ end of T-INT2 might have been critical for mediation of the observed long-range chromatin interactions.
The Sp1 cluster in the T-INT2 region is essential for functional activity of WT-hTERT promoter in CRC
To verify whether the Sp1 cluster in T-INT2 region functionally regulates hTERT expression, we removed a ∼560-bp region within T-INT2, which contained five Sp1 binding motifs, resulting in the T-INT2-Sp1-KO genotype in HCT116 and DLD1 cells (Figure 4A). In multiple isogenic clones of HCT116T-INT2-Sp1-KO and DLD1T-INT2-Sp1-KO cells, hTERT expression was dramatically reduced compared with HCT116T-INT2-Sp1-WT and DLD1T-INT2-Sp1-WT cells where this cluster was intact, suggesting that the Sp1 cluster in T-INT2 is essential for functional regulation of hTERT expression via the WT-hTERT promoter (Figure 4B, C). We performed ATAC-seq in HCT116T-INT2-Sp1-WT and HCT116T-INT2-Sp1-KO cells to determine if the T-INT2 region regulates the compactness of the hTERT promoter. As compared to the HCT116T-INT2-Sp1-WT clones, we observed a significant reduction in open chromatin formed at the hTERT promoter in two independent HCT116T-INT2-Sp1-KO clones (Supplementary Figure S4A). As a control, we tested Gapdh and Clptm1l promoters (Supplementary Figure S4B, C) and observed no difference in ATAC signals for these regions. As a control, we showed that there is no significant difference in Sp1 and JunD levels in isogenic lines suggesting that the reduction of hTERT expression is due to lack of the T-INT2 long range chromatin interaction but not because of Sp1 or JunD protein levels (Supplementary Figure S4D). We conducted an anti-H3K4Me3 ChIP-qPCR experiment and observed a reduction in active histone marks in the WT-hTERT promoter of HCT116T-INT2-Sp1-KO cells relative to HCT116T-INT2-Sp1-WT cells (Figure 4D). ChIP with an anti-Sp1 antibody demonstrated that removal of the Sp1 cluster in the T-INT2 region reduced Sp1 occupancy on the WT-hTERT proximal promoter in HCT116T-INT2-Sp1-KO cells (Figure 4E). We also confirmed that, in the absence of the JunD-CTCF region, Sp1 occupancy also diminished in the T-INT2 region in HCT116JunD-CTCF-KO cells (Supplementary Figure S5A), suggesting that Sp1 binding in the proximal WT-hTERT promoter was dependent on its interaction with distal Sp1 subunits bound to the T-INT2 region (Figure 2D and 4E). These results indicated that the Sp1 cluster in T-INT2 is essential and specific for regulating the WT-hTERT promoter (shown in model Figure 4A). As a result of the reduction in hTERT gene expression, we also observed a significant loss of telomerase activity in the HCT116T-INT2-Sp1-KO cells (Figure 4F).
Figure 4.

The Sp1 cluster in the T-INT2 region is essential for functional activity of WT-hTERT promoter in CRC. (A) Schematic representation of the wild-type (WT)-hTERT promoter and the long-range chromatin interaction region T-INT2. The hTERT promoter is shown in two parts: the proximal promoter (up to −1 kb) and long-distance elements. The inactive T-INT1 is shown as transparent, and the active T-INT2 region shown as red, with the chromosome coordinates (chr5:1 417 891–1 425 309). The discontinuous line indicates the CRISPR/Cas9-targeted Sp1 cluster in T-INT2 (T-INT2-SP1). (B, C) Comparison of hTERT expression levels in parental, HCT116T-INT2-SP1-WT, HCT116T-INT2-SP1-KO and DLD1T-INT2-SP1-WT, DLD1T-INT2-SP1-KO cells. Genotyping PCR results are shown for the HCT116T-INT2-SP1-WT and DLD1 T-INT2-SP1-WT (1100 bp) and HCT116T-INT2-SP1-KO and DLD1T-INT2-SP1-KO (540 bp) clones. (D, E) ChIP was performed in HCT116T-INT2-SP1-WT and HCT116T-INT2-SP1-KO cells using antibodies against H3K4Me3 and Sp1 followed by qPCR with primers specific to the proximal hTERT promoter region. Enrichment was calculated by the % input method. (F) Telomerase activity was measured by RT-qPCR in HCT116T-INT2-SP1-WT and HCT116T-INT2-SP1-KO cells. (G, H) HCT116T-INT2-SP1-WT and HCT116T-INT2-SP1-KO cells were transplanted into NOD/SCID mice and allowed to form tumours. After 24 days tumours were harvested and analysed. (H) Tumour volume was measured at indicated time points (n = 4). Error bars indicate mean ± SD of three independent experiments. P-values were calculated by Student's t-test (*P < 0.05; **P < 0.01).
To explore the in vivo functional outcome, we implanted HCT116T-INT2-Sp1-WT and HCT116T-INT2-Sp1-KO isogenic cells into NOD/SCID mice. Removal of the Sp1 cluster in the T-INT2 region in the HCT116T-INT2-Sp1-KO cells resulted in a significant dampening of hTERT expression (Figure 4B) and tumour growth (Figure 4G, H) relative to that seen in the HCT116T-INT2-Sp1-WT cells. These results suggested that Sp1 molecules bound to T-INT2 and the proximal promoter regions were key mediators of a long-range interaction that regulated hTERT expression and cell growth in cancer cells with the WT-hTERT promoter (Seen in model Figure 4A)
These results suggest that the long-range chromatin interaction mediated by T-INT2 contributes to an open chromatin structure at the hTERT promoter and this chromatin interaction allows binding of the master transcription factor Sp1 to the hTERT promoter, driving hTERT expression in cancer cells harboring the WT-hTERT promoter.
High β-catenin levels initiate a JunD-mediated cascade of events leading to WT-hTERT expression in CRC
To understand how JunD levels increase in CRC and what is the trigger for reactivation of hTERT, we investigated β-catenin stabilization that occurs due to APC/β-catenin mutations in CRC. Mutations in APC are highly prevalent in colon cancer and they lead to the activation of several oncogenic signaling pathways (39) via the upregulation of β-catenin protein (40). Analysis of fresh CRC patient samples revealed a significant correlation between β-catenin and JunD expression (Supplementary Figure S5B, C). To validate the mechanical link between β-catenin and JunD, we analyzed HCT116 and HT29 lines expressing high and low levels of β-catenin, respectively (Supplementary Figure S5D). Knock-down of β-catenin with two different shRNAs reduced hTERT expression in HCT116 cells while not altering the Sp1 levels (Supplementary Figure S5E–G). Similar to JunD, β-catenin depletion led to hTERT-T-INT2 chromatin interaction (Supplementary Figure S5H). Interestingly, silencing β-catenin in HT29 cells did not have any impact on hTERT expression (Supplementary Figure S5I, J); possibly due to very low level of β-catenin in the basal condition (Supplementary Figure S5D). However, knock-down of APC increased β-catenin stability and enhanced JunD protein expression and increased hTERT expression significantly in HT29 cells (Supplementary Figure S5K–M). In addition, we confirmed that APC mediated β-catenin stability also increased Sp1 occupancy on the WT-hTERT proximal promoter (Supplementary Figure S5N).
Salinomycin inhibits JunD-mediated interaction between T-INT2 and proximal WT-hTERT promoter in CRCs
To identify potential inhibitors which can be used to disrupt specific chromatin structure that can inhibit WT-hTERT expression in a cancer cell specific manner, we analysed drug screening datasets on preclinical cell lines (35 CRC cell lines using 4603 drugs using DepMap (19). We analysed the correlation between JunD expression levels and cell viability upon drug treatments (Figure 5A). Salinomycin, an antibiotic widely used against gram-positive bacteria, has been shown to have anti-cancer effects against hepatocellular carcinoma and CRC (41–45), and reduce JunD levels in cancer cells (41), was found as a hit in the screen. Significantly, a comparison of high (top 30%) and low (bottom 30%) JunD expressing CRC lines revealed significantly higher efficacy of salinomycin in inhibiting hTERT expression in CRCs with high JunD expression levels (Figure 5B). Analysis of salinomycin efficacy as a function of hTERT promoter status also revealed higher efficacy in cells with WT-hTERT promoter (Supplementary Figure S6A, B). Consistent with these results, salinomycin showed poorer efficacy in bladder cancer lines where Mut-hTERT promoters are predominant drivers of hTERT (Supplementary Figure S6C). These results indicate that salinomycin sensitivity is possibly specific to CRC. It most likely works due to its direct effect on JunD expression levels, which controls WT-hTERT transcription via the aforementioned mechanisms. For confirmation of this mechanism, we treated isogenic HCT116JunD-CTCF-WT, HCT116JunD-CTCF-KO and DLD1JunD-CTCF-WT, DLD1JunD-CTCF-WT cells with salinomycin for 24 h and observed a more pronounced reduction of hTERT expression in the HCT116JunD-CTCF-WT and DLD1JunD-CTCF-WT cells as compared to HCT116JunD-CTCF-KO and DLD1JunD-CTCF-KO counterparts (Figure 5C–H), suggesting that salinomycin reduced hTERT expression mainly via its action of JunD mediated cascade initiated due to its binding on the JunD-CTCF region described above. Notably, salinomycin treatment disrupted the interaction between T-INT2 and the WT-hTERT proximal promoter (Supplementary Figure S6D), and specifically inhibited hTERT expression in cells with WT-hTERT promoter (Figure 5E). Having observed that salinomycin showed JunD dependent hTERT inhibition, we tested its action using low and high JunD expressing PDCs. Compared to low JunD expressing PDCs, salinomycin caused a significant reduction in hTERT expression in cells with high JunD expressing PDCs (Figure 5F, G). Furthermore, we observed a dramatic reduction of cell growth in high JunD expressing patient-derived primary CRC cells (Figure 5H, I). Our results suggested that elevated levels of JunD in CRCs directly initiate and contribute to the activation of the WT-hTERT promoter by increasing the binding of CTCF in a region located ∼140 kb upstream of the hTERT promoter and ∼20 kb upstream of the T-INT2 region. This binding facilitates the bending of the chromatin such that Sp1 dimers in the proximal hTERT promoter (chr5: 1 295 067-1 295 177) can bind Sp1 dimers at the T-INT2 region (chr5: 1 417 932-1 418 217) thereby stabilizing this long-range interaction and leading to productive transcription (Figure 6).
Figure 5.

Salinomycin treatment inhibits long-range chromatin interaction and hTERT expression in colon cancer cells. (A) Prediction analysis of drugs potency in CRC cell lines. Plot shows ranked drug compounds based on correlation of JunD mRNA expression with cell viability in drug response assay. (B) Violin plot shows comparison of cell viability in salinomycin treated CRC cell lines separated by JunD mRNA expression levels. (C, D) HCT116JunD-CTCF-WT, HCT116JunD-CTCF-KO, DLD1JunD-CTCF-WT and DLD1JunD-CTCF-KO cells were treated with DMSO or salinomycin (2 mM) for 24 h. hTERT gene expression was measured by RT-qPCR. Actin was used as a control. The graph shows the mean values of three independent HCT116JunD-CTCF-WT, HCT116JunD-CTCF-KO, DLD1JunD-CTCF-WT and DLD1JunD-CTCF-KO clones. (E) Gene expression analysis of hTERT is shown by RT-qPCR analysis in HCT116–146C and HCT116–146C>T cells treated with or without salinomycin for 24 h. (F, G) Patient-derived colon cancer cells CRC1489 and 1258 (Low-JunD) and CRC1846 and CRC1707 (high-JunD) were treated with or without salinomycin for indicated concentrations and time points. The graph shows the hTERT gene expression levels measured by RT-qPCR. (H, I) Graph shows growth rate of the CRC1846 and CRC1707 cells treated with salinomycin by the CCK8 assay. Error bars indicate the mean ± SD of three experiments. p-values were calculated by Student's t-test (*P < 0.05; **P < 0.01; ***P < 0.001).
Figure 6.

Stepwise hTERT reactivation mechanism in colorectal cancer. Oncogenic mutations in APC and beta-catenin enhance JunD levels in colon cancer. Increased JunD enhances occupancy of CTCF on the JunD-CTCF region that mediates the formation of long-range chromatin interaction between T-INT2 and hTERT, stabilizing Sp1 on the hTERT promoter. This 3D chromatin organization increases the accessibility of the WT-hTERT promoter and enables Pol2 to transcribe the hTERT gene.
DISCUSSION
To circumvent undesirable cytotoxicity of telomerase enzymatic inhibitors, which block hTERT action in cancer cells and stem cells alike, it is crucial to decipher mechanisms that specifically drive hTERT reactivation in cancer cells. Transcriptional reactivation of hTERT is the rate-limiting step in tumorigenesis, which makes it a valuable candidate for cancer therapy. Reactivation of hTERT occurs by two distinct mechanisms based on the status of its promoter. Mut-hTERT promoter reactivation is initiated by binding of specific transcription factors on the de novo cancer cell-specific mutant site. Still, the cascade of events that ensue upon binding transcription factors to this mutant site are not fully understood. We had previously identified a long-range chromatin interaction between the proximal Mut-hTERT promoters and a genomic region T-INT1 located 260kb upstream that was required for reactivation of hTERT subsequent to the binding of factors to the mutant site (7). Tetramerization of GABPα/β subunits tethered between T-INT1 and the de novo ETS site in the proximal Mut-hTERT promoter was found to be essential for creating and stabilizing this long-range chromatin interaction, which promoted the opening of the chromatin around the Mut-hTERT promoter via recruitment of chromatin modifiers like BRD4, eventually leading to hTERT expression (7,46). Before these studies, the use of non-chromatinised plasmid-based reporters had led us to believe that hTERT promoter is regulated only by proximal elements less than 300 bp from the start of transcription (47–49). This study (7) changed the paradigm that hTERT promoter was regulated by chromatin regions way beyond the 300 bp that researchers studied using reporter gene assays (47–49). This information, on one hand, added to a key academic understanding of how hTERT reactivation is regulated in physiology, but importantly, on the other hand, also opened opportunities for targetting hTERT reactivation using transcriptional and epigenetic machinery that operates way beyond the proximal hTERT promoter.
How does one begin to unravel the mechanistic details of hTERT reactivation in further detail? With the advent of genome editing methods, it was possible to edit the hTERT loci on chromosome 5 and gain information on hTERT regulation in the true chromatin context in the endogenous loci. However, unlike with Mut-hTERT promoters, where the introduction of single residue changes in the genome allows for making reagents for studying reactivation of hTERT, there are no such defined events yet discovered that lead to reactivation of WT-hTERT promoters in vivo. However, the knowledge gained from our studies on Mut-hTERT promoter could be a starting point in deciphering that hTERT promoters are not driven by elements just proximal to the basal promoter but require distal regions which have not been interpreted previously. Hence in this study, we set out to identify previously uncharacterized molecular and epigenetic drivers of WT-hTERT which drive hTERT reactivation in 70% of cancers without hTERT promoter mutations.
Unlike stem cells and induced pluripotent stem cells, where 4 factors (Oct4, Sox2, Klf4 and c-Myc) keep WT-hTERT promoter active (50,51), many cancers such as CRC are driven by defined oncogenic alterations which function in a step wise manner. Hence, we reasoned that using CRC as a model to study WT-hTERT reactivation would be ideal as it would allow us the ability to toggle the signals (oncogenes or known alterations in CRC like APC) that lead eventually to activation of hTERT, which is the rate-limiting step in transformation. The rationale for such an approach was also based on our findings that specific chromatin regions such as the T-INT1 could be defined as key drivers of Mut-hTERT promoters would allow the designing of different classes of drugs that target the 2 distinct hTERT reactivation mechanisms with minimal cytotoxicity. In the step-wise transformation model in CRC, KRAS and APC mutations lead to the accumulation of β-catenin and activation of other oncogenic pathways driven by AP-1 transcription family members (52,53). We found that the expression of JunD, an AP-1 family member, was highly correlated with hTERT expression (Figure 1A and B). JunD upregulation in CRCs, regulates the transcription of target genes by recruitment of epigenetic factors, including CBP/p300, which are known to unwind chromatin, enabling other co-factors and epigenetic mediators like CTCF to bind to nearby regions (21,24,25). Integrative analysis of ChIP-seq (for JunD and CTCF) and ATAC-seq (in CRC and healthy colon tissues) data identified an accessible chromatin interaction region specific to CRC cells with high expression of hTERT (and JunD). JunD occupancy in this region augmented CTCF enrichment. CTCF is one of the epigenetic molecules that regulate the 3D dynamics of chromatin, and it mediates around 1800 interchromosomal and intrachromosomal interactions (54). CTCF and cohesin work together to mediate long-range interactions (55). Our results demonstrated that JunD-mediated CTCF enrichment in the JunD-CTCF region altered the chromatin structure and regulated long-range chromatin interaction between the hTERT promoter and a genomic region (T-INT2) located ∼120 kb upstream of the hTERT promoter. Interaction between the T-INT2 region and the hTERT promoter was stabilized by Sp1 tetramerization and this formation was crucial for WT-hTERT reactivation, telomerase activity and transformation. Furthermore, the T-INT2 region was specific for reactivation of the WT-hTERT promoter, as deletion of this region did not affect the activity of Mut-hTERT promoters (Supplementary Figure S1M, N).
Sp1 is a critical transcription factor for regulating promoters like hTERT that lack TATA boxes (56–58). However, given that the long-range chromatin interaction that activates the WT-hTERT promoter depends on Sp1 bridging, why do healthy somatic cells with abundant Sp1 not employ this exact mechanism to keep hTERT activated all the time? Our results showed that in cancers with the WT-hTERT promoter, reactivation of the promoter involved two co-occurring steps: a) elevation of JunD levels, enabling CTCF to bind its target site and alter the chromatin structure to mediate T-INT2 and proximal hTERT chromatin interaction; and b) Sp1–Sp1 multimerization by chromatin looping, enhancing Sp1 occupancy on the hTERT proximal promoter, increasing active histone mark enrichment and decreasing promoter compactness. Crucially, our data suggested that accumulation of beta-catenin during the step-wise CRC transformation process leads to enhanced JunD levels, which was likely to be the initial trigger that invoked a complex string of molecular events that activated the WT-hTERT promoter. This multi-layered sequence of events explains why normal cells, despite expressing abundant Sp1, have no hTERT expression. Indeed, the transformation of healthy cells into cancer requires multiple genetic and metabolic alterations leading to hTERT reactivation (59,60). Therefore, hTERT promoter activity is tightly regulated in somatic cells to limit replicative capacity. In somatic cells with long telomeres, TRF2 facilitates a subtelomere loop with the hTERT promoter that suppresses hTERT promoter activity and blocks transcription machinery to transcribe the hTERT gene (61). Therefore, how cells switch from the suppressive subtelomeric chromatin interaction to activating hTERT-T-INT2 must be further investigated.
Our results also illuminated commonalities and differences in the hTERT reactivation mechanisms between cancer cells with WT-hTERT and Mut-hTERT promoters. We found that regardless of mutational status, hTERT promoters are regulated epigenetically by long-range chromatin interactions that are mediated by tetramerization of transcription factors in cancer cells. However, the distal regulatory regions that WT-hTERT and Mut-hTERT promoters interact with and the transcription factors that tetramerize for the formation of respective long-range interaction along with co-regulators are significantly different in both promoters. Such differences allow for precise targeting of cancers based on their hTERT promoter status. As proof of principle, we have shown here that Salinomycin, an FDA-approved drug, could be used to disrupt oncogenic chromatin structures in CRC lines to switch off WT-hTERT promoters. On the other hand, in our previous study (7), we have demonstrated that cancers with Mut-hTERT promoters could be targeted specifically with BRD4 inhibitors.
Overall, our results suggest that understanding how transcription factors function in the 3D chromosomal environment offers great therapeutic potential, as chromatin interactions are largely specific to particular cell types and genes (62). Direct inhibition of molecules like hTERT and Sp1, which have dual roles in cancer progression and normal cellular homeostasis, has routinely failed because of intolerable cytotoxicity. Therefore, our results suggest the potential for new opportunities to identify inhibition strategies that can be effectively implemented to target cancer cells specifically without causing toxicity to stem cells.
DATA AVAILABILITY
The data are available in the GEO open-access repository (GSE163077).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Stephanie Conos and Dr Michelle Shuling Ong for editing and proofreading the manuscript. We thank Dorcas Hei for the scientific illustrations (Models were created with BioRender.com). Editorial assistance was provided by Dr R. Phillips of Insight Editing London. Primary CRC models were generated and kindly gifted by Dr Xiaoqian Zhang, Shumei Chia and Jiamin Loo, Genome Institute of Singapore. We sincerely thank them for their contributions.
Author contributions: SCA and VT conceived the study and designed the experiments. SCA performed the experiments with the help of QFN, CHTC, ZE-S. and JLL. JYHC performed the expression correlation, 4C and ATAC-seq analysis, with the help of KC and LA under the supervision of MJF. SA performed and analysed the in vivo tumour-growth experiments. CC, IBT and RD contributed with the primary patient samples and PDX analyses. SCA and VT directed the study and SCA wrote the paper with the input of all authors.
Contributor Information
Semih Can Akıncılar, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore.
Joelle Yi Heng Chua, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore.
Qin Feng Ng, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore.
Claire Hian Tzer Chan, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore.
Zahra Eslami-S, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore.
Kaijing Chen, Cancer Science Institute of Singapore, National University of Singapore, 117599, Singapore.
Joo-Leng Low, Laboratory of Precision Oncology and Cancer Evolution, Genome Institute of Singapore, A*STAR, 138672, Singapore.
Surendar Arumugam, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore.
Luay Aswad, Cancer Science Institute of Singapore, National University of Singapore, 117599, Singapore.
Clarinda Chua, Genome Institute of Singapore, Agency for Science, Technology and Research (A*STAR), 138672, Singapore; Department of Medical Oncology, National Cancer Centre Singapore, 169610, Singapore.
Iain Beehuat Tan, Genome Institute of Singapore, Agency for Science, Technology and Research (A*STAR), 138672, Singapore; Department of Medical Oncology, National Cancer Centre Singapore, 169610, Singapore.
Ramanuj DasGupta, Laboratory of Precision Oncology and Cancer Evolution, Genome Institute of Singapore, A*STAR, 138672, Singapore.
Melissa Jane Fullwood, Cancer Science Institute of Singapore, National University of Singapore, 117599, Singapore; School of Biological Sciences, Nanyang Technological University, 637551, Singapore.
Vinay Tergaonkar, Division of Cancer Genetics and Therapeutics, Laboratory of NFκB Signaling, Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A*STAR), 138673, Singapore; Department of Pathology, Yong Loo Lin School of Medicine, National University of Singapore (NUS), 119074, Singapore; Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore (NUS), 117596, Singapore.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation under its Competitive Research Programme [NRF-CRP17-2017-02 to V.T.]; Agency for Science Technology and Research, Singapore (A*STAR) for funding and supporting the Tergaonkar laboratory and this project; NRF Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative; Singapore Ministry of Education Academic Research Fund Tier 2 grant [MOET2EP30120-0009 to M.J.F.]. Funding for open access charge: The V.T. laboratory is supported by the National Research Foundation-Competitive Research Programme [NRF-CRP17-2017-02]; IMCB A*STAR.
Conflict of interest statement. None declared.
REFERENCES
- 1. Shay J.W., Wright W.E.. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005; 26:867–874. [DOI] [PubMed] [Google Scholar]
- 2. Hiyama E., Hiyama K.. Telomere and telomerase in stem cells. Br J. Cancer. 2007; 96:1020–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akincilar S.C., Chan C.H.T., Ng Q.F., Fidan K., Tergaonkar V.. Non-canonical roles of canonical telomere binding proteins in cancers. Cell Mol. Life Sci. 2021; 78:4235–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vinagre J., Almeida A., Populo H., Batista R., Lyra J., Pinto V., Coelho R., Celestino R., Prazeres H., Lima L.et al.. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013; 4:2185. [DOI] [PubMed] [Google Scholar]
- 5. Huang F.W., Hodis E., Xu M.J., Kryukov G.V., Chin L., Garraway L.A.. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013; 339:957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horn S., Figl A., Rachakonda P.S., Fischer C., Sucker A., Gast A., Kadel S., Moll I., Nagore E., Hemminki K.et al.. TERT promoter mutations in familial and sporadic melanoma. Science. 2013; 339:959–961. [DOI] [PubMed] [Google Scholar]
- 7. Akincilar S.C., Khattar E., Boon P.L., Unal B., Fullwood M.J., Tergaonkar V.. Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 2016; 6:1276–1291. [DOI] [PubMed] [Google Scholar]
- 8. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B.et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krijger P.H.L., Geeven G., Bianchi V., Hilvering C.R.E., de Laat W.. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods. 2020; 170:17–32. [DOI] [PubMed] [Google Scholar]
- 15. Cao F., Zhang Y., Cai Y., Animesh S., Zhang Y., Akincilar S.C., Loh Y.P., Li X., Chng W.J., Tergaonkar V.et al.. Chromatin interaction neural network (ChINN): a machine learning-based method for predicting chromatin interactions from DNA sequences. Genome Biol. 2021; 22:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geeven G., Teunissen H., de Laat W., de Wit E.. peakC: a flexible, non-parametric peak calling package for 4C and Capture-C data. Nucleic Acids Res. 2018; 46:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Akincilar S.C., Wu L., Ng Q.F., Chua J.Y.H., Unal B., Noda T., Chor W.H.J., Ikawa M., Tergaonkar V.. NAIL: an evolutionarily conserved lncRNA essential for licensing coordinated activation of p38 and NFkappaB in colitis. Gut. 2021; 70:1857–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghosh A., Saginc G., Leow S.C., Khattar E., Shin E.M., Yan T.D., Wong M., Zhang Z., Li G., Sung W.K.et al.. Telomerase directly regulates NF-kappaB-dependent transcription. Nat. Cell Biol. 2012; 14:1270–1281. [DOI] [PubMed] [Google Scholar]
- 19. Corsello S.M., Nagari R.T., Spangler R.D., Rossen J., Kocak M., Bryan J.G., Humeidi R., Peck D., Wu X., Tang A.A.et al.. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer. 2020; 1:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akincilar S.C., Low K.C., Liu C.Y., Yan T.D., Oji A., Ikawa M., Li S., Tergaonkar V.. Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett. 2015; 589:974–984. [DOI] [PubMed] [Google Scholar]
- 21. Wang H., Birkenbach M., Hart J.. Expression of Jun family members in human colorectal adenocarcinoma. Carcinogenesis. 2000; 21:1313–1317. [PubMed] [Google Scholar]
- 22. Eide P.W., Bruun J., Lothe R.A., Sveen A.. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017; 7:16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rowland T.J., Dumbovic G., Hass E.P., Rinn J.L., Cech T.R.. Single-cell imaging reveals unexpected heterogeneity of telomerase reverse transcriptase expression across human cancer cell lines. Proc. Natl Acad. Sci. U.S.A. 2019; 116:18488–18497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Toufaily C., Lokossou A.G., Vargas A., Rassart E., Barbeau B.. A CRE/AP-1-like motif is essential for induced syncytin-2 expression and fusion in human trophoblast-like model. PLoS One. 2015; 10:e0121468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barnes P.J., Adcock I.M.. Transcription factors and asthma. Eur. Respir J. 1998; 12:221–234. [DOI] [PubMed] [Google Scholar]
- 26. Cubenas-Potts C., Corces V.G.. Architectural proteins, transcription, and the three-dimensional organization of the genome. FEBS Lett. 2015; 589:2923–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Toh L., Lau P., Wang X.. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J. Biol. Chem. 2012; 287:32494–32511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khattar E., Tergaonkar V.. Transcriptional Regulation of Telomerase Reverse Transcriptase (TERT) by MYC. Front. Cell Dev. Biol. 2017; 5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goueli B.S., Janknecht R.. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol. Cell Biol. 2004; 24:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vageli D., Ioannou M.G., Koukoulis G.K.. Transcriptional activation of hTERT in breast carcinomas by the Her2-ER81-related pathway. Oncol Res. 2009; 17:413–423. [DOI] [PubMed] [Google Scholar]
- 31. Oh S.T., Kyo S., Laimins L.A.. Telomerase activation by human papillomavirus type 16 E6 protein: induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001; 75:5559–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kyo S., Takakura M., Taira T., Kanaya T., Itoh H., Yutsudo M., Ariga H., Inoue M.. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000; 28:669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mastrangelo I.A., Courey A.J., Wall J.S., Jackson S.P., Hough P.V.. DNA looping and Sp1 multimer links: a mechanism for transcriptional synergism and enhancement. Proc. Natl Acad. Sci. U.S.A. 1991; 88:5670–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H.et al.. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014; 42:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deshane J., Kim J., Bolisetty S., Hock T.D., Hill-Kapturczak N., Agarwal A.. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010; 285:16476–16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nolis I.K., McKay D.J., Mantouvalou E., Lomvardas S., Merika M., Thanos D.. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl Acad. Sci. U.S.A. 2009; 106:20222–20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kieffer-Kwon K.R., Nimura K., Rao S.S.P., Xu J., Jung S., Pekowska A., Dose M., Stevens E., Mathe E., Dong P.et al.. Myc regulates chromatin decompaction and nuclear architecture during B cell activation. Mol. Cell. 2017; 67:566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akincilar S.C., Unal B., Tergaonkar V.. Reactivation of telomerase in cancer. Cell Mol. Life Sci. 2016; 73:1659–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fuchs S.Y., Ougolkov A.V., Spiegelman V.S., Minamoto T.. Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle. 2005; 4:1522–1539. [DOI] [PubMed] [Google Scholar]
- 40. Yang J., Zhang W., Evans P.M., Chen X., He X., Liu C.. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J. Biol. Chem. 2006; 281:17751–17757. [DOI] [PubMed] [Google Scholar]
- 41. Xu L., Wang T., Meng W.Y., Wei J., Ma J.L., Shi M., Wang Y.G.. Salinomycin inhibits hepatocellular carcinoma cell invasion and migration through JNK/JunD pathway-mediated MMP9 expression. Oncol Rep. 2015; 33:1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang F., He L., Dai W.Q., Xu Y.P., Wu D., Lin C.L., Wu S.M., Cheng P., Zhang Y., Shen M.et al.. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS One. 2012; 7:e50638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang F., Dai W., Wang Y., Shen M., Chen K., Cheng P., Zhang Y., Wang C., Li J., Zheng Y.et al.. The synergistic in vitro and in vivo antitumor effect of combination therapy with salinomycin and 5-fluorouracil against hepatocellular carcinoma. PLoS One. 2014; 9:e97414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Al Dhaheri Y., Attoub S., Arafat K., Abuqamar S., Eid A., Al Faresi N., Iratni R. Salinomycin induces apoptosis and senescence in breast cancer: upregulation of p21, downregulation of survivin and histone H3 and H4 hyperacetylation. Biochim. Biophys. Acta. 2013; 1830:3121–3135. [DOI] [PubMed] [Google Scholar]
- 45. Skeberdyte A., Sarapiniene I., Aleksander-Krasko J., Stankevicius V., Suziedelis K., Jarmalaite S.. Dichloroacetate and Salinomycin Exert a Synergistic Cytotoxic Effect in Colorectal Cancer Cell Lines. Sci. Rep. 2018; 8:17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shanmugam R., Ozturk M.B., Low J.L., Akincilar S.C., Chua J.Y.H., Thangavelu M.T., Periyasamy G., DasGupta R., Tergaonkar V.. Genome-wide screens identify specific drivers of mutant hTERT promoters. Proc. Natl Acad. Sci. U.S.A. 2022; 119:e2105171119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shlyueva D., Stampfel G., Stark A.. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014; 15:272–286. [DOI] [PubMed] [Google Scholar]
- 48. Glubb D.M., Shi W., Beesley J., Fachal L., Pritchard J.L., McCue K., Barnes D.R., Antoniou A.C., Dunning A.M., Easton D.F.et al.. Candidate causal variants at the 8p12 breast cancer risk locus regulate DUSP4. Cancers (Basel). 2020; 12:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dekker J., Misteli T.. Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol. 2015; 7:a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takasawa K., Arai Y., Yamazaki-Inoue M., Toyoda M., Akutsu H., Umezawa A., Nishino K.. DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced pluripotent stem cells. Hum. Cell. 2018; 31:78–86. [DOI] [PubMed] [Google Scholar]
- 51. Stadtfeld M., Nagaya M., Utikal J., Weir G., Hochedlinger K.. Induced pluripotent stem cells generated without viral integration. Science. 2008; 322:945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Korinek V., Barker N., Morin P.J., van Wichen D., de Weger R., Kinzler K.W., Vogelstein B., Clevers H.. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275:1784–1787. [DOI] [PubMed] [Google Scholar]
- 53. Morin P.J., Sparks A.B., Korinek V., Barker N., Clevers H., Vogelstein B., Kinzler K.W.. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997; 275:1787–1790. [DOI] [PubMed] [Google Scholar]
- 54. Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F.et al.. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011; 43:630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim S., Yu N.K., Kaang B.K.. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015; 47:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu J., Lee W., Jiang C., Keller E.B.. Start site selection by Sp1 in the TATA-less human Ha-ras promoter. J. Biol. Chem. 1994; 269:5391–5402. [PubMed] [Google Scholar]
- 57. Kollmar R., Sukow K.A., Sponagle S.K., Farnham P.J.. Start site selection at the TATA-less carbamoyl-phosphate synthase (glutamine-hydrolyzing)/aspartate carbamoyltransferase/dihydroorotase promoter. J. Biol. Chem. 1994; 269:2252–2257. [PubMed] [Google Scholar]
- 58. Blake M.C., Jambou R.C., Swick A.G., Kahn J.W., Azizkhan J.C.. Transcriptional initiation is controlled by upstream GC-box interactions in a TATAA-less promoter. Mol. Cell Biol. 1990; 10:6632–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Khattar E., Maung K.Z.Y., Chew C.L., Ghosh A., Mok M.M.H., Lee P., Zhang J., Chor W.H.J., Cildir G., Wang C.Q.et al.. Rap1 regulates hematopoietic stem cell survival and affects oncogenesis and response to chemotherapy. Nat. Commun. 2019; 10:5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ozturk M.B., Li Y., Tergaonkar V.. Current insights to regulation and role of telomerase in human diseases. Antioxidants (Basel). 2017; 6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim W., Ludlow A.T., Min J., Robin J.D., Stadler G., Mender I., Lai T.P., Zhang N., Wright W.E., Shay J.W.. Regulation of the human telomerase gene TERT by telomere position effect-over long distances (TPE-OLD): implications for aging and cancer. PLoS Biol. 2016; 14:e2000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Babu D., Fullwood M.J.. 3D genome organization in health and disease: emerging opportunities in cancer translational medicine. Nucleus. 2015; 6:382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are available in the GEO open-access repository (GSE163077).